
Abstract
Prostate cancer (PCa) is one of the commonest cancers in men and a major cause of cancer-related mortality. Family history is the strongest known risk factor for developing PCa. This is illustrated by the observation that a man with one close relative (such as a father or a brother) with PCa has approximately twice the risk of developing PCa when compared to a man with no family history. A man with two close male relatives affected has a fivefold increase in lifetime risk. This degree of relative risk and the increase in its magnitude indicate a strong genetic component to disease development. However, unlike other cancers such as breast, ovarian, and colonic cancers, the search for mutations in candidate genes is proving to be more elusive. Uncovering the genes that predispose to PCa among families where disease is clustered has been the objective of many research groups over the past 15 years. Epidemiological and twin studies support a role for the genetic predisposition to PCa. Familial cancer loci have been identified, but discovery of the genes that cause familial prostate cancer (FPC) remains largely elusive. Unraveling the genetics of PCa is challenging and is likely to involve the analysis of numerous predisposing factors, which may be manifestations of multiple mutagenic pathways. Increased familial risk of prostate cancer could be due to the inheritance of multiple moderate-risk genetic variants. Although the study of hereditary prostate cancer (HPC) has increased our understanding of its genetic etiology, many issues remain largely unresolved. This difficulty with identification of PCa predisposition genes may be due to a number of reasons. PCa, in terms of total prevalence, is a very common condition, and it may not be far wide of the mark to say that the majority of prostates in the Western world will eventually harbor some cancer cells. The disease varies significantly in the spectrum of aggressiveness. We do not know, with absolute conviction, which patients who have been diagnosed with PCa require treatment. It is against this quandary that genetics could play its influence. PCa is diagnosed in the later years of life; therefore, obtaining DNA samples from living affected men for more than one generation is often not possible, and linkage in large pedigrees may be unfeasible. The presence within high-risk pedigrees of phenocopies (individuals with PCa but without the genetic alteration) weakens the linkage results. The genetic heterogeneity of this complex disease (the fact that different pedigrees may be due to different genetic mutations) and the uncertainty regarding the optimal genetic model could render linkage results inaccurate, making gene identification difficult.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


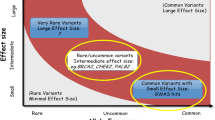
Keywords
- Prostate Cancer
- Dizygotic Twin
- Predisposition Gene
- Nijmegen Breakage Syndrome
- Hereditary Prostate Cancer
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Prostate cancer (PCa) is one of the commonest cancers in men and a major cause of cancer-related mortality. Family history is the strongest known risk factor for developing PCa. This is illustrated by the observation that a man with one close relative (such as a father or a brother) with PCa has approximately twice the risk of developing PCa when compared to a man with no family history. A man with two close male relatives affected has a fivefold increase in lifetime risk. This degree of relative risk and the increase in its magnitude indicate a strong genetic component to disease development. However, unlike other cancers such as breast, ovarian, and colonic cancers, the search for mutations in candidate genes is proving to be more elusive. Uncovering the genes that predispose to PCa among families where disease is clustered has been the objective of many research groups over the past 15 years. Epidemiological and twin studies support a role for the genetic predisposition to PCa. Familial cancer loci have been identified, but discovery of the genes that cause familial prostate cancer (FPC) remains largely elusive. Unraveling the genetics of PCa is challenging and is likely to involve the analysis of numerous predisposing factors, which may be manifestations of multiple mutagenic pathways. Increased familial risk of prostate cancer could be due to the inheritance of multiple moderate-risk genetic variants. Although the study of hereditary prostate cancer (HPC) has increased our understanding of its genetic etiology, many issues remain largely unresolved. This difficulty with identification of PCa predisposition genes may be due to a number of reasons. PCa, in terms of total prevalence, is a very common condition, and it may not be far wide of the mark to say that the majority of prostates in the Western world will eventually harbor some cancer cells. The disease varies significantly in the spectrum of aggressiveness. We do not know, with absolute conviction, which patients who have been diagnosed with PCa require treatment. It is against this quandary that genetics could play its influence. PCa is diagnosed in the later years of life; therefore, obtaining DNA samples from living affected men for more than one generation is often not possible, and linkage in large pedigrees may be unfeasible. The presence within high-risk pedigrees of phenocopies (individuals with PCa but without the genetic alteration) weakens the linkage results. The genetic heterogeneity of this complex disease (the fact that different pedigrees may be due to different genetic mutations) and the uncertainty regarding the optimal genetic model could render linkage results inaccurate, making gene identification difficult.
Significant linkage in FPC was first published by a group from Johns Hopkins University, USA [1]. They reported linkage at a locus on chromosome 1q24-25, which they named hereditary prostate cancer 1 (HPC1). Since then, several large linkage studies have taken place, and the results of many different groups have uncovered new loci and challenged others [2–5]. To this date, research on PCa linkage has reported genotyping data in over 1,600 families. There are numerous contradictory studies reporting or refuting linkage within a multitude of areas in the genome, and this challenges our understanding of the genetic basis of this disease. This is in contrast from the search for a familial breast cancer predisposition gene in which analysis of linkage in select regions revealed a site where the BRCA1 gene was situated [6]. This demonstrates that the genetic predisposition to PCa is highly complex, probably involving numerous predisposition genes and that a high proportion of high-risk families may not be attributable to a single high-risk gene. Conventional linkage may not be the best method of predisposition gene identification in this disease because of genetic heterogeneity whereby various familial clusters are due to different genes. This chapter addresses the current evidence that supports a genetic component to the etiology of PCa and attempts to put into context the diverse findings that have been implicated with the development of HPC. It explores why understanding the genetics of PCa has been so difficult. Lastly, management strategies of men with HPC are discussed.
Evidence for Genetic Etiology
Current evidence for the genetic etiology of PCa can be grouped into epidemiological evidence, case–control studies, cohort studies, and twin studies.
Epidemiological Evidence
In the 1950s–1960s, it was noted that the risk of PCa development in relatives of sufferers was higher than the population average [7, 8]. Large families have been observed, in which prostate cancers seemed to cluster. Early observations were made in large families studied in Utah [9, 10]. To further explore the evidence for a familial component to PCa development, case control, cohort, and twin studies have been reported.
Case–Control Studies
Case–control studies can be divided into two broad strategies. One strategy compares prostate cancer incidence in first-degree relatives of PCa patients (cases) with the incidence in relatives of cancer-free individuals (controls). The second strategy compares the percentage of PCa cases vs. controls with a family history of the disease [7–9, 11–26]. These studies indicated that the relative risks (RR) among first-degree relatives of affected individuals range from 0.64 to 11.00-fold [27–29]. With the exception of the RR of 0.64 [11], in a study, which was carried out on a small sample set of 39 families, 15 of these 16 studies reported an RR of 1.76 or more. This RR increases further when more than one relative is affected. Steinberg et al. [15] demonstrated that the RR with an affected first-degree relative was 2.0, with a second-degree relative was 1.7, but with both first- and second-degree relatives combined, RR rose markedly to 8.8. Additionally, the RR increased as the number of family members increased, with RRs of 2.2, 4.9, and 10.9 observed for 1, 2, and 3 additional affected relatives besides the proband, respectively [15]. This is robust evidence for the involvement of a genetic component in familial disease as these increases in RR are too large to be dismissed solely as an environmental effect. Further evidence of a genetic influence is demonstrated by the observation that the RR to relatives increases as the age of the proband decreases [9, 30]. A brother of a proband with PCa at the age of 50 has a 1.9-fold higher risk of developing prostate cancer compared with a brother of a man diagnosed with the disease at the age of 70 [30].
Cohort Studies
Cohort studies attempt to avoid potential bias through probing an unselected population. Goldgar et al. [31] showed an FPC RR of 2.21 in first-degree relatives of 6,350 probands from an unselected population from the Utah Population Database. Likewise, Gronberg et al. [32] found an RR of 1.70 from their study involving 5,496 sons of Swedish men from Cancer Registry data.
Twin Studies
These have demonstrated an increased RR in mono compared with dizygotic twins of just over 3- to 6-fold [33]. Page et al. [34] studied 15,924 male twin pairs and found pair-wise concordance (twin pairs where both men were affected) rates among monozygotic twins was 15.7 %, while for dizygotic twins the frequency was 3.7 % (p = <0.001). Proband-wise concordance (number of concordant affected twins divided by total number of affected twins) was 27.1 % for monozygotic twins and 7.1 % for dizygotic twins, which gives a risk ratio of 3.8. Similar results were noted in a Finnish study [35]. A further study concluded that up to 42 % of PCa risk could be attributable to heritable factors [36]. The absolute risk of prostate cancer for twins diagnosed up to the age of 75 was found to be sixfold higher for mono- vs. dizygotic twins (18 % vs. 3 %). It also demonstrated a statistically significant reduction in time interval between the age at diagnosis for monozygotic twins compared with dizygotic twins (5.7 years vs. 8.8 years; p = 0.04).
