
Abstract
Copper (Cu) and iron (Fe) are redox active metals essential for the regulation of cellular pathways that are fundamental for brain function, including neurotransmitter synthesis and release, neurotransmission, and protein turnover. Cu and Fe are tightly regulated by sophisticated homeostatic systems that tune the levels and localization of these redox active metals. The regulation of Cu and Fe necessitates their coordination to small organic molecules and metal chaperone proteins that restrict their reactions to specific protein centres, where Cu and Fe cycle between reduced (Fe2+, Cu+) and oxidised states (Fe3+, Cu2+). Perturbation of this regulation is evident in the brain affected by neurodegeneration. Here we review the evidence that links Cu and Fe dyshomeostasis to neurodegeneration as well as the promising preclinical and clinical studies reporting pharmacological intervention to remedy Cu and Fe abnormalities in the treatment of Alzheimer’s disease (AD), Parkinson’s disease (PD) and Amyotrophic lateral sclerosis (ALS).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The brain has a high oxidative metabolic activity and relatively modest antioxidant defence and hence it is particularly susceptible to oxidative stress that can be exacerbated by the presence of redox active metals, copper (Cu) and iron (Fe), that are broadly distributed in the brain [1,2,3]. Cu and Fe control essential processes of the central nervous system (CNS), including neurotransmitter synthesis, myelin production, oxygen transportation and synaptic signalling [4, 5]. This is achieved through structural and catalytic regulation of enzymes, receptors and protein channels [4,5,6]. The redox properties of Cu and Fe are required for most of these cellular functions, yet are a source for the formation of reactive oxygen species (ROS). ROS promotes neurodegeneration through the oxidation, misfolding, and aggregation of essential proteins. Further, ROS can damage other macromolecules, including lipids that are converted to lipid peroxides, which are key players of ferroptosis, a new type of cell death that can affect neurons [7]. To prevent oxidative stress, and subsequent cell death, cells have several mechanisms to control/repair oxidative damage and ‘scavenge’ free radicals. To prevent Cu- and Fe-mediated ROS formation, the CNS has an array of proteins and small molecules that store, transport and deliver Cu and Fe safely to where they are required. During ageing the human and mouse brain display an accumulation of both metals, which indicates that their regulation is affected by age-dependent factors [8]. This imbalance is pronounced in neurogenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD), and Amyotrophic lateral sclerosis (ALS). In this review we discuss the current understanding of brain homeostasis of Cu and Fe, their impact on cell death pathways, the implications for neurodegenerative diseases and therapeutic approaches that aim to regulate these essential biologically active metals to mitigate/prevent neurodegeneration.
Cu metabolism in CNS
Cu in plasma is mainly bound to ceruloplasmin (CP) (Fig. 1), with a small proportion associated with albumin, transcuprein and amino acids [9, 10]. Cu is transported to the brain through the blood–brain-barrier (BBB), and to a lesser extent through the blood–cerebrospinal fluid barrier (BCB) mostly as free ion [9]. Cu uptake in neurons is mediated by copper-transporter 1 (CTR1) and once in the cytosol, copper binds to chaperones ATOX-1 (antioxidant 1 copper chaperone 1), CCS (copper chaperone for SOD1) or COX17 (cytochrome c oxidase copper chaperone) that deliver copper to specific enzymes. Excess Cu is chelated by metallothioneins and glutathione (GSH; millimolar concentration) to prevent redox activity and ROS production [11, 12]. The P-Type ATPases, ATP7A and ATP7b are localized predominantly in the membrane of the Golgi apparatus where they load Cu to cuproenzymes including ceruloplasmin and dopamine β-hydroxylase [13, 14]. In response to increases in intracellular Cu, ATP7A translocates Cu across the plasma membrane [15, 16]. Transfer of Cu from ATOX-1 to ATP7A/ATP7B increases its ATPase activity while in its Cu free form, ATOX1 is able to retrieve Cu from these ATPases and reduce their activity.

Cu/Fe transport and metabolism. The majority of Cu and Fe in blood circulation is bound to ceruloplasmin and transferrin, respectively. Brain Cu uptake occurs via the copper transporter-1 (CTR-1) expressed in the cell surface of blood capillary endothelial cells (BCEC). The P-type ATPases, ATP7A and ATP7B, are efflux pumps that traffic to the membrane in response to increases in intracellular Cu and mediate Cu export. Once in the cytosol Cu binds to chaperones (ATOX1, CCS and COX17) that aid delivery of Cu to cupro-biomolecules including ATP7A/B, Superoxide dismutase-1 (SOD1) and Cytochrome C oxidase, respectively. Fe is transported to brain in association with transferrin that binds to transferrin receptor-1 on the surface of BCECs. This complex is endocytosed. In endosomes, Fe dissociates and converted to its ferrous form (Fe3+ ⇒ Fe2+) by the reductase STEAP3. Fe is transported from the endosomes into the cytosol by the divalent metal transporter-1 (DMT1) where it again associates with transferrin. Fe is taken up by the mitochondria, where it is essential for the respiratory chain and biosynthesis of heme and iron–sulfur clusters, by transferrin receptor 2. The main exporter of intracellular Fe is ferroportin-1. Upon export, Fe is oxidised by the ferroxidases ceruloplasmin and hephaestin. APP facilitates Fe export via binding to ferroportin-1
Fe metabolism in CNS
Fe uptake by neurons occurs following binding of Tf to transferrin receptor 1 (TfR1) located on the surface of brain capillary endothelial cells (BCECs) (Fig. 1). The Tf-TfR1 complex is internalised by endocytosis into endosomes where low pH leads to dissociation of Fe from Tf. In endosomes, ferric iron is reduced to ferrous iron via the reductase STEAP3 [17] and is transported across the endosomal membrane to the cytosol via DMT1 [18]. Once in the cytoplasm Fe can be utilised for neuronal function and metabolism while excess iron can be complexed to ferritin and stored or expelled from the neuron by ferroportin 1 and oxidased by circulating or astrocyte-bound CP, prior to loading onto extracellular Tf [19].
Non-transferrin bound iron (NTBI) can be directly transported across the cell membrane by ZIP8 and ZIP14, which are members of the ZRT/IRT-like (ZIP) solute carrier family [20,21,22]. Similar to DMT1, ZIP14 is capable of transporting Tf bound Fe from the endosome to the cytoplasm [22]. ZIP8 and ZIP14 are newly identified iron transporters which may have distinct biological roles. For instance, ZIP8/ZIP14 iron transporting activity is maximal at above pH 7, while DMT1 functions efficiently at pH 5.5 [20, 21, 23]. Indeed, in rat hippocampal neurons ZIP8 is the major NTBI transporter in contrast to DMT1 which primarily transports Tf bound Fe [24].
Poly (rC)-binding proteins (PCBP) deliver Fe to ferritin for storage and to proteins that require non-heme iron as a cofactor, including hypoxia-inducible factor (HIF) prolyl and asparaginyl hydroxylases and deoxyhypusine hydroxylase [25,26,27].
Iron regulatory proteins (IRPs) are responsive to intracellular iron levels and bind to iron-responsive element (IRE) on mRNA transcripts corresponding to proteins involved in homeostatic regulation of iron. For instance, IRP1 and IRP2 are RNA-binding proteins that alter translation of ferritin, ferroportin and TfR mRNA [28, 29]. Cytosolic Fe can bind to IRPs and induce a conformational change that would prevent interaction with IRE motifs in mRNA. Conversely, a decrease in Fe enables IRPs to freely bind IRE and modify translation. Transcripts of proteins involved in storage or export of iron, such as ferritin and ferroportin, harbour IRE on the stem-loop structures of 5′-untranslated region (UTR) and IRP binding prevents their translation and promotes their degradation. Conversely, transcripts of iron import proteins, such as TfR1 and DMT1, harbor IRE on their 3′-UTR and are thus stabilised and translated during iron-depleted conditions.
Storage and buffering of Fe is essential to prevent ROS production and oxidative damage. Ferritin is the major iron storage protein in glia and neurons, whilst neuromelanin captures large amounts of iron in certain neuronal populations (i.e. dopaminergic neurons of substantia nigra) for long-term iron storage [30]. Fe can be released and re-used from ferritin through its autophagic-lysosomal degradation (ferritinophagy) [31]. In addition, heme-oxygenase 1 can catalyse the degradation of heme to ferrous Fe in order to maintain Fe homeostasis [32].
Alzheimer’s disease (AD)
AD is the most common type of dementia in the elderly affecting nearly 50 million people globally with this number predicted to double every 20 years, making it a major health concern for the ageing population [33,34,35]. The pathology of AD includes the aggregation of extracellular amyloid-β (Aβ) peptide and intracellular neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau [36, 37]. In AD, synaptic and neuronal loss are observed in the hippocampus, frontal cortex, temporal lobe, parietal lobe and cingulate gyrus, leading to impaired memory, language, problem-solving ability and thus decline in cognition [38,39,40]. The accumulation of Cu and Fe is consistently observed in the amyloid plaques of AD patient brains [41, 42]. Magnetic resonance image (MRI) studies revealed elevated Fe in the hippocampus of AD patients correlated to decreased integrity of the hippocampus, which is most likely due to loss of myelination [43]. The mechanisms by which these metals impact on disease have been intensively studied in the past decade. Although it has yet to be determined whether accumulation of these metals precedes or is a consequence of pathology, it is clear that they impact on multiple factors that contribute to disease.
Cu and Fe in AD
Amyloid plaques accumulate Cu and Fe [41, 44,45,46,47,48]. Cu and Fe deposit at the core and rims of senile plaques and co-localise with Aβ [41, 45, 49]. Moreover, these metals are elevated in the neuropil of AD patients [41, 50]. At senile plaques, Cu and Fe contribute to pathology by promoting ROS production and protein misfolding/aggregation, which will be discussed below. In contrast, there is evidence of an overall brain metal deficiency in AD [51]. Meta-analysis indicates that Cu is significantly reduced in the AD brain [52]. These changes may be associated with dysregulation of copper metabolism in blood [53]. An accumulation of Fe is observed in AD affected regions including parietal cortex, motor cortex and hippocampus [43, 54,55,56]. Thus both Fe and Cu dyshomeostasis is observed in pathologically relevant regions of AD brains.
Redox active metals, oxidative stress and protein aggregation
Both Cu and Fe are redox active metals. Fe is focally incorporated into the core and halo regions of senile plaques and NFTs where it can generate free radicals via Fenton reaction (Fe2+ + H2O2 → Fe3+ + OH− + ·OH). In the Fenton reaction Fe2+ ions are stoichiometrically oxidised by H2O2 to Fe3+ and a hydroxyl radical (·OH), the most reactive ROS [47, 57, 58]. Histochemical studies have indicated that Fe can bind to Aβ; however, the formation of ROS following this interaction is not yet clear [47]. It is worth noting that Fe3+ does not form a stable complex with Aβ, which quickly precipitates [59].
