
Abstract
The endogenous cannabinoid system is an important regulatory system involved in physiological homeostasis. Endocannabinoid signaling is known to modulate neural development, immune function, metabolism, synaptic plasticity, and emotional state. Accumulating evidence also implicates brain endocannabinoid signaling in the processing of natural and drug-induced reward states and dysregulated endocannabinoid signaling in the etiology of aberrant reward function and drug addiction. In this chapter, we discuss the influence of endocannabinoid signaling on the rewarding and motivational effects of natural rewards such as food, sex, and social interaction, as well as evidence demonstrating an endocannabinoid influence in the rewarding effects of abused drugs. The effects of long-term drug consumption on endocannabinoid signaling are discussed, along with evidence that the resultant dysregulation of endocannabinoid function contributes to various aspects of drug dependence and addiction including physical symptoms of drug withdrawal, increased stress responsivity, negative affective states, dysregulated synaptic plasticity, dysregulated extinction of drug-related memories, relapse to drug taking, and impaired cognitive function. Lastly, consideration is given to the role for dysregulated endocannabinoid signaling in pathological food reward and eating disorders.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Present knowledge of the endogenous cannabinoid system (ECS) derives from decades of research demonstrating that the effects of Cannabis are mediated by cannabinoid receptors in the brain and the subsequent identification and characterization of the endogenous ligands for these receptors. Although our understanding of the nature and breadth of endocannabinoid (eCB) influences is constantly expanding, it is now acknowledged that the brain ECS plays a prominent role in modulating brain reward function and the maintenance of emotional homeostasis. This chapter examines the evidence for an eCB influence in the positive reinforcing effects of natural rewards and drugs of abuse, as well as dysregulation of ECS function that may influence or contribute to aberrant synaptic plasticity, negative emotional states, and impaired learning and memory processes resulting from long-term drug exposure that sustains compulsive drug consumption characteristic of the addicted state.
1 Neurobiology of Reward
The brain reward system plays an important role in our survival as a species. In addition to homeostatic factors that drive behavior, the hedonic or pleasurable effects produced by eating, exercise, and sexual activity provide important motivational effects that increase the likelihood of future engagement in these critical activities (known as positive reinforcement). In a similar manner, though less commonly considered, the reward system participates in negative hedonic responses in which aversive or unpleasant events (sickness, bodily harm) increase the likelihood of behaviors that will relieve or avoid these negative states (negative reinforcement). Accordingly, proper reward system function is critical for the maintenance of behaviors that promote survival.
Our understanding of the neurobiological substrates for reward and reinforcement is derived from studies in the 1950s by Olds and Milner who demonstrated that rodents will work to receive electrical stimulation in various brain regions (Olds and Milner 1954). Although stimulation of many brain sites supports operant behavior, the most sensitive sites involve the trajectory of the medial forebrain bundle that connects the ventral tegmental area (VTA) to the basal forebrain, including the nucleus accumbens (NAc) in the ventral striatum. Substantial subsequent evidence has demonstrated a critical involvement of mesocorticolimbic dopamine (DA) signaling in the mediation of reward and reinforcement, suggesting that activation of this system induces general behavioral activation (Le Moal and Simon 1991), promotes goal-directed behavior (Salamone et al. 2007), and increases incentive salience to environmental stimuli (Robinson and Berridge 1993). Functional neuroimaging studies in humans demonstrate that the mesocorticolimbic system is activated by natural rewards including the sight of appetizing food and orgasm (Komisaruk et al. 2004; Volkow et al. 2011), and rodent studies demonstrate that food consumption, sex, and exercise each increase NAc DA signaling (Bassareo and Di Chiara 1999a; Fiorino et al. 1997; Freed and Yamamoto 1985; Hattori et al. 1994). PET studies in humans have shown that psychostimulants, nicotine, alcohol, and marijuana each increase DA in the dorsal and ventral striatum and that this is associated with the subjective rewarding effects of the drugs (Tomasi and Volkow 2013). Similarly, rodent studies demonstrate that all addictive drugs possess the ability to increase mesocorticolimbic DA and that this contributes to the positive reinforcing effects of these substances (particularly psychostimulants and nicotine) (Di Chiara and Bassareo 2007; Koob and Volkow 2010). Substantial evidence suggests that DA-independent signaling in the mesolimbic system also substantially impacts reward processing, including cholinergic, opioid, glutamatergic, and GABAergic signaling (Koob 1992; Nestler 2005). The NAc receives input from the amygdala, frontal cortex, and hippocampus that may be converted to motivational actions through its connections with the extrapyramidal motor system. In particular, the central nucleus of the amygdala (CeA) appears to play a role in mediating/modulating the positive reinforcing effects of abused drugs (Koob and Volkow 2010), and the ventral pallidum/substantia innominata, which receives a major innervation from the NAc, is also critically involved in modulating the positive reinforcing effects of both natural and drug rewards (Koob and Volkow 2010).
In general, NAc DA levels are decreased by many aversive conditions such as unavoidable shock, medial hypothalamic stimulation, chronic pain, and certain patterns of over- or undereating (Umberg and Pothos 2011). Numerous studies have also demonstrated deficiencies in NAc DA during withdrawal from addictive drugs (Umberg and Pothos 2011; Koob and Volkow 2010). Further, dysregulation of key neurochemical elements involved in stress responses mediated by the CeA, bed nucleus of the stria terminalis (BNST), frontal cortex, and medial shell of the NAc plays a major role in the negative reinforcing effects of withdrawal from highly palatable food and drugs of abuse (Koob and Volkow 2010; Koob et al. 2014; Blasio et al. 2013; Iemolo et al. 2013). Signaling systems involved in these processes include dysregulation of both pro-stress (corticotropin-releasing factor (CRF), dynorphin) and anti-stress (NPY, nociceptin) systems.
Thus, reward processing is mediated in large part through an interconnected network of structures that includes the VTA, NAc, ventral pallidum, CeA, BNST, and prefrontal cortex (PFC). In addition to the well-known involvement of mesocorticolimbic DA, reward processing is also heavily influenced by many other systems including glutamate, GABA, opioid peptides, and stress-related signaling systems.
2 The Endogenous Cannabinoid System (ECS)
Endocannabinoids are neuroactive lipids that participate in a range of physiological processes including reward, motivation, emotional homeostasis, pain processing, neuroprotection, and synaptic plasticity contributing to learning and memory. For the purposes of this chapter, a superficial overview of the main ECS components is provided below, and readers are referred to more in-depth coverage of this topic provided by several excellent recently published reviews (Lu and Mackie 2015; Hillard 2015; Mechoulam and Parker 2013).
In addition to the endocannabinoid lipid moieties themselves, the ECS is comprised of G protein-coupled receptors and metabolic enzymes for the synthesis and degradation of the ligands. Two major types of cannabinoid receptor have been characterized and cloned: CB1 and CB2. CB1 receptors (CB1Rs) are the most abundant G protein-coupled receptor expressed in the adult brain, with particularly dense expression in each of the interconnected structures involved in reward (Glass et al. 1997; Wang et al. 2003b; Herkenham et al. 1991) where they exert widespread modulatory influences on excitatory and inhibitory signaling in a manner that influences reward processing (Sidhpura and Parsons 2011; Panagis et al. 2014). In particular, eCBs play a prominent role in fine-tuning the activity of the VTA-NAc DA projection and its influence on approach and avoidance behaviors that govern reward acquisition (Hernandez and Cheer 2015). CB2 receptors (CB2Rs) are mainly expressed by immune cells with recent evidence also suggesting CB2R expression in neurons, glia, and endothelial cells in the brain (Atwood and Mackie 2010). CB1R and CB2R are coupled to similar transduction systems primarily through Gi or Go proteins. CB1Rs directly inhibit the release of GABA, glutamate, and acetylcholine that produce widespread effects on neural signaling across many neurotransmitter systems.
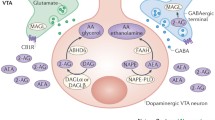
At present the best characterized endocannabinoid ligands are N-arachidonylethanolamide (anandamide, AEA) and 2-arachidonoylglycerol (2-AG). Due to their lipid nature, AEA and 2-AG are not stored in vesicles but are synthesized on an “on demand” basis by cleavage from membrane precursors and immediate release through Ca2+-dependent mechanisms. AEA is derived from the phospholipid precursor N-arachidonoyl phosphatidylethanolamine (NAPE), and while the precise mechanisms for AEA formation are not known, a very likely candidate is through N-acyl-phosphatidylethanolamine phospholipase D (NAPE-PLD). 2-AG derives primarily from the hydrolytic metabolism of 1,2-diacylglycerol (DAG) by sn-1-selective DAG lipases (DAGLα, DAGLβ). AEA is primarily catabolized through fatty acid amide hydrolase 1 (FAAH1), and 2-AG is catabolized through monoacylglycerol lipase (MAGL) and to a lesser extent α,β-hydrolase 6 (ABHD6), cyclooxygenase-2 (COX-2), and FAAH1. The eCB catabolic enzymes have distinct cellular anatomical locations with MAGL localized predominantly in presynaptic terminals and FAAH1 to the postsynaptic domain of neurons. AEA and 2-AG each exert agonist activity at CB1R and CB2R. AEA binds with slightly higher affinity to CB1R vs. CB2R, and like Δ9-tetrahydrocannabinol (Δ9-THC, the main psychoactive component of the Cannabis plant), AEA exhibits low efficacy as an agonist at both receptors, producing submaximal signaling upon binding. 2-AG binds with essentially equal affinity at CB1R and CB2R and exhibits greater agonist efficacy than AEA. AEA and 2-AG also exhibit agonist properties at several secondary receptors including peroxisome proliferator-activated receptors (PPARs), GPR55, and GPR119, and AEA exerts potent agonist effects at transient receptor potential ion channels including TRPV1.
3 Rewarding Effects of Altered eCB Signaling
Both exogenous AEA and exogenous 2-AG increase extracellular DA levels in the NAc in a CB1-dependent manner (Solinas et al. 2006), and substantial evidence demonstrates a strong eCB influence in fine-tuning the activity of midbrain DA cells (Melis and Pistis 2012). Given this influence, and the rewarding effects produced by natural and synthetic CB1 agonists, there has been considerable interest in the influence of eCB signaling in the modulation of brain reward function.
Several preclinical studies have investigated whether enhanced eCB signaling itself produces rewarding effects, and much of this has compared eCB-induced behaviors with those produced by exogenous CB1 agonists. The metabolically stable AEA analogs (R)-methanandamide, O-1812, and AM1346 each fully generalize to Δ9-THC in drug discrimination tests (Burkey and Nation 1997; Jarbe et al. 2006; Alici and Appel 2004; Solinas et al. 2007), and exogenous AEA itself also produces Δ9-THC-like discriminative effects when administered following FAAH inhibition (Solinas et al. 2007; Vann et al. 2009; Wiley et al. 2014). Because neither AEA nor FAAH inhibition produces Δ9-THC-like responding in their own right (Burkey and Nation 1997; Wiley et al. 1997, 2014; Gobbi et al. 2005; Solinas et al. 2007), these findings suggest that administration of exogenous AEA produces Δ9-THC-like interoceptive effects only under conditions of reduced hydrolytic clearance. In the absence of MAGL inhibition, exogenous 2-AG also does not substitute for Δ9-THC in discrimination tests (Wiley et al. 2014), though interestingly the MAGL inhibitor JZL184 produces partial generalization to Δ9-THC even without exogenously administered 2-AG (Wiley et al. 2014; Long et al. 2009b). However, these effects of JZL184 may result from concurrent MAGL and FAAH inhibition based on evidence that dual inhibition of MAGL and FAAH fully substitutes for Δ9-THC in discrimination tests (Wiley et al. 2014; Long et al. 2009b), and the more selective MAGL inhibitor KML29 does not generalize to Δ9-THC (Ignatowska-Jankowska et al. 2014). The Δ9-THC-like discriminative properties of enhanced eCB signaling are all blocked by CB1 antagonism. Collectively, these findings indicate that systemic administration of eCBs produces CB1-dependent Δ9-THC-like discriminative stimulus effects, though combined enhancement of AEA and 2-AG signaling is most effective for producing Δ9-THC-like effects. Neither exogenously administered AEA nor selective FAAH, MAGL, or dual FAAH/MAGL inhibition produces rewarding effects in the conditioned place preference model (Mallet and Beninger 1998; Gobbi et al. 2005; Gamage et al. 2015), and FAAH inhibitors and putative eCB transport inhibitors do not alter brain stimulation reward thresholds (Vlachou et al. 2006). Drugs that inhibit eCB hydrolysis do not support operant self-administration behavior in rodents, though exogenous AEA, the metabolically stable AEA analog methanandamide, and exogenous 2-AG do support operant self-administration behavior by squirrel monkeys through a CB1R-reliant mechanism (Justinova et al. 2005, 2008a, 2011). Moreover, the eCB clearance inhibitors URB597, AM404, and VDM11 support operant self-administration behavior by squirrel monkeys with a prior history of nicotine, cocaine, or AEA self-administration (Justinova et al. 2015; Schindler et al. 2016). However, unlike drugs with prominent abuse liability, these eCB clearance inhibitors do not increase mesolimbic DA release (Solinas et al. 2007; Murillo-Rodriguez et al. 2013; Justinova et al. 2015), and it remains to be determined whether these compounds will be self-administered by drug-naïve monkeys or other species. Collectively, these findings suggest that administration of exogenous eCBs can produce positive reinforcing effects in a variety of paradigms indexing brain reward, though most evidence suggests that pharmacological compounds that enhance eCB tone (e.g., selective FAAH or MAGL clearance inhibitors) do not produce substantial rewarding effects.
4 Endocannabinoid Influence on Natural Rewards
4.1 Food Reward
It is well known that palatable foods can lead people to eat, even when satiated. As such, eating can be motivated not only by hunger or homeostatic processes but also by hedonics. The ECS plays a prominent homeostatic role in controlling metabolic functions such as energy balance and food intake (Silvestri and Di Marzo 2013), and growing evidence also demonstrates an important ECS influence in the hedonic or rewarding properties of food intake. Both exogenous and endogenous CB1 agonists reduce the latency to feed in pre-satiated or free-feeding animals, increase the amount of food consumed, and increase an animal’s’ willingness to work for food reward (Kirkham and Williams 2001; Farrimond et al. 2011; Gallate and McGregor 1999; Kirkham et al. 2002; Williams and Kirkham 1999; Martinez-Gonzalez et al. 2004; Solinas and Goldberg 2005). Manipulations of CB1 tone appear to preferentially influence the motivation for highly palatable rewards compared with standard isocaloric chow or neutral or mildly aversive rewards (Guegan et al. 2013; Dipatrizio and Simansky 2008; Shinohara et al. 2009). For example, studies on the microstructure of fluid consumption and taste reactivity demonstrate that CB1 receptor agonists increase the hedonic effects of palatable rewards such as sucrose (Higgs et al. 2003; Jarrett et al. 2005, 2007; Mahler et al. 2007). Conversely, CB1 receptor antagonism preferentially reduces the consumption of palatable and/or high-fat foods vs. standard chow (Simiand et al. 1998; South et al. 2007; Mathes et al. 2008), reduces the conditioned reinforcing effects produced by palatable substances such as sucrose or chocolate (Chaperon et al. 1998), and reduces the motivation for palatable foods indexed under progressive ratio schedules of reinforcement (Gallate and McGregor 1999; Gallate et al. 1999; Solinas and Goldberg 2005).
The neural pathways contributing to the hedonic aspects of feeding include the mesocorticolimbic system (Kalivas and Volkow 2005; Nestler 2005). Food-related visual or olfactory cues increase mesolimbic DA signaling in both humans (Macht and Mueller 2007) and laboratory animals (Hajnal et al. 2004; Rada et al. 2005; Sclafani et al. 1998) in part through CB1-reliant mechanisms (Melis et al. 2007). The mesocorticolimbic system influences feeding behavior through interconnections with hypothalamic nuclei (Berridge et al. 2010), and many of the hypothalamic signaling molecules that modulate homeostatic feeding behavior are also present in mesocorticolimbic structures such as the NAc, VTA, and PFC (Horvath and Diano 2004; Kelley et al. 2005). In fact, an influence of feeding-related hormones on eCB production (Di Marzo et al. 2001; Bermudez-Silva et al. 2012) and signaling (Edwards and Abizaid 2016) has been described, and this mechanism may play a prominent role in the regulation of consummatory behaviors. Fasting increases AEA and 2-AG levels in the limbic forebrain and hypothalamus of rodents, an effect that is ameliorated following feeding (Kirkham et al. 2002; Hanus et al. 2003). Doses of CB1 agonists that induce food consumption in satiated rodents activate the corticostriatal-hypothalamic pathway (including the NAc) (Dodd et al. 2009), and food consumption is increased by direct infusion of eCBs or FAAH inhibitors into the hypothalamic nuclei (Anderson-Baker et al. 1979; Jamshidi and Taylor 2001; Verty et al. 2005) or NAc shell (Kirkham et al. 2002; Soria-Gomez et al. 2007). In particular, the NAc shell has been identified as a critical locus for the eCB modulation of the hedonic properties of food (Berridge et al. 2010; Mahler et al. 2007), and intra-NAc eCB infusions increase neural activity in hypothalamic nuclei through a CB1-dependent mechanism (Soria-Gomez et al. 2007). Furthermore, transgenic mice overexpressing MAGL in the forebrain (MGL-Tg mice) do not form a conditioned place preference for high-fat foods, indicating that reductions in 2-AG may impede high-fat food reward (Wei et al. 2016). Collectively, these findings demonstrate that eCB signaling in the CNS plays a role in the motivation for and hedonic response to food consumption. Interestingly, there is emerging evidence that in addition to sweet taste receptors (T1R2/T1R3), sweet-sensitive taste cells in the peripheral system also express CB1 receptors (Jyotaki et al. 2010), and both AEA and 2-AG increase gustatory nerve responses to sweet solutions (Yoshida et al. 2010). Thus, the ECS modulates food palatability via both central and peripheral pathways.
4.2 Sexual Reward
CB1 receptors are densely expressed in the neural circuits that regulate sexual behavior, and substantial evidence indicates that both exogenous and endogenous cannabinoids can alter sexual behavior, though the effects are often highly divergent in males and females (Gorzalka et al. 2010; Lopez 2010). With regard to the hedonic aspects of sexual behavior, the majority of studies indicate a facilitatory effect of Cannabis on subjective indices of sexual arousal in women including desire, orgasmic function, and pleasure/enjoyment/satisfaction (Koff 1974; Dawley et al. 1979; Halikas et al. 1982; Klein et al. 2012). Similarly, low-dose Cannabis exposure increases sexual desire in males though simultaneously hindering erectile functioning. Rodent studies have revealed both facilitatory and inhibitory effects of cannabinoid agonists and eCB clearance inhibition (e.g., FAAH inhibitors) on female sexual receptivity and proceptivity and a general inhibitory effect on male sexual performance (Gorzalka et al. 2010; Lopez 2010; Canseco-Alba and Rodriguez-Manzo 2016), though the relative influence of these manipulations on the motivational vs. performance aspects of sexual behavior is not clear. In the most direct evaluation of an eCB influence on sexual motivation, Klein and colleagues observed that physiological indices of sexual arousal in women are associated with decreased serum AEA levels, and subjective indices of arousal are associated with decreases in serum levels of both AEA and 2-AG (Klein et al. 2012). However, while these indices reflect an ECS involvement in the anticipatory/motivational effects produced by erotic visual stimuli, it remains unclear whether endocannabinoid signaling is altered by sexual activity itself and, if so, whether this results in a facilitation or attenuation of brain reward processing. Endocannabinoid signaling may also influence sexual reward indirectly through modulation of emotional state and/or sensation perception, though these possibilities have not been explored.
4.3 Social Interaction
Cooperative playing and social interaction are rewarding activities in both humans and rodents (Tabibnia and Lieberman 2007; Douglas et al. 2004). In rodents, social play is enhanced following inhibition of FAAH or putative eCB transporters (with VDM11) but diminished following administration of exogenous CB1 agonists such as WIN 55,212-2 (Trezza and Vanderschuren 2008a, b, 2009). This highlights the distinct effects produced by selectively enhancing the effects of eCB signaling in selected synapses, wherein eCB formation is evoked (e.g., eCB clearance inhibition) vs. widespread CB1 activation by exogenous agonists that engage CB1 signaling in circuits not normally activated by a given behavior (Trezza et al. 2010). The hedonic aspects of social play are mediated through both opioid (mu opioid in particular) and CB1 mechanisms, and the CB1 influence appears to involve subsequent increases in DA signaling (Trezza et al. 2010). Interestingly, social reward is influenced by opioid and cannabinoid interactions in a manner similar to drug and food reward (Fattore et al. 2004; Solinas and Goldberg 2005).
5 eCB Influence on the Acute Rewarding Effects of Drugs of Abuse
There is substantial evidence implicating the ECS in the motivational effects produced by several classes of abused drugs including ethanol, nicotine, opiates, and psychostimulants. Much of this is based on the influence of CB1 receptor signaling on drug-induced behaviors, though some studies have evaluated manipulations of eCB tone on drug reinforcement.
In general, CB1 receptors exert a facilitatory influence on the rewarding effects of several classes of abused drugs (Table 1). Rodent studies employing the conditioned place preference and operant self-administration paradigms demonstrate that CB1 agonists increase the motivational and rewarding effects of alcohol (Colombo et al. 2002; Wang et al. 2003a; Gallate et al. 1999; Malinen and Hyytia 2008; Getachew et al. 2011), nicotine (Valjent et al. 2002; Gamaleddin et al. 2012b), and opiates (such as morphine and heroin) (Manzanedo et al. 2004; Solinas et al. 2005). In contrast, diminished CB1 receptor signaling (either genetic receptor deletion or pharmacological inhibition) attenuates the motivational and rewarding effects of alcohol (Houchi et al. 2005; Thanos et al. 2005; Hungund et al. 2003; Naassila et al. 2004; Arnone et al. 1997; Wang et al. 2003a; Freedland et al. 2001; Gallate and McGregor 1999; Colombo et al. 1998; Gessa et al. 2005), nicotine (Cohen et al. 2002; Shoaib 2008; Cossu et al. 2001; Simonnet et al. 2013), and opiates (Chaperon et al. 1998; Navarro et al. 2001; Singh et al. 2004; Martin et al. 2000; Ledent et al. 1999; Cossu et al. 2001; Caille and Parsons 2003; De Vries et al. 2003; Solinas et al. 2003). The CB1 influence on alcohol and nicotine reward is mediated in part, through modulation of the mesolimbic DA response to these drugs as CB1 receptor antagonism blocks the excitation of VTA DA cells produced by alcohol and nicotine, thereby attenuating the effects of these drugs on NAc DA release (Cohen et al. 2002; Hungund et al. 2003; Perra et al. 2005; Cheer et al. 2007). Blockade of CB1 receptors in the VTA decreases both alcohol and nicotine self-administration (Malinen and Hyytia 2008; Alvarez-Jaimes and Parsons 2009; Caille et al. 2007; Simonnet et al. 2013), and antagonism of NAc CB1 receptors similarly reduces alcohol consumption (Alvarez-Jaimes and Parsons 2009; Caille et al. 2007). However, while nicotine reward is critically dependent on the mesolimbic DA system (D’Souza and Markou 2011), the motivational and rewarding effects of alcohol and opiates are less DA dependent (Koob 2013; Shippenberg and Elmer 1998; Pettit et al. 1984; Gerrits and Van Ree 1996; Platt et al. 2001) with CB1R modulation of the rewarding effects of these drugs likely involving non-dopaminergic mechanisms. Indeed, although CB1 receptor deletion results in attenuated opiate-induced increases in NAc DA (Mascia et al. 1999), acute CB1 receptor blockade does not alter opiate-induced increases in NAc DA (Caille and Parsons 2003, 2006; Tanda et al. 1997). Rather, CB1 receptor antagonism may modulate opiate reward through DA-independent attenuation of opiate-induced reductions in ventral pallidal GABA signaling (Caille and Parsons 2006).
The effects of CB1 receptor manipulations on psychostimulant reward are less consistent. In contrast to the facilitation of ethanol, nicotine, and opiate reward, CB1 agonists reduce both cocaine-induced facilitation of brain stimulation reward and cocaine self-administration by rats (Vlachou et al. 2003; Fattore et al. 1999). Further, CB1 receptor inactivation does not affect psychostimulant-induced place conditioning (Martin et al. 2000; Houchi et al. 2005), psychostimulant self-administration (Cossu et al. 2001; Tanda et al. 2000; Fattore et al. 1999; De Vries et al. 2001; Caille and Parsons 2006; Filip et al. 2006; Caille et al. 2007; Lesscher et al. 2005), cocaine-induced increases in nucleus accumbens dopamine (Caille and Parsons 2003; Soria et al. 2005), or cocaine-induced enhancement of brain stimulation reward (Vlachou et al. 2003; Xi et al. 2008). However, some reports indicate that CB1 inactivation attenuates cocaine and amphetamine self-administration (Soria et al. 2005; Xi et al. 2008; Vinklerova et al. 2002), blocks cocaine-induced increases in NAc DA signaling (Li et al. 2009; Cheer et al. 2007), and decreases cocaine-induced enhancement of brain stimulation reward sensitivity (Xi et al. 2008). In general, the inconsistent and generally subtle effects reported suggest that CB1 receptors exert a relatively modest influence over psychostimulant-induced reward.
Recent evidence in mice also implicates CB2Rs in the modulation of drug reward. CB2R agonists reduce cocaine-induced CPP and cocaine self-administration in wild-type mice but not CB2R knockout mice (Xi et al. 2011; Ignatowska-Jankowska et al. 2013; Zhang et al. 2014a), and cocaine self-administration is also reduced by CB2R overexpression in the brain (Aracil-Fernandez et al. 2012). CB2R agonists also reduce alcohol-induced CPP and alcohol consumption (Al Mansouri et al. 2014). Mouse VTA DA neurons express CB2R mRNA and receptor immunostaining, and CB2R agonists decrease the activity of these neurons in wild-type but not CB2R knockout mice (Zhang et al. 2014b). Surprisingly, CB2R agonists facilitate nicotine-induced CPP (Ignatowska-Jankowska et al. 2013), and CB2R inactivation reduces both nicotine-induced CPP and nicotine self-administration in mice (Navarrete et al. 2013; Ignatowska-Jankowska et al. 2013). In contrast, CB2R antagonism does not alter cocaine or nicotine self-administration in rats but attenuates cocaine-induced and conditioned locomotion and reinstatement of cocaine-seeking behavior (without altering nicotine-induced drug seeking) (Blanco-Calvo et al. 2014; Adamczyk et al. 2012b; Gamaleddin et al. 2012b). Further, a recent study reported the CB2R agonism reduces cocaine consumption by rats, but not mice, and produces opposite effects on the motivation for cocaine in these species (Zhang et al. 2014a). These distinctions may result from species differences in the splicing and expression of CB2R genes, possibly conferring distinct CB2R structure, function, or pharmacology (Zhang et al. 2014a).
6 Evidence that Drugs of Abuse Increase Brain eCB Levels
The inhibitory influence of CB1 receptor antagonism on drug reward has led to the hypothesis that abused drugs increase brain eCB formation possibly resulting in aberrant eCB signaling following long-term drug exposure. Several observations support these hypotheses.