Segregation Analyses
“Segregation analyses” is a method that studies the structure of familial clusters and describes the likely mode of inheritance, age-specific cumulative risk (penetrance), and allele frequency of genetic predisposition to a disease. Carter et al. [30], using such analyses, suggested that early-onset PCa (<55 years) may be due to a rare autosomal dominant highly penetrant allele, which could account for up to 43 % of disease in this age group and up to 9 % of PCa in men aged up to 85 years. Alleles for such a rare autosomal dominant gene were predicted to exist at a frequency of 0.003 and to cause a cumulative risk of disease of 88 % by 85 years of age compared with 5 % for noncarriers. Other researchers have arrived at similar conclusions but have suggested a commoner allele frequency and a lower penetrance of about 67 % (Gronberg et al. [32], allele frequency 0.0167; Schaid et al. [37], allele frequency 0.006). A recessive or X-linked model is suggested by some studies, which noted higher risks to brothers of PCa cases compared with fathers [38, 39]. Ewis et al. [40] report an odds ratio of 2.04 (p = 0.02) for allele C of dYs19 in Japanese prostate cancer patients, while other alleles of this region were protective against the disease (allele D, OR 0.26 p = 0.002). The Y chromosome (father-to-son transmission) is therefore also implicated. It is possible that a mixture of several models coexist, giving rise to age-related risks [41]. Dominantly inherited risk allele(s) may predispose to early-onset disease, and a recessive or X-linked model could account for its later onset [42].
Molecular Analysis Evidence: Linkage Studies (Genome-Wide Scans)
Linkage analysis is a gene-localizing technique that looks for co-segregation of a disease in sizeable, high-risk families, with disease-causing genetic mutations. Linkage analysis has been used to successfully map many familial cancer loci, e.g., colorectal cancer, breast/ovarian cancer, and melanoma [43]. Initially, linkage analysis helps to pinpoint the region within which a disease-causing locus may lie by analyzing co-inheritance of polymorphic stretches of DNA, e.g., microsatellite markers. The sequencing of the human genome will facilitate the use of single nucleotide polymorphisms (SNPs). As these are more common than polymorphic runs of DNA sequence, a denser linkage maps can be determined. Once a region of linkage is identified, then candidate gene mutation analysis within the region can be undertaken to identify the disease-causing mutation.
Candidate Gene Analysis Evidence
The search for genetic markers of disease susceptibility often utilizes the candidate gene approach. Here, a gene is targeted based on the properties and metabolic pathways of its protein product. PCa cases were noted to be clustered among breast cancer families as far back as the 1990s [44, 45]. The relative risk (RR) of PCa development in male carriers of mutations in the breast cancer predisposition genes BRCA1 and BRCA2 is increased. The RR with respect to BRCA1 was found to be 3.33 [46] and 1.82 in a further analysis by the BCLC [47]. That of BRCA2 was found to be 4.65. The RR is higher in men with PCa diagnosed before 65 years (RR 7.33), with an estimated cumulative incidence by the age of 70 of 7.5–33.0 %. A founder mutation 999del5 in BRCA2 has been identified in study carried out in Iceland. This mutation is reported to confer a cumulative PCa risk to carriers of 7.6 % by the age of 70 [48]. Sixty seven percent of men who had the mutation all developed advanced PCa associated with a high mortality rate [49], implying that BRCA2 predisposes to more aggressive disease. A report in a Swedish family carrying the BRCA2 mutation 6051delA [50] adds weight to the evidence that such mutations are pathogenic. A mutation screen of BRCA1 and BRCA2 genes was conducted by Gayther et al. [51]. Two germline deleterious BRCA2 mutations were discovered. A study conducted by Edwards et al. [52] on 263 men aged <55 at diagnosis discovered six pathogenic mutations located outside the ovarian cancer cluster region in the gene, implying a genotype/phenotype correlation that accounted for 2 % of PCa at this young age. This equated to an RR of 23 by the age of 60 and conferred an absolute risk of PCa of 1.3 and 10 % by the age 55 and 65, respectively. This supports the notion that BRCA2 is a high-risk PCa gene. Two recent studies have reported an increased risk of prostate cancer associated with the Ashkenazi founder mutations in the BRCA genes, lending further evidence to these data [53, 54]. Subsequently, germline mutations have been found in the NBS gene in the Slavic population at a higher frequency in PCa cases than controls [55] and in the CHEK2 gene [56]. This implies that PCa predisposition may in some instances be due to mutations in genes in the DNA repair pathway, that in the homozygous form give rise to a severe phenotype (in the case of BRCA2 this would be Fanconi’s anemia D2 and in the case of NBS would be Nijmegen Breakage Syndrome), but in the heterozygous form, would increase the risk of PCa development.
Genome Searches
The running of a large number of microsatellites, typically in the region of 400, has many terms: genome-wide search, genome-wide scan, or genome-wide screen – and can conveniently be abbreviated to GWS. Numerous linkage analysis experiments have been conducted across the genome to identify prostate cancer susceptibility loci. The ACTANE (Anglo-Canadian-Texan-Australian-Norwegian-EU Biomed) group has focused on the collection of early-onset clinically detected disease. This is because the disease manifests 10 years later on average than PSA-detected disease, and therefore these men would have had a raised PSA level at even earlier age and may therefore be highly predisposed genetically [28]. Thus far, several GWS have been published for PCa [1, 3, 5, 57–72]. The significant results are as follows.
1q23-24: HPC1 and the RNASEL Data
The first GWS identified a locus named HPC1 (hereditary prostate cancer 1) at 1q24-25. A group from Johns Hopkins University, Baltimore, carried out the study in 91 North American and Swedish families, and their report suggested that up to 34 % of families could be linked to this locus [1]. Several other groups have since either confirmed [73–76] or refuted [57, 58, 60, 64, 77, 78] the original report. Goode et al. [64] and Goddard et al. [79] found evidence of genetic linkage in families with more aggressive PCa. A meta-analysis conducted by Xu et al. [80] representing many groups within the International Consortium for Prostate Cancer Genetics (ICPCG) reported data obtained on 772 families and reported that a lower estimate of 6 % of all families was linked to 1q24-25. A more extensive analysis concluded that HPC1 might have a role in a subset of families with numerous young-onset cases, particularly among Afro-Caribbean men. Carpten et al. [81] subsequently found mutations in the cell proliferation and apoptosis-regulating gene RNASEL which was in this region. Of 8 families that were linked to the 1q region, two had germline mutations; one was a stop Glu265Ter (E265X) termination codon, but the other was a missense mutation. Neither segregated with the disease. Some, but not all, further reports have shown RNASEL mutations to be associated with PCa risk but with a much lower relative risk than would have been predicted by the linkage evidence. Rokman et al. [82] showed that the Glu265X in RNASEL was present 4.5-fold more often in affected family members compared with controls. Other groups have found that RNASEL may confer much smaller PCa risks or have found no mutations at all in PCa cases; therefore, it is likely to be low-penetrance PCa cancer gene that is at odds with the linkage evidence [83, 84]. This suggests that either the linkage results are misleading or that a highly penetrant HPC1 exists but is still to be discovered.
Other Loci and Candidates from GWS
Other loci have unfortunately had a similar history to that described above, namely, loci are identified that have significant logarithm of the odds (LOD) scores, and candidate genes have mutations described therein which are subsequently refuted or whose risks fall on further detailed analysis [85, 86].
Other Significant Loci
PCaP (1q42.2-43; Berthon et al. [57]) – this was a locus that was identified in the German/French population but not corroborated by other researchers. CAPB (1p36; Gibbs et al. [59]) – a locus which is associated with primary brain tumor and PCa which on further analysis seemed more associated with young-onset PCa than brain tumor [87]. A locus has been described on chromosome 16q in sibling pairs by Suarez et al. [58] and one on 20q (HPC20) by Berry et al. [63], but these are yet to be independently confirmed. Another locus has been described on the long arm of chromosome X (HPCX; Xq27-28) by Xu et al. [88]). This has been corroborated by some other researchers, but the gene has yet to be identified. There are also loci that are implicated in the development of more aggressive disease, e.g., 7q, 19q [89–91]. Eight GWS have been published recently in one issue of the Prostate (ACTANE Consortium [72]; Lange et al. [65]; Schleutker et al. [66]; Cunningham et al. [67]; Xu et al. [68]; Wiklund et al. [69]; Janer et al. [70]; Witte et al. [71], Dec 2003). A summary of these was published in a review by Easton et al. [5]. The conclusion of these GWS to date is that there are numerous loci suggested by the GWS from various groups which are not consistently replicated by independent groups on study of further PCa families. This implies that there is considerable genetic heterogeneity in PCa. The possibility that PCa is due to a combination of low penetrance means that more common genetic variants may be entertained when large families are rare and it is difficult to locate predisposition genes by linkage. Candidate studies of polymorphisms are presently under way in PCa. There is currently no uniform pattern of polymorphisms that confers markedly increased risk from the data. The most consistent polymorphisms to date that confer a moderately increased risk are in the SRD5A2, GSTP1, and the AR genes [92–102].
