
Abstract
Pollen, an extremely reduced bi-cellular or tri-cellular male reproductive structure of flowering plants, serves as a model for numerous studies covering a wide range of developmental and physiological processes. The pollen development and subsequent progamic phase represent two fragile and vital phases of plant ontogenesis, and pollen was among the first singular plant tissues thoroughly characterised at the transcriptomic level. Here we present an overview of high-throughput tools applied in pollen research on numerous plant species. Transcriptomics, being the first experimental approach used, has provided and continues providing valuable information about global and specific gene expression and its dynamics. However, the proteome does not fully reflect the transcriptome, namely, because post-transcriptional regulatory levels, especially translation, mRNA storage and protein modifications, are active during male gametophyte development and during progamic phase. Transcriptomics therefore should be complemented by other -omic tools to get more realistic insight, most importantly proteomics and other specialised approaches mapping the involvement of regulatory RNAs and protein post-translational modifications as well as experiments designed to identify the subsets of total -omes like translatome, secretome or allergome.
Jan Fíla and Lenka Záveská Drábková contributed equally to this work.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Reproduction is one of the most important processes performed by living organisms on Earth, which enables the survival of species. Plant reproduction has several important aspects, some of which differ from animal reproduction. In plants (Archaeplastida), there are two altering generations that differ in their ploidy. The diploid sporophyte produces haploid spores by meiosis, and the spores give rise to haploid gametophyte, in which gametes are produced by mitosis (unlike animals, gametes of which are formed by meiosis). Two haploid gametes fuse together, forming a diploid zygote, from which a new sporophyte generation develops.
The ancestors of higher plants, Streptophyta, spent a vast majority of their lives as haploid gametophytes (reviewed in Qiu et al. 2012). Their zygote that was formed by two fusing gametes immediately underwent meiosis and gave rise to four haploid spores. In their descendants, meiosis was delayed, and a multicellular diploid generation was formed by several rounds of zygote mitosis. During subsequent evolution, one of the generations tended to be reduced. In bryophytes (Bryophyta sensu lato)—i.e. mosses, hornworts and liverworts—gametophyte dominates, and sporophyte is dependent on it. On the other hand, sporophyte is dominant in ferns (Monilophyta) and lycophytes (Lycopodiophyta), but they still form gametophytes as an independent generation. Later, in gymnosperms (Gymnospermae) and angiosperms (Angiospermae), gametophytes were notably reduced. In gymnosperms, the male gametophyte contains 4–40 cells, whilst the female gametophyte comprises several thousands of cells. Angiosperm gametophytes are even more reduced, having male gametophyte composed of 2–3 cells, whereas female gametophyte comprises typically 7 cells with 8 nuclei (although there exist several other alternative arrangements with different number of cells in the embryo sac; Reiser and Fischer 1993).
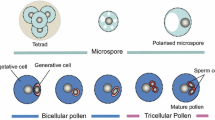
The mature angiosperm plants belong to diploid sporophyte generation. Angiosperms produce spores, gametophytes and gametes of separate sexes—male and female. The male gametophyte is formed inside the anthers of a flower; two initials differentiate from the sporophytic tissue in the anther—tapetal initial (which gives rise to tapetum) and pollen mother cell (microsporocyte; see McCormick 1993). A pollen mother cell divides into four microsporocytes by meiosis. The microspore tetrad is first connected by callose, which is later digested by the activity of callases (enzymes digesting callose) released by the tapetum. The freed microspores increase their size and vacuolise, and their nuclei migrate to the periphery (McCormick 1993; Borg and Twell 2010). The microspores then undergo highly asymmetric pollen mitosis I (PMI) leading to the production of a large vegetative cell and a small generative cell. The generative cell, the origin of male germline, is then engulfed by the vegetative cell and migrates into its cytoplasm (Berger and Twell 2011). The asymmetry of PMI is of key importance, which was proven by the serious phenotypic defects of Arabidopsis thaliana mutants gemini pollen 1 (Park et al. 1998) or two-in-one (Oh et al. 2005). The generative cell undergoes one more round of cell division, pollen mitosis II (PMII), which occurs either before mature pollen is discharged or afterwards. Two sperm cells are formed from a generative cell by PMII. Consequently, the mature pollen can be shed as bi-cellular or tri-cellular (Brewbaker 1967).
Initially, the ancestral state of angiosperm pollen grains was inferred as bi-cellular, because all ancient woody Magnoliales shed this pollen type. Moreover, this bi-cellular pollen is phylogenetically widespread, and it was believed that tri-cellular pollen was restricted to aquatics, grasses and some herbs (Elfving 1879; Strasburger 1884). More recent investigations revealed that pollen tri-cellularity is not irreversible and that tri-cellular lineages diversify slowly and sometimes reverse to bi-cellular lineages. This reflects a linkage between the evolution of sporophyte lifestyle and developmental lability of male gametophyte (Williams et al. 2014a). The ancient aquatic plant Ceratophyllum and several monocot lineages, such as Araceae, Alismataceae and Nymphaeaceae, support the tri-cellular ancestry of pollen, which in this case represents a selective advantage over bi-cellular pollen. Williams et al. (2014a) proposed that bi-cellular pollen evolved secondarily from tri-cellular ancestors during shifts away from rapid life cycle or from limited reproduction. In total, thirteen orders of angiosperms shed prevalently tri-cellular pollen (Fig. 10.1). In the orders with bi-cellular pollen, about 2–44% species produce also tri-cellular pollen.

Distribution of tri-cellular and bi-cellular pollen within angiosperms. Interrelationships are based on APG IV (APG IV, 2016). Percentages summarise all taxa with tree species matches from Williams et al. (2014a). Tri-cellular pollen is dominant in blue-coloured orders. Boxes indicate that some of the omics is already done for the order
The simultaneous presence of both pollen types in one species is very uncommon. For instance, the coexistence of bi- and tri-cellular pollen grains at the same time is in early-divergent angiosperm Annona cherimola (Magnoliales, Annonaceae). There, the production of the actual pollen type depends on environmental factors such as temperature and humidity during the pollen maturation (Lora et al. 2009).
Upon reaching the stylar papillary cells, pollen grain rehydration and activation occurs (Vogler et al. 2015). Later on, pollen tube growth through the female connective tissues starts processes, which are accompanied by reciprocal communication of pollen tube and pistil tissues (Hafidh et al. 2014, 2016b; Higashiyama 2015, see also Chap. 8). The pollen tube delivers two sperm cells (male haploid gametes) to the embryo sac. One sperm cell fuses with the egg cell (female haploid gamete) giving rise to a diploid zygote, which represents a start of a novel diploid sporophyte generation. The zygote subsequently undergoes several rounds of mitotic divisions giving rise to embryo and a new plant. The second sperm cell fuses with the central nucleus of the embryo sac, giving rise to the triploid endosperm. The double fertilisation is typical in angiosperms and was reviewed in more detail by Raghavan (2003).
In this chapter, we will discuss how various -omic techniques notably broadened the wealth of information about male gametophyte development. There were over hundred -omic studies performed on male gametophyte that were published so far (Table 10.1). Of these studies, transcriptomics represented the dominant experimental approach with 51% of all -omics experiments followed by proteomics (26%). The remaining 23% of experiments were shared by the identification and analyses of translatome, miRNAome, methylome, phosphoproteome, allergome, secretome and metabolome. Phylogenetically, most main orders (altogether 15) representing all major groups of seed plants are covered but to a different extent according to the distribution of model species. Therefore, the majority of information (75% experiments) was gathered on four well-distributed orders, Poales (Monocots, Commelinids, key models Oryza sativa and Zea mays), Brassicales (Rosids, key model species Arabidopsis thaliana), Solanales (Asterids, key models Nicotiana tabacum and Solanum lycopersicum) and Liliales (Monocots, key model Lilium longiflorum). Moreover, these model species were subjected to a combination of several -omic approaches, which enabled the mutual comparison of various -omic datasets of different origins.
2 Transcriptomics
Transcriptomics was the first -omics technique applied to the male gametophyte (Becker et al. 2003; Honys and Twell 2003; Lee and Lee 2003) and so far transcriptomic profiles of at least one male gametophyte developmental stage were published for 22 seed plant species, 21 of which were angiosperms (Table 10.2).
Transcriptomic studies underwent development from sequencing of cDNA or EST libraries through serial analysis of gene expression (SAGE) and microarray-based studies to the most recent deep sequencing technologies (RNAseq). Sanger sequencing of cDNA or EST libraries is relatively low-throughput method, expensive and generally not quantitative. To overcome these limitations, tag-based methods were developed, including SAGE, cap analysis of gene expression (CAGE) and massively parallel signature sequencing (MPSS). These approaches are high throughput and measure precise gene expression levels. However, most of them are based on Sanger sequencing, and a significant portion of the short tags cannot be uniquely mapped to the reference genome. Moreover, only a portion of the transcripts can be analysed, and gene isoforms are generally indistinguishable from each other. These disadvantages limited the application of traditional sequencing technology in annotating the structure of transcriptomes (Wang et al. 2009). DNA microarrays started to appear during the late 1990s; however the first article was published by Schena et al. (1995). Gene chip technique brought the quantum leap in gene expression studies and became standard because of its well-established sample preparation and data analysis protocols, rapid turnaround time, wealth of archived data and data-mining methodologies. However, the use of expression microarrays is limited, as they require fabrication, and alongside their rigidity, they also depend on prior knowledge of genes and gene sequences (Loraine et al. 2013). Recently, the development of novel DNA deep sequencing technologies such as RNAseq has started to be a dominant technique, mainly because it requires only a little a priori knowledge of the genome, and therefore it enables transcriptome studies in non-model plant species. It allows both mapping and quantifying transcriptomes.
The majority of male gametophytic transcriptomics studies are still based on microarray analyses representing 33 experiments in 11 species (Table 10.2). Affymetrix has been the most commonly used platform, but a significant share of experiments used alternative Agilent (Zea mays, Nicotiana tabacum) and Roche NimbleGen (Cryptomeria japonica, Arabidopsis thaliana, Vitis vinifera) platforms. However, the number of model systems investigated by RNAseq is boosting; it currently reached 23 experiments in 15 species. Since all transcriptomic studies employing RNAseq were published in last few years, they are responsible for the recent massive increase in plant species with analysed pollen transcriptomes, especially among models, genomes of which have not been sequenced and annotated yet. For example, Rutley and Twell (2015) mentioned in their review only 10 angiosperm species. There is a limited overlap of species with male gametophyte being analysed on both platforms—microarrays and RNAseq. There were only five key models—Arabidopsis thaliana, Oryza sativa, Zea mays, Solanum lycopersicum and Lilium longiflorum (Table 10.2). Of them, four provided sufficient wealth of information, because the microarray transcriptome profiling of L. longiflorum pollen was achieved on a custom cDNA microarray. However, such overlap was sufficient for the comparisons of both platforms. Not surprisingly, RNAseq enabled the identification of larger transcriptome fraction mainly due to the absence of probes for numerous genes on microarrays and as a result of continuous refined genome annotations leading to further reduction of reliable gene models. However, such increase of the number of identified genes was not dramatic. The number of genes expressed in Arabidopsis mature pollen was calculated 6044 (Rutley and Twell 2015) as an average value from the range of 3954–7235 genes published in original mature pollen microarray-based datasets (Borges et al. 2008; Honys and Twell 2004; Pina et al. 2005; Qin et al. 2009; Schmid et al. 2005; Wang et al. 2008). On the contrary, of the 5525 annotated protein-coding loci that have no corresponding probe set on the Affymetrix ATH1 microarray, 451 genes were identified as expressed in pollen by RNAseq with normalised expression values of 5 reads per million (RPM) or greater (Loraine et al. 2013). Similarly, RNAseq transcriptomes of Zea mays mature pollen comprised 13,418 (Davidson et al. 2011) or 14,591 genes (Chettoor et al. 2014) in comparison to 10,539 genes previously identified using Agilent 44K maize microarray (Ma et al. 2008). Higher sensitivity is not the only advantage of RNAseq, the sequencing of non-exonic transcripts allowed for the first time the broad view on the alternative splicing in Arabidopsis pollen including the discovery of novel pollen-specific splicing patterns (Loraine et al. 2013). For the same reason, the over-representation of transposable element-related transcripts was observed in maize pollen, although to a lesser extent than in embryo sac transcriptomes sequenced in parallel (Chettoor et al. 2014). Similar pattern was detected for transcripts encoding small signalling peptides of DEFENSIN/LURE (DEFL) family since probes for small peptide genes were often omitted from earlier microarray studies (Chettoor et al. 2014).
Not surprisingly, 49 out of 63 datasets (77%) related to mature pollen (Table 10.2). Only a fraction of experiments included also pollen developmental stages. Full pollen development including at least four developmental stages is available for three species—Arabidopsis thaliana, Oryza sativa and Nicotiana tabacum (Bokvaj et al. 2015; Honys and Twell 2004; Wei et al. 2010). Considering less developmental stages sufficient for the evaluation of transcriptome dynamics throughout pollen development would lead to the addition of four more species—Zea mays (meiocytes and pollen; Chettoor et al. 2014; Dukowic-Schulze et al. 2014; Xu et al. 2012), Lilium longiflorum (microspores and mature pollen; Okada et al. 2007), Brassica napus (microspores and mature pollen; Whittle et al. 2010) and Fragaria vesca (microspores and mature pollen; Hollender et al. 2014). The inclusion of progamic phase changed the list of analysed species. More than one time point of pollen germination and in vitro pollen tube growth was analysed in four species—Arabidopsis thaliana (germinating pollen and pollen tubes; Wang et al. 2008), Lilium longiflorum (hydrated pollen, germinating pollen and pollen tubes; Lang et al. 2015; Obermeyer et al. 2013), Nicotiana tabacum (4h and 24h pollen tubes; Hafidh et al. 2012a, b) and Pyrus bretschneideri (hydrated pollen and pollen tubes; Zhou et al. 2016). Only one time point of progamic phase (germinating pollen) was analysed in Oryza sativa (Wei et al. 2010). Finally, only in vitro pollen tube transcriptome without the reference mature pollen sample is available for Camellia sinensis (Wang et al. 2016). The quantification of transcriptome dynamics showed similar expression pattern throughout pollen development and progamic phase in all species analysed. In general, the complexity of male gametophyte transcriptome was lower than that of any sporophytic tissue analysed. It reached its maximum in early developmental stages and was subsequently reduced until mature pollen reaching only 61% in A. thaliana and N. tabacum and even 46% in O. sativa of the maximum value in the male gametophyte. (Honys and Twell 2004; Wei et al. 2010; Peng et al. 2012; Bokvaj et al. 2015; Rutley and Twell 2015). During progamic phase, the size of pollen tube transcriptomes remained relatively stable, similar to that of mature pollen or slightly larger increasing usually only by 0.1–3% in comparison to mature pollen in Pyrus bretschneideri, Arabidopsis thaliana, Oryza sativa and Nicotiana tabacum (Qin et al. 2009; Wei et al. 2010; Hafidh et al. 2012a, b; Zhou et al. 2016). Therefore, there was no apparent difference between fast growing tri-cellular pollen tubes and less advanced but metabolically more active bi-cellular pollen tubes. The only exception was another Arabidopsis study, in which the transcriptome complexity in 4h pollen tubes increased by 24% (Wang et al. 2008).