Early studies reported that chronic alcohol exposure increases 2-AG and AEA formation in human neuroblastoma cells and cultured rodent neurons (Basavarajappa and Hungund 1999; Basavarajappa et al. 2000, 2003). Subsequent evaluations of postmortem brain tissue eCB content clearly demonstrate alcohol-induced alterations in 2-AG and AEA production, though substantial inconsistencies among studies make it difficult to draw clear conclusions on the direction of change and regional nature of the effects (Vinod et al. 2006, 2012; Gonzalez et al. 2002, 2004b; Malinen et al. 2009; Rubio et al. 2007). In vivo microdialysis studies in rats demonstrate that voluntary alcohol consumption robustly increases nucleus accumbens 2-AG but not AEA levels (Caille et al. 2007), while forced alcohol administration decreases AEA levels and induces more modest increases in 2-AG (Ferrer et al. 2007; Alvarez-Jaimes et al. 2009). This provides initial evidence that the volitional nature of drug exposure (e.g., voluntary vs. forced administration) may differentially impact eCB responses. The effects of alcohol on brain eCB production may also be region specific as alcohol-induced disruptions of extracellular eCB levels and mRNA expression of their associated enzymes are consistently observed in striatal regions (Caille et al. 2007; Ceccarini et al. 2013; Ferrer et al. 2007; Alvarez-Jaimes et al. 2009; Henricks et al. 2016) but not frontal cortical areas (Alvarez-Jaimes and Parsons 2009), and ethanol self-administration is reduced by localized antagonism of CB1 receptors in the VTA and NAc shell but not PFC of outbred rats (Malinen and Hyytia 2008; Alvarez-Jaimes and Parsons 2009; Caille et al. 2007) (though disruptions in eCB processing have been observed in cortical regions of rats selectively bred for high alcohol consumption (Hansson et al. 2007) and in mice following long-term alcohol exposure (Vinod et al. 2006)).
Δ9-THC and synthetic CB1 agonists also induce region-specific alterations in brain tissue eCB content. For example, chronic Δ9-THC increases AEA (but not 2-AG) in limbic forebrain tissue and increases 2-AG (but not AEA) in the hippocampus, brainstem, and cerebellar tissue (Di Marzo et al. 2000; Gonzalez et al. 2004a; Castelli et al. 2007). In vivo microdialysis studies demonstrate that acute CB1 agonist exposure increases 2-AG but decreases AEA levels in the rat hypothalamus (Bequet et al. 2007), and preliminary evidence from human clinical research studies also points to cannabinoid-induced increases in 2-AG and decrements in AEA production (Leweke et al. 2007; Morgan et al. 2013).
Chronic nicotine exposure also induces region-specific disruptions in brain tissue eCB content, with increased AEA (but not 2-AG) levels evident in limbic forebrain and caudate but decreased AEA and 2-AG levels in the cortex (Gonzalez et al. 2002). In vivo microdialysis studies demonstrate nicotine-induced increases in both 2-AG and AEA in the VTA and a sensitization of nicotine-induced 2-AG, but not AEA, formation results from chronic nicotine exposure (Buczynski et al. 2013). This enhancement of nicotine-induced VTA 2-AG formation results in diminished inhibitory constraint of VTA cell excitability, thereby enhancing nicotine-induced elevations in mesolimbic DA levels (Buczynski et al. 2016). Interestingly, while VTA 2-AG levels are increased by both voluntary self-administration and response-independent forced nicotine administration, VTA AEA levels are increased only by voluntary self-administration. Together with evidence of distinct eCB responses to self-administered vs. forced alcohol exposure (Caille et al. 2007; Ferrer et al. 2007; Alvarez-Jaimes et al. 2009), these observations suggest that brain eCB production can be influenced by activity of neural systems involved in the motivation for drug consumption, in addition to drug-related pharmacological effects.
Opiates also induce differential effects on brain 2-AG and AEA levels. Chronic morphine dose dependently reduces tissue 2-AG content in the striatum, cortex, hippocampus, hypothalamus, and limbic forebrain, while increased AEA content is observed in these regions (Vigano et al. 2003, 2004). Consistently, in vivo microdialysis studies demonstrate that heroin self-administration significantly increases interstitial AEA levels but decreases interstitial 2-AG levels in the nucleus accumbens (Caille et al. 2007).
In general, psychostimulants induce more subtle alterations in brain eCB levels as compared with the drugs described above (Zlebnik and Cheer 2016). Early studies reported that acute high-dose cocaine administration induces a subtle but significant increase in forebrain 2-AG content in mice (Patel et al. 2003), while chronic cocaine induces comparably subtle decreases in forebrain 2-AG content in rats (Gonzalez et al. 2002), though neither 2-AG nor AEA content is altered in other regions following acute or chronic cocaine exposure (Patel et al. 2003; Gonzalez et al. 2002). Similarly, cocaine self-administration does not alter either 2-AG or AEA content in nucleus accumbens microdialysates (Caille et al. 2007). However, recent studies provide some evidence of psychostimulant-induced alterations in eCB levels and processing. For example, cocaine self-administration is reported to induce increased cerebellar AEA content; decreased 2-AG content in the frontal cortex, hippocampus, and cerebellum; and an increased ratio of DAGLa/MAGL expression in the hippocampus (Bystrowska et al. 2014; Rivera et al. 2013). Further, sensitization of the motor-activating effects of cocaine is associated with altered eCB metabolic enzyme expression in the cerebellum (Palomino et al. 2014), and detoxified cocaine addicts present significant increases in plasma AEA and decreases in plasma 2-AG content (Pavon et al. 2013).
These collective findings indicate that cannabinoids, ethanol, nicotine, and opiates can alter brain eCB content, consistent with substantial evidence of a CB1 receptor influence on the behavioral effects produced by these drugs. Although there is relatively less information on psychostimulant-induced alterations in brain eCB content, the modest effects that have been reported are consistent with the subtle disruptions in psychostimulant-induced behaviors observed following CB1 receptor antagonism.
eCBs are rapidly degraded, and thus strategies that reduce eCB clearance have been employed as a means to further investigate the eCB influence on drug reward. Most investigations have focused on the effects of FAAH inhibition because selective tools for inhibiting MAGL and other eCB clearance enzymes were not available until recently. Such studies have shed light on important species differences that confound the overall conclusions that can be made from existing data. For example, FAAH inhibition in mice increases nicotine reward in the CPP paradigm (Merritt et al. 2008; Muldoon et al. 2013), though in rats, FAAH inhibition prevents nicotine-induced CPP, diminishes nicotine self-administration, and blunts nicotine-induced increases in NAc DA release (Scherma et al. 2008). The potentiation of nicotine reward in mice by FAAH inhibition is CB1R mediated, whereas the reduction in nicotine reward in rats results from activation of PPAR-α by non-cannabinoid lipids such as oleoylethanolamide (OEA) and palmitoylethanolamide (PEA) that are hydrolytically cleared by FAAH (Melis and Pistis 2014). FAAH inhibition also produces distinct species-related alterations in alcohol consumption, with increased intake observed in mice but not rats (Blednov et al. 2007; Vinod et al. 2008; Hansson et al. 2007; Cippitelli et al. 2008). The mechanisms underlying these differences are not understood. Brain region-specific disruptions in FAAH activity may be an important factor in regard to alcohol reward as inhibition of FAAH activity specifically in the PFC results in increased alcohol consumption, and rats selectively bred for high alcohol intake and preference are characterized by reduced FAAH activity specifically in the PFC (Hansson et al. 2007; Cippitelli et al. 2008). The effects of FAAH inhibition on opiate and psychostimulant reward have primarily been studied in rats. FAAH inhibition does not alter morphine- or cocaine-induced disruptions in VTA DA cell firing or the self-administration of either drug (Justinova et al. 2008a; Luchicchi et al. 2010). However, FAAH inhibition diminishes cocaine-induced alterations in NAc medium-spiny neuron activity (Luchicchi et al. 2010), and this may contribute to enhanced sensitization of both cocaine-induced motor activity and mesolimbic dopamine responses following repeated cocaine exposure (Mereu et al. 2013). Other studies have investigated the effects of putative eCB transport inhibitors such as AM404 and VDM11, and the findings thus far suggest that these compounds produce subtle and inconsistent effects on nicotine and cocaine reward (Scherma et al. 2012; Gamaleddin et al. 2011, 2013; Vlachou et al. 2008).
While growing evidence implicates ECS influences in the modulation of acute drug reward, additional efforts are needed to further clarify the nature of eCB disruptions caused by different classes of abused drugs and the neural mechanisms through which these eCB influences are mediated. Selective inhibitors of 2-AG clearance have recently been developed, but studies are still in their infancy, so there are presently no published reports on the effects of enhanced 2-AG tone on drug reward and related physiological events. As such there remains a substantial gap of knowledge given the prominent 2-AG influence on neural signaling and plasticity related to both drug and natural rewards. Nevertheless, the role of the CB1R in drug reward is unequivocal. Although there is evident complexity related to the effects produced by eCB clearance inhibition (producing discrete modulation of eCB tone in specific synapses/circuits as compared with broad CB1R activation by exogenous CB1R agonists), the extant evidence strongly supports an eCB influence on the sensitivity to and motivation for several drugs of abuse.
7 Overview of Drug Dependence, Addiction, and Withdrawal
The transition from intermittent and controlled drug use to compulsive forms of drug seeking and drug taking that characterize addiction is influenced by a number of factors. Substantial evidence implicates genetic influences in the development of substance use disorders and pathological forms of eating, sexual behavior, and gambling (Ducci and Goldman 2012). A crucial role for epigenetic mechanisms driving lasting changes in addiction-related gene expression is also increasingly recognized (Nestler 2014). Additionally, long-term drug exposure induces lasting neuroadaptations that alter the motivational mechanisms that propel drug seeking and use. Although initial drug use is motivated by the hedonic effects of drug consumption, prolonged drug exposure results in progressive blunting of reward system function that can motivate consumption of increasingly larger amounts of drug. Evidence suggests this results in part from decreased function of the mesolimbic DA system and its output to the ventral pallidum. Escalated frequency and amount of drug consumption lead to a dependent state wherein negative affective symptoms emerge during drug abstinence (e.g., dysphoria, anxiety, irritability). These negative emotional states arise from the recruitment of stress-signaling systems within the extended amygdala (such as corticotropin-releasing factor and dynorphin) and dysregulation of systems that constrains these responses (such as neuropeptide Y and nociceptin) (Koob and Volkow 2010). Renewed drug consumption alleviates these negative affective states, and this is conceptualized to contribute to compulsive drug use through negative reinforcement mechanisms (Koob and Volkow 2010). Superimposed on these processes is a dysregulation of corticostriatal mechanisms contributing to stimulus response learning, conditioned reinforcement, and incentive motivation resulting in a narrowed focus on drug seeking at the expense of natural rewards (Kalivas and Volkow 2005; Everitt et al. 2008). eCBs exert prominent modulatory influence in the extended amygdala and corticostriatal circuits implicated in the etiology of addiction, and as described below, increasing evidence suggests that preexisting genetic influences on the ECS and/or drug-induced dysregulation of eCB function may participate in the development and maintenance of addiction. The following sections consider the consequences of chronic drug exposure on eCB signaling within the reward circuitry and related disruptions in synaptic plasticity, affective state, and learning and memory mechanisms related to extinction and relapse.
8 Evidence of Altered ECS Function Following Chronic Drug Exposure
It stands to reason that chronic Cannabis use induces dysregulation of brain cannabinoid receptors' availability and function. In vivo PET imaging studies in humans have revealed downregulation of brain CB1R binding that correlates with the duration of prior Cannabis use, with particularly robust decrements observed in the temporal lobe, anterior and posterior cingulate cortex, and nucleus accumbens (Hirvonen et al. 2013; Ceccarini et al. 2015). These decrements in CB1R availability appear to progressively recover with prolonged Cannabis abstinence. A similar profile of decreased brain CB1R function and posttreatment recovery has been consistently reported in rodents given chronic CB1R agonist exposure (Breivogel et al. 1999; Sim et al. 1996). Recent experiments employing stochastic optical reconstruction microscopy (STORM) demonstrate that chronic exposure to clinically relevant doses of Δ9-THC results in a startling loss of CB1Rs on terminals of perisomatically projecting GABA interneurons in the mouse hippocampus and internalization of the remaining CB1Rs (Dudok et al. 2015). The resulting deficits in inhibitory CB1R control over hippocampal GABA release persisted during several weeks of Δ9-THC abstinence, and this may underlie the enduring loss of hippocampal LTP in rodents and memory deficits in humans evident following chronic cannabinoid exposure (Puighermanal et al. 2012). Surprisingly little is known of the effect of chronic cannabinoid exposure on other facets of ECS function. Chronic cannabinoid exposure increases enzymatic clearance of AEA and reduces brain tissue AEA content in rodents (Di Marzo et al. 2000; Schlosburg et al. 2009), and frequent Cannabis smokers present decreased AEA and increased 2-AG levels in the blood (Leweke et al. 2007; Morgan et al. 2013), though increased serum AEA levels are evident following at least 6 months of Cannabis abstinence (Muhl et al. 2014). The contribution of these disruptions to Cannabis use disorder and related physiological and behavioral disruptions is presently unexplored. However, as discussed below, eCBs provide important homeostatic constraint over emotional state (Lutz 2009) and sleep function (Murillo-Rodriguez et al. 2011), and it’s conceivable that Δ9-THC-induced impairment of eCB signaling contributes to negative emotional states and sleep disturbances present during protracted Cannabis abstinence (Budney et al. 2001; Gates et al. 2015).
Chronic exposure to non-cannabinoid drugs also disrupts eCB signaling and processing. Chronic alcohol exposure in rodents alters eCB-related gene expression in a manner sensitive to the intermittent nature of alcohol exposure and post-alcohol abstinence period (Serrano et al. 2012) and downregulates CB1R expression and function (Mitrirattanakul et al. 2007; Ceccarini et al. 2013). Postmortem studies of alcohol-dependent humans also demonstrate disrupted CB1R expression in the ventral striatum and cortical regions (Vinod et al. 2010), and in vivo imaging studies demonstrate decreased CB1R availability in heavy drinking alcoholics that persist for at least 1 month of abstinence (Hirvonen et al. 2013; Ceccarini et al. 2014) (but see Neumeister et al. (2012)). Although a potential contribution of CNR1 gene variants to these observations cannot be excluded, a common interpretation based on animal studies is that these CB1R adaptations in alcoholic humans are a consequence of prolonged alcohol-induced increases in brain eCB levels. This is supported by evidence of transient recovery (and perhaps eventual upregulation) of CB1R function in humans during protracted alcohol abstinence (Vinod et al. 2006; Mitrirattanakul et al. 2007). In rodents, chronic nicotine exposure induces distinct age-related disruptions in CB1R binding, with increased levels evident in the PFC, VTA, and hippocampus of adolescent but not adult rats (Werling et al. 2009), and increased hippocampal and decreased striatal CB1R binding in adult rats during protracted nicotine abstinence (Marco et al. 2007). Few studies have investigated altered CB1R binding following chronic opiate or psychostimulant exposure, but findings in rodents implicate impaired CB1R function in the development and expression of opiate dependence (Rubino et al. 1997; Cichewicz et al. 2001) and demonstrate that chronic cocaine increases CB1R binding in dorsal striatum, NAc, and cortical areas (Adamczyk et al. 2012a). Interestingly, detoxified cocaine addicts present significant increases in plasma AEA and decreases in plasma 2-AG content (Pavon et al. 2013), but the functional consequence of these disturbances is not known. Overall, accruing data suggests that long-term exposure to a variety of drug classes compromises eCB processing and CB1R expression and function. As discussed below, these perturbations may contribute to aberrant neural signaling during acute and protracted drug abstinence.
9 eCB Influence on Physical Withdrawal Symptoms
In dependent individuals, drug abstinence is associated with transient physical or somatic symptoms that can persist for several days. The intensity of these symptoms varies between drug classes, with severe somatic withdrawal signs evident during abstinence from opiates and alcohol and substantially less severe symptoms during abstinence from nicotine, cocaine, and cannabinoids (West and Gossop 1994). Early conceptualizations of addiction are focused on the relief from somatic withdrawal symptoms as the motivational basis for drug use by dependent individuals (Wikler 1948; Dole 1965; Dole et al. 1966), though more recent evidence suggests that alleviation of somatic withdrawal is not a major factor contributing to relapse (Heilig et al. 2010; Hershon 1977). In fact the greatest susceptibility to drug relapse typically occurs well after the abatement of somatic withdrawal symptoms. Nonetheless, there is value in understanding the mechanisms contributing to somatic withdrawal symptoms for the development of palliative therapies for dependent individuals and avoidance of medically serious conditions associated with acute drug detoxification.
Chronic alcohol exposure results in significant downregulation of CB1 receptor expression and function in many brain regions (Basavarajappa et al. 1998; Basavarajappa and Hungund 1999; Vinod et al. 2006, 2010; Moranta et al. 2006; Mitrirattanakul et al. 2007; Ceccarini et al. 2013, 2014; Hirvonen et al. 2013; Neumeister et al. 2012) and leads to disruptions in brain tissue AEA and 2-AG content (Vinod et al. 2006, 2012; Gonzalez et al. 2002, 2004b; Malinen et al. 2009; Rubio et al. 2007). Clinical research has revealed a correlation between the severity of alcohol withdrawal symptoms and polymorphisms in the gene encoding CB1 receptors (Schmidt et al. 2002), and alcohol withdrawal severity is increased in CB1 receptor knockout mice (Naassila et al. 2004). Accordingly, it is possible that impaired CB1 signaling contributes to the intensity of physical symptoms of alcohol withdrawal. In this regard, it is notable that FAAH knockout mice exhibit significantly diminished alcohol withdrawal-related seizures relative to wild-type mice (Vinod et al. 2008; Blednov et al. 2007). The somatic symptoms of alcohol withdrawal, including seizure activity, result primarily from excessive excitatory glutamate signaling (De Witte et al. 2003). Interestingly, AEA modulates NMDA receptor function (Hampson et al. 1998) and reduces NMDA- and electroshock-induced seizures (Hayase et al. 2001; Wallace et al. 2002). Therefore, enhancement of AEA tone may provide protection against alcohol withdrawal-related seizures, though more research is necessary to support this possibility.
Chronic nicotine exposure does not appear to alter brain CB1 receptor expression (Gonzalez et al. 2002), and precipitated somatic withdrawal signs in nicotine-dependent mice are not altered by CB1 receptor deletion or moderate doses of the CB1 receptor antagonist SR141716A (Castane et al. 2002, 2005; Cossu et al. 2001; Balerio et al. 2004; Merritt et al. 2008). In contrast, the Manzanares group observed that somatic indices of nicotine withdrawal are absent in CB2 knockout mice and are attenuated in WT mice by treatment with the CB2 antagonist AM630 (Navarrete et al. 2013), though no genotypic differences in somatic withdrawal were evident following a shorter duration of nicotine exposure (7 days vs. 14 days) (Ignatowska-Jankowska et al. 2013). This suggests a potential CB2 influence in the somatic symptoms of withdrawal following long-term nicotine exposure. Acute Δ9-THC administration decreases somatic symptoms of nicotine withdrawal (Balerio et al. 2004, 2006). Moreover, nicotine-dependent MAGL knockout mice exhibit diminished precipitated withdrawal signs, and the MAGL inhibitor JZL184 dose-dependently reduces somatic and aversive signs of precipitated nicotine withdrawal in dependent mice through a CB1 receptor-dependent mechanism (Muldoon et al. 2015). This same study also points to an association between MAGL gene polymorphisms and the severity of nicotine withdrawal symptoms in humans. In contrast, acute FAAH inhibition does not diminish somatic nicotine withdrawal symptoms in rats (Cippitelli et al. 2011), while somatic symptoms of withdrawal are worsened in mice by FAAH inhibition, though this may occur through non-CB1 mechanisms (Merritt et al. 2008). Collectively these findings suggest that enhanced CB1 receptor signaling resulting from MAGL inhibition may provide therapeutic benefit for the somatic symptoms of nicotine withdrawal.
Chronic opiate exposure alters CB1 receptor expression and function (Rubino et al. 1997; Cichewicz et al. 2001; Smith et al. 2007), and this appears to play a role in the development and expression of opiate dependence. For example, the somatic symptoms of opiate withdrawal are significantly reduced in CB1 knockout mice or WT mice receiving chronic CB1 antagonist treatment concurrent with chronic morphine administration (Ledent et al. 1999; Lichtman et al. 2001; Mas-Nieto et al. 2001; Rubino et al. 2000), and it has long been recognized that Δ9-THC effectively reduces the intensity of somatic symptoms of opiate withdrawal (Bhargava 1976; Hine et al. 1975). These findings suggest that chronic opiate exposure dysregulates CB1 receptor or eCB function in a manner that contributes to elicitation of somatic symptoms during drug withdrawal. Consistent with this hypothesis, administration of Δ9-THC, exogenous AEA, or exogenous 2-AG attenuates the intensity of somatic withdrawal symptoms (Vela et al. 1995; Yamaguchi et al. 2001; Gamage et al. 2015), and high doses of the selective MAGL inhibitor JZL184 significantly reduce all indices of somatic withdrawal in opiate-dependent mice, while the selective FAAH inhibitor PF-3845 reduces only a subset of withdrawal responses (Ramesh et al. 2011, 2013; Gamage et al. 2015). Perhaps more importantly, a novel dual inhibitor exhibiting >100-fold greater potency at FAAH vs. MAGL (SA-57) (Niphakis et al. 2012) significantly reduces somatic indices of precipitated withdrawal (Gamage et al. 2015). This is an important observation given that high-dose JZL184 exposure elicits some cannabimimetic effects (Long et al. 2009a), and repeated high-dose JZL184 administration induces functional CB1 receptor tolerance and results in cannabinoid dependence (Schlosburg et al. 2010); these effects may limit the clinical viability of selective MAGL inhibition for treatment of opiate withdrawal. In contrast, SA-57 doses that efficaciously reduce opiate withdrawal do not produce cannabimimetic effects (Ramesh et al. 2013), and the moderate MAGL inhibition induced by this compound is not likely to induce CB1 receptor downregulation or cannabinoid dependence (Kinsey et al. 2013; Ghosh et al. 2013). However, a caveat to these findings is that while SA-57 attenuates somatic aspects of withdrawal, its administration does not prevent the aversive aspects of withdrawal as measured by the conditioned place aversion assay in mice (Gamage et al. 2015). Nonetheless, these collective findings suggest that modest pharmacological enhancement of eCB signaling may provide palliative effects for the physical symptoms of opiate withdrawal.
10 Stress Responsivity
Substantial clinical and preclinical data indicate that various forms of stress are involved in the etiology and maintenance of addiction. High levels of stress often precede the development of substance use disorders (Jose et al. 2000; Richman et al. 1996; Rospenda et al. 2000), and stress-related increases in glucocorticoid signaling increases the acquisition of drug self-administration by rodents (Goeders 2002; Goeders and Guerin 1996; Koob and Kreek 2007; Vengeliene et al. 2003). Consistently, evidence suggests that both stress- and drug-induced activation of the hypothalamic-pituitary-adrenal (HPA) axis sensitizes the function of reward pathways through glucocorticoid mechanisms (Piazza and Le Moal 1998; Rouge-Pont et al. 1995, 1998; Tidey and Miczek 1997). However, long-term drug use results in a blunting of reward system function, increased influence of CNS stress systems, and dysregulation of the HPA axis that is theorized to induce an allostatic shift in motivational influence that propels an escalation of drug consumption (Koob and Kreek 2007; Le Moal 2009; Koob and Le Moal 1997).
It is now well established that eCB signaling serves a homeostatic role in the constraint of HPA axis activation (Cota 2008; Gorzalka et al. 2008; Morena et al. 2016; also see chapter “Endocannabinoids, Stress, and Negative Affect” that details this function in greater depth). Stress-induced glucocorticoid secretion increases eCB production in several regions implicated in drug dependence and addiction including the amygdala, hippocampus, PFC, hypothalamic nuclei, and periaqueductal gray (Di et al. 2003, 2005, 2009; Hill et al. 2010a; Hohmann et al. 2005; Patel et al. 2004, 2005). Strong evidence demonstrates that eCB-mediated CB1 activation in these and other regions constrains stress-induced HPA axis activation and resultant increases in glucocorticoid secretion (Barna et al. 2004; Cota et al. 2007; Di et al. 2003; Manzanares et al. 1999; Patel et al. 2004; Uriguen et al. 2004; Wade et al. 2006). In contrast, stress-induced reductions in AEA content are consistently observed in the amygdala, PFC, and hippocampus (Hill et al. 2008, 2009; Patel et al. 2005; Rademacher and Hillard 2007), and this is theorized to contribute to stress responsivity through a release of tonic HPA axis inhibition (Patel et al. 2004). Regardless of the specific mechanism that prevails, it is clear that eCB signaling participates in the constraint of HPA axis activation and may contribute to the termination of stress responses.
In this regard, it is conceivable that disrupted eCB-mediated plasticity induced by long-term drug exposure contributes to a persistent dysregulation of HPA axis function and increases stress sensitivity associated with addiction (Koob and Kreek 2007; Le Moal 2009; Koob and Le Moal 1997). Because stress exposure is implicated in the development of addiction (Goeders 2002; Goeders and Guerin 1996; Jose et al. 2000; Koob and Kreek 2007; Richman et al. 1996; Rospenda et al. 2000; Vengeliene et al. 2003), it is notable that stress can alter eCB-mediated plasticity in addiction-related brain regions (Natividad et al. 2017). For example, prolonged stress exposure impairs eCB-mediated DSI, LTD, and fEPSPs in the NAc (Wang et al. 2010), attenuates eCB-mediated DSE and DSI in the PVN (Wamsteeker et al. 2010), and enhances 2-AG-mediated DSI in the basolateral amygdala (Patel et al. 2009). Similarly, establishment of conditioned fear increases the efficacy of DSE and DSI in the central nucleus of the amygdala (Kamprath et al. 2011).
11 Affective Disruptions
In addition to transient somatic symptoms (West and Gossop 1994), withdrawal from most drugs of abuse is associated with increased negative affective symptoms such as anxiety and depression (Alling et al. 1982; Coffey et al. 2000; Janiri et al. 2005; Nunes and Levin 2004; Nunes et al. 2004). Withdrawal-related negative affective states can persist for months and in some cases years during protracted abstinence, and the severity of these symptoms is closely associated with susceptibility to relapse (Annis et al. 1998; Miller and Harris 2000). Moreover, there is a prevalent comorbidity between affective disorders and substance use disorders, and affective disruption may be an antecedent to addiction (Bruijnzeel et al. 2004; Castle 2008; Conway et al. 2006; Pani et al. 2010; Schuckit 2006). As discussed in detail in chapter “Lipid Mediators in the Regulation of Emotions, Memory and Cognitive Functions” of this book, substantial evidence implicates the ECS in the regulation of affective state, and dysfunctional eCB signaling is associated with increased anxiety and depression. In light of the evidence for drug-induced changes in brain eCB levels and eCB-mediated synaptic regulation, it is conceivable that dysregulated eCB function contributes to affective disturbances associated with drug dependence and protracted withdrawal. Several studies have begun to investigate an involvement of eCB signaling in withdrawal-related affective disruptions (or the efficacy of eCB manipulations as treatments for these disruptions), and as described below, these studies have predominantly focused on anxiety-like behavior that accompanies withdrawal from chronic exposure to drugs of abuse and highly palatable food.
