
Abstract
Alzheimer’s disease (AD) is the most common form of adult neurode-generation and is characterised by progressive loss of cognitive function leading to death. The neuropathological hallmarks include extracellular amyloid plaque accumulation in affected regions of the brain, formation of intraneuronal neurofibrillary tangles, chronic neuroinflammation, oxidative stress, and abnormal biometal homeostasis. Of the latter, major changes in copper (Cu) levels and localisation have been identified in AD brain, with accumulation of Cu in amyloid deposits, together with deficiency of Cu in some brain regions. The amyloid precursor protein (APP) and the amyloid beta (Aβ) peptide both have Cu binding sites, and interaction with Cu can lead to potentially neurotoxic outcomes through generation of reactive oxygen species. In addition, AD patients have systemic changes to Cu metabolism, and altered Cu may also affect neuroinflammatory outcomes in AD. Although we still have much to learn about Cu homeostasis in AD patients and its role in disease aetiopathology, therapeutic approaches for regulating Cu levels and interactions with Cu-binding proteins in the brain are currently being developed. This review will examine how Cu is associated with pathological changes in the AD brain and how these may be targeted for therapeutic intervention.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Copper
- Alzheimer’s disease
- Ceruloplasmin
- Reactive oxygen species
- Amyloid precursor protein
- Tau
- Neuroinflammation
- Clioquinol
- PBT-2
Background
Alzheimer’s disease (AD) is a neurodegenerative disorder that is the most common form of dementia, affecting approximately 47 million people worldwide (Prince et al. 2015). Advanced ageing is a notable risk factor for AD, whereby almost 50% of cases are found in individuals older than 85 years. Fewer than 5% of cases are due to genetic mutations with the remaining 95% resulting from occurrence without known familial genetic mutation (Ceccom et al. 2012). With advancements in the medical industry and prolonged life expectancy, the prevalence of AD is predicted to increase immensely (Ballard et al. 2011). AD patients typically display symptoms of early memory loss, personality and behavioural changes and deficits in sensory and motor functions. Macroscopically, the brain of AD patients presents synaptic and neuronal loss resulting in brain atrophy, affecting regions including the entorhinal cortex, hippocampus, basal forebrain and amygdala.
The complex aetiology of AD has been widely studied but is yet to be fully understood. It is associated with the key hallmarks of extracellular amyloid peptide accumulation, intracellular tau hyperphosphorylation, neuroinflammation and oxidative stress. In recent years there has been a substantial focus on the role of transition metals, particularly iron (Fe), zinc (Zn) and copper (Cu) in AD aetiology. These metals bind to amyloid-β peptide (Aβ), accelerate Aβ aggregation and consequently promote neurotoxic plaque formation. Fe and Cu are also likely to be involved in promotion of oxidative stress and neuroinflammatory changes in the AD brain (Choo et al. 2013; Pratico 2008; Sayre et al. 2008). In addition, cognitive decline in AD has been associated with the interference of the processing and function of the amyloid-β precursor protein (APP) and the phosphorylation and aggregation of the microtubule-associated protein (MAP), tau, both of which are associated with altered metal homeostasis (Crouch et al. 2009).
Copper Homeostasis
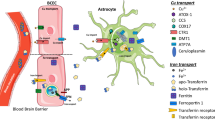
Although there are key roles for a number of metals in AD, this review will focus on Cu. This metal is a key trace element, necessary for all oxygen-requiring processes. Cu is concentrated at high levels in the brain for metabolic needs and additional functions. Specifically, Cu is an essential cofactor that readily binds to enzymes, shifting between the Cu(II) and Cu(I) oxidative states (Hung et al. 2010). The redox capacity of Cu is biochemically important for biological energy metabolism (cytochrome c oxidase), iron metabolism (ceruloplasmin), antioxidative activity (copper zinc superoxide dismutase, SOD1), neurotransmitter synthesis (dopamine-β-monooxygenase), neuropeptide synthesis (peptidylglycine-α-amdinating enzyme) and neuronal myelination (Davies et al. 2013; Scheiber et al. 2014). As a consequence of its redox activity, Cu can also induce oxidative stress through the production of reactive oxygen species (ROS) and its ability to bind with molecular oxygen (McCord and Fridovich 1969). Tight regulation of Cu therefore exists through the duodenal absorption, uptake and excretion from cells and sequestration within cells in order to prevent both excess Cu accumulation and Cu deficiency. This process further prevents abnormal Cu-oxygen interactions (Kaden et al. 2011). The trafficking and transportation of Cu is highly important for the maintenance of Cu homeostasis. In plasma, ceruloplasmin (Cp), albumin and transcuprein are major Cu-binding proteins (Choo et al. 2013). Subcellular Cu transportation involves the Ctr1 protein for the transit into cells across the cell membrane and the Cu ATP7a/b transporters for Cu exportation. In addition, cytosolic Cu chaperones aid delivery and include the Cox17 system in the mitochondria (Amaravadi et al. 1997), copper chaperone for SOD (CCS) (Culotta et al. 1997) and the ATOX1 system in trans-Golgi network (Klomp et al. 1997). These critical cellular mechanisms are essential for maintaining normal neuronal health and function.
Copper in the Brain
The brain contains approximately 7.3% of total body Cu content (Hung et al. 2010). The brain possesses disproportionately low levels of antioxidants, thereby making it highly susceptible to oxidative stress induced by the redox nature of Cu (Hung et al. 2013). Cu homeostasis and transport must therefore be tightly regulated in the brain in order to maintain neuronal health. In the cortex and hippocampus, Cu is released into the synaptic cleft of glutamatergic synapses as an essential component of neuronal transmission (Bush and Tanzi 2008; Zheng et al. 2010). Upon synaptic depolarisation, Cu and Zn are released into the synaptic cleft at micromolar concentrations estimated at approximately 15 μM (Hung et al. 2010; Kardos et al. 1989), whereby excitatory and inhibitory neurotransmission can be modulated. Free ionic Cu is released at NMDA-responsive synapses, and Cu efflux is thought to be associated with the activation of NMDA receptors. This release of Cu may act as a post-translational mechanism to modulate the extracellular s-nitrosylation of NMDA receptors (Bush and Tanzi 2008). Impaired Cu regulation could promote the glutamatergic dysfunction that is present in the AD brain (Ayton et al. 2015). Cu also has the ability to block GABAergic and AMPAergic neurotransmission on rat olfactory bulb neurons (Trombley and Shepherd 1996) and AMPAergic neurotransmission on rat cortical neurons (Weiser and Wienrich 1996). However, more recently it has been established that Cu acts as more than just a negative modulator of neurotransmission. Following 3 hours of Cu exposure, AMPAergic neurotransmission has been seen to increase. Cu therefore demonstrates a unique biphasic mechanism in neurotransmission. Furthermore, it has been demonstrated that Cu has acute inhibitory effects on long-term potentiation (Opazo et al. 2014).