Pollen development is tightly regulated primarily at the level of transcription; it is under the control of at least two successive global gene expression programmes, early and late. The switch point between both developmental programmes occurs after pollen mitosis I in both tri-cellular (A. thaliana, Twell et al. 2006) and bi-cellular (N. tabacum, Honys et al., unpublished data) pollen. The initiation of the late programme therefore more likely reflects the progress of pollen maturation rather than the timing of pollen mitosis II (Hafidh et al. 2012a; Rutley and Twell 2015), supporting the uniqueness of the late male gametophytic transcriptome (Honys and Twell 2004) as shown also by principal component analyses in several species (Tang et al. 2010; Russell et al. 2012; Bokvaj et al. 2015; Rutley and Twell 2015). Generally, genes involved in cell cycle control and transcription regulation were expressed in both early and late male gametophyte transcriptomes, however, with variable expression of individual transcription factor (TF) genes and gene families, like MYB/MYB related, AP2-EREBP, C2H2, bHLH, MADS, bZIP, WRKY and TCP. On the contrary, the gene ontology (GO) category of protein synthesis/translation was over-represented in early developmental stages, whereas cell wall synthesis, cytoskeleton, signalling, protein turnover and localisation were upregulated closer to pollen maturation and in growing pollen tubes (Twell et al. 2006; Wei et al. 2010; Hafidh et al. 2012a; Costa et al. 2013, Zhou et al. 2016).
Comparative and developmental transcriptomic studies served as an information background for follow-up research including reverse genetic screens for male gametophytic transcription factors (Reňák et al. 2012), F-box proteins (Ikram et al. 2014), signalling proteins (Chen et al. 2014) and numerous functional studies. Unlike them, transcriptomic studies comparing wild-type and mutant pollen were rare, and besides the search for genetic interactions in pollen tubes deficient in two arabinogalactan protein-coding genes, agp6 and agp11 (Costa et al. 2013), they aimed to identify the transcriptional networks that regulate cell differentiation and define cell-specific functions during pollen development (Verelst et al. 2007b; Gibalová et al. 2009).
The comparison of wild-type and agp6/agp11 double-mutant pollen tubes revealed 1022 differentially expressed genes (14.7% of the pollen tube transcriptome), almost equally distributed among upregulated and downregulated sets. GO categorisation of these genes was similar as in other late pollen transcriptomes; however, the over-representation of several protein groups (F-box proteins, receptor-like protein kinases, protein chaperones and proteins involved in calcium signalling) highlighted the interactions of AGP6 and AGP11 with members of the pollen tube endosome machinery enabling the recycling of AGPs to perform their signalling role (Costa et al. 2013).
MADS-domain transcription factors play key roles in the development of higher eukaryotes functioning as higher-order complexes. Five members of the MIKC* subgroup of the MADS-box family (AGL30, AGL65, AGL66, AGL94 and AGL104) were strongly upregulated in late stages of Arabidopsis pollen development (Pina et al. 2005) and were shown to form several heterodimeric complexes preferentially binding MEF2-type CArG-box sequence motifs (consensus CTA(A/T)4TAG) also over-represented in promoters of late pollen-expressed genes (Verelst et al. 2007a). Transcription profiling of double, triple (Verelst et al. 2007b) and even quadruple (Adamczyk and Fernandez 2009) mutants deficient in several combinations of MIKC* genes revealed the intriguing complexity of MADS-box TF network directing cellular differentiation during pollen maturation, a process that is essential for male reproductive fitness in flowering plants (Verelst et al. 2007b; Adamczyk and Fernandez 2009). Interestingly, the importance of MIKC* MADS-box TFs ZmMADS2 for maize pollen development was demonstrated even earlier on (Schreiber et al. 2004). The functional conservation of MIKC* MADS-box complexes in Arabidopsis and rice indicated that the function of heterodimeric MIKC* protein complexes in pollen development has been conserved since the divergence of monocots and eudicots, roughly 150 million years ago (Liu et al. 2013).
Basic leucine zipper (bZIP) transcription factors act as homo- or heterodimers and these effector-type TFs control many aspects of plant development including reproduction. Pollen-expressed TF AtbZIP34 is active during late stages of male reproductive development with a complex sporophytic and gametophytic mode of action. Transcription profiling of atbzip34 mutant pollen led to the finding that AtbZIP34 regulon comprises membrane-associated transporters and proteins involved in lipid metabolism and cell wall synthesis (Gibalová et al. 2009).
Transcription profiling was used also to unravel the regulon of male germline-specific R2R3-MYB transcription factor, DUO POLLEN1 (DUO1, Durbarry et al. 2005; Rotman et al. 2005), playing an important role in sperm cell differentiation. However, Borg et al. (2011) adopted different strategies as they analysed the transcriptomes of seedlings with ectopically expressed DUO1 in an estradiol-inducible manner and identified 63 candidate targets. Moreover, DUO1 was shown to directly regulate its target promoters through binding to the canonical MYB sites (Borg et al. 2011). The role of two DUO1 target genes DAZ1 and DAZ2 has been characterised; they both encode EARFootnote 1 motif-containing C2H2-type zinc finger proteins that are important for both generative cell division and DUO1-dependent germ cell differentiation (Borg et al. 2014).
In connection with bi- and tri-cellular pollen types in angiosperms, DUO1 may have been involved in the control of the timing of generative-cell division during the evolution of the pollen type (Hafidh et al. 2012b; Rotman et al. 2005). In order to illustrate complicated processes and interrelationships in plant protein families, we adopted the DUO1 gene as an example for the demonstration of frequently used genomic tools for studying the roles of specific proteins in the cell and to briefly summarise current knowledge and line out the distribution of different orthologs within angiosperms. Up to 2015, the lack of knowledge has hindered the comprehension of the origin and evolutionary history of MYB gene family across plants. It was reported that the intron patterns of R2R3-MYB transcription factors were greatly conserved in model higher plants (Matus et al. 2008; Du et al. 2012). However, the prevalence of R2R3-MYBs was quite different indicating that their introns were established in the common ancestor of land plants. Moreover, intron patterns in algae were variable and different from land plants. These findings suggested that algae and land plant lineages used different splicing patterns. Du et al. (2015) confirmed that R2R3-MYBs were older than 3R-MYBs which may be evolutionarily derived from R2R3-MYBs via intragenic domain duplication. The interesting feature is that intron patterns of land plant R2R3-MYBs were exclusively conserved within each subfamily. Phylogenetic relationships of DUO1 and related MYB-family transcription factors expressed in pollen from selected plant species including basal angiosperms are shown in Fig. 10.2. Members of different orders tend to cluster together within a given clade indicating that clades could have been expanded after divergence from their common ancestor (Du et al. 2015). This finding suggests the common origin of family members. All dicots clustered in their own clades and are separated from monocots and basal angiosperms, which exhibit lineage-specific expansion. The resolution within main orders is species dependent and possibly suffers from missing data (i.e. representatives from many groups across the phylogenetic tree that are not available yet) and long-branch attraction artefacts. Alternatively, the species-specific R2R3-MYBs may represent genomic relics that evolved independently as Du et al. (2015) adumbrated. These results show that there may be more lineage-specific subfamilies in DUO1 gene, and their evolution history could be solved in a future by using a more representative set of species and by combination with the expression -omics studies.

Phylogenetic relationships of pollen-specific DUO POLLEN 1 gene. The evolutionary history was inferred by using the maximum likelihood method based on the JTT matrix-based model. The tree with the highest log likelihood (−2637.8067) is shown. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The tree was divided into five main phylogenetic subgroups with bootstrap support >80% shown in different colours
It has been established that pollen tube growth in vitro and in vivo differ and that the directional pollen tube growth is greatly influenced by pistil tissues (Palanivelu and Preuss 2006; Palanivelu and Tsukamoto 2012). Therefore, pollen tubes growing in vitro and in vivo were compared in Lilium longiflorum (Huang et al. 2011). Similar analysis including also mature pollen was performed in Olea europaea (Carmona et al. 2015; Iaria et al. 2016). To characterise the changes in gene expression of A. thaliana pollen tubes growing in vivo and pistil-activated pollen tubes, the semi-in vivo approach was applied (Qin et al. 2009). It was shown that the pistil activation induced the expression of 1254 genes (18% of the overall transcriptome), many of which were pollen specific. None of these genes were active in in vitro growing pollen tubes. Similar analysis was performed on A. thalianasemi-in vivo pollen tubes growing under the influence of isolated ovules that induced the expression of 719 genes (Chen et al. 2014). In both studies, genes with potential function in signalling, pollen tube growth/cell extension and transcription were over-represented among the pistil-activated genes, including three MYB transcription factors (MYB97, MYB101, MYB120, Qin et al. 2009) that were later shown to play a crucial role during pollen tube differentiation required for sperm release (Leydon et al. 2013; Liang et al. 2013). Eighteen candidate genes for pollen tube guidance including TIR-NBS-LRRFootnote 2 proteins, DEFL proteins, protein kinases and receptor-like protein kinases were selected for reverse genetic screen, however, only with limited success, due to the functional redundancy in these large gene families (Chen et al. 2014).
Both cell types forming mature pollen–‘somatic’ vegetative cell and generative cell/sperm cells as the male germline–have very different cell fates that are reflected in their gene expression. Therefore, several experiments compared the transcriptomes of isolated male gametes with either whole mature pollen or even with isolated vegetative nuclei. In plants dispersing tri-cellular pollen, mature pollen grains were used. The first, although limited sperm cell transcriptome obtained by EST library sequencing, was published for Zea mays (Engel et al. 2003). In Plumbago zeylanica, producing dimorphic sperm cells, their individual transcriptomes were compared to that of vegetative nuclei (Gou et al. 2009), whereas in Arabidopsis thaliana (Borges et al. 2008) and Oryza sativa (Russell et al. 2012; Anderson et al. 2013), sperm cell transcriptomes were compared to intact mature pollen grains. Similar analysis was performed in Lilium longiflorum that releases bi-cellular pollen and therefore the transcriptome of generative cell was studied (Okada et al. 2006, 2007). To isolate sperm cells of plants producing bi-cellular pollen, Nicotiana tabacum pollen was germinated semi-in vivo, sperm cells were collected from pollen tubes emerging from cut pistils and their transcriptome was obtained by conventional EST sequencing (Xin et al. 2011). Seven studies of sperm cells from six plant species covered both dicots and monocots as well as plants producing bi- and tri-cellular pollen, providing us with an ample material for comparisons. The identified fractions of individual sperm cell transcriptomes varied according to the method used, from around 600 (Plumbago zeylanica, EST sequencing, Gou et al. 2009) to tens of thousands genes (Oryza sativa, RNAseq, Anderson et al. 2013). Such numbers clearly prove that sperm cells were definitely not transcriptionally and metabolically inactive entities. In all species, sperm cell transcriptomes were different not only from sporophytic control datasets but also from the transcriptomes of whole pollen/vegetative nuclei (i.e. Russell et al. 2012) as visualised also by PCA analyses (Borges et al. 2008; Russell et al. 2012). Moreover, there were striking differences between the transcriptomes of both types of dimorphic Plumbago zeylanica sperm cells, destined to fuse with egg cell and central cell, respectively (Gou et al. 2009). Interesting similarities were found in the presumed function of sperm cell-expressed proteins including the significant fraction of proteins of unknown function. Sperm cell transcriptomes were enriched in GO categories of cell cycle proteins, membrane-associated proteins, proteins involved in signal transduction, protein destination, ubiquitin-mediated proteolysis and epigenetic modifications (Engel et al. 2003; Okada et al. 2006, 2007; Borges et al. 2008; Xin et al. 2011; Russell et al. 2012; Anderson et al. 2013). On the contrary, genes for RNAi machinery were downregulated (Russell et al. 2012; Anderson et al. 2013). The fact that sperm cell transcriptomes of A. thaliana, N. tabacum and Z. mays shared only 0.3% genes (7.6% of genes were shared by at least two of three datasets, Xin et al. 2011) pointed out either the possibility that still only limited transcriptome fractions were identified and/or that there were more significant differences between sperm cells of dicots and monocots as well as those formed in bi- and tri-cellular pollen than originally expected. In any case, this fact is stimulating for further exciting research.
Finally, transcriptome profiles of gametophyte generation are available also for a few species of bryophytes and ferns—Physcomitrella patens (O’Donoghue et al. 2013; Ortiz-Ramirez et al. 2016; Xiao et al. 2011), Tortula ruralis (Oliver et al. 2004), Marchantia polymorpha (Higo et al. 2016) and Pteridium aquilinum (Der et al. 2011). However, since we concentrate solely on seed plants, gymnosperms and angiosperms, these datasets are not present in Table 10.2.
3 Proteomics
Transcriptomic analyses have provided and continue providing valuable information about global and specific gene expression and its dynamics. Transcriptomic data, however, do not provide complete information about gene expression since the proteome does not fully reflect the transcriptome (de Groot et al. 2007). This is especially true for systems with high level of translational regulation, such as the male gametophyte. The principal reason is that it is not technically feasible for transcriptomics to take into account the possible contribution of post-transcriptional regulatory levels of gene expression (Keene 2007). The effect of splicing (Collins 2011; Lorkovic and Barta 2004; Reddy et al. 2012) including the identification of potentially alternatively spliced transcripts (Kazan 2003; Sanchez et al. 2011; Xing and Li 2011) can be studied with the complete tilling gene chip (Whole-Genome ChIP Tilling Array ATH6, Roche-NimbleGen Systems, Inc.) or, ideally, by RNAseq (Loraine et al. 2013). However, further post-transcriptional regulatory levels, especially translation and mRNA storage, are active during male gametophyte development (Hafidh et al. 2011, 2016a). For all above reasons, it remains necessary to complement transcriptomics with proteomic data to get more realistic insight.