Recent Findings of the UK GWAS and Potential Clinical Role
Eeles et al. previously conducted a genome-wide association study in which 541,129 SNPs were genotyped in 1,854 PCa cases with clinically detected disease and in 1,894 controls. They then extended the study to evaluate potential correlations in a second stage in which they genotyped 43,671 SNPs in 3,650 PCa cases and 3,940 controls and in a further stage involving an additional 16,229 cases and 14,821 controls from 21 studies. They identified seven new PCa susceptibility loci on chromosomes 2, 4, 8, 11, and 22 (with P = 1.6 × 10(−8) to P = 2.7 × 10(−33)) [103]. It is possible that the seven novel genetic loci found could contain several potential candidate genes, which could contribute to PCa development and progression. A key association was found on chromosome 10, just 2 bp away from the transcription start site of the microseminoprotein B (MSMB) gene. MSMB encodes PSP94, a member of the immunoglobulin-binding factor family made by epithelial cells of the prostate and secreted into seminal plasma. Loss of expression of PSP94 is linked with recurrence after radical prostatectomy. This seems to suggest that this SNP may be causally related to disease risk [104]. Therefore, PSP94 could be a future screening target especially as it can be found in blood. There is a region on chromosome 7 that the gene LMTK2 (also known as BREK) which codes for a signaling protein [105] and could act as a novel target for drug therapy. The chromosome 19 hit contains kallikrein genes KLK2 and KLK3 which code for the proteins hK2 and PSA, respectively. There is evidence that hK2 may be useful for PCa screening and prognosis [106]. Twenty-four SNPs in the KLK3 (PSA) gene have subsequently been evaluated in men from five studies, and no association was reported with PCa risk [107]. Eeles et al. looked at the variation in KLK genes, PSA, and risk of PCa. In the first stage of a study, they used controls selected for low PSA levels. Stage 2 controls were not selected for a low PSA. However, they still found an association. Following this a study involving 13 groups worldwide where the controls were not selected for a low PSA level, still showed an association of the chromosome 19 SNP (between KLK2 and KLK3) with PCa risk [108]. The chromosome 6 association is in intron 5 of SLC22A3, one of the organic cation transporter (OCT) genes. These have been shown to be critical for elimination of some drugs and toxins [109]. Many genes are near the SNP of interest on the X chromosome. The NUDT10 and NUDT11 genes encode enzymes that determine the rate of phosphorylation in DNA repair, stress responses, and apoptosis [110].
Other PCa GWAS
Two other groups of researchers, CGEMS (USA) [111] and deCODE Group (Iceland) [112], published their PCa GWAS at the same time as the UK GWS. Both confirmed previously reported associations at chromosomes 8q and 17q. CGEMS found similar hits to UK GWS on chromosomes 10 and 11. They additionally found novel tagSNP associations on chromosomes 7 and 10. The deCODE team found a novel region on chromosome 2p15 in their population. Duggan et al. in another GWAS investigated aggressive PCas that were defined by having at least one of the following: stage T3/T4, N+, M+, Grade III, Gleason score ≥8, or preoperative serum PSA of at least 50 ng mL−1. This group reported a different association on chromosome 9 located within the DAB2IP gene, which encodes a novel Ras GTPase-activating protein [113]. More recently, Sun et al. from the same group identified a second independent risk locus in chromosome 17q12 within the HNF1B gene [114].
Prostate Cancer Predisposition Gene Discovery
There are many uncertainties in the area of genetic predisposition that are currently taxing researchers in this area. These include (a) what is the optimal genetic model for PCa? (b) are there different predisposition genes in different populations? and (c) how much concordance is there between various groups for the putative loci? The results of future large-scale multicenter studies will ultimately answer these questions. It is entirely possible that the studies undertaken thus far are underpowered and pooling of data may improve the chances of finding genuine underlying linkage. This is the aim of the creation of groups such as the International Consortium of Prostate Cancer Genetics (ICPCG). Groups undertaking linkage analyses worldwide collaborate within the umbrella of this consortium. In 2000, via a meta-analysis, the ICPCG found that the 1q24 locus may contribute to about 6 % of PCa families and was more commonly found in larger prostate cancer clusters whose average age of onset was <65 years [80]. Current data indicates that progression to clinical disease is more likely following a raised PSA and occurs a median time of 10 years after the PSA has risen [115]. In theory, patients in families that are diagnosed with clinically detected disease may have different set of predisposing genes to those involved in PSA screen detected patients. At present, whether this is true, this is unknown. On the issue of genetic heterogeneity for linkage, i.e., presence of more than one PCa predisposition genes, it has been shown that two percent of early-onset cases have deleterious mutations in the BRCA2 gene and that a further small percentage is due to NBS and CHEK2 mutations. Yet models suggest that up to 43 % of such cases may harbor a predisposition gene [30]. This indicates that there are further PCa susceptibility genes that are yet to be discovered. In an age when the majority of monogenic human disease genes have been identified, a particular challenge for human geneticists will be resolving complex polygenic and multifactorial diseases. It is likely that the majority of genetic predisposition to PCa will follow this model where there exist many rather than one PCa predisposition gene per family.
Clinical Management Concepts for HPC
The question of whether a genetic change influencing PCa causation is associated with factors altering treatment is an important consideration. Recent reports are contradictory. Carefully documented multicentered, prospective family history data collection and outcome analysis are crucial to improving our understanding of this condition. The current management issues surrounding HPC involve several considerations: (i) the degree of biological aggressiveness of HPC, (ii) whether HPC per se is an independent predictor of treatment outcome, (iii) whether there is a difference in the survival curves between sporadic and HPC, and (iv) the outcome patterns in those patients treated various radical treatments, i.e., prostatectomy vs. radiotherapy by family history.
Determining the Degree of Biological Aggressiveness
Walsh initially observed that there was no significant difference between phenotypes of sporadic, familial, and HPC undergoing radical prostatectomy with respect to clinical stage, pre-op PSA, PSA density, prostate weight, Gleason score, pathologic stage, or tumor histology [115]. This was challenged by subsequent observation that patients with localized PCa who reported a positive family history tended to have a worse outcome at 3 and 5 years following treatment, be it radiation therapy or surgery, than those with sporadic cancers [116]. This was then again refuted by three subsequent studies that found no difference in the aggressiveness of HPC compared to sporadic disease [117–119]. This area therefore remains unresolved.
Is HPC an Independent Predictor of Treatment Outcome?
Kupelian et al. [120] first indicated that a positive family history for PCa correlates with treatment outcome, in a sizeable unselected series of patients, suggesting that familial PCa may have a more aggressive course than nonfamilial PCa.
Is There a Survival Differences Between Sporadic and HPC?
No significant differences in either overall or cause-specific survival were found between sporadic, familial, and HPC patients [121]. Present treatment guidelines do not differ based on presence or absence of FPC.
Should Men with a Family History of Prostate Cancer Avoid Conservative Treatment?
Based on current evidence, there is a rationale for genetic screening of men at risk once the genes responsible for prostate cancer are identified. The American Urological Association (AUA) recommends that men who are at high risk for developing PCa such as men with a family history of disease or men of African-American descent commence routine PCa screening at the age of 40 [122], whereas The American Cancer Society recommends that men receive PSA or digital rectal examination testing annually at the age of 50 or earlier if they have a family history of the disease or are of African-American descent [123]. In outcomes in HPC men treated with radiotherapy vs. radical prostatectomy, Hanlon et al. [124] found no significant difference in biochemical failure rates between carefully matched men with and without a family history of PCa. This backs other studies that failed to show an elevated risk of failure after definitive therapy for clinically localized PCa in men with either combined hereditary and familial and patients with the sporadic form of the disease.