Cu cycles between its oxidative states (Cu+ and Cu2+) when bound to Aβ and this interaction catalysis the formation of superoxide (as an intermediate product), H2O2 and ·OH in vitro [60,61,62,63,64]. As a consequence of metal-catalysed ROS production, Aβ peptides are susceptible to oxidation damage. Relevant to pathology, studies have shown that oxidising Aβ increases its propensity to aggregate and form β-sheet rich fibrils of Aβ42 [65]. In addition, oxidation of Aβ alters the way it interacts with metals. For example, Cu binding to oxidised Aβ produces further ROS [66]. In the AD brain, ROS leads to oxidation damage that impacts the function of proteins, perturbs lipid membranes and damages DNA [67,68,69]. The evidence of oxidative stress includes increased levels of carbonylated proteins, increased 4-HNE (a by-product of lipid peroxidation) and oxidised DNA bases [1, 70,71,72,73,74].
Cu and Fe are required for the activity of antioxidant enzymes superoxide dismutase 1 (SOD1) and catalase, respectively. These enzymes catalyse the decomposition of ROS species, superoxide anion and H2O2, respectively. Therefore, storage, transport and regulation of these metals are crucial for cell survival.
Together with promoting oxidative stress, Cu potentiates the aggregation and misfolding of Aβ. Cu2+ ions bind with high affinity to Aβ peptides [75,76,77,78,79] [80]. This interaction increases the proportion of β-sheet and α-helix structures, which promotes Aβ aggregation [81]. In turn, Cu bound to amyloid has increased cell toxicity [81, 82].
The role of Fe in promoting Aβ aggregation and/or misfolding is still under debate. For instance, studies have shown that Fe2+ and Fe3+ ions promote Aβ aggregation and annular protofibrils whilst slowing the production of ordered cross-β fibrils [83,84,85,86,87]. In contrast, another study demonstrated that Fe promotes the formation of shorter and less ordered Aβ aggregates, which are more neurotoxic [87]. Other studies found that Fe binds to Aβ with low affinity and does not seem to co-localize with β-amyloid extracted from plaques [83,84,85]. Further studies are required to clarify Fe role in Aβ aggregation/misfolding.
As mentioned in “Alzheimer’s disease (AD)”, neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau are another hallmark pathological marker of AD. Cu accumulates in NFTs and binds to tau protein increasing its aggregation in vitro [46, 88,89,90,91]. In vivo, Cu enhances tau phosphorylation in transgenic mice models of AD (APPswe, PS1, P301L tau) [92, 93].
APP and metal homeostasis
Amyloid precursor protein (APP) is central to AD pathology as its processing by secretases (β- and γ-secretases) leads to production of the Aβ peptide (Fig. 2). Fe and Cu regulate the expression of the APP, which regulates the levels of Cu and Fe. APP has Cu binding sites, which probably enable APP to sense and control Cu levels and location [94,95,96,97]. Overexpression of APP in cells and in transgenic mice leads to a reduction in intracellular Cu [98, 99]. Conversely, the knockdown of APP increased Cu in primary cortical neurons [100]. APP and its product sAPPα have a role in Fe export by binding and stabilising ferroportin on the plasma membrane to facilitate Fe efflux from neurons [101, 102]. In addition, the mRNA APP transcript possesses an IRE in the 5′-UTR. In low cellular Fe conditions, IRP1 binds the IRE of APP mRNA inhibiting its translation and in-turn downregulating APP-mediated stabilisation of FPN, further inhibiting Fe efflux [103, 104]. The role of APP in Fe export is further supported by a study that showed that APP knockout mice display accumulation of brain Fe at 12 months of age [101]. This indicates that in the healthy brain APP may play an important role in Fe homeostasis.

APP trafficking and processing. APP is synthesized in the endoplasmic reticulum (ER) and trafficked through the trans-Golgi Network (TGN) to the cell membrane via secretory vesicles. Along route, APP is processed by α-, β- and γ-secretases via two distinct pathways. In the non-amyloidogenic pathway (A) α-secretase at the PM cleaves APP to release an N-terminal domain (sAPPα). The C-terminal fragment (CT83) can then be internalized and cleaved by γ-secretases to produce APP intracellular domain (AICD) or CTFγ and p3 peptide. In the amyloidogenic pathway APP is cleaved by β-secretase to release sAPPβ that can be further processed by the γ-secretase complex to produce CTFγ and Abeta peptides (Aβ). Secretases are present in various locations and thus APP processing occurs in various compartments. PM plasma membrane
Cu is linked to the processing, trafficking and regulation of the APP. Whether Cu increases or decreases the production of Aβ is unclear. Cu promotes APP expression, increases its localization at the cell surface and reduces its endocytosis [105, 106]. Cu enhances APP dimerization and increases extracellular release of Aβ [107]. In contrast, in cell and cell-free assays Cu was shown to reduce the levels of Aβ interacting with the γ-secretase complex [108]. Borchardt et al. also reported that copper decreased Aβ with a concomitant increase in the secretion of the ectodomain (sAPP-α) [109]. In AD patients, Cu deficiency is linked to enhanced Aβ production following induction of the amyloidogenic processing of APP, although the molecular mechanism by which Cu mediates this effect is unclear [110].
Therapeutic potentials of targeting brain Cu and Fe levels
Till date majority of AD therapeutic strategies focus on clearance of amyloid or limiting the production of Aβ species [111] [112, 113]. However, none of these strategies, including those that have achieved a reduction in amyloid, have resulted in cognitive improvement [112, 113]. Therefore, there is a need for alternative therapeutic approaches for AD. Modifying metal homeostasis pharmacologically in AD patients has been considered as a promising alternative.
Brain Cu deficiency is a feature of AD and small molecules that deliver Cu to neurons have been explored as a therapeutic. Clioquinol (CQ), a bioavailable anti-parasitic agent, has been trialled as a potential AD therapeutic as an ionophore as it is able to redistribute Cu (and Zn) from extracellular senile plaques to Cu-deficient neurons [114] (Table 1). In animal models of AD, oral administration of CQ for 9 weeks reduced amyloid burden and improved cognitive performance [115, 116]. Furthermore, in an Aβ-injection model, CQ inhibited Aβ aggregation, redox toxicity and reduced neuronal cell loss [117, 118]. In a small human phase II clinical trial, CQ was able to reduce Aβ deposition in patients with mild disease and prevented clinical deterioration [119].
Following these studies, PBT2 (8-hydroxyquinoline derivative) was generated (Table 1). Treatment of PBT2 to an AD mouse model showed greater efficacy compared to CQ as seen by improved cognitive function and decreased Aβ burden [120]. Promising results were also obtained in a phase 2a clinical trial (78 AD patients), 12 weeks of treatment led to a dose-dependent lowering of Aβ in CSF and improved 2 measures of executive function at the highest dose (250 mg) [121, 122]. However, follow-up studies with increased trial period (12 months) failed to show an improvement in cognition and amyloid burden [123].
The copper-containing bis(thiosemicarbazone) compound CuIIGTSM has neuroprotective properties in cell culture and in AD transgenic mouse models [124] (Table 1). CuIIGTSM lowers Aβ levels, GSK3β activity and phosphorylated levels of tau, whilst in APP/PS1 transgenic mice it improves cognitive performance (shown by Y-maze) [125, 126]. The related complex, CuIIATSM, was not effective in AD mouse models, but has therapeutic benefits in four PD mouse models and ALS mouse models (discussed below).
Historically, the first intervention that tested iron chelation in AD was using desferrioxamine (DFO) in a 2-year single-blind phase 2 clinical trial. In the trial 48 patients were randomised to receive intramuscular DFO (5 times/week, 125 mg twice daily), an oral placebo or no treatment. This intervention is reported to slow cognitive decline by 50% [127]. To improve delivery of DFO, which shows low BBB penetrance, an intranasal version was tested in an AD mouse model [128]. Similarly, intranasal desferrioxamine improved cognition, decreased GSK activity, oxidative stress and soluble Aβ species [128]. A further study demonstrated a lowering of tau hyperphosphorylation by inhibition of CDK5 and GSK3 [129]. Other iron chelators tested in animal models include Deferasirox [130] and M30 [131], both of which showed improvement in pathology, reduction in Aβ and tangles, as well as improved cognitive performance.
The chelator deferiprone (DFP) has been evaluated as a therapeutic agent for a number of neurogenerative disease: AD, PD and ALS (the latter two will be discussed below). In neurons DFP reduced Aβ toxicity in cell culture [132], whilst in a rabbit AD animal model, DFP reduced Aβ burden and tau phosphorylation [133]. Following these results, a Phase 2 study is currently being undertaken for AD (NCT03234686).
Parkinson’s disease
PD is a chronic motor neurodegenerative disease characterised by the accumulation of the α-synuclein protein in Lewy bodies and the loss of monoaminergic neurons. Although neurodegeneration is evident throughout the nigrostriatal tract, most neuronal loss occurs in the substantia nigra pars compacta (SNc), which is composed of dopaminergic neurons. To a lesser extent, neurodegeneration occurs throughout the basal ganglia extending to olfactory bulbs, sympathetic ganglia and dopaminergic neurons in the gut [134]. Loss of dopaminergic neurons and the consequent reduction of the neurotransmitter dopamine leads to motor symptoms including dyskinesia, instability, tremors and rigidity with an age of onset around 55 years and disease progressing for about 20 years [135]. Neuronal death, has been attributed to mitochondrial dysfunction, dopamine toxicity, oxidative stress, and misfolding and oligomerization of α-synuclein [136]. To date the most effective treatment is l-3,4-dihydroxyphenylalanine (l-DOPA), the precursor of dopamine. l-DOPA treatment is able to alleviate some of the motor symptoms but fails to prevent the progression of the disease with degeneration continuing and clinical symptoms worsening. Numerous studies have reported perturbation of Fe and Cu in PD which plays a multi-factorial role in pathogenesis.
Cu and Fe dyshomeostasis in PD
PD patients have exhibited elevated Cu levels in the cerebral spinal fluid which has been associated with oxidative stress and protein conformational changes [137, 138]. In addition, there is a high Cu level in PD blood serum, with levels positively correlating with disease severity [139]. Conversely, Cu levels and Cu bound to neuromelanin in the SN and degenerating regions of the PD brain are diminished [140, 141]. Lower Cu levels in SN could be due to impaired Cu transport in PD. Indeed, there is a reduction in expression of CTR1 in the SN of PD patients [140, 142, 143]. As a consequence of reduced levels of Cu, SOD1 is hypometallated in PD [139]. This may affect the activity of SOD1 which is essential for removal of ROS. Metallothionein stores and sequesters Cu and by doing so prevents oxidative stress. The brain-specific isoform MT3 is reduced in PD, which may be associated with susceptibility to oxidative stress [144].