Copper Levels in the Brain in AD
Dyshomeostasis of Cu levels in the brain is a common characteristic in neurodegenerative diseases, including AD. Studies have indicated that mis-localisation of Cu can be observed in the brain of AD patients, and some regions may be in excess while others Cu deficient. This Cu imbalance has extensive effects on neuronal function and is significantly linked to cognitive deficits and AD pathology (Mao et al. 2012; Rembach et al. 2013; Squitti 2014). It has been widely demonstrated that Cu accumulates within Aβ plaques, where Cu levels have been observed at ~390 μM. This is a substantial increase when compared to the brain of normal age-matched control patients, where Cu is found at a concentration of ~79 μM (Mot et al. 2011). Moreover, tissue surrounding the Aβ plaque (high in Cu) has been demonstrated to present with substantially lower levels of Cu, engendering local Cu deficiency (Zheng et al. 2010; Mot et al. 2011). Rembach et al. (2013) established that Cu levels in the frontal cortex are significantly lower than age-matched healthy controls, specifically confined to the soluble fraction. Post-mortem examination is required to measure Cu concentration in the brain and detect Aβ plaques (Hung et al. 2013; Rembach et al. 2013). In order to determine Cu levels in living patients, an ancillary method must be used (e.g. serum Cu) that may in the future be a diagnostic tool for AD (Wang et al. 2015).
A large proportion of the literature has focussed on measuring Cu levels in the serum and cerebrospinal fluid (CSF) of living AD patients. Cu transport in the serum can be found in the form of non-ceruloplasmin-bound Cu (non-Cp-Cu) or bound to Cp or albumin. It is the uptake of the free Cu ion that passes the brain barriers (the blood-brain barrier (BBB) and the blood-cerebrospinal fluid barrier) and is distributed to the CSF and brain parenchyma. The choroid plexus tightly regulates the transportation of Cu into the CSF, whereas movement into the parenchyma may appear more readily in the cerebral capillaries (Choi and Zheng 2009). Meta-analysis of Cu serum levels has indicated that AD patients display higher Cu serum levels (particularly non-Cp-Cu) compared to healthy non-diseased controls (Wang et al. 2015; Squitti 2012; Ventriglia et al. 2012). Interestingly, analysis revealed that Cu levels in the CSF showed no discrepancy between AD patients and controls (Bucossi et al. 2011). The reason for differential plasma Cu but not CSF Cu in AD patients compared to healthy controls is not yet understood.
Copper and Amyloid Aggregation
The aggregation of Aβ is thought to be a major contributor in AD pathology, which may be explained by the amyloid cascade hypothesis (Henry et al. 2010). The amyloid hypothesis suggests that aggregated and oligomerised Aβ is the major contributing factor to synaptic and neuronal degeneration in AD. Amyloid deposition and subsequent senile plaque formation can occur through age-related changes in amyloid generation and clearance and are also induced by mutations in genes such as amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2). Originally it was believed that plaque formation led to neuronal cell death and AD (Reitz 2012). However, over recent years this hypothesis has been modified to suggest that it is the soluble aggregated forms of Aβ and not the endpoint plaques per se that drive neuronal death. With many clinical trials currently focussed on inhibition or removal of amyloid, the veracity of the amyloid hypothesis will soon be established.
Aβ peptides are metabolic products, generated by the proteolysis of APP (Henry et al. 2010). APP is a ubiquitously expressed, transmembrane glycoprotein that accumulates at nerve terminals (Buxbaum et al. 1998) and is thought to be involved in axonal transport, vesicular trafficking, cell adhesion, neuronal survival, apoptosis and perhaps protein folding and degradation (Hung et al. 2010; Cottrell et al. 2005). APP is approximately 110–140 kDa in size, and heterogeneity emerges due to alternative exon splicing and post-translational modifications (Selkoe 2001). Two Cu-binding domains exist within APP, one localised in the Aβ region (Fig. 1) and the other in the N-terminus (Fig. 2) (Hung et al. 2010). The ligands His147, His151, Tyr168 and Met170 are required for high-affinity Cu binding to the N-terminal domain (Barnham et al. 2003; Kong et al. 2007) (Fig. 2). Cu(II) reductase activity is present within the Cu-binding domain of APP, which may further contribute to free radical formation and is sufficient to promote copper-mediated neurotoxicity (Hung et al. 2010).

Schematic of amyloid beta peptide showing amino acid residues involved in binding of copper. Secretase cleavage sites are also shown

Schematic of amyloid precursor protein (APP) showing different domains including the N-terminal copper-binding domain. GFLD growth factor-like domain, CuBD copper-binding domain, RERMS amino acid sequence associated with growth-promoting activity, CAPPD central APP domain, AICD APP intracellular domain, PM plasma membrane
The enzymatic cleavage of APP occurs via either the amyloidogenic or non-amyloidogenic pathways. The non-amyloidogenic pathway requires α-secretase and γ-secretase, whereas amyloidogenic processing of APP occurs by β-secretase and γ-secretase (Hung et al. 2010; Ayton et al. 2015). The activity of β-secretase-BACE1 in the amyloidogenic pathway is regulated by Cu. Cu binds to BACE1 in the Cu(I) binding site present in the C-terminal domain of BACE1, whereby BACE1 mRNA expression is upregulated and hence decreases β-cleavage of APP. This upregulation and increased activity of BACE1 has been observed in copper-deficient fibroblasts (Cater et al. 2008). Therefore, altered levels of intracellular Cu may influence APP metabolism. Increased intracellular Cu elevated the secretion of the α-cleaved APP, whereas the β-cleaved APP production and subsequent secretion were increased in Cu-deficient cells (Cater et al. 2008). Copper has therefore been shown to modulate APP not only via the Cu-binding domain but also through its processing and hence control of Aβ production. Regulation of Aβ subsequently effects Aβ-induced neurotoxicity (Barnham et al. 2003).
