
Abstract
Plant microRNAs (miRNAs) act as negative regulators of gene expression by slicing target transcripts or inhibiting translation. A number of miRNAs play important roles in development. In order to investigate the potential function of miRNAs during male gametogenesis in rice, we obtained both gene and small RNA expression profiles by combining microarray and high-throughput sequencing technologies. From the microarray datasets, 2,925 male gametophyte-specific genes were identified, including 107 transcription factors and three significant Argonaute genes (AGO12, AGO13, and AGO17). From the sRNA-Seq datasets, 104 unique miRNAs (miRus) were identified, including 47 known miRus and 57 novel miRus; interestingly, most of the new miRus are pollen-specific and not conserved among species. Furthermore, an interactive network of miRNA-target was constructed based on the two datasets. By employing enrichment analysis, the miRNA-regulated targets were found to be involved in both the up and down pathways, but predominantly in the down pathways, including 37 GO biological processes and 32 KEGG pathways. These findings indicate that miRNAs play a broad regulatory role during male gametophyte development in rice.
Similar content being viewed by others
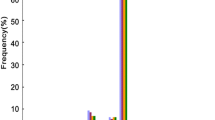
Avoid common mistakes on your manuscript.
Introduction
Plant miRNAs act as negative regulators that mediate both transcriptional gene silencing (TGS) and posttranscriptional gene silencing (PTGS) (Huntzinger and Izaurralde 2011). In PTGS, miRNA have near perfect pairing to a sequence-specific target and repress the target gene expression primarily through mRNA cleavage (Ghildiyal and Zamore 2009). Recent studies suggest that non-perfect miRNA-target pairing could inhibit mRNA translation, which may be more common in plants (Brodersen et al. 2008; Eckardt 2009). In TGS, miRNA can suppress gene expression by mediating the DNA methylation of target genes in a few cases (Wu et al. 2010). A number of miRNAs are currently known to play crucial roles in a variety of developmental processes, such as in leaf, shoot and root development, as well as in flowering time, patterning and polarity, vascular and plastid development, and so on (Jover-Gil et al. 2005; Chuck et al. 2009; Zhu and Helliwell 2011; Huijser and Schmid 2011). However, most previous studies focused on the plant sporophyte, and little is known about the role of miRNA in male gametophyte development.
In flowering plants, the male gametophyte (or pollen grain) plays a vital role in plant fertility and fruit production. In the anther, the diploid pollen mother cells (PMCs) undergo meiosis to form a tetrad of haploid microspores. Male gametophyte development begins from the free microspore stage. The microspore enlarges and then undergoes an asymmetric mitosis to form two cells: the vegetative cell and the generative cell. The larger vegetative cell does not divide again but eventually forms the pollen tube. The smaller generative cell becomes completely enclosed within the cytoplasm of the vegetative cell. In grasses and crucifers, the generative cell then undergoes a second mitosis to form two sperm cells. Gametophyte development is complete when the sperm cells are released into the embryo sac for double fertilization (McCormick 1993; Pua et al. 2010). During the past decade, several genes involved in the regulation of pollen development have been identified (Suzuki 2009; Wilson and Zhang 2009; Borg et al. 2009), and whole transcriptomic analyses have indicated that the male gametophyte has a complex gene expression pattern, which is quite distinct compared to the sporophyte (Honys and Twell 2004; Borges et al. 2008; Wei et al. 2010; Fujita et al. 2010; Deveshwar et al. 2011). Recently, in Arabidopsis thaliana, a number of small RNA pathway genes during the male gametophyte development have been observed using microarray and RT-PCR analysis (Chambers and Shuai 2009; Grant-Downton et al. 2009a); more than 80 miRNA families have been detected in mature pollen by 454 and Illumina sequencing (Grant-Downton et al. 2009b; Wilson and Zhang 2009; Borges et al. 2011). Furthermore, a few confirmed target genes have been demonstrated to be involved in the regulation of pollen development (Grant-Downton et al. 2009b). Despite all this, however, the regulatory activity of miRNAs in male gametophytes remains unclear, and little systematic analysis has been undertaken in rice (Fujioka et al. 2008; Ron et al. 2010; Van Ex et al. 2011).
A comprehensive analysis of the integration between gene and miRNA profiles will facilitate a better understanding of the diverse regulation of miRNAs. Here, by applying Affymetrix microarray technology, we obtained the genes expression profiles of three pollen development stages: uninucleate microspore (UNM/Pa), bicellular pollen (BCP/Pb) and tricellular pollen (TCP/Pc). Comparing microarray data in the genome-scale, we found a common characteristic between rice and Arabidopsis that abundant genes were down-regulated in the transition stage from BCP to TCP stage. In addition, various miRNAs pathway were detected in developing pollen. Thus, we extracted the small RNAs (sRNAs) for deep sequencing, and identified 104 miRNAs in TCP stage. We then predicted these miRNA targets from the expressed pollen genes, and a complex miRNA-target interaction network was shown. To reveal the miRNAs function, the GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were used, and we found the targets involved in both the down and up pathways, especially in the most down pathways. Our findings indicated that miRNAs play a broad regulatory role during male gametophyte development in rice.
Materials and methods
Pollen isolation and purification
Rice (Oryza sativa L. cv. Guichao No. 2) was cultured under natural conditions. When fresh rice spikelet from microspores or two-cell stage (Gou et al. 2001) were put into an extraction buffer containing 15% sucrose, mature pollens into 45% one, they were broken by Tissue-Tearor gently. Followed by filtration through 150, 50 and 30 μm nylon filter, the pollen on the last one was harvested. The different discontinuous sucrose gradient centrifugation (15%/25%/50% for uninucleate microspore, 15%/35%/60% for bicellular pollen, and 45%/60% for tricellular pollen) was carried out corresponding to three stages of rice pollen. After centrifuged at 2,000 rpm for 5 min, pollen was collected from the specific interphase which was the second layer of the first two and the bottom of the last one. Followed by centrifugation of 2,000 rpm at 4°C for 5 min, the supernatant was discarded while the resulting pollen precipitation were resuspended by moderate extraction buffer and stored at −80°C.
A drop of iodine-potassium iodine (IKI) was added to the pollen to reveal the presence of starch, and the material was immediately observed suing light microscope. The purity of the isolated fractions was determined by light microscopy and 4′, 6-diaminophenylindole (DAPI) staining, and the viability was assessed by fluorescein 3′, 6′-diacetate (FDA) treatment (Eady et al. 1995).
Purification of RNA
The total RNAs of pollen in different stages were isolated using the RNeasy Plant Kit (Qiagen) for Affymetrix Rice Genome Arrays hybridization, and all procedures were carried out according to the technical manual.
The total RNA of TCP was extracted by MiRVana™ RNA Isolation Kit (Ambion) for Illumina GA2 sequencing as described in previous research (Mi et al. 2008).
The microarray data and small RNA sequence data from this study have been submitted to NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under SuperSeries accession nos. GSE29212.
Microarray data analysis
Before performing data analysis on the high-through data, we have built a local database for annotation and integrating different platform experimental data. In the database, the MSU Rice Genome Annotation 6.1 (http://rice.plantbiology.msu.edu) was used as reference, and the Affymetrix Rice Genome Array Probe Set ID was mapped to the gene locus ID and covered 93.7% non-TE related genes (Fig. 1a; Ouyang et al. 2007). The gene’s signal value was used in subsequent data analyses instead of the probe pair’s. Some extra annotations were also added, as shown in Supplemental Table S1.