Chemoprevention Trials
PCa chemoprevention is the judicious administration of agents that impair one or more steps in prostatic carcinogenesis. The principle aspects of chemoprevention include agents, their molecular targets, strategic endpoint biomarkers, their critical pathways, and cohorts identified by genetic and acquired risk factors [125]. The identification of genetic susceptibility loci would enable a cohort of men at high risk of developing PCa to be identified to serve as subjects for chemoprevention trials. If such trials yield favorable outcomes, they could potentially lead to a recommendation for preventative therapy in genetic mutation carriers. Several putative chemopreventive agents are currently being trailed. Results of a large population-based, randomized phase III trial demonstrated that finasteride might prevent PCa. However, the paper indicated that only low-grade tumors were prevented and in fact the number of high-grade tumors was significantly higher in the finasteride arm. Clarke et al. [126] studied the possible effect of supplemental dietary selenium on the change in the incidence of PCa. They found that although selenium confers no protective benefit on the primary study endpoint of squamous and basal cell carcinomas of the skin, the selenium-treated group in their series had substantially lower incidence of PCa as a secondary endpoint. Further investigations are clearly warranted. Initial data seem to suggest at least some benefit with the use of other agents may potential confer preventative effect. These include vitamin E, vitamin D, other 5-alpha-reductase inhibitors, cyclooxygenase-2 inhibitors, lycopene, and green tea. Some of which are being tested in new large-scale phase III clinical trials [127]. The Selenium and Vitamin E Cancer Prevention Trial (SELECT) is an intergroup phase III clinical trial that aims to test the efficacy of selenium and vitamin E alone and in combination in the prevention of prostate cancer [128]. The emergence of new powerful techniques such as proteomic analysis of tissue-based and secreted proteins [129] and gene chip cDNA microarrays for multiplex gene expression profiling is likely to facilitate the identification of new molecular targets, cohorts at risk, and the design of appropriate combination trials.
Targeted Screening
Several controversies surround the management of the relatives of PCa patients. Targeted screening involves monitoring serum PSA levels in relatives of young- or early-onset PCa or families with multiple cases. The optimal age at which screening should be initiated is yet to be determined. The sub-30- and sub-40-year-old groups would not be screened by the majority of clinicians, and many would commence screening either at age 5 years younger than youngest age at diagnosis of a relative or 40 years but not normally younger than this. Targeted screening studies have demonstrated a higher proportion of raised PSA levels in relatives of cases in families compared with sporadic cases. In a screening study of PCa, among high-risk families [130], it was shown that previously unsuspected and clinically relevant cancers were found in 24 % of a total of 34 first-degree relatives compared to the approximately 1 (3 %) expected (p < 0.01). The study emphasized the paramount importance of thorough screening in first-degree relatives of prostate cancer patients. The first targeted screening study based on BRCA1/2 genotype started in 2003 (the IMPACT study; Tischkowitz and Eeles, 2003) [131]. In 2011, the team published the findings of one wing of their study involving 300 men (205 mutation carriers; 89 BRCA1, 116 BRCA2, and 95 controls) over 33 months. At the baseline screen (year 1), 7.0 % (21/300) underwent a prostate biopsy. PCa was diagnosed in ten individuals, a prevalence of 3.3 %. The positive predictive value of PSA screening in this cohort was 47·6 % (10/21). One PCa was diagnosed at year 2. Of the 11 PCas diagnosed, 9 were in mutation carriers, 2 in controls, and 8 were found to be clinically significant. Thus, suggesting that the positive predictive value of PSA screening in BRCA mutation carriers is high. Furthermore, it showed that screening seems to detect clinically significant PCa. The findings of this study support the rationale for continued screening in such “high-risk” men [132].
Future Perspectives
With the recent exponential increase in the development and improvement of techniques involving proteomics, there has been increased optimism in the prospect of finding clinically relevant candidate genes, gene clusters, and signaling pathways. This would potentially lead to better diagnostic and/or more specific targeted therapeutic plans in the management of sufferers of PCa [133].
The current ability to tally and compare genome-wide expression profiles in tissue samples could potentially shed light on the molecular pathology toward PCa detection and monitoring of disease progression and/or recurrence. Early gene expression signature studies were hindered by the inherent limitations of bioinformatic tools. It is anticipated that the validity of molecular signatures of PCa will ultimately be proven by cross-validation on novel datasets and direct coupling of these to prospective and translational studies [134]. Sun et al., in an attempt to predict PCa recurrence based on molecular signatures, conducted a computational analysis of gene expression profile data obtained from 79 cases. Of these, 39 were classified as having disease recurrence. At the 90 % sensitivity level, a novel-derived prognostic genetic signature achieved 85 % specificity. The results were compared to a clinically validated postoperative nomogram. The study was purported to be the first reported genetic signature to outperform a clinically used postoperative nomogram. They demonstrated the feasibility of utilizing gene expression information for potential PCa prognosis [135].
PCa inheritance following a simple Mendelian pattern may be identified in the families of probands with early-onset cases. Currently, the only clinically applicable measure to try to reduce PCa mortality in families with hereditary disease is screening, which aims to diagnose the disease when it is still at a curable stage. The precise mechanism of how gene mutations contribute to an increased susceptibility for PCa remains elusive, but the finding of germline mutations in the BRCA2, CHEK2, and NBS1 genes suggest that a proportion may occur due to mutations in the DNA repair pathway. This has ramifications on treatment of such individuals with DNA-damaging agents. It is most likely that the cause of the majority of PCa cases will be multifactorial and will involve environmental and genetic factors. The recent exponential advances in understanding the clinical genetics of PCa offer great optimism toward optimizing the management of PCa. From a clinical genetics point of view, this could usher with it a new paradigm in the way we manage PCa.
References
Smith JR, Freije D, Carpten JD, Gronberg H, Xu J, Isaacs SD, Brownstein MJ, Bova GS, Guo H, Bujnovszky P, Nusskern DR, Damber JE, Bergh A, Emanuelsson M, Kallioniemi OP, Walker-Daniels J, Bailey-Wilson JE, Beaty TH, Meyers DA, Walsh PC, Collins FS, Trent JM, Isaacs WB. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996;274(5291):1371–4.
Eeles RA, the UK Familial Prostate Study Coordinating Group and the CRC/BPG UK Familial Prostate Cancer Study Collaborators. Genetic predisposition to prostate cancer. Prostate Cancer Prostatic Dis. 1999;2(1):9–15.
Ostrander EA, Stanford JL. Genetics of prostate cancer: too many loci, too few genes. Am J Hum Genet. 2000;67(6):1367–75. Epub 2000 Nov 07. Review.
Simard J, Dumont M, Labuda D, Sinnett D, Meloche C, El-Alfy M, Berger L, Lees E, Labrie F, Tavtigian SV. Prostate cancer susceptibility genes: lessons learned and challenges posed. Endocr Relat Cancer. 2003;10(2):225–59. Review.
Easton DF, Schaid DJ, Whittemore AS, Isaacs WJ, the International Consortium for Prostate Cancer Genetics. Where are the prostate cancer genes? A summary of eight genome wide searches. Prostate. 2003;57(4):261–9.
Hall JM, Lee MK, Newman B, Morrow JE, Anderson LA, Huey B, King MC. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990;250(4988):1684–9.
Morganti G, Gianferrari L, Cresseri A, Arrigoni G, Lovati G. Clinico-statistical and genetic research on neoplasms of the prostate. Acta Genet Stat Med. 1957;6(2):1956–304. French.
Woolf CM. An investigation of the familial aspects of carcinoma of the prostate. Cancer. 1960;13:739–44.
Cannon L, Bishop DT, Skolnick M, Hunt S, Lyon JL, Smart CR. Genetic epidemiology of prostate cancer in the Utah Mormon genealogy. Cancer Surv. 1982;1:47–69.
Cannon-Albright L, Eeles RA. Progress in prostate cancer. Nat Genet. 1995;9(4):336–8.
Steele R, Lees RE, Kraus AS, Rao C. Sexual factors in the epidemiology of cancer of the prostate. J Chronic Dis. 1971;24(1):29–37.
Krain LS. Some epidemiologic variables in prostatic carcinoma in California. Prev Med. 1974;3(1):154–9.
Schuman LM, Mandel J, Blackard C, Bauer H, Scarlett J, McHugh R. Epidemiologic study of prostatic cancer: preliminary report. Cancer Treat Rep. 1977;61(2):181–6.
Meikle AW, Smith JA, West DW. Familial factors affecting prostatic cancer risk and plasma sex-steroid levels. Prostate. 1985;6(2):121–8.
Steinberg GD, Carter BS, Beaty TH, Childs B, Walsh PC. Family history and the risk of prostate cancer. Prostate. 1990;17(4):337–47.
Fincham SM, Hill GB, Hanson J, Wijayasinghe C. Epidemiology of prostatic cancer: a case–control study. Prostate. 1990;17(3):189–206.
Spitz MR, Currier RD, Fueger JJ, Babaian RJ, Newell GR. Familial patterns of prostate cancer: a case–control analysis. J Urol. 1991;146(5):1305–7.
Ghadirian P, Cadotte M, Lacroix A, Perret C. Family aggregation of cancer of the prostate in Quebec: the tip of the iceberg. Prostate. 1991;19(1):43–52.