In both healthy and AD patients, heterogeneous forms of Aβ have been identified. Extensive research has focussed on the 40-amino acid form of Aβ (Aβ1–40) and the Aβ species ending with a C-terminal residue of 42 (Aβ1–42). Aβ1–40 accounts for ~90% of secreted Aβ, and the remaining 10% of total Aβ secreted from cells is mostly Aβ1–42, which aggregates more readily to form plaque deposits (Citron et al. 1996; Small and McLean 1999; White et al. 2006a). Extracellular plaque formation is primarily composed of aggregated Aβ peptides, both insoluble fibrillar Aβ and soluble Aβ oligomers. Aβ1–40 is the predominant soluble isoform of Aβ (Ahuja et al. 2015) with increasing evidence suggesting a close relationship with cognitive impairment (Lue et al. 1999). Alternatively, Aβ1–42 has a higher propensity to aggregate, which could enhance toxic outcomes. Lue et al. (1999) interestingly espouse that soluble Aβ (or 1–40, 1–42) has the ability to impact a wider area of neurons and synapses in comparison to insoluble Aβ and, thus, may have a greater role in neurotoxicity.
Metals such as Cu, Fe and Zn play a significant role in the formation of soluble Aβ oligomers, and in particular Cu(II)-Aβ interactions are a major driver of peptide aggregation. The oligomer species of Aβ generated is dependent on the molar ratio of Cu(II) to Aβ. At sub-equimolar ratios, amyloid-like aggregates form that are highly stable and resistant to sodium dodecyl sulphate (SDS). Conversely, at supra-equimolar ratios of Cu(II) to Aβ, soluble, less stable, neurotoxic oligomers are formed. Cu(II)-induced aggregates may be spontaneously formed at Cu(II)/peptide ratios of 0.25:1; however, amyloid formation is not subsequently accelerated as compared to Aβ1–42 in the absence of Cu(II) (Smith et al. 2007). The formation of a His bridge between Cu(II) ions leads to the generation of His-bridged Aβ oligomers that are highly toxic (Smith et al. 2006; Huang et al. 2004). His modification has been demonstrated to reduce the amount of Cu-mediated Aβ1–40 aggregation, but aggregation is not entirely abolished. These data suggest that the interaction with non-His residues on the peptide is partially responsible for the aggregation of this particular form of Aβ (Atwood et al. 1998). The tendency of Aβ peptides to self-aggregate is dependent on the specific oligomer of Aβ, as analysis has proposed that peptide precipitation may be mediated by the high-affinity Cu(II) binding site present in Aβ1–42 oligomers. In comparison, Aβ1–40 is less self-aggregating and may be explained by the presence of a lower-affinity Cu(II) binding site (Atwood et al. 2000). Conclusively, it is well established that nucleated aggregation, where aggregation follows from a pre-aggregated seed, is considerably accelerated in the presence of Cu(II) (Huang et al. 2004) and heightened by mild acidic conditions similar to that in AD brains (Atwood et al. 1998).
Cu and Tau
Tau is a microtubule-associated protein (MAP) expressed abundantly in the CNS, predominantly in neurons, and at lower levels in astrocytes and oligodendrocytes (LoPresti et al. 1995). Normal tau plays a significant role in axonal microtubule (MT) organisation, specifically axonal growth and development of neuronal polarity. This is achieved through axonal stabilising of MTs by promoting MT assembly. In addition, recent findings established a multifunctional role of tau, recognising its interaction with synaptic vesicles as a key factor in neurotransmission (Hung et al. 2013; de Calignon et al. 2012; Liu et al. 2012; Pooler et al. 2013). The regulation of tau is controlled by specific protein kinases and phosphatases, which mediate post-translational phosphorylation (Higuchi et al. 2002). Highly phosphorylated tau aggregates, sequestering normal tau and disrupting MTs. This characteristic of tau links directly to the aetiology and pathogenesis of neurodegenerative diseases such as AD (Khlistunova et al. 2006).
A relationship between Cu and tau has been established to suggest that the promoter of the tau gene (MAPT) may be regulated by the Cu-responsive transcription factor, Sp1 (Heicklen-Klein and Ginzburg 2000; Song et al. 2008). Sp1 is also a key regulator of BACE1 processing of APP to produce Aβ (Christensen et al. 2004). Overexpression of tau inhibits kinesin-dependent transport of peroxisomes, increasing the vulnerability of cells to oxidative stress and hence degeneration. Additionally, inhibiting APP transport into axons and dendrites induces cell body accumulation of APP (Stamer et al. 2002). Therefore, a clear link between Cu-responsive overregulation of tau and APP may be important. However, critical evidence in the literature demonstrates that despite the role of tau in APP and Aβ-induced cognitive decline, Aβ accumulation most likely precedes and drives the accumulation of tau neurofibrillary tangles (NFT) (Götz et al. 2001; Hu et al. 2008; Lewis et al. 2001).
Structural analysis of tau protein postulates that the microtubule-binding domain (MBD) comprises of four highly conserved 18-amino-acid repeats, R1, R2, R3 and R4. The MBD is well recognised for its role in the formation of paired helical filaments (PHFs), promoting tau aggregation and formation of NFTs in vitro. Neurotoxicity to neurons may be induced by abnormal tau aggregation; hence, several studies have been conducted to determine the aggregation process. More specifically, the relationship between these repeat regions and Cu(II) has been investigated (Ma et al. 2005, 2006; Zhou et al. 2007). Cu(II) binding to repeat regions is a pH-dependent and stoichiometrically determined process. The R2 and R3 peptides adopt a monomeric α-helical structure in the presence of Cu(II). The helical structure induces PHF formation that aggregates to form NFTs (Ma et al. 2005, 2006). Furthermore, R3 peptide may additionally form a β-sheet structure in 1 mol eq. of Cu(II). Interestingly, Cu(II) binding to R1 peptide has been shown to delay the onset and level of R1 aggregation. It is suggested that Cu(II) coordination affects the electrostatic surface of R1 peptide, hence regulating in vivo aggregation of tau protein (Zhou et al. 2007). The present literature therefore suggests that Cu(II) binding to repeat of the MBD influences peptide aggregation, hence encapsulating an important feature of AD, the presence of NFTs. Moreover, tau demonstrates redox activity when bound to Cu inappropriately, which further contributes to oxidative stress (Hung et al. 2010; Su et al. 2007). R2 peptides have the capacity to reduce Cu(II) to Cu(I), favouring the generation of hydrogen peroxide (H2O2) and hence subsequent ROS (Su et al. 2007).
Cu and Oxidative Stress in AD
Oxidative stress is a predominant feature in AD and ageing brains, whereby conditions of dyshomeostasis lead to the generation of ROS. ROS production can be facilitated by the high redox nature of Cu. Oxidative stress may therefore be induced through participation in Fenton and Haber-Weiss reactions, Aβ-Cu(II) binding, diminished glutathione (GSH) levels and reduced expression of Cu-dependent enzymes (Hung et al. 2010; Mot et al. 2011; Halliwell and Gutteridge 1984). ROS may interact with biomolecules and engender irreversible oxidative modification such as lipid peroxidation, protein oxidation and nucleic acid cleavage leading to cellular impairment (Hung et al. 2010; Halliwell and Gutteridge 1984).