Soon after the first pollen transcriptomic analyses appeared in the last decade, the initial studies of pollen proteome were published. Surprisingly, the first plant species with published pollen proteome, though very incomplete, was a gymnosperm, Pinus strobus (Fernando 2005). Since then, numerous angiosperm species followed, for example, Arabidopsis thaliana (Grobei et al. 2009; Holmes-Davis et al. 2005; Noir et al. 2005; Sheoran et al. 2006; Zou et al. 2009), Oryza sativa (Dai et al. 2006, 2007), Solanum lycopersicum (Lopez-Casado et al. 2012; Sheoran et al. 2007), Lilium longiflorum (Pertl et al. 2009), Brassica napus (Sheoran et al. 2009a), Quercus ilex (Valero Galvan et al. 2012), and Helianthus annuus (Ghosh et al. 2015).
The pioneering proteomic studies were based on the excision of intact proteins in the isolated spots acquired from gels after 2-D gel electrophoresis (2-DE), which were protease-treated and analysed by mass spectrometry. Therefore, it resulted in only a very limited fraction of the total proteome, comprising usually several hundred proteins, almost invariably fewer than 1000. Therefore, 2-D gel-based proteomics has much lower coverage than transcriptomics and a little overlap between individual experiments. For example, three first published Arabidopsis pollen proteomic datasets identified 135 (Holmes-Davis et al. 2005), 121 (Noir et al. 2005) and 96 (Sheoran et al. 2006) proteins, respectively (Fig. 10.3). Considering overlaps between these datasets, 2-DE proteomics enabled the identification of 267 mature pollen proteins. Affymetrix ATH1 gene chip harboured probes for 237 of these proteins representing only very limited fraction of 13,977 genes active during pollen development (Honys and Twell 2004), of which 6044 were identified in mature pollen (Rutley and Twell 2015). Of identified 267 mature pollen proteins, 200 (75%) were found only in a single study, whilst only 18 proteins (7%) were identified by all three groups. This very small coverage of the pollen proteome is not surprising. One can assume that proteins found by more authors were encoded by most strongly expressed genes. Indeed, all 18 genes encoding these proteins were among the most abundant in pollen transcriptome. They also belonged to the functional categories containing usually only a limited number of typically very highly expressed genes: energy metabolism (8), stress response (4), synthesis and metabolism of the cell walls (2), cytoskeleton (2), protein synthesis (1), and metabolism (1). The result was also influenced by protein extraction protocol used by different groups that affects the composition of purified proteome fraction drastically, as independently demonstrated in tobacco pollen (Fíla et al. 2011).

Quantification of Arabidopsis thaliana transcriptomic and proteomic studies
Characterisation of although a limited part of the pollen proteome enabled the functional categorisation and comparison of pollen transcriptome and proteome. Again, there is significantly higher proportion of functional categories grouping abundant proteins into proteomic datasets. On the contrary, the obvious variability between individual 2-DE-analysed pollen proteomes is not as high as might be expected from their variability. As an exception, stress-related proteins and proteins of unknown function differ significantly. However, especially here, the possible influence of the downstream processing of the biological material cannot be excluded. In comparison to transcriptome, there is higher proportion of proteins involved in massive processes of general and energy metabolism and of protein synthesis and metabolism. Similar trends can be seen in the reference datasets characterising Oryza sativa pollen proteome (Dai et al. 2006, 2007), another model species with tri-cellular pollen. In this respect, the original Pinus strobus pollen proteome (Fernando 2005) is significantly different, not only because pine is a gymnosperm, in which male gametophyte structure and development differ from the angiosperms, but at least partly due to the significantly smaller size of identified proteome fraction and the lack of known genomic sequence of any gymnosperm at the time of publication. Therefore, there was particularly high fraction of unknown proteins in Pinus strobus proteome.
A fundamental breakthrough not only in pollen research was the introduction of gel-free proteomic techniques, which increased the efficiency of peptide and protein identification by one order. Gel-free techniques also enabled more accurate quantification of protein abundance. However, only a limited number of plant species were used for gel-free pollen proteome characterisation (Table 10.2). The first shotgun proteomic study identified 3465 proteins in Arabidopsis pollen (Grobei et al. 2009) that represented almost 13-times enlargement of a known fraction of the mature pollen proteome including the vast majority of proteins previously identified by 2-DE techniques. Such extension was also reflected in the functional categories, to which the identified proteins belonged, which were closer to the transcriptomic studies including the significant representation of the stable structural proteins (Fig. 10.3). Comparison of proteomic and transcriptomic datasets showed that the vast majority of 2928 (85% of described proteome) genes identified in pollen by proteomic and transcriptomic approaches was encoded by transcripts present already in early stages of male gametophyte development (Grobei et al. 2009; Honys and Twell 2004), thus representing an independent confirmation of the extent of translational regulation of gene expression in pollen (Honys et al. 2000, 2009; Honys and Twell 2004). Finally, 537 pollen proteins (15% of pollen proteome) had not been described in any transcriptomic study known at that time which made proteomics an attractive method for gene expression studies complementary to transcriptomics.
Similarly to transcriptomics, proteomic studies also aimed at the characterisation of the proteome dynamics during the pollen development and pollen tube growth. The list of species is shorter containing only Solanum lycopersicum (Chaturvedi et al. 2013), Nicotiana tabacum (Ischebeck et al. 2014) representing angiosperms and Picea wilsonii (Chen et al. 2012) as a representative of gymnosperms. Chen et al. (2012) investigated the influence of the limited nutrient supply on pollen tube growth, namely, the deficiency of sucrose, calcium and boron. In total, 166 proteins and 42 phosphoproteins (see also next section) were identified by LC-MS/MS as differentially regulated. Such number, low for gel-free approach, was mainly caused by the lack of conifer genome sequence data at that time. The identified proteins were involved in a variety of signalling pathways, providing new insights into the multifaceted mechanism of nutrient function including indicated nutrient-specific effects (Chen et al. 2012). Remaining two studies provided the most comprehensive male gametophytic proteomic datasets at the moment; they covered numerous stages of pollen development of related Solanaceae species providing evidence for developmentally controlled processes that might help to prepare the cells for specific developmental programmes and environmental stresses. In Solanum lycopersicum, five pollen developmental stages were compared—microsporocytes, tetrads, microspores, polarised microspores and mature pollen (Chaturvedi et al. 2013). In Nicotiana tabacum, the covered period was even broader—diploid microsporocytes, meiosis, tetrads, microspores, polarised microspores, bi-cellular pollen, mature pollen and pollen tubes, altogether eight stages (Ischebeck et al. 2014). In tomato, 1821 proteins were identified (Chaturvedi et al. 2013), whereas the tobacco analysis led to the identification of 3817 protein groups (Ischebeck et al. 2014). In both species, principal component analyses (Chaturvedi et al. 2013; Ischebeck et al. 2014) provided a similar picture as those resulted from transcriptomic studies (Bokvaj et al. 2015; Rutley and Twell 2015) and demonstrated that pollen development is highly controlled sequential process also at the proteome level. From the predicted functions, energy-related proteins are upregulated during the later stages of tomato pollen development. It indicates that pollen germination depends upon presynthesised proteins in mature pollen. In contrast, heat stress-related proteins are highly abundant in very early developmental stages, suggesting a dominant role in stress protection (Chaturvedi et al. 2013). Similar observations were made in tobacco, where the early developmental stages were enriched also with ribosomal and other translation-related proteins (Ischebeck et al. 2014).
In several cases, the individual studies targeted specific cell types, namely the generative or sperm cells in Lilium davidii (Zhao et al. 2013), and Oryza sativa (Abiko et al. 2013). In rice, sperm cell gel-free proteome was compared to the whole mature pollen grain. Of 2179 sperm cell-expressed proteins, 77 were preferentially present in the male gametes (Abiko et al. 2013). The comprehensive study by Zhao et al. (2013) employed 2-D DIGE followed by MALDI-TOF/TOF mass spectrometry to identify 101 proteins differentially expressed in lily generative and sperm cells. These proteins are involved in diverse cellular and metabolic processes, with preferential involvement in the metabolism, cell cycle, signalling, the ubiquitin/proteasome pathway, and chromatin remodelling, i.e. similar categories as revealed by transcriptomics. Impressively, almost all proteins in ubiquitin-mediated proteolysis and the cell cycle were upregulated in sperm cells, whereas those in chromatin remodelling and stress response were downregulated (Zhao et al. 2013).
Other studies were devoted to specialised cellular compartments, including membranes (Lilium longiflorum, Pertl et al. (2009); Lilium davidii, Han et al. (2010); and Solanum lycopersicum; Paul et al. (2016)), nuclei (Yang et al. 2016) and messenger ribonucleoprotein (mRNP) complexes (Honys et al. 2009). Other studies focused on specific protein groups (e.g. allergens) or proteins characterised by specific post-translational modifications, notably phosphorylation (Fíla et al. 2012, 2016; Mayank et al. 2012, see below). In all species analysed, membrane proteomes confirmed the presence of expected membrane-associated proteins on/in plasma membrane as well as endomembranes (Pertl et al. 2009; Han et al. 2010; Paul et al. 2016). In Lilium longiflorum, the differences in abundance of various protein types were observed in both membrane fractions in mature pollen and in several time points of pollen tube growth. For example, increase in the abundance of proteins involved in cytoskeleton, carbohydrate, energy metabolism, as well as ion transport was observed before pollen germination (10–30 min), whereas proteins involved in membrane/protein trafficking, signal transduction, stress response and protein biosynthesis decreased in abundance during this time (Pertl et al. 2009). Similar proteins were identified in membrane proteomes of two tomato cultivars, and the presence of proteins corresponding to energy-related pathways (glycolysis and Krebs cycle) enabled to present a hypothetical model of energy reservoir of the male gametophyte (Paul et al. 2016). Lilium davidii pollen and pollen tubes plasma membrane proteome fraction comprised also proteins of translational apparatus and DNA/RNA-binding proteins with preferential occurrence of ribosomal proteins. The identification of these proteins probably resulted from the presence of cytoskeleton-binding polysomes anchored to the plasma membrane via actin filaments or targeted to lipid rafts (Han et al. 2010). The association of translation apparatus and RNA-storage particles with the actin cytoskeleton was observed also in tobacco pollen and pollen tubes (Honys et al. 2009) where the protein composition of large ribonucleoprotein particles (EPPs) was studied. EPP complexes are formed in immature pollen where they contain translationally silent mRNAs. Although massively activated at the early progamic phase, they also serve as a long-term storage of mRNA transported along with the translational machinery to the tip region. Since EPPs contain ribosomal subunits, rRNAs and a set of mRNAs, they were hypothesised to represent well-organised machinery devoted to mRNA storage, transport and subsequent controlled activation resulting in protein synthesis, processing and localisation, extremely useful in fast tip-growing pollen tube. Expression of vast majority of the closest orthologues of EPP proteins also in Arabidopsis male gametophyte further extended this concept from tobacco to Arabidopsis, the model species with advanced tri-cellular pollen (Honys et al. 2009). The last cellular compartments analysed for the proteomic perspective were the vegetative, generative and sperm cell nuclei of Lilium davidii (Yang et al. 2016). The profiling of histone variants of all five histone families in all three cell types revealed 92 identities representing 32 histone variants. Generative and sperm cells had almost identical histone profiles and similar histone H3 modification patterns, significantly different from those of vegetative nuclei. These results suggested that differential histone programmes, important for the identity establishment and differentiation of the male germline, may be established following the asymmetric division (Yang et al. 2016).
To summarise, it is obvious that proteomics studied a broader spectrum of species (including plants not representing the classical models) compared to microarray transcriptomics since proteomics is not limited by sequenced genomic DNA of the particular species. The EST sequences or protein sequences from related species can be used instead. It also highlighted post-transcriptional levels of gene expression that could not be addressed by transcriptomics.
4 Phosphoproteomics
Pollen rehydration and activation is accompanied by two main regulatory mechanisms of gene expression. The first one is represented by translation regulation. A notable part of stored, translationally regulated transcripts are localised in EDTA/puromycin-resistant particles (EPPs) in tobacco (Nicotiana tabacum, Honys et al. 2000, 2009). The transcripts inside these complexes are stored in a translationally silent form in mature pollen, whereas upon pollen rehydration, the mRNAs are being de-repressed and translated. Since most growing processes inside the pollen tube are localised to the tip, EPP complexes are transported towards the pollen tube tip (Honys et al. 2009). The second mechanism of gene expression regulation is phosphorylation, which is one of the most dynamic post-translational modifications of proteins. Protein phosphorylation in reaction to rehydration was revealed not only in Nicotiana tabacum male gametophyte (Fíla et al. 2016) but also in rehydrated plants of the xerophyte Craterostigma plantagineum (Röhrig et al. 2008) and in the cells in Zea mays leaf growing zone (Bonhomme et al. 2012). Large-scale studies of protein phosphorylation usually employ various enrichment protocols to enable the identification of phosphorylated peptides in the total complex protein crude extract. The enrichment can be either carried out at the level of intact proteins or alternatively from the peptide mixture acquired after cleavage of the complex total protein crude extract by a specific protease (Fíla and Honys 2012). Both these phosphoproteomic approaches showed their advantages as well as limitations.
The first phosphoproteomic study performed on male gametophyte was that of Arabidopsis thaliana mature pollen (Mayank et al. 2012, Table 10.3). This study applied a combination of three phosphopeptide-enriching protocols: immobilised metal affinity chromatography (IMAC), metal oxide/hydroxide affinity chromatography (MOAC) and sequential elution from IMAC (SIMAC). The study presented collectively 962 phosphopeptides carrying 609 phosphorylation sites that belonged to 598 phosphoproteins. From the functional point of view, most identified phosphoproteins took part in the regulation of protein metabolism and function, metabolism, protein fate, protein binding, signal transduction and cellular transport. Several kinases were also among the identified phosphoproteins, particularly AGCFootnote 3 protein kinases, Ca2+-dependent protein kinases and sucrose non-fermenting protein kinases 1.