In tobacco-dependent individuals, smoking cessation leads to withdrawal symptoms that persist for several months (Hughes 2007; Hughes et al. 1991; Markou 2008). Symptoms such as dysphoria/aversion, anxiety, and irritability contribute to negative reinforcement mechanisms that perpetuate nicotine addiction (Hughes et al. 1991; Koob and Volkow 2010; Piper et al. 2011; Allen et al. 2008; Rose et al. 2010), and the severity of these symptoms is a predictor of increased relapse risk (Piasecki et al. 2003a, b, c; al’Absi et al. 2004). Because eCB signaling is implicated in the initiation and maintenance of nicotine self-administration, it is possible that persistent disruptions in eCB signaling contribute to the negative affective states of nicotine withdrawal. However, few studies have investigated this possibility.
The Maldonado group reported that CB1 knockout mice exhibit more robust anxiety-like behavior during nicotine withdrawal than similarly treated wild-type mice do (Bura et al. 2010). However, nicotine-naïve CB1 knockout mice also exhibit greater anxiety-like behavior than wild types (Bura et al. 2010; Haller et al. 2002; Martin et al. 2002; Uriguen et al. 2004), and this innate phenotype clouds the determination as to whether or not withdrawal-related anxiety-like behavior is exacerbated by CB1 receptor deletion. In rats, significant increases in anxiety-like behavior are evident after 36 h of spontaneous nicotine withdrawal, and this is dose-dependently reversed by treatment with the FAAH inhibitor URB597 at doses that do not alter anxiety-like behavior in nicotine-naïve rats (Cippitelli et al. 2011). This is temporally correlated with significant increases in AEA content in the amygdala, hypothalamus, and prefrontal cortical tissue, though no significant alterations in tissue 2-AG content are evident regardless of region analyzed (Cippitelli et al. 2011). It is somewhat surprising that FAAH inhibition ameliorates anxiety-like behavior at a time when AEA levels appear to be increased in brain regions known to modulate anxiety-like behaviors. However, while increased AEA content in these regions may reflect a compensatory response to withdrawal, it is possible the effect magnitude is insufficient to fully constrain withdrawal-related disturbances underlying increased anxiety. Thus, the bolstering of this response through FAAH inhibition provides more efficacious reversal of withdrawal-related neural dysfunction (Cippitelli et al. 2011). It is worth noting, however, that disruptions in postmortem tissue AEA content may not provide a precise index of signaling-competent AEA levels in vivo (Buczynski and Parsons 2010), and a more clear in vivo view of withdrawal-related disruptions in regional eCBs may require further study. Nonetheless, these findings provide compelling evidence that acute FAAH inhibition ameliorates increased anxiety-like states during protracted nicotine withdrawal.
The putative eCB uptake inhibitor AM404 has been found to attenuate nicotine withdrawal-related increases in immobility in the Porsolt forced swim test (Mannucci et al. 2011). Increased immobility in this test is often interpreted as a form of despair or hopelessness, and this paradigm is commonly used as a screen for antidepressant efficacy. Accordingly, withdrawal-related increases in immobility in the Porsolt test may reflect a form of depressive-like behavior, and the effects of AM404 may thus reflect a beneficial effect of enhanced eCB signaling on withdrawal-related depressive-like states (Mannucci et al. 2011). However, AM404 also dose-dependently reduced immobility in nicotine-naïve mice in this study, and as such a preferential influence of this eCB clearance inhibitor on withdrawal-related behaviors is unclear. Moreover, modeling depressive-like behavior in rodents is best accomplished through the use of several paradigms (Abelaira et al. 2013; Yan et al. 2010), and further work is clearly needed to evaluate possible eCB influences on nicotine withdrawal-related depression-like behavior.
Post-traumatic stress disorder (PTSD) is an anxiety disorder with particularly high prevalence among individuals with alcohol use disorders (Kessler et al. 1995). The fear-potentiated startle (FPS) paradigm is often used to model PTSD-like behaviors (Grillon 2002; Hijzen et al. 1995). Rodent lines selectively bred for high alcohol consumption exhibit significantly greater FPS than corollary lines bred for low alcohol consumption (McKinzie et al. 2000; Barrenha and Chester 2007), and these rodent lines may thus provide models for characterizing mechanisms contributing to the anxiety, alcohol use disorders, and intersection of these pathologies. In this regard, the nonselective FAAH inhibitor LY2183240 selectively reduces FPS in high alcohol-preferring mice but not low alcohol-preferring mice (Powers et al. 2010). However, LY2183240 effects on FPS were evident only following repeated FPS testing and not upon first exposure to the FPS paradigm, suggesting that FAAH inhibition facilitates the extinction of learned fear responses in high alcohol-preferring mice, consistent with substantial evidence that FAAH inhibition generally accelerates extinction of aversive memories (Gunduz-Cinar et al. 2013). LY2183240 also enhanced the expression of alcohol-induced conditioned place preference without altering alcohol consumption itself. This suggests that FAAH inhibition influences memory-related processes regulating the expression of conditioned fear and conditioned alcohol reward in animals genetically predisposed toward high alcohol consumption. Recent evidence also points to the efficacy of the more selective FAAH inhibitor URB597 for attenuating anxiety-like behavior evident in rats following acute, high-dose alcohol administration (Cippitelli et al. 2008). In this study, URB597 did not alter alcohol self-administration, cue-induced alcohol-seeking, or stress-induced alcohol-seeking behavior. Collectively, these findings suggest that FAAH inhibition may be beneficial for reducing alcohol-related anxiety-like behavior, though further investigation is warranted, particularly evaluations of persistent anxiety-like behavior associated with protracted withdrawal in alcohol-dependent subjects.
In humans, the chronic use of the NMDA glutamate receptor antagonist phencyclidine (PCP) evokes both positive and negative symptoms of schizophrenia and many of the characteristic cognitive deficits associated with the disease (Murray 2002). In rats, withdrawal from sub-chronic PCP exposure is associated with reduced social interaction (Lee et al. 2005; Seillier and Giuffrida 2009; Seillier et al. 2013), a phenotype often interpreted as an anxiety-like behavior (Lapiz-Bluhm et al. 2008) and reminiscent of negative symptoms of schizophrenia. PCP withdrawal is associated with reduced AEA content in the amygdala and prefrontal cortex (Seillier and Giuffrida 2009; Seillier et al. 2013), and withdrawal-related reductions in social interaction are reversed by the FAAH inhibitor URB597 through a CB1-reliant mechanism (Seillier and Giuffrida 2009; Seillier et al. 2013).
Similar to drug addiction, eating disorders and pathological obesity are conceptualized as chronic relapsing conditions with alternating periods of abstinence (e.g., dieting) and relapse (compulsive overeating) (Corwin and Grigson 2009; Cottone et al. 2009a; Epstein and Shaham 2010; Johnson and Kenny 2010; Parylak et al. 2011). Rats given scheduled intermittent access to highly palatable sucrose food exhibit a withdrawal state upon forced abstinence from the palatable food, including increased anxiety-like behavior and decreased motivation for standard lab food (Cottone et al. 2008, 2009a, b). These effects are most evident during early abstinence and abate within 2–3 days. Moreover, these animals exhibit robust excessive food consumption of the palatable food when given renewed access. The negative affective state during abstinence and the excessive consumption upon renewed access to palatable food are each mediated by increased CRF1 signaling in the amygdala (Iemolo et al. 2013), similar to withdrawal-related negative affective states and compulsive drug consumption evident in drug-dependent subjects (Koob et al. 2014; Koob 2010). This suggests that dysregulation of amygdalar signaling is a common factor contributing to pathological motivation for and consumption of food and drugs. Interestingly, 2-AG content is significantly increased in the CeA following 4 days’ abstinence from palatable food, and this occurs in conjunction with increased CeA CB1 mRNA and CB1 protein expression (Blasio et al. 2013). Moreover, either systemic or intra-CeA administration of the CB1 antagonist rimonabant precipitates an anxiogenic-like state in animals with a history of palatable food consumption but not in rats given only with standard lab chow. This suggests that increased 2-AG-mediated CB1 signaling in CeA counteracts a withdrawal-like state in rats during abstinence from palatable food (Blasio et al. 2013).
12 Synaptic Plasticity
Substantial evidence demonstrates that exposure to most drugs of abuse dysregulates synaptic plasticity mechanisms in a variety of brain regions involved in the evolution of addiction (Hyman et al. 2006; Luscher and Malenka 2011). Acute exposure to alcohol, nicotine, morphine, or cocaine induces transient loss of LTP at inhibitory GABAergic synapses in the VTA (Niehaus et al. 2010; Liu et al. 2005; Melis et al. 2002; Nugent et al. 2007) and enhanced excitatory strength onto VTA DA neurons (Borgland et al. 2004; Faleiro et al. 2004; Saal et al. 2003). This tandem potentiation of excitatory transmission and loss of inhibitory LTP at VTA synapses serves to heighten midbrain DA cell excitability, increasing dopaminergic signaling throughout the mesocorticolimbic system.
As described in detail in the chapter titled “Endocannabinoid-Dependent Synaptic Plasticity in Striatum”, eCBs and CB1 receptors are implicated in several forms of synaptic plasticity involving presynaptic expression mechanisms (Lovinger 2008; Heifets and Castillo 2009). Both short- and long-term forms of eCB-mediated synaptic plasticity occur in many brain regions critically involved in various stages of the addiction cycle including the NAc, VTA, amygdala, prefrontal cortex, hippocampus, and dorsal striatum. Accordingly, drug-induced disruption of these mechanisms may contribute to aberrations in learning, memory, and affective state that propel addiction. Although a full description of these processes is provided in “Endocannabinoid-Dependent Synaptic Plasticity in Striatum”, it is worth noting here that exposure to several classes of abused drugs dysregulates eCB-mediated forms of synaptic plasticity. For example, eCB-mediated LTD of excitatory and inhibitory signaling in the rodent NAc and hippocampus is abolished following a 7-day treatment with either Δ9-THC or the synthetic CB1 agonist WIN 55,212-2 (Hoffman et al. 2003; Mato et al. 2004), correlating with decreased sensitivity to CB1 inhibition of both excitatory and inhibitory signaling.
Both acute and chronic alcohol exposure substantially reduces a CB1-dependent form of plasticity that results in long-lasting disinhibition of striatal output neurons (Clarke and Adermark 2010; Adermark et al. 2011) and reduces eCB-mediated LTD at inhibitory striatal synapses (though alcohol-induced disruption of eCB-LTD at excitatory synapses has not been observed) (Clarke and Adermark 2010). Because the dorsal striatum is involved in reward-guided learning and habitual behavior (Volkow et al. 2007; Yin et al. 2008), it is possible that these alcohol-induced disruptions in eCB-LTD contribute to maladaptive habitual behavior that contributes to addiction. Consistent with this hypothesis, recent work by the Lovinger lab demonstrates that chronic and intermittent alcohol exposure increases 2-AG levels in the dorsolateral striatum (DLS), leading to a compensatory downregulation of CB1 receptor signaling and loss of CB1-mediated LTD at excitatory synapses (DePoy et al. 2013). This was associated with enhancement of DLS-mediated learning processes that may contribute to habitual behaviors.
Cocaine exposure results in the loss of eCB-LTD of evoked excitatory transmission in the NAc (Fourgeaud et al. 2004) and facilitated eCB-LTD of GABAergic signaling at VTA DA synapses (Pan et al. 2008; Fourgeaud et al. 2004; Liu et al. 2005). Collectively, these cocaine-induced disruptions in eCB-LTD result in imbalanced mesolimbic DA function characterized by diminished inhibitory control over VTA DA cell bodies and heightened excitatory signaling in the NAc. Chronic cocaine exposure also results in disruption of eCB-LTD of excitatory transmission in the bed nucleus of the stria terminalis (BNST) (Grueter et al. 2006), a stress-responsive structure wherein excitatory transmission is critically involved in mediating stress-reward interactions and anxiety-like behavior (Delfs et al. 2000; McElligott and Winder 2009). The BNST sends substantial projections to the VTA, and accordingly disruption of BNST plasticity likely influences motivational responses to stress.
Little is known regarding the potential influence of opiate or nicotine exposure on eCB-mediated forms of synaptic plasticity. However, a recent report demonstrated that a history of nicotine self-administration induces CB1-dependent LTP at synapses of infralimbic cortex afferents to the BNST (Reisiger et al. 2014). This perturbation may have relevance to many consequences of chronic nicotine exposure, and clear evidence is provided that this neuroplastic change contributes to cue-induced reinstatement of nicotine-seeking behavior (in an animal model of relapse).
As noted above, stress is implicated in the development of addiction (Koob and Kreek 2007), and it is therefore notable that stress can alter eCB-mediated plasticity in addiction-related brain regions. Prolonged exposure to unpredictable stress impairs eCB-mediated DSI, LTD, and fEPSPs in the NAc (Wang et al. 2010), while repeated restraint stress reduces eCB-mediated DSE and DSI in the rat PVN (Wamsteeker et al. 2010) and increases 2-AG-mediated DSI in the mouse basolateral amygdala (Patel et al. 2009). Similarly, establishment of conditioned fear increases the efficacy of eCB-mediated DSE and DSI in the central nucleus of the amygdala (Kamprath et al. 2011). Further, it has recently been shown that acute restraint stress induces a switch from eCB-mediated LTD to eCB-mediated LTP at synapses of mPFC afferents to the BNST (Glangetas et al. 2013). As previously discussed, the ECS participates in a negative feedback system that limits the expression of anxiety under stressful circumstances and contributes to the suppression of aversive memories. These processes are mediated in part through eCB-mediated synaptic plasticity in the amygdalar nuclei (Kamprath et al. 2011; Lafenetre et al. 2007; Viveros et al. 2007), and disruptions of this eCB-mediated plasticity may contribute to its dysregulated affect (including increased anxiety) associated with protracted drug abstinence.
13 Extinction and Relapse
The ECS plays a prominent role in learning and memory processes (Hashimotodani et al. 2007; Heifets and Castillo 2009; Marsicano and Lafenetre 2009), and because eCB signaling is disrupted by most drugs of abuse (see above sections), a role for the ECS in learning and memory components of addiction may be hypothesized. As previously reviewed, CB1 receptors play an important role in the conditioned rewarding effects of alcohol (Houchi et al. 2005; Hungund et al. 2003; Thanos et al. 2005), opiates (Chaperon et al. 1998; Martin et al. 2000; Navarro et al. 2001; Singh et al. 2004), and nicotine (Castane et al. 2002; Forget et al. 2005, 2006; Le Foll and Goldberg 2004; Merritt et al. 2008), with a lesser CB1 influence reported for the conditioning effects of psychostimulants (Houchi et al. 2005; Martin et al. 2000). Although these behaviors are generally interpreted in the context of drug reward, a CB1 receptor influence on the associative learning aspects of drug exposure is also likely, which as discussed below may have relevance to the persistent reactivity to drug-related memories that characterize addiction.
13.1 Drug Seeking (Relapse)
Drug exposure produces powerful interoceptive effects, and memory of these effects increases the likelihood of continued drug use. With continued drug use, these interoceptive effects become associated with environmental cues to the extent where these drug-associated cues alone can induce craving and thereby propel drug use. This form of drug-related memory is also causal in relapse to drug use following periods of abstinence (Carter and Tiffany 1999; McLellan et al. 2000). Several factors are believed to be causal in drug relapse including craving induced by environments or situations previously associated with drug use (e.g., conditioning factors), acute exposure to the drug itself or a pharmacologically related agent during abstinence (e.g., drug priming), and stressful events.
Recently developed animal models of drug seeking demonstrate an important influence of cannabinoid signaling in the reinstatement of extinguished drug-seeking and drug-taking behaviors. For example, Δ9-THC and various synthetic CB1 agonists reinstate drug seeking for cannabinoids (Justinova et al. 2008b; Spano et al. 2004), opioids (De Vries et al. 2003; Fattore et al. 2003, 2005), ethanol (Lopez-Moreno et al. 2004; McGregor et al. 2005), nicotine (Biala and Budzynska 2008; Gamaleddin et al. 2012a), and cocaine (De Vries et al. 2001). Conversely, CB1 receptor antagonism attenuates drug-seeking behavior associated with a variety of abused substances. Rimonabant (SR141716A) significantly reduces operant responding for conditioned cues previously associated with Δ9-THC (Justinova et al. 2008b), heroin (De Vries et al. 2003; Fattore et al. 2003; 2005), nicotine (Cohen et al. 2005; De Vries et al. 2005; Diergaarde et al. 2008), and ethanol (Cippitelli et al. 2005; Economidou et al. 2006). CB1 antagonism also reduces excessive ethanol intake exhibited following periods of abstinence (an animal model of relapse-like alcohol consumption) (Gessa et al. 2005), and the novel CB1 antagonist SLV330 reduces both drug- and cue-induced reinstatement of ethanol- and nicotine-seeking behavior (de Bruin et al. 2011). Studies employing site-specific antagonist infusions have uncovered important contributions of CB1 receptors in the PFC and NAc shell in cue-induced reinstatement of both heroin- and nicotine-seeking behavior, and CB1 receptors in the BLA appear to contribute to cue-induced nicotine-seeking, but not heroin-seeking, behavior (Alvarez-Jaimes et al. 2008; Kodas et al. 2007). Although CB1 receptor inactivation produces subtle and inconsistent effects on psychostimulant self-administration as compared with other drugs of abuse, substantial evidence indicates a strong CB1 receptor influence on the reinstatement of psychostimulant-seeking behavior. CB1 antagonism attenuates both drug-primed and cue-induced reinstatement of cocaine-seeking (De Vries et al. 2001; Filip et al. 2006; Xi et al. 2006; Adamczyk et al. 2012b) and methamphetamine-seeking behavior in rats (Anggadiredja et al. 2004; Boctor et al. 2007). Further, both drug- and cue-induced reinstatement of cocaine and methamphetamine seeking is attenuated by the negative allosteric CB1 modulator ORG27569 (Jing et al. 2014). Interestingly, although footshock-induced reinstatement of cocaine seeking is not blocked by either rimonabant or the CB1 antagonist AM251 (De Vries et al. 2001; Kupferschmidt et al. 2012a), AM251 blocks the reinstatement of cocaine seeking induced by forced swim stress and exogenous CRF administration (Vaughn et al. 2012; Kupferschmidt et al. 2012a), suggesting a CB1 influence on some forms of stress-induced reinstatement of drug seeking. These latter findings may relate specifically to an AM251-induced modulation of the effects produced by the stress peptide CRF, given that AM251 also attenuates the anxiogenic effects of both cocaine withdrawal (an effect mediated by increased CRF signaling (DeVries and Pert 1998)) and exogenously administered CRF (Kupferschmidt et al. 2012b). However, the mechanisms for this putative interaction are not clear, and these findings are at odds with evidence that CB1 signaling generally suppresses the physiological and behavioral responses to stress (see discussion below).
Thus, CB1 receptor signaling is implicated in drug-seeking behavior for a variety of abused substances that differ substantially in pharmacodynamic mechanisms including cannabinoids, opioids, alcohol, nicotine, and psychostimulants. Moreover, CB1 receptor antagonism blocks both cue- and “prime”-induced reinstatements of seeking behavior for nondrug rewards such as sucrose and corn oil ((De Vries et al. 2005; Ward et al. 2009), but see Xi et al. (2006)). Accordingly, CB1 receptor signaling appears to participate in the modulation of conditioned reward in general.
Few studies have characterized the influence of eCB signaling on drug-seeking behavior through more direct manipulations of eCB processing such as the inhibition of hydrolytic clearance mechanisms. Three studies have found that both drug-primed and cue-induced nicotine- and cocaine-seeking behaviors are reduced by URB597 and the less selective FAAH inhibitor PMSF (Forget et al. 2009; Scherma et al. 2008; Adamczyk et al. 2009). This is somewhat surprising given that CB1 receptor stimulation enhances both nicotine- and cocaine-seeking behavior (Biala and Budzynska 2008; De Vries et al. 2001). However, because tonic eCB production is believed to be low, the inhibition of eCB clearance is anticipated to preferentially amplify eCB signaling in circuits/synapses activated by a given stimulus (in this case, drug-seeking behavior), rather than producing more widespread indiscriminate CB1 activation as is produced by exogenous CB1 agonists. Moreover, FAAH hydrolyzes a large variety of fatty acid moieties (Ahn et al. 2008), and it is conceivable that the effects of FAAH inhibition on drug seeking are mediated through actions of non-cannabinoid lipids. In this regard, it’s notable that the putative eCB uptake inhibitor VDM11 also attenuates both nicotine- and cue-induced nicotine-seeking behavior (Gamaleddin et al. 2011). This latter finding is of interest as unlike URB597, VDM11 elevates AEA levels with reduced effects on non-cannabinoid FAAH substrates that may influence behavior through PPARα signaling (De Petrocellis et al. 2000; van der Stelt et al. 2006). Similar to the effects of VDM11, the putative eCB uptake inhibitor AM404 dose-dependently attenuates both nicotine- and cue-induced nicotine-seeking behavior, though AM404 did not alter nicotine self-administration itself (Gamaleddin et al. 2013). In contrast to the effects on nicotine- and cocaine-seeking behavior, neither URB597 nor AM404 alters either cue- or stress-induced reinstatement of alcohol-seeking behavior (Cippitelli et al. 2007; 2008).
Recent data demonstrate that detoxified alcoholics present significantly lower baseline plasma AEA levels as compared with nondependent social drinkers (Mangieri et al. 2009). Further, a significant negative relationship between resting plasma AEA levels and the severity of cue-induced craving was evident in social drinkers but not alcoholics. In social drinkers, alcohol-related cues significantly increased both craving and plasma AEA levels, and the relative magnitude of cue-induced increases in AEA was significantly correlated with the severity of craving. However, cue-induced changes in AEA were indexed by a percentage change from baseline, and thus this index is substantially influenced by baseline AEA levels (e.g., the same absolute change in AEA content will be reflected as a greater percentage change in subjects with lower vs. higher baseline AEA levels). Accordingly, the positive correlation between cue-induced changes in plasma AEA and craving intensity further supports the importance of baseline AEA levels in cue-induced craving in social drinkers. Interestingly, although alcohol-related cues elicited more intense craving in alcoholics vs. social drinkers, alcoholics did not present significant cue-induced increases in plasma AEA. Based on these observations, it was concluded that cue-induced increases in plasma AEA contributes to interoceptive signaling that moderates the desire for alcohol in social drinkers. It may also be concluded that deficits in plasma AEA levels confer increased susceptibility to cue-induced alcohol craving. While cue-induced elevations in plasma AEA may contribute to craving responses, it is curious that elevations in AEA were not evident in alcoholics who also presented the greatest level of cue-induced craving. It is conceivable that cue-induced increases in plasma AEA reflect an adaptive response, and its absence in alcoholics somehow contributes to excessive craving responses to alcohol-related cues. Further evaluations in humans are clearly needed to investigate these issues.
13.2 Extinction Learning
The potent motivational effects of drug-related cues create substantial difficulties during periods of attempted drug abstinence and are causal in the reinstatement of drug intake (e.g., relapse) (Carter and Tiffany 1999). One approach for lessening the motivational impact of drug-associated cues is through extinction learning, wherein a subject learns that a drug-associated cue no longer has predictive value. However, extinction therapy for addiction is generally ineffective for reducing relapse in both humans (Conklin and Tiffany 2002) and rodents (Crombag and Shaham 2002), and it is conceivable that this results from disruptions in the learning mechanisms that override the original association memory (Bouton 2004; Rescorla 1996). The ECS plays a prominent role in memory extinction, particularly the extinction of aversive memory. Substantial evidence demonstrates that CB1 signaling blockade results in impaired extinction of cued fear memory, contextual fear memory, and fear-potentiated startle and spatial memory under aversive mildly stressful conditions (Marsicano et al. 2002; Suzuki et al. 2004; Chhatwal et al. 2005, 2009; Niyuhire et al. 2007; Varvel et al. 2005; Varvel and Lichtman 2002; Pamplona et al. 2006, 2008). Moreover, recent findings demonstrate that a line of mice selectively bred for high alcohol consumption exhibits excessive fear-potentiated startle responses and that the nonselective FAAH inhibitor LY2183240 facilitates extinction of fear-potentiated startle in these mice but not mice bred for low alcohol consumption (Powers et al. 2010). This suggests that FAAH inhibition influences memory-related processes regulating the expression of conditioned fear in animals genetically predisposed toward high alcohol consumption. However, it has consistently been reported that CB1 inactivation does not alter the extinction of behaviors motivated by appetitive and non-aversive memories (Holter et al. 2005; Niyuhire et al. 2007; Ward et al. 2007; Harloe et al. 2008; Hernandez and Cheer 2011). Because aversive memory may be involved in relapse to drug taking (Kaplan et al. 2011; Quirk and Gehlert 2003; Stewart and Kushner 2001), deficient eCB signaling following long-term drug exposure may contribute to the limited efficacy of extinction therapy for addiction.
13.3 Amygdala: Cortical Communication as a Possible Mechanism for eCB Modulation of Drug-Seeking Behavior
Considerable evidence highlights the importance of functional communication between the basolateral amygdala and prefrontal cortex (BLA->PFC) as a mediator of emotionally salient learning, memory, and synaptic plasticity. Distinct subpopulations of neurons in the PFC control the processing of aversive fear-related memory (Laviolette et al. 2005; Lauzon et al. 2009; Laviolette and Grace 2006) and rewarding and appetitive memory (Bishop et al. 2011; Sun et al. 2011), and both of these subpopulations are influenced by CB1 modulation of BLA inputs (Tan et al. 2014). Growing evidence demonstrates that excessive activation of CB1 receptors in either the BLA or PFC leads to amplification of aversive emotional information, while deficient CB1 signaling in these areas blunts the salience of normally aversive events while enhancing the salience of sub-reward threshold stimuli (Tan et al. 2010, 2014; Laviolette and Grace 2006). These processes appear to be mediated through direct interactions with subcortical DAergic motivational systems arising from the VTA, and through these interactions, cortical CB1 receptors are capable of modulating reward processing, even the switching of emotional valence from normally rewarding conditioned stimuli to aversive stimuli (Ahmad et al. 2013). The specific contribution of these mechanisms in the pathologically elevated motivational salience to drug-predictive cues and/or motivational effects of aversive memory contributing to drug seeking and relapse remains to be characterized (Tan et al. 2014). However, the potential involvement of these mechanisms not only in addiction but also in abnormal emotional learning and memory in other neuropsychiatric conditions is worth noting.
14 Cognitive Function
Impairments in cognitive processing are hallmarks of addiction. The progression from initial, intermittent, controlled drug use to compulsive drug seeking and consumption in the addicted state is conceptualized as a progressive deterioration of executive control over behavior (primarily reliant on frontal-cortical and hippocampal function) and the emergence of habitual or compulsive behaviors that drive drug seeking and drug taking (mediated in large part through dorsal striatal signaling) (Everitt and Robbins 2005; Jentsch and Taylor 1999). Endocannabinoids exert prominent control of frontal-cortical, hippocampal, and striatal synaptic signaling, and growing evidence implicates dysregulated eCB signaling in both aberrant executive function and habit-based behaviors driven by dorsal striatal signaling.