Fe levels are elevated in various regions of the PD brain, with marked loading occurring at SNc, as observed in post-mortem brain tissue and in living patients [145,146,147,148]. In the early stages of PD, neuronal loss is restricted to the SNc [149]. Various factors most likely act together in the PD brain to bring about Fe accumulation. These factors include: increased permeability or dysfunction of the BBB; increased pro-inflammatory state; increased lactoferrin receptors in neurons and microvessels and increased expression of DMT1 in dopamine neurons; altered iron transport by Tf-TfR type 2 and mutations in genes relevant to Fe transport and binding [72, 150,151,152,153]. Decreases in ferritin levels have been reported in the SN that could be due to sustained IRP1 activity observed in post-mortem PD brain and may impact iron storage [154]. Moreover, ferroportin is downregulated in several neurotoxin-based mice models of PD including MPTP and 6-Hydroxydopamine (6-OHDA) [141]. Low ferritin levels and high Fe levels are indicative of Fe loading and provide an environment susceptible to oxidative damage.
Cu and Fe promote misfolding and aggregation of α-synuclein
The physiological functions of α-synuclein are yet to be fully elucidated. Studies have linked its function to maintaining synaptic function, neurotransmitter storage and neurotransmitter release within the synapse. However, its aggregation is a pathological hallmark of neurodegenerative diseases coined synucleinopathies. In PD, misfolded α-synuclein forms the major constituent of Lewy bodies (LB), which are intracellular inclusions found in the SNc, locus coeruleus, raphe nucleus and dorsal motor nucleus of the vagus [155, 156]. The propensity of α-synuclein to aggregate and oligomerize is highly dependent on post-translational modifications including acetylation, phosphorylation, oxidation, nitration, ubiquitination and truncation. Relevant to pathology, nitrated-α-synuclein is neurotoxic, prone to aggregation and highly enriched in LB [157]. Injection of nitrated-α-synuclein directly into the SNc of rats recapitulated many of the pathological features of PD [158]. In addition, PD patients exhibit nitrosative stress as detected by increased levels of nitrotyrosine in blood and brain [159]. Nitrosative stress occurs when reactive nitrogen radicals, namely peroxynitrite (ONOO−), modifies a wide range of cellular elements including tyrosine nitration. Peroxynitrite is produced via a non-enzymatic, pH dependent reaction of nitric oxide and superoxide, both of which are ROS present during oxidative stress.
α-Synuclein has multiple Cu binding specific sites, located on the N-terminal (residues 1, 5 and 50) and on the C-terminal (residues 107–140) [160]. These metal–protein interactions promote α-synuclein fibrillation and aggregation [161,162,163], which is modulated by post-translational modifications, including acetylation [164] and phosphorylation [165]. In addition, Cu association with α-synuclein alters the redox properties of Cu which leads to oxidation of biomolecules such as ascorbic acid, which produces H2O2, and antioxidants such as GSH [166]. This sets a chain reaction where H2O2 catalyses the oxidation of dopamine. In normal physiological conditions, most of α-synuclein is post-translationally modified by N-terminus acetylation (α-synucleinNAc). Cu bound to α-synucleinNAc activates oxygen resulting in intermolecular (Y39–Y39) cross-linking within the fibrillar core and intramolecular cross-linking with the C-terminal region. These interactions induced by Cu binding suggest Cu plays a role in attenuating fibrillar elongation [167].
Elevated Fe load observed in the SNc also affects the aggregation of α-synuclein. Ferrous and ferric iron can both bind to α-synuclein, with the latter promoting its aggregation [168]. Phosphorylated α-synuclein at residue S129 has been identified within Lewy bodies and shown to have a stronger binding affinity to ferrous iron [165]. In vitro studies have shown that ferric iron can catalyse the conversion of α-synuclein from the α-helix to the β-sheet conformation present in Lewy bodies [162]. Interactions between iron and α-synuclein and its potential role in PD have been recently reviewed by Chen et al. [169].
Fe, dopamine and PD
The SNc is more vulnerable to oxidative stress due to its natural high Fe content and the production of dopamine [170]. One of the mechanisms by which Fe contributes to pathology is by deleterious interactions with dopamine. Dopamine (DA) is the most important catecholamine neurotransmitter in the brain and its signalling through dopaminergic neurons governs reward, cognition and motor functions. However, despite its essential signalling function it is susceptible to oxidation that can potentially lead to neurotoxic species. In the presence of oxygen, both Cu2+ and Fe3+ can catalyse the oxidation of dopamine. Since labile Fe accumulates in the SNc, it is likely to contribute to dopamine oxidation. The by-products of these reactions include toxic semiquinone radical (DASQ) or quinone species (DAQ) of dopamine as well as O2−, H2O2 and hydroxyl radicals that can be formed following intricate chain reactions [171]. These end products can promote oxidative stress and mitochondrial dysfunction as well as induce and stabilise oligomeric α-synuclein formation [172, 173]. In addition, Cu and Fe can promote the enzymatic deamination of DA by monoamine oxidase (MAO) that generates dihydroxyphenylacetic acid (DOPAC) and H2O2 which can lead to further production of ROS [174]. Therefore, in a microenvironment which promotes oxidative stress (i.e. Fe and Cu accumulation) and dopamine oxidation can be neurotoxic causing damage to axon terminals and their synaptic vesicles.
Therapeutic potential of metal complexes and chelators in PD
Neurotoxin mouse models have been used to study PD pathogenesis and validating potential therapeutic targets. The neurotoxins 1-methyl-4-phenylpyridinium (MPTP) and 6-hydroxydopamine (6-OHDA) specifically target and drive the death of dopaminergic neurons. Following administration of these neurotoxins there is an approximate 50% decrease in dopaminergic neurons in the SNc and mice develop motor deficits comparable to those observed in late stage of PD in humans. These models have been used to study copper complexed diacetyl-bis(4-methylthiosemicarbazonato)copperII [CuIIatsm] and a range of iron chelators (DFO, deferasirox and DFP) as potential therapeutics (Table 1) [157,158,159,160].
The copper complex CuIIatsm has been tested as a therapeutic for PD and ALS (discussed below). CuIIatsm is a neutral, lipophilic and thermodynamically stable complex that has had a number of pharmacological uses including as an anti-tumour agent [175, 176]. In more recent years, CuIIatsm has been used as a PET imaging agent for labelling hypoxic tissues in CNS and peripheral tissues [177,178,179,180]. CuIIatsm is a small molecular weight (Mw = 321) complex, capable of crossing the BBB [176]. Moreover, it is a stable compound with low toxicity, and hence an ideal therapeutic candidate [175]. Administration of CuIIatsm leads to retention in hypoxic tissue due to mitochondrial impairment in the respiratory transport chain, which has been reported in PD. Furthermore, Ikawa et al. reported that CuIIatsm accumulated in the striatum and the more the disease progressed the more accumulation was observed [180].
To date CuIIatsm has been tested in various mouse models of PD, all of which showed promising results, specifically improved motor and cognitive function, improved dopamine metabolism and rescue of dopaminergic neuronal cell loss in SNc [181]. A major mode of action of CuIIatsm is the scavenging of peroxynitrite, inhibiting related toxicity and preventing the nitration and oligomerization of α-synuclein [181]. In addition, CuIIatsm treatment may elevate tyrosine hydroxylase (TH), the enzyme that catalyses the production of dopamine by hydroxylation of l-tyrosine to form its precursor L-DOPA. In this study the levels of dopamine doubled in response to treatment but were not restored to the level of the control [181]. However, the increased dopamine levels were sufficient to significantly improve motor skills. CuIIatsm was also able to increase the expression of vesicular monamine transporter 2 (VMAT2), which is a protein responsible for packaging dopamine into vesicles prior to its release from the synapse. Hence, CuIIatsm is not only able to increase dopamine production, but also place it in the compartment required for signalling.
Another therapeutic strategy is to target Fe overload seen in the SNc of PD brain. Fe chelators including DFO, deferasirox and DFP, have shown benefits in attenuating PD clinical symptoms (e.g. reducing dopaminergic neuronal loss and hydroxyl radical formation) [182]. All three of these are FDA approved pharmaceuticals for the treatment of Fe overload in thalassemia major and are well tolerated [183, 184]. DFP has been investigated for its therapeutic potential in cell culture and animal models as well as in pilot human clinical trials [132, 182, 185, 186]. Importantly, DFP crosses the BBB and has little impact on the systemic level of Fe and haematological indices partially due to its ability to deliver chelated metal to apotransferrin [187]. The ability of DFP to scavenge labile Fe limits ROS production [186, 187]. In the human neuroblastoma catecholaminergic cell line (SH-SY5Y), DFP showed neuroprotection against the neurotoxin MPTP [132], whilst in the 6-OHDA mouse PD model it attenuated dopaminergic neuronal loss and increased dopamine content in the SNc [182]. Most importantly, DFP has been tested in pilot human clinical trials with promising results [185]. DFP was able to reduce iron levels in specific brain regions, specifically the caudate nucleus and dentate nucleus (as assessed by MRI) with no generalized removal of Fe from the brain. Devos et al. [185] reported that PD patients have improved motor performance following DFP treatment [185]. However, Martin-Bastido reported a trend toward improvement in PD patients administered with DFP, which did not reach significance [185, 188]. These studies pave the way to larger clinical trials which further explore DFP dosage and longer treatment times.
Amyotrophic lateral sclerosis (ALS)
ALS is an aggressive neurodegenerative disease characterised by a rapid neuromuscular deterioration caused by the death of motor neurons in the cerebral cortex, brainstem and spinal cord [189]. In addition, there is a loss of axons in the lateral columns of the spinal cord. With no efficient treatment available to stop muscle weakness, wasting and paralysis and respiratory breakdown, ALS leads to death 3–5 years post-diagnosis [190]. Currently the only FDA approved pharmaceutical for ALS, Riluzole, is able to extend survival by 3 months and has no effect on muscle weakness [191]. The majority (90–95%) of cases of ALS are sporadic with a small proportion linked to familial linked genetic mutations; 25% of which are mutations of the superoxide dismutase (SOD1) gene. Mutations in SOD1, of which there are over 160 thus far, result in SOD1 misfolding and aggregation [192]. However, it is important to note that clinical symptoms and pathology are indistinguishable between familiar and sporadic ALS, suggesting common aetiology. The aetiology of ALS, however, remains largely unclear. As per AD and PD, pathology can be attributed to multiple factors which can be occurring simultaneously and feedback on each other. ALS is linked to glutamate excitotoxicity, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress and deposition of toxic ubiquitinated neuronal inclusions composed of both transactive response DNA binding protein 43 kDa (TDP-43) and fused in sarcoma (FUS) [193,194,195,196].