Cu is thought to play a key causative role in oxidative stress-induced neurodegeneration by Fenton and Haber-Weiss reactions, where it directly catalyses the formation of ROS (Hung et al. 2010; Ahuja et al. 2015). This is a two-step process involving the reaction between cupric ion (Cu(II)) and superoxide anion radical (O2 •- ) or biological reductants such as ascorbic acid or glutathione (GSH) to produce the reduced cuprous ion (Cu(I)). Cu(I) in the presence of H2O2 can catalyse the formation of highly unstable hydroxyl (•OH) radicals (Ahuja et al. 2015; Halliwell and Gutteridge 1984; Barbusiński 2009). Reactive •OH radicals may react with biomolecules close to the site of formation, exacerbating oxidative stress in this region (Jomova and Valko 2011). In addition, the capacity of Cu to induce DNA damage and oxidation of bases is induced by ROS production via the Fenton reaction (Moriwaki et al. 2008):
Amyloid deposits present in AD patients exhibit high levels of copper and oxidative stress markers. Cu(II) noticeably potentiates Aβ neurotoxicity, promoting the greatest toxic effect for Aβ1–42 compared to Aβ1–40 (Jomova and Valko 2011; Cuajungco et al. 2000). This is based on the peptide’s ability to reduce Cu(II) to Cu(I) and therefore mediate O2-dependent H2O2 production. The redox potential of Cu hence plays a significant role in exacerbating and facilitating Aβ-induced oxidative stress and subsequent neuronal death in AD (Huang et al. 1999). Interestingly, further oxidative damage may be mediated by dityrosine cross-linking between Aβ peptides. Cross-linking can be induced through direct attack of •OH radicals on Aβ peptides (Barnham et al. 2004; Galeazzi et al. 1999; White et al. 2006b), and the oxidative environment causes the accumulation of multi-protein aggregates that have a greater resistance to clearance (White et al. 2006b; Perry et al. 2002).
Elevated levels of Cu diminish GSH, an antioxidant present in cells. GSH is a substrate for enzymes that remove ROS. Dyshomeostasis of Cu and hence reduced levels of GSH may induce an oxidative environment that enhances the production and cytotoxic effects of ROS (Ahuja et al. 2015).
In AD patients, brain tissue can present with elevated Cu levels as well as Cu-deficient regions. The expression of Cu-dependent enzymes (SOD1 and ATOX1) was considerably reduced in multiple microarray studies, reinforcing that neurons may be Cu deficient (Myhre et al. 2013). In addition to its antioxidant role, SOD1 also has anti-inflammatory functions through ROS detoxification. Therefore, reduced expression and hence activity of SOD1 would heighten toxic ROS accumulation and exacerbate both oxidative stress and chronic inflammation (Choo et al. 2013).
Cu and Inflammation
Inflammation is a significant pathological factor present in neurodegenerative diseases such as AD. Inflammation is normally a protective response in the brain involving interactions between cells and mediators to prevent cell injury. The resident macrophages of the brain parenchyma, microglia, exhibit protective effects such as monitoring the local microenvironment and responding to disturbances. Slight disruptions in the microenvironment cause morphological and functional changes in microglia, which may further contribute to neuroinflammation present in the brain (Bamberger et al. 2003; Minghetti 2005). During such periods of dyshomeostasis in AD, inflammation may be triggered by the accumulation of abnormal protein aggregates or by pro- and anti-inflammatory cytokine imbalances (Minghetti 2005; Wyss-Coray and Mucke 2002). Copper can play a central role in toxic and protective inflammatory reactions (Choo et al. 2013).
The binding of Cu with Aβ induces Aβ deposition and neurotoxicity through ROS generation (Barnham et al. 2004). The production of ROS alone contributes to the neurotoxic inflammatory environment. Additionally, the presence of activated inflammatory cells, such as microglia, surrounding Aβ plaques contributes significantly to the chronic inflammatory response (Choo et al. 2013; Dickson et al. 1988; McGeer et al. 1989; Rozemuller et al. 1989). Microglia participate in the clearance of senile plaques through phagocytosis or Aβ plaque degradation (DeWitt et al. 1998; Shaffer et al. 1995). The ultimate outcome of this ‘beneficial’ microglial response is unknown. Paradoxically, phagocytosis of Aβ may stimulate immune activation and release of pro-inflammatory mediators (Choo et al. 2013). Interestingly, astrocytes are closely associated with microglial phagocytosis and prevent clearance and slow degradation of amyloid plaque materials. These materials can therefore persist and further contribute to AD (DeWitt et al. 1998; Shaffer et al. 1995). Additionally, within close vicinity of the amyloid plaques, increased levels of complement, cytokines, chemokines and free radicals have been observed, promoting a self-propagating toxic cycle leading to neurodegeneration in AD (Minghetti 2005).
Cu displays both pro- and anti-inflammatory characteristics. It is well established that Cu induces peripheral secretion of IL-6 (Schmalz et al. 1998) and IL-8, the latter specifically through NF-κB activation (Kennedy et al. 1998). However, little evidence exists depicting the direct relationship of Cu to neuroinflammation. Current literature has reported that cholesterol and Cu may synergistically interact to generate Cu-induced neurotoxicity through oxidative stress-mediated apoptosis in AD (Choo et al. 2013; Lu et al. 2006). Conversely, it has been suggested that Cu can contribute to the development of an anti-inflammatory microglial phenotype (M2). A specific study indicated that the BV2 microglial cell line exposed to LPS induced an inflammatory phenotype (M1); however, the combination of both Cu(I) and LPS may lead to a shift towards an M2-like phenotype. Cu alone, in the absence of LPS, has not shown any effect on either M1 or M2 phenotypes. The studies propose that the shift from M1 to M2 is due to the redox state of NO, which may be due to Cu(I). Furthermore, the absence of NO is proposed to be a factor in the adoption of the M2 microglial state (Choo et al. 2013; Bamberger et al. 2003).
Non-ceruloplasmin Cu in Plasma
Non-ceruloplasmin Cu (non-Cp-Cu) may also be referred to as “free Cu” and is simply defined as serum Cu not bound to ceruloplasmin (Cp). It is well established that AD patients display elevated levels of non-Cp-Cu (Squitti et al. 2005). Furthermore, this increase may be representative of total serum copper (Squitti 2014). Free Cu levels correlate with cognitive function (measured by Mini-Mental State Examination (MMSE)), and higher level of free Cu is a predictor of severe cognitive decline, worsening MMSE outcomes, in patients with AD (Salustri et al. 2010; Squitti et al. 2009). Findings suggest that non-Cp-Cu may be a predictor for the progression of mild cognitive impairment to AD (Squitti et al. 2014). Additionally, alterations in electroencephalographic (EEG) rhythms have been observed in patients with elevated non-Cp-Cu, more specifically the slowing of cortical EEG rhythms (Zappasodi et al. 2008).