Endocannabinoid signaling regulates neural activity, structural plasticity, and functional output of the medial prefrontal cortex (McLaughlin et al. 2014) and is implicated in several PFC-mediated cognitive functions including attentional set shifting (Klugmann et al. 2011) and reversal learning in the Morris water maze (Varvel and Lichtman 2002; Lee et al. 2014). CB1 receptor antagonism or deletion results in shorter and less complex dendrites of neurons in layer II/III of the prelimbic cortex (Hill et al. 2011; Lee et al. 2014) and impairments in cognitive flexibility that resemble behaviors in mPFC-lesioned animals (Lacroix et al. 2002; Lee et al. 2014). Conversely, genetic deletion or pharmacological inhibition of FAAH enhances the acquisition of the Barnes maze task (Wise et al. 2009), enhances memory acquisition in a passive avoidance task (Mazzola et al. 2009), and facilitates acquisition and extinction of a spatial memory task (Varvel et al. 2007). Physical activity increases circulating AEA levels in both humans and rodents (Sparling et al. 2003; Hill et al. 2010b; Raichlen et al. 2013; Heyman et al. 2012), and this may contribute to the beneficial effects of exercise on hippocampal cell proliferation, neurogenesis, and synaptic plasticity (Hill et al. 2010b; Wolf et al. 2010; Madronal et al. 2012). Treadmill running increases spatial working memory through a CB1-reliant mechanism, and similar working memory enhancement is observed in sedentary animals treated with URB597 (Ferreira-Vieira et al. 2014).
Despite growing evidence for an eCB influence in cognitive functions mediated by the mPFC and hippocampus, few studies have investigated disruptions in these mechanisms following long-term drug exposure. Drug-induced impairments in recognition memory may result from altered hippocampal CB1 receptor function, and blockade of CB1 receptors during drug exposure has been shown to prevent deficits in recognition memory produced by chronic morphine, MDMA, and nicotine exposure (Nawata et al. 2010; Vaseghi et al. 2012, 2013; Saravia et al. 2016). Regarding drug-induced disruptions in frontal cortical function, the Woodward lab recently demonstrated that alcohol withdrawal is associated with deficient CB1-mediated inhibition of GABAergic signaling in layers II/III of the PFC and possible increases in CB1-mediated inhibition of GABAergic signaling in layers V/VI (Pava and Woodward 2014), suggesting a role for dysregulated CB1 signaling in altered cortical network activity that may underlie impaired cognitive function in detoxified alcoholics.
15 Food Addiction and Eating Disorders
Food addiction is defined as “a loss of control over food intake,” and growing evidence demonstrates that food and drug addiction have overlapping neuroadaptations in the mesolimbic system (Lutter and Nestler 2009). Indeed, eating disorders such as binge eating disorder (BED), anorexia nervosa (AN), and bulimia nervosa (BN) are hypothesized to derive in part from aberrant brain reward function (Kaye 2008; Marco et al. 2012; Scherma et al. 2014; Stoving et al. 2009). Similar to drug addiction, food addiction is defined in part by compulsive eating, continued aberrant feeding behavior despite negative consequences, and unsuccessful attempts to “normalize” dysfunctional eating (Cassin and von Ranson 2007; Gearhardt et al. 2012). The influence of cultural pressures and stress in these disorders is well documented, though there are also strong genetic contributions to these diseases (Kaye 2008; Klump et al. 2001). Furthermore, it is estimated that more than 50% of individuals with BED meet the criteria for food addiction (Cassin and von Ranson 2007; Gearhardt et al. 2012).
Several findings suggest an ECS involvement in eating disorders, including aberrant reward processing associated with these conditions (Table 2). For example, abnormal eating behaviors like self-starvation (in AN) or binge eating (in BN or BED) become rewarding in their own right, and this may be influenced by disrupted eCB function (Monteleone et al. 2012; Cervino et al. 2009). Patients with AN or BED (but not BN) present with increased blood AEA levels as compared with controls, though no changes in blood 2-AG levels are associated with these disorders (Monteleone et al. 2005). Moreover, blood AEA levels are significantly and inversely correlated with plasma leptin levels in women with AN, consistent with the inhibitory influence of leptin on AEA synthesis (Di Marzo et al. 2001) and leptin deficiency in these eating disorders. It should be noted, however, that although plasma eCB levels provide a potential index of disrupted eCB processing, they do not directly reflect dynamic changes in eCB signaling in the brain or other tissues. A CNR1 polymorphism is associated with vulnerability to AN (Siegfried et al. 2004) (but see Muller et al. (2008)), CB1 mRNA levels are increased in the blood of AN and BN patients (Frieling et al. 2009), and increased brain CB1 binding has been observed in patients with AN and BN (Gerard et al. 2011). Specifically, AN and BN patients exhibit increased CB1 availability in the bilateral insular cortex (a region critically involved in reward and emotional processing as well as interoception), and increased CB1 availability is evident in the inferior frontal and temporal cortices of AN patients (regions involved in emotional processing and executive function). An association between the C385A polymorphism in the gene encoding FAAH (leading to diminished FAAH activity) and the CNR1 polymorphism rs1049353 has been observed in higher frequency in patients with AN and BN (Monteleone et al. 2009) with a synergistic effect of these polymorphisms evident in patients with AN. The C385A polymorphism is also significantly associated with obesity (Monteleone et al. 2008; Sipe et al. 2005), though the relative contributions of possible disruptions in metabolism and hedonic mechanisms have not been determined. An association between a nonsynonymous CNR2 polymorphism and both AN and BN has also recently been reported (Ishiguro et al. 2010).
Limited research has been conducted on the effects of CB manipulations in animal models of eating disorders. Sub-chronic Δ9-THC treatment (0.5–2 mg/kg/day) was found to increase food intake and reduce body weight loss in an activity-based rat model of anorexia nervosa (Verty et al. 2005), though these effects were not evident in similar studies conducted in mice (Lewis and Brett 2010). Conversely, CB1 receptor antagonism selectively reduces binge-like vs. normal patterns of consumption for highly palatable sweet and high-fat foods (Scherma et al. 2013). It is possible this latter finding involves a mesolimbic DA mechanism in light of evidence that binge-like consumption of palatable or high-fat foods elicits increased NAc DA release upon repeated bouts of consumption (Rada et al. 2005; Liang et al. 2006), while the DAergic response to normal patterns of consumption abates upon repeated exposure to palatable foods (Bassareo and Di Chiara 1999b). Regarding clinical studies in humans, two small initial trials assessing the efficacy of Δ9-THC treatments for AN failed to demonstrate significant benefits on weight gain, though some beneficial effects on depression and perfectionism were evident (for review see Stoving et al. 2009). However, a more recent and larger trial demonstrated that a 4-week treatment with dronabinol induced a small but significant weight gain in women suffering from severe-enduring AN (Andries et al. 2014). Further, a recent randomized, placebo-controlled, double-blind trial in 289 obese subjects with binge eating disorders demonstrated that a 6-month treatment with rimonabant significantly reduced binge eating and led to significant weight loss with modest presentation of adverse psychiatric side effects (Pataky et al. 2013).
16 Conclusions and Future Directions
Endocannabinoid signaling participates in the mediation and modulation of both natural reward (such as food consumption, exercise, sex, and social interaction) and drug-induced reward. Most natural rewards and drugs of abuse alter brain eCB levels, and there is a robust CB1R influence on the motivation for both natural and drug rewards. Chronic drug use results in a neuroadaptive downregulation of CB1R and/or CB2R availability and function and may also lead to disruptions in eCB biosynthesis and clearance. The resulting deficit in eCB signaling may contribute to increased stress responsivity, increased negative affect, impaired extinction of drug-related memories, and drug seeking/craving that are known contributors to relapse. Recent animal studies suggest that eCB clearance inhibitors may have therapeutic potential for ameliorating these behavioral abnormalities related to addiction disorders. This therapeutic approach may present fewer unwanted behavioral effects than that produced by exogenous cannabinoids, given that eCBs are generally produced in a synapse-specific manner, and as such, eCB clearance inhibition may preferentially facilitate eCB signaling in specific circuits engaged by distinct stimuli (e.g., stress, drug-associated cues, etc.).
There are a number of notable gaps in our understanding of the eCB influence on reward and addiction. The ECS plays a prominent role in neuronal guidance and brain development (Maccarrone et al. 2014), and as such, disruptions in eCB function at an early age likely have substantial consequences for adult brain function. This is underscored by increasing evidence of the long-term consequences of prenatal or adolescent cannabinoid exposure (Hurd et al. 2014; Calvigioni et al. 2014). Though the effects of early-life exposure to non-cannabinoid drugs are well studied, the specific contributions of persistent drug-induced disruptions in eCB signaling on adult neural function and behavior are not understood. Robust bidirectional interactions between the ECS and sex hormones are now recognized (Gorzalka and Dang 2012), but few studies have characterized possible sex differences in the eCB influence on reward function, addiction, stress, and cognitive processing. There are also substantial limitations in the interpretation and replication of genetic analyses of the eCB influence in addiction due to heterogeneity of the populations studied, drug class, polysubstance use, and even drug use phenotypes examined. Large-scale future studies across different populations and drug classes will be critical to understand the relative impact and causal nature of ECS-related genetic mutations in the vulnerability to addictive disorders. Filling these gaps of knowledge is critical given the important need for scientific data to help guide current discussions and changes being made in marijuana legalization policies.
References
al’Absi M, Hatsukami D, Davis GL, Wittmers LE (2004) Prospective examination of effects of smoking abstinence on cortisol and withdrawal symptoms as predictors of early smoking relapse. Drug Alcohol Depend 73(3):267–278. doi:10.1016/j.drugalcdep.2003.10.014
Abelaira HM, Reus GZ, Quevedo J (2013) Animal models as tools to study the pathophysiology of depression. Revista brasileira de psiquiatria 35(Suppl 2):S112–S120. doi:10.1590/1516-4446-2013-1098
Adamczyk P, McCreary AC, Przegalinski E, Mierzejewski P, Bienkowski P, Filip M (2009) The effects of fatty acid amide hydrolase inhibitors on maintenance of cocaine and food self-administration and on reinstatement of cocaine-seeking and food-taking behavior in rats. J Physiol Pharmacol 60(3):119–125
Adamczyk P, Faron-Gorecka A, Kusmider M, Dziedzicka-Wasylewska M, Papp M, Filip M (2012a) Long-lasting increase in [(3)H]CP55,940 binding to CB1 receptors following cocaine self-administration and its withdrawal in rats. Brain Res 1451:34–43. doi:10.1016/j.brainres.2012.02.052
Adamczyk P, Miszkiel J, McCreary AC, Filip M, Papp M, Przegalinski E (2012b) The effects of cannabinoid CB1, CB2 and vanilloid TRPV1 receptor antagonists on cocaine addictive behavior in rats. Brain Res 1444:45–54. doi:10.1016/j.brainres.2012.01.030
Adermark L, Jonsson S, Ericson M, Soderpalm B (2011) Intermittent ethanol consumption depresses endocannabinoid-signaling in the dorsolateral striatum of rat. Neuropharmacology 61(7):1160–1165. doi:10.1016/j.neuropharm.2011.01.014
Ahmad T, Lauzon NM, de Jaeger X, Laviolette SR (2013) Cannabinoid transmission in the prelimbic cortex bidirectionally controls opiate reward and aversion signaling through dissociable kappa versus mu-opiate receptor dependent mechanisms. J Neurosci 33(39):15642–15651. doi:10.1523/JNEUROSCI.1686-13.2013
Ahn K, McKinney MK, Cravatt BF (2008) Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev 108(5):1687–1707. doi:10.1021/cr0782067
Al Mansouri S, Ojha S, Al Maamari E, Al Ameri M, Nurulain SM, Bahi A (2014) The cannabinoid receptor 2 agonist, beta-caryophyllene, reduced voluntary alcohol intake and attenuated ethanol-induced place preference and sensitivity in mice. Pharmacol Biochem Behav 124:260–268. doi:10.1016/j.pbb.2014.06.025
Alici T, Appel JB (2004) Increasing the selectivity of the discriminative stimulus effects of delta 9-tetrahydrocannabinol: complete substitution with methanandamide. Pharmacol Biochem Behav 79(3):431–437. doi:10.1016/j.pbb.2004.08.020
Allen SS, Bade T, Hatsukami D, Center B (2008) Craving, withdrawal, and smoking urges on days immediately prior to smoking relapse. Nicotine Tob Res 10(1):35–45. doi:10.1080/14622200701705076
Alling C, Balldin J, Bokstrom K, Gottfries CG, Karlsson I, Langstrom G (1982) Studies on duration of a late recovery period after chronic abuse of ethanol. A cross-sectional study of biochemical and psychiatric indicators. Acta Psychiatr Scand 66(5):384–397
Alvarez-Jaimes L, Parsons LH (2009) Regional influence of CB1 receptor signaling on ethanol self-administration by rats. Open Neuropsychopharmacol 2:77–85
Alvarez-Jaimes L, Polis I, Parsons LH (2008) Attenuation of cue-induced heroin-seeking behavior by cannabinoid CB1 antagonist infusions into the nucleus accumbens core and prefrontal cortex, but not basolateral amygdala. Neuropsychopharmacology 33(10):2483–2493. doi:10.1038/sj.npp.1301630
Alvarez-Jaimes L, Stouffer DG, Parsons LH (2009) Chronic ethanol treatment potentiates ethanol-induced increases in interstitial nucleus accumbens endocannabinoid levels in rats. J Neurochem 111(1):37-48. doi:10.1111/j.1471-4159.2009.06301.x
Anderson-Baker WC, McLaughlin CL, Baile CA (1979) Oral and hypothalamic injections of barbiturates, benzodiazepines and cannabinoids and food intake in rats. Pharmacol Biochem Behav 11(5):487–491
Andries A, Frystyk J, Flyvbjerg A, Stoving RK (2014) Dronabinol in severe, enduring anorexia nervosa: a randomized controlled trial. Int J Eat Disord 47(1):18–23. doi:10.1002/eat.22173
Anggadiredja K, Nakamichi M, Hiranita T, Tanaka H, Shoyama Y, Watanabe S, Yamamoto T (2004) Endocannabinoid system modulates relapse to methamphetamine seeking: possible mediation by the arachidonic acid cascade. Neuropsychopharmacology 29(8):1470–1478. doi:10.1038/sj.npp.1300454
Annis HM, Sklar SM, Moser AE (1998) Gender in relation to relapse crisis situations, coping, and outcome among treated alcoholics. Addict Behav 23(1):127–131
Aracil-Fernandez A, Trigo JM, Garcia-Gutierrez MS, Ortega-Alvaro A, Ternianov A, Navarro D, Robledo P, Berbel P, Maldonado R, Manzanares J (2012) Decreased cocaine motor sensitization and self-administration in mice overexpressing cannabinoid CB(2) receptors. Neuropsychopharmacology 37(7):1749–1763. doi:10.1038/npp.2012.22
Arnone M, Maruani J, Chaperon F, Thiebot MH, Poncelet M, Soubrie P, Le Fur G (1997) Selective inhibition of sucrose and ethanol intake by SR 141716, an antagonist of central cannabinoid (CB1) receptors. Psychopharmacology 132(1):104–106
Atwood BK, Mackie K (2010) CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol 160(3):467–479. doi:10.1111/j.1476-5381.2010.00729.x
Balerio GN, Aso E, Berrendero F, Murtra P, Maldonado R (2004) Delta9-tetrahydrocannabinol decreases somatic and motivational manifestations of nicotine withdrawal in mice. Eur J Neurosci 20(10):2737–2748. doi:10.1111/j.1460-9568.2004.03714.x
Balerio GN, Aso E, Maldonado R (2006) Role of the cannabinoid system in the effects induced by nicotine on anxiety-like behaviour in mice. Psychopharmacology 184(3-4):504–513. doi:10.1007/s00213-005-0251-9
Barna I, Zelena D, Arszovszki AC, Ledent C (2004) The role of endogenous cannabinoids in the hypothalamo-pituitary-adrenal axis regulation: in vivo and in vitro studies in CB1 receptor knockout mice. Life Sci 75(24):2959–2970. doi:10.1016/j.lfs.2004.06.006
Barrenha GD, Chester JA (2007) Genetic correlation between innate alcohol preference and fear-potentiated startle in selected mouse lines. Alcohol Clin Exp Res 31(7):1081–1088. doi:10.1111/j.1530-0277.2007.00396.x
Basavarajappa BS, Hungund BL (1999) Chronic ethanol increases the cannabinoid receptor agonist anandamide and its precursor N-arachidonoyl phosphatidylethanolamine in SK-N-SH cells. J Neurochem 72(2):522–528
Basavarajappa BS, Cooper TB, Hungund BL (1998) Chronic ethanol administration down-regulates cannabinoid receptors in mouse brain synaptic plasma membrane. Brain Res 793(1-2):212–218
Basavarajappa BS, Saito M, Cooper TB, Hungund BL (2000) Stimulation of cannabinoid receptor agonist 2-arachidonylglycerol by chronic ethanol and its modulation by specific neuromodulators in cerebellar granule neurons. Biochim Biophys Acta 1535(1):78–86
Basavarajappa BS, Saito M, Cooper TB, Hungund BL (2003) Chronic ethanol inhibits the anandamide transport and increases extracellular anandamide levels in cerebellar granule neurons. Eur J Pharmacol 466(1-2):73–83
Bassareo V, Di Chiara G (1999a) Differential responsiveness of dopamine transmission to food-stimuli in nucleus accumbens shell/core compartments. Neuroscience 89(3):637–641
Bassareo V, Di Chiara G (1999b) Modulation of feeding-induced activation of mesolimbic dopamine transmission by appetitive stimuli and its relation to motivational state. Eur J Neurosci 11(12):4389–4397
Bequet F, Uzabiaga F, Desbazeille M, Ludwiczak P, Maftouh M, Picard C, Scatton B, Le Fur G (2007) CB1 receptor-mediated control of the release of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. Eur J Neurosci 26(12):3458–3464. doi:10.1111/j.1460-9568.2007.05900.x
Bermudez-Silva FJ, Cardinal P, Cota D (2012) The role of the endocannabinoid system in the neuroendocrine regulation of energy balance. J Psychopharmacol 26(1):114–124. doi:10.1177/0269881111408458
Berridge KC, Ho CY, Richard JM, DiFeliceantonio AG (2010) The tempted brain eats: pleasure and desire circuits in obesity and eating disorders. Brain Res 1350:43–64. doi:10.1016/j.brainres.2010.04.003
Bhargava HN (1976) Effect of some cannabinoids on naloxone-precipitated abstinence in morphine-dependent mice. Psychopharmacology 49(3):267–270
Biala G, Budzynska B (2008) Calcium-dependent mechanisms of the reinstatement of nicotine-conditioned place preference by drug priming in rats. Pharmacol Biochem Behav 89(1):116–125. doi:10.1016/j.pbb.2007.12.005
Bishop SF, Lauzon NM, Bechard M, Gholizadeh S, Laviolette SR (2011) NMDA receptor hypofunction in the prelimbic cortex increases sensitivity to the rewarding properties of opiates via dopaminergic and amygdalar substrates. Cereb Cortex 21(1):68–80. doi:10.1093/cercor/bhq060
Blanco-Calvo E, Rivera P, Arrabal S, Vargas A, Pavon FJ, Serrano A, Castilla-Ortega E, Galeano P, Rubio L, Suarez J, Rodriguez de Fonseca F (2014) Pharmacological blockade of either cannabinoid CB1 or CB2 receptors prevents both cocaine-induced conditioned locomotion and cocaine-induced reduction of cell proliferation in the hippocampus of adult male rat. Front Integr Neurosci 7:106. doi:10.3389/fnint.2013.00106
Blasio A, Iemolo A, Sabino V, Petrosino S, Steardo L, Rice KC, Orlando P, Iannotti FA, Di Marzo V, Zorrilla EP, Cottone P (2013) Rimonabant precipitates anxiety in rats withdrawn from palatable food: role of the central amygdala. Neuropsychopharmacology 38(12):2498–2507. doi:10.1038/npp.2013.153
Blednov YA, Cravatt BF, Boehm SL 2nd, Walker D, Harris RA (2007) Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology 32(7):1570–1582. doi:10.1038/sj.npp.1301274
Boctor SY, Martinez JL Jr, Koek W, France CP (2007) The cannabinoid CB1 receptor antagonist AM251 does not modify methamphetamine reinstatement of responding. Eur J Pharmacol 571(1):39–43. doi:10.1016/j.ejphar.2007.06.004
Borgland SL, Malenka RC, Bonci A (2004) Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci 24(34):7482–7490. doi:10.1523/JNEUROSCI.1312-04.2004
Bouton ME (2004) Context and behavioral processes in extinction. Learn Mem 11(5):485–494. doi:10.1101/lm.78804
Breivogel CS, Childers SR, Deadwyler SA, Hampson RE, Vogt LJ, Sim-Selley LJ (1999) Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J Neurochem 73(6):2447–2459
Bruijnzeel AW, Repetto M, Gold MS (2004) Neurobiological mechanisms in addictive and psychiatric disorders. Psychiatr Clin North Am 27(4):661–674. doi:10.1016/j.psc.2004.06.005
Buczynski MW, Parsons LH (2010) Quantification of brain endocannabinoid levels: methods, interpretations and pitfalls. Br J Pharmacol 160(3):423–442. doi:10.1111/j.1476-5381.2010.00787.x
Buczynski MW, Polis IY, Parsons LH (2013) The volitional nature of nicotine exposure alters anandamide and oleoylethanolamide levels in the ventral tegmental area. Neuropsychopharmacology 38(4):574–584. doi:10.1038/npp.2012.210
Buczynski MW, Herman MA, Hsu KL, Natividad LA, Irimia C, Polis IY, Pugh H, Chang JW, Niphakis MJ, Cravatt BF, Roberto M, Parsons LH (2016) Diacylglycerol lipase disinhibits VTA dopamine neurons during chronic nicotine exposure. Proc Natl Acad Sci USA 113(4):1086–1091. doi:10.1073/pnas.1522672113
Budney AJ, Hughes JR, Moore BA, Novy PL (2001) Marijuana abstinence effects in marijuana smokers maintained in their home environment. Arch Gen Psychiatry 58(10):917–924
Bura SA, Burokas A, Martin-Garcia E, Maldonado R (2010) Effects of chronic nicotine on food intake and anxiety-like behaviour in CB(1) knockout mice. Eur Neuropsychopharmacol 20(6):369–378. doi:10.1016/j.euroneuro.2010.02.003
Burkey RT, Nation JR (1997) (R)-methanandamide, but not anandamide, substitutes for delta 9-THC in a drug-discrimination procedure. Exp Clin Psychopharmacol 5(3):195–202
Bystrowska B, Smaga I, Frankowska M, Filip M (2014) Changes in endocannabinoid and N-acylethanolamine levels in rat brain structures following cocaine self-administration and extinction training. Prog Neuropsychopharmacol Biol Psychiatry 50:1–10. doi:10.1016/j.pnpbp.2013.12.002
Caille S, Parsons LH (2003) SR141716A reduces the reinforcing properties of heroin but not heroin-induced increases in nucleus accumbens dopamine in rats. Eur J Neurosci 18(11):3145–3149
Caille S, Parsons LH (2006) Cannabinoid modulation of opiate reinforcement through the ventral striatopallidal pathway. Neuropsychopharmacology 31(4):804–813. doi:10.1038/sj.npp.1300848
Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH (2007) Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci 27(14):3695–3702. doi:10.1523/JNEUROSCI.4403-06.2007
Calvigioni D, Hurd YL, Harkany T, Keimpema E (2014) Neuronal substrates and functional consequences of prenatal cannabis exposure. Eur Child Adolesc Psychiatry 23(10):931–941. doi:10.1007/s00787-014-0550-y
Canseco-Alba A, Rodriguez-Manzo G (2016) Intra-VTA anandamide infusion produces dose-based biphasic effects on male rat sexual behavior expression. Pharmacol Biochem Behav 150-151:182–189. doi:10.1016/j.pbb.2016.11.004
Carter BL, Tiffany ST (1999) Cue-reactivity and the future of addiction research. Addiction 94(3):349–351
Cassin SE, von Ranson KM (2007) Is binge eating experienced as an addiction? Appetite 49(3):687–690. doi:10.1016/j.appet.2007.06.012