Cu, SOD1 and ALS
Although other genes have been linked to ALS, SOD1 mutations have been the most extensively studied. Moreover, transgenic mice models expressing ALS SOD1 mutations have proven to be robust animal models. SOD1 transgenic mice develop similar disease progression to the human ALS disease and thus ideal to study pathology and to test candidate therapeutics. SOD1 is a metalloenzyme that plays an important role in preventing oxidative stress by scavenging reactive oxygen species. It is a homodimer which is active and highly stable following post-translational modifications, including coordination with Zn and Cu, disulphide bond formation and dimerization [197,198,199]. In contrast, the metal-free form (apoSOD1) is unstable and prone to aggregation. In vitro, apoSOD1 is toxic to motor neurons due to production of peroxynitrite and oxidation damage [200]. SOD1 mutations linked to ALS increase misfolding and aggregation of the protein as well as alter the coordination to Zn and Cu. Thus, ALS mutations are not a loss of function (i.e. no effect on its anti-oxidant function) effect but rather a gain of toxic function. Studies in ALS mice models (e.g. SOD1G37R) demonstrate a high level of metal-deficient SOD1 in spinal cord tissue implicating the metals in pathogenesis, specifically in protein misfolding and aggregation. Misfolding of SOD during biosynthesis has been shown to occur in vitro in the presence of high labile Zn2+, which aberrantly binds to the protein in both the Zn and Cu binding sites forming a di-zinc SOD1. The di-Zinc SOD1 misfolding protein has a greater propensity to aggregate [201]. This is relevant as ALS mice models have elevated levels of labile Zn in the brain and spinal cord.
Copper homeostasis is also perturbed in ALS SOD1 mice models. Transgenic mice models carrying different SOD1 mutations have elevated Cu levels in the spinal cord [202, 203]. In the case of the G93A SOD1 mice model, Cu accumulation precedes the presentation of clinical symptoms, implicating high Cu as a characteristic pathology [203]. Cu imbalance is attributed to disruption of Cu metabolism. SOD1 transgenic mice have shown an increase in the levels of CTR1 and a decrease in the copper efflux protein ATP7A in the spinal cord tissue. Moreover, high levels of metallothioneins (MT), which have high affinity to Cu, are present in the spinal cord SOD1G93A [204]. Paradoxically, SOD1 extracted from ALS transgenic mice is in a copper-free form despite the high levels of intracellular Cu [205]. Given the importance of SOD1 metallation, therapeutics that increase the pool of holoSOD1 have been trialled.
Fe and ALS
In ALS, oxidative damage has been detected in neurons within organelle compartments (e.g. Golgi, ER and mitochondria) and by the presence of elevated oxidative stress biomarkers in bodily fluids including urine and CSF [206,207,208,209,210]. Much of this oxidative stress has been attributed to the breakdown of Fe metabolism in ALS. A number of MRI studies in ALS patients have shown an abnormal Fe accumulation in the motor cortex [211,212,213,214,215]. In the CSF of ALS patients, but not healthy aged-matched control, an elevation of inappropriate Fe ligands was detected, which increased iron redox activity and ·OH production [216].
Fe metabolism impairment and consequent dyshomeostasis has been reported in ALS patients and SOD1 transgenic mice models (both SOD1G37R and SOD1G93A). In SODG37R mice, there is differential expression of Fe regulatory proteins (i.e. DMT1, TfR1, ferroportin, ferritin and CP) detected at an advanced stage of the disease (12 months of age). These proteins were highly expressed rostrally in comparison to the caudal (lumbar) region. The net effect is Fe loading and correlated with Fe deposition and neuronal loss in the spinal cord of these mice [217]. Jeong et al. also reported an increase in mitochondrial ferritin (mitFtn) in the ventral horn motor neurons and astrocytes in comparison to age-matched controls [217]. Mitochondria is the main site of ROS production and, therefore, Fe trapped in this organelle by mitFTn will lead to further oxidation damage and ROS. Similarly, SODG93A mice display Fe loading with 180% increase in Fe in the spinal cord in mice at 120 days of age in comparison to controls [218]. These mice also have elevated expression of TfR1 protein, which is the main protein responsible for Fe uptake in neurons. Consequently, SOD1G93A mice have increased uptake of Fe as seen by Fe loading localised to spinal cord. In cell culture, SH-SY5Y neuroblastoma cells stably expressing human wildtype-SOD1 or SOD1 containing the G93A mutation also present increased TfR1, ferritin and DMT1 at the mRNA level. Similar to animal models, an increase in mitFtn1 and 2 was observed in stable cells. Although isolated mitochondria failed to show Fe loading, SOD1G93A expressing cells showed a significant increase in Fe comparison to WT cells [218].
Numerous studies have reported high serum ferritin in ALS patients compared to healthy controls [219,220,221,222,223]. This accords with other studies in ALS patients that report a lowering of serum Tf [223, 224]. Indeed, serum ferritin has been considered as a candidate for biomarker of disease aggravation [221], and high levels of serum ferritin (indicative of high body Fe levels) correlate to poor prognosis [222, 223]. Nadjar et al. conducted a study testing 694 ALS patients and 297 healthy controls and found high ferritin and lower transferrin levels (although Tf had increased saturation) in serum [223]. Tf can exist in an iron-free form (apotransferrin) or bound to two ferric iron atoms (holo or diferric transferrin) and thus high levels of saturated transferrin would correlate with Fe loading. Remarkably, the levels of ferritin correlated to survival time in ALS patients with high expression leading to reduced lifespan by 300 days compared to patients with low serum ferritin [223]. Similar results were observed by MRI, which reported Fe accumulation in the motor cortex [224]. Electron paramagnetic resonance (EPR) spectrometry studies have shown high levels of “inappropriate liganded Fe” in CNS. In other words, there are high levels of bidentate or tridentate ligands (i.e. labile Fe) that possess free Fe coordination sites that increase Fe solubility and promote Fe redox cycling and ROS production [216].
Fe dyshomeostasis may thus contribute significantly to the oxidative damage observed in ALS and is a consequence of changes in the levels of regulatory proteins in favour of Fe loading. Studies suggest that further accumulation of Fe can occur as a consequence of chronic neuroinflammation which occurs in ALS patients and SOD1 transgenic mice models of ALS [218, 225]. Microglia and astrocytes, which express high ferritin, scavenge excess Fe and are consequently protected from oxidation damage. Inflammation modifies the expression of Fe-regulating hormone hepcidin and also ferritin that would in turn downregulate expression of Fe efflux protein Fpn, leading to Fe accumulation. The process of demyelination also promotes accumulation of Fe. In the brain the majority of Fe is stored as non-heme iron in oligodendrocytes and myelin. Fe plays an important role as a cofactor for enzymes important for myelin production. In ALS, oligodendrocytes start to degenerate in the ALS and this precedes the loss of neurons [226].
Therapeutic potentials of targeting brain Cu and Fe levels
Oral administration of CuIIatsm to numerous SOD1 ALS mice models led to improved clinical symptoms and prolonged survival (Table 1). The mode of action of CuIIatsm may be different from that observed in the context of PD, and may involve decreasing the pool of Cu-deficient SOD1 and increasing the pool of holoSOD1 by mediating the in vivo transfer of Cu to SOD1 [205]. Thus, CuIIatsm administration may be associated with a range of therapeutic benefits in a range of neurodegenerative disorders. Specifically, CuIIatsm reduces death of motor neurons, decreases the level of oxidatively modified proteins (including protein nitration and carbonylation), reduces cytosolic accumulation of TDP43 both in vitro and in vivo and attenuates astrocyte and microglial activation. CuIIatsm acts as anti-oxidant by scavenging peroxynitrites [227]. Peroxynitrite (produced by superoxide and nitric oxide) is a highly toxic ROS reported to play a role in ALS pathology. Peroxynitrite can (1) nitrate tyrosine or nitrosate cysteine residues in proteins, (2) inactivate cellular proteins including SOD2 and (3) induce apoptosis in motor neurons. Overall, CuIIatsm is able to prolong survival and slow progression of motor deficits in a variety of transgenic mice models which include SODG93A, SOD1G37R and SODG93A × CCS double transgenic mice [228,229,230,231], the latter of which is a mouse model with pronounced ALS pathology featuring enhanced mitochondrial dysfunction and a severely shortened lifespan (mice die with 30–50 days of birth) [231]. The expression of CCS and mutant SOD1 reduces copper import into mitochondrial and reduces the activity of cytochrome c, a key player of the respiratory chain [232, 233]. It is Cu complexed to ATSM that has therapeutic benefits, as the ATSM moiety alone did not show any efficacy in ALS transgenic mice, emphasizing the role of CuIIatsm in metal delivery [230]. To date, CuIIatsm has shown the most promising therapeutic outcome in ALS. Understanding the mode of action by which CuIIatsm exerts its numerous therapeutic benefits would pave the way to designing new generation therapeutics.
Targeting Fe dyshomeostasis by the use of chelators has shown promising results. Structurally diverse Fe chelators have extended lifespan and reduced neuronal loss. Jeong et al. studied the clinical benefits of the lipholic Fe chelator SIH, which increase lifespan by 5 weeks and reduced the loss of motor neurons [217]. In a later study, SOD1G39A transgenic mice were treated with the Fe chelators VK-28 and M30, which extended life span and reduced spinal cord motor neuron loss. These effects can be attributed to the reduction of elevated iron levels, transferrin receptor expression, decreased ROS and suppressed microglial and astrocytic activation in the spinal cord [218]. Subsequent studies report that M30 and other structurally similar chelators work in part by inducing expression of hypoxia-inducible factor-1α (HIF-α). HIF-α is a transcriptional activator that induces the expression of a variety of genes involved in neuroprotection including erythropoietin (EPO), vascular endothelial growth factor (VEGF), enolase I and inducible nitric oxidase synthase (iNOS) in NSC-34 cells and rat embryonic cortical neurons [234, 235]. Of particular interest is the induction of VEGF at the spinal cord which has been shown to be neuroprotective. ALS mouse models treated with VEGF therapy showed lower motor neuron death and extending survival [236, 237]. Moreover, VEGF gene polymorphisms have been linked to the development of sporadic ALS [238, 239]. The clinical Fe chelator, DFO, also induces VEGF expression in non-ALS models [240].
DFP has undergone a 1-year pilot clinical trial in a small group of (23) ALS patients. Low dose of DFP (30 mg/kg/day) showed a good safety, lowered ALS functioning rating scale and BMI (body mass index) after 3 months of treatment in comparison to the previous 3-month treatment-free period. DFP reduced Fe concentration in cervical spinal cord, medulla oblongata and motor cortex but did not alter Fe levels in other regions outside the motor system [241]. In addition, oxidative markers and neurofilament light chains were reduced in the CSF following treatment [241]. This therapeutic strategy has been coined ‘conservative Fe chelation’. DFP is an ideal conservative Fe chelator as at low doses it can cross membranes, decrease regional Fe accumulation and re-deploy it to transferrin to be distributed throughout the body and by doing so avoiding anaemia [185]. The therapeutic benefits and efficacy of DFP are being further investigated in a double-blind, placebo-controlled, multicentre French study with an estimated enrolment of 210 participants and a 12 month period (NCT03293069).