The next male gametophyte of angiosperms, which was subjected to phosphoproteomic techniques, was tobacco (Nicotiana tabacum; Fíla et al. 2012, 2016, Table 10.3). The former study identified 139 phosphoprotein candidates from mature pollen and pollen grains activated in vitro for 30 min. In order to improve the number of unambiguously positioned phosphorylation sites, titanium dioxide phosphopeptide enrichment was performed on trypsin-digested mature pollen crude extract, which led to the identification of 51 more phosphorylation sites localised in the phosphoproteins already identified in mature pollen giving a total of 52 unambiguous phosphorylation sites. In order to understand the processes during pollen grain activation and the start of pollen tube growth, pollen grains activated in vitro for 5 min were also taken into consideration in the subsequent study (Fíla et al. 2016). To increase the probability of phosphoprotein identification, phosphopeptide-enriching MOAC with titanium dioxide matrix was applied exclusively. In the mentioned three stages of tobacco male gametophyte, 471 phosphopeptides were identified, which carried 432 unambiguously determined phosphorylation sites. These phosphorylated peptides were assigned to 301 phosphoproteins. The phosphopeptide enrichment of the three stages increased notably the number of identified phosphorylation sites. The dominant functions were transcription, protein synthesis, protein destination and storage and signal transduction. It is also worth mentioning that almost one fifth of identified phosphopeptides was put into categories with unknown function or unclear classification. These results are in agreement with tobacco male gametophyte proteome, where approx. fifteen percent of male gametophyte-specific proteins were of unknown classification. The unknown phosphoproteins represent likely male gametophyte-specific or male gametophyte-enriched proteins, function of which will be probably important for regulation of pollen activation and pollen tube growth. A notable part of the identified phosphopeptides showed a significant regulatory trend in the progamic phase of male gametophyte. Most of the regulated peptides were shown to be exclusive for mature pollen grains. The only alternative study that considered other stages than mature pollen was performed with pollen tubes from Picea wilsonii (Chen et al. 2012). However, it differed in two ways from the above studies: (1) a gymnosperm was studied instead of an angiosperm, and (2) the proteome and phosphoproteome of Picea wilsonii pollen tubes were studied not from the developmental point of view but as a reaction to growth media lacking sucrose or Ca2+ ions (which serve as important nutrients for pollen tube growth). The Picea wilsonii study thus revealed 166 proteins and 42 phosphoproteins playing their roles in signalling of media deficiency.
Most recently, Zea mays became the first monocot with a published mature pollen phosphoproteome (Chao et al. 2016), but again, no other gametophyte stages were studied (Table 10.3). Despite this, the maize pollen phosphoproteomic dataset became the largest one since it presented 4638 phosphopeptides, which belonged to 2257 phosphoproteins. These phosphorylated peptides led to the identification of 5291 phosphorylation sites with many multiply phosphorylated phosphopeptides and also carrying more than one phosphorylation site. The dominant molecular functions were ion binding, kinase activity, transmembrane transporter activity, oxidoreductase activity, and DNA binding, whilst the enriched biological processes were represented by protein posttranslational modification, cell organisation, signalling G-proteins, calcium signalling, abiotic stress, protein targeting, and RNA–RNA binding.
The functional categories of the identified phosphopeptides were quite similar in male gametophytes of all studied species. Since pollen tube tip growth requires several cellular mechanisms, such as small GTPase signalling, ion gradient formation, cytoskeleton organisation and transport of secretory vesicles (Palanivelu and Preuss 2000; Šamaj et al. 2006), dominant phosphoproteins common to all datasets belonged to at least some of these categories. Moreover, in tobacco, the protein synthesis category included proteins likely responsible for translation regulation, including EPP particles (Honys et al. 2009). The highest number of proteins responsible for the actual regulatory processes was identified in the maize phosphoproteome, including male sterility-associated proteins together with proteins influencing maize productivity (Chao et al. 2016). The number of proteins identified in tobacco could be influenced by the fact that the identifications of the second tobacco male gametophyte phosphoproteome relied on expressed sequence tags (EST sequences) that were acquired mainly from sporophyte tissues and thus could lack gametophyte-specific proteins.
The phosphoproteomic datasets are usually analysed whether they contain any over-represented sequence context surrounding the phosphorylation site. However, such a motif over-representation compared to the background dataset remains speculative, and the rare sequence motifs could remain undetectable. Moreover, the link between a particular kinase and target protein still remains to be experimentally proven. In Arabidopsis thaliana pollen phosphoproteome (Mayank et al. 2012), only serine-phosphorylated peptides were subjected to motif search, and only two motifs were identified: xxxxxxS*Pxxxxx and xxxRxxS*xxxxxx (phosphorylated amino acid is indicated by an asterisk behind the one-letter code). The prolyl-directed phosphorylation is usually mediated by mitogen-activated protein kinases and/or cyclin-dependent protein kinases, whereas the latter basic motif is recognised by Ca2+-dependent protein kinases (Lee et al. 2011). Later, the motif search in the second tobacco male gametophyte phosphoproteome (Fíla et al. 2016) enabled the identification of five motifs with central phosphoserine (particularly xxxxxxS*Pxxxxx, xxxRxxS*xxxxxx, xxxKxxS*xxxxxx, xxxxxxS*DxExxx, and xxxxxxS*xDDxxx) but also one with a phosphothreonine in the middle (xxxxxxT*Pxxxxx). The prolyl-directed phosphorylation (regardless whether a serine or a threonine occupied the middle position of the motif) was mediated by mitogen-activated protein kinases and/or cyclin-dependent protein kinases (Lee et al. 2011). Then, there were two alkaline and two acidic motifs. The former motifs were represented by an arginine or a lysine on the third position before the actual phosphorylated serine, which were recognised by Ca2+-dependent protein kinases and Ca2+-dependent protein kinases–sucrose-non-fermenting protein kinases (CDPK–SnRK) (Lee et al. 2011). The latter, acidic motifs can be in principle merged to one phosphorylation motif, xxxxxxS*(D/E)(D/E)(D/E)xxx, which was reported to be targeted by casein kinase 2 (CK2) (Lee et al. 2011). The broadest spectrum of kinase motifs was identified in maize mature pollen phosphoproteome (Chao et al. 2016), but several motifs were in principle shared with tobacco and Arabidopsis. However, these shared motifs were more specified in maize by other amino acid positions around the phosphorylation site so one phosphorylation site was actually split into more similar motifs differing in the specified position(s). In spite of more identified phosphorylation motifs in the most recent study, it still had common phosphorylation motifs with tobacco and Arabidopsis pollen phosphoproteomes. It is likely that pollen activation will bear similarities across various angiosperm species. Chao and colleagues thus identified 23 phosphoserine motifs and 4 phosphothreonine motifs, which were further sorted into 4 groups: 8 phosphorylation motifs were considered as prolyl-directed phosphorylation, 5 motifs were alkaline and 4 were acidic. The remaining ten phosphorylation motifs were collected in the group ‘others’. Several phosphorylation motifs (mainly from the group ‘others’) were considered as novel, and these motifs can represent the male gametophyte-specific regulatory pathways. Finally, it should be noted that all mentioned phosphorylation motifs were acquired by in silico data search, and it will be required to perform additional experiments in order to link a particular protein kinase with its target protein(s).
5 Specialised Pollen -Omics
To make the list of pollen -omics complete, we cannot leave out more specialised studies covering generally only limited number of model species. Of them, translatomics, methylomics and miRNAomics are based on transcriptomic approaches since they characterised specialised RNA populations functionally related to mRNA fate in the cytoplasm and translation. Likewise, pollen allergome and secretome were identified by proteomic techniques, whereas metabolomics employs a different set of techniques which will be discussed elsewhere in this book (see Chap. 12).
The study of Lin et al. (2014) represents the first and so far the only attempt to identify and characterise the subset of actively translated transcripts in in vivo-growing pollen tubes of Arabidopsis thaliana. The authors adopted elegant solution for the isolation of polysome-RNA complexes from pollen tubes growing through tiny Arabidopsis pistils; they were affinity purified via HIS6-FLAG dual-epitope tagged ribosomal protein RPL18 expressed under pollen vegetative cell-specific promoter LAT52. The comparison with in vitro-cultivated pollen tubes revealed over 500 transcripts specifically enriched in in vivo-elongating pollen tubes including transcripts encoding proteins involved in micropylar guidance, pollen tube burst and repulsion of multiple pollen tubes in embryo sac (Lin et al. 2014). Although the similar functional categorisation of genes upregulated in in vivo translatome (Lin et al. 2014), semi-in vivo transcriptome (Qin et al. 2009) and pollen–pistil interaction-induced transcriptome (Boavida et al. 2011), there was only very little overlap at the level of individual genes. However, it is difficult to conclude, whether such differences reflected a different nature of de novo transcription during in vivo and semi-in vivo pollen tube growth or only a subset of induced transcripts is being actively translated.
MicroRNAs (miRNAs) represent only a small portion of transcriptome, but they play an important role in post-transcriptional regulation of gene expression, mRNA cleavage, mRNA destabilisation through poly(A) tail shortening and translation inhibition (Brodersen et al. 2008; Carthew and Sontheimer 2009). Therefore, the identification of pollen miRNAs and their targets and especially the dynamics of miRNAome is of key importance for understanding the fine modulation of gene expression in the male gametophyte and in the process of the male germline differentiation. Of the seven studies published so far, one was devoted to gymnosperm Pinus taeda (Quinn et al. 2014) and the remaining six to three angiosperm model plants Arabidopsis thaliana (Chambers and Shuai 2009; Grant-Downton et al. 2009b; Borges et al. 2011), Oryza sativa (Peng et al. 2012; Wei et al. 2011), and Zea mays (Li et al. 2013). In these studies, 24-nt miRNAs represented the most abundant class. Alongside the identification of known and novel miRNA families, these studies also identified their putative mRNA targets, and in few cases, they even demonstrated the regulatory function of the respective miRNAs.
Chambers and Shuai (2009) profiled the expression of 70 known miRNAs in Arabidopsis mature pollen using miRCURY microarray and the comparison of their expression with transcriptomic profiles of their putative targets indicated the activity of several candidate miRNAs in pollen. The first large-scale study employing de novo sequencing was, as usual, conducted on Arabidopsis mature pollen and pollen miRNAome was compared to that of leaves (Grant-Downton et al. 2009b). Out of 33 miRNA families identified in pollen, expression of 17 was validated by RT-PCR, and most of them were found to be enriched in the male gametophyte with three (miR157, ath-MIR2939, and miR845) being putatively pollen specific. Moreover, the study reported, for the first time, the presence of trans-acting siRNAs in pollen (Grant-Downton et al. 2009b). Borges et al. (2011) analysed miRNA populations sequenced by Slotkin et al. (2009) with a special interest in miRNAs active in the male germline. They confirmed most of the previously identified pollen-expressed miRNAs (with the exception of miR776, Grant-Downton et al. 2009b) and found even higher representation of miRNA families both in mature pollen (75 families) and in sperm cells (83) including 25 potentially novel miRNAs processed in sperm cells and pollen. Of them, miR159 was particularly interesting, since it was highly enriched in sperm cells and was predicted to be involved in the regulation of DUO1 (Palatnik et al. 2007, Grant-Downton et al. 2009a). miR156 and miR158 represented other candidates for the role in the male germline as they were enriched in sperm cells and likely to associate with sperm cell-enriched ARGONAUTE 5 (see Borges et al. 2011). Rice studies (Peng et al. 2012; Wei et al. 2011) described the miRNAome dynamics during male gametophyte development in stages previously used for the transcriptome analysis (Wei et al. 2010). The authors identified numerous known and novel miRNAs, often pollen-enriched, and showed the correlation of their expression profiles with their potential targets (Wei et al. 2011). In maize, the comparison of miRNA populations isolated from mature pollen, in vitro-cultivated-pollen tubes and non-pollinated as well as pollinated silks identified 56 miRNAs (40 conserved and 16 novel) differentially expressed between pollen and pollen tubes and 38 miRNAs (30 conserved and 8 novel) showing differential expression pattern between mature non-pollinated and pollinated silks. The analyses of these miRNAs and their potential targets (predominantly auxin signal transduction and transcription regulation) also showed the participation of miRNA pathway in the regulation of pollen–pistil interactions (Li et al. 2013). In loblolly pine, miRNA populations were compared between mature and germinating pollen and 47 miRNAs (23% of 208 identified in total) representing 22 families were upregulated and downregulated (14 and 8 families, respectively) in germinated pollen. Together with other similarities and differences with Arabidopsis and rice pollen miRNA populations, it highlighted that the microRNA pathway is active also during pollen germination in gymnosperms (Quinn et al. 2014).
The methylome sequencing of haploid cell types during male gametogenesis highlighted the differential methylation patterns in vegetative and sperm cells (Calarco et al. 2012, Ibarra et al. 2012). Plant male germline retains symmetric DNA methylation, whereas the asymmetric methylation is lost there. On the contrary, asymmetric DNA methylation is restored in vegetative cells and during post-fertilisation embryo development. This de novo CHH methylation is a result of the activity of DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) and employs 24-nt siRNAs. Differential genome reprogramming in pollen contributes to epigenetic inheritance, imprinting and transposon silencing (Calarco et al. 2012).
Of the two specialised proteomics-based techniques, allergomics was applied to a wider selection of plant species covering five orders of monocots and dicots (Table 10.2). For the obvious relation to pollen allergenicity, the selection lacks the usual models but concentrates mainly on wind-pollinated plants producing large amounts of pollen, predominantly grasses (Abou Chakra et al. 2012; Campbell et al. 2015; Kao et al. 2005; Schmidt et al. 2010; Schulten et al. 2013) and ragweed (Ambrosia spp.; Bordas-Le Floch et al. 2015; Zhao et al. 2016). Therefore, all studies were performed on mature pollen and were gel based. The purified proteins were separated by 2-DE, human IgE-binding proteins were identified, excised and analysed by mass spectrometry. Most of the major pollen allergen families were found in both dicot and monocot pollen—profilin, expansin, berberine bridge enzyme, pectate lyase, Ole e 1, cytochrome C and group 5/6 grass allergen (ribonuclease) families followed by enolase, EF hand, polygalacturonase, pathogenesis-related and prolamin families (Abou Chakra et al. 2012; Bordas-Le Floch et al. 2015; Campbell et al. 2015; Kao et al. 2005; Schmidt et al. 2010; Schulten et al. 2013; Zhao et al. 2016). In Phleum pratense, the major grass allergens were confirmed also in pollen cytoplasmic granules released from pollen grains that may represent respirable vectors of allergens (Abou Chakra et al. 2012).