In rice microarray dataset, a total of 54 CEL files obtained from GEO (GSE6893 and GSE7951) and 6 CEL files of our experiments were imported into AffylmGUI (Wettenhall et al. 2006), and the data were normalized using the Robust Multiarray Averaging (RMA) method (Irizarry et al. 2003). The GEO accession numbers and sample group descriptions are listed in Supplemental Table S2. After averaging expression values of probe sets in repeat arrays, signal-values (log2 scale) for all probe sets were loaded into local MySQL database. Meanwhile, the dChip software was used to analysis each microarray group for detection call, respectively, and each probe set was assigned a “P” (present) or “A” (absent) using the Model-Based Expression Indexes (MBEI) algorithm (Li 2008) and the results was loaded into local database. The probe set have been mapped to corresponding genes loci to identify expressed genes (see Supplemental Table S16), and average mapped probe values (signal-value ≥ 6.5 and with present call) represent the genes expression values. Genes with expression valves ≥ 6.5 were collected as expressed gene list in each group. To identify the stage enriched or specific expressed genes, we used the following criteria: (1) statistically significant differential expression, P value <0.01 (Student T test); (2) genes signal-value > 7; (3) genes signal-value had a 2.5-fold (log2 scale) greater than mean (the average of all groups, except the test group). The Arabidopsis microarray datasets were also processed by the same methods and all CEL files were downloaded from GEO database (for detailed see Supplemental Table S3).
sRNA-Seq data analysis
The GEO accession numbers and description of our TCP sRNA-Seq and the 7 published sRNA-Seq datasets (GSM278571, GSM278572, GSM455965, GSM520640, GSM489087, GSM361264, and GSM417540) were shown in Supplemental Table S4.
Each raw datasets was processed following the steps: (1) raw reads were removed 5′- and 3′-adapter and converted to tags (distinct sRNA sequences) by a custom Perl script; (2) tags were mapped to MSU version 6.1 rice nuclear and organellar genome (http://rice.plantbiology.msu.edu/) with two mismatches allowed, and output all mapped loci, using the SOAPaligner software (Li et al. 2009); (3) according to the locus number and reads abundance, the tags were filtered and then loaded into local MySQL database (Supplemental Table S4); (4) tags loci were annotated based on genome structure and function using Perl-DBI query; (5) overlapped tags loci were clustered as a cluster, and neighboring clusters were merged into a unit (Supplemental Fig. 7A). Similar with tags, the clusters and units were annotated.
miRNAs and siRNAs identification
The methods for identification were shown in the Supplemental Figs. 7–8. When multiple sRNA-Seq data were used for comparison analysis, the raw data should been normalized. In a sRNA-Seq dataset i ∈ (1, …, n), tag normalized-count = tag raw-count ∗ NF i , and normalization factor (NF) was measured using the following equation:
where the n represents the number of sRNA-Seq datasets; R represents the sum of mapped reads/hits (hits divide by reads); T represents the sum of mapped tags.
Targets prediction
First, miRNA sequences were aligned to TCP expressed genes using RNAhybrid (Kruger and Rehmsmeier 2006) with the permissive options: (1) permit one internal loop, one bulge loop; (2) minimum free energy (MFE) < −30 kcal/mol; (3) constrain perfect base pairing (containing G:U pair) from position 8 to 12 relative to the 5′-end of the miRNA. Secondly, the miRNA-target alignments were scored and filtered using a Perl script according to the following five criteria: (1) No more than one single-nucleotide bulge or gap, all of which were considered as mismatch for scoring; (2) From position 2 to 13, each mismatch was assessed for a penalty of +2 while G:U pairs was assessed for a penalty of +1, and total penalty was no more than 5; (3) From position 14 to 19, each mismatch was assessed for a penalty of +1 while G:U pairs was assessed for a penalty of +0.5; (4) Other position, each mismatch or G:U pairs was for assessed a penalty of +0.5; (5) From position 2 to 19, mismatches was not more than four, total mismatches including G:U wobble were not more than seven; (6) The total score was no more than [length(miRNA)-9)]*0.5 + 6. These criteria were based on the previous research methods (Fahlgren and Carrington 2009) and experimental results (Wang et al. 2009). Finally, potential targets were also predicted using Target-align (Xie and Zhang 2010) with permitted 5 max mismatches, and the common targets were retained. In addition, a few high reliable but non-shared targets were also retained (Supplemental Table S12).
Enrichment analysis
ClueGO was used to cluster and visualize GO/KEGG terms corresponding to a list of genes, or compare between two functional clusters (Bindea et al. 2009). From our local rice genome annotation database, the genes annotation of GO terms (BP, CC, MF) and KEGG pathway were extracted and created to corresponding document files as ClueGO annotation sources. The new annotations files could cover more rice genes, because three rice GO sources (MSU6.1, Affymetrix and Amigo) have been integrated (Supplemental Table S1). The ClueGO analysis parameters in this paper, have been described in the figures legend, unmentioned parameters use the default settings.
Reverse transcriptase-PCR
TCP RNA (1 μg) was transformed into cDNA in a 20 μl reaction containing 1 μl Quant Reverse Transcritase (TIANGEN, BeiJing, CN), 2 μl dNTP (2.5 mM each), 2 μl Oligo dT primer (10 μM), and 2 μl 10× RT mix. One microliter of cDNA sample was used for subsequent PCR reactions using gene-specific primers with the following cycle conditions: 94°C, 30 s; 55°C, 30 s; 72°C, 1 min for 35 cycles. All primers used to detect AGOs and pre-miRNAs in the experiment were shown in Supplemental Fig. 10B2.
Liquid northern hybridization
Liquid northern hybridization of miRNAs was performed according to previous research (Wang et al. 2010). A series of oligo-DNAs matching with detected miRNAs were labeled at 5′ terminals with fluorescein isothiocyanate (synthesized by Takara Biotechnology, see the Supplemental Fig. 10A2). Fifteen microgrammes of miRNA were mixed with 5 μl Hybridization Buffer (30 mmol l−1 Sodium phosphate buffer (pH 8.0), 0.3 mol l−1 of NaCl, 10 mmol l−1 of EDTA) and 10 pmol probes, incubated at 42°C for 60 min, followed by 10 U exonuclease I (NEB) for 2 h for digestion non-hybridized sequences. Samples were separated on a 15% Non-denaturing PAGE gel. After the run completed, the gel was moved onto a Dark Reader transilluminator (Clare Chemical Research, Inc.) for observation and photography with a Canon A650IS digital camera.
Quantitative real-time RT-PCR analysis
The expression levels of the checked genes were examined by real-time quantitative RT-PCR. Total RNAs were prepared from pollen of different development stages, and converted to cDNAs using iScript™ cDNA Synthesis Kit (Bio-Rad, http://www.bio-rad.com) primed by oligo(dT) and random primer. The cDNAs were then used as templates for real-time PCR with gene-specific primers as listed in Supplemental Table S17. Real-time PCR was performed using SsoFast™ EvaGreen Supermix (Bio-Rad) on CFX96TM Real-time System (Bio-Rad). The checked genes were detected in parallel and used as the internal control. The 2−ΔΔCT method was used to analyze the relative transcript levels (Livak and Schmittgen 2001).
RLM-5′ RACE
The 5′ rapid amplification of cDNA ends (RACE) assay was used to identify products of miRNA directed cleavage in vivo (Llave et al. 2002). According to the manufacturer’s instructions, the First Choice®RLM-RACE Kit (Ambion) was used to clone the cleavage products. Two micrograms of total RNA isolated from the tricellular pollen were ligated to a 5′ RACE adapter followed by RT-PCR. After nested PCR, the fragments obtained from 5′ RACE experiments were cloned into the pMD19-T simple vector, and individual clones were selected for DNA sequencing. The outer and inner primers specific for verified genes were listed in Supplemental Table S17.
Results
Isolation and purification of pollen grains for hybridization and sequencing
We isolated and purified UNM, BCP and TCP (see “Materials and methods) using the modified method by our lab (Gou et al. 2001). Cytological observation and fluorescein-3′, 6′-diacetate (FDA) staining showed that both the purity and viability were over 90% for each stage (Supplemental Fig. 1). The total RNAs of UNM, BCP and TCP were extracted and hybridized respectively using 57 K Affymetrix Rice Genome Array to obtain the mRNA profiling of developing pollen. For each stage, two biological replicates derived from independent plant populations were performed, and the gene expression profiles of the replicates arrays were found to be significantly and positively correlated (r = 0.99), suggesting that the microarray data are reliable. To obtain the sRNA profiling of TCP, total RNAs from the TCP were prepared and 13–40 nt RNAs were excised for Illumina GA2 high-throughput sequencing. Over 13.7 million raw reads with error rates below 0.2% were acquired.