Fe, ferroptosis and neurodegeneration
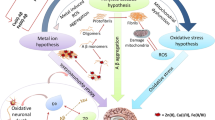
Ferroptosis is a newly identified cell death pathway triggered by the built up of lipid peroxides which is biochemically and morphologically distinct from other cell death processes such as apoptosis, necrosis and autophagy (Fig. 3) [242]. However, it has been associated with neurodegeneration and pathological cell death [243, 244]. Not surprisingly, there is evidence of ferroptosis in AD, PD and ALS, which will be discussed below [244].

Ferroptosis is an iron-dependent cell death resulting from a build-up of lipid peroxides. Glutathione peroxidase 4 (GPX4) is considered a critical regulator of ferroptosis through its ability to mitigate lipid peroxidation. Iron is critical for the execution of ferroptotic death as iron chelation can rescue cells from ferroptosis
Lethal levels of lipid peroxidation lead to ferroptosis which is iron-dependent. Ferroptosis can be prevented by inducing a lipid repair system involving glutathione and GPX4. GPX4 (glutathione peroxidase 4) converts lipid hydroperoxides (L-OOH) to lipid alcohols (L-OH) [245]. Ferroptosis can be suppressed by iron chelators, lipophilic antioxidants, inhibitors of lipid peroxidation and depletion of polyunsaturated fatty acids (PUFAs) which are the substrates of pro-ferroptotic lipid peroxidation products catalysed by ACSL4 (Acyl-CoA Synthetase Long-Chain Family Member 4) [246]. Although the key players that orchestrate ferroptosis have been identified the role of Fe in this process remains to be fully elucidated. Due its redox activity, Fe may directly oxidise lipids via Fenton chemistry [247]. Alternatively, Fe could impact lipid peroxidation via its coordination to iron-dependent oxidases such as lipoxygenase. Lipoxygenase is iron-containing enzyme catalysing dioxygenation of polyunsaturated fatty acids (PUFA) in lipids which can activate ferroptosis [248]. Ferroptosis relies on the bioavailability of intracellular iron via its uptake or through liberation from degradation of ferritin or catabolism of heme. Cells can be made resistant to ferroptosis by manipulating the expression of proteins involved in Fe metabolism including TfR1, ferritin and Tf. Fe chelators can also rescue cells from ferroptosis, emphasizing the role of Fe in ferroptosis [242, 249, 250]. Ferritinophagy, which leads to the release of Fe and an increase in labile Fe, increases sensitivity to ferroptosis [251].
The lack of apoptotic cell death markers in the AD brain suggests that the loss of neurons is due to an alternate cell death process. There is, however, an elevation of Fe in the AD brain and high ferritin CSF is an indicator of cognitive decline in patients. Lipid peroxidation is reported to be an early event in AD pathology [252, 253]. Depletion of glutathione, which is required for GPX4 activity, was evident in the frontal cortex and hippocampus and correlated with cognitive decline [254]. In the AD mouse APP/PS1, administration of deuterated PUFAs (D-PUFA) which can delay ferroptosis not only reduced lipid peroxidation but also reduced amyloid burden [246, 255]. Moreover, a conditional neuronal GPX4 (in forebrain) knockout mouse model exhibited AD-like cognitive impairment, hippocampal neurodegeneration, lipid peroxidation and neuroinflammation [256]. These studies emphasize the role of ferroptosis in AD neurodegeneration. Importantly, inhibitors of ferroptosis have shown protection from neurodegeneration in AD. Thus, preventing/delaying ferroptosis may be of therapeutic benefit in AD.
Ferroptosis markers including lipid peroxidation, elevated Fe and depleted glutathione have been reported as part of PD pathology. In mouse models of PD iron chelators have shown efficiency in alleviating motor symptoms [185, 257]. One mechanism by which iron chelators function is by enhancing GPX activity in the CSF [185]. In addition, the antioxidant and inhibitor of ferroptosis, N-acetylcysteine (NAC), enhances brain glutathione and partially protects from neurodegeneration in PD mouse models. NAC has also been in used in 3-month short-term phase II clinical trial (NCTO2445651) with promising results including a reduced loss of dopaminergic neurons in the caudate and putamen of PD patients, who also had significant improvement in clinical symptoms [258]. Do Van et al. recently reported that the ferroptosis inhibitor, ferrostatin 1, reduced cell loss and improved behavioural impairments in the MPTP-intoxicated mouse model [259].
In the case of ALS, patients demonstrate enhanced lipid peroxidation in serum and CSF as well as decreased glutathione levels in the motor cortex, implicating ferroptosis in neuropathogenesis [260]. A recent study has shown that markers of ferroptosis in ALS are associated with clinical decline [261]. As discussed above, DFP (an Fe chelator and a ferroptosis inhibitor) enhanced the life span of SOD1G86R mouse ALS model and has shown clinical benefits in a small cohort of ALS patients [241].
The role of copper on ferroptosis has not been explored exhaustively. A possible link between copper and ferroptosis is at the level of glutathione regulation. Therefore, copper-dependent glutathione regulation should affect GPX4 activity and thus ferroptosis. However, a recent report indicates that copper can promote cell death by an independent cell death pathway, named cuproptosis [262]. Further studies are required to confirm whether cuproptosis and ferroptosis are truly independent mechanisms of cell death and their role in neurodegenerative diseases.
Concluding remarks
Despite differences in the clinical presentation and the CNS regions affected, AD, PD and ALS have key features in common: oxidative stress and protein misfolding/oligomerization linked to increased neurotoxicity. The normal ageing brain is characterised by changes in metal levels which are exacerbated in neurodegenerative diseases. Thus, it is not surprising that ageing is the main risk factor for AD and PD. Throughout the past decade a large body of research has focused on determining the role of biological metals in neurodegeneration which appears to be multi-factorial. Strong evidence from various research groups demonstrate the integral role that Cu and Fe have in various aspects of neurodegeneration. Most importantly, preclinical and clinical studies have demonstrated the therapeutic potential of metal modifying complexes and chelators in their ability to reduce neurodegeneration and improve clinical symptoms (Table 1).
Abbreviations
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- ALS:
-
Amyotrophic lateral sclerosis
- ATOX-1:
-
Antioxidant 1 copper chaperone 1
- BBB:
-
Blood–brain-barrier
- BCB:
-
Blood–cerebrospinal fluid barrier
- BCECs:
-
Brain capillary endothelial cells
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- CP:
-
Ceruloplasmin
- CQ:
-
Clioquinol
- CTR1:
-
Copper-transporter 1
- CCS:
-
Copper chaperone for SOD1
- COX17:
-
Cytochrome c oxidase copper chaperone
- DMT1:
-
Divalent metal transporter 1
- DFP:
-
Deferiprone
- GSH:
-
Glutathione
- 6-OHDA:
-
6-Hydroxydopamine
- IRE:
-
Iron-responsive element
- IRPs:
-
Iron regulatory proteins
- LB:
-
Lewy bodies
- L-DOPA:
-
l-3,4-Dihydroxyphenylalanine
- MRI:
-
Magnetic resonance image
- NFTs:
-
Neurofibrillary tangles
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- SNc:
-
Substantia nigra pars compacta
- SOD1:
-
Superoxide dismutase 1
- Tf:
-
Transferrin
- TfR1:
-
Transferrin receptor 1
- UTR:
-
Untranslated region
References
Halliwell B (2006) J Neurochem 97:1634–1658
Butterfield DA, Halliwell B (2019) Nat Rev Neurosci 20:148–160
Bush AI (2003) Trends Neurosci 26:207–214
Gaier ED, Eipper BA, Mains RE (2013) J Neurosci Res 91:2–19
Belaidi AA, Bush AI (2016) J Neurochem 139(Suppl 1):179–197
Opazo CM, Greenough MA, Bush AI (2014) Front Aging Neurosci 6:143
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA (2017) Cell 171:273–285
Angelova DF, Brown DR (2015) Metals 5:2070–2092
Choi BS, Zheng W (2009) Brain Res 1248:14–21
Meyer LA, Durley AP, Prohaska JR, Harris ZL (2001) J Biol Chem 276:36857–36861
Maryon EB, Molloy SA, Kaplan JH (2013) Am J Physiol Cell Physiol 304:C768–C779
Tapia L, Gonzalez-Aguero M, Cisternas MF, Suazo M, Cambiazo V, Uauy R, Gonzalez M (2004) Biochem J 378:617–624
La Fontaine S, Mercer JF (2007) Arch Biochem Biophys 463:149–167
Linz R, Lutsenko S (2007) J Bioenerg Biomembr 39:403–407
Greenough M, Pase L, Voskoboinik I, Petris MJ, O’Brien AW, Camakaris J (2004) Am J Physiol Cell Physiol 287:C1463–C1471
Pase L, Voskoboinik I, Greenough M, Camakaris J (2004) Biochem J 378:1031–1037
Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD (2005) Nat Genet 37:1264–1269