Sexual reproduction in plants requires extensive cell–cell communication at many stages and at many levels including pollen–pistil interaction involving female sporophytic and gametophytic tissues that ends by the direct communication between male and female gametes preceding fertilisation. The extreme compactness of flower tissues as well as the need to separate individual cells made research in this area immensely difficult, and so far, most of the information came from the female side (reviewed by Kessler and Grossniklaus 2011, see also Chap. 8). The attempts to characterise the male–female crosstalk at the global scale first represented transcriptomic and proteomic studies of complex female tissues typically before and after pollination. Most of these studies also studied the phenomenon of pollen (in)compatibility. However, before that, it was interesting to analyse the pollination interface. Sang et al. (2012) analysed the differences in the proteomes of wet and dry stigmas in Nicotiana tabacum (wet) and Zea mays (dry) and compared them with the exudates from wet tobacco stigmas. With 177 identified proteins, tobacco stigmatic exudates were richer in proteins than stigmatic exudates of Lilium longiflorum and Olea europaea comprising 51 and 57 proteins, respectively (Rejon et al. 2013). Similarly, Nazemof et al. (2014) identified proteins involved in Triticale stigma development. However, these studies had only limited coverage because of the use of gel-based proteomics. Most recently, protein composition of ovular secretes, pollination drops, on female cones of two closely related gymnosperm species—Cephalotaxus koreana and C. sinensis—was similar to that of other gymnosperms including gnetophytes and contained mainly defence-related proteins and carbohydrate-modifying enzymes (Pirone-Davies et al. 2016). Therefore, a deeper insight was achieved through transcriptomics when transcriptomes of stigmatic papillary cells in three Brassicaceae species, Arabidopsis thaliana, A. halleri and Brassica rapa, showed great degree of similarity. Fifty-eight percent of papilla-expressed genes were shared by all three species (Osaka et al. 2013), and only a minor fraction of expressed genes was species specific. Interestingly, gene expression in Arabidopsis papillar cells does not seem to be much influenced by the pollination, since 77% of genes were active in non-pollinated papillar cells as well as in these cells after the pollination with compatible and incompatible pollen (Matsuda et al. 2014).
Another step forward represented the comparison of non-pollinated and pollinated pistils studied at both transcriptomic and proteomic levels. Proteome differences caused by pistil pollination were studied in Glycine max (Li et al. 2012), Oryza sativa (Li et al. 2016), Prunus armeniaca (Feng et al. 2006), Solanum pennellii (Chalivendra et al. 2013) and, interestingly, also in basal angiosperm Liriodendron chinense (Li et al. 2014). Transcriptomic studies comprised Arabidopsis thaliana (Boavida et al. 2011), Olea europaea (Carmona et al. 2015; Iaria et al. 2016), Oryza sativa (Li et al. 2016), Citrus clementina (Caruso et al. 2012) and the direct comparison of self-compatible Solanum pimpinellifolium and self-incompatible Solanum chilense (Zhao et al. 2015). These studies confirmed that not only pollination but also cross- versus self-pollination induced novel gene expression and protein synthesis.
For the characterisation of proteins directly involved in male–female interactions, it became necessary to analyse proteins secreted from both main players (see also Chap. 8). The comparison of apoplastic proteins isolated from Arabidopsis thaliana mature pollen and pollen tubes cultivated in vitro for 6 h enabled the identification of 71 novel proteins expressed after pollen germination. Of them, 50 proteins were secreted; they were involved in cell wall modification and remodelling, protein metabolism and signal transduction (Ge et al. 2011). This study provided the first insight into pollen-secreted proteins functioning in pollen germination and pollen tube growth. However, the use of DIGE limited the number of identified proteins. More importantly, it was already shown that the contact with female tissues significantly changed and enriched pollen tube gene expression (Qin et al. 2009; Lin et al. 2014). Therefore, it became necessary to evaluate the situation in vivo or semi-in vivo. Recently, such secretomes were published for two related species. Proteomic analysis of Solanum chacoense ovule exudates isolated by tissue-free gravity-extraction method enabled the identification of 305 ovule-secreted proteins, 58% of which appeared to be ovule specific (Liu et al. 2015). Similarly, gel-free proteomics was used to characterise the secretome of Nicotiana tabacum semi-in vivo cultivated pistil-activated pollen tubes (Hafidh et al. 2016b). Here, 801 proteins were identified with high frequency of small proteins <20 kDa. Interestingly, the majority (57%) of pollen tube-secreted proteins lacked signal peptide and were shown to be secreted unconventionally. This study not only highlighted a potential mechanism for unconventional secretion of pollen tube proteins but also indicated their potential functions in pollen tube guidance towards ovules for sexual reproduction. Hafidh et al. (2016b) demonstrated that the knockdown of unconventionally secreted translationally controlled tumour protein (TCTP) in Arabidopsis thaliana pollen tubes caused their poor navigation to the target ovule and low transmission of the mutant allele through the male. Unconventional protein secretion was described also in the ovules (Liu et al. 2015) but to a much smaller extent. The combination of both datasets, although obtained from different species, represents a significant contribution to our current efforts for dissecting possible mechanism for cell–cell communication between the pollen tube and female reproductive cells.
6 Conclusion and Perspective
The introduction of -omics techniques brought a very notable insight into the research of male gametophyte development, its dynamics and regulation. The past two decades and recent few years in particular faced the outburst of novel -omics techniques. However, transcriptomics still represents the main source of information and usually the first choice mainly due to its robustness, established infrastructure and data processing pipelines. Both microarrays and RNAseq offer big gene coverage, but the microarrays depend on a priori knowledge of sequence information and cannot display, for instance, splicing variants and/or shortened or altered transcripts. Although these issues are circumvented by deep sequencing technologies, the majority of data was achieved by the microarrays. That is why mostly male gametophytes of model plants were studied and only a limited number of alternative species have a known pollen transcriptome. Nevertheless, no transcriptomic technique solved the issue of the correlation of the abundance of transcripts to corresponding proteins. Therefore, proteomics is considered more reflecting gene regulation since it proves the presence of a protein. The original in-gel techniques showed limited gene coverage since they usually identified no more than 1000 proteins with a little overlap between independent experiments. On the other hand, because proteomics does not rely on the knowledge of genomic sequences, male gametophyte proteomes of non-model plants were studied. The versatility of proteomic approaches also enabled specialised studies like specific sets of proteins (membrane proteome, secretome, allergome) as well as functional and regulatory post-translational modifications, usually on limited samples only. Of them, phosphoproteome was studied in male gametophyte, namely, in three angiosperms—Arabidopsis thaliana, tobacco and maize. In tobacco, phosphoproteome dynamics during pollen activation was studied. In order to evaluate the regulation of early phases of pollen activation in bi- and tri-cellular species, it would be interesting, although challenging, to perform similar experiments on the tri-cellular species to acquire properly activated pollen, but the novel information brought by these approaches is definitely worth the effort.
In spite of the growing list of proteins with known function, pollen -omics revealed many proteins with unknown function or unclear classification that will surely deserve a further investigation by the subsequent functional studies. These unknowns likely represent pollen-specific proteins or proteins with a notable function related to male gametophyte. Integration of phylogenetic comparative methods with the analysis of genomic data enables to design experiments and generate new insights concerning the origin and structure of the genomes. The phylogenomic approach based on sequence similarities can identify gene duplications—orthologs vs. paralogs, infer evolutionary rate variation among taxa and separate sequence convergences from shared origins. The prediction of gene function can be improved by incorporating the evolutionary history of the genes themselves and reconstructing their historical sequence and function using a phylogenetic framework. However, the majority of available plant genome sequences originate from crop plants that make deeper phylogenomic analyses still unfeasible. In the future, with more data available, the comparison of gene families among species from a phylogenetically different group of plants will allow a comprehensive study of pollen-specific genes and gene family evolution in plants.
Perhaps the most interesting trend in modern work has been a move towards synthesis. During the past two decades, various high-throughput plant -omic studies have revealed a boom due to technological advancement. Numerous articles focused not only on model plants but also other organisms. However, some of these analyses were carried out with expertise on the bioinformatics field, but with minimal biological relevance. The most interesting part of the research started with integration of data from various resources as well as the combination of these two fields.
Notes
- 1.
EAR motif is short Ethylene-responsive element binding factor-associated Amphiphilic Repression motif present in plant transcriptional repressors that mediates transcriptional repression by the association with corepressors responsible for chromatin modification (Kagale and Rozwadowski 2011).
- 2.
TIR-NBS-LRR is a large receptor subfamily, a part of the ‘R’ gene superfamily implicated in pathogen recognition. TIR-NBS-LRR proteins contain N-terminal domain with toll/interleukin-1 receptor homology (TIR), nucleotide binding site (NBS) and leucine-rich repeat (LRR) domains (Meyers et al. 2003).
- 3.
AGC kinases represent the subgroup of serine/threonine protein kinases named after three representative families, the cyclic guanosine monophosphate (cAMP)-dependent protein kinase (PKA), the cyclic guanosine monophosphate (cGMP)-dependent protein kinase (PKG) and the protein kinase C (PKC) families (Pearce et al. 2010).
Abbreviations
- 2-D DIGE:
-
two dimensional fluorescence difference gel electrophoresis
- 2-DE:
-
two dimensional gel electrophoresis
- bHLH:
-
basic helix-loop-helix transcription factor
- bZIP TF:
-
basic leucine zipper transcription factor
- cAMP:
-
cyclic adenosine monophosphate
- CAGE:
-
cap analysis of gene expression
- cGMP:
-
cyclic guanosine monophosphate
- DEFL protein:
-
defensin-like family protein
- EAR motif:
-
ethylene-responsive element binding factor-associated amphiphilic repression motif
- EPP:
-
EDTA/puromycin-resistant particle
- GO:
-
gene ontology
- IMAC:
-
immobilized metal affinity chromatography
- LC–MS/MS:
-
liquid chromatography–tandem mass spectrometry
- MADS-box TF:
-
family of transcription factors containing conserved MADS DNA-binding domain
- MALDI–TOF/TOF:
-
matrix-assisted laser desorption/ionization–time-of-flight tandem mass spectrometry
- MIKC* type proteins:
-
subfamily of MADS-box proteins with conserved domain structure, where the MADS (M) domain is followed by Intervening (I), Keratin-like (K) and C-terminal domains
- MOAC:
-
metal oxide/hydroxide affinity chromatography
- MPSS:
-
massively parallel signature sequencing
- mRNP:
-
messenger ribonucleoprotein particle
- MYB family proteins:
-
transcription factor protein family characterised by the presence of MYB (myeloblastosis) DNA-binding domain
- PKA:
-
cAMP-dependent protein kinase
- PKC:
-
protein kinase C
- PKG:
-
cGMP-dependent protein kinase
- R2R3-MYB:
-
MYB-protein subfamily characterised by the R2R3-type MYB domain
- RNAseq:
-
RNA deep sequencing technologies
- RPM:
-
reads per million
- SAGE:
-
serial analysis of gene expression
- SIMAC:
-
sequential elution from IMAC
- TCTP:
-
translationally controlled tumour protein
- TF:
-
transcription factor
References
Abiko M, Furuta K, Yamauchi Y, Fujita C, Taoka M, Isobe T, Okamoto T (2013) Identification of proteins enriched in rice egg or sperm cells by single-cell proteomics. PLoS One 8:e69578. doi:10.1371/journal.pone.0069578
Abou Chakra OR, Sutra JP, Demey Thomas E, Vinh J, Lacroix G, Poncet P, Senechal H (2012) Proteomic analysis of major and minor allergens from isolated pollen cytoplasmic granules. J Proteome Res 11:1208–1216
Adamczyk BJ, Fernandez DE (2009) MIKC* MADS domain heterodimers are required for pollen maturation and tube growth in Arabidopsis. Plant Physiol 149:1713–1723