The liver plays a central role in Cu storage, Cu movement and Cu coordination into Cp. It has been hypothesised that free Cu may arise from impaired transfer of Cu into the secretory pathway of hepatocytes. Moreover, research has been undertaken to determine whether there is a correlation between free Cu and liver function in AD. A negative correlation was found between free copper and markers of liver function. This study had multiple limitations and hence further investigation is required (Squitti et al. 2007). In addition, genetic defects associated with the Cu efflux pump, ATP7b, may cause altered loading of Cu into Cp. The transmembrane domain of the ATP7b ionic pump is associated with AD and increases the amount of non-Cp-Cu released into the circulation (Squitti 2012; Squitti et al. 2008).
A substantial number of meta-analyses have been conducted on Cu levels and specifically non-Cp-Cu levels. These meta-analyses in fact have demonstrated that the fraction of non-Cp-Cu in circulation is increased and as a whole Cu dyshomeostasis consists of decreased Cu in the brain (Schrag et al. 2011) and an increase in the blood (Wang et al. 2015). These studies undoubtedly support the correlation between altered Cu levels and AD pathogenesis. However, limited research has been conducted on the direct mechanisms in which elevated non-Cp-Cu levels effects AD patients.
Therapeutic Considerations
As eluded to in this review, metal ion dyshomeostasis is central to AD pathogenesis and hence has been a target for therapeutic interventions. Currently there is no clinical cure for AD. However, the development and investigation of therapeutics such as dietary Cu supplementation, Cu chelation and Cu complexes have been considered in AD.
Dietary supplementation of Cu has been studied as a therapeutic approach for AD. The brains of APP transgenic mice have lower levels of Cu and display reduced activity of Cu/Zn-SOD1 in comparison to wild-type mice. Following oral treatment of Cu, APP transgenic mice exhibited restored SOD1 activity to normal levels and an increase in bioavailable brain Cu levels and decreased Aβ1–40 and Aβ1–42. These mice did not present a premature death phenotype (Bayer et al. 2003). It was suspected that in AD patients Cu intake may stabilise cognition. A randomised, double-blinded, placebo-controlled phase II clinical trial in patients with mild AD was conducted to evaluate the efficacy of oral Cu supplementation for 12 months. AD patients were treated with either Cu-(II)-orotate-dihydrate (8 mg Cu daily) or the placebo, and no significant differences were observed in primary outcome measures. The results demonstrated that although long-term oral intake of 8 mg Cu is well tolerated by AD patients, it is not therapeutic and has no effect on AD progression (Kessler et al. 2008). However, it should be noted that if Cu regulation is abnormal in AD (as supported by the evidence discussed here), then supplementation with dietary Cu is unlikely to have any impact. To overcome this, therapeutics are needed that bypass faulty Cu-handling processes.
Clioquinol (CQ; 5-chloro-7-iodo-8-hydroxyquinoline) (Fig. 3) is a small lipophilic, metal-protein attenuating compound (MPAC) that has demonstrated therapeutic potential in neurodegenerative diseases such as AD (Di Vaira et al. 2004). Conflicting hypotheses exist with regard to the approach CQ undertakes to interfere with metal homeostasis. Initially CQ was regarded as a metal chelator, suggesting that it may lead to alteration of Cu or Zn levels in specific diseased brains regions (Hegde et al. 2009; Treiber et al. 2004). CQ was later considered a metal-protein attenuating compound (MPAC), a substance that may influence and restore metal ion homeostasis without greatly effecting overall Cu regulation (Grossi et al. 2009). However, it is now termed an ionophore, acting as a Cu carrier facilitating Cu transport across membranes (White et al. 2006b; Grossi et al. 2009; Filiz et al. 2008; Caragounis et al. 2007). The beneficial effects of CQ were reported in a nine-week study of oral CQ administration in AD mice. Results depicted reduced Aβ deposition and improved cognitive performance (Cherny et al. 2001). In a phase II clinical trial of CQ, 32 patients were recruited in this double-blind, placebo-controlled, parallel-group randomised study. The CQ group demonstrated improved cognitive performance and a decline in plasma Aβ1–42 concentration. However, in this study the cognitive benefit of CQ was only illustrated in the more severely affected subjects (Ritchie et al. 2003). Following some controversial published data on CQ, it has recently been withdrawn from human clinical studies (Hegde et al. 2009). Furthermore, a derivative of CQ, another hydroxyquinoline ligand (PBT-2) (Fig. 3) has been clinically tested in a human phase IIa double-blinded trial that demonstrated lowered CSF Aβ1–42 levels and improvement in two areas of a neuropsychological test battery (Lannfelt et al. 2008). PBT-2 and CQ have the ability to block H2O2 generation through the Aβ-Cu complex. PBT-2 can also decrease interstitial brain Aβ and improve cognitive performance to a greater degree than CQ. In addition, PBT-2 has outperformed CQ as an ionophore and shows increased BBB permeability (Adlard et al. 2008). Additional large and longer studies are required to further determine the beneficial effects of PBT-2 on AD patients.

Schematic structure of the metal ionophores clioquinol (CQ) and PBT-2
Cu complexes of bis(thiosemicarbazone) (BTSC) are able to bind Cu(II) and Zn(II) to form stable, lipophilic complexes that cross membranes and specifically the BBB (Duncan and White 2012; Green et al. 1988). In addition, Cu complexes of BTSCs (Cu(II)(btsc)s) have been investigated as a potential therapeutic of AD. Treatment of APP-CHO cells with Cu(II)(btsc)s specifically, glyoxalbis(N (4)-methyl-3-thiosemicarbazonato)copper(II) (Cu(II)(gtsm)) demonstrated increased intracellular Cu levels and a reduction in secreted Aβ levels (Donnelly et al. 2008). Furthermore, neurotoxic pathways are regulated by glycogen synthase kinase 3β (GSK3β) and have been targeted as a therapeutic in AD. Treatment with Cu(II)(gtsm) in APP/PS1 transgenic AD mice resulted in reduced active GSK3β, lower abundance of Aβ trimers and phosphorylated tau and conclusively reversed cognitive deficits in the APP/PS1 transgenic mice (Crouch et al. 2009). Additionally, despite pyrrolidine dithiocarbamate (PDTC) being classified as a Cu-chelating compound, it has also demonstrated in APP/PS1 mice to down regulate the GSK3β pathway and thus improve spatial learning. This study also presented that Cu levels in the brain increased with PDTC treatment and moreover reduced tau phosphorylation. It remains unknown whether PDTC binds and prevents metal binding of Aβ (Malm et al. 2007).