Castane A, Valjent E, Ledent C, Parmentier M, Maldonado R, Valverde O (2002) Lack of CB1 cannabinoid receptors modifies nicotine behavioural responses, but not nicotine abstinence. Neuropharmacology 43(5):857–867
Castane A, Berrendero F, Maldonado R (2005) The role of the cannabinoid system in nicotine addiction. Pharmacol Biochem Behav 81(2):381–386. doi:10.1016/j.pbb.2005.01.025
Castelli MP, Paola Piras A, D’Agostino A, Pibiri F, Perra S, Gessa GL, Maccarrone M, Pistis M (2007) Dysregulation of the endogenous cannabinoid system in adult rats prenatally treated with the cannabinoid agonist WIN 55,212-2. Eur J Pharmacol 573(1-3):11–19. doi:10.1016/j.ejphar.2007.06.047
Castle DJ (2008) Anxiety and substance use: layers of complexity. Expert Rev Neurother 8(3):493–501. doi:10.1586/14737175.8.3.493
Ceccarini J, Casteels C, Koole M, Bormans G, Van Laere K (2013) Transient changes in the endocannabinoid system after acute and chronic ethanol exposure and abstinence in the rat: a combined PET and microdialysis study. Eur J Nucl Med Mol Imaging 40(10):1582–1594. doi:10.1007/s00259-013-2456-1
Ceccarini J, Hompes T, Verhaeghen A, Casteels C, Peuskens H, Bormans G, Claes S, Van Laere K (2014) Changes in cerebral CB1 receptor availability after acute and chronic alcohol abuse and monitored abstinence. J Neurosci 34(8):2822–2831. doi:10.1523/JNEUROSCI.0849-13.2014
Ceccarini J, Kuepper R, Kemels D, van Os J, Henquet C, Van Laere K (2015) [18F]MK-9470 PET measurement of cannabinoid CB1 receptor availability in chronic cannabis users. Addict Biol 20(2):357–367. doi:10.1111/adb.12116
Cervino C, Vicennati V, Pasquali R, Pagotto U (2009) Feeding disorders and obesity. Curr Top Behav Neurosci 1:373–385. doi:10.1007/978-3-540-88955-7_15
Chaperon F, Soubrie P, Puech AJ, Thiebot MH (1998) Involvement of central cannabinoid (CB1) receptors in the establishment of place conditioning in rats. Psychopharmacology (Berl) 135(4):324–332
Cheer JF, Wassum KM, Sombers LA, Heien ML, Ariansen JL, Aragona BJ, Phillips PE, Wightman RM (2007) Phasic dopamine release evoked by abused substances requires cannabinoid receptor activation. J Neurosci 27(4):791–795. doi:10.1523/JNEUROSCI.4152-06.2007
Chhatwal JP, Davis M, Maguschak KA, Ressler KJ (2005) Enhancing cannabinoid neurotransmission augments the extinction of conditioned fear. Neuropsychopharmacology 30(3):516–524. doi:10.1038/sj.npp.1300655
Chhatwal JP, Gutman AR, Maguschak KA, Bowser ME, Yang Y, Davis M, Ressler KJ (2009) Functional interactions between endocannabinoid and CCK neurotransmitter systems may be critical for extinction learning. Neuropsychopharmacology 34(2):509–521. doi:10.1038/npp.2008.97
Cichewicz DL, Haller VL, Welch SP (2001) Changes in opioid and cannabinoid receptor protein following short-term combination treatment with delta(9)-tetrahydrocannabinol and morphine. J Pharmacol Exp Ther 297(1):121–127
Cippitelli A, Bilbao A, Hansson AC, del Arco I, Sommer W, Heilig M, Massi M, Bermudez-Silva FJ, Navarro M, Ciccocioppo R, de Fonseca FR (2005) Cannabinoid CB1 receptor antagonism reduces conditioned reinstatement of ethanol-seeking behavior in rats. Eur J Neurosci 21(8):2243–2251. doi:10.1111/j.1460-9568.2005.04056.x
Cippitelli A, Bilbao A, Gorriti MA, Navarro M, Massi M, Piomelli D, Ciccocioppo R, Rodriguez de Fonseca F (2007) The anandamide transport inhibitor AM404 reduces ethanol self-administration. Eur J Neurosci 26(2):476–486. doi:10.1111/j.1460-9568.2007.05665.x
Cippitelli A, Cannella N, Braconi S, Duranti A, Tontini A, Bilbao A, Defonseca FR, Piomelli D, Ciccocioppo R (2008) Increase of brain endocannabinoid anandamide levels by FAAH inhibition and alcohol abuse behaviours in the rat. Psychopharmacology (Berl) 198(4):449–460. doi:10.1007/s00213-008-1104-0
Cippitelli A, Astarita G, Duranti A, Caprioli G, Ubaldi M, Stopponi S, Kallupi M, Sagratini G, Rodriguez de Fonseca F, Piomelli D, Ciccocioppo R (2011) Endocannabinoid regulation of acute and protracted nicotine withdrawal: effect of FAAH inhibition. PLoS One 6(11):e28142. doi:10.1371/journal.pone.0028142
Clarke RB, Adermark L (2010) Acute ethanol treatment prevents endocannabinoid-mediated long-lasting disinhibition of striatal output. Neuropharmacology 58(4–5):799–805. doi:10.1016/j.neuropharm.2009.12.006
Coffey SF, Dansky BS, Carrigan MH, Brady KT (2000) Acute and protracted cocaine abstinence in an outpatient population: a prospective study of mood, sleep and withdrawal symptoms. Drug Alcohol Depend 59(3):277–286
Cohen C, Perrault G, Voltz C, Steinberg R, Soubrie P (2002) SR141716, a central cannabinoid (CB(1)) receptor antagonist, blocks the motivational and dopamine-releasing effects of nicotine in rats. Behav Pharmacol 13(5–6):451–463
Cohen C, Perrault G, Griebel G, Soubrie P (2005) Nicotine-associated cues maintain nicotine-seeking behavior in rats several weeks after nicotine withdrawal: reversal by the cannabinoid (CB1) receptor antagonist, rimonabant (SR141716). Neuropsychopharmacology 30(1):145–155. doi:10.1038/sj.npp.1300541
Colombo G, Agabio R, Fa M, Guano L, Lobina C, Loche A, Reali R, Gessa GL (1998) Reduction of voluntary ethanol intake in ethanol-preferring sP rats by the cannabinoid antagonist SR-141716. Alcohol Alcohol 33(2):126–130
Colombo G, Serra S, Brunetti G, Gomez R, Melis S, Vacca G, Carai MM, Gessa L (2002) Stimulation of voluntary ethanol intake by cannabinoid receptor agonists in ethanol-preferring sP rats. Psychopharmacology 159(2):181–187. doi:10.1007/s002130100887
Conklin CA, Tiffany ST (2002) Applying extinction research and theory to cue-exposure addiction treatments. Addiction 97(2):155–167
Conway KP, Compton W, Stinson FS, Grant BF (2006) Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry 67(2):247–257
Corwin RL, Grigson PS (2009) Symposium overview--Food addiction: fact or fiction? J Nutr 139(3):617–619. doi:10.3945/jn.108.097691
Cossu G, Ledent C, Fattore L, Imperato A, Bohme GA, Parmentier M, Fratta W (2001) Cannabinoid CB1 receptor knockout mice fail to self-administer morphine but not other drugs of abuse. Behav Brain Res 118(1):61–65
Cota D (2008) The role of the endocannabinoid system in the regulation of hypothalamic-pituitary-adrenal axis activity. J Neuroendocrinol 20(Suppl 1):35–38. doi:10.1111/j.1365-2826.2008.01673.x
Cota D, Steiner MA, Marsicano G, Cervino C, Herman JP, Grubler Y, Stalla J, Pasquali R, Lutz B, Stalla GK, Pagotto U (2007) Requirement of cannabinoid receptor type 1 for the basal modulation of hypothalamic-pituitary-adrenal axis function. Endocrinology 148(4):1574–1581. doi:10.1210/en.2005-1649
Cottone P, Sabino V, Steardo L, Zorrilla EP (2008) Intermittent access to preferred food reduces the reinforcing efficacy of chow in rats. Am J Physiol Regul Integr Comp Physiol 295(4):R1066–R1076. doi:10.1152/ajpregu.90309.2008
Cottone P, Sabino V, Roberto M, Bajo M, Pockros L, Frihauf JB, Fekete EM, Steardo L, Rice KC, Grigoriadis DE, Conti B, Koob GF, Zorrilla EP (2009a) CRF system recruitment mediates dark side of compulsive eating. Proc Natl Acad Sci USA 106(47):20016–20020. doi:10.1073/pnas.0908789106
Cottone P, Sabino V, Steardo L, Zorrilla EP (2009b) Consummatory, anxiety-related and metabolic adaptations in female rats with alternating access to preferred food. Psychoneuroendocrinology 34(1):38–49. doi:10.1016/j.psyneuen.2008.08.010
Crombag HS, Shaham Y (2002) Renewal of drug seeking by contextual cues after prolonged extinction in rats. Behav Neurosci 116(1):169–173
Dawley HH Jr, Winstead DK, Baxter AS, Gay JR (1979) An attitude survey of the effects of marijuana on sexual enjoyment. J Clin Psychol 35(1):212–217
de Bruin NM, Lange JH, Kruse CG, Herremans AH, Schoffelmeer AN, van Drimmelen M, De Vries TJ (2011) SLV330, a cannabinoid CB(1) receptor antagonist, attenuates ethanol and nicotine seeking and improves inhibitory response control in rats. Behav Brain Res 217(2):408–415. doi:10.1016/j.bbr.2010.11.013
De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V (2000) Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett 483(1):52–56
De Vries TJ, Shaham Y, Homberg JR, Crombag H, Schuurman K, Dieben J, Vanderschuren LJ, Schoffelmeer AN (2001) A cannabinoid mechanism in relapse to cocaine seeking. Nat Med 7(10):1151–1154. doi:10.1038/nm1001-1151
De Vries TJ, Homberg JR, Binnekade R, Raaso H, Schoffelmeer AN (2003) Cannabinoid modulation of the reinforcing and motivational properties of heroin and heroin-associated cues in rats. Psychopharmacology 168(1-2):164–169. doi:10.1007/s00213-003-1422-1
De Vries TJ, de Vries W, Janssen MC, Schoffelmeer AN (2005) Suppression of conditioned nicotine and sucrose seeking by the cannabinoid-1 receptor antagonist SR141716A. Behav Brain Res 161(1):164–168. doi:10.1016/j.bbr.2005.02.021
De Witte P, Pinto E, Ansseau M, Verbanck P (2003) Alcohol and withdrawal: from animal research to clinical issues. Neurosci Biobehav Rev 27(3):189–197
Delfs JM, Zhu Y, Druhan JP, Aston-Jones G (2000) Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature 403(6768):430–434. doi:10.1038/35000212
DePoy L, Daut R, Brigman JL, MacPherson K, Crowley N, Gunduz-Cinar O, Pickens CL, Cinar R, Saksida LM, Kunos G, Lovinger DM, Bussey TJ, Camp MC, Holmes A (2013) Chronic alcohol produces neuroadaptations to prime dorsal striatal learning. Proc Natl Acad Sci USA 110(36):14783–14788. doi:10.1073/pnas.1308198110
DeVries AC, Pert A (1998) Conditioned increases in anxiogenic-like behavior following exposure to contextual stimuli associated with cocaine are mediated by corticotropin-releasing factor. Psychopharmacology 137(4):333–340
Di S, Malcher-Lopes R, Halmos KC, Tasker JG (2003) Nongenomic glucocorticoid inhibition via endocannabinoid release in the hypothalamus: a fast feedback mechanism. J Neurosci 23(12):4850–4857
Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG (2005) Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol 569(Pt 3):751–760. doi:10.1113/jphysiol.2005.097477
Di S, Maxson MM, Franco A, Tasker JG (2009) Glucocorticoids regulate glutamate and GABA synapse-specific retrograde transmission via divergent nongenomic signaling pathways. J Neurosci 29(2):393–401. doi:10.1523/JNEUROSCI.4546-08.2009
Di Chiara G, Bassareo V (2007) Reward system and addiction: what dopamine does and doesn’t do. Curr Opin Pharmacol 7(1):69–76. doi:10.1016/j.coph.2006.11.003
Di Marzo V, Berrendero F, Bisogno T, Gonzalez S, Cavaliere P, Romero J, Cebeira M, Ramos JA, Fernandez-Ruiz JJ (2000) Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J Neurochem 74(4):1627–1635
Di Marzo V, Goparaju SK, Wang L, Liu J, Batkai S, Jarai Z, Fezza F, Miura GI, Palmiter RD, Sugiura T, Kunos G (2001) Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 410(6830):822–825. doi:10.1038/35071088
Diergaarde L, de Vries W, Raaso H, Schoffelmeer AN, De Vries TJ (2008) Contextual renewal of nicotine seeking in rats and its suppression by the cannabinoid-1 receptor antagonist Rimonabant (SR141716A). Neuropharmacology 55(5):712–716. doi:10.1016/j.neuropharm.2008.06.003
Dipatrizio NV, Simansky KJ (2008) Inhibiting parabrachial fatty acid amide hydrolase activity selectively increases the intake of palatable food via cannabinoid CB1 receptors. Am J Physiol Regul Integr Comp Physiol 295(5):R1409–R1414. doi:10.1152/ajpregu.90484.2008
Dodd GT, Stark JA, McKie S, Williams SR, Luckman SM (2009) Central cannabinoid signaling mediating food intake: a pharmacological-challenge magnetic resonance imaging and functional histology study in rat. Neuroscience 163(4):1192–1200. doi:10.1016/j.neuroscience.2009.07.022
Dole VP (1965) Thoughts on narcotics addiction. Bull NY Acad Med 41:211–213
Dole VP, Nyswander ME, Kreek MJ (1966) Narcotic blockade. Arch Intern Med 118(4):304–309
Douglas LA, Varlinskaya EI, Spear LP (2004) Rewarding properties of social interactions in adolescent and adult male and female rats: impact of social versus isolate housing of subjects and partners. Dev Psychobiol 45(3):153–162. doi:10.1002/dev.20025
D’Souza MS, Markou A (2011) Neuronal mechanisms underlying development of nicotine dependence: implications for novel smoking-cessation treatments. Addict Sci Clin Pract 6(1):4–16
Ducci F, Goldman D (2012) The genetic basis of addictive disorders. Psychiatr Clin North Am 35(2):495–519. doi:10.1016/j.psc.2012.03.010
Dudok B, Barna L, Ledri M, Szabo SI, Szabadits E, Pinter B, Woodhams SG, Henstridge CM, Balla GY, Nyilas R, Varga C, Lee SH, Matolcsi M, Cervenak J, Kacskovics I, Watanabe M, Sagheddu C, Melis M, Pistis M, Soltesz I, Katona I (2015) Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat Neurosci 18(1):75–86. doi:10.1038/nn.3892
Economidou D, Mattioli L, Cifani C, Perfumi M, Massi M, Cuomo V, Trabace L, Ciccocioppo R (2006) Effect of the cannabinoid CB1 receptor antagonist SR-141716A on ethanol self-administration and ethanol-seeking behaviour in rats. Psychopharmacology 183(4):394–403. doi:10.1007/s00213-005-0199-9
Edwards A, Abizaid A (2016) Driving the need to feed: insight into the collaborative interaction between ghrelin and endocannabinoid systems in modulating brain reward systems. Neurosci Biobehav Rev 66:33–53. doi:10.1016/j.neubiorev.2016.03.032
Epstein DH, Shaham Y (2010) Cheesecake-eating rats and the question of food addiction. Nat Neurosci 13(5):529–531. doi:10.1038/nn0510-529
Everitt BJ, Robbins TW (2005) Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8(11):1481–1489. doi:10.1038/nn1579
Everitt BJ, Belin D, Economidou D, Pelloux Y, Dalley JW, Robbins TW (2008) Review. Neural mechanisms underlying the vulnerability to develop compulsive drug-seeking habits and addiction. Philos Trans R Soc Lond B Biol Sci 363(1507):3125–3135. doi:10.1098/rstb.2008.0089
Faleiro LJ, Jones S, Kauer JA (2004) Rapid synaptic plasticity of glutamatergic synapses on dopamine neurons in the ventral tegmental area in response to acute amphetamine injection. Neuropsychopharmacology 29(12):2115–2125. doi:10.1038/sj.npp.1300495
Farrimond JA, Mercier MS, Whalley BJ, Williams CM (2011) Cannabis sativa and the endogenous cannabinoid system: therapeutic potential for appetite regulation. Phytother Res 25(2):170–188. doi:10.1002/ptr.3375
Fattore L, Martellotta MC, Cossu G, Mascia MS, Fratta W (1999) CB1 cannabinoid receptor agonist WIN 55,212-2 decreases intravenous cocaine self-administration in rats. Behav Brain Res 104(1-2):141–146
Fattore L, Spano MS, Cossu G, Deiana S, Fratta W (2003) Cannabinoid mechanism in reinstatement of heroin-seeking after a long period of abstinence in rats. Eur J Neurosci 17(8):1723–1726
Fattore L, Cossu G, Spano MS, Deiana S, Fadda P, Scherma M, Fratta W (2004) Cannabinoids and reward: interactions with the opioid system. Crit Rev Neurobiol 16(1-2):147–158
Fattore L, Deiana S, Spano SM, Cossu G, Fadda P, Scherma M, Fratta W (2005) Endocannabinoid system and opioid addiction: behavioural aspects. Pharmacol Biochem Behav 81(2):343–359. doi:10.1016/j.pbb.2005.01.031
Ferreira-Vieira TH, Bastos CP, Pereira GS, Moreira FA, Massensini AR (2014) A role for the endocannabinoid system in exercise-induced spatial memory enhancement in mice. Hippocampus 24(1):79–88. doi:10.1002/hipo.22206
Ferrer B, Bermudez-Silva FJ, Bilbao A, Alvarez-Jaimes L, Sanchez-Vera I, Giuffrida A, Serrano A, Baixeras E, Khaturia S, Navarro M, Parsons LH, Piomelli D, Rodriguez de Fonseca F (2007) Regulation of brain anandamide by acute administration of ethanol. Biochem J 404(1):97–104. doi:10.1042/BJ20061898
Filip M, Golda A, Zaniewska M, McCreary AC, Nowak E, Kolasiewicz W, Przegalinski E (2006) Involvement of cannabinoid CB1 receptors in drug addiction: effects of rimonabant on behavioral responses induced by cocaine. Pharmacol Rep 58(6):806–819
Fiorino DF, Coury A, Phillips AG (1997) Dynamic changes in nucleus accumbens dopamine efflux during the Coolidge effect in male rats. J Neurosci 17(12):4849–4855
Forget B, Hamon M, Thiebot MH (2005) Cannabinoid CB1 receptors are involved in motivational effects of nicotine in rats. Psychopharmacology 181(4):722–734. doi:10.1007/s00213-005-0015-6
Forget B, Barthelemy S, Saurini F, Hamon M, Thiebot MH (2006) Differential involvement of the endocannabinoid system in short- and long-term expression of incentive learning supported by nicotine in rats. Psychopharmacology 189(1):59–69. doi:10.1007/s00213-006-0525-x
Forget B, Coen KM, Le Foll B (2009) Inhibition of fatty acid amide hydrolase reduces reinstatement of nicotine seeking but not break point for nicotine self-administration--comparison with CB(1) receptor blockade. Psychopharmacology (Berl) 205(4):613–624. doi:10.1007/s00213-009-1569-5
Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, Manzoni OJ (2004) A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci 24(31):6939–6945. doi:10.1523/JNEUROSCI.0671-04.2004
Freed CR, Yamamoto BK (1985) Regional brain dopamine metabolism: a marker for the speed, direction, and posture of moving animals. Science 229(4708):62–65
Freedland CS, Sharpe AL, Samson HH, Porrino LJ (2001) Effects of SR141716A on ethanol and sucrose self-administration. Alcohol Clin Exp Res 25(2):277–282
Frieling H, Albrecht H, Jedtberg S, Gozner A, Lenz B, Wilhelm J, Hillemacher T, de Zwaan M, Kornhuber J, Bleich S (2009) Elevated cannabinoid 1 receptor mRNA is linked to eating disorder related behavior and attitudes in females with eating disorders. Psychoneuroendocrinology 34(4):620–624. doi:10.1016/j.psyneuen.2008.10.014
Gallate JE, McGregor IS (1999) The motivation for beer in rats: effects of ritanserin, naloxone and SR 141716. Psychopharmacology (Berl) 142(3):302–308
Gallate JE, Saharov T, Mallet PE, McGregor IS (1999) Increased motivation for beer in rats following administration of a cannabinoid CB1 receptor agonist. Eur J Pharmacol 370(3):233–240
Gamage TF, Ignatowska-Jankowska BM, Muldoon PP, Cravatt BF, Damaj MI, Lichtman AH (2015) Differential effects of endocannabinoid catabolic inhibitors on morphine withdrawal in mice. Drug Alcohol Depend 146:7–16. doi:10.1016/j.drugalcdep.2014.11.015
Gamaleddin I, Guranda M, Goldberg SR, Le Foll B (2011) The selective anandamide transport inhibitor VDM11 attenuates reinstatement of nicotine seeking behaviour, but does not affect nicotine intake. Br J Pharmacol 164(6):1652–1660. doi:10.1111/j.1476-5381.2011.01440.x
Gamaleddin I, Wertheim C, Zhu AZ, Coen KM, Vemuri K, Makryannis A, Goldberg SR, Le Foll B (2012a) Cannabinoid receptor stimulation increases motivation for nicotine and nicotine seeking. Addict Biol 17(1):47–61. doi:10.1111/j.1369-1600.2011.00314.x
Gamaleddin I, Zvonok A, Makriyannis A, Goldberg SR, Le Foll B (2012b) Effects of a selective cannabinoid CB2 agonist and antagonist on intravenous nicotine self administration and reinstatement of nicotine seeking. PLoS One 7(1):e29900. doi:10.1371/journal.pone.0029900
Gamaleddin I, Guranda M, Scherma M, Fratta W, Makriyannis A, Vadivel SK, Goldberg SR, Le Foll B (2013) AM404 attenuates reinstatement of nicotine seeking induced by nicotine-associated cues and nicotine priming but does not affect nicotine- and food-taking. J Psychopharmacol 27(6):564–571. doi:10.1177/0269881113477710
Gates P, Albertella L, Copeland J (2015) Cannabis withdrawal and sleep: a systematic review of human studies. Subst Abus. doi:10.1080/08897077.2015.1023484
Gearhardt AN, White MA, Masheb RM, Morgan PT, Crosby RD, Grilo CM (2012) An examination of the food addiction construct in obese patients with binge eating disorder. Int J Eat Disord 45(5):657–663. doi:10.1002/eat.20957
Gerard N, Pieters G, Goffin K, Bormans G, Van Laere K (2011) Brain type 1 cannabinoid receptor availability in patients with anorexia and bulimia nervosa. Biol Psychiatry 70(8):777–784. doi:10.1016/j.biopsych.2011.05.010
Gerrits MA, Van Ree JM (1996) Effect of nucleus accumbens dopamine depletion on motivational aspects involved in initiation of cocaine and heroin self-administration in rats. Brain Res 713(1-2):114–124
Gessa GL, Serra S, Vacca G, Carai MA, Colombo G (2005) Suppressing effect of the cannabinoid CB1 receptor antagonist, SR147778, on alcohol intake and motivational properties of alcohol in alcohol-preferring sP rats. Alcohol Alcohol 40(1):46–53. doi:10.1093/alcalc/agh114
Getachew B, Hauser SR, Dhaher R, Katner SN, Bell RL, Oster SM, McBride WJ, Rodd ZA (2011) CB1 receptors regulate alcohol-seeking behavior and alcohol self-administration of alcohol-preferring (P) rats. Pharmacol Biochem Behav 97(4):669–675. doi:10.1016/j.pbb.2010.11.006
Ghosh S, Wise LE, Chen Y, Gujjar R, Mahadevan A, Cravatt BF, Lichtman AH (2013) The monoacylglycerol lipase inhibitor JZL184 suppresses inflammatory pain in the mouse carrageenan model. Life Sci 92(8–9):498–505. doi:10.1016/j.lfs.2012.06.020
Glangetas C, Girard D, Groc L, Marsicano G, Chaouloff F, Georges F (2013) Stress switches cannabinoid type-1 (CB1) receptor-dependent plasticity from LTD to LTP in the bed nucleus of the stria terminalis. J Neurosci 33(50):19657–19663. doi:10.1523/JNEUROSCI.3175-13.2013
Glass M, Dragunow M, Faull RL (1997) Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience 77(2):299–318
Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D (2005) Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA 102(51):18620–18625. doi:10.1073/pnas.0509591102
Goeders NE (2002) Stress and cocaine addiction. J Pharmacol Exp Ther 301(3):785–789
Goeders NE, Guerin GF (1996) Role of corticosterone in intravenous cocaine self-administration in rats. Neuroendocrinology 64(5):337–348
Gonzalez S, Cascio MG, Fernandez-Ruiz J, Fezza F, Di Marzo V, Ramos JA (2002) Changes in endocannabinoid contents in the brain of rats chronically exposed to nicotine, ethanol or cocaine. Brain Res 954(1):73–81
Gonzalez S, Fernandez-Ruiz J, Di Marzo V, Hernandez M, Arevalo C, Nicanor C, Cascio MG, Ambrosio E, Ramos JA (2004a) Behavioral and molecular changes elicited by acute administration of SR141716 to Delta9-tetrahydrocannabinol-tolerant rats: an experimental model of cannabinoid abstinence. Drug Alcohol Depend 74(2):159–170. doi:10.1016/j.drugalcdep.2003.12.011
Gonzalez S, Valenti M, de Miguel R, Fezza F, Fernandez-Ruiz J, Di Marzo V, Ramos JA (2004b) Changes in endocannabinoid contents in reward-related brain regions of alcohol-exposed rats, and their possible relevance to alcohol relapse. Br J Pharmacol 143(4):455–464. doi:10.1038/sj.bjp.0705963
Gorzalka BB, Dang SS (2012) Minireview: Endocannabinoids and gonadal hormones: bidirectional interactions in physiology and behavior. Endocrinology 153(3):1016–1024. doi:10.1210/en.2011-1643
Gorzalka BB, Hill MN, Hillard CJ (2008) Regulation of endocannabinoid signaling by stress: implications for stress-related affective disorders. Neurosci Biobehav Rev 32(6):1152–1160. doi:10.1016/j.neubiorev.2008.03.004
Gorzalka BB, Hill MN, Chang SC (2010) Male-female differences in the effects of cannabinoids on sexual behavior and gonadal hormone function. Horm Behav 58(1):91–99. doi:10.1016/j.yhbeh.2009.08.009
Grillon C (2002) Startle reactivity and anxiety disorders: aversive conditioning, context, and neurobiology. Biol Psychiatry 52(10):958–975
Grueter BA, Gosnell HB, Olsen CM, Schramm-Sapyta NL, Nekrasova T, Landreth GE, Winder DG (2006) Extracellular-signal regulated kinase 1-dependent metabotropic glutamate receptor 5-induced long-term depression in the bed nucleus of the stria terminalis is disrupted by cocaine administration. J Neurosci 26(12):3210–3219. doi:10.1523/JNEUROSCI.0170-06.2006
Guegan T, Cutando L, Ayuso E, Santini E, Fisone G, Bosch F, Martinez A, Valjent E, Maldonado R, Martin M (2013) Operant behavior to obtain palatable food modifies neuronal plasticity in the brain reward circuit. Eur Neuropsychopharmacol 23(2):146–159. doi:10.1016/j.euroneuro.2012.04.004
Gunduz-Cinar O, Hill MN, McEwen BS, Holmes A (2013) Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends Pharmacol Sci 34(11):637–644. doi:10.1016/j.tips.2013.08.008
Hajnal A, Smith GP, Norgren R (2004) Oral sucrose stimulation increases accumbens dopamine in the rat. Am J Physiol Regul Integr Comp Physiol 286(1):R31–R37. doi:10.1152/ajpregu.00282.2003