Anderson SN, Johnson CS, Jones DS, Conrad LJ, Gou X, Russell SD, Sundaresan V (2013) Transcriptomes of isolated Oryza sativa gametes characterized by deep sequencing: evidence for distinct sex-dependent chromatin and epigenetic states before fertilization. Plant J 76:729–741
Aya K, Suzuki G, Suwabe K, Hobo T, Takahashi H, Shiono K, Yano K, Tsutsumi N, Nakazono M, Nagamura Y, Matsuoka M, Watanabe M (2011) Comprehensive network analysis of anther-expressed genes in rice by the combination of 33 laser microdissection and 143 spatiotemporal microarrays. PLoS One 6:e26162. doi:10.1371/journal.pone.0026162
Barranca M, Fontana S, Taverna S, Duro G, Zanella-Cleon I, Becchi M, De Leo G, Alessandro R (2010) Proteomic analysis of Parietaria judaica pollen and allergen profiling by an immunoproteomic approach. Biotechnol Lett 32:565–570
Becker JD, Boavida LC, Carneiro J, Haury M, Feijo JA (2003) Transcriptional profiling of Arabidopsis tissues reveals the unique characteristics of the pollen transcriptome. Plant Physiol 133:713–725
Becker JD, Takeda S, Borges F, Dolan L, Feijó JA (2014) Transcriptional profiling of Arabidopsis root hairs and pollen defines an apical cell growth signature. BMC Plant Biol 14. doi:10.1186/s12870-014-0197-3
Berger F, Twell D (2011) Germline specification and function in plants. Annu Rev Plant Biol 62:461–484
Boavida LC, Borges F, Becker JD, Feijo JA (2011) Whole genome analysis of gene expression reveals coordinated activation of signaling and metabolic pathways during pollen-pistil interactions in Arabidopsis. Plant Physiol 155:2066–2080
Bokvaj P, Hafidh S, Honys D (2015) Transcriptome profiling of male gametophyte development Nicotiana tabacum. Genomics Data 3:106–111
Bonhomme L, Valot B, Tardieu F, Zivy M (2012) Phosphoproteome dynamics upon changes in plant water status reveal early events associated with rapid growth adjustment in maize leaves. Mol Cell Proteomics 11:957–972
Bordas-Le Floch V, Le Mignon M, Bouley J, Groeme R, Jain K, Baron-Bodo V, Nony E, Mascarell L, Moingeon P (2015) Identification of novel short ragweed pollen allergens using combined transcriptomic and immunoproteomic approaches. PLoS One 10:e0136258
Borg M, Twell D (2010) Life after meiosis: patterning the angiosperm male gametophyte. Biochem Soc Trans 38:577–582
Borg M, Brownfield L, Khatab H, Sidorova A, Lingaya M, Twell D (2011) The R2R3 MYB transcription factor DUO1 activates a male germline-specific regulon essential for sperm cell differentiation in Arabidopsis. Plant Cell 23:534–549
Borg M, Rutley N, Kagale S, Hamamura Y, Gherghinoiu M, Kumar S, Sari U, Esparza-Franco MA, Sakamoto W, Rozwadowski K, Higashiyama T, Twell D (2014) An EAR-dependent regulatory module promotes male germ cell division and sperm fertility in Arabidopsis. Plant Cell 26:2098–2113
Borges F, Gomes G, Gardner R, Moreno N, McCormick S, Feijo JA, Becker JD (2008) Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol 148:1168–1181
Borges F, Pereira PA, Slotkin RK, Martienssen RA, Becker JD (2011) MicroRNA activity in the Arabidopsis male germline. J Exp Bot 62:1611–1620
Brewbaker JL (1967) Distribution and phylogenetic significance of binucleate and trinucleate pollen grains in angiosperms. Am J Bot 54:1069–1083
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190
Bryce M, Drews O, Schenk MF, Menzel A, Estrella N, Weichenmeier I, Smulders MJ, Buters J, Ring J, Gorg A, Behrendt H, Traidl-Hoffmann C (2010) Impact of urbanization on the proteome of birch pollen and its chemotactic activity on human granulocytes. Int Arch Allergy Immunol 151:46–55
Calarco JP, Borges F, Donoghue MTA, Van Ex F, Jullien PE, Lopes T, Gardner R, Berger F, Feijó J, Becker JD, Martienssen RA (2012) Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 151:194–205
Campbell BC, Gilding EK, Timbrell V, Guru P, Loo D, Zennaro D, Mari A, Solley G, Hill MM, Godwin ID, Davies JM (2015) Total transcriptome, proteome, and allergome of Johnson grass pollen, which is important for allergic rhinitis in subtropical regions. J Allergy Clin Immunol 135:133–142
Carmona R, Zafra A, Seoane P, Castro AJ, Guerrero-Fernández D, Castillo-Castillo T, Medina-García A, Cánovas FM, Aldana-Montes JF, Navas-Delgado I, Alché JD, Claros MG (2015) ReprOlive: a database with linked data for the olive tree (Olea europaea L.) reproductive transcriptome. Front Plant Sci 6. doi:10.3389/fpls.2015.00625
Carthew RW, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655
Caruso M, Merelo P, Distefano G, La Malfa S, Lo Piero AR, Tadeo FR, Talon M, Gentile A (2012) Comparative transcriptome analysis of stylar canal cells identifies novel candidate genes implicated in the self-incompatibility response of Citrus clementina. BMC Plant Biol 12:20. doi:10.1186/1471-2229-12-20
Chalivendra SC, Lopez-Casado G, Kumar A, Kassenbrock AR, Royer S, Tovar-Mendez A, Covey PA, Dempsey LA, Randle AM, Stack SM, Rose JK, McClure B, Bedinger PA (2013) Developmental onset of reproductive barriers and associated proteme changes in stigma/styles of Solanum pennellii. J Exp Bot 64:265–279
Chambers C, Shuai B (2009) Profiling microRNA expression in Arabidopsis pollen using microRNA array and real-time PCR. BMC Plant Biol 9. doi:10.1186/1471-2229-9-87
Chao Q, Gao ZF, Wang YF, Li Z, Huang XH, Wang YC, Mei YC, Zhao BG, Li L, Jiang YB, Wang BC (2016) The proteome and phosphoproteome of maize pollen uncovers fertility candidate proteins. Plant Mol Biol 91:287–304
Chaturvedi P, Ischebeck T, Egelhofer V, Lichtscheidl I, Weckwerth W (2013) Cell-specific analysis of the tomato pollen proteome from pollen mother cell to mature pollen provides evidence for developmental priming. J Proteome Res 12:4892–4903
Chen T, Wu X, Chen Y, Li X, Huang M, Zheng M, Baluška F, Šamaj J, Lin J (2009) Combined proteomic and cytological analysis of Ca2+-calmodulin regulation in Picea meyeri pollen tube growth. Plant Physiol 149:1111–1126
Chen C, Farmer AD, Langley RJ, Mudge J, Crow JA, May GD, Huntley J, Smith AG, Retzel EF (2010) Meiosis-specific gene discovery in plants: RNAseq applied to isolated Arabidopsis male meiocytes. BMC Plant Biol 10:280. doi:10.1186/1471-2229-10-280
Chen Y, Liu P, Hoehenwarter W, Lin J (2012) Proteomic and phosphoproteomic analysis of Picea wilsonii pollen development under nutrient limitation. J Proteome Res 11:4180–4190
Chen Y, Zou M, Cao Y (2014) Transcriptome analysis of the Arabidopsis semi-in vivo pollen tube guidance system uncovers a distinct gene expression profile. J Plant Biol 57(2):93–105
Chettoor AM, Givan SA, Cole RA, Coker CT, Unger-Wallace E, Vejlupkova Z, Vollbrecht E, Fowler JE, Evans MS (2014) Discovery of novel transcripts and gametophytic functions via RNAseq analysis of maize gametophytic transcriptomes. BMC Plant Biol 15:414
Collins LJ (2011) Spliceosomal RNA infrastructure: the network of splicing components and their regulation by miRNAs. Adv Exp Med Biol 722:86–102
Costa M, Nobre MS, Becker JD, Masiero S, Amorim MI, Pereira LG, Coimbra S (2013) Expression-based and co-localization detection of arabinogalactan protein 6 and arabinogalactan protein 11 interactors in Arabidopsis pollen and pollen tubes. BMC Plant Biol 13. doi:10.1186/1471-2229-13-7
Dai S, Li L, Chen T, Chong K, Xue Y, Wang T (2006) Proteomic analyses of Oryza sativa mature pollen reveal novel proteins associated with pollen germination and tube growth. Proteomics 6:2504–2529
Dai S, Chen T, Chong K, Xue Y, Liu S, Wang T (2007) Proteomics identification of differentially expressed proteins associated with pollen germination and tube growth reveals characteristics of germinated Oryza sativa pollen. Mol Cell Proteomics 6:207–230
Davidson RM, Hansey CN, Gowda M, Childs KL, Lin H, Vaillancourt B, Sekhon RS, de Leon N, Kaeppler SM, Jiang N, Buell CR (2011) Utility of RNA sequencing for analysis of maize reproductive transcriptomes. Plant Genome J 4:191
de Groot MJ, Daran-Lapujade P, van Breukelen B, Knijnenburg TA, de Hulster EA, Reinders MJ, Pronk JT, Heck AJ, Slijper M (2007) Quantitative proteomics and transcriptomics of anaerobic and aerobic yeast cultures reveals post-transcriptional regulation of key cellular processes. Microbiology 153:3864–3878
Der JP, Barker MS, Wickett NJ, de Pamphilis CW, Wolf PG (2011) De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genomics 12. doi:10.1186/1471-2164-12-99
Du H, Feng B-R, Yang S-S, Huang Y-B, Tang Y-X (2012) The R2R3-MYB transcription factor gene family in maize. PLoS One 7:e37463. doi:10.1371/journal.pone.0037463
Du H, Liang Z, Zhao S, Nan MG, Tran LS, Lu K, Huang YB, Li JN (2015) The evolutionary history of R2R3-MYB proteins across 50 eukaryotes: new insights into subfamily classification and expansion. Sci Rep 5:11037. doi:10.1038/srep11037
Dukowic-Schulze S, Sundararajan A, Mudge J, Ramaraj T, Farmer AD, Wang M, Sun Q, Pillardy J, Kianian SF, Retzel EF, Pawloski WP, Chen C (2014) The transcriptome landscape of early maize meiosis. BMC Plant Biol 14:18. doi:10.1186/1471-2229-14-118
Durbarry A, Vizir I, Twell D (2005) Male germ line development in Arabidopsis. duo pollen mutants reveal gametophytic regulators of generative cell cycle progression. Plant Physiol 137(1):297–307
El Kelish A, Zhao F, Heller W, Dumer J, Winkler JB, Behdendt H, Traidl-Hoffmann C, Horres R, Pfeiffer M, Ernst D (2014) Ragweed (Ambrosia artemisiifolia) pollen allergenicity: SuperSAGE transcriptomic analysis upon elevated CO2 and drought stress. BMC Plant Biol 14:176
Elfving F (1879) Studien über die Pollenkörner der Angiospermen. Jenaische Zeitschrift für Naturwissenschaft 13:1–28
Engel ML, Chaboud A, Dumas C, McCormick S (2003) Sperm cells of Zea mays have a complex complement of mRNAs. Plant J 34:697–707
Fasoli M, Dal Santo S, Zenoni S, Tornielli GB, Farina L, Zamboni A, Porceddu A, Venturini L, Bicego M, Murino V, Ferrarini A, Delledonne M, Pezzotti M (2012) The grapevine expression atlas reveals a deep transcriptome shift driving the entire plant into a maturation program. Plant Cell 24:3489–3505
Feng J, Chen X, Yuan Z, He T, Zhang L, Wu Y, Liu W, Liang Q (2006) Proteome comparison following self- and across-pollination in self-incompatible apricot (Prunus armeniaca L.) Protein J 25:328–335
Fernando DD (2005) Characterization of pollen cube development in Pinus strobus (Eastern white pine) through proteomic analysis of differentially expressed proteins. Proteomics 5:4917–4926
Fíla J, Honys D (2012) Enrichment techniques employed in phosphoproteomics. Amino Acids 43:1025–1047
Fíla J, Čapková V, Feciková J, Honys D (2011) Impact of homogenization and protein extraction conditions on the obtained tobacco pollen proteomic patterns. Biol Plant 55:499–506
Fíla J, Matros A, Radau S, Zahedi RP, Čapková V, Mock H-P, Honys D (2012) Revealing phosphoproteins playing role in tobacco pollen activated in vitro. Proteomics 12:3229–3250
Fíla J, Radau S, Matros A, Hartmann A, Scholz U, Feciková J, Mock HP, Čapková V, Zahedi RP, Honys D (2016) Phosphoproteomics profiling of tobacco mature pollen and pollen activated in vitro. Mol Cell Proteomics 15:1338–1350
Frank G, Pressman E, Ophir R, Althan L, Shaked R, Freedman M, Shen S, Firon N (2009) Transcriptional profiling of maturing tomato (Solanum lycopersicum L.) microspores reveals the involvement of heat shock proteins, ROS scavengers, hormones, and sugars in the heat stress response. J Exp Bot 60:3891–3908
Futamura N, Ujino-Ihara T, Nishiguchi M, Kanamori H, Yoshimura K, Sakaguchi M, Shinohara K (2006) Analysis of expressed sequence tags from Cryptomeria japonica pollen reveals novel pollen-specific transcripts. Tree Physiol 26:1517–1528
Ge W, Song Y, Zhang C, Zhang Y, Burlingame AL, Guo Y (2011) Proteomic analyses of apoplastic proteins from germinating Arabidopsis thaliana pollen. Biochim Biophys Acta 1814:1964–1973
Ghosh N, Sircar G, Saha B, Pandey N, Gupta Bhattacharya S (2015) Search for allergens from the pollen proteome of sunflower (Helianthus annuus L.): a major sensitizer for respiratory allergy patients. PLoS One 10:e0138992. doi:10.1371/journal.pone.0138992
Gibalová A, Reňák D, Matczuk K, Dupl’áková N, Cháb D, Twell D, Honys D (2009) AtbZIP34 is required for Arabidopsis pollen wall patterning and the control of several metabolic pathways in developing pollen. Plant Mol Biol 70:581–601
Gou X, Yuan T, Wei X, Russell SD (2009) Gene expression in the dimorphic sperm cells of Plumbago zeylanica: transcript profiling, diversity, and relationship to cell type. Plant J 60:33–47
Grant-Downton R, Hafidh S, Twell D, Dickinson HG (2009a) Small RNA pathways are present and functional in the angiosperm male gametophyte. Mol Plant 2:500–512
Grant-Downton R, Le Trionnaire G, Schmid R, Rodriguez-Enriquez J, Hafidh S, Mehdi S, Twell D, Dickinson H (2009b) MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. BMC Genomics 10. doi:10.1186/1471-2164-10-643
Grobei MA, Qeli E, Brunner E, Rehrauer H, Zhang R, Roschitzki B, Basler K, Ahrens CH, Grossniklaus U (2009) Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res 19:1786–1800
Haerizadeh F, Wong CE, Bhalla PL, Gresshoff PM, Singh MB (2009) Genomic expression profiling of mature soybean (Glycine max) pollen. BMC Plant Biol 9. doi:10.1186/1471-2229-9-25
Hafidh S, Čapková V, Honys D (2011) Safe keeping the message: mRNP complexes tweaking after transcription. Adv Exp Med Biol 722:118–136
Hafidh S, Breznenová K, Honys D (2012a) De novo post-pollen mitosis II tobacco pollen tube transcriptome. Plant Signal Behav 7:918–921