Future Directions and Conclusions
The present review gives an insight into the role of Cu in the complex neurodegenerative disorder, AD. Considerable evidence exists to demonstrate that altered Cu homeostasis in the brain is a key factor in AD. This is likely to involve mis-localisation of Cu rather than excess or deficiency per se. Increased Cu may exacerbate some subtypes, but mostly Cu changes are part of the disease, leading to loss of Cu function in key cell types and Cu-mediated toxicity in other cells or locations including amyloid aggregation. Copper-based therapeutics may be developed for Cu chelation and need to progress to address Aβ-Cu interactions, control tau-Cu and decrease oxidative stress and neuroinflammation in the brain. Ultimately, further research should contribute to ameliorating the deleterious effects of Cu dyshomeostasis in AD patients.
References
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Aβ. Neuron. 2008;59(1):43–55.
Ahuja A, Dev K, Tanwar RS, Selwal KK, Tyagi PK. Copper mediated neurological disorder: visions into amyotrophic lateral sclerosis, Alzheimer and Menkes disease. J Trace Elem Med Biol. 2015;29:11–23.
Amaravadi R, Glerum DM, Tzagoloff A. Isolation of a cDNA encoding the human homolog of COX17, a yeast gene essential for mitochondrial copper recruitment. Hum Genet. 1997;99(3):329–33.
Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NME, Romano DM, et al. Dramatic aggregation of Alzheimer Aβ by Cu (II) is induced by conditions representing physiological acidosis. J Biol Chem. 1998;273(21):12817–26.
Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, et al. Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42. J Neurochem. 2000;75(3):1219–33.
Ayton S, Lei P, Bush AI. Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics. 2015;12(1):109–20.
Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer’s disease. Lancet (London, England). 2011;377(9770):1019–31.
Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar β-amyloid mediates microglial activation. J Neurosci. 2003;23(7):2665–74.
Barbusiński K. Fenton reaction-controversy concerning the chemistry. Ecolog Chem Eng Sci. 2009;16(3):347–58.
Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, et al. Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain a regulator of neuronal copper homeostasis. J Biol Chem. 2003;278(19):17401–7.
Barnham KJ, Haeffner F, Ciccotosto GD, Curtain CC, Tew D, Mavros C, et al. Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J. 2004;18(12):1427–9.
Bayer TA, Schäfer S, Simons A, Kemmling A, Kamer T, Tepests R, et al. Dietary Cu stabilizes brain superoxide dismutase 1 activity and reduces amyloid Aβ production in APP23 transgenic mice. Proc Natl Acad Sci. 2003;100(24):14187–92.
Bucossi S, Ventriglia M, Panetta V, Salustri C, Pasqualetti P, Mariani S, et al. Copper in Alzheimer’s disease: a meta-analysis of serum,plasma, and cerebrospinal fluid studies. J Alzheimer’s Dis. 2011;24(1):175–85.
Bush AI, Tanzi RE. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics. 2008;5(3):421–32.
Buxbaum JD, Thinakaran G, Koliatsos V, O’Callahan J, Slunt HH, Price DL, et al. Alzheimer amyloid protein precursor in the rat hippocampus: transport and processing through the perforant path. J Neurosci. 1998;18(23):9629–37.
Caragounis A, Du T, Filiz G, Laughton KM, Volitakis I, Sharples RA, et al. Differential modulation of Alzheimer’s disease amyloid beta-peptide accumulation by diverse classes of metal ligands. Biochem J. 2007;407(3):435–50.
Cater MA, McInnes KT, Li Q-X, Volitakis I, La Fontaine S, Mercer JF, et al. Intracellular copper deficiency increases amyloid-β secretion by diverse mechanisms. Biochem J. 2008;412(1):141–52.
Ceccom J, Coslédan F, Halley H, Francès B, Lassalle JM, Meunier B. Copper chelator induced efficient episodic memory recovery in a non-transgenic Alzheimer’s mouse model. PLoS One. 2012;7(8):e43105.
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30(3):665–76.
Choi B-S, Zheng W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009;1248:14–21.
Choo XY, Alukaidey L, White AR, Grubman A. Neuroinflammation and copper in Alzheimer’s disease. Int J Alzheimers Dis. 2013;2013:145345.
Christensen MA, Zhou W, Qing H, Lehman A, Philipsen S, Song W. Transcriptional regulation of BACE1, the β-amyloid precursor protein β-secretase, by Sp1. Mol Cell Biol. 2004;24(2):865–74.
Citron M, Diehl TS, Gordon G, Biere AL, Seubert P, Selkoe DJ. Evidence that the 42-and 40-amino acid forms of amyloid β protein are generated from the β-amyloid precursor protein by different protease activities. Proc Natl Acad Sci. 1996;93(23):13170–5.
Cottrell BA, Galvan V, Banwait S, Gorostiza O, Lombardo CR, Williams T, et al. A pilot proteomic study of amyloid precursor interactors in Alzheimer’s disease. Ann Neurol. 2005;58(2):277–89.
Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, et al. Increasing Cu bioavailability inhibits Abeta oligomers and tau phosphorylation. Proc Natl Acad Sci U S A. 2009;106(2):381–6.
Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, et al. Evidence that the β-amyloid plaques of Alzheimer’s disease represent the redox-silencing and entombment of Aβ by zinc. J Biol Chem. 2000;275(26):19439–42.
Culotta VC, Klomp LW, Strain J, Casareno RLB, Krems B, Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272(38):23469–72.
Davies KM, Hare DJ, Cottam V, Chen N, Hilgers L, Halliday G, et al. Localization of copper and copper transporters in the human brain. Metallomics Integ Biometal Sci. 2013;5(1):43–51.
de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73(4):685–97.
DeWitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol. 1998;149(2):329–40.
Di Vaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, Zatta P. Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: structural characterization of its zinc (II) and copper (II) complexes. Inorg Chem. 2004;43(13):3795–7.
Dickson DW, Farlo J, Davies P, Crystal H, Fuld P, Yen S-H. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am J Pathol. 1988;132(1):86.
Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, et al. Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide. J Biol Chem. 2008;283(8):4568–77.
Duncan C, White AR. Copper complexes as therapeutic agents. Metallomics. 2012;4(2):127–38.
Filiz G, Price KA, Caragounis A, Du T, Crouch PJ, White AR. The role of metals in modulating metalloprotease activity in the AD brain. Eur Biophys J. 2008;37(3):315–21.