Gene expression features during male gametophyte development in rice
A total of 21 diverse organ/stage transcriptome data were normalized and used for comparison analysis (Supplemental Table S2; see “Materials and methods”). First, we measured the number of expressed genes in each sample (Supplemental Fig. 2A). An overview of the statistical results clearly showed that the transcriptional activity in each male gametophyte stage was lower than those in other organs/stages. Next, we identified organ-/stage-specific genes, and their number, which progressively increased from the Pa to Pc stage in the male gametophyte and were higher than those of other organs (Supplemental Fig. 2B). Both results are consistent with previous research (Wei et al. 2010; Fujita et al. 2010), and similar results are shown for Arabidopsis male gametophyte (Supplemental Table S3; Supplemental Fig. 3).
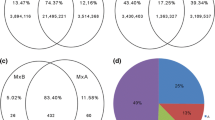
In our microarray data, a total of 13,663 genes, which covered 35.1% genes on the Affymetrix Rice Array, were detected during the gametophyte development (Fig. 1a). Among the expressed genes, 10,901, 9,930 and 7,674 genes clusters were detected at the Pa, Pb, and Pc stages respectively, respectively, whereas 5,425 genes clusters were shared among all stages (Fig. 1b). In addition, a total of 2,925 (21.4%) stage-specific genes were identified in the developing gametophyte (Fig. 1c), which were consistent with previous research (Fujita et al. 2010). The stage-specific genes contained 107 transcription factors (TFs) that were divided into 41 families (Supplemental Fig. 4). These genes and the TFs expressed distribution in different stages, suggesting that they play an important role in the gametophyte development (Supplemental Fig. 2C).

Gene expression features during the male gametophyte development. Three pollen development stages: uninucleate microspore (UNM/Pa), bicellular pollen (BCP/Pb) and tricellular pollen (TCP/Pc). a Distribution of the number of gene types. Pro represents protein-coding genes with known function (for details of other types see Supplemental Table S1). REPP and TPP types are TE-related genes. Blue MSU 6.1 genome annotation. Red Affymetrix Rice Genome Array mapped genes, 74.4% probe sets were mapped to 38025 MSU gene loci covering 65.6% of the genome. Green expressed genes in male gametophyte of rice, which covers 35.1% of the genes on the array. b A Venn diagram showing the number of genes detected across the gametophyte development stages. c A scatter plot showing the distribution of the pollen-specific genes in each stage. The X-axis shows Pc versus Pb with a log2 fold-change; the Y-axis shows Pb versus Pa whit a log2 fold-change. d Dynamic changes in the expressed genes during the male gametophyte development in rice. Pb2a: Pb versus Pa, comparison of expressed genes; Pb2a-spe: Pb versus Pa, comparison of the gametophyte-specific genes. Pc2b and Pc2b-spe are similar. In Pb2a, non-changed: gene signal-value > 6.5 in both stages; down-regulated: log2 fold-change < −1.5 and Pb gene signal-value > 6.5; up-regulated: log2 fold-change > 1.5 and Pa genes signal-value > 6.5. Other is similar
In order to show the dynamic changes of these genes, we compared adjacent stage of the male gametophyte. To our surprise, large-scale changes in gene expression emerged during the pollen development, and this phenomenon was not found among other development stages. Here, 21.4% (2,510/11,709) of genes were up-regulated and 28.2% (3,306/11,709) were down-regulated from the Pa to Pc stage, whereas up-regulated genes decreased to 11.1% (1,086/9,772) and down-regulated genes increased to 40.9% (3,998/9,772) from the Pb to Pc stage (Fig. 1d). Obvious changes were also observed by analyzing stage-specific genes (Fig. 1d). The same analysis was performed in Arabidopsis, as shown in Supplemental Fig. 3C, there is a high correlation between the transcriptomes of Pa and Pb, and large-scale gene expression changes occurred from Pb to Pc stages. The results were consistent with original study (Honys and Twell 2004). Through the comparison, we found a common characteristic that abundant transcript populations sharp reduce and shift from Pb to Pc stages in rice and Arabidopsis, which may be the result of miRNA-mediated regulation.
To evaluate our array data, 13 pollen stage-specific genes were randomly selected for real-time quantitative reverse transcription RT-PCR (qRT-PCR) analyses. As shown in Supplemental Fig. 5, their expression patterns detected by qRT-PCR were in good correlation with those obtained by array. The results indicate that expression profiles observed by the Affymetrix microarray are highly reliable.
miRNA pathway gene expression profiles during male gametophyte development
To explore the role of miRNAs in the male gametophyte development, we first detected the miRNAs pathway genes on our microarray. The rice genome contains 19 copies of the Argonaute (AGO) family and 8 copies of Dicer-like (DCL) family. In addition, another 11 miRNA-related genes were collected (Park et al. 2002; Dong et al. 2008; Kapoor et al. 2008; Laubinger et al. 2008; Voinnet 2009; Vazquez et al. 2010). The expression profiles of these genes are shown in Fig. 2a–c. At least 11 AGO genes, 7 DCL genes and 8 miRNA-related genes were reliably expressed (log2 > 7) in the gametophyte development process, including 6 AGO (AGO1A, AGO2, AGO4A, AGO13, AGO16, and AGO17), 2 DCL (DCL2A and DCL2B) and 2 miRNA-related genes (CBP20 and XRNa) were continuously expressed to the Pc stage.

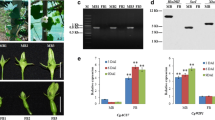
Microarray-based expression analysis of Argonaute (AGO), Dicer-like (DCL), RNA-dependent RNA polymerase (RDR), and miRNA processing-related genes in rice. Each column represents a microarray sample: vegetative tissues (7R 7-day Root, 7S 7-day Seeding, Lm Mature Leaf, Ly Young Leaf), Sa Shoot Apical Meristem, six stages of panicle development (P1–P6), five stages of seed development (S1–S5) and reproductive tissues (St Stigma, Ov Ovary, Pa UNM, Pb BCP, Pc TCP). For details see Supplemental Table S2. The color bar in bottom represents log2 expression values, expressed gene cutoff at 6.5 (log2 scale). a AGO family genes. b DCL family genes. c miRNA pathway-related genes. d RDR family genes. e AGO12, AGO13 and AGO17 RT-PCR products amplified from TCP cDNA in rice
According to previous studies, we found two known rice miRNA pathways existing in male gametophyte: the DCL1-AGO1-dependent PTGS pathway (Liu et al. 2005; Wu et al. 2009) and the DCL3-AGO4-dependent DNA methylation pathway (Wu et al. 2010). Whilst, the pri-miRNA/pre-RNA processing related proteins, namely, CBP20, ABH1 (CBP80), SE, DDL, HYL1, and the miRNA methylation and transport proteins HEN1 and HASTY, as well as small RNA degradation-related XRN and SDN proteins also were detected in developing male gametophyte. Focusing on the Pb and Pc stages, the high expressed AGO1A and AGO4A may be the main effectors of gene silencing (Fig. 2a). As AGO1 not only cleaves transcripts guided by miRNAs but also leaded miRNAs to direct translational repression (Brodersen et al. 2008; Lanet et al. 2009), it is probable that both mechanisms operate in rice pollen.