Halikas J, Weller R, Morse C (1982) Effects of regular marijuana use on sexual performance. J Psychoactive Drugs 14(1-2):59–70. doi:10.1080/02791072.1982.10471911
Haller J, Bakos N, Szirmay M, Ledent C, Freund TF (2002) The effects of genetic and pharmacological blockade of the CB1 cannabinoid receptor on anxiety. Eur J Neurosci 16(7):1395–1398
Hampson AJ, Bornheim LM, Scanziani M, Yost CS, Gray AT, Hansen BM, Leonoudakis DJ, Bickler PE (1998) Dual effects of anandamide on NMDA receptor-mediated responses and neurotransmission. J Neurochem 70(2):671–676
Hansson AC, Bermudez-Silva FJ, Malinen H, Hyytia P, Sanchez-Vera I, Rimondini R, Rodriguez de Fonseca F, Kunos G, Sommer WH, Heilig M (2007) Genetic impairment of frontocortical endocannabinoid degradation and high alcohol preference. Neuropsychopharmacology 32(1):117–126. doi:10.1038/sj.npp.1301034
Hanus L, Avraham Y, Ben-Shushan D, Zolotarev O, Berry EM, Mechoulam R (2003) Short-term fasting and prolonged semistarvation have opposite effects on 2-AG levels in mouse brain. Brain Res 983(1-2):144–151
Harloe JP, Thorpe AJ, Lichtman AH (2008) Differential endocannabinoid regulation of extinction in appetitive and aversive Barnes maze tasks. Learn Mem 15(11):806–809. doi:10.1101/lm.1113008
Hashimotodani Y, Ohno-Shosaku T, Kano M (2007) Endocannabinoids and synaptic function in the CNS. Neuroscientist 13(2):127–137. doi:10.1177/1073858406296716
Hattori S, Naoi M, Nishino H (1994) Striatal dopamine turnover during treadmill running in the rat: relation to the speed of running. Brain Res Bull 35(1):41–49
Hayase T, Yamamoto Y, Yamamoto K (2001) Protective effects of cannabinoid receptor agonists against cocaine and other convulsant-induced toxic behavioural symptoms. J Pharmacy Pharmacol 53(11):1525–1532
Heifets BD, Castillo PE (2009) Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol 71:283–306. doi:10.1146/annurev.physiol.010908.163149
Heilig M, Egli M, Crabbe JC, Becker HC (2010) Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol 15(2):169–184. doi:10.1111/j.1369-1600.2009.00194.x
Henricks AM, Berger AL, Lugo JM, Baxter-Potter LN, Bieniasz KV, Craft RM, McLaughlin RJ (2016) Sex differences in alcohol consumption and alterations in nucleus accumbens endocannabinoid mRNA in alcohol-dependent rats. Neuroscience 335:195–206. doi:10.1016/j.neuroscience.2016.08.032
Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991) Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11(2):563–583
Hernandez G, Cheer JF (2011) Extinction learning of rewards in the rat: is there a role for CB1 receptors? Psychopharmacology 217(2):189–197. doi:10.1007/s00213-011-2275-7
Hernandez G, Cheer JF (2015) To act or not to act: endocannabinoid/dopamine interactions in decision-making. Front Behav Neurosci 9:336. doi:10.3389/fnbeh.2015.00336
Hershon HI (1977) Alcohol withdrawal symptoms and drinking behavior. J Stud Alcohol 38(5):953–971
Heyman E, Gamelin FX, Goekint M, Piscitelli F, Roelands B, Leclair E, Di Marzo V, Meeusen R (2012) Intense exercise increases circulating endocannabinoid and BDNF levels in humans--possible implications for reward and depression. Psychoneuroendocrinology 37(6):844–851. doi:10.1016/j.psyneuen.2011.09.017
Higgs S, Williams CM, Kirkham TC (2003) Cannabinoid influences on palatability: microstructural analysis of sucrose drinking after delta(9)-tetrahydrocannabinol, anandamide, 2-arachidonoyl glycerol and SR141716. Psychopharmacology (Berl) 165(4):370–377. doi:10.1007/s00213-002-1263-3
Hijzen TH, Houtzager SW, Joordens RJ, Olivier B, Slangen JL (1995) Predictive validity of the potentiated startle response as a behavioral model for anxiolytic drugs. Psychopharmacology (Berl) 118(2):150–154
Hill MN, Carrier EJ, McLaughlin RJ, Morrish AC, Meier SE, Hillard CJ, Gorzalka BB (2008) Regional alterations in the endocannabinoid system in an animal model of depression: effects of concurrent antidepressant treatment. J Neurochem 106(6):2322–2336. doi:10.1111/j.1471-4159.2008.05567.x
Hill MN, McLaughlin RJ, Morrish AC, Viau V, Floresco SB, Hillard CJ, Gorzalka BB (2009) Suppression of amygdalar endocannabinoid signaling by stress contributes to activation of the hypothalamic-pituitary-adrenal axis. Neuropsychopharmacology 34(13):2733–2745. doi:10.1038/npp.2009.114
Hill MN, Karatsoreos IN, Hillard CJ, McEwen BS (2010a) Rapid elevations in limbic endocannabinoid content by glucocorticoid hormones in vivo. Psychoneuroendocrinology 35(9):1333–1338. doi:10.1016/j.psyneuen.2010.03.005
Hill MN, Titterness AK, Morrish AC, Carrier EJ, Lee TT, Gil-Mohapel J, Gorzalka BB, Hillard CJ, Christie BR (2010b) Endogenous cannabinoid signaling is required for voluntary exercise-induced enhancement of progenitor cell proliferation in the hippocampus. Hippocampus 20(4):513–523. doi:10.1002/hipo.20647
Hill MN, Hillard CJ, McEwen BS (2011) Alterations in corticolimbic dendritic morphology and emotional behavior in cannabinoid CB1 receptor-deficient mice parallel the effects of chronic stress. Cereb Cortex 21(9):2056–2064. doi:10.1093/cercor/bhq280
Hillard CJ (2015) The endocannabinoid signaling system in the CNS: a primer. Int Rev Neurobiol 125:1–47. doi:10.1016/bs.irn.2015.10.001
Hine B, Friedman E, Torrelio M, Gershon S (1975) Morphine-dependent rats: blockade of precipitated abstinence by tetrahydrocannabinol. Science 187(4175):443–445
Hirvonen J, Zanotti-Fregonara P, Umhau JC, George DT, Rallis-Frutos D, Lyoo CH, Li CT, Hines CS, Sun H, Terry GE, Morse C, Zoghbi SS, Pike VW, Innis RB, Heilig M (2013) Reduced cannabinoid CB1 receptor binding in alcohol dependence measured with positron emission tomography. Mol Psychiatry 18(8):916–921. doi:10.1038/mp.2012.100
Hoffman AF, Oz M, Caulder T, Lupica CR (2003) Functional tolerance and blockade of long-term depression at synapses in the nucleus accumbens after chronic cannabinoid exposure. J Neurosci 23(12):4815–4820
Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435(7045):1108–1112. doi:10.1038/nature03658
Holter SM, Kallnik M, Wurst W, Marsicano G, Lutz B, Wotjak CT (2005) Cannabinoid CB1 receptor is dispensable for memory extinction in an appetitively-motivated learning task. Eur J Pharmacol 510(1-2):69–74. doi:10.1016/j.ejphar.2005.01.008
Horvath TL, Diano S (2004) The floating blueprint of hypothalamic feeding circuits. Nat Rev Neurosci 5(8):662–667. doi:10.1038/nrn1479
Houchi H, Babovic D, Pierrefiche O, Ledent C, Daoust M, Naassila M (2005) CB1 receptor knockout mice display reduced ethanol-induced conditioned place preference and increased striatal dopamine D2 receptors. Neuropsychopharmacology 30(2):339–349. doi:10.1038/sj.npp.1300568
Hughes JR (2007) Effects of abstinence from tobacco: valid symptoms and time course. Nicotine Tob Res 9(3):315–327. doi:10.1080/14622200701188919
Hughes JR, Gust SW, Skoog K, Keenan RM, Fenwick JW (1991) Symptoms of tobacco withdrawal. A replication and extension. Arch Gen Psychiatry 48(1):52–59
Hungund BL, Szakall I, Adam A, Basavarajappa BS, Vadasz C (2003) Cannabinoid CB1 receptor knockout mice exhibit markedly reduced voluntary alcohol consumption and lack alcohol-induced dopamine release in the nucleus accumbens. J Neurochem 84(4):698–704
Hurd YL, Michaelides M, Miller ML, Jutras-Aswad D (2014) Trajectory of adolescent cannabis use on addiction vulnerability. Neuropharmacology 76(Pt B):416–424. doi:10.1016/j.neuropharm.2013.07.028
Hyman SE, Malenka RC, Nestler EJ (2006) Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29:565–598. doi:10.1146/annurev.neuro.29.051605.113009
Iemolo A, Blasio A, St Cyr SA, Jiang F, Rice KC, Sabino V, Cottone P (2013) CRF-CRF1 receptor system in the central and basolateral nuclei of the amygdala differentially mediates excessive eating of palatable food. Neuropsychopharmacology 38(12):2456–2466. doi:10.1038/npp.2013.147
Ignatowska-Jankowska BM, Muldoon PP, Lichtman AH, Damaj MI (2013) The cannabinoid CB2 receptor is necessary for nicotine-conditioned place preference, but not other behavioral effects of nicotine in mice. Psychopharmacology (Berl) 229(4):591–601. doi:10.1007/s00213-013-3117-6
Ignatowska-Jankowska BM, Ghosh S, Crowe MS, Kinsey SG, Niphakis MJ, Abdullah RA, Tao Q, ST ON, Walentiny DM, Wiley JL, Cravatt BF, Lichtman AH (2014) In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol 171 (6):1392-1407. doi:10.1111/bph.12298
Ishiguro H, Carpio O, Horiuchi Y, Shu A, Higuchi S, Schanz N, Benno R, Arinami T, Onaivi ES (2010) A nonsynonymous polymorphism in cannabinoid CB2 receptor gene is associated with eating disorders in humans and food intake is modified in mice by its ligands. Synapse 64(1):92–96. doi:10.1002/syn.20714
Jamshidi N, Taylor DA (2001) Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol 134(6):1151–1154. doi:10.1038/sj.bjp.0704379
Janiri L, Martinotti G, Dario T, Reina D, Paparello F, Pozzi G, Addolorato G, Di Giannantonio M, De Risio S (2005) Anhedonia and substance-related symptoms in detoxified substance-dependent subjects: a correlation study. Neuropsychobiology 52(1):37–44. doi:10.1159/000086176
Jarbe TU, Lamb RJ, Liu Q, Makriyannis A (2006) Discriminative stimulus functions of AM-1346, a CB1R selective anandamide analog in rats trained with Delta9-THC or (R)-methanandamide (AM-356). Psychopharmacology (Berl) 188(3):315–323. doi:10.1007/s00213-006-0517-x
Jarrett MM, Limebeer CL, Parker LA (2005) Effect of Delta9-tetrahydrocannabinol on sucrose palatability as measured by the taste reactivity test. Physiol Behav 86(4):475–479. doi:10.1016/j.physbeh.2005.08.033
Jarrett MM, Scantlebury J, Parker LA (2007) Effect of delta9-tetrahydrocannabinol on quinine palatability and AM251 on sucrose and quinine palatability using the taste reactivity test. Physiol Behav 90(2-3):425–430. doi:10.1016/j.physbeh.2006.10.003
Jentsch JD, Taylor JR (1999) Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology 146(4):373–390
Jing L, Qiu Y, Zhang Y, Li JX (2014) Effects of the cannabinoid CB(1) receptor allosteric modulator ORG 27569 on reinstatement of cocaine- and methamphetamine-seeking behavior in rats. Drug Alcohol Depend 143:251–256. doi:10.1016/j.drugalcdep.2014.08.004
Johnson PM, Kenny PJ (2010) Dopamine D2 receptors in addiction-like reward dysfunction and compulsive eating in obese rats. Nat Neurosci 13(5):635–641. doi:10.1038/nn.2519
Jose BS, van Oers HA, van de Mheen HD, Garretsen HF, Mackenbach JP (2000) Stressors and alcohol consumption. Alcohol Alcohol 35(3):307–312
Justinova Z, Solinas M, Tanda G, Redhi GH, Goldberg SR (2005) The endogenous cannabinoid anandamide and its synthetic analog R(+)-methanandamide are intravenously self-administered by squirrel monkeys. J Neurosci 25(23):5645–5650. doi:10.1523/JNEUROSCI.0951-05.2005
Justinova Z, Mangieri RA, Bortolato M, Chefer SI, Mukhin AG, Clapper JR, King AR, Redhi GH, Yasar S, Piomelli D, Goldberg SR (2008a) Fatty acid amide hydrolase inhibition heightens anandamide signaling without producing reinforcing effects in primates. Biol Psychiatry 64(11):930–937. doi:10.1016/j.biopsych.2008.08.008
Justinova Z, Munzar P, Panlilio LV, Yasar S, Redhi GH, Tanda G, Goldberg SR (2008b) Blockade of THC-seeking behavior and relapse in monkeys by the cannabinoid CB(1)-receptor antagonist rimonabant. Neuropsychopharmacology 33(12):2870–2877. doi:10.1038/npp.2008.21
Justinova Z, Yasar S, Redhi GH, Goldberg SR (2011) The endogenous cannabinoid 2-arachidonoylglycerol is intravenously self-administered by squirrel monkeys. J Neurosci 31(19):7043–7048. doi:10.1523/JNEUROSCI.6058-10.2011
Justinova Z, Panlilio LV, Moreno-Sanz G, Redhi GH, Auber A, Secci ME, Mascia P, Bandiera T, Armirotti A, Bertorelli R, Chefer SI, Barnes C, Yasar S, Piomelli D, Goldberg SR (2015) Effects of fatty acid amide hydrolase (FAAH) inhibitors in non-human primate models of nicotine reward and relapse. Neuropsychopharmacology. doi:10.1038/npp.2015.62
Jyotaki M, Shigemura N, Ninomiya Y (2010) Modulation of sweet taste sensitivity by orexigenic and anorexigenic factors. Endocr J 57(6):467–475
Kalivas PW, Volkow ND (2005) The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry 162(8):1403–1413. doi:10.1176/appi.ajp.162.8.1403
Kamprath K, Romo-Parra H, Haring M, Gaburro S, Doengi M, Lutz B, Pape HC (2011) Short-term adaptation of conditioned fear responses through endocannabinoid signaling in the central amygdala. Neuropsychopharmacology 36(3):652–663. doi:10.1038/npp.2010.196
Kaplan GB, Heinrichs SC, Carey RJ (2011) Treatment of addiction and anxiety using extinction approaches: neural mechanisms and their treatment implications. Pharmacol Biochem Behav 97(3):619–625. doi:10.1016/j.pbb.2010.08.004
Kaye W (2008) Neurobiology of anorexia and bulimia nervosa. Physiol Behav 94(1):121–135. doi:10.1016/j.physbeh.2007.11.037
Kelley AE, Baldo BA, Pratt WE, Will MJ (2005) Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiol Behav 86(5):773–795. doi:10.1016/j.physbeh.2005.08.066
Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB (1995) Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry 52(12):1048–1060
Kinsey SG, Wise LE, Ramesh D, Abdullah R, Selley DE, Cravatt BF, Lichtman AH (2013) Repeated low-dose administration of the monoacylglycerol lipase inhibitor JZL184 retains cannabinoid receptor type 1-mediated antinociceptive and gastroprotective effects. J Pharmacol Exp Ther 345(3):492–501. doi:10.1124/jpet.112.201426
Kirkham TC, Williams CM (2001) Endogenous cannabinoids and appetite. Nutr Res Rev 14(1):65–86. doi:10.1079/NRR200118
Kirkham TC, Williams CM, Fezza F, Di Marzo V (2002) Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol 136(4):550–557. doi:10.1038/sj.bjp.0704767
Klein C, Hill MN, Chang SC, Hillard CJ, Gorzalka BB (2012) Circulating endocannabinoid concentrations and sexual arousal in women. J Sex Med 9(6):1588–1601. doi:10.1111/j.1743-6109.2012.02708.x
Klugmann M, Goepfrich A, Friemel CM, Schneider M (2011) AAV-mediated overexpression of the CB1 receptor in the mPFC of adult rats alters cognitive flexibility, social behavior, and emotional reactivity. Front Behav Neurosci 5:37. doi:10.3389/fnbeh.2011.00037
Klump KL, Miller KB, Keel PK, McGue M, Iacono WG (2001) Genetic and environmental influences on anorexia nervosa syndromes in a population-based twin sample. Psychol Med 31(4):737–740
Kodas E, Cohen C, Louis C, Griebel G (2007) Cortico-limbic circuitry for conditioned nicotine-seeking behavior in rats involves endocannabinoid signaling. Psychopharmacology (Berl) 194(2):161–171. doi:10.1007/s00213-007-0813-0
Koff WC (1974) Marijuana and sexual activity. J Sex Res 10(3):194–204. doi:10.1080/00224497409550850
Komisaruk BR, Whipple B, Crawford A, Liu WC, Kalnin A, Mosier K (2004) Brain activation during vaginocervical self-stimulation and orgasm in women with complete spinal cord injury: fMRI evidence of mediation by the vagus nerves. Brain Res 1024(1–2):77–88. doi:10.1016/j.brainres.2004.07.029
Koob GF (1992) Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci 13(5):177–184
Koob GF (2010) The role of CRF and CRF-related peptides in the dark side of addiction. Brain Res 1314:3–14. doi:10.1016/j.brainres.2009.11.008
Koob GF (2013) Theoretical frameworks and mechanistic aspects of alcohol addiction: alcohol addiction as a reward deficit disorder. Curr Top Behav Neurosci 13:3–30. doi:10.1007/7854_2011_129
Koob G, Kreek MJ (2007) Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry 164(8):1149–1159. doi:10.1176/appi.ajp.2007.05030503
Koob GF, Le Moal M (1997) Drug abuse: hedonic homeostatic dysregulation. Science 278(5335):52–58
Koob GF, Volkow ND (2010) Neurocircuitry of addiction. Neuropsychopharmacology 35(1):217–238. doi:10.1038/npp.2009.110
Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW Jr, George O (2014) Addiction as a stress surfeit disorder. Neuropharmacology 76(Pt B):370–382. doi:10.1016/j.neuropharm.2013.05.024
Kupferschmidt DA, Klas PG, Erb S (2012a) Cannabinoid CB1 receptors mediate the effects of corticotropin-releasing factor on the reinstatement of cocaine seeking and expression of cocaine-induced behavioural sensitization. Br J Pharmacol 167(1):196–206. doi:10.1111/j.1476-5381.2012.01983.x
Kupferschmidt DA, Newman AE, Boonstra R, Erb S (2012b) Antagonism of cannabinoid 1 receptors reverses the anxiety-like behavior induced by central injections of corticotropin-releasing factor and cocaine withdrawal. Neuroscience 204:125–133. doi:10.1016/j.neuroscience.2011.07.022
Lacroix L, White I, Feldon J (2002) Effect of excitotoxic lesions of rat medial prefrontal cortex on spatial memory. Behav Brain Res 133(1):69–81
Lafenetre P, Chaouloff F, Marsicano G (2007) The endocannabinoid system in the processing of anxiety and fear and how CB1 receptors may modulate fear extinction. Pharmacol Res 56(5):367–381. doi:10.1016/j.phrs.2007.09.006
Lapiz-Bluhm MD, Bondi CO, Doyen J, Rodriguez GA, Bedard-Arana T, Morilak DA (2008) Behavioural assays to model cognitive and affective dimensions of depression and anxiety in rats. J Neuroendocrinol 20(10):1115–1137. doi:10.1111/j.1365-2826.2008.01772.x
Lauzon NM, Bishop SF, Laviolette SR (2009) Dopamine D1 versus D4 receptors differentially modulate the encoding of salient versus nonsalient emotional information in the medial prefrontal cortex. J Neurosci 29(15):4836–4845. doi:10.1523/JNEUROSCI.0178-09.2009
Laviolette SR, Grace AA (2006) Cannabinoids potentiate emotional learning plasticity in neurons of the medial prefrontal cortex through basolateral amygdala inputs. J Neurosci 26(24):6458–6468. doi:10.1523/JNEUROSCI.0707-06.2006
Laviolette SR, Lipski WJ, Grace AA (2005) A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input. J Neurosci 25(26):6066–6075. doi:10.1523/JNEUROSCI.1168-05.2005
Le Foll B, Goldberg SR (2004) Rimonabant, a CB1 antagonist, blocks nicotine-conditioned place preferences. Neuroreport 15(13):2139–2143
Le Moal M (2009) Drug abuse: vulnerability and transition to addiction. Pharmacopsychiatry 42(Suppl 1):S42–S55. doi:10.1055/s-0029-1216355
Le Moal M, Simon H (1991) Mesocorticolimbic dopaminergic network: functional and regulatory roles. Physiol Rev 71(1):155–234
Ledent C, Valverde O, Cossu G, Petitet F, Aubert JF, Beslot F, Bohme GA, Imperato A, Pedrazzini T, Roques BP, Vassart G, Fratta W, Parmentier M (1999) Unresponsiveness to cannabinoids and reduced addictive effects of opiates in CB1 receptor knockout mice. Science 283(5400):401–404
Lee PR, Brady DL, Shapiro RA, Dorsa DM, Koenig JI (2005) Social interaction deficits caused by chronic phencyclidine administration are reversed by oxytocin. Neuropsychopharmacology 30(10):1883–1894. doi:10.1038/sj.npp.1300722
Lee TT, Filipski SB, Hill MN, McEwen BS (2014) Morphological and behavioral evidence for impaired prefrontal cortical function in female CB1 receptor deficient mice. Behav Brain Res 271:106–110. doi:10.1016/j.bbr.2014.05.064
Lesscher HM, Hoogveld E, Burbach JP, van Ree JM, Gerrits MA (2005) Endogenous cannabinoids are not involved in cocaine reinforcement and development of cocaine-induced behavioural sensitization. Eur Neuropsychopharmacol 15(1):31–37. doi:10.1016/j.euroneuro.2004.04.003
Leweke FM, Giuffrida A, Koethe D, Schreiber D, Nolden BM, Kranaster L, Neatby MA, Schneider M, Gerth CW, Hellmich M, Klosterkotter J, Piomelli D (2007) Anandamide levels in cerebrospinal fluid of first-episode schizophrenic patients: impact of cannabis use. Schizophr Res 94(1–3):29–36. doi:10.1016/j.schres.2007.04.025
Lewis DY, Brett RR (2010) Activity-based anorexia in C57/BL6 mice: effects of the phytocannabinoid, Delta9-tetrahydrocannabinol (THC) and the anandamide analogue, OMDM-2. Eur Neuropsychopharmacol 20(9):622–631. doi:10.1016/j.euroneuro.2010.04.002
Li X, Hoffman AF, Peng XQ, Lupica CR, Gardner EL, Xi ZX (2009) Attenuation of basal and cocaine-enhanced locomotion and nucleus accumbens dopamine in cannabinoid CB1-receptor-knockout mice. Psychopharmacology (Berl) 204(1):1–11. doi:10.1007/s00213-008-1432-0
Liang NC, Hajnal A, Norgren R (2006) Sham feeding corn oil increases accumbens dopamine in the rat. Am J Physiol Regul Integr Comp Physiol 291(5):R1236–R1239. doi:10.1152/ajpregu.00226.2006
Lichtman AH, Sheikh SM, Loh HH, Martin BR (2001) Opioid and cannabinoid modulation of precipitated withdrawal in delta(9)-tetrahydrocannabinol and morphine-dependent mice. J Pharmacol Exp Ther 298(3):1007–1014
Liu QS, Pu L, Poo MM (2005) Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature 437(7061):1027–1031. doi:10.1038/nature04050
Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF (2009a) Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5(1):37–44. doi:10.1038/nchembio.129
Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF (2009b) Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci USA 106(48):20270–20275. doi:10.1073/pnas.0909411106
Lopez HH (2010) Cannabinoid-hormone interactions in the regulation of motivational processes. Horm Behav 58(1):100–110. doi:10.1016/j.yhbeh.2009.10.005
Lopez-Moreno JA, Gonzalez-Cuevas G, Rodriguez de Fonseca F, Navarro M (2004) Long-lasting increase of alcohol relapse by the cannabinoid receptor agonist WIN 55,212-2 during alcohol deprivation. J Neurosci 24(38):8245–8252. doi:10.1523/JNEUROSCI.2179-04.2004
Lovinger DM (2008) Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol 184:435–477. doi:10.1007/978-3-540-74805-2_14
Lu HC, Mackie K (2015) An introduction to the endogenous cannabinoid system. Biol Psychiatry. doi:10.1016/j.biopsych.2015.07.028
Luchicchi A, Lecca S, Carta S, Pillolla G, Muntoni AL, Yasar S, Goldberg SR, Pistis M (2010) Effects of fatty acid amide hydrolase inhibition on neuronal responses to nicotine, cocaine and morphine in the nucleus accumbens shell and ventral tegmental area: involvement of PPAR-alpha nuclear receptors. Addict Biol 15(3):277–288. doi:10.1111/j.1369-1600.2010.00222.x
Luscher C, Malenka RC (2011) Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron 69(4):650–663. doi:10.1016/j.neuron.2011.01.017
Lutter M, Nestler EJ (2009) Homeostatic and hedonic signals interact in the regulation of food intake. J Nutr 139(3):629–632. doi:10.3945/jn.108.097618
Lutz B (2009) Endocannabinoid signals in the control of emotion. Curr Opin Pharmacol 9(1):46–52. doi:10.1016/j.coph.2008.12.001
Maccarrone M, Guzman M, Mackie K, Doherty P, Harkany T (2014) Programming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat Rev Neurosci 15(12):786–801. doi:10.1038/nrn3846
Macht M, Mueller J (2007) Immediate effects of chocolate on experimentally induced mood states. Appetite 49(3):667–674. doi:10.1016/j.appet.2007.05.004
Madronal N, Gruart A, Valverde O, Espadas I, Moratalla R, Delgado-Garcia JM (2012) Involvement of cannabinoid CB1 receptor in associative learning and in hippocampal CA3-CA1 synaptic plasticity. Cereb Cortex 22(3):550–566. doi:10.1093/cercor/bhr103
Mahler SV, Smith KS, Berridge KC (2007) Endocannabinoid hedonic hotspot for sensory pleasure: anandamide in nucleus accumbens shell enhances ‘liking’ of a sweet reward. Neuropsychopharmacology 32(11):2267–2278. doi:10.1038/sj.npp.1301376
Malinen H, Hyytia P (2008) Ethanol self-administration is regulated by CB1 receptors in the nucleus accumbens and ventral tegmental area in alcohol-preferring AA rats. Alcohol Clin Exp Res 32(11):1976–1983. doi:10.1111/j.1530-0277.2008.00786.x
Malinen H, Lehtonen M, Hyytia P (2009) Modulation of brain endocannabinoid levels by voluntary alcohol consumption in alcohol-preferring AA rats. Alcohol Clin Exp Res 33(10):1711–1720. doi:10.1111/j.1530-0277.2009.01008.x
Mallet PE, Beninger RJ (1998) Delta9-tetrahydrocannabinol, but not the endogenous cannabinoid receptor ligand anandamide, produces conditioned place avoidance. Life Sci 62(26):2431–2439
Mangieri RA, Hong KI, Piomelli D, Sinha R (2009) An endocannabinoid signal associated with desire for alcohol is suppressed in recently abstinent alcoholics. Psychopharmacology (Berl) 205(1):63–72. doi:10.1007/s00213-009-1518-3