Hafidh S, Breznenová K, Růžička P, Feciková J, Čapková V, Honys D (2012b) Comprehensive analysis of tobacco pollen transcriptome unveils common pathways in polar cell expansion and underlying heterochronic shift during spermatogenesis. BMC Plant Biol 12:24. doi:10.1186/1471-2229-12-24
Hafidh S, Potěšil D, Fíla J, Feciková J, Čapková V, Zdráhal Z, Honys D (2014) In search of ligands and receptors of the pollen tube: the missing link in pollen tube perception. Biochem Soc Trans 42:388–394
Hafidh S, Fíla J, Honys D (2016a) Male gametophyte development and function in angiosperms: a general concept. Plant Reprod 29:31–51
Hafidh S, Potěšil D, Fíla J, Čapková V, Zdráhal Z, Honys D (2016b) Quantitative proteomics of the tobacco pollen tube secretome identifies novel pollen tube guidance proteins important for fertilization. Genome Biol 17:81. doi:10.1186/s13059-016-0928-x
Han B, Chen S, Dai S, Yang N, Wang T (2010) Isobaric tags for relative and absolute quantification-based comparative proteomics reveals the features of plasma membrane-associated proteomes of pollen grains and pollen tubes from Lilium davidii. J Integrative Plant Biol 52:1043–1058
Higashiyama T (2015) The mechanism and key molecules involved in pollen tube guidance. Annu Rev Plant Biol 66:393–413
Higo A, Niwa M, Yamato KT, Yamada L, Sawada H, Sakamoto T, Kurata T, Shirakawa M, Endo M, Shigenobu S, Yamaguchi K, Ishizaki K, Nishihama R, Kohchi T, Araki T (2016) Transcriptional framework of male gametogenesis in the liverwort Marchantia polymorpha L. Plant Cell Physiol 57:325–338
Hirano K, Aya K, Hobo T, Sakakibara H, Kojima M, Shim RA, Hasegawa Y, Ueguchi-Tanaka M, Matsuoka M (2008) Comprehensive transcriptome analysis of phytohormone biosynthesis and signaling genes in microspore/pollen and tapetum of rice. Plant Cell Physiol 49:1429–1450
Hobo T, Suwabe K, Aya K, Suzuki G, Yano K, Ishimizu T, Fujita M, Kikuchi S, Hamada K, Miyano M, Fujioka T, Kaneko F, Kazama T, Mizuta Y, Takahashi H, Shiono K, Nakazono M, Tsutsumi N, Nagamura Y, Kurata N, Watanabe M, Matsuoka M (2008) Various spatiotemporal expression profiles of anther-expressed genes in rice. Plant Cell Physiol 49:1417–1428
Hollender CA, Kang C, Darwish O, Geretz A, Matthews BF, Slovin J, Alkharouf N, Liu Z (2014) Floral transcriptomes in woodland strawberry uncover developing receptacle and anther gene networks. Plant Physiol 165:1062–1075
Holmes-Davis R, Tanaka CK, Vensel WH, Hurkman WJ, McCormick S (2005) Proteome mapping of mature pollen of Arabidopsis thaliana. Proteomics 5:4864–4884
Honys D, Twell D (2003) Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol 132:640–652
Honys D, Twell D (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol 5. doi:10.1186/gb-2004-5-11-r85
Honys D, Combe JP, Twell D, Čapková V (2000) The translationally repressed pollen-specific ntp303 mRNA is stored in non-polysomal mRNPs during pollen maturation. Sex Plant Reprod 13:135–144
Honys D, Reňák D, Feciková J, Jedelský PL, Nebesářova J, Dobrev P, Čapková V (2009) Cytoskeleton-associated large RNP complexes in tobacco male gametophyte (EPPs) are associated with ribosomes and are involved in protein synthesis, processing, and localization. J Proteome Res 8:2015–2031
Huang J-C, Chang L-C, Wang M-L, Guo C-L, Chung M-C, Jauh G-Y (2011) Identification and exploration of pollen tube small proteins encoded by pollination-induced transcripts. Plant Cell Physiol 52:1546–1559
Iaria D, Chiappetta A, Muzzalupo I (2016) De novo transcriptome sequencing of Olea europaea L. to identify genes involved in the development of the pollen tube. Sci World J 2016:4305252. doi:10.1155/2016/4305252
Ibarra CA, Feng X, Schoft VK, Hsieh TF, Uzawa R, Rodrigues JA, Zemach A, Chumak N, Machlicova A, Nishimura T, Rojas D, Fischer RL, Tamaru H, Zilberman D (2012) Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 337:1360–1364
Ikram S, Durandet M, Vesa S, Pereira S, Guerche P, Bonhomme S (2014) Functional redundancy and/or ongoing pseudogenization among F-box protein genes expressed in Arabidopsis male gametophyte. Plant Reprod 27:95–107
Ischebeck T, Valledor L, Lyon D, Gingl S, Nagler M, Meijon M, Egelhofer V, Weckwerth W (2014) Comprehensive cell-specific protein analysis in early and late pollen development from diploid microsporocytes to pollen tube growth. Mol Cell Proteomics 13:295–310
Kagale S, Rozwadowski K (2011) EAR motif-mediated transcriptional repression in plants. Epigenetics 6:141–146
Kanter U, Heller W, Durner J, Winkler JB, Engel M, Behrendt H, Holzinger A, Braun P, Hauser M, Ferreira F, Mayer K, Pfeifer M, Ernst D (2013) Molecular and immunological characterization of ragweed (Ambrosia artemisiifolia L.) pollen after exposure of the plants to elevated ozone over a whole growing season. PLoS One 8. doi:10.1371/journal.pone.0061518
Kao SH, Su SN, Huang SW, Tsai JJ, Chow LP (2005) Sub-proteome analysis of novel IgE-binding proteins from Bermuda grass pollen. Proteomics 5:3805–3813
Kazan K (2003) Alternative splicing and proteome diversity in plants: the tip of the iceberg has just emerged. Trends Plant Sci 8:468–471
Keene JD (2007) RNA regulons: coordination of post-transcriptional events. Nat Rev Genet 8:533–543
Kessler SA, Grossniklaus U (2011) She’s the boss: signaling in pollen tube reception. Curr Opin Plant Biol 14:622–627
Lang V, Usadel B, Obermeyer G (2015) De novo sequencing and analysis of the lily pollen transcriptome: an open access data source for an orphan plant species. Plant Mol Biol 87:69–80
Lee JY, Lee DH (2003) Use of serial analysis of gene expression technology to reveal changes in gene expression in Arabidopsis pollen undergoing cold stress. Plant Physiol 132:517–529
Lee TY, Bretana NA, Lu CT (2011) PlantPhos: using maximal dependence decomposition to identify plant phosphorylation sites with substrate site specificity. BMC Bioinformatics 12:13. doi:10.1186/1471-2105-12-261
Leydon AR, Beale KM, Woroniecka K, Castner E, Chen J, Horgan C, Palanivelu R, Johnson MA (2013) Three MYB transcription factors control pollen tube differentiation required for sperm release. Curr Biol 23:1209–1214
Li J, Chen J, Zhang Z, Pan Y (2008) Proteome analysis of tea pollen (Camellia sinensis) under different storage conditions. Agric Food Chem 56:7535–7544
Li M, Sha A, Zhou X, Yang P (2012) Comparative proteomic analyses reveal the changes of metabolic features in soybean (Glycine max) pistils upon pollination. Sex Plant Reprod 25:281–291
Li XM, Sang YL, Zhao XY, Zhang XS (2013) High-throughput sequencing of small RNAs from pollen and silk and characterization of miRNAs as candidate factors involved in pollen-silk interactions in maize. PLoS One 8:e72852. doi:10.1371/journal.pone.0072852
Li M, Wang K, Wang X, Yang P (2014) Morphological and proteomic analysis reveal the role of pistil under pollination in Liriodendron chinense (Hemsl.) Sarg. PLoS One 9:e99970. doi:10.1371/journal.pone.0099970
Li M, Wang K, Li S, Yang P (2016) Exploration of rice pistil responses during early post-pollination through a combined proteomic and transcriptomic analysis. J Proteomics 131:214–226
Liang Y, Tan Z-M, Zhu L, Niu Q-K, Zhou J-J, Li M, Chen L-Q, Zhang X-Q, Ye D (2013) MYB97, MYB101 and MYB120 function as male factors that control pollen tube-synergid interaction in Arabidopsis thaliana fertilization. PLoS Genet 9. doi:10.1371/journal.pgen.1003933
Lin S-Y, Chen P-W, Chuang M-H, Juntawong P, Bailey-Serres J, Jauh G-Y (2014) Profiling of translatomes of in vivo-grown pollen tubes reveals genes with roles in micropylar guidance during pollination in Arabidopsis. Plant Cell 26:602–618
Liu Y, Cui S, Wu F, Yan S, Lin X, Du X, Chong K, Schilling S, Theissen G, Meng Z (2013) Functional conservation of MIKC*-type MADS box genes in Arabidopsis and rice pollen maturation. Plant Cell 25:1288–1303
Liu Y, Joly V, Dorion S, Rivoal J, Matton DP (2015) The plant ovule secretome: a different view toward pollen-pistil interactions. J Proteome Res 14:4763–4775
Lopez-Casado G, Covey PA, Bedinger PA, Mueller LA, Thannhauser TW, Zhang S, Fei Z, Giovannoni JJ, Rose JK (2012) Enabling proteomic studies with RNA-Seq: the proteome of tomato pollen as a test case. Proteomics 12:761–774
Lora J, Herrero M, Hormaza JI (2009) The coexistence of bicellular and tricellular pollen in Annona cherimola (Annonaceae): implications for pollen evolution. Am J Bot 96:802–808
Loraine AE, McCormick S, Estrada A, Patel K, Qin P (2013) RNA-Seq of Arabidopsis pollen uncovers novel transcription and alternative splicing. Plant Physiol 162(2):1092–1109
Loraine AE, Blakley IC, Jagadeesan S, Harper J, Miller G, Firon N (2015) Analysis and visualization of RNAseq expression data using RStudio, Bioconductor, and Integrated Genome Browser. Methods Mol Biol 1284:481–501
Lorkovic ZJ, Barta A (2004) Compartmentalization of the splicing machinery in plant cell nuclei. Trends Plant Sci 9:565–568
Luo M, Taylor JM, Spriggs A, Zhang H, Wu X, Russell S, Singh M, Koltunow A (2011) A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet 7:e1002125. doi:10.1371/journal.pgen.1002125
Ma J, Skibbe DS, Fernandes J, Walbot V (2008) Male reproductive development: gene expression profiling of maize anther and pollen ontogeny. Genome Biol 9. doi:10.1186/gb-2008-9-12-r181
Mani BM, Huerta-Ocampo JA, Garcia-Sanchez JR, Barrera-Pacheco A, de la Rosa AP, Teran LM (2015) Identification of Ligustrum lucidum pollen allergens using a proteomics approach. Biochem Biophys Res Commun 468:788–792
Matsuda T, Matsushima M, Nabemoto M, Osaka M, Sakazono S, Masuko-Suzuki H, Takahashi H, Nakazono M, Iwano M, Takayama S, Shimizu KK, Okumura K, Suzuki G, Watanabe M, Suwabe K (2014) Transcriptional characteristics and differences in Arabidopsis stigmatic papilla cells pre- and post-pollination. Plant Cell Physiol 56:663–673
Matus JT, Aquea F, Arce-Johnson P (2008) Analysis of the grape MYB R2R3 subfamily reveals expanded wine quality-related clades and conserved gene structure organization across Vitis and Arabidopsis genomes. BMC Plant Biol 8:83. doi:10.1186/1471-2229-8-83
Mayank P, Grossman J, Wuest S, Boisson-Dernier A, Roschitzki B, Nanni P, Nuehse T, Grossniklaus U (2012) Characterization of the phosphoproteome of mature Arabidopsis pollen. Plant J 72:89–101
McCormick S (1993) Male gametophyte development. Plant Cell 5:1265–1275
Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW (2003) Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15:809–834
Miyazaki S, Murata T, Sakurai-Ozato N, Kubo M, Demura T, Fukuda H, Hasebe M (2009) ANXUR1 and 2, sister genes to FERONIA/SIRENE, are male factors for coordinated fertilization. Curr Biol 19:1327–1331
Nazemof N, Couroux P, Rampitsch C, Xing T, Robert LS (2014) Proteomic profiling reveals insights into Triticeae stigma development and function. J Exp Bot 65:6069–6080
Noir S, Brautigam A, Colby T, Schmidt J, Panstruga R (2005) A reference map of the Arabidopsis thaliana mature pollen proteome. Biochem Biophys Res Commun 337:1257–1266
O’Donoghue MT, Chater C, Wallace S, Gray JE, Beerling DJ, Fleming AJ (2013) Genome-wide transcriptomic analysis of the sporophyte of the moss Physcomitrella patens. J Exp Bot 64:3567–3581
Obermeyer G, Fragner L, Lang V, Weckwerth W (2013) Dynamic adaption of metabolic pathways during germination and growth of lily pollen tubes after inhibition of the electron transport chain. Plant Physiol 162:1822–1833
Oh SA, Johnson A, Smertenko A, Rahman D, Park SK, Hussey PJ, Twell D (2005) A divergent cellular role for the FUSED kinase family in the plant-specific cytokinetic phragmoplast. Curr Biol 15:2107–2111
Ohr H, Bui AQ, Le BH, Fischer RL, Choi Y (2007) Identification of putative Arabidopsis DEMETER target genes by GeneChip analysis. Biochem Biophys Res Commun 364:856–860
Okada T, Bhalla PL, Singh MB (2006) Expressed sequence tag analysis of Lilium longiflorum generative cells. Plant Cell Physiol 47:698–705
Okada T, Singh MB, Bhalla PL (2007) Transcriptome profiling of Lilium longiflorum generative cells by cDNA microarray. Plant Cell Rep 26:1045–1052
Oliver MJ, Dowd SE, Zaragoza J, Mauget SA, Payton PR (2004) The rehydration transcriptome of the desiccation-tolerant bryophyte Tortula ruralis: transcript classification and analysis. BMC Genomics 5:89. doi:10.1186/1471-2164-5-89
Ortiz-Ramirez C, Hernandez-Coronado M, Thamm A, Catarino B, Wang M, Dolan L, Feijo JA, Becker JD (2016) A transcriptome atlas of Physcomitrella patens provides insights into the evolution and development of land plants. Mol Plant 9:205–220
Osaka M, Matsuda T, Sakazono S, Masuko-Suzuki H, Maeda S, Sewaki M, Sone M, Takahashi H, Nakazono M, Iwano M, Takayama S, Shimizu KK, Yano K, Lim YP, Suzuki G, Suwabe K, Watanabe M (2013) Cell type-specific transcriptome of Brassicaceae stigmatic papilla cells from a combination of laser microdissection and RNA sequencing. Plant Cell Physiol 54:1894–1906
Palanivelu R, Preuss D (2000) Pollen tube targeting and axon guidance: parallels in tip growth mechanisms. Trends Cell Biol 10:517–524
Palanivelu R, Preuss D (2006) Distinct short-range ovule signals attract or repel Arabidopsis thaliana pollen tubes in vitro. BMC Plant Biol 6:7. doi:10.1186/1471-2229-6-7
Palanivelu R, Tsukamoto T (2012) Pathfinding in angiosperm reproduction: pollen tube guidance by pistils ensures successful double fertilization. Wiley Interdiscip Rev Dev Biol 1:96–113
Palatnik JF, Wollmann H, Schommer C, Schwab R, Boisbouvier J, Rodriguez R, Warthmann N, Allen E, Dezulian T, Huson D, Carrington JC, Weigel D (2007) Sequence and expression differences underlie functional specialization of Arabidopsis microRNAs miR159 and miR319. Dev Cell 13:115–125