Moos T, Morgan EH (2004) J Neurochem 88:233–245
Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L (2014) Lancet Neurol 13:1045–1060
Pinilla-Tenas JJ, Sparkman BK, Shawki A, Illing AC, Mitchell CJ, Zhao N, Liuzzi JP, Cousins RJ, Knutson MD, Mackenzie B (2011) Am J Physiol Cell Physiol 301:C862–C871
Wang CY, Jenkitkasemwong S, Duarte S, Sparkman BK, Shawki A, Mackenzie B, Knutson MD (2012) J Biol Chem 287:34032–34043
Zhao N, Gao J, Enns CA, Knutson MD (2010) J Biol Chem 285:32141–32150
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA (1997) Nature 388:482–488
Ji C, Kosman DJ (2015) J Neurochem 133:668–683
Frey AG, Nandal A, Park JH, Smith PM, Yabe T, Ryu MS, Ghosh MC, Lee J, Rouault TA, Park MH, Philpott CC (2014) Proc Natl Acad Sci USA 111:8031–8036
Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, Subramanian P, Hsu E, Natarajan N, Nandal A, Stemmler TL, Philpott CC (2013) J Biol Chem 288:17791–17802
Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, Philpott CC (2011) Cell Metab 14:647–657
Pantopoulos K (2004) Ann N Y Acad Sci 1012:1–13
Zhang DL, Ghosh MC, Rouault TA (2014) Front Pharmacol 5:124
Double KL, Gerlach M, Schunemann V, Trautwein AX, Zecca L, Gallorini M, Youdim MB, Riederer P, Ben-Shachar D (2003) Biochem Pharmacol 66:489–494
Asano T, Komatsu M, Yamaguchi-Iwai Y, Ishikawa F, Mizushima N, Iwai K (2011) Mol Cell Biol 31:2040–2052
Nitti M, Piras S, Brondolo L, Marinari UM, Pronzato MA, Furfaro AL (2018) Int J Mol Sci 19:2260–2280
Zhang YW, Thompson R, Zhang H, Xu H (2011) Mol Brain 4:3
Unzeta M, Esteban G, Bolea I, Fogel WA, Ramsay RR, Youdim MB, Tipton KF, Marco-Contelles J (2016) Front Neurosci 10:205
Wimo A, Jonsson L, Bond J, Prince M, Winblad B, I. Alzheimer Disease (2013) Alzheimers Dement 9:1–11
Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR (2000) Brain Res Brain Res Rev 33:95–130
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Proc Natl Acad Sci USA 82:4245–4249
Davies P, Maloney AJ (1976) Lancet 2:1403
Drachman DA, Leavitt J (1974) Arch Neurol 30:113–121
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR (1982) Science 215:1237–1239
Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR (1998) J Neurol Sci 158:47–52
Pithadia AS, Lim MH (2012) Curr Opin Chem Biol 16:67–73
Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G (2013) J Alzheimers Dis 37:127–136
Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR (2003) Biochemistry 42:2768–2773
Miller LM, Wang Q, Telivala TP, Smith RJ, Lanzirotti A, Miklossy J (2006) J Struct Biol 155:30–37
Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA (2000) J Neurochem 74:270–279
Smith MA, Harris PL, Sayre LM, Perry G (1997) Proc Natl Acad Sci USA 94:9866–9868
Deibel MA, Ehmann WD, Markesbery WR (1996) J Neurol Sci 143:137–142
Zatta P, Drago D, Bolognin S, Sensi SL (2009) Trends Pharmacol Sci 30:346–355
Bishop GM, Robinson SR, Liu Q, Perry G, Atwood CS, Smith MA (2002) Dev Neurosci 24:184–187
Roberts BR, Ryan TM, Bush AI, Masters CL, Duce JA (2012) J Neurochem 120(Suppl 1):149–166
Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM (2011) Prog Neurobiol 94:296–306
Schrag M, Mueller C, Zabel M, Crofton A, Kirsch WM, Ghribi O, Squitti R, Perry G (2013) Neurobiol Dis 59:100–110
Ayton S, James SA, Bush AI (2017) Cell Chem Biol 24:1192–1194
Hwang EJ, Kim HG, Kim D, Rhee HY, Ryu CW, Liu T, Wang Y, Jahng GH (2016) Med Phys 43:4718
Luo Z, Zhuang X, Kumar D, Wu X, Yue C, Han C, Lv J (2013) PLoS One 8:e76203
Connor JR, Menzies SL, St Martin SM, Mufson EJ (1992) J Neurosci Res 31:75–83
Meadowcroft MD, Connor JR, Smith MB, Yang QX (2009) J Magn Reson Imaging 29:997–1007
Valensin D, Migliorini C, Valensin G, Gaggelli E, La Penna G, Kozlowski H, Gabbiani C, Messori L (2011) Inorg Chem 50:6865–6867
Dikalov SI, Vitek MP, Mason RP (2004) Free Radic Biol Med 36:340–347
Guilloreau L, Combalbert S, Sournia-Saquet A, Mazarguil H, Faller P (2007) ChemBioChem 8:1317–1325
Nadal RC, Rigby SE, Viles JH (2008) Biochemistry 47:11653–11664
Nakamura M, Shishido N, Nunomura A, Smith MA, Perry G, Hayashi Y, Nakayama K, Hayashi T (2007) Biochemistry 46:12737–12743
Reybier K, Ayala S, Alies B, Rodrigues JV, Bustos Rodriguez S, La Penna G, Collin F, Gomes CM, Hureau C, Faller P (2016) Angew Chem Int Ed Engl 55:1085–1089
Taniguchi A, Sasaki D, Shiohara A, Iwatsubo T, Tomita T, Sohma Y, Kanai M (2014) Angew Chem Int Ed Engl 53:1382–1385
Cheignon C, Faller P, Testemale D, Hureau C, Collin F (2016) Metallomics 8:1081–1089
Butterfield DA, Reed T, Newman SF, Sultana R (2007) Free Radic Biol Med 43:658–677
Selfridge JE, Lezi E, Lu J, Swerdlow RH (2013) Neurobiol Dis 51:3–12
Zhao Y, Zhao B (2013) Oxid Med Cell Longev 2013:316523
Gabbita SP, Lovell MA, Markesbery WR (1998) J Neurochem 71:2034–2040
Hardas SS, Sultana R, Clark AM, Beckett TL, Szweda LI, Murphy MP, Butterfield DA (2013) Redox Biol 1:80–85
Markesbery WR, Lovell MA (1998) Neurobiol Aging 19:33–36
Mecocci P, MacGarvey U, Beal MF (1994) Ann Neurol 36:747–751
Sayre LM, Zelasko DA, Harris PL, Perry G, Salomon RG, Smith MA (1997) J Neurochem 68:2092–2097
Atwood CS, Scarpa RC, Huang X, Moir RD, Jones WD, Fairlie DP, Tanzi RE, Bush AI (2000) J Neurochem 75:1219–1233
Barritt JD, Viles JH (2015) J Biol Chem 290:27791–27802
Drew SC (2017) Front Neurosci 11:317
Mital M, Wezynfeld NE, Fraczyk T, Wiloch MZ, Wawrzyniak UE, Bonna A, Tumpach C, Barnham KJ, Haigh CL, Bal W, Drew SC (2015) Angew Chem Int Ed Engl 54:10460–10464
Sarell CJ, Syme CD, Rigby SE, Viles JH (2009) Biochemistry 48:4388–4402
Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI (2004) J Biol Inorg Chem 9:954–960
Dai XL, Sun YX, Jiang ZF (2006) Acta Biochim Biophys Sin (Shanghai) 38:765–772
Sarell CJ, Wilkinson SR, Viles JH (2010) J Biol Chem 285:41533–41540
Greenough MA, Camakaris J, Bush AI (2013) Neurochem Int 62:540–555
Grundke-Iqbal I, Fleming J, Tung YC, Lassmann H, Iqbal K, Joshi JG (1990) Acta Neuropathol 81:105–110
Quintana C, Bellefqih S, Laval JY, Guerquin-Kern JL, Wu TD, Avila J, Ferrer I, Arranz R, Patino C (2006) J Struct Biol 153:42–54
Bolognin S, Messori L, Drago D, Gabbiani C, Cendron L, Zatta P (2011) Int J Biochem Cell Biol 43:877–885
Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, Hider R, Marciniak SJ, Lomas DA, Crowther DC (2011) J Biol Chem 286:4248–4256
Ma Q, Li Y, Du J, Liu H, Kanazawa K, Nemoto T, Nakanishi H, Zhao Y (2006) Peptides 27:841–849
Ma QF, Li YM, Du JT, Kanazawa K, Nemoto T, Nakanishi H, Zhao YF (2005) Biopolymers 79:74–85
Martic S, Rains MK, Kraatz HB (2013) Anal Biochem 442:130–137
Soragni A, Zambelli B, Mukrasch MD, Biernat J, Jeganathan S, Griesinger C, Ciurli S, Mandelkow E, Zweckstetter M (2008) Biochemistry 47:10841–10851
Kitazawa M, Cheng D, Laferla FM (2009) J Neurochem 108:1550–1560
Voss K, Harris C, Ralle M, Duffy M, Murchison C, Quinn JF (2014) Transl Neurodegener 3:24
Faller P, Hureau C (2009) Dalton Trans. https://doi.org/10.1039/b813398k:1080-1094
Hesse L, Beher D, Masters CL, Multhaup G (1994) FEBS Lett 349:109–116
Bush AI, Multhaup G, Moir RD, Williamson TG, Small DH, Rumble B, Pollwein P, Beyreuther K, Masters CL (1993) J Biol Chem 268:16109–16112
Miller Y, Ma B, Nussinov R (2010) Proc Natl Acad Sci USA 107:9490–9495
Treiber C, Simons A, Strauss M, Hafner M, Cappai R, Bayer TA, Multhaup G (2004) J Biol Chem 279:51958–51964
Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li QX (2002) J Biol Chem 277:44670–44676
Bellingham SA, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, Cappai R, Camakaris J (2004) J Neurochem 91:423–428
Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI (2010) Cell 142:857–867
Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA (2014) PLoS One 9:e114174
Cho HH, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT (2010) J Biol Chem 285:31217–31232
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR (2002) J Biol Chem 277:45518–45528
Acevedo KM, Hung YH, Dalziel AH, Li QX, Laughton K, Wikhe K, Rembach A, Roberts B, Masters CL, Bush AI, Camakaris J (2011) J Biol Chem 286:8252–8262
Hou P, Liu G, Zhao Y, Shi Z, Zheng Q, Bu G, Xu H, Zhang YW (2015) Neurobiol Aging 36:1310–1315
Noda Y, Asada M, Kubota M, Maesako M, Watanabe K, Uemura M, Kihara T, Shimohama S, Takahashi R, Kinoshita A, Uemura K (2013) Neurosci Lett 547:10–15
Gerber H, Wu F, Dimitrov M, Osuna GM, Fraering PC (2017) J Biol Chem 292:3751–3767
Borchardt T, Camakaris J, Cappai R, Masters CL, Beyreuther K, Multhaup G (1999) Biochem J 344(Pt 2):461–467
Bayer TA, Schafer S, Simons A, Kemmling A, Kamer T, Tepest R, Eckert A, Schussel K, Eikenberg O, Sturchler-Pierrat C, Abramowski D, Staufenbiel M, Multhaup G (2003) Proc Natl Acad Sci USA 100:14187–14192