Mannucci C, Navarra M, Pieratti A, Russo GA, Caputi AP, Calapai G (2011) Interactions between endocannabinoid and serotonergic systems in mood disorders caused by nicotine withdrawal. Nicotine Tob Res 13(4):239–247. doi:10.1093/ntr/ntq242
Manzanares J, Corchero J, Fuentes JA (1999) Opioid and cannabinoid receptor-mediated regulation of the increase in adrenocorticotropin hormone and corticosterone plasma concentrations induced by central administration of delta(9)-tetrahydrocannabinol in rats. Brain Res 839(1):173–179
Manzanedo C, Aguilar MA, Rodriguez-Arias M, Navarro M, Minarro J (2004) Cannabinoid agonist-induced sensitisation to morphine place preference in mice. Neuroreport 15(8):1373–1377
Marco EM, Granstrem O, Moreno E, Llorente R, Adriani W, Laviola G, Viveros MP (2007) Subchronic nicotine exposure in adolescence induces long-term effects on hippocampal and striatal cannabinoid-CB1 and mu-opioid receptors in rats. Eur J Pharmacol 557(1):37–43. doi:10.1016/j.ejphar.2006.11.013
Marco EM, Romero-Zerbo SY, Viveros MP, Bermudez-Silva FJ (2012) The role of the endocannabinoid system in eating disorders: pharmacological implications. Behav Pharmacol 23(5-6):526–536. doi:10.1097/FBP.0b013e328356c3c9
Markou A (2008) Review. Neurobiology of nicotine dependence. Philos Trans R Soc Lond B Biol Sci 363(1507):3159–3168. doi:10.1098/rstb.2008.0095
Marsicano G, Lafenetre P (2009) Roles of the endocannabinoid system in learning and memory. Curr Top Behav Neurosci 1:201–230. doi:10.1007/978-3-540-88955-7_8
Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418(6897):530–534. doi:10.1038/nature00839
Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O (2000) Cocaine, but not morphine, induces conditioned place preference and sensitization to locomotor responses in CB1 knockout mice. Eur J Neurosci 12(11):4038–4046
Martin M, Ledent C, Parmentier M, Maldonado R, Valverde O (2002) Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology 159(4):379–387. doi:10.1007/s00213-001-0946-5
Martinez-Gonzalez D, Bonilla-Jaime H, Morales-Otal A, Henriksen SJ, Velazquez-Moctezuma J, Prospero-Garcia O (2004) Oleamide and anandamide effects on food intake and sexual behavior of rats. Neurosci Lett 364(1):1–6. doi:10.1016/j.neulet.2004.03.080
Mascia MS, Obinu MC, Ledent C, Parmentier M, Bohme GA, Imperato A, Fratta W (1999) Lack of morphine-induced dopamine release in the nucleus accumbens of cannabinoid CB(1) receptor knockout mice. Eur J Pharmacol 383(3):R1–R2
Mas-Nieto M, Pommier B, Tzavara ET, Caneparo A, Da Nascimento S, Le Fur G, Roques BP, Noble F (2001) Reduction of opioid dependence by the CB(1) antagonist SR141716A in mice: evaluation of the interest in pharmacotherapy of opioid addiction. Br J Pharmacol 132(8):1809–1816. doi:10.1038/sj.bjp.0703990
Mathes CM, Ferrara M, Rowland NE (2008) Cannabinoid-1 receptor antagonists reduce caloric intake by decreasing palatable diet selection in a novel dessert protocol in female rats. Am J Physiol Regul Integr Comp Physiol 295(1):R67–R75. doi:10.1152/ajpregu.00150.2008
Mato S, Chevaleyre V, Robbe D, Pazos A, Castillo PE, Manzoni OJ (2004) A single in-vivo exposure to delta 9THC blocks endocannabinoid-mediated synaptic plasticity. Nat Neurosci 7(6):585–586. doi:10.1038/nn1251
Mazzola C, Medalie J, Scherma M, Panlilio LV, Solinas M, Tanda G, Drago F, Cadet JL, Goldberg SR, Yasar S (2009) Fatty acid amide hydrolase (FAAH) inhibition enhances memory acquisition through activation of PPAR-alpha nuclear receptors. Learn Mem 16(5):332–337. doi:10.1101/lm.1145209
McElligott ZA, Winder DG (2009) Modulation of glutamatergic synaptic transmission in the bed nucleus of the stria terminalis. Prog Neuropsychopharmacol Biol Psychiatry 33(8):1329–1335. doi:10.1016/j.pnpbp.2009.05.022
McGregor IS, Dam KD, Mallet PE, Gallate JE (2005) Delta9-THC reinstates beer- and sucrose-seeking behaviour in abstinent rats: comparison with midazolam, food deprivation and predator odour. Alcohol Alcohol 40(1):35–45. doi:10.1093/alcalc/agh113
McKinzie DL, Sajdyk TJ, McBride WJ, Murphy JM, Lumeng L, Li TK, Shekhar A (2000) Acoustic startle and fear-potentiated startle in alcohol-preferring (P) and -nonpreferring (NP) lines of rats. Pharmacol Biochem Behav 65(4):691–696
McLaughlin RJ, Hill MN, Gorzalka BB (2014) A critical role for prefrontocortical endocannabinoid signaling in the regulation of stress and emotional behavior. Neurosci Biobehav Rev 42:116–131. doi:10.1016/j.neubiorev.2014.02.006
McLellan AT, Lewis DC, O’Brien CP, Kleber HD (2000) Drug dependence, a chronic medical illness: implications for treatment, insurance, and outcomes evaluation. JAMA 284(13):1689–1695
Mechoulam R, Parker LA (2013) The endocannabinoid system and the brain. Annu Rev Psychol 64:21–47. doi:10.1146/annurev-psych-113011-143739
Melis M, Pistis M (2012a) Hub and switches: endocannabinoid signalling in midbrain dopamine neurons. Philos Trans R Soc Lond B Biol Sci 367(1607):3276–3285. doi:10.1098/rstb.2011.0383
Melis M, Pistis M (2012b) Hub and switches: endocannabinoid signalling in midbrain dopamine neurons. Philos Trans R Soc Lond B Biol Sci 367(1607):3276–3285. doi:10.1098/rstb.2011.0383
Melis M, Pistis M (2014) Targeting the interaction between fatty acid ethanolamides and nicotinic receptors: therapeutic perspectives. Pharmacol Res 86:42–49. doi:10.1016/j.phrs.2014.03.009
Melis M, Camarini R, Ungless MA, Bonci A (2002) Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22(6):2074–2082
Melis T, Succu S, Sanna F, Boi A, Argiolas A, Melis MR (2007) The cannabinoid antagonist SR 141716A (Rimonabant) reduces the increase of extra-cellular dopamine release in the rat nucleus accumbens induced by a novel high palatable food. Neurosci Lett 419(3):231–235. doi:10.1016/j.neulet.2007.04.012
Mereu M, Tronci V, Chun LE, Thomas AM, Green JL, Katz JL, Tanda G (2013) Cocaine-induced endocannabinoid release modulates behavioral and neurochemical sensitization in mice. Addict Biol 20(4):91–103. doi:10.1111/adb.12080
Merritt LL, Martin BR, Walters C, Lichtman AH, Damaj MI (2008) The endogenous cannabinoid system modulates nicotine reward and dependence. J Pharmacol Exp Ther 326(2):483–492. doi:10.1124/jpet.108.138321
Miller WR, Harris RJ (2000) A simple scale of Gorski’s warning signs for relapse. J Stud Alcohol 61(5):759–765
Mitrirattanakul S, Lopez-Valdes HE, Liang J, Matsuka Y, Mackie K, Faull KF, Spigelman I (2007) Bidirectional alterations of hippocampal cannabinoid 1 receptors and their endogenous ligands in a rat model of alcohol withdrawal and dependence. Alcohol Clin Exp Res 31(5):855–867. doi:10.1111/j.1530-0277.2007.00366.x
Monteleone P, Matias I, Martiadis V, De Petrocellis L, Maj M, Di Marzo V (2005) Blood levels of the endocannabinoid anandamide are increased in anorexia nervosa and in binge-eating disorder, but not in bulimia nervosa. Neuropsychopharmacology 30(6):1216–1221. doi:10.1038/sj.npp.1300695
Monteleone P, Tortorella A, Martiadis V, Di Filippo C, Canestrelli B, Maj M (2008) The cDNA 385C to A missense polymorphism of the endocannabinoid degrading enzyme fatty acid amide hydrolase (FAAH) is associated with overweight/obesity but not with binge eating disorder in overweight/obese women. Psychoneuroendocrinology 33(4):546–550. doi:10.1016/j.psyneuen.2008.01.004
Monteleone P, Bifulco M, Di Filippo C, Gazzerro P, Canestrelli B, Monteleone F, Proto MC, Di Genio M, Grimaldi C, Maj M (2009) Association of CNR1 and FAAH endocannabinoid gene polymorphisms with anorexia nervosa and bulimia nervosa: evidence for synergistic effects. Genes Brain Behav 8(7):728–732. doi:10.1111/j.1601-183X.2009.00518.x
Monteleone P, Piscitelli F, Scognamiglio P, Monteleone AM, Canestrelli B, Di Marzo V, Maj M (2012) Hedonic eating is associated with increased peripheral levels of ghrelin and the endocannabinoid 2-arachidonoyl-glycerol in healthy humans: a pilot study. J Clin Endocrinol Metab 97(6):E917–E924. doi:10.1210/jc.2011-3018
Moranta D, Esteban S, Garcia-Sevilla JA (2006) Ethanol desensitizes cannabinoid CB1 receptors modulating monoamine synthesis in the rat brain in vivo. Neurosci Lett 392(1–2):58–61. doi:10.1016/j.neulet.2005.08.061
Morena M, Patel S, Bains JS, Hill MN (2016) Neurobiological interactions between stress and the endocannabinoid system. Neuropsychopharmacology 41(1):80–102. doi:10.1038/npp.2015.166
Morgan CJ, Page E, Schaefer C, Chatten K, Manocha A, Gulati S, Curran HV, Brandner B, Leweke FM (2013) Cerebrospinal fluid anandamide levels, cannabis use and psychotic-like symptoms. Br J Psychiatry 202(5):381–382. doi:10.1192/bjp.bp.112.121178
Muhl D, Kathmann M, Hoyer C, Kranaster L, Hellmich M, Gerth CW, Faulhaber J, Schlicker E, Leweke FM (2014) Increased CB2 mRNA and anandamide in human blood after cessation of cannabis abuse. Naunyn Schmiedebergs Arch Pharmacol 387(7):691–695. doi:10.1007/s00210-014-0984-2
Muldoon PP, Lichtman AH, Parsons LH, Damaj MI (2013) The role of fatty acid amide hydrolase inhibition in nicotine reward and dependence. Life Sci 92(8–9):458–462. doi:10.1016/j.lfs.2012.05.015
Muldoon PP, Chen J, Harenza JL, Abdullah RA, Sim-Selley LJ, Cravatt BF, Miles MF, Chen X, Lichtman AH, Damaj MI (2015) Inhibition of monoacylglycerol lipase reduces nicotine withdrawal. Br J Pharmacol 172(3):869–882. doi:10.1111/bph.12948
Muller TD, Reichwald K, Bronner G, Kirschner J, Nguyen TT, Scherag A, Herzog W, Herpertz-Dahlmann B, Lichtner P, Meitinger T, Platzer M, Schafer H, Hebebrand J, Hinney A (2008) Lack of association of genetic variants in genes of the endocannabinoid system with anorexia nervosa. Child Adolesc Psychiatr Ment Health 2(1):33. doi:10.1186/1753-2000-2-33
Murillo-Rodriguez E, Poot-Ake A, Arias-Carrion O, Pacheco-Pantoja E, Fuente-Ortegon Ade L, Arankowsky-Sandoval G (2011) The emerging role of the endocannabinoid system in the sleep-wake cycle modulation. Cent Nerv Syst Agents Med Chem 11(3):189–196
Murillo-Rodriguez E, Palomero-Rivero M, Millan-Aldaco D, Di Marzo V (2013) The administration of endocannabinoid uptake inhibitors OMDM-2 or VDM-11 promotes sleep and decreases extracellular levels of dopamine in rats. Physiol Behav 109:88–95. doi:10.1016/j.physbeh.2012.11.007
Murray JB (2002) Phencyclidine (PCP): a dangerous drug, but useful in schizophrenia research. J Psychol 136(3):319–327. doi:10.1080/00223980209604159
Naassila M, Pierrefiche O, Ledent C, Daoust M (2004) Decreased alcohol self-administration and increased alcohol sensitivity and withdrawal in CB1 receptor knockout mice. Neuropharmacology 46(2):243–253
Natividad LA, Buczynski MW, Herman MA, Kirson D, Oleata CS, Irimia C, Polis I, Ciccocioppo R, Roberto M, Parsons LH (2017) Constitutive increases in amygdalar corticotropin-releasing factor and fatty acid amide hydrolase drive an anxious phenotype. Biol Psychiatry. doi:10.1016/j.biopsych.2017.01.005
Navarrete F, Rodriguez-Arias M, Martin-Garcia E, Navarro D, Garcia-Gutierrez MS, Aguilar MA, Aracil-Fernandez A, Berbel P, Minarro J, Maldonado R, Manzanares J (2013) Role of CB2 cannabinoid receptors in the rewarding, reinforcing, and physical effects of nicotine. Neuropsychopharmacology 38(12):2515–2524. doi:10.1038/npp.2013.157
Navarro M, Carrera MR, Fratta W, Valverde O, Cossu G, Fattore L, Chowen JA, Gomez R, del Arco I, Villanua MA, Maldonado R, Koob GF, Rodriguez de Fonseca F (2001) Functional interaction between opioid and cannabinoid receptors in drug self-administration. J Neurosci 21(14):5344–5350
Nawata Y, Hiranita T, Yamamoto T (2010) A cannabinoid CB(1) receptor antagonist ameliorates impairment of recognition memory on withdrawal from MDMA (Ecstasy). Neuropsychopharmacology 35(2):515–520. doi:10.1038/npp.2009.158
Nestler EJ (2005) Is there a common molecular pathway for addiction? Nat Neurosci 8(11):1445–1449. doi:10.1038/nn1578
Nestler EJ (2014) Epigenetic mechanisms of drug addiction. Neuropharmacology 76(Pt B):259–268. doi:10.1016/j.neuropharm.2013.04.004
Neumeister A, Normandin MD, Murrough JW, Henry S, Bailey CR, Luckenbaugh DA, Tuit K, Zheng MQ, Galatzer-Levy IR, Sinha R, Carson RE, Potenza MN, Huang Y (2012) Positron emission tomography shows elevated cannabinoid CB1 receptor binding in men with alcohol dependence. Alcohol Clin Exp Res 36(12):2104–2109. doi:10.1111/j.1530-0277.2012.01815.x
Niehaus JL, Murali M, Kauer JA (2010) Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci 32(1):108–117. doi:10.1111/j.1460-9568.2010.07256.x
Niphakis MJ, Johnson DS, Ballard TE, Stiff C, Cravatt BF (2012) O-hydroxyacetamide carbamates as a highly potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem Neurosci 3(5):418–426. doi:10.1021/cn200089j
Niyuhire F, Varvel SA, Thorpe AJ, Stokes RJ, Wiley JL, Lichtman AH (2007) The disruptive effects of the CB1 receptor antagonist rimonabant on extinction learning in mice are task-specific. Psychopharmacology 191(2):223–231. doi:10.1007/s00213-006-0650-6
Nugent FS, Penick EC, Kauer JA (2007) Opioids block long-term potentiation of inhibitory synapses. Nature 446(7139):1086–1090. doi:10.1038/nature05726
Nunes EV, Levin FR (2004) Treatment of depression in patients with alcohol or other drug dependence: a meta-analysis. JAMA 291(15):1887–1896. doi:10.1001/jama.291.15.1887
Nunes EV, Sullivan MA, Levin FR (2004) Treatment of depression in patients with opiate dependence. Biol Psychiatry 56(10):793–802. doi:10.1016/j.biopsych.2004.06.037
Olds J, Milner P (1954) Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47(6):419–427
Palomino A, Pavon FJ, Blanco-Calvo E, Serrano A, Arrabal S, Rivera P, Alen F, Vargas A, Bilbao A, Rubio L, Rodriguez de Fonseca F, Suarez J (2014) Effects of acute versus repeated cocaine exposure on the expression of endocannabinoid signaling-related proteins in the mouse cerebellum. Front Integr Neurosci 8:22. doi:10.3389/fnint.2014.00022
Pamplona FA, Prediger RD, Pandolfo P, Takahashi RN (2006) The cannabinoid receptor agonist WIN 55,212-2 facilitates the extinction of contextual fear memory and spatial memory in rats. Psychopharmacology 188(4):641–649. doi:10.1007/s00213-006-0514-0
Pamplona FA, Bitencourt RM, Takahashi RN (2008) Short- and long-term effects of cannabinoids on the extinction of contextual fear memory in rats. Neurobiol Learn Mem 90(1):290–293. doi:10.1016/j.nlm.2008.04.003
Pan B, Hillard CJ, Liu QS (2008) Endocannabinoid signaling mediates cocaine-induced inhibitory synaptic plasticity in midbrain dopamine neurons. J Neurosci 28(6):1385–1397. doi:10.1523/JNEUROSCI.4033-07.2008
Panagis G, Mackey B, Vlachou S (2014) Cannabinoid regulation of brain reward processing with an emphasis on the role of CB1 receptors: a step back into the future. Front Psychiatry 5:92. doi:10.3389/fpsyt.2014.00092
Pani PP, Maremmani I, Trogu E, Gessa GL, Ruiz P, Akiskal HS (2010) Delineating the psychic structure of substance abuse and addictions: should anxiety, mood and impulse-control dysregulation be included? J Affect Disord 122(3):185–197. doi:10.1016/j.jad.2009.06.012
Parylak SL, Koob GF, Zorrilla EP (2011) The dark side of food addiction. Physiol Behav 104(1):149–156. doi:10.1016/j.physbeh.2011.04.063
Pataky Z, Gasteyger C, Ziegler O, Rissanen A, Hanotin C, Golay A (2013) Efficacy of rimonabant in obese patients with binge eating disorder. Exp Clin Endocrinol Diabetes 121(1):20–26. doi:10.1055/s-0032-1329957
Patel S, Rademacher DJ, Hillard CJ (2003) Differential regulation of the endocannabinoids anandamide and 2-arachidonylglycerol within the limbic forebrain by dopamine receptor activity. J Pharmacol Exp Ther 306(3):880–888. doi:10.1124/jpet.103.054270
Patel S, Roelke CT, Rademacher DJ, Cullinan WE, Hillard CJ (2004) Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology 145(12):5431–5438. doi:10.1210/en.2004-0638
Patel S, Cravatt BF, Hillard CJ (2005) Synergistic interactions between cannabinoids and environmental stress in the activation of the central amygdala. Neuropsychopharmacology 30(3):497–507. doi:10.1038/sj.npp.1300535
Patel S, Kingsley PJ, Mackie K, Marnett LJ, Winder DG (2009) Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhances short-term endocannabinoid signaling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology 34(13):2699–2709. doi:10.1038/npp.2009.101
Pava MJ, Woodward JJ (2014) Chronic ethanol alters network activity and endocannabinoid signaling in the prefrontal cortex. Front Integr Neurosci 8:58. doi:10.3389/fnint.2014.00058
Pavon FJ, Araos P, Pastor A, Calado M, Pedraz M, Campos-Cloute R, Ruiz JJ, Serrano A, Blanco E, Rivera P, Suarez J, Romero-Cuevas M, Pujadas M, Vergara-Moragues E, Gornemann I, Torrens M, de la Torre R, Rodriguez de Fonseca F (2013) Evaluation of plasma-free endocannabinoids and their congeners in abstinent cocaine addicts seeking outpatient treatment: impact of psychiatric co-morbidity. Addict Biol 18(6):955–969. doi:10.1111/adb.12107
Perra S, Pillolla G, Melis M, Muntoni AL, Gessa GL, Pistis M (2005) Involvement of the endogenous cannabinoid system in the effects of alcohol in the mesolimbic reward circuit: electrophysiological evidence in vivo. Psychopharmacology (Berl) 183(3):368–377. doi:10.1007/s00213-005-0195-0
Pettit HO, Ettenberg A, Bloom FE, Koob GF (1984) Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats. Psychopharmacology (Berl) 84(2):167–173
Piasecki TM, Jorenby DE, Smith SS, Fiore MC, Baker TB (2003a) Smoking withdrawal dynamics: I. Abstinence distress in lapsers and abstainers. J Abnorm Psychol 112(1):3–13
Piasecki TM, Jorenby DE, Smith SS, Fiore MC, Baker TB (2003b) Smoking withdrawal dynamics: II. Improved tests of withdrawal-relapse relations. J Abnorm Psychol 112(1):14–27
Piasecki TM, Jorenby DE, Smith SS, Fiore MC, Baker TB (2003c) Smoking withdrawal dynamics: III. Correlates of withdrawal heterogeneity. Exp Clin Psychopharmacol 11(4):276–285. doi:10.1037/1064-1297.11.4.276
Piazza PV, Le Moal M (1998) The role of stress in drug self-administration. Trends Pharmacol Sci 19(2):67–74
Piper ME, Cook JW, Schlam TR, Jorenby DE, Baker TB (2011) Anxiety diagnoses in smokers seeking cessation treatment: relations with tobacco dependence, withdrawal, outcome and response to treatment. Addiction 106(2):418–427. doi:10.1111/j.1360-0443.2010.03173.x
Platt DM, Rowlett JK, Spealman RD (2001) Discriminative stimulus effects of intravenous heroin and its metabolites in rhesus monkeys: opioid and dopaminergic mechanisms. J Pharmacol Exp Ther 299(2):760–767
Powers MS, Barrenha GD, Mlinac NS, Barker EL, Chester JA (2010) Effects of the novel endocannabinoid uptake inhibitor, LY2183240, on fear-potentiated startle and alcohol-seeking behaviors in mice selectively bred for high alcohol preference. Psychopharmacology (Berl) 212(4):571–583. doi:10.1007/s00213-010-1997-2
Puighermanal E, Busquets-Garcia A, Maldonado R, Ozaita A (2012) Cellular and intracellular mechanisms involved in the cognitive impairment of cannabinoids. Philos Trans R Soc Lond B Biol Sci 367(1607):3254–3263. doi:10.1098/rstb.2011.0384
Quirk GJ, Gehlert DR (2003) Inhibition of the amygdala: key to pathological states? Ann NY Acad Sci 985:263–272
Rada P, Avena NM, Hoebel BG (2005) Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience 134(3):737–744. doi:10.1016/j.neuroscience.2005.04.043
Rademacher DJ, Hillard CJ (2007) Interactions between endocannabinoids and stress-induced decreased sensitivity to natural reward. Prog Neuropsychopharmacol Biol Psychiatry 31(3):633–641. doi:10.1016/j.pnpbp.2006.12.013
Raichlen DA, Foster AD, Seillier A, Giuffrida A, Gerdeman GL (2013) Exercise-induced endocannabinoid signaling is modulated by intensity. Eur J Appl Physiol 113(4):869–875. doi:10.1007/s00421-012-2495-5
Ramesh D, Ross GR, Schlosburg JE, Owens RA, Abdullah RA, Kinsey SG, Long JZ, Nomura DK, Sim-Selley LJ, Cravatt BF, Akbarali HI, Lichtman AH (2011) Blockade of endocannabinoid hydrolytic enzymes attenuates precipitated opioid withdrawal symptoms in mice. J Pharmacol Exp Ther 339(1):173–185. doi:10.1124/jpet.111.181370
Ramesh D, Gamage TF, Vanuytsel T, Owens RA, Abdullah RA, Niphakis MJ, Shea-Donohue T, Cravatt BF, Lichtman AH (2013) Dual inhibition of endocannabinoid catabolic enzymes produces enhanced antiwithdrawal effects in morphine-dependent mice. Neuropsychopharmacology 38(6):1039–1049. doi:10.1038/npp.2012.269
Reisiger AR, Kaufling J, Manzoni O, Cador M, Georges F, Caille S (2014) Nicotine self-administration induces CB1-dependent LTP in the bed nucleus of the stria terminalis. J Neurosci 34(12):4285–4292. doi:10.1523/JNEUROSCI.3149-13.2014
Rescorla RA (1996) Preservation of Pavlovian associations through extinction. Q J Exp Psychol B Comp Physiol Psychol 49(3):245–258
Richman JA, Flaherty JA, Rospenda KM (1996) Perceived workplace harassment experiences and problem drinking among physicians: broadening the stress/alienation paradigm. Addiction 91(3):391–403
Rivera P, Miguens M, Coria SM, Rubio L, Higuera-Matas A, Bermudez-Silva FJ, de Fonseca FR, Suarez J, Ambrosio E (2013) Cocaine self-administration differentially modulates the expression of endogenous cannabinoid system-related proteins in the hippocampus of Lewis vs. Fischer 344 rats. Int J Neuropsychopharmacol 16(6):1277–1293. doi:10.1017/S1461145712001186
Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain research Brain research reviews 18(3):247–291
Rose JE, Salley A, Behm FM, Bates JE, Westman EC (2010) Reinforcing effects of nicotine and non-nicotine components of cigarette smoke. Psychopharmacology 210(1):1–12. doi:10.1007/s00213-010-1810-2
Rospenda KM, Richman JA, Wislar JS, Flaherty JA (2000) Chronicity of sexual harassment and generalized work-place abuse: effects on drinking outcomes. Addiction 95(12):1805–1820. doi:10.1080/09652140020011117
Rouge-Pont F, Marinelli M, Le Moal M, Simon H, Piazza PV (1995) Stress-induced sensitization and glucocorticoids. II. Sensitization of the increase in extracellular dopamine induced by cocaine depends on stress-induced corticosterone secretion. J Neurosci 15(11):7189–7195
Rouge-Pont F, Deroche V, Le Moal M, Piazza PV (1998) Individual differences in stress-induced dopamine release in the nucleus accumbens are influenced by corticosterone. Eur J Neurosci 10(12):3903–3907
Rubino T, Tizzoni L, Vigano D, Massi P, Parolaro D (1997) Modulation of rat brain cannabinoid receptors after chronic morphine treatment. Neuroreport 8(15):3219–3223
Rubino T, Massi P, Vigano D, Fuzio D, Parolaro D (2000) Long-term treatment with SR141716A, the CB1 receptor antagonist, influences morphine withdrawal syndrome. Life Sci 66(22):2213–2219
Rubio M, McHugh D, Fernandez-Ruiz J, Bradshaw H, Walker JM (2007) Short-term exposure to alcohol in rats affects brain levels of anandamide, other N-acylethanolamines and 2-arachidonoyl-glycerol. Neurosci Lett 421(3):270–274. doi:10.1016/j.neulet.2007.05.052
Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37(4):577–582
Salamone JD, Correa M, Farrar A, Mingote SM (2007) Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology 191(3):461–482. doi:10.1007/s00213-006-0668-9
Saravia R, Flores A, Plaza-Zabala A, Busquets-Garcia A, Pastor A, de la Torre R, Di Marzo V, Marsicano G, Ozaita A, Maldonado R, Berrendero F (2016) cb1 cannabinoid receptors mediate cognitive deficits and structural plasticity changes during nicotine withdrawal. Biol Psychiatry. doi:10.1016/j.biopsych.2016.07.007
Scherma M, Panlilio LV, Fadda P, Fattore L, Gamaleddin I, Le Foll B, Justinova Z, Mikics E, Haller J, Medalie J, Stroik J, Barnes C, Yasar S, Tanda G, Piomelli D, Fratta W, Goldberg SR (2008) Inhibition of anandamide hydrolysis by cyclohexyl carbamic acid 3′-carbamoyl-3-yl ester (URB597) reverses abuse-related behavioral and neurochemical effects of nicotine in rats. J Pharmacol Exp Ther 327 (2):482–490. doi:10.1124/jpet.108.142224
Scherma M, Justinova Z, Zanettini C, Panlilio LV, Mascia P, Fadda P, Fratta W, Makriyannis A, Vadivel SK, Gamaleddin I, Le Foll B, Goldberg SR (2012) The anandamide transport inhibitor AM404 reduces the rewarding effects of nicotine and nicotine-induced dopamine elevations in the nucleus accumbens shell in rats. Br J Pharmacol 165(8):2539–2548. doi:10.1111/j.1476-5381.2011.01467.x