Galeazzi L, Ronchi P, Franceschi C, Giunta S. In vitro peroxidase oxidation induces stable dimers of beta-amyloid (1-42) through dityrosine bridge formation. Amyloid. 1999;6(1):7–13.
Götz J, Chen F, Van Dorpe J, Nitsch R. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293(5534):1491–5.
Green MA, Klippenstein DL, Tennison JR. Copper(II) bis(thiosemicarbazone) complexes as potential tracers for evaluation of cerebral and myocardial blood flow with PET. J Nucl Med. 1988;29(9):1549–57.
Grossi C, Francese S, Casini A, Rosi MC, Luccarini I, Fiorentini A, et al. Clioquinol decreases amyloid-β burden and reduces working memory impairment in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis. 2009;17(2):423–40.
Halliwell B, Gutteridge J. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J. 1984;219(1):1.
Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, et al. Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis. 2009;17(3):457–68.
Heicklen-Klein A, Ginzburg I. Tau promoter confers neuronal specificity and binds Sp1 and AP-2. J Neurochem. 2000;75(4):1408–18.
Henry W, Querfurth H, LaFerla F. Mechanisms of disease Alzheimer’s disease. New Engl J Med. 2010;362:329–44.
Higuchi M, Lee VM-Y, Trojanowski JQ. Tau and axonopathy in neurodegenerative disorders. NeuroMolecular Med. 2002;2(2):131–50.
Hu M, Waring JF, Gopalakrishnan M, Li J. Role of GSK-3beta activation and alpha7 nAChRs in Abeta(1-42)-induced tau phosphorylation in PC12 cells. J Neurochem. 2008;106(3):1371–7.
Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, et al. Cu (II) potentiation of Alzheimer Aβ neurotoxicity correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem. 1999;274(52):37111–6.
Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Aβ peptides. J Biol Inorg Chem. 2004;9(8):954–60.
Hung YH, Bush AI, Cherny RA. Copper in the brain and Alzheimer’s disease. J Biol Inorg Chem. 2010;15(1):61–76.
Hung YH, Bush AI, La Fontaine S. Links between copper and cholesterol in Alzheimer’s disease. Front Physiol. 2013;4:111.
Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283(2):65–87.
Kaden D, Bush AI, Danzeisen R, Bayer TA, Multhaup G. Disturbed copper bioavailability in Alzheimer’s disease. Int J Alzheimers Dis. 2011;2011:345614.
Kardos J, Kovacs I, Hajos F, Kalman M, Simonyi M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett. 1989;103(2):139–44.
Kennedy T, Ghio AJ, Reed W, Samet J, Zagorski J, Quay J, et al. Copper-dependent inflammation and nuclear factor-kappaB activation by particulate air pollution. Am J Respir Cell Mol Biol. 1998;19(3):366–78.
Kessler H, Bayer TA, Bach D, Schneider-Axmann T, Supprian T, Herrmann W, et al. Intake of copper has no effect on cognition in patients with mild Alzheimer’s disease: a pilot phase 2 clinical trial. J Neural Transm. 2008;115(8):1181–7.
Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, et al. Inducible expression of Tau repeat domain in cell models of tauopathy aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281(2):1205–14.
Klomp LW, Lin S-J, Yuan DS, Klausner RD, Culotta VC, Gitlin JD. Identification and functional expression of HAH1, a novel human gene involved in copper homeostasis. J Biol Chem. 1997;272(14):9221–6.
Kong G-W, Adams JJ, Cappai R, Parker MW. Structure of Alzheimer’s disease amyloid precursor protein copper-binding domain at atomic resolution. Acta Crystallogr Sect F: Struct Biol Cryst Commun. 2007;63(10):819–24.
Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7(9):779–86.
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–91.
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302.
LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci. 1995;92(22):10369–73.
Lu J, Zheng Y-L, Wu D-M, Sun D-X, Shan Q, Fan S-H. Trace amounts of copper induce neurotoxicity in the cholesterol-fed mice through apoptosis. FEBS Lett. 2006;580(28–29):6730–40.
Lue L-F, Kuo Y-M, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155(3):853–62.
Ma QF, Li YM, Du JT, Kanazawa K, Nemoto T, Nakanishi H, et al. Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers. 2005;79(2):74–85.
Ma Q, Li Y, Du J, Liu H, Kanazawa K, Nemoto T, et al. Copper binding properties of a tau peptide associated with Alzheimer’s disease studied by CD, NMR, and MALDI-TOF MS. Peptides. 2006;27(4):841–9.
Malm TM, Iivonen H, Goldsteins G, Keksa-Goldsteine V, Ahtoniemi T, Kanninen K, et al. Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden. J Neurosci. 2007;27(14):3712–21.
Mao X, Ye J, Zhou S, Pi R, Dou J, Zang L, et al. The effects of chronic copper exposure on the amyloid protein metabolisim associated genes’ expression in chronic cerebral hypoperfused rats. Neurosci Lett. 2012;518(1):14–8.
McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem. 1969;244(22):6049–55.
McGeer PL, Akiyama H, Itagaki S, McGeer EG. Immune system response in Alzheimer’s disease. Can J Neurol Sci. 1989;16(4 Suppl):516–27.
Minghetti L. Role of inflammation in neurodegenerative diseases. Curr Opin Neurol. 2005;18(3):315–21.
Moriwaki H, Osborne MR, Phillips DH. Effects of mixing metal ions on oxidative DNA damage mediated by a Fenton-type reduction. Toxicol In Vitro. 2008;22(1):36–44.
Mot AI, Wedd AG, Sinclair L, Brown DR, Collins SJ, Brazier MW. Metal attenuating therapies in neurodegenerative disease. Expert Rev Neurother. 2011;11(12):1717–45.
Myhre O, Utkilen H, Duale N, Brunborg G, Hofer T. Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases: possible impact of environmental exposures. Oxidative Med Cell Longev. 2013;2013:726954.
Opazo CM, Greenough MA, Bush AI. Copper: from neurotransmission to neuroproteostasis. Front Aging Neurosci. 2014;6:143.
Perry G, Cash AD, Smith MA. Alzheimer disease and oxidative stress. Biomed Res Int. 2002;2(3):120–3.
Pooler AM, Phillips EC, Lau DH, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013;14(4):389–94.
Pratico D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: lights and shadows. Ann N Y Acad Sci. 2008;1147:70–8.
Prince M, Wimo A, Guerchet M, Ali G, Wu Y, Prina M. World Alzheimer report 2015 [Internet]. London. 2015. Available from: http://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf
Reitz C. Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis. 2012;2012:369808.