Whittemore AS, Wu AH, Kolonel LN, John EM, Gallagher RP, Howe GR, West DW, Teh CZ, Stamey T. Family history and prostate cancer risk in black, white, and Asian men in the United States and Canada. Am J Epidemiol. 1995;141(8):732–40.
Hayes RB, Liff JM, Pottern LM, Greenberg RS, Schoenberg JB, Schwartz AG, Swanson GM, Silverman DT, Brown LM, Hoover RN, et al. Prostate cancer risk in U.S. blacks and whites with a family history of cancer. Int J Cancer. 1995;60(3):361–4.
Isaacs SD, Kiemeney LA, Baffoe-Bonnie A, Beaty TH, Walsh PC. Risk of cancer in relatives of prostate cancer probands. J Natl Cancer Inst. 1995;87(13):991–6.
Keetch DW, Rice JP, Suarez BK, Catalona WJ. Familial aspects of prostate cancer: a case control study. J Urol. 1995;154(6):2100–2.
Lesko SM, Rosenberg L, Shapiro S. Family history and prostate cancer risk. Am J Epidemiol. 1996;144(11):1041–7.
Ghadirian P, Howe GR, Hislop TG, Maisonneuve P. Family history of prostate cancer: a multi-center case–control study in Canada. Int J Cancer. 1997;70(6):679–81.
Glover Jr FE, Coffey DS, Douglas LL, Russell H, Cadigan M, Tulloch T, Wedderburn K, Wan RL, Baker TD, Walsh PC. Familial study of prostate cancer in Jamaica. Urology. 1998;52(3):441–3.
Bratt O, Kristoffersson U, Lundgren R, Olsson H. Familial and hereditary prostate cancer in southern Sweden. A population-based case–control study. Eur J Cancer. 1999;35(2):272–7.
Eeles RA, Dearnaley DP, Ardern-Jones A, Shearer RJ, Easton DF, Ford D, Edwards S, Dowe A, 105 collaborators. Familial prostate cancer: the evidence and the Cancer Research Campaign/British Prostate Group (CRC/BPG) UK Familial Prostate Cancer Study. Br J Urol. 1997;79 Suppl 1:8–14. Review.
Singh R, Eeles RA, Durocher F, Simard J, Edwards S, Badzioch M, Kote-Jarai Z, Teare D, Ford D, Dearnaley D, Ardern-Jones A, Murkin A, Dowe A, Shearer R, Kelly J, Labrie F, Easton D, Narod SA, Tonin PN, Foulkes WD. High risk genes predisposing to prostate cancer development-do they exist? Prostate Cancer Prostatic Dis. 2000;3(4):241–7.
Johns LE, Houlston RS. A systematic review and meta-analysis of familial prostate cancer risk. BJU Int. 2003;91(9):789–94. Review.
Carter BS, Beaty TH, Steinberg GD, Childs B, Walsh PC. Mendelian inheritance of familial prostate cancer. Proc Natl Acad Sci U S A. 1992;89(8):3367–71.
Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst. 1994;86(21):1600–8.
Gronberg H, Damber L, Damber JE. Familial prostate cancer in Sweden. A nationwide register cohort study. Cancer. 1996;77(1):138–43.
Ahlbom A, Lichtenstein P, Malmstrom H, Feychting M, Hemminki K, Pedersen NL. Cancer in twins: genetic and nongenetic familial risk factors. J Natl Cancer Inst. 1997;89(4):287–93.
Page WF, Braun MM, Partin AW, Caporaso N, Walsh P. Heredity and prostate cancer: a study of World War II veteran twins. Prostate. 1997;33(4):240–5.
Verkasalo PK, Kaprio J, Koskenvuo M, Pukkala E. Genetic predisposition, environment and cancer incidence: a nationwide twin study in Finland, 1976–1995. Int J Cancer. 1999;83(6):743–9.
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343(2):78–85.
Schaid DJ, McDonnell SK, Blute ML, Thibodeau SN. Evidence for autosomal dominant inheritance of prostate cancer. Am J Hum Genet. 1998;62(6):1425–38.
Narod SA, Dupont A, Cusan L, Diamond P, Gomez JL, Suburu R, Labrie F. The impact of family history on early detection of prostate cancer. Nat Med. 1995;1(2):99–101.
Monroe KR, Yu MC, Kolonel LN, Coetzee GA, Wilkens LR, Ross RK, Henderson BE. Evidence of an X-linked or recessive genetic component to prostate cancer risk. Nat Med. 1995;1(8):827–9.
Ewis AA, Lee J, Naroda T, Sasahara K, Sano T, Kagawa S, Iwamoto T, Nakahori Y. Linkage between prostate cancer incidence and different alleles of the human Y-linked tetranucleotide polymorphism DYS19. J Med Invest. 2002;49(1–2):56–60.
Cui J, Staples MP, Hopper JL, English DR, McCredie MR, Giles GG. Segregation analyses of 1,476 population-based Australian families affected by prostate cancer. Am J Hum Genet. 2001;68(5):1207–18. Epub 2001 Apr 11.
Conlon EM, Goode EL, Gibbs M, Stanford JL, Badzioch M, Janer M, Kolb S, Hood L, Ostrander EA, Jarvik GP, Wijsman EM. Oligogenic segregation analysis of hereditary prostate cancer pedigrees: evidence for multiple loci affecting age at onset. Int J Cancer. 2003;105(5):630–5.
Eeles RA, Cannon-Albright L. Familial prostate cancer and its management. In: Eeles RA, Easton DF, Ponder BAJ, Eng C, editors. Genetic predisposition to cancer. 2nd ed. London: Arnold; 2004.
Tulinius H, Olafsdottir GH, Sigvaldason H, Tryggvadottir L, Bjarnadottir K. Neoplastic diseases in families of breast cancer patients. J Med Genet. 1994;31(8):618–21.
Anderson DE, Badzioch MD. Familial breast cancer risks. Effects of prostate and other cancers. Cancer. 1993;72:114–9.
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast cancer linkage consortium. Lancet. 1994;343:692–5.
Thompson D, Easton DF, the Breast Cancer Linkage Consortium. Cancer incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–65.
Thorlacius S, Struewing JP, Hartge P, Olafsdottir GH, Sigvaldason H, Tryggvadottir L, Wacholder S, Tulinius H, Eyfjord JE. Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet. 1998;352:1337–9.
Sigurdsson S, Thorlacius S, Tomasson J, Tryggvadottir L, Benediktsdottir K, Eyfjord JE, Jonsson E. BRCA2 mutation in Icelandic prostate cancer patients. J Mol Med. 1997;75:758–61.
Gronberg H, Ahman AK, Emanuelsson M, Bergh A, Damber JE, Borg A. BRCA2 mutation in a family with hereditary prostate cancer. Genes Chromosomes Cancer. 2001;30:299–301.
Gayther SA, de Foy KA, Harrington P, Pharoah P, Dunsmuir WD, Edwards SM, Gillett C, Ardern-Jones A, Dearnaley DP, Easton DF, Ford D, Shearer RJ, Kirby RS, Dowe AL, Kelly J, Stratton MR, Ponder BA, Barnes D, Eeles RA. The frequency of germ-line mutations in the breast cancer predisposition genes BRCA1 and BRCA2 in familial prostate cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res. 2000;60:4513–8.
Edwards SM, Kote-Jarai Z, Meitz J, Hamoudi R, Hope Q, Osin P, Jackson R, Southgate C, Singh R, Falconer A, Dearnaley DP, Ardern-Jones A, Murkin A, Dowe A, Kelly J, Williams S, Oram R, Stevens M, Teare DM, Ponder BA, Gayther SA, Easton DF, Eeles RA, Cancer Research UK/British Prostate Group UK Familial Prostate Cancer Study Collaborators; British Association of Urological Surgeons Section of Oncology. Two percent of men with early-onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet. 2003;72(1):1–12. Epub 2002 Dec 09.
Kirchhoff T, Kauff ND, Mitra N, Nafa K, Huang H, Palmer C, Gulati T, Wadsworth DSE, Robson ME, Ellis NA, Offit K. BRCA mutations and risk of prostate cancer in Ashkenazi Jews. Clin Cancer Res. 2004;10(9):2918–21.
Giusti RM, Rutter JL, Duray PH, Freedman LS, Konichezky M, Fisher-Fischbein J, Greene MH, Maslansky B, Fischbein A, Gruber SB, Rennert G, Ronchetti RD, Hewitt SM, Struewing JP, Iscovich J. A twofold increase in BRCA mutation related prostate cancer among Ashkenazi Israelis is not associated with distinctive histopathology. J Med Genet. 2003;40(10):787–92.
Cybulski C, Gorski B, Debniak T, Gliniewicz B, Mierzejewski M, Masojc B, Jakubowska A, Zlowocka E, Sikorski A, Narod SA, Lubinski J. NBS1 is a prostate cancer susceptibility gene. Cancer Res. 2004;64(4):1215–9.