Park SK, Howden R, Twell D (1998) The Arabidopsis thaliana gametophytic mutation gemini pollen1 disrupts microspore polarity, division asymmetry and pollen cell fate. Development 125:3789–3799
Paul P, Chaturvedi P, Selymesi M, Ghatak A, Mesihovic A, Scharf KD, Weckwerth W, Simm S, Schleiff E (2016) The membrane proteome of male gametophyte in Solanum lycopersicum. J Proteomics 131:48–60
Pearce LR, Komander D, Alessi DR (2010) The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11:9–22
Peng H, Chun J, Ai TB, Tong YA, Zhang R, Zhao MM, Chen F, Wang SH (2012) MicroRNA profiles and their control of male gametophyte development in rice. Plant Mol Biol 80:85–102
Pertl H, Schulze WX, Obermeyer G (2009) The pollen organelle membrane proteome reveals highly spatial-temporal dynamics during germination and tube growth of lily pollen. J Proteome Res 8:5142–5152
Pina C, Pinto F, Feijo JA, Becker JD (2005) Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiol 138:744–756
Pirone-Davies C, Prior N, von Aderkas P, Smith D, Hardie D, Friedman WE, Mathews S (2016) Insights from the pollination drop proteome and the ovule transcriptome of Cephalotaxus at the time of pollination drop production. Ann Bot 117:973–984
Qin Y, Leydon AR, Manziello A, Pandey R, Mount D, Denic S, Vasic B, Johnson MA, Palanivelu R (2009) Penetration of the stigma and style elicits a novel transcriptome in pollen tubes, pointing to genes critical for growth in a pistil. PLoS Genet 5. doi:10.1371/journal.pgen.1000621
Qiu YL, Taylor AB, McManus HA (2012) Evolution of the life cycle in land plants. J Syst Evol 50:171–194
Quinn CR, Iriyama R, Fernando DD (2014) Expression patterns of conserved microRNAs in the male gametophyte of loblolly pine (Pinus taeda). Plant Reprod 27:69–78
Raghavan V (2003) Some reflections on double fertilization, from its discovery to the present. New Phytol 159:565–583
Reddy AS, Day IS, Gohring J, Barta A (2012) Localization and dynamics of nuclear speckles in plants. Plant Physiol 158:67–77
Reiser L, Fischer RL (1993) The ovule and the embryo sac. Plant Cell 5:1291–1301
Rejon JD, Delalande F, Schaeffer-Reiss C, Carapito C, Zienkiewicz K, de Dios AJ, Isabel Rodriguez-Garcia M, Van Dorsselaer A, Jesus Castro A (2013) Proteomics profiling reveals novel proteins and functions of the plant stigma exudate. J Exp Bot 64:5695–5705
Reňák D, Dupľáková N, Honys D (2012) Wide-scale screening of T-DNA lines for transcription factor genes affecting male gametophyte development in Arabidopsis. Sex Plant Reprod 25:39–60
Rodriguez de Francisco L, Romero-Rodriguez MC, Navarro-Cerrillo RM, Minino V, Perdomo O, Jorrin-Novo JV (2016) Characterization of the orthodox Pinus occidentalis seed and pollen proteomes by using complementary gel-based and gel-free approaches. J Proteomics 143:382–389
Röhrig H, Colby T, Schmidt J, Harzen A, Facchinelli F, Bartels D (2008) Analysis of desiccation-induced candidate phosphoproteins from Craterostigma plantagineum isolated with a modified metal oxide affinity chromatography procedure. Proteomics 8:3548–3560
Rotman N, Durbarry A, Wardle A, Yang WC, Chaboud A, Faure JE, Berger F, Twell D (2005) A novel class of MYB factors controls sperm-cell formation in plants. Curr Biol 15:244–248
Russell SD, Bhalla PL, Singh MB (2008) Transcriptome-based examination of putative pollen allergens of rice (Oryza sativa ssp. japonica). Mol Plant 1:751–759
Russell SD, Gou X, Wong CE, Wang X, Yuan T, Wei X, Bhalla PL, Singh MB (2012) Genomic profiling of rice sperm cell transcripts reveals conserved and distinct elements in the flowering plant male germ lineage. New Phytol 195:560–573
Rutley N, Twell D (2015) A decade of pollen transcriptomics. Plant Reprod 28:73–89
Saha B, Sircar G, Pandey N, Gupta Bhattacharya S (2015) Mining novel allergens from coconut pollen employing manual de novo sequencing and homology-driven proteomics. J Proteome Res 14:4823–4833
Šamaj J, Muller J, Beck M, Böhm N, Menzel D (2006) Vesicular trafficking, cytoskeleton and signalling in root hairs and pollen tubes. Trends Plant Sci 11:594–600
Sanchez SE, Petrillo E, Kornblihtt AR, Yanovsky MJ (2011) Alternative splicing at the right time. RNA Biol 8:954–959
Sanetomo R, Hosaka K (2013) Pollen transcriptome analysis of Solanum tuberosum (2n=4x=48), S-demissum (2n=6x=72), and their reciprocal F-1 hybrids. Plant Cell Rep 32:623–636
Sang YL, Xu M, Ma FF, Chen H, Xu XH, Gao XQ, Zhang XS (2012) Comparative proteomic analysis reveals similar and distinct features of proteins in dry and wet stigmas. Proteomics 12:1983–1998
Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470
Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37:501–506
Schmidt H, Gelhaus C, Nebendahl M, Janssen O, Petersen A (2010) Characterization of Phleum pratense pollen extracts by 2-D DIGE and allergen immunoreactivity. Proteomics 10:4352–4362
Schreiber DN, Bantin J, Dresselhaus T (2004) The MADS box transcription factor ZmMADS2 is required for anther and pollen maturation in maize and accumulates in apoptotic bodies during anther dehiscence. Plant Physiol 134:1069–1079
Schulten V, Greenbaum JA, Hauser M, McKinney DM, Sidney J, Kolla R, Lindestam Arlehamn CS, Oseroff C, Alam R, Broide DH, Ferreira F, Grey HM, Sette A, Peters B (2013) Previously undescribed grass pollen antigens are the major inducers of T helper 2 cytokine-producing T cells in allergic individuals. Proc Natl Acad Sci U S A 110:3459–3464
Shen Y, Venu RC, Nobuta K, Wu X, Notibala V, Demirci C, Meyers BC, Wang GL, Ji G, Li QQ (2011) Transcriptome dynamics through alternative polyadenylation in developmental and environmental responses in plants revealed by deep sequencing. Genome Res 21:1478–1486
Sheoran IS, Sproule KA, Olson DJH, Ross ARS, Sawhney VK (2006) Proteome profile and functional classification of proteins in Arabidopsis thaliana (Landsberg erecta) mature pollen. Sex Plant Reprod 19:185–196
Sheoran IS, Ross ARS, Olson DJH, Sawhney VK (2007) Proteomic analysis of tomato (Lycopersicon esculentum) pollen. J Exp Bot 58:3525–3535
Sheoran IS, Pedersen EJ, Ross ARS, Sawhney VK (2009a) Dynamics of protein expression during pollen germination in canola (Brassica napus). Planta 230:779–793
Sheoran IS, Ross ARS, Olson DJH, Sawhney VK (2009b) Differential expression of proteins in the wild type and 7B-1 male-sterile mutant anthers of tomato (Solanum lycopersicum): a proteomic analysis. J Proteomics 71:624–636
Slotkin RK, Vaughn M, Borges F, Tanurdzic M, Becker JD, Feijo JA, Martienssen RA (2009) Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 136:461–472
Sousa R, Osório H, Duque L, Ribeiro H, Cruz A, Anbreu I (2014) Identification of Plantago lanceolata pollen allergens using an immunoproteomic approach. J Investig Allergol Clin Immunol 24:177–183
Strasburger E (1884) Neue Untersuchungen uber den Befruchtungsvorgang bei den Phanerogamen als Grundlage fur eine Theorie der Zeugung. Gustav Fischer, Jena
Suwabe K, Suzuki G, Takahashi H, Shiono K, Endo M, Yano K, Fujita M, Masuko H, Saito H, Fujioka T, Kaneko F, Kazama T, Mizuta Y, Kawagishi-Kobayashi M, Tsutsumi N, Kurata N, Nakazono M, Watanabe M (2008) Separated transcriptomes of male gametophyte and tapetum in rice: validity of a laser microdissection (LM) microarray. Plant Cell Physiol 49:1407–1416
Tang X, Zhang ZY, Zhang WJ, Zhao XM, Li X, Zhang D, Liu QQ, Tang WH (2010) Global gene profiling of laser-captured pollen mother cells indicates molecular pathways and gene subfamilies involved in rice meiosis. Plant Physiol 154:1855–1870
Tran F, Penniket C, Patel RV, Provart NJ, Laroche A, Rowland O, Robert LS (2013) Developmental transcriptional profiling reveals key insights into Triticeae reproductive development. Plant J 74:971–988
Tsubomura M, Kurita M, Watanabe A (2016) Determination of male strobilus developmental stages by cytological and gene expression analyses in Japanese cedar (Cryptomeria japonica). Tree Physiol 36:653–666
Twell D, Oh S-A, Honys D (2006) Pollen development, a genetic and transcriptomic view. In: Malhó R (ed) Plant cell monographs: the pollen tube, vol 3. Springer, Berlin, pp 15–45
Valero Galvan J, Valledor L, Gonzalez Fernandez R, Navarro Cerrillo RM, Jorrin-Novo JV (2012) Proteomic analysis of Holm oak (Quercus ilex subsp. ballota [Desf.] Samp.) pollen. J Proteomics 75:2736–2744
Verelst W, Saedler H, Muenster T (2007a) MIKC* MADS-protein complexes bind motifs enriched in the proximal region of late pollen-specific Arabidopsis promoters. Plant Physiol 143:447–460
Verelst W, Twell D, de Folter S, Immink R, Saedler H, Muenster T (2007b) MADS-complexes regulate transcriptome dynamics during pollen maturation. Genome Biol 8. doi:10.1186/gb-2007-8-11-r249
Vogler F, Konrad SSA, Sprunck S (2015) Knockin’ on pollen’s door: live cell imaging of early polarization events in germinating Arabidopsis pollen. Front Plant Sci 6. doi:10.3389/fpls.2015.00246
Wang Y, Zhang W-Z, Song L-F, Zou J-J, Su Z, Wu W-H (2008) Transcriptome analyses show changes in gene expression to accompany pollen germination and tube growth in Arabidopsis. Plant Physiol 148:1201–1211
Wang Z, Gerstein M, Snyder M (2009) RNAseq: a revolutionary tool for transcriptomics. Nat Rev Genet 10:57–63
Wang W, Sheng X, Shu Z, Li D, Pan J, Ye X, Chang P, Li X, Wang Y (2016) Combined cytological and transcriptomic analysis reveals a nitric oxide signaling pathway involved in cold-inhibited Camellia sinensis pollen tube growth. Front Plant Sci 7:456. doi:10.3389/fpls.2016.00456
Wei LQ, Xu WY, Deng ZY, Su Z, Xue Y, Wang T (2010) Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. BMC Genomics 11:338. doi:10.1186/1471-2164-11-338
Wei LQ, Yan LF, Wang T (2011) Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol 12. doi:10.1186/gb-2011-12-6-r53
Whittle CA, Malik MR, Li R, Krochko JE (2010) Comparative transcript analyses of the ovule, microspore, and mature pollen in Brassica napus. Plant Mol Biol 72:279–299
Williams JH, Taylor ML, O’Meara BC (2014a) Repeated evolution of tricellular (and bicellular) pollen. Am J Bot 101:559–571
Williams JS, Der JP, de Pamphilis CW, Kao TH (2014b) Transcriptome analysis reveals the same 17 S-locus F-box genes in two haplotypes of the self-incompatibility locus of Petunia inflata. Plant Cell 26:2873–2888
Wu J, Shahid MQ, Guo H, Yin W, Chen Z, Wang L, Liu X, Lu Y (2014) Comparative cytological and transcriptomic analysis of pollen development in autotetraploid and diploid rice. Plant Reprod 27:181–196
Xiao L, Wang H, Wan P, Kuang T, He Y (2011) Genome-wide transcriptome analysis of gametophyte development in Physcomitrella patens. BMC Plant Biol 11:177
Xin H-P, Peng X-B, Ning J, Yan T-T, Ma L-G, Sun M-X (2011) Expressed sequence-tag analysis of tobacco sperm cells reveals a unique transcriptional profile and selective persistence of paternal transcripts after fertilization. Sex Plant Reprod 24:37–46
Xing D, Li QQ (2011) Alternative polyadenylation and gene expression regulation in plants. Wiley Interdiscip Rev RNA 2:445–458
Xu XH, Chen H, Sang YL, Wang F, Ma JP, Gao X-Q, Zhang XS (2012) Identification of genes specifically or preferentially expressed in maize silk reveals similarity and diversity in transcript abundance of different dry stigmas. BMC Genomics:13. doi:10.1186/1471-2164-13-294
Yang H, Yang N, Wang T (2016) Proteomic analysis reveals the differential histone programs between male germline cells and vegetative cells in Lilium davidii. Plant J 85:660–674
Zhang X-M, Zhao L, Larson-Rabin Z, Li D-Z, Guo Z-H (2012) De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PLoS One 7. doi:10.1371/journal.pone.0042082
Zhang H, Egger RL, Kelliher T, Morrow D, Fernandes J, Nan GL, Walbot V (2014) Transcriptomes and proteomes define gene expression progression in pre-meiotic maize anthers. G3 (Bethesda) 4:993–1010
Zhao X, Yang N, Wang T (2013) Comparative proteomic analysis of generative and sperm cells reveals molecular characteristics associated with sperm development and function specialization. J Proteome Res 12:5058–5071
Zhao P, Zhang L, Zhao L (2015) Dissection of the style’s response to pollination using transcriptome profiling in self-compatible (Solanum pimpinellifolium) and self-incompatible (Solanum chilense) tomato species. BMC Plant Biol 15:119. doi:10.1186/s12870-015-0492-7
Zhao F, Elkelish A, Durner J, Lindermayr C, Winkler JB, Ruёff F, Behrendt H, Traidl-Hoffmann C, Holzinger A, Kofler W, Braun P, von Toerne C, Hauck SM, Ernst D, Frank U (2016) Common ragweed (Ambrosia artemisiifolia L.): allergenicity and molecular characterization of pollen after plant exposure to elevated NO2. Plant Cell Environ 39:147–164
Zhou H, Yin H, Chen J, Liu X, Gao Y, Wu J, Zhang S (2016) Gene-expression profile of developing pollen tube of Pyrus bretschneideri. Gene Expr Patterns 20:11–21
Zou J, Song L, Zhang W, Wang Y, Ruan S, Wu W-H (2009) Comparative proteomic analysis of Arabidopsis mature pollen and germinated pollen. J Integr Plant Biol 51:438–455
Acknowledgements
The authors gratefully acknowledge the financial support from the Czech Science Foundation (grants no. 15–16050S and 17-23183S).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Fíla, J., Záveská Drábková, L., Gibalová, A., Honys, D. (2017). When Simple Meets Complex: Pollen and the -Omics. In: Obermeyer, G., Feijó, J. (eds) Pollen Tip Growth. Springer, Cham. https://doi.org/10.1007/978-3-319-56645-0_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-56645-0_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-56644-3
Online ISBN: 978-3-319-56645-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)