Rembach A, Hare DJ, Lind M, Fowler CJ, Cherny RA, McLean C, et al. Decreased copper in Alzheimer’s disease brain is predominantly in the soluble extractable fraction. Int J Alzheimers Dis. 2013;2013:623241.
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Aβ amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60(12):1685–91.
Rozemuller JM, Eikelenboom P, Pals ST, Stam FC. Microglial cells around amyloid plaques in Alzheimer’s disease express leucocyte adhesion molecules of the LFA-1 family. Neurosci Lett. 1989;101(3):288–92.
Salustri C, Barbati G, Ghidoni R, Quintiliani L, Ciappina S, Binetti G, et al. Is cognitive function linked to serum free copper levels? A cohort study in a normal population. Clin Neurophysiol. 2010;121(4):502–7.
Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21(1):172–88.
Scheiber IF, Mercer JF, Dringen R. Metabolism and functions of copper in brain. Prog Neurobiol. 2014;116:33–57.
Schmalz G, Schuster U, Schweikl H. Influence of metals on IL-6 release in vitro. Biomaterials. 1998;19(18):1689–94.
Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM. Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog Neurobiol. 2011;94(3):296–306.
Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66.
Shaffer LM, Dority MD, Gupta-Bansal R, Frederickson RC, Younkin SG, Brunden KR. Amyloid β protein (Aβ) removal by neuroglial cells in culture. Neurobiol Aging. 1995;16(5):737–45.
Small DH, McLean CA. Alzheimer’s disease and the amyloid β protein. J Neurochem. 1999;73(2):443–9.
Smith DP, Smith DG, Curtain CC, Boas JF, Pilbrow JR, Ciccotosto GD, et al. Copper-mediated amyloid-β toxicity is associated with an intermolecular histidine bridge. J Biol Chem. 2006;281(22):15145–54.
Smith DP, Ciccotosto GD, Tew DJ, Fodero-Tavoletti MT, Johanssen T, Masters CL, et al. Concentration dependent Cu2+ induced aggregation and Dityrosine formation of the Alzheimer’s disease amyloid-β peptide. Biochemistry. 2007;46(10):2881–91.
Song I-S, Chen HH, Aiba I, Hossain A, Liang ZD, Klomp LW, et al. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol Pharmacol. 2008;74(3):705–13.
Squitti R. Copper dysfunction in Alzheimer’s disease: from meta-analysis of biochemical studies to new insight into genetics. J Trace Elem Med Biol. 2012;26(2):93–6.
Squitti R. Copper subtype of Alzheimer’s disease (AD): meta-analyses, genetic studies and predictive value of non-ceruloplasmim copper in mild cognitive impairment conversion to full AD. J Trace Elem Med Biol. 2014;28(4):482–5.
Squitti R, Pasqualetti P, Dal Forno G, Moffa F, Cassetta E, Lupoi D, et al. Excess of serum copper not related to ceruloplasmin in Alzheimer disease. Neurology. 2005;64(6):1040–6.
Squitti R, Ventriglia M, Barbati G, Cassetta E, Ferreri F, Dal Forno G, et al. ‘Free’ copper in serum of Alzheimer’s disease patients correlates with markers of liver function. J Neural Transm. 2007;114(12):1589–94.
Squitti R, Quattrocchi CC, Salustri C, Rossini PM. Ceruloplasmin fragmentation is implicated in ‘free’ copper deregulation of Alzheimer disease. Prion. 2008;2(1):23–7.
Squitti R, Bressi F, Pasqualetti P, Bonomini C, Ghidoni R, Binetti G, et al. Longitudinal prognostic value of serum “free” copper in patients with Alzheimer disease. Neurology. 2009;72(1):50–5.
Squitti R, Ghidoni R, Siotto M, Ventriglia M, Benussi L, Paterlini A, et al. Value of serum nonceruloplasmin copper for prediction of mild cognitive impairment conversion to Alzheimer disease. Ann Neurol. 2014;75(4):574–80.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow E-M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156(6):1051–63.
Su X-Y, Wu W-H, Huang Z-P, Hu J, Lei P, Yu C-H, et al. Hydrogen peroxide can be generated by tau in the presence of Cu (II). Biochem Biophys Res Commun. 2007;358(2):661–5.
Treiber C, Simons A, Strauss M, Hafner M, Cappai R, Bayer TA, et al. Clioquinol mediates copper uptake and counteracts copper efflux activities of the amyloid precursor protein of Alzheimer’s disease. J Biol Chem. 2004;279(50):51958–64.
Trombley PQ, Shepherd GM. Differential modulation by zinc and copper of amino acid receptors from rat olfactory bulb neurons. J Neurophysiol. 1996;76(4):2536–46.
Ventriglia M, Bucossi S, Panetta V, Squitti R. Copper in Alzheimer’s disease: a meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis. 2012;30(4):981–4.
Wang Z-X, Tan L, Wang H-F, Ma J, Liu J, Tan M-S, et al. Serum iron, zinc, and copper levels in patients with Alzheimer’s disease: a replication study and meta-analyses. J Alzheimers Dis. 2015;47(3):565–81.
Weiser T, Wienrich M. The effects of copper ions on glutamate receptors in cultured rat cortical neurons. Brain Res. 1996;742(1–2):211–8.
White AR, Barnham KJ, Bush AI. Metal homeostasis in Alzheimer’s disease. Expert Rev Neurother. 2006a;6(5):711–22.
White AR, Du T, Laughton KM, Volitakis I, Sharples RA, Xilinas ME, et al. Degradation of the Alzheimer disease amyloid β-peptide by metal-dependent up-regulation of metalloprotease activity. J Biol Chem. 2006b;281(26):17670–80.
Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35(3):419–32.
Zappasodi F, Salustri C, Babiloni C, Cassetta E, Del Percio C, Ercolani M, et al. An observational study on the influence of the APOE-ε4 allele on the correlation between ‘free’copper toxicosis and EEG activity in Alzheimer disease. Brain Res. 2008;1215:183–9.
Zheng Z, White C, Lee J, Peterson TS, Bush AI, Sun GY, et al. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J Neurochem. 2010;114(6):1630–8.
Zhou L-X, Du J-T, Zeng Z-Y, Wu W-H, Zhao Y-F, Kanazawa K, et al. Copper (II) modulates in vitro aggregation of a tau peptide. Peptides. 2007;28(11):2229–34.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Mathys, Z.K., White, A.R. (2017). Copper and Alzheimer’s Disease. In: Aschner, M., Costa, L. (eds) Neurotoxicity of Metals. Advances in Neurobiology, vol 18. Springer, Cham. https://doi.org/10.1007/978-3-319-60189-2_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-60189-2_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-60188-5
Online ISBN: 978-3-319-60189-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)