Dong X, Wang L, Taniguchi K, Wang X, Cunningham JM, McDonnell SK, Qian C, Marks AF, Slager SL, Peterson BJ, Smith DI, Cheville JC, Blute ML, Jacobsen SJ, Schaid DJ, Tindall DJ, Thibodeau SN, Liu W. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet. 2003;72(2):270–80. Epub 2003 Jan 17.
Berthon P, Valerie A, Cohen-Akenine A, Drelon E, Paiss T, Wohr G, Latil A, Millasseau P, Mellah I, Cohen N, Blanche H, Bellane-Chantelot C, Demenais F, Teillac P, Le Duc A, de Petriconi R, Hautmann R, Chumakov I, Bachner L, Maitland NJ, Lidereau R, Vogel W, Fournier G, Mangin P, Cussenot O, et al. Predisposing gene for early-onset prostate cancer, localized on chromosome 1q42.2-43. Am J Hum Genet. 1998;62(6):1416–24.
Suarez BK, Lin J, Burmester JK, Broman KW, Weber JL, Banerjee TK, Goddard KA, Witte JS, Elston RC, Catalona WJ. A genome screen of multiplex sibships with prostate cancer. Am J Hum Genet. 2000;66(3):933–44.
Gibbs M, Stanford JL, McIndoe RA, Jarvik GP, Kolb S, Goode EL, Chakrabarti L, Schuster EF, Buckley VA, Miller EL, Brandzel S, Li S, Hood L, Ostrander EA. Evidence for a rare prostate cancer prostate cancer-susceptibility locus at chromosome 1p36. Hum Genet. 1999;64(3):776–87.
Berry R, Schaid DJ, Smith JR, French AJ, Schroeder JJ, McDonnell SK, Peterson BJ, Wang ZY, Carpten JD, Roberts SG, Tester DJ, Blute ML, Trent JM, Thibodeau SN. Linkage analyses at the chromosome 1 loci 1q24-25 (HPC1), 1q42. 2–43 (PCAP), and 1p36 (CAPB) in families with hereditary prostate cancer. Am J Hum Genet. 2000;66(2):539–46.
Tavtigian SV, Simrad J, Teng DH, Abtin V, Baumgard M, Beck A, Camp NJ, Carillo AR, Chen Y, Dayananth P, Desrochers M, Dumont M, Farnham JM, Frank D, Frye C, Ghaffari S, Gupte JS, Hu R, Iliev D, Janecki T, Kort EN, Laity KE, Leavitt A, Leblanc G, McArthur-Morrison J, Pederson A, Penn B, Peterson KT, Reid JE, Richards S, Schroeder M, Smith R, Snyder SC, Swedlund B, Swensen J, Thomas A, Tranchant M, Woodland AM, Labrie F, Skolnick MH, Neuhausen S, Rommens J, Cannon-Albright LA. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27(2):172–80.
Hsieh CL, Oakley-Girvan I, Gallagher RP, Wu AH, Kolonel LN, Teh CZ, Halpern J, West DW, Paffenbarger Jr RS, Whittemore AS. Re: prostate cancer susceptibility locus on chromosome 1q: a confirmatory study. J Natl Cancer Inst. 1997;89(24):1893–4.
Berry R, Schroeder JJ, French AJ, McDonnell SK, Peterson BJ, Cunningham JM, Thibodeau SN, Schaid DJ. Evidence for a prostate cancer – susceptibility locus on chromosome 20. Am J Hum Genet. 2000;67(1):82–91. Epub 2000 May 16.
Goode EL, Stanford JL, Chakrabarti L, Gibbs M, Kolb S, McIndoe RA, Buckley VA, Schuster EF, Neal CL, Miller EL, Brandzel S, Hood L, Ostrander EA, Jarvik GP. Linkage analysis of 150 high-risk prostate cancer families at 1q24-25. Genet Epidemiol. 2000;18(3):251–75.
Lange EM, Gillanders EM, Davis CC, Brown WM, Campbell JK, Jones M, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, Markey C, Giri V, Dimmer JB, Montie JE, Trent JM, Cooney KA. Genome-wide scan for prostate cancer susceptibility genes using families from the University of Michigan prostate cancer genetics project finds evidence for linkage on chromosome 17 near BRCA1. Prostate. 2003;57(4):326–34.
Schleutker J, Baffoe-Bonnie AB, Gillanders E, Kainu T, Jones MP, Freas-Lutz D, Markey C, Gildea D, Riedesel E, Albertus J, Gibbs Jr KD, Matikainen M, Koivisto PA, Tammela T, Bailey-Wilson JE, Trent JM, Kallioniemi OP. Genome-wide scan for linkage in Finnish hereditary prostate cancer (HPC) families identifies novel susceptibility loci at 11q14 and 3p25-26. Prostate. 2003;57(4):280–9.
Cunningham JM, McDonnell SK, Marks A, Hebbring S, Anderson SA, Peterson BJ, Slager S, French A, Blute ML, Schaid DJ, Thibodeau SN, Mayo Clinic, Rochester, Minnesota. Genome linkage screen for prostate cancer susceptibility loci: results from the Mayo Clinic Familial Prostate Cancer Study. Prostate. 2003;57(4):335–46.
Xu J, Gillanders EM, Isaacs SD, Chang BL, Wiley KE, Zheng SL, Jones M, Gildea D, Riedesel E, Albertus J, Freas-Lutz D, Markey C, Meyers DA, Walsh PC, Trent JM, Isaacs WB. Genome-wide scan for prostate cancer susceptibility genes in the Johns Hopkins hereditary prostate cancer families. Prostate. 2003;57(4):320–5.
Wiklund F, Gillanders EM, Albertus JA, Bergh A, Damber JE, Emanuelsson M, Freas-Lutz DL, Gildea DE, Goransson I, Jones MS, Jonsson BA, Lindmark F, Markey CJ, Riedesel EL, Stenman E, Trent JM, Gronberg H. Genome-wide scan of Swedish families with hereditary prostate cancer: suggestive evidence of linkage at 5q11. 2 and 19p13.3. Prostate. 2003;57(4):290–7.
Janer MFD, Stanford JL, Badzioch MD, Kolb S, Deutsch K, Peters MA, Goode EL, Welti R, DeFrance HB, Iwasaki L, Li S, Hood L, Ostrander EA, Jarvik GP. Genomic scan of 254 hereditary prostate cancer families. Prostate. 2003;57(4):309–19.
Witte JSSB, Thiel B, Lin J, Yu A, Banerjee TK, Burmester JK, Casey G, Catalona WJ. Genome-wide scan of brothers: replication and fine mapping of prostate cancer susceptibility and aggressiveness loci. Prostate. 2003;57(4):298–308.
The International ACTANE Consortium. Results of a genome-wide linkage analysis in prostate cancer families ascertained through the ACTANE consortium. Prostate. 2003;57(4):270–9.
Gronberg H, Smith J, Emanuelsson M, Jonsson BA, Bergh A, Carpten J, Isaacs W, Xu J, Meyers D, Trent J, Damber JE. In Swedish families with hereditary prostate cancer, linkage to the HPC1 locus on chromosome 1q24-25 is restricted to families with early-onset prostate cancer. Am J Hum Genet. 1999;65(1):134–40.
Cooney KA, McCarthy JD, Lange E, Huang L, Miesfeldt S, Montie JE, Oesterling JE, Sandler HM, Lange K. Prostate cancer susceptibility locus on chromosome 1q: a confirmatory study. J Natl Cancer Inst. 1997;89(13):955–9.
Neuhausen SL, Farnham JH, Kort E, Tavtigian SV, Skolnick MH, Cannon-Albright LA. Prostate cancer susceptibility locus HPC1 in Utah high-risk pedigrees. Hum Mol Genet. 1999;8(13):2437–42.
Xu J, Zheng SL, Chang B, Smith JR, Carpten JD, Stine OC, Isaacs SD, Wiley KE, Henning L, Ewing C, Bujnovszky P, Bleeker ER, Walsh PC, Trent JM, Meyers DA, Isaacs WB. Linkage of prostate cancer susceptibility loci to chromosome 1. Hum Genet. 2001;108(4):335–45. Epub 28 March 2001.
McIndoe RA, Stanford JL, Gibbs M, Jarvik GP, Brandzel S, Neal CL, Li S, Gammack JT, Gay AA, Goode EL, Hood L, Ostrander EA. Linkage analysis of 49 high-risk families does not support a common familial prostate cancer-susceptibility gene at 1q24-25. Am J Hum Genet. 1997;61(2):347–53.