However, the functions of most AGO members in rice remain unknown (Mallory and Vaucheret 2010). A comparison microarray analysis indicated that AGO12, AGO13, and AGO17 are male gametophyte-specific (AGO12 and AGO17 are specific for Pa and Pb stages, AGO13 is specific for Pb and Pc stages), and that AGO14 and AGO18 are specific for female reproductive organs (stigma and ovary) (Fig. 2a). These findings have not previously been reported and studied, and the patterns that multi-AGO specifically expressed in the male germ line are different from Arabidopsis that only AGO5 was enriched (Borges et al. 2011). In order to evaluate the accuracy of microarray, RT-PCR was used to inspect the expression of three male gametophyte-specific AGOs. The results corresponded to the microarray analysis (Fig. 2e). Interestingly, the rice AGO12, AGO13, AGO14, and MEL1 and Arabidopsis AGO5 were clustered into the MEL1 group, as evidenced by phylogenetic analysis (Kapoor et al. 2008), and the rice MEL1 has been proved to enrich expression in the germ line (Nonomura et al. 2007). Furthermore, the MEL1 groups, AGO17, and AGO18 share a common lineage with AGO1 group (Kapoor et al. 2008), and the adjacent phylogenetic relationship suggests that those gametophyte-specific AGOs may be involved in miRNA activities. Our finding implies that those AGOs play an important role in the sexual reproduction of rice by binding special miRNAs.
Aside from miRNAs, we also detected the siRNAs pathway RNA-dependent RNA polymerases (RDR) on our microarray. The rice genome contains 5 RDR genes, in which only RDR2 was expressed credibly (log2 > 7) at the Pa stage (Fig. 2d). This means that those RDR-related endo-siRNA pathways are either absent or in a low active state between the Pb and Pc stages. These results are different from those of Arabidopsis, and previous research has indicated that Arabidopsis RDR2-RDR6 genes are all expressed in mature pollen, as confirmed by RT-PCR (Chambers and Shuai 2009; Grant-Downton et al. 2009a).
Small RNA expression features in tricellular pollen
After confirming the existence of the miRNA pathways, we excised 13–40 nt RNA from TCP for high-throughput sequencing and over obtained thirteen million raw reads (short sequences). According to our sRNA-Seq data processing flow (see “Materials and methods), a total of 2,032,599 tags (distinct reads) were loaded into MySQL database for further analysis, which covered 90.4% of total mapped reads and 70.5% of raw reads. In order to compare with other tissues, we have collected and processed 7 published sRNA-Seq datasets in different stages/organs of rice (Supplemental Table S4).
Analyzing the size distribution of the mapped reads and tags can illustrate the diversity and abundance of sRNAs. In the TCP small RNAs (TP-sRNAs) library, there are five read peaks (16, 19, 21–22, 24, and 33–36 nt), but only two tag peaks (21–22, 24 nt) (Fig. 3a). Indeed, the sRNA size for sequencing was inconsistent in different datasets, thus we examined the distribution of 18–28 nt reads in all libraries (Supplemental Fig. 6A). The results indicated that the 21 and 24 nt peaks are the common features of all tissue libraries and consistent with the size distribution expected for miRNAs and endo-siRNAs (Zhu et al. 2008; Mi et al. 2008; Wu et al. 2010). A distinctly high proportion of reads in 19 nt was observed in TP-sRNAs (Supplemental Fig. 6A), furthermore, most of the reads in the 16, 19, 33–36 nt size have not been reported so far. In all libraries, credible expressed tags within 18–28 nt were used for intersection analysis (Fig. 3b). The results revealed that TCP has high various and specific sRNA population; 58,138 credible tags were specifically expressed in TCP, which account for 76.6% of the total TP-sRNA tags (75,868, read-counts ≥ 5). 8M-sRNA is a mixed tissues library, which includes flowering panicle tissue; therefore, it has the biggest intersection with TP-sRNAs.

Small RNA expression features in tricellular pollen. a Length distribution of sRNAs in TCP. The reads and tags come from the data that have been mapped in the genome; the unknown reads/tags are mapped in the genome with unknown RNA locus [see (d)]. b Tags distribution in different datasets. The 18–28 nt size tags (read-counts ≥ 5) within all libraries are used for statistical analysis; only the top 13 in the intersection list are displayed. The 8 sRNA libraries are: G1, grains with 1–5 days after fertilization (DAF); G2, 6–10 DAF grains; 3 W, 3-week seeding; 3W2, another 3-week seeding; 4 W, 4-week seeding; FL, flag leaf; TP, TCP; 8 M, 8 mix tissues. For details see Supplemental Table S4. c Hits-length distribution of sRNAs in TCP. The hits represent the number of tag’s locus in the genome; the data (hits ≤ 50) that were loaded into MySQL database are used for statistical analysis, and the data (hits > 50) come from mapped tags with read-counts over 10. d The reads of the series “miRNA” come from those mapped on the pre-miRNAs locus; the “other” series contain snRNA, snoRNA, intron and SRP; the intergenics, genes and gene-antisence series are considered as sRNAs with unknown function. The sRNA-Seq datasets labels see (b)
The number of read distributions based on loci (hits) number can reflect the sRNA biogenetic feature. Approximately 31.0% reads were derived from single hits, 35.2% reads from 2 to 6 hits, 30.7% reads from 7 to 50 hits and 3.1% from hits > 50, and reads with hits > 50 were dominantly plotted at the 21–22 and 24 nt size (Fig. 3c). Compared with other libraries, the reads from the multi-loci had a high percentage of total sRNAs in TCP, but tags percentages are similar across all libraries (Supplemental Fig. 6B). Those analyses indicated that the multi-loci sRNAs had a high expression in TCP.
Next, we annotated sRNAs based on their genomic locations and functions (Fig. 3d). Statistical analysis demonstrated that the TP-sRNA library contained about 52.8% of tRNAs, 4.2% of rRNAs, 1.0% of other known sRNAs and only 0.7% of the miRNAs; 40.8% of unknown sRNAs were also detected. The size distribution of unknown reads is also shown in Fig. 3a. The 16, 19, and 33–36 nt size sRNA peaks disappeared, indicating that most of these sRNAs are fragments of tRNAs. Based on the annotation of genome the repeat marker, the TP-sRNAs were annotated, showing that more than half of the total and unknown sRNAs were derived from the DNA repeat area (Supplemental Fig. 6C).
Overall, TCP has specific sRNA population and expression profiles. The phenomenon that most of tRNAs were degraded and vast genes were down regulated implied that the metabolic rate decrease dramatically during the ripening process of TCP. Meanwhile, a number of novel miRNAs may be concealed in the abundance of unknown sRNAs and involved in these processes.
Identification of novel miRNAs and their expression features in tricellular pollen
Recently, many miRNAs have been identified by employing small RNA deep sequencing and exhaustive bioinformatics analysis in rice (Zhu et al. 2008; Lacombe et al. 2008; Sunkar et al. 2008; Lu et al. 2008). However, the limited overlap between the sRNAs datasets from multiple tissues suggested that many tissue-specific miRNAs remain undiscovered (Fig. 3b). Here, we presented a new algorithm for identifying novel miRNAs and endo-siRNAs (see “Materials and methods). Based on our prediction model, the potential pre-miRNAs could be mainly classified into three types: “True+”, “True”, and “Like”. The “True+” and “True” types contain the miRNA/miRNA* duplex, but “Like” lacks miRNA* confirmation; however, it could be annotated and supported by other sRNA-seq datasets (Supplemental Figs. 7–8). The three types were scored according to the identification criteria (Meyersa et al. 2008), and were confirmed as a veritable pre-miRNAs by a cutoff value.
Here, more than 10 raw read-counts were considered as actual miRNAs. We identified 57 novel unique miRNAs (miRu) derived from 39 pre-miRNAs (Supplemental Table S5; Supplemental File F1), and 42 known miRus which derived from 55 trusty pre-miRNAs (Supplemental Table S6). Before predicting, a total of 47 known miRus were detected in TP-sRNAs, which indicated that the current approach could achieve 89.4% sensitivity. In the novel pre-miRNAs list, 9 “True+”, 12 “True” and 18 “Like” types were found, and 10 “Like” types were confirmed by other sRNA libraries containing the miRNA/miRNA* duplex (Supplemental Table S5). Obviously, while mining novel miRNAs, the methods that relying on multi-sRNA-Seq datasets were more advantageous and reliable than relying on only one set. By this method, 45 novel pre-miRNAs containing 57 miRNAs were identified from the other seven sRNA libraries (Supplemental File F2). Meanwhile, we also identified 36 pre-siRNAs from the TP-sRNA datasets, including 7 TCP-specific pre-siRNAs (Supplemental Table S7); all of them were distributed in the intergenic region and 25.0% were derived from the repeat region. Most pre-siRNAs were 24 nt clusters, and only two were 21 nt clusters. The 24 nt siRNAs likely to be involved in heterochromatin formation in the repeated regions of the genome, as well as the silencing of other regions (Le Trionnaire et al. 2010; Bourc’his and Voinnet 2010). However, the number of 24 and 21 nt clusters expressed relatively low in the TCP stage compared with other tissue (Johnson et al. 2009), which was consistent with the results of the RDR gene expression analysis based on the microarray (Fig. 2d).