Scherma M, Fattore L, Satta V, Businco F, Pigliacampo B, Goldberg SR, Dessi C, Fratta W, Fadda P (2013) Pharmacological modulation of the endocannabinoid signalling alters binge-type eating behaviour in female rats. Br J Pharmacol 169(4):820–833. doi:10.1111/bph.12014
Scherma M, Fattore L, Castelli MP, Fratta W, Fadda P (2014) The role of the endocannabinoid system in eating disorders: neurochemical and behavioural preclinical evidence. Curr Pharm Des 20(13):2089–2099
Schindler CW, Scherma M, Redhi GH, Vadivel SK, Makriyannis A, Goldberg SR, Justinova Z (2016) Self-administration of the anandamide transport inhibitor AM404 by squirrel monkeys. Psychopharmacology. doi:10.1007/s00213-016-4211-3
Schlosburg JE, Carlson BL, Ramesh D, Abdullah RA, Long JZ, Cravatt BF, Lichtman AH (2009) Inhibitors of endocannabinoid-metabolizing enzymes reduce precipitated withdrawal responses in THC-dependent mice. AAPS J 11(2):342–352. doi:10.1208/s12248-009-9110-7
Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13(9):1113–1119. doi:10.1038/nn.2616
Schmidt LG, Samochowiec J, Finckh U, Fiszer-Piosik E, Horodnicki J, Wendel B, Rommelspacher H, Hoehe MR (2002) Association of a CB1 cannabinoid receptor gene (CNR1) polymorphism with severe alcohol dependence. Drug Alcohol Depend 65(3):221–224
Schuckit MA (2006) Comorbidity between substance use disorders and psychiatric conditions. Addiction 101(Suppl 1):76–88. doi:10.1111/j.1360-0443.2006.01592.x
Sclafani A, Bodnar RJ, Delamater AR (1998) Pharmacology of food conditioned preferences. Appetite 31(3):406. doi:10.1006/appe.1998.0211
Seillier A, Giuffrida A (2009) Evaluation of NMDA receptor models of schizophrenia: divergences in the behavioral effects of sub-chronic PCP and MK-801. Behav Brain Res 204(2):410–415. doi:10.1016/j.bbr.2009.02.007
Seillier A, Martinez AA, Giuffrida A (2013) Phencyclidine-induced social withdrawal results from deficient stimulation of cannabinoid CB(1) receptors: implications for schizophrenia. Neuropsychopharmacology 38(9):1816–1824. doi:10.1038/npp.2013.81
Serrano A, Rivera P, Pavon FJ, Decara J, Suarez J, Rodriguez de Fonseca F, Parsons LH (2012) Differential effects of single versus repeated alcohol withdrawal on the expression of endocannabinoid system-related genes in the rat amygdala. Alcohol Clin Exp Res 36(6):984–994. doi:10.1111/j.1530-0277.2011.01686.x
Shinohara Y, Inui T, Yamamoto T, Shimura T (2009) Cannabinoid in the nucleus accumbens enhances the intake of palatable solution. Neuroreport 20(15):1382–1385. doi:10.1097/WNR.0b013e3283318010
Shippenberg TS, Elmer GI (1998) The neurobiology of opiate reinforcement. Crit Rev Neurobiol 12(4):267–303
Shoaib M (2008) The cannabinoid antagonist AM251 attenuates nicotine self-administration and nicotine-seeking behaviour in rats. Neuropharmacology 54(2):438–444. doi:10.1016/j.neuropharm.2007.10.011
Sidhpura N, Parsons LH (2011) Endocannabinoid-mediated synaptic plasticity and addiction-related behavior. Neuropharmacology 61(7):1070–1087. doi:10.1016/j.neuropharm.2011.05.034
Siegfried Z, Kanyas K, Latzer Y, Karni O, Bloch M, Lerer B, Berry EM (2004) Association study of cannabinoid receptor gene (CNR1) alleles and anorexia nervosa: differences between restricting and binging/purging subtypes. Am J Med Genet B Neuropsychiatr Genet 125B(1):126–130. doi:10.1002/ajmg.b.20089
Silvestri C, Di Marzo V (2013) The endocannabinoid system in energy homeostasis and the etiopathology of metabolic disorders. Cell Metab 17(4):475–490. doi:10.1016/j.cmet.2013.03.001
Sim LJ, Hampson RE, Deadwyler SA, Childers SR (1996) Effects of chronic treatment with delta9-tetrahydrocannabinol on cannabinoid-stimulated [35S]GTPgammaS autoradiography in rat brain. J Neurosci 16(24):8057–8066
Simiand J, Keane M, Keane PE, Soubrie P (1998) SR 141716, a CB1 cannabinoid receptor antagonist, selectively reduces sweet food intake in marmoset. Behav Pharmacol 9(2):179–181
Simonnet A, Cador M, Caille S (2013) Nicotine reinforcement is reduced by cannabinoid CB1 receptor blockade in the ventral tegmental area. Addict Biol 18(6):930–936. doi:10.1111/j.1369-1600.2012.00476.x
Singh ME, Verty AN, McGregor IS, Mallet PE (2004) A cannabinoid receptor antagonist attenuates conditioned place preference but not behavioural sensitization to morphine. Brain Res 1026(2):244–253. doi:10.1016/j.brainres.2004.08.027
Sipe JC, Waalen J, Gerber A, Beutler E (2005) Overweight and obesity associated with a missense polymorphism in fatty acid amide hydrolase (FAAH). Int J Obes (Lond) 29(7):755–759. doi:10.1038/sj.ijo.0802954
Smith PA, Selley DE, Sim-Selley LJ, Welch SP (2007) Low dose combination of morphine and delta9-tetrahydrocannabinol circumvents antinociceptive tolerance and apparent desensitization of receptors. Eur J Pharmacol 571(2-3):129–137. doi:10.1016/j.ejphar.2007.06.001
Solinas M, Goldberg SR (2005) Motivational effects of cannabinoids and opioids on food reinforcement depend on simultaneous activation of cannabinoid and opioid systems. Neuropsychopharmacology 30(11):2035–2045. doi:10.1038/sj.npp.1300720
Solinas M, Panlilio LV, Antoniou K, Pappas LA, Goldberg SR (2003) The cannabinoid CB1 antagonist N-piperidinyl-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl) -4-methylpyrazole-3-carboxamide (SR-141716A) differentially alters the reinforcing effects of heroin under continuous reinforcement, fixed ratio, and progressive ratio schedules of drug self-administration in rats. J Pharmacol Exp Ther 306(1):93–102. doi:10.1124/jpet.102.047928
Solinas M, Panlilio LV, Tanda G, Makriyannis A, Matthews SA, Goldberg SR (2005) Cannabinoid agonists but not inhibitors of endogenous cannabinoid transport or metabolism enhance the reinforcing efficacy of heroin in rats. Neuropsychopharmacology 30(11):2046–2057. doi:10.1038/sj.npp.1300754
Solinas M, Justinova Z, Goldberg SR, Tanda G (2006) Anandamide administration alone and after inhibition of fatty acid amide hydrolase (FAAH) increases dopamine levels in the nucleus accumbens shell in rats. J Neurochem 98(2):408–419. doi:10.1111/j.1471-4159.2006.03880.x
Solinas M, Tanda G, Justinova Z, Wertheim CE, Yasar S, Piomelli D, Vadivel SK, Makriyannis A, Goldberg SR (2007) The endogenous cannabinoid anandamide produces delta-9-tetrahydrocannabinol-like discriminative and neurochemical effects that are enhanced by inhibition of fatty acid amide hydrolase but not by inhibition of anandamide transport. J Pharmacol Exp Ther 321(1):370–380. doi:10.1124/jpet.106.114124
Soria G, Mendizabal V, Tourino C, Robledo P, Ledent C, Parmentier M, Maldonado R, Valverde O (2005) Lack of CB1 cannabinoid receptor impairs cocaine self-administration. Neuropsychopharmacology 30(9):1670–1680. doi:10.1038/sj.npp.1300707
Soria-Gomez E, Matias I, Rueda-Orozco PE, Cisneros M, Petrosino S, Navarro L, Di Marzo V, Prospero-Garcia O (2007) Pharmacological enhancement of the endocannabinoid system in the nucleus accumbens shell stimulates food intake and increases c-Fos expression in the hypothalamus. Br J Pharmacol 151(7):1109–1116. doi:10.1038/sj.bjp.0707313
South T, Deng C, Huang XF (2007) AM 251 and beta-Funaltrexamine reduce fat intake in a fat-preferring strain of mouse. Behav Brain Res 181(1):153–157. doi:10.1016/j.bbr.2007.03.028
Spano MS, Fattore L, Cossu G, Deiana S, Fadda P, Fratta W (2004) CB1 receptor agonist and heroin, but not cocaine, reinstate cannabinoid-seeking behaviour in the rat. Br J Pharmacol 143(3):343–350. doi:10.1038/sj.bjp.0705932
Sparling PB, Giuffrida A, Piomelli D, Rosskopf L, Dietrich A (2003) Exercise activates the endocannabinoid system. Neuroreport 14(17):2209–2211. doi:10.1097/01.wnr.0000097048.56589.47
Stewart SH, Kushner MG (2001) Introduction to the special issue on “Anxiety sensitivity and addictive behaviors”. Addict Behav 26(6):775–785
Stoving RK, Andries A, Brixen K, Flyvbjerg A, Horder K, Frystyk J (2009) Leptin, ghrelin, and endocannabinoids: potential therapeutic targets in anorexia nervosa. J Psychiatr Res 43(7):671–679. doi:10.1016/j.jpsychires.2008.09.007
Sun N, Chi N, Lauzon N, Bishop S, Tan H, Laviolette SR (2011) Acquisition, extinction, and recall of opiate reward memory are signaled by dynamic neuronal activity patterns in the prefrontal cortex. Cereb Cortex 21(12):2665–2680. doi:10.1093/cercor/bhr031
Suzuki A, Josselyn SA, Frankland PW, Masushige S, Silva AJ, Kida S (2004) Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J Neurosci 24(20):4787–4795. doi:10.1523/JNEUROSCI.5491-03.2004
Tabibnia G, Lieberman MD (2007) Fairness and cooperation are rewarding: evidence from social cognitive neuroscience. Ann NY Acad Sci 1118:90–101. doi:10.1196/annals.1412.001
Tan H, Lauzon NM, Bishop SF, Bechard MA, Laviolette SR (2010) Integrated cannabinoid CB1 receptor transmission within the amygdala-prefrontal cortical pathway modulates neuronal plasticity and emotional memory encoding. Cereb Cortex 20(6):1486–1496. doi:10.1093/cercor/bhp210
Tan H, Ahmad T, Loureiro M, Zunder J, Laviolette SR (2014) The role of cannabinoid transmission in emotional memory formation: implications for addiction and schizophrenia. Front Psychiatry 5:73. doi:10.3389/fpsyt.2014.00073
Tanda G, Pontieri FE, Di Chiara G (1997) Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 276(5321):2048–2050
Tanda G, Munzar P, Goldberg SR (2000) Self-administration behavior is maintained by the psychoactive ingredient of marijuana in squirrel monkeys. Nat Neurosci 3(11):1073–1074. doi:10.1038/80577
Thanos PK, Dimitrakakis ES, Rice O, Gifford A, Volkow ND (2005) Ethanol self-administration and ethanol conditioned place preference are reduced in mice lacking cannabinoid CB1 receptors. Behav Brain Res 164(2):206–213. doi:10.1016/j.bbr.2005.06.021
Tidey JW, Miczek KA (1997) Acquisition of cocaine self-administration after social stress: role of accumbens dopamine. Psychopharmacology 130(3):203–212
Tomasi D, Volkow ND (2013) Striatocortical pathway dysfunction in addiction and obesity: differences and similarities. Crit Rev Biochem Mol Biol 48(1):1–19. doi:10.3109/10409238.2012.735642
Trezza V, Vanderschuren LJ (2008a) Bidirectional cannabinoid modulation of social behavior in adolescent rats. Psychopharmacology (Berl) 197(2):217–227. doi:10.1007/s00213-007-1025-3
Trezza V, Vanderschuren LJ (2008b) Cannabinoid and opioid modulation of social play behavior in adolescent rats: differential behavioral mechanisms. Eur Neuropsychopharmacol 18(7):519–530. doi:10.1016/j.euroneuro.2008.03.001
Trezza V, Vanderschuren LJ (2009) Divergent effects of anandamide transporter inhibitors with different target selectivity on social play behavior in adolescent rats. J Pharmacol Exp Ther 328(1):343–350. doi:10.1124/jpet.108.141069
Trezza V, Baarendse PJ, Vanderschuren LJ (2010) The pleasures of play: pharmacological insights into social reward mechanisms. Trends Pharmacol Sci 31(10):463–469. doi:10.1016/j.tips.2010.06.008
Umberg EN, Pothos EN (2011) Neurobiology of aversive states. Physiol Behav 104(1):69–75. doi:10.1016/j.physbeh.2011.04.045
Uriguen L, Perez-Rial S, Ledent C, Palomo T, Manzanares J (2004) Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology 46(7):966–973. doi:10.1016/j.neuropharm.2004.01.003
Valjent E, Mitchell JM, Besson MJ, Caboche J, Maldonado R (2002) Behavioural and biochemical evidence for interactions between delta 9-tetrahydrocannabinol and nicotine. Br J Pharmacol 135(2):564–578. doi:10.1038/sj.bjp.0704479
van der Stelt M, Mazzola C, Esposito G, Matias I, Petrosino S, De Filippis D, Micale V, Steardo L, Drago F, Iuvone T, Di Marzo V (2006) Endocannabinoids and beta-amyloid-induced neurotoxicity in vivo: effect of pharmacological elevation of endocannabinoid levels. Cell Mol Life Sci 63(12):1410–1424. doi:10.1007/s00018-006-6037-3
Vann RE, Warner JA, Bushell K, Huffman JW, Martin BR, Wiley JL (2009) Discriminative stimulus properties of delta9-tetrahydrocannabinol (THC) in C57Bl/6J mice. Eur J Pharmacol 615(1-3):102–107. doi:10.1016/j.ejphar.2009.05.010
Varvel SA, Lichtman AH (2002) Evaluation of CB1 receptor knockout mice in the Morris water maze. J Pharmacol Exp Ther 301(3):915–924
Varvel SA, Anum EA, Lichtman AH (2005) Disruption of CB(1) receptor signaling impairs extinction of spatial memory in mice. Psychopharmacology 179(4):863–872. doi:10.1007/s00213-004-2121-2
Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH (2007) Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology 32(5):1032–1041. doi:10.1038/sj.npp.1301224
Vaseghi G, Rabbani M, Hajhashemi V (2012) The CB(1) receptor antagonist, AM281, improves recognition loss induced by naloxone in morphine withdrawal mice. Basic Clin Pharmacol Toxicol 111(3):161–165. doi:10.1111/j.1742-7843.2012.00881.x
Vaseghi G, Rabbani M, Hajhashemi V (2013) The effect of AM281, a cannabinoid antagonist, on memory performance during spontaneous morphine withdrawal in mice. Res Pharm Sci 8(1):59–64
Vaughn LK, Mantsch JR, Vranjkovic O, Stroh G, Lacourt M, Kreutter M, Hillard CJ (2012) Cannabinoid receptor involvement in stress-induced cocaine reinstatement: potential interaction with noradrenergic pathways. Neuroscience 204:117–124. doi:10.1016/j.neuroscience.2011.08.021
Vela G, Ruiz-Gayo M, Fuentes JA (1995) Anandamide decreases naloxone-precipitated withdrawal signs in mice chronically treated with morphine. Neuropharmacology 34(6):665–668
Vengeliene V, Siegmund S, Singer MV, Sinclair JD, Li TK, Spanagel R (2003) A comparative study on alcohol-preferring rat lines: effects of deprivation and stress phases on voluntary alcohol intake. Alcohol Clin Exp Res 27(7):1048–1054. doi:10.1097/01.ALC.0000075829.81211.0C
Verty AN, McGregor IS, Mallet PE (2005) Paraventricular hypothalamic CB(1) cannabinoid receptors are involved in the feeding stimulatory effects of Delta(9)-tetrahydrocannabinol. Neuropharmacology 49(8):1101–1109. doi:10.1016/j.neuropharm.2005.03.025
Vigano D, Grazia Cascio M, Rubino T, Fezza F, Vaccani A, Di Marzo V, Parolaro D (2003) Chronic morphine modulates the contents of the endocannabinoid, 2-arachidonoyl glycerol, in rat brain. Neuropsychopharmacology 28(6):1160–1167. doi:10.1038/sj.npp.1300117
Vigano D, Valenti M, Cascio MG, Di Marzo V, Parolaro D, Rubino T (2004) Changes in endocannabinoid levels in a rat model of behavioural sensitization to morphine. Eur J Neurosci 20(7):1849–1857. doi:10.1111/j.1460-9568.2004.03645.x
Vinklerova J, Novakova J, Sulcova A (2002) Inhibition of methamphetamine self-administration in rats by cannabinoid receptor antagonist AM251. J Psychopharmacol 16(2):139–143
Vinod KY, Yalamanchili R, Xie S, Cooper TB, Hungund BL (2006) Effect of chronic ethanol exposure and its withdrawal on the endocannabinoid system. Neurochem Int 49(6):619–625. doi:10.1016/j.neuint.2006.05.002
Vinod KY, Sanguino E, Yalamanchili R, Manzanares J, Hungund BL (2008) Manipulation of fatty acid amide hydrolase functional activity alters sensitivity and dependence to ethanol. J Neurochem 104(1):233–243. doi:10.1111/j.1471-4159.2007.04956.x
Vinod KY, Kassir SA, Hungund BL, Cooper TB, Mann JJ, Arango V (2010) Selective alterations of the CB1 receptors and the fatty acid amide hydrolase in the ventral striatum of alcoholics and suicides. J Psychiatr Res 44(9):591–597. doi:10.1016/j.jpsychires.2009.11.013
Vinod KY, Maccioni P, Garcia-Gutierrez MS, Femenia T, Xie S, Carai MA, Manzanares J, Cooper TB, Hungund BL, Colombo G (2012) Innate difference in the endocannabinoid signaling and its modulation by alcohol consumption in alcohol-preferring sP rats. Addict Biol 17(1):62–75. doi:10.1111/j.1369-1600.2010.00299.x
Viveros MP, Marco EM, Llorente R, Lopez-Gallardo M (2007) Endocannabinoid system and synaptic plasticity: implications for emotional responses. Neural Plast 2007:52908. doi:10.1155/2007/52908
Vlachou S, Nomikos GG, Panagis G (2003) WIN 55,212-2 decreases the reinforcing actions of cocaine through CB1 cannabinoid receptor stimulation. Behav Brain Res 141(2):215–222
Vlachou S, Nomikos GG, Panagis G (2006) Effects of endocannabinoid neurotransmission modulators on brain stimulation reward. Psychopharmacology (Berl) 188(3):293–305. doi:10.1007/s00213-006-0506-0
Vlachou S, Stamatopoulou F, Nomikos GG, Panagis G (2008) Enhancement of endocannabinoid neurotransmission through CB1 cannabinoid receptors counteracts the reinforcing and psychostimulant effects of cocaine. Int J Neuropsychopharmacol 11(7):905–923. doi:10.1017/S1461145708008717
Volkow ND, Fowler JS, Wang GJ, Swanson JM, Telang F (2007) Dopamine in drug abuse and addiction: results of imaging studies and treatment implications. Arch Neurol 64(11):1575–1579. doi:10.1001/archneur.64.11.1575
Volkow ND, Wang GJ, Baler RD (2011) Reward, dopamine and the control of food intake: implications for obesity. Trends Cogn Sci 15(1):37–46. doi:10.1016/j.tics.2010.11.001
Wade MR, Degroot A, Nomikos GG (2006) Cannabinoid CB1 receptor antagonism modulates plasma corticosterone in rodents. Eur J Pharmacol 551(1–3):162–167. doi:10.1016/j.ejphar.2006.08.083
Wallace MJ, Martin BR, DeLorenzo RJ (2002) Evidence for a physiological role of endocannabinoids in the modulation of seizure threshold and severity. Eur J Pharmacol 452(3):295–301
Wamsteeker JI, Kuzmiski JB, Bains JS (2010) Repeated stress impairs endocannabinoid signaling in the paraventricular nucleus of the hypothalamus. J Neurosci 30(33):11188–11196. doi:10.1523/JNEUROSCI.1046-10.2010
Wang L, Liu J, Harvey-White J, Zimmer A, Kunos G (2003a) Endocannabinoid signaling via cannabinoid receptor 1 is involved in ethanol preference and its age-dependent decline in mice. Proc Natl Acad Sci USA 100(3):1393–1398. doi:10.1073/pnas.0336351100
Wang X, Dow-Edwards D, Keller E, Hurd YL (2003b) Preferential limbic expression of the cannabinoid receptor mRNA in the human fetal brain. Neuroscience 118(3):681–694
Wang W, Sun D, Pan B, Roberts CJ, Sun X, Hillard CJ, Liu QS (2010) Deficiency in endocannabinoid signaling in the nucleus accumbens induced by chronic unpredictable stress. Neuropsychopharmacology 35(11):2249–2261. doi:10.1038/npp.2010.99
Ward SJ, Walker EA, Dykstra LA (2007) Effect of cannabinoid CB1 receptor antagonist SR141716A and CB1 receptor knockout on cue-induced reinstatement of Ensure and corn-oil seeking in mice. Neuropsychopharmacology 32(12):2592–2600. doi:10.1038/sj.npp.1301384
Ward SJ, Rosenberg M, Dykstra LA, Walker EA (2009) The CB1 antagonist rimonabant (SR141716) blocks cue-induced reinstatement of cocaine seeking and other context and extinction phenomena predictive of relapse. Drug Alcohol Depend 105(3):248–255. doi:10.1016/j.drugalcdep.2009.07.002
Wei D, Lee D, Li D, Daglian J, Jung KM, Piomelli D (2016) A role for the endocannabinoid 2-arachidonoyl-sn-glycerol for social and high-fat food reward in male mice. Psychopharmacology (Berl) 233(10):1911–1919. doi:10.1007/s00213-016-4222-0
Werling LL, Reed SC, Wade D, Izenwasser S (2009) Chronic nicotine alters cannabinoid-mediated locomotor activity and receptor density in periadolescent but not adult male rats. Int J Dev Neurosci 27(3):263–269. doi:10.1016/j.ijdevneu.2008.12.008
West R, Gossop M (1994) Overview: A comparison of withdrawal symptoms from different drug classes. Addiction 89(11):1483–1489
Wikler A (1948) Recent progress in research on the neurophysiologic basis of morphine addiction. Am J Psychiatry 105(5):329–338. doi:10.1176/ajp.105.5.329
Wiley JL, Golden KM, Ryan WJ, Balster RL, Razdan RK, Martin BR (1997) Evaluation of cannabimimetic discriminative stimulus effects of anandamide and methylated fluoroanandamide in rhesus monkeys. Pharmacol Biochem Behav 58(4):1139–1143
Wiley JL, Walentiny DM, Wright MJ Jr, Beardsley PM, Burston JJ, Poklis JL, Lichtman AH, Vann RE (2014) Endocannabinoid contribution to Delta(9)-tetrahydrocannabinol discrimination in rodents. Eur J Pharmacol 737:97–105. doi:10.1016/j.ejphar.2014.05.013
Williams CM, Kirkham TC (1999) Anandamide induces overeating: mediation by central cannabinoid (CB1) receptors. Psychopharmacology (Berl) 143(3):315–317
Wise LE, Harloe JP, Lichtman AH (2009) Fatty acid amide hydrolase (FAAH) knockout mice exhibit enhanced acquisition of an aversive, but not of an appetitive, Barnes maze task. Neurobiol Learn Mem 92(4):597–601. doi:10.1016/j.nlm.2009.06.001
Wolf SA, Bick-Sander A, Fabel K, Leal-Galicia P, Tauber S, Ramirez-Rodriguez G, Muller A, Melnik A, Waltinger TP, Ullrich O, Kempermann G (2010) Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun Signal 8:12. doi:10.1186/1478-811X-8-12
Xi ZX, Gilbert JG, Peng XQ, Pak AC, Li X, Gardner EL (2006) Cannabinoid CB1 receptor antagonist AM251 inhibits cocaine-primed relapse in rats: role of glutamate in the nucleus accumbens. J Neurosci 26(33):8531–8536. doi:10.1523/JNEUROSCI.0726-06.2006
Xi ZX, Spiller K, Pak AC, Gilbert J, Dillon C, Li X, Peng XQ, Gardner EL (2008) Cannabinoid CB1 receptor antagonists attenuate cocaine’s rewarding effects: experiments with self-administration and brain-stimulation reward in rats. Neuropsychopharmacology 33(7):1735–1745. doi:10.1038/sj.npp.1301552
Xi ZX, Peng XQ, Li X, Song R, Zhang HY, Liu QR, Yang HJ, Bi GH, Li J, Gardner EL (2011) Brain cannabinoid CB(2) receptors modulate cocaine’s actions in mice. Nat Neurosci 14(9):1160–1166. doi:10.1038/nn.2874
Yamaguchi T, Hagiwara Y, Tanaka H, Sugiura T, Waku K, Shoyama Y, Watanabe S, Yamamoto T (2001) Endogenous cannabinoid, 2-arachidonoylglycerol, attenuates naloxone-precipitated withdrawal signs in morphine-dependent mice. Brain Res 909(1-2):121–126
Yan HC, Cao X, Das M, Zhu XH, Gao TM (2010) Behavioral animal models of depression. Neurosci Bull 26(4):327–337. doi:10.1007/s12264-010-0323-7
Yin HH, Ostlund SB, Balleine BW (2008) Reward-guided learning beyond dopamine in the nucleus accumbens: the integrative functions of cortico-basal ganglia networks. Eur J Neurosci 28(8):1437–1448. doi:10.1111/j.1460-9568.2008.06422.x
Yoshida R, Ohkuri T, Jyotaki M, Yasuo T, Horio N, Yasumatsu K, Sanematsu K, Shigemura N, Yamamoto T, Margolskee RF, Ninomiya Y (2010) Endocannabinoids selectively enhance sweet taste. Proc Natl Acad Sci USA 107(2):935–939. doi:10.1073/pnas.0912048107
Zhang HY, Bi GH, Li X, Li J, Qu H, Zhang SJ, Li CY, Onaivi ES, Gardner EL, Xi ZX, Liu QR (2014a) Species differences in cannabinoid receptor 2 and receptor responses to cocaine self-administration in mice and rats. Neuropsychopharmacology 40(4):1037–1051. doi:10.1038/npp.2014.297
Zhang HY, Gao M, Liu QR, Bi GH, Li X, Yang HJ, Gardner EL, Wu J, Xi ZX (2014b) Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc Natl Acad Sci USA 111(46):E5007–E5015. doi:10.1073/pnas.1413210111
Zlebnik NE, Cheer JF (2016) Drug-induced alterations of endocannabinoid-mediated plasticity in brain reward regions. J Neurosci 36(40):10230–10238. doi:10.1523/JNEUROSCI.1712-16.2016
Acknowledgments
This work was supported by grants from the US National Institutes of Health (NIH): AA020404, AA006420, AA022249 and AA017447 to LHP, and T32 AA 7456-33 and F32 AA025257-01 to SAL. This is manuscript number 29294 from The Scripps Research Institute. This chapter is dedicated in loving memory of Dr. Loren H Parsons. He was an exceptional mentor and compassionate friend who contributed heavily to endocannabinoid research. He has left a strong legacy for the scientific community to build on and will be sorely missed.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Laredo, S.A., Marrs, W.R., Parsons, L.H. (2017). Endocannabinoid Signaling in Reward and Addiction: From Homeostasis to Pathology. In: Melis, M. (eds) Endocannabinoids and Lipid Mediators in Brain Functions. Springer, Cham. https://doi.org/10.1007/978-3-319-57371-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-57371-7_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-57369-4
Online ISBN: 978-3-319-57371-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)