Hardy JA, Higgins GA (1992) Science 256:184–185
Masaldan S, Belaidi AA, Ayton S, Bush AI (2019) Pharmaceuticals (Basel) 12:93
Nikseresht S, Bush AI, Ayton S (2019) Br J Pharmacol 176:3622–3635
Li C, Wang J, Zhou B (2010) J Alzheimers Dis 21:1249–1262
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI (2001) Neuron 30:665–676
Grossi C, Francese S, Casini A, Rosi MC, Luccarini I, Fiorentini A, Gabbiani C, Messori L, Moneti G, Casamenti F (2009) J Alzheimers Dis 17:423–440
LeVine H 3rd, Ding Q, Walker JA, Voss RS, Augelli-Szafran CE (2009) Neurosci Lett 465:99–103
Mancino AM, Hindo SS, Kochi A, Lim MH (2009) Inorg Chem 48:9596–9598
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL (2003) Arch Neurol 60:1685–1691
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI (2008) Neuron 59:43–55
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RE, Cummings JL, Herd CM, Bush AI (2010) J Alzheimers Dis 20:509–516
Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW, PES Group (2008) Lancet Neurol 7:779–786
Villemagne VL, Rowe CC, Barnham KJ, Cherny R, Woodward M, Bozinosvski S, Salvado O, Bourgeat P, Perez K, Fowler C, Rembach A, Maruff P, Ritchie C, Tanzi R, Masters CL (2017) Alzheimers Dement (N Y) 3:622–635
Bica L, Liddell JR, Donnelly PS, Duncan C, Caragounis A, Volitakis I, Paterson BM, Cappai R, Grubman A, Camakaris J, Crouch PJ, White AR (2014) PLoS One 9:e90070
Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, Perez KA, Nurjono M, Caragounis A, Du T, Laughton K, Volitakis I, Bush AI, Li QX, Masters CL, Cappai R, Cherny RA, Donnelly PS, White AR, Barnham KJ (2009) Proc Natl Acad Sci USA 106:381–386
Donnelly PS, Caragounis A, Du T, Laughton KM, Volitakis I, Cherny RA, Sharples RA, Hill AF, Li QX, Masters CL, Barnham KJ, White AR (2008) J Biol Chem 283:4568–4577
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF (1991) Lancet 337:1304–1308
Hanson LR, Fine JM, Renner DB, Svitak AL, Burns RB, Nguyen TM, Tuttle NJ, Marti DL, Panter SS, Frey WH 2nd (2012) Drug Deliv Transl Res 2:160–168
Guo C, Wang P, Zhong ML, Wang T, Huang XS, Li JY, Wang ZY (2013) Neurochem Int 62:165–172
Banerjee P, Sahoo A, Anand S, Bir A, Chakrabarti S (2016) J Alzheimers Dis 49:681–693
Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Weinreb O (2012) Antioxid Redox Signal 17:860–877
Molina-Holgado F, Gaeta A, Francis PT, Williams RJ, Hider RC (2008) J Neurochem 105:2466–2476
Prasanthi JR, Schrag M, Dasari B, Marwarha G, Dickson A, Kirsch WM, Ghribi O (2012) J Alzheimers Dis 30:167–182
Lang AE, Lozano AM (1998) N Engl J Med 339:1130–1143
Jankovic J (2008) J Neurol Neurosurg Psychiatry 79:368–376
Schulz JB (2008) J Neurol 255(Suppl 5):3–7
Hozumi I, Hasegawa T, Honda A, Ozawa K, Hayashi Y, Hashimoto K, Yamada M, Koumura A, Sakurai T, Kimura A, Tanaka Y, Satoh M, Inuzuka T (2011) J Neurol Sci 303:95–99
Pall HS, Williams AC, Blake DR, Lunec J, Gutteridge JM, Hall M, Taylor A (1987) Lancet 2:238–241
Arnal N, Cristalli DO, de Alaniz MJ, Marra CA (2010) Brain Res 1319:118–130
Davies KM, Bohic S, Carmona A, Ortega R, Cottam V, Hare DJ, Finberg JP, Reyes S, Halliday GM, Mercer JF, Double KL (2014) Neurobiol Aging 35:858–866
Torsdottir G, Kristinsson J, Sveinbjornsdottir S, Snaedal J, Johannesson T (1999) Pharmacol Toxicol 85:239–243
Davies KM, Mercer JF, Chen N, Double KL (2016) Clin Sci (Lond) 130:565–574
Montes S, Rivera-Mancia S, Diaz-Ruiz A, Tristan-Lopez L, Rios C (2014) Oxid Med Cell Longev 2014:147251
Howells C, West AK, Chung RS (2010) FEBS J 277:2931–2939
Dexter DT, Wells FR, Agid F, Agid Y, Lees AJ, Jenner P, Marsden CD (1987) Lancet 2:1219–1220
Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD (1989) J Neurochem 52:1830–1836
Hirsch EC, Brandel JP, Galle P, Javoy-Agid F, Agid Y (1991) J Neurochem 56:446–451
Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MB (1989) J Neurochem 52:515–520
Double KL, Reyes S, Werry EL, Halliday GM (2010) Prog Neurobiol 92:316–329
Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG (2009) Mol Neurodegener 4:47
Gerlach M, Double KL, Youdim MB, Riederer P (2006) J Neural Transm Suppl 70:133–142
Mills E, Dong XP, Wang F, Xu H (2010) Future Med Chem 2:51–64
Ward RJ, Colivicchi MA, Allen R, Schol F, Lallemand F, de Witte P, Ballini C, Corte LD, Dexter D (2009) J Neurochem 111:1119–1128
Dexter DT, Carayon A, Javoy-Agid F, Agid Y, Wells FR, Daniel SE, Lees AJ, Jenner P, Marsden CD (1991) Brain 114(Pt 4):1953–1975
Conway KA, Harper JD, Lansbury PT (1998) Nat Med 4:1318–1320
Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Science 276:2045–2047
Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuron 34:521–533
Yu Z, Xu X, Xiang Z, Zhou J, Zhang Z, Hu C, He C (2010) PLoS One 5:e9956
Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB, Gollomp SM, Ischiropoulos H, Lee VM, Trojanowski JQ (2000) Am J Pathol 157:1439–1445
Abeyawardhane DL, Heitger DR, Fernandez RD, Forney AK, Lucas HR (2019) ACS Chem Neurosci 10:1402–1410
Paik SR, Shin HJ, Lee JH, Chang CS, Kim J (1999) Biochem J 340(Pt 3):821–828
Uversky VN, Li J, Fink AL (2001) J Biol Chem 276:44284–44296
Rasia RM, Bertoncini CW, Marsh D, Hoyer W, Cherny D, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO (2005) Proc Natl Acad Sci USA 102:4294–4299
Mason RJ, Paskins AR, Dalton CF, Smith DP (2016) Biochemistry 55:4737–4741
Lu Y, Prudent M, Fauvet B, Lashuel HA, Girault HH (2011) ACS Chem Neurosci 2:667–675
Wang C, Liu L, Zhang L, Peng Y, Zhou F (2010) Biochemistry 49:8134–8142
Abeyawardhane DL, Fernandez RD, Heitger DR, Crozier MK, Wolver JC, Lucas HR (2018) J Am Chem Soc 140:17086–17094
Peng Y, Wang C, Xu HH, Liu YN, Zhou F (2010) J Inorg Biochem 104:365–370
Chen B, Wen X, Jiang H, Wang J, Song N, Xie J (2019) Free Radic Biol Med 141:253–260
Zecca L, Stroppolo A, Gatti A, Tampellini D, Toscani M, Gallorini M, Giaveri G, Arosio P, Santambrogio P, Fariello RG, Karatekin E, Kleinman MH, Turro N, Hornykiewicz O, Zucca FA (2004) Proc Natl Acad Sci USA 101:9843–9848
Cobley JN, Fiorello ML, Bailey DM (2018) Redox Biol 15:490–503
Munoz P, Cardenas S, Huenchuguala S, Briceno A, Couve E, Paris I, Segura-Aguilar J (2015) Toxicol Sci 145:37–47
Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L (2017) Prog Neurobiol 155:96–119
Maker HS, Weiss C, Silides DJ, Cohen G (1981) J Neurochem 36:589–593
Wada K, Fujibayashi Y, Tajima N, Yokoyama A (1994) Biol Pharm Bull 17:701–704
Fodero-Tavoletti MT, Villemagne VL, Paterson BM, White AR, Li QX, Camakaris J, O’Keefe G, Cappai R, Barnham KJ, Donnelly PS (2010) J Alzheimers Dis 20:49–55
Dearling JL, Packard AB (2010) Nucl Med Biol 37:237–243
Fujibayashi Y, Taniuchi H, Yonekura Y, Ohtani H, Konishi J, Yokoyama A (1997) J Nucl Med 38:1155–1160
Vavere AL, Lewis JS (2007) Dalton Trans. https://doi.org/10.1039/b705989b:4893-4902
Ikawa M, Okazawa H, Tsujikawa T, Matsunaga A, Yamamura O, Mori T, Hamano T, Kiyono Y, Nakamoto Y, Yoneda M (2015) Neurology 84:2033–2039
Hung LW, Villemagne VL, Cheng L, Sherratt NA, Ayton S, White AR, Crouch PJ, Lim S, Leong SL, Wilkins S, George J, Roberts BR, Pham CL, Liu X, Chiu FC, Shackleford DM, Powell AK, Masters CL, Bush AI, O’Keefe G, Culvenor JG, Cappai R, Cherny RA, Donnelly PS, Hill AF, Finkelstein DI, Barnham KJ (2012) J Exp Med 209:837–854
Dexter DT, Statton SA, Whitmore C, Freinbichler W, Weinberger P, Tipton KF, Della Corte L, Ward RJ, Crichton RR (2011) J Neural Transm (Vienna) 118:223–231
Cohen AR, Galanello R, Piga A, De Sanctis V, Tricta F (2003) Blood 102:1583–1587
Galanello R, Campus S (2009) Acta Haematol 122:155–164
Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, Jonneaux A, Ryckewaert G, Garcon G, Rouaix N, Duhamel A, Jissendi P, Dujardin K, Auger F, Ravasi L, Hopes L, Grolez G, Firdaus W, Sablonniere B, Strubi-Vuillaume I, Zahr N, Destee A, Corvol JC, Poltl D, Leist M, Rose C, Defebvre L, Marchetti P, Cabantchik ZI, Bordet R (2014) Antioxid Redox Signal 21:195–210
Glickstein H, El RB, Shvartsman M, Cabantchik ZI (2005) Blood 106:3242–3250
Sohn YS, Mitterstiller AM, Breuer W, Weiss G, Cabantchik ZI (2011) Br J Pharmacol 164:406–418
Martin-Bastida A, Lao-Kaim NP, Loane C, Politis M, Roussakis AA, Valle-Guzman N, Kefalopoulou Z, Paul-Visse G, Widner H, Xing Y, Schwarz ST, Auer DP, Foltynie T, Barker RA, Piccini P (2017) Eur J Neurol 24:357–365
Saberi S, Stauffer JE, Schulte DJ, Ravits J (2015) Neurol Clin 33:855–876
Pratt AJ, Getzoff ED, Perry JJ (2012) Degener Neurol Neuromuscul Dis 2:1–14
Bensimon G, Lacomblez L, Meininger V (1994) N Engl J Med 330:585–591
Sreedharan J, Brown RH Jr (2013) Ann Neurol 74:309–316
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) Biochem Biophys Res Commun 351:602–611
Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK (2008) Neurobiol Dis 30:400–407