Statistical analysis (Supplemental Fig. 9) with the TCP miRNA sets revealed that most of them are 21 and 24 nt. The 21 nt miRNAs displays a strong preference for initiating with a 5′ terminal nucleotide U, which have been confirmed to primarily bind to rice AGO1 (Mi et al. 2008; Wu et al. 2009); the 24 nt miRNAs preferentially initiate with a 5′ A or U, which have been confirmed to bind to rice AGO4 and AGO16 (Wu et al. 2010), and the feature 5′ terminal nucleotide obviously different from 24 nt siRNA. However, four 20-nt miRNAs have highest abundance of reads, and all of them have a C at the 3′ terminus.
Compared with other sRNAs libraries, the abundance and variety of known miRus are relatively low in the TP-sRNAs (Supplemental Table S8; Fig. 3d), which is consistent with previous studies on mature Arabidopsis pollen (Chambers and Shuai 2009). To identify the pollen-specific expression of miRNAs in the TP-sRNAs, 104 expressed miRus mentioned above were used for hierarchical clustering analysis (Fig. 4). Here, the known miRus were designated osa-miRu001 to osa-miRu324, and the novel miRus were named new-miRu001 to new-miRu057 in the TP library (Supplemental Table S6). The profiles showed that most of the novel miRNAs were pollen-specific. In contrast, only two known miRNAs were pollen-specific. These are osa-miRu207 and osa-miRu208, both of which are derived from osa-MIR1874, which is specific expression in panicles in previous studies (Lu et al. 2008). Furthermore, the clustering tree of eight samples showed significantly correlated expressions between adjacent stages of development, such as G1 to G2 and 3W to 4W (Fig. 4). Another interesting question is whether TCP miRNAs could be delivered to early grains by double fertilization. This problem has been discussed in previous research (Borges et al. 2011). In the current study, TP (TCP) and G1 shared little in common (Fig. 4), which demonstrates that most TCP miRNAs perform their functions in pollen.

Hierarchical clustering analyses of 104 miRus. Each row represents a unique miRNA (miRu) and each column represents a smRNA-Seq sample. Only the 104 pollen-expressed miRus were included (raw read-counts > 9), and miRNAs expression intensity shown using normalized read-counts which were log2 transformed. Green low expression, red high expression. Sample of TP designates tricellular pollen, other details see Supplemental Table S4. The hierarchical clustering was generated using Euclidean distance, and visualized using the program MeV4 (Saeed et al. 2003). The miRus can be grouped into two major clusters: (1) housekeeping miRNAs; (2) tissue-specific miRNAs. The group “TCP” represents TP-specific miRNAs
We wondered whether these expressed miRNAs in rice pollen are conserved. At first, 57 rice new-miRus were used for BLAST searching miRBase-16, and 4 conserved new-miRus were identified (Supplemental Table S6). Next, we compared 104 rice miRus with the 75 Arabidopsis miRus which expressed in mature pollen (Chambers and Shuai 2009). The results indicated that 57.9% (11/19) conserved miRNA families were expressed in both rice and Arabidopsis pollen; however, the MIR396 family was only expressed in rice pollen, and MIR158, MIR171_1, MIR172, MIR390, MIR397, MIR399, and MIR408 were only expressed in Arabidopsis. In addition, 28 known miRus and 56 new-miRus were rice-specific, and 43 known miRus were Arabidopsis-specific (Supplemental Table S9). To further identify conservation levels of 104 rice miRus in different species, we performed genome-wide searches requiring less than two mismatches in eight plant species, the results show that approximately 35.6% of these miRus are conserved across two species (excluding rice). Interestingly, only 14 out of 41 pollen-specific miRus were conserved across at least one species (Supplemental Table S6), suggesting that these rice-specific MIRNA may be arose relatively recently in evolutionary time (Cuperus et al. 2011).
We reported 39 new miRNA precursors in rice TCP, of which 6 can yield multiple distinct miRNA/miRNA* duplexes from different positions and 4 can yield distinct miR-5p and miR-3p (Supplemental File F2; Meyersa et al. 2008). Interestingly, these multiple miRNA precursors included one known osa-MIR1874, and 90% are pollen-specific expression (Supplemental Table S6). Comparison with known MIRNA annotation showed that a high proportion (33% in other sRNAs libraries, 39% in the TP-sRNAs library) of new MIRNA belonged to the intronic category. In TCP miRNAs, 22 intronic MIRNA (39%) were found, of which 6 MIRNA host genes were pollen-specific (Supplemental Table S11), suggesting that they have a synergistic or antagonistic regulatory effect (Lutter et al. 2010).
To validate the sequencing and predicted results, we have performed 5 miRNA liquid Northern hybridization (Wang et al. 2010) and 6 pre-miRNA RT-PCR analyses. Distinct bands around 21 nt were observed for 5 miRNA cases, and 6 pre-miRNA were detected by RT-PCR, indicating a 100% validation of our results (Supplemental Fig. 10).
Analysis of miRNA targets in the male gametophyte
Recent studies show that some miRNAs may inhibit the translation by binding non-perfectly to target mRNAs (Brodersen et al. 2008; Montgomery et al. 2008). Although a few computational tools and rules have been developed to predict plant miRNA targets [miRU (Zhang 2005); Helper tools (Moxon et al. 2008); TAPIR (Bonnet et al. 2010) and psRNATarget (Dai and Zhao 2011)], these programs have a strictly limited criteria. Here, we selected RNAhybrid (Kruger and Rehmsmeier 2006) and Target-align (Xie and Zhang 2010) to predict miRNA targets, since both tools have more flexible criteria and can get more potential candidates (see “Materials and methods).
A total of 1,061 potential target genes were identified from the gametophyte genes list, of which 90% targets were shared by both predicted tools (Supplemental Table S12). In order to evaluate the predicted results, 198 verifiable miRNA-target alignments were scored using our penalty rules (see “Materials and methods). These targets come from three rice mRNA degradome libraries where mRNAs cleaved by miRNAs were identified (Wu et al. 2009; Li et al. 2010; Zhou et al. 2010); 81.4% of penalty scores was distributed at 1–5 (Supplemental Fig. 12A) and 97.5% of alignment have no more than 4 mismatches. In contrast, among our prediction targets, 82.2% of penalty scores was distributed between 4.5 and 6.5 (Fig. 5a) and 86.2% of alignment have no more than 4 mismatches (Supplemental Table S12). The results suggest that most potential targets were regulated through translational repression rather than cleavage. To test our hypothesis, we investigated the changes in target gene expression during the Pc and Pb (Pbc) stages (Fig. 5b). In total, 745 targets expressed in Pbc stages, including 295 down-regulated, 98 up-regulated and 352 unchanged. The proportion of the down-regulated genes in the targets genes (39.6%) has not differed much than that in the total expressed genes (40.9%), which showed that miRNA-mediated degradation of target mRNAs was not obvious and most targets may be were regulated by translational repression.