Eeles RA, Durocher F, Edwards S, Teare D, Badzioch M, Hamoudi R, Gill S, Biggs P, Dearnaley D, Ardern-Jones A, Dowe A, Shearer R, McLennan DL, Norman RL, Ghadirian P, Aprikian A, Ford D, Amos C, King TM, Labrie F, Simard J, Narod SA, Easton D, Foulkes WD. Linkage analysis of chromosome 1q markers in 136 prostate cancer families. The Cancer Research Campaign/British Prostate Group U.K. Familial Prostate Cancer Study Collaborators. Am J Hum Genet. 1998;62(3):653–8.
Goddard KA, Witte JS, Suarez BK, Catalona WJ, Olson JM. Model-free linkage analysis with covariates confirms linkage of prostate cancer to chromosomes 1 and 4. Am J Hum Genet. 2001;68(5):1197–206. Epub 2001 Apr 13.
Xu J. Combined analysis of hereditary prostate cancer linkage to 1q24-25: results from 772 hereditary prostate cancer families from the International Consortium for Prostate Cancer Genetics. Am J Hum Genet. 2000;66(3):945–57. Erratum in: Am J Hum Genet 2000, 67 (2): 541–542.
Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, Faruque M, Moses T, Ewing C, Gillanders E, Hu P, Bujnovszky P, Makalowska I, Baffoe-Bonnie A, Faith D, Smith J, Stephan D, Wiley K, Brownstein M, Gildea D, Kelly B, Jenkins R, Hostetter G, Matikainen M, Schleutker J, Klinger K, Connors T, Xiang Y, Wang Z, De Marzo A, Papadopoulos N, Kallioniemi OP, Burk R, Meyers D, Gronberg H, Meltzer P, Silverman R, Bailey-Wilson J, Walsh P, Isaacs W, Trent J. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30(2):181–4. Epub 2002 Jan 22.
Rokman AI, Konen T, Seppala EH, Nupponen N, Autio V, Mononen N, Bailey-Wilson J, Trent J, Carpten J, Matikainen MP, Koivisto PA, Tammela TL, Kallioniemi OP, Schleutker J. Germline alterations of the RNASEL gene, a candidate HPC1 gene at 1q25, in patients and families with prostate cancer. Am J Hum Genet. 2002;70(5):1299–304. Epub 2002 Apr 08. Erratum in: Am J Hum Genet 2002 Jul, 71 (1): 215.
Casey G, Neville PJ, Plummer SJ, Xiang Y, Krumroy LM, Klein EA, Catalona WJ, Nupponen N, Carpten JD, Trent JM, Silverman RH, Witte JS. RNASEL Arg462Gln variant is implicated in up to 13 % of prostate cancer cases. Nat Genet. 2002;32(4):581–3. Epub 2002 Nov 04.
Chen H, Griffen AR, Wu YQ, Tomsho LP, Zuhlke KA, Lange EM, Gruber SB, Cooney KA. RNASEL mutations in hereditary prostate cancer. J Med Genet. 2003;40(3):e21.
Wang L, McDonnel SK, Cunningham JM, Hebbring S, Jacobsen SJ, Cerhan JR, Slager SL, Blute ML, Schaid DJ, Thibodeau SN. No association of germline alteration of MSR1 with prostate cancer risk. Nat Genet. 2003;35(2):128–9. Epub 2003 Sep 07.
Meitz JC, Edwards SM, Easton DF, Murkin A, Ardern-Jones A, Jackson RA, Williams S, Dearnaley DP, Stratton MR, Houlston RS, Eeles RA, Cancer Research UK/BPG UK Familial Prostate Cancer Study Collaborators. HPC2/ELAC2 polymorphisms and prostate cancer risk: analysis by age of onset of disease. Br J Cancer. 2002;87(8):905–8.
Badzioch M, Eeles R, Leblanc G, Foulkes WD, Giles G, Edwards S, Goldgar D, Hopper JL, Bishop DT, Moller P, Heimdal K, Easton D, Simard J. Suggestive evidence for a site specific prostate cancer gene on chromosome 1p36. The CRC/BPG UK Familial Prostate Cancer Study Coordinators and Collaborators. The EU Biomed Collaborators. J Med Genet. 2000;37(12):947–9.
Xu J, Meyers D, Freije D, Isaacs S, Wiley K, Nusskern D, Ewing C, Wilkens E, Bujnovszky P, Bova GS, Walsh P, Isaacs W, Schleutker J, Matikainen M, Tammela T, Visakorpi T, Kallioniemi OP, Berry R, Schaid D, French A, McDonnell S, Schroeder J, Blute M, Thibodeau S, Trent J, et al. Evidence for a prostate cancer susceptibility locus on the X chromosome. Nat Genet. 1998;20(2):175–9.
Witte JS, Goddard KA, Conti DV, Elston RC, Lin J, Suarez BK, Broman KW, Burmester JK, Weber JL, Catalona WJ. Genomewide scan for prostate cancer-aggressiveness loci. Am J Hum Genet. 2000;67(1):92–9. Epub 2000 May 24.
Slager SL, Schaid DJ, Cunningham JM, McDonnell SK, Marks AF, Peterson BJ, Hebbring SJ, Anderson S, French AJ, Thibodeau SN. Confirmation of linkage of prostate cancer aggressiveness with chromosome 19q. Am J Hum Genet. 2003;72(3):759–62. Epub 2003 Jan 30.
Neville PJ, Conti DV, Krumroy LM, Catalona WJ, Suarez BK, Witte JS, Casey G. Prostate cancer aggressiveness locus on chromosome segment 19q12-q13. 1 Identified by linkage and allelic imbalance studies. Genes Chromosomes Cancer. 2003;36(4):332–9.
Irvine RA, Yu MC, Ross RK, Coetzee GA. The CAG and GGC microsatellites of the androgen receptor gene are in linkage disequilibrium in men with prostate cancer. Cancer Res. 1995;55:1937–40.
Hardy DO, Scher HI, Bogenreider T, Sabbatini P, Zhang ZF, Nanus DM, Catterall JF. Androgen receptor CAG repeat lengths in prostate cancer: correlation with age of onset. J Clin Endocrinol Metab. 1996;81:4400–5.
Ingles SA, Ross RK, Yu MC, Irvine RA, La Pera G, Haile RW, Coetzee GA. Association of prostate cancer risk with genetic polymorphisms in vitamin D receptor and androgen receptor. J Natl Cancer Inst. 1997;89:166–70.
Stanford JL, Just JJ, Gibbs M, Wicklund KG, Neal CL, Blumenstein BA, Ostrander EA. Polymorphic repeats in the androgen receptor gene: molecular markers of prostate cancer risk. Cancer Res. 1997;57:1194–8.
Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Dahl D, Brufsky A, Talcott J, Hennekens CH, Kantoff PW. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A. 1997;94:3320–3.
Hakimi JM, Schoenberg MP, Rondinelli RH, Piantadosi S, Barrack ER. Androgen receptor variants with short glutamine or glycine repeats may identify unique subpopulations of men with prostate cancer. Clin Cancer Res. 1997;3:1599–608.
Miller EA, Stanford JL, Hsu L, Noonan E, Ostrander EA. Polymorphic repeats in the androgen receptor gene in high-risk sibships. Prostate. 2001;48:200–5.
Hsing AW, Gao YT, Wu G, Wang X, Deng J, Chen YL, Sesterhenn IA, Mostofi FK, Benichou J, Chang C. Polymorphic CAG and GGN repeat lengths in the androgen receptor gene and prostate cancer risk: a population-based case–control study in China. Cancer Res. 2000;60:5111–6.
Edwards SM, Badzioch MD, Minter R, Hamoudi R, Collins N, Ardern-Jones A, Dowe A, Osborne S, Kelly J, Shearer R, Easton DF, Saunders GF, Dearnaley DP, Eeles RA. Androgen receptor polymorphisms: association with prostate cancer risk, relapse and overall survival. Int J Cancer. 1999;84:458–65.
Makridakis NM, Ross RK, Pike MC, Crocitto LE, Kolonel LN, Pearce CL, Henderson BE, Reichardt JK. Association of missense substitution in SRD5A2 gene with prostate cancer in African-American and Hispanic men in Los Angeles, USA. Lancet. 1999;354:975–8.
Kote-Jarai Z, Easton D, Edwards SM, Jefferies S, Durocher F, Jackson RA, Singh R, Ardern-Jones A, Murkin A, Dearnaley DP, Shearer R, Kirby R, Houlston R, Eeles R, CRC/BPG UK Familial Prostate Cancer Study Collaborators. Relationship between glutathione S-transferase M1, P1 and Tpolymorphisms and early onset prostate cancer. Pharmacogenetics. 2001;11:325–30.