Barber SC, Shaw PJ (2010) Free Radic Biol Med 48:629–641
Rothstein JD (2009) Ann Neurol 65(Suppl 1):S3–S9
Banci L, Bertini I, Boca M, Girotto S, Martinelli M, Valentine JS, Vieru M (2008) PLoS One 3:e1677
Culotta VC, Yang M, O’Halloran TV (2006) Biochim Biophys Acta 1763:747–758
Valentine JS, Doucette PA, Zittin Potter S (2005) Annu Rev Biochem 74:563–593
Beckman JS, Carson M, Smith CD, Koppenol WH (1993) Nature 364:584
Leal SS, Cristovao JS, Biesemeier A, Cardoso I, Gomes CM (2015) Metallomics 7:333–346
Tokuda E, Okawa E, Ono S (2009) J Neurochem 111:181–191
Tokuda E, Okawa E, Watanabe S, Ono S, Marklund SL (2013) Neurobiol Dis 54:308–319
Gong YH, Elliott JL (2000) Exp Neurol 162:27–36
Roberts BR, Lim NK, McAllum EJ, Donnelly PS, Hare DJ, Doble PA, Turner BJ, Price KA, Lim SC, Paterson BM, Hickey JL, Rhoads TW, Williams JR, Kanninen KM, Hung LW, Liddell JR, Grubman A, Monty JF, Llanos RM, Kramer DR, Mercer JF, Bush AI, Masters CL, Duce JA, Li QX, Beckman JS, Barnham KJ, White AR, Crouch PJ (2014) J Neurosci 34:8021–8031
Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH Jr, Beal MF (1997) J Neurochem 69:2064–2074
Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M, Bellmunt MJ, Ferrer I, Pamplona R, Portero-Otin M (2007) Brain 130:3111–3123
Morimoto S, Kuzuhara S, Kokubo Y (2009) Mov Disord 24:123–126
Nakagomi S, Barsoum MJ, Bossy-Wetzel E, Sutterlin C, Malhotra V, Lipton SA (2008) Neurobiol Dis 29:221–231
Smith RG, Henry YK, Mattson MP, Appel SH (1998) Ann Neurol 44:696–699
Cheung G, Gawel MJ, Cooper PW, Farb RI, Ang LC, Gawal MJ (1995) Radiology 194:263–270
Hecht MJ, Fellner F, Fellner C, Hilz MJ, Neundorfer B, Heuss D (2002) J Neurol Sci 199:59–65
Santillo AF, Skoglund L, Lindau M, Eeg-Olofsson KE, Tovi M, Engler H, Brundin RM, Ingvast S, Lannfelt L, Glaser A, Kilander L (2009) Alzheimer Dis Assoc Disord 23:298–300
Yasui M, Ota K, Garruto RM (1993) Neurotoxicology 14:445–450
Kwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, Butman JA, Chen L, Bayat E, Russell J, Siddique T, Duyn JH, Rouault TA, Floeter MK (2012) PLoS One 7:e35241
Ignjatovic A, Stevic Z, Lavrnic D, Nikolic-Kokic A, Blagojevic D, Spasic M, Spasojevic I (2012) Amyotroph Lateral Scler 13:357–362
Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S (2009) J Neurosci 29:610–619
Wang Q, Zhang X, Chen S, Zhang X, Zhang S, Youdium M, Le W (2011) Neurodegener Dis 8:310–321
Goodall EF, Haque MS, Morrison KE (2008) J Neurol 255:1652–1656
Qureshi M, Brown RH Jr, Rogers JT, Cudkowicz ME (2008) Open Neurol J 2:51–54
Yu J, Wang N, Qi F, Wang X, Zhu Q, Lu Y, Zhang H, Che F, Li W (2018) Biomed Rep 9:333–338
Ikeda K, Hirayama T, Takazawa T, Kawabe K, Iwasaki Y (2012) Intern Med 51:1501–1508
Nadjar Y, Gordon P, Corcia P, Bensimon G, Pieroni L, Meininger V, Salachas F (2012) PLoS One 7:e45034
Mitchell RM, Simmons Z, Beard JL, Stephens HE, Connor JR (2010) Muscle Nerve 42:95–103
McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG (1993) Glia 7:84–92
Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE (2013) Nat Neurosci 16:571–579
Soon CP, Donnelly PS, Turner BJ, Hung LW, Crouch PJ, Sherratt NA, Tan JL, Lim NK, Lam L, Bica L, Lim S, Hickey JL, Morizzi J, Powell A, Finkelstein DI, Culvenor JG, Masters CL, Duce J, White AR, Barnham KJ, Li QX (2011) J Biol Chem 286:44035–44044
Hilton JB, Mercer SW, Lim NK, Faux NG, Buncic G, Beckman JS, Roberts BR, Donnelly PS, White AR, Crouch PJ (2017) Sci Rep 7:42292
McAllum EJ, Roberts BR, Hickey JL, Dang TN, Grubman A, Donnelly PS, Liddell JR, White AR, Crouch PJ (2015) Neurobiol Dis 81:20–24
Vieira FG, Hatzipetros T, Thompson K, Moreno AJ, Kidd JD, Tassinari VR, Levine B, Perrin S, Gill A (2017) IBRO Rep 2:47–53
Williams JR, Trias E, Beilby PR, Lopez NI, Labut EM, Bradford CS, Roberts BR, McAllum EJ, Crouch PJ, Rhoads TW, Pereira C, Son M, Elliott JL, Franco MC, Estevez AG, Barbeito L, Beckman JS (2016) Neurobiol Dis 89:1–9
Son M, Elliott JL (2014) J Neurol Sci 336:1–7
Son M, Leary SC, Romain N, Pierrel F, Winge DR, Haller RG, Elliott JL (2008) J Biol Chem 283:12267–12275
Avramovich-Tirosh Y, Bar-Am O, Amit T, Youdim MB, Weinreb O (2010) Curr Alzheimer Res 7:300–306
Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB (2009) FASEB J 23:3766–3779
Storkebaum E, Lambrechts D, Dewerchin M, Moreno-Murciano MP, Appelmans S, Oh H, Van Damme P, Rutten B, Man WY, De Mol M, Wyns S, Manka D, Vermeulen K, Van Den Bosch L, Mertens N, Schmitz C, Robberecht W, Conway EM, Collen D, Moons L, Carmeliet P (2005) Nat Neurosci 8:85–92
Wang Y, Mao XO, Xie L, Banwait S, Marti HH, Greenberg DA, Jin K (2007) J Neurosci 27:304–307
Lysogorskaia EV, Abramycheva N, Zakharova MN, Illarioshkin SN (2012) Zh Nevrol Psikhiatr Im S S Korsakova 112:42–45
Stalmans I, Lambrechts D, De Smet F, Jansen S, Wang J, Maity S, Kneer P, von der Ohe M, Swillen A, Maes C, Gewillig M, Molin DG, Hellings P, Boetel T, Haardt M, Compernolle V, Dewerchin M, Plaisance S, Vlietinck R, Emanuel B, de Gittenberger Groot AC, Scambler P, Morrow B, Driscol DA, Moons L, Esguerra CV, Carmeliet G, Behn-Krappa A, Devriendt K, Collen D, Conway SJ, Carmeliet P (2003) Nat Med 9:173–182
Beerepoot LV, Shima DT, Kuroki M, Yeo KT, Voest EE (1996) Cancer Res 56:3747–3751
Moreau C, Danel V, Devedjian JC, Grolez G, Timmerman K, Laloux C, Petrault M, Gouel F, Jonneaux A, Dutheil M, Lachaud C, Lopes R, Kuchcinski G, Auger F, Kyheng M, Duhamel A, Perez T, Pradat PF, Blasco H, Veyrat-Durebex C, Corcia P, Oeckl P, Otto M, Dupuis L, Garcon G, Defebvre L, Cabantchik ZI, Duce J, Bordet R, Devos D (2018) Antioxid Redox Signal 29:742–748
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR (2012) Cell 149:1060–1072
Conrad M, Angeli JP, Vandenabeele P, Stockwell BR (2016) Nat Rev Drug Discov 15:348–366
Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C (2019) Free Radic Biol Med 133:221–233
Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C (1982) Biochim Biophys Acta 710:197–211
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR (2016) Proc Natl Acad Sci USA 113:E4966–E4975
Shah R, Shchepinov MS, Pratt DA (2018) ACS Cent Sci 4:387–396
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao G, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M (2017) Nat Chem Biol 13:91–98
Reed JC, Pellecchia M (2012) Cell 149:963–965
Yang WS, Stockwell BR (2008) Chem Biol 15:234–245
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC (2014) Nature 509:105–109
Pratico D, Sung S (2004) J Alzheimers Dis 6:171–175
Reed TT, Pierce WM, Markesbery WR, Butterfield DA (2009) Brain Res 1274:66–76
Mandal PK, Saharan S, Tripathi M, Murari G (2015) Biol Psychiatry 78:702–710
Raefsky SM, Furman R, Milne G, Pollock E, Axelsen P, Mattson MP, Shchepinov MS (2018) Neurobiol Aging 66:165–176
Hambright WS, Fonseca RS, Chen L, Na R, Ran Q (2017) Redox Biol 12:8–17
Weinreb O, Mandel S, Youdim MBH, Amit T (2013) Free Radic Biol Med 62:52–64
Monti DA, Zabrecky G, Kremens D, Liang TW, Wintering NA, Cai J, Wei X, Bazzan AJ, Zhong L, Bowen B, Intenzo CM, Iacovitti L, Newberg AB (2016) PLoS One 11:e0157602
Van Do B, Gouel F, Jonneaux A, Timmerman K, Gele P, Petrault M, Bastide M, Laloux C, Moreau C, Bordet R, Devos D, Devedjian JC (2016) Neurobiol Dis 94:169–178
Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH (2004) Neurology 62:1758–1765
Devos D, Moreau C, Kyheng M, Garcon G, Rolland AS, Blasco H, Gele P, Timothee Lenglet T, Veyrat-Durebex C, Corcia P, Dutheil M, Bede P, Jeromin A, Oeckl P, Otto M, Meninger V, Danel-Brunaud V, Devedjian JC, Duce JA, Pradat PF (2019) Sci Rep 9:2918
Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W, Thiru P, Reidy M, Kugener G, Rossen J, Kocak M, Kory N, Tsherniak A, Santagata S, Whitesell L, Ghobrial IM, Markley JL, Lindquist S, Golub TR (2019) Nat Chem Biol 15:681–689
Acknowledgements
The Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. This work was supported by the Australian Research Council (AIB, CO), the National Health and Medical Research Council and the CRC for Mental Health (AIB). Figures 1 and 2 were compiled using images from Servier Medical art.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
Prof. Bush is a shareholder in Prana Biotechnology Ltd, Cogstate Ltd, Brighton Biotech LLC, Grunbiotics Pty Ltd, Eucalyptus Pty Ltd, and Mesoblast Ltd. He is a paid consultant for, and has a profit share interest in, Collaborative Medicinal Development Pty Ltd.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Acevedo, K., Masaldan, S., Opazo, C.M. et al. Redox active metals in neurodegenerative diseases. J Biol Inorg Chem 24, 1141–1157 (2019). https://doi.org/10.1007/s00775-019-01731-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-019-01731-9