Analysis of miRNA targets during the bicellular (Pb) to tricellular (Pc) stages. a Distribution of the penalty scores for the predicted miRNA-target alignment. miRNAs and the targets are come from the expressed genes during gametophyte development; 42 known miRNAs (osa-miRNA) have 709 alignments, and 42 new-miRNAs have 543 alignments. In total, 1,061 potential target genes were identified. The alignments were scored according to our methods (details shown in the “Materials and methods” section). b Scatter plot of target genes for Pc versus Pb. 98 up-regulated (U) genes are shown in red, 295 down-regulated (D) genes shown in light blue, 352 unchanged genes (M) are shown in blue, and 316 the unexpressed genes (N) are shown in green. The red lines represent the gene expression threshold (6.5) of genes. Up- and down-regulated genes were identified based on the 1.5-fold threshold change. The yellow box plots show the intensity of expressed genes. c Pie chart showing the gene type distribution of 745 targets in the Pb and Pc stages. TFs transcription factors, EC enzyme; other abbreviations are shown in Supplemental Table S1. d Categorization of the Pbc’s target genes by the GO molecular function. In total 745 potential targets, 483 genes were annotated by GO MF. The GO terms were enriched at depth three with P value <0.05 (hypergeometric test), and 8 terms including 324 target genes were retained
Next, we employed 5′ rapid amplification of cDNA ends (5′ RACE) (Llave et al. 2002) and degradome data analysis approaches to validate the predicted miRNA targets (Addo-Quaye et al. 2008; German et al. 2008). In total, 17 targets were confirmed by the rice young panicle degradome sequencing dataset downloaded from NCBI (accession nos. GSM476257, Zhou et al. 2010). On the other hand, 12 potential targets were randomly selected for 5′ RACE confirmation, but only 3 target cleavage products were detected and one of them shared by degradome (Supplemental Fig. S11).
Among the identified targets, several miRNA targets are especially noteworthy. First, five putative auxin response factor (ARF) (LOC_Os10g33940, LOC_Os02g06910, LOC_Os04g57610, LOC_Os06g46410, LOC_Os12g41950) were confirmed that they were cleaved by MIR160 (osa-miRu002, and osa-miRu041) and miR167 (miRu007 and miRu049). In our array dataset, there are 14 ARF genes are high expressed prior to Pb stage, but most of them afterwards suddenly drop below detectable levels. Interestingly, among them 10 ARF genes were predicted to be targeted by miRNAs, indicating ARF is a key effecter of miRNA-mediated silencing in developing pollen (Supplemental Table S16). Second, two nuclear transcription factor Y (NFY) subunit (LOC_Os03g07880, and LOC_Os03g48970) were confirmed as targets of osa-miR169c (osa-miRu053) (Li et al. 2010; Zhou et al. 2010). The NFY gene is a conserved transcription factor that binds with high specificity to CCAAT motifs in the promoters of a variety of genes (Romier et al. 2003). Different NFY subunits combinations may control various aspects of rice growth and development through tissue specific expression (Thirumurugan et al. 2008). This indicates that miRNAs can regulate genes expression indirectly by silencing some key transcription factors. Third, two important genes: elongation factor Tu (LOC_Os03g44780) (Weijland et al. 1992) and polymerase Rpb1, domain 2 (LOC_Os02g05880) (Cramer et al. 2001) were confirmed to be cleaved respectively by pollen-specific new-miRu043 and new-miRu029. This indicates that miRNAs can directly regulate genes expression at both transcription and translation level.
Given that these miRNAs were found in the Pc stages of gametophyte and mainly carried out their function in the Pbc stages, subsequent studies were focused on Pbc’s targets. First, these targets were classified based on the genes types (Fig. 5c). Approximately 7% of the targets were transcription factors (TFs), 3% were kinases (KE), 22% were enzymes (EC), and most were expressed proteins (Pro known function, 46%; Pro-u unknown function, 17%) that belong to classes different from the previously verifiable targets (Li et al. 2010; Zhou et al. 2010). Among these TFs, 13 were specifically expressed in pollen, which were targeted by miRNAs and affected pollen development and specific genes controlling metabolism. A list of these TFs and miRNAs was shown in Supplemental Table S13. For example, LOC_Os12g06480 (PHD-finger family protein) and its homologous genes AT1G43770 are specifically expressed in Pb stage and down regulated in Pc stage both in rice and Arabidopsis (Supplemental Table S16). The PHD proteins exist universally in the nucleus, and their functions tend to lie in the control of chromatin or transcription (Bienz 2006). In rice, it was targeted by new-miRu007 with few mismatches, indicating that the down-regulation was mediated by miRNA. However, the functions of most of these TFs are unknown, and these data will be available for further analysis.
To gain more information on these Pbc’s targets, GO molecular functions (MF) were extracted and enriched. As a result, a total of 8 significantly over-represented GO categories were obtained, which contained 44% target genes (Fig. 5d). Furthermore, the Pbc’s target genes and all the expressed genes in the percentage of chips covering genes were compared using the GO MF categories (Supplemental Fig. 12B). As a whole, the analysis indicated that miRNAs regulate a large variety of genes in many dominative categories, such as nucleotide/nucleic-acid/protein/ion binding. In addition, a number of small categories, such as ligase activity, transmembrane and substrate-specific transporter activity, more tended to be regulated. In contrast, genes with transferase activity, hydrolase activity, oxidoreductase activity, ribosomal structural constituents, and so on, showed no signs of regulation by miRNAs.
In the Pbc stages, 87 miRus targeted 745 genes with 870 target sites. Obviously, the interaction between miRNAs and their targets have many-to-many relationships; for instance, most of miRNAs have multiple target genes, and 14.6% of the target genes were targeted by multiple miRNAs. A bipartite graph basal network model was constructed to show global network of miRNA-target interaction (Supplemental Fig. 13).
GO terms and KEGG pathway enrichment analysis of targets
After collecting all necessary information on the miRNAs and targets, it is necessary for us to understand what biological process are the target genes involved in the pollen development. To handle this problem, the GO term and the KEGG pathway annotation were used to enrich and classify the given genes into specific informative groups.
First, using the GO biological process (BP) as annotation sources, 745 Pbc’s target genes were extracted from the miRNA-target interaction network and enriched with network-based approaches using Cytoscape with the ClueGO plug-in (Jiang et al. 2007). The results were shown in Supplemental Fig. 14. The enrichment network contained 152 target genes, of which 87.5% were clustered into 14 biological process groups, and 29 GO terms displayed statistical significance (P < 0.001). Remarkably, all of 29 significant GO terms were enriched into one group: nitrogen compound biosynthesis group that correlates with ion transport, ATP and nucleoside metabolic process. Thus, 62 target members from this group were extracted for further analysis and then integrated into the GO MF and GO cellular compartments (CC) for ClueGO networking (Fig. 6a). The results demonstrated that most of these targets are involved in energy-coupled transmembrane transport; for instance, the proton-transporting V-type H+-ATPase (V-ATPase) complex has 14 protein subunits in rice annotation, 6 of which are targeted by miRNAs. The vacuolar V-ATPase is involved in regulating pH homeostasis and driving coupled transport, and it is essential for Golgi organization and development of the male gametophyte (Dettmer et al. 2005; Beyenbach and Wieczorek 2006). Other research also suggested that the V-ATPase has a potential function in regulating plasma membrane morphology and dynamics (Baxter et al. 2005; Baars et al. 2007).

Enrichment analyses of miRNA targets. a The GO enriched network analysis of selected target genes. Total of 81 targets genes were extracted from “cellular nitrogen compound biosynthetic” group that were shown in Supplemental Fig. 14. The nodes represent individual GO terms (quadrate showing GO cellular compartment and circular showing GO molecular function), and the edges represent the functional relationships between processes. The node size represents the term enrichment significance (largest node: P < 0.001). Nodes of the same color represent a function-related group, and terms not in groups are shown in white. The network was clustered and visualized with ClueGO (kappa score: 0.4, GO leave: 4–8, using all GO evidence codes). b Statistics of miRNA-mediated biological processes. When the percentage of common genes between the GO/pathway and the target is above 10%, the biological process is significantly regulated by miRNAs (Supplemental Tables S14–S15). c Statistics of biological processes associated with miRNA regulation. miRNAs were listed when the sum of their significantly related terms (GO BP and KEGG pathway) was greater than 2
Of course, not all genes had annotations in each category. In the Pbc stages, the GO BP, MF, and CC annotations for all targets genes were 50, 65, and 54 respectively. Given that 58% of the rice targets are homologous with Arabidopsis pollen expressed genes (Supplemental Table S16), these orthologous genes in Arabidopsis were analyzed according to the method mentioned above. The results shown that many pollen-specific processes, such as pollen development, calcium ion transport, lipid modification and cell cycle cytokinesis, were annotated. Meanwhile, some process enrichments were more significant than rice, such as posttranscriptional regulation of gene expression, regulation of signal transduction, and response to auxin stimulus (Supplemental Fig. 15).