Koopmeiners JS, Karyadi DM, Kwon EM, Stern MC, Corral R, Joshi AD, Shahabi A, McDonnell SK, Sellers TA, Pow-Sang J, Chambers S, Aitken J, Gardiner RA, Batra J, Kedda MA, Lose F, Polanowski A, Patterson B, Serth J, Meyer A, Luedeke M, Stefflova K, Ray AM, Lange EM, Farnham J, Khan H, Slavov C, Mitkova A, Cao G, UK Genetic Prostate Cancer Study. Collaborators/British Association of Urological Surgeons’ Section of Oncology; UK ProtecT Study Collaborators; PRACTICAL Consortium, Easton DF. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat Genet. 2009;41(10):1116–21. Epub 2009 Sep 20.
Reeves JR, Dulude H, Panchal C, Daigneault L, Ramnani DM. Prognostic value of prostate secretory protein of 94 amino acids and its binding protein after radical prostatectomy. Clin Cancer Res. 2006;12:6018–22.
Kawa S, Fujimoto J, Tezuka T, Nakazawa T, Yamamoto T. Involvement of BREK, a serine/threonine kinase enriched in brain, in NGF signalling. Genes Cells. 2004;9:219–32.
Steuber T, Helo P, Lilja H. Circulating biomarkers for prostate cancer. World J Urol. 2007;25:111–9.
Ahn J, Berndt SI, Wacholder S, Kraft P, Kibel AS, et al. Variation in KLK genes, prostate-specific antigen and risk of prostate cancer. Nat Genet. 2008;40:1032–4.
Eeles R, Giles G, Neal D, Muir K, Easton DF, for the PRACTICAL Consortium. Reply to ‘variation in KLK genes, prostate-specific antigen and risk of prostate cancer’. Nat Genet. 2008;40:1035–6.
Verhaagh S, Schweifer N, Barlow DP, Zwart R. Cloning of the mouse and human solute carrier 22a3 (Slc22a3/SLC22A3) identifies a conserved cluster of three organic cation transporters on mouse chromosome 17 and human 6q26-q27. Genomics. 1999;55:209–18.
Hidaka K, Caffrey JJ, Hua L, Zhang T, Falck JR, et al. An adjacent pair of human NUDT genes on chromosome X are preferentially expressed in testis and encode two new isoforms of diphosphoinositol polyphosphate phosphohydrolase. J Biol Chem. 2002;277:32730–8.
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–5.
Gudmundsson J, Sulem P, Rafnar T, Bergthorsson JT, Manolescu A, et al. Common sequence variants on 2p15 and Xp11.22 confer susceptibility to prostate cancer. Nat Genet. 2008;40:281–3.
Duggan D, Zheng SL, Knowlton M, Benitez D, Dimitrov L, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J Natl Cancer Inst. 2007;99:1836–44.
Sun J, Zheng SL, Wiklund F, Isaacs SD, Purcell LD, et al. Evidence for two independent prostate cancer risk-associated loci in the HNF1B gene at 17q12. Nat Genet. 2008;40:1153–5.
Parkes C, Wald NJ, Murphy P, George L, Watt HC, Kirby R, Knekt P, Helzlsouer KJ, Tuomilehto J. Prospective observational study to assess value of prostate specific antigen as screening test for prostate cancer. BMJ. 1995;311(7016):1340–3.
Walsh PC. Hereditary Prostate Cancer, podium talk at the annual meeting of the American Society of Clinical Oncology. 1996.
Kupelian PA, Klein EA, Witte JS, Kupelian VA, Suh JH. Familial prostate cancer: a different disease? J Urol. 1997;158(6):2197–201.
Valeri A, Azzouzi R, Drelon E, Delannoy A, Mangin P, Fournier G, Berthon P, Cussenot O. Early-onset hereditary prostate cancer is not associated with specific clinical and biological features. Prostate. 2000;45(1):66–71.
Bova GS, Partin AW, Isaacs SD, Carter BS, Beaty TL, Isaacs WB, Walsh PC. Biological aggressiveness of hereditary prostate cancer: long-term evaluation following radical prostatectomy. J Urol. 1998;160(3 Pt 1):660–3.
Kupelian PA, Kupelian VA, Witte JS, Macklis R, Klein EA. Family history of prostate cancer in patients with localized prostate cancer: an independent predictor of treatment outcome. J Clin Oncol. 1997;15:1478.
Cussenot O, Valeri A, Berthon P, Fournier G, Mangin P. Hereditary prostate cancer and other genetic predispositions to prostate cancer. Urol Int. 1998;60 Suppl 2:30–4; discussion 35. Review.
American Urological Association. Prostate cancer awareness for men: a doctor’s guide for patients. 2001: 4–5.
Cancer reference information: can prostate cancer be found early? American Cancer Society. 2001. http://www.cancer.org/index.
Hanlon AL, Hanks GE. Patterns of inheritance and outcome in patients treated with external beam radiation for prostate cancer. Urology. 1998;52(5):735–8.
Lieberman R. Chemoprevention of prostate cancer: current status and future directions. Cancer Metastasis Rev. 2002;21(3–4):297–309.
Clark LC, Dalkin B, Krongrad A, Combs Jr GF, Turnbull BW, Slate EH, Witherington R, Herlong JH, Janosko E, Carpenter D, Borosso C, Falk S, Rounder J. Related articles, links. Decreased incidence of prostate cancer with selenium supplementation: results of a double-blind cancer prevention trial. Br J Urol. 1998;81(5):730–4.
Klein EA, Thompson IM. Update on chemoprevention of prostate cancer. Curr Opin Urol. 2004;14(3):143–9.
Klein EA. Clinical models for testing chemopreventative agents in prostate cancer and overview of SELECT: the Selenium and Vitamin E Cancer Prevention Trial. Recent Results Cancer Res. 2003;163:212–25; discussion 264–6. Review.
Kommu S, Sharifi R, Edwards S, Eeles R. Proteomics and urine analysis – a potential promising new tool in urology. BJU Int. 2004;93(9):1172–3.
McWhorter WP, Hernandez AD, Meikle AW, Terreros DA, Smith Jr JA, Skolnick MH, Cannon-Albright LA, Eyre HJ. A screening study of prostate cancer in high risk families. J Urol. 1992;148(3):826–8.
Tischkowitz M, Eeles R, IMPACT study. Identification of Men with genetic predisposition to prostate cancer and its clinical treatment collaborators. Mutations in BRCA1 and BRCA2 and predisposition to prostate cancer. Lancet. 2003;362(9377):80; author reply 80.
Mitra AV, Bancroft EK, Barbachano Y, Page EC, Foster CS, Jameson C, Mitchell G, Lindeman GJ, Stapleton A, Suthers G, Evans DG, Cruger D, Blanco I, Mercer C, Kirk J, Maehle L, Hodgson S, Walker L, Izatt L, Douglas F, Tucker K, Dorkins H, Clowes V, Male A, Donaldson A, Brewer C, Doherty R, Bulman B, Osther PJ, Salinas M, Eccles D, Axcrona K, Jobson I, Newcombe B, Cybulski C, Rubinstein WS, Buys S, Townshend S, Friedman E, Domchek S, Ramon Y, Cajal T, Spigelman A, Teo SH, Nicolai N, Aaronson N, Ardern-Jones A, Bangma C, Dearnaley D, Eyfjord J, Falconer A, Grönberg H, Hamdy F, Johannsson O, Khoo V, Kote-Jarai Z, Lilja H, Lubinski J, Melia J, Moynihan C, Peock S, Rennert G, Schröder F, Sibley P, Suri M, Wilson P, Bignon YJ, Strom S, Tischkowitz M, Liljegren A, Ilencikova D, Abele A, Kyriacou K, van Asperen C, Kiemeney L, IMPACT Study Collaborators, Easton DF, Eeles RA. Targeted prostate cancer screening in men with mutations in BRCA1 and BRCA2 detects aggressive prostate cancer: preliminary analysis of the results of the IMPACT study. BJU Int. 2011;107(1):28–39. doi:10.1111/j.1464-410X.2010.09648.x. Epub 2010 Sep 14.
Blueggel M, Chamrad D, Meyer HE. Bioinformatics in proteomics. Curr Pharm Biotechnol. 2004;5(1):79–88. Review.
Ein-Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci U S A. 2006;103(15):5923–8. Epub 2006 Apr 3.
Sun Y, Goodison S. Optimizing molecular signatures for predicting prostate cancer recurrence. Prostate. 2009;69(10):1119–27.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag London
About this chapter
Cite this chapter
Kommu, S.S., Bishara, S., Edwards, S.M. (2013). Prostate Cancer Genetics. In: Tewari, A. (eds) Prostate Cancer: A Comprehensive Perspective. Springer, London. https://doi.org/10.1007/978-1-4471-2864-9_11
Download citation
DOI: https://doi.org/10.1007/978-1-4471-2864-9_11
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-2863-2
Online ISBN: 978-1-4471-2864-9
eBook Packages: MedicineMedicine (R0)