Although the above analysis results showed that miRNAs were involved in many important metabolic and development processes, whether or not miRNAs play an important role in such remains to be determined. To further explore this problem, the down-regulated and up-regulated genes enriched cluster were compared with targets enriched cluster using ClueGO compare analysis function. First, we focused on the biological process in which 1,186 up-regulated genes were involved in the Pbc stages (Fig. 1d). After the comparison, an information list of the enriched network was produced by ClueGO, and the GO terms, whose percentage of associated genes was set at over 30%, were selected; redundant terms were not listed. As shown in Table 1, 4 terms representing up-regulated processes were included, 2 of which were associated with miRNAs. For instance, term GO:0032312 (regulation of ARF GTPase activity) mediated intracellular signal transduction, including 6 genes (4 up-regulated genes and 2 targets genes) which cover 46% of the term associated genes of the whole genome. Next, we aimed to the biological process in which 3,398 down-regulated genes were involved (Fig. 1d). As we expected, many significant down-regulated processes emerged in the Pbc stages. When the percentage of associated genes was set to over 30%, after the removal of redundant terms, 42 obviously down-regulated GO terms were detected, 88% of which were associated with miRNAs (Supplemental Table S14). It is noteworthy that a number of amino acid biosynthetic processes were obviously down-regulated, Such as GO:0000105 (histidine biosynthetic), GO:0006573 (valine metabolic), GO:0009064 (glutamine family amino acid metabolic), GO:009086 (methionine metabolic), and GO:0009309 (amine biosynthetic), 4 of which were associated with the miRNAs regulation. Another significant down-regulated process was related to transcription and translation, including GO:0006357 (regulation of transcription from RNA polymerase II promoter), GO:0006412 (translation), GO:0042255 (ribosome assembly), and GO:0043039 (tRNA aminoacylation), all of them were associated with miRNAs. In addition, the monosaccharide, starch, glycerophospholipid, lipid, nucleotide, and so on, most of those metabolic processes associated with miRNAs and were down-regulated observably. The results of both enrichment analyses reflected physiological cellular changes in male gametophyte development. Generally, most biological processes were observed to be rapidly down-regulated when pollen started to ripen, and most changes may be due to miRNA-mediated negative regulation.
In order to further confirm our findings, the KEGG enrichment networks were also used for comparison analysis between down-regulated genes and target genes. Although only 20% of the genes were annotated, most of them were enzymes associated with metabolism. The results are shown in Supplemental Table S15. A total of 41 pathways appeared to be overrepresented, of which 78% were associated with miRNA regulation. In these miRNA-related pathways, 30% were significantly related to miRNA (Fig. 6b), which contained more than 10% common genes between the compared clusters, such as osa00071 (Fatty acid metabolism), a total 19 down-regulated genes were found, of which 2 genes were miRNA target genes. Similar to the findings in the previous GO enrichment, the expression of amino acid biosynthetic genes was likely weakened. These results were also reflected by the aminoacyl-tRNA biosynthesis and ribosome related pathways.
For each important miRNA, the number of related processes that were significantly down regulated was listed (Fig. 6c). Surprisingly, the pollen-specific new-miRu43 is shown as a very key regulator which participated in 14 GO and KEGG pathways in developing pollen. The other two key pollen-specific miRNAs, new-miRu001 and new-miRu004, both derived from a common precursor new-MIR3001 (Supplemental File F1), and mediated the regulation of 8 GO and KEGG pathways. The remaining 5 are known as osa-miRu004 (osa-miR164b, f), osa-miRu045 (osa-miR164d), osa-miRu138 (osa-miR444c.1), osa-miRu095 (osa-miR444d.2), and osa-miRu137 (osa-miR444b.2). The two MIR164 family members are also expressed in Arabidopsis mature pollen (Supplemental Table S9), suggesting that they may have conserved target sites in the homologous genes and be involved in regulating same metabolic pathways. Interestingly, there are 3 members in MIR444 family, which previously be proved as a characteristic of monocots plant (Cuperus et al. 2011). This indicates that MIR444 has evolved specific functions in regulating rice male gametophyte development.
Discussion
Here we report a synthetic analysis integrating both genes and miRNAs transcriptome data during pollen development in rice. By microarray analysis, miRNA-pathway profiling, miRNAs mining, targets prediction, and enrichment analysis of target functions, four significant findings were determined in this study. First, a common characteristic was found in rice and Arabidopsis, whose vast number of genes were down-regulated from the BCP to TCP stage. Second, miRNA-pathways were found to exist in the developing pollen, and three pollen-specific and miRNA-related AGO proteins were identified. These findings suggest that a specific miRNA population is present. Third, 104 unique miRNAs in TCP were identified, 57 of which are novel and derived from 39 pre-miRNAs. Most of the novel pre-miRNAs were non-conserved, and exhibited multi-miRNA yields and intronic biogenesis. Finally, functional analysis of the targets demonstrated that most of the down-regulated pathways result from miRNAs-mediated PTGS.
Our analysis of rice microarray data was not only compared different tissues of rice but also rice with Arabidopsis. Although transcript profiling of the male gametophyte had been characterized both in rice and Arabidopsis (Honys and Twell 2004; Wei et al. 2010), a comparison between the two species has rarely been conducted. The comparison analysis also indicated that numerous changes in gene expression occurred from the UMP to BCP stage, but the results obtained were different from those of Arabidopsis, in which only a few changes in gene expression change occurred. Although both species have more than 55.8% of homologous gene, the reason behind the different expression patterns observed between them remains unknown. Besides, a total of 2,925 pollen stage-enriched or pollen-specific genes, including 107 TFs, were identified, but the interactions between these TFs and the pollen-specific miRNAs have not been analyzed in detail. A closely related regulation network (Qiu et al. 2010) that plays an important role in the control of pollen development is assumed to exist.
The analysis of sRNA-Seq data (Illumina platform) also included a comparison of multiple datasets that contain almost all of the current submitted data of rice in the GEO database. A comprehensive analysis demonstrated that TCP has various and unique sRNA populations; in particular, it includes a large amount of tRNA fragments that are not random degradation products. Although most tRNA function-related pathways are down-regulated by miRNAs, such as aminoacyl-tRNA biosynthesis, translational initiation, and amino acid biosynthesis, no evidence of the degradation of tRNAs as being directly related to these pathways was detected (Phizicky and Hopper 2010). Recent studies have shown that some tRNA-derived small RNAs (tsRNAs) are generated by Dicer-dependent cleavage (Babiarz et al. 2008) and associated with the Piwi protein (Couvillion et al. 2010); however, their function remains incompletely understood.
In this study, we presented a new algorithm used for identifying novel miRNAs. Multiple datasets support and multi-criterion scoring will provide more evidence of the absence of miRNA* confirmation and can identify more miRNAs than previous methods (Zhu et al. 2008). Moreover, we compared TCP expressed miRNAs between rice and Arabidopsis. The results indicate that only 11 conserved rice miRNA families that are homologous to the miRNAs expressed in Arabidopsis exist, most of pollen miRNAs are species specificity. In addition, the RDR family is a key biosynthetic gene family of siRNA; however, the current study shows that RDR family genes exhibit different expression patterns in rice and Arabidopsis. Taken together, these features suggest that sRNA-mediated regulations have different functions in rice and Arabidopsis pollen development.
A total of 745 target genes were predicted during the BCP and TCP stages using our deliberative filtered rules. By integrating GO annotation sources, relatively detailed information on rice genes was obtained, and GO enriched network analysis shown that most metabolic pathways related to miRNA-mediated regulation. Significantly, 16 GO BP and 14 KEGG pathways were obviously regulated by miRNA (Fig. 7). A very interesting question is whether the pollen-specific targets are signally regulated by pollen-specific miRNAs, and the relationship is shown in Fig. 7. There are 12 pollen-specific miRNAs involved in 73.4% down-regulated pathways, of which 7 pathways only are targeted by pollen-specific miRNAs, and only protein litigation process has high proportion of pollen-specific genes. These observations indicate that this specific–specific interaction is not apparent. Furthermore, although most down-regulations were related to miRNA, whether or not the expression level is consistent between the mRNAs and the protein remains unknown (Grobei et al. 2009). Besides, most miRNAs have low expression levels, such that a new method of investigating the efficiency of miRNA regulation must be designed, and the mechanism of miRNA-mediated silencing must be confirmed by other experiments.

Obvious down-regulated biological processes mediated by miRNAs in male gametophyte of rice. A total of 16 GO biological processes and 14 KEGG pathways were listed in Fig. 6b and Supplemental Tables S14–S15, and 12 specific miRNAs and 24 nonspecific miRNAs are obviously involved in regulation of these down-regulated processes. Top color bar show stage-specific genes fractions (total genes divided by stage-specific genes in each node). Line width of edge represents the number of miRNA (values range from 1 to 5)
In conclusion, our results indicated that the rice has a number of pollen-specific miRNA populations that they play an important role in controlling the phase transition from BCP to TCP in rice. It is worth noting that when we finished our manuscript, we noticed a related but independent work has just been published (Wei et al. 2011). Their analysis of miRNA includes three stages of data, thus the miRNAs dynamic relationship can be revealed in developing pollen. However, the data integration analysis between microarray-based genes and miRNAs haven’t been considered, so the functions of miRNA in control of pollen development were still lack the systematic analysis. On the other hand, in our study, the methods of miRNA and target gene identification have been innovated and improved rather than the published method, thus more depth and valuable information can be discovered. Moreover, with improvements in GO annotation, future analyses are expected to reveal more miRNA-based regulatory pathways.
Abbreviations
- miRNA:
-
MicroRNA
- miRus:
-
Unique miRNAs
- AGO:
-
Argonaute
- TGS:
-
Transcriptional gene silencing
- PTGS:
-
Posttranscriptional gene silencing
- UMP/Pa:
-
Uninucleate microspores
- BCP/Pb:
-
Bicellular pollen
- TCP/Pc:
-
Tricellular pollen
- RDR:
-
RNA-dependent RNA polymerases
- DCL:
-
Dicer-like protein
- siRNA:
-
Small interfering RNA
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
References
Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ (2008) Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 18:758–762
Baars TL, Petri S, Peters C, Mayer A (2007) Role of the V-ATPase in regulation of the vacuolar fission-fusion equilibrium. Mol Biol Cell 18:3873–3882
Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R (2008) Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev 22:2773–2785
Baxter IR, Young JC, Armstrong G, Foster N, Bogenschutz N, Cordova T, Peer WA, Hazen SP, Murphy AS, Harper JF (2005) A plasma membrane H+-ATPase is required for the formation of proanthocyanidins in the seed coat endothelium of Arabidopsis thaliana. Proc Natl Acad Sci USA 102:2649–2654
Beyenbach KW, Wieczorek H (2006) The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol 209:577–589
Bienz M (2006) The PHD finger, a nuclear protein-interaction domain. Trends Biochem Sci 31:35–40
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman WH, Pages F, Trajanoski Z, Galon J (2009) ClueGO: a Cytoscape plug-into decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25:1091–1093
Bonnet E, He Y, Billiau K, Van de Peer Y (2010) TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26:1566–1568
Borg M, Brownfield L, Twell D (2009) Male gametophyte development: a molecular perspective. J Exp Bot 60:1465–1478
Borges F, Gomes G, Gardner R, Moreno N, McCormick S, Feijo JA, Becker JD (2008) Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol 148:1168–1181
Borges F, Pereira PA, Slotkin RK, Martienssen RA, Becker JD (2011) MicroRNA activity in the Arabidopsis male germline. J Exp Bot 62:1611–1620
Bourc’his D, Voinnet O (2010) A small-RNA perspective on gametogenesis, fertilization, and early zygotic development. Science 330:617–622
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190
Chambers C, Shuai B (2009) Profiling microRNA expression in Arabidopsis pollen using microRNA array and real-time PCR. BMC Plant Biol 9:87
Chuck G, Candela H, Hake S (2009) Big impacts by small RNAs in plant development. Curr Opin Plant Biol 12:81–86
Couvillion MT, Sachidanandam R, Collins K (2010) A growth-essential Tetrahymena Piwi protein carries tRNA fragment cargo. Genes Dev 24:2742–2747
Cramer P, Bushnell DA, Kornberg RD (2001) Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science 292:1863–1876
Cuperus JT, Fahlgren N, Carrington JC (2011) Evolution and functional diversification of MIRNA genes. Plant Cell 23:431–442
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
Dettmer J, Schubert D, Calvo-Weimar O, Stierhof YD, Schmidt R, Schumacher K (2005) Essential role of the V-ATPase in male gametophyte development. Plant J 41:117–124
Deveshwar P, Bovill WD, Sharma R, Able JA, Kapoor S (2011) Analysis of anther transcriptomes to identify genes contributing to meiosis and male gametophyte development in rice. BMC Plant Biol 11:78
Dong Z, Han MH, Fedoroff N (2008) The RNA-binding proteins HYL1 and SE promote accurate in vitro processing of pri-miRNA by DCL1. Proc Natl Acad Sci USA 105:9970–9975
Eady C, Lindsey K, Twell D (1995) The significance of microspore division and division symmetry for vegetative cell-specific transcription and generative cell differentiation. Plant Cell 7:65–74
Eckardt NA (2009) Investigating translational repression by microRNAs in Arabidopsis. Plant Cell 21:1624
Fahlgren N, Carrington JC (2009) miRNA target prediction in plants. Methods Mol Biol 592:51–57
Fujioka T, Kaneko F, Kazama T, Suwabe K, Suzuki G, Makino A, Mae T, Endo M, Kawagishi-Kobayashi M, Watanabe M (2008) Identification of small RNAs in late developmental stage of rice anthers. Genes Genet Syst 83:281–284
Fujita M, Horiuchi Y, Ueda Y, Mizuta Y, Kubo T, Yano K, Yamaki S, Tsuda K, Nagata T, Niihama M, Kato H, Kikuchi S, Hamada K, Mochizuki T, Ishimizu T, Iwai H, Tsutsumi N, Kurata N (2010) Rice expression atlas in reproductive development. Plant Cell Physiol 51:2060–2081
German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, De Paoli E, Lu C, Schroth G, Meyers BC, Green PJ (2008) Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol 26:941–946
Ghildiyal M, Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10:94–108
Gou XP, Xu Y, Tang L, Yan F, Chen F (2001) Representative cDNA library from isolated rice sperm cells. Acta Botanica Sinica 43:1093–1096
Grant-Downton R, Hafidh S, Twell D, Dickinson HG (2009a) Small RNA pathways are present and functional in the angiosperm male gametophyte. Mol Plant 2:500–512
Grant-Downton R, Le Trionnaire G, Schmid R, Rodriguez-Enriquez J, Hafidh S, Mehdi S, Twell D, Dickinson H (2009b) MicroRNA and tasiRNA diversity in mature pollen of Arabidopsis thaliana. Bmc Genomics 10:643
Grobei MA, Qeli E, Brunner E, Rehrauer H, Zhang R, Roschitzki B, Basler K, Ahrens CH, Grossniklaus U (2009) Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res 19:1786–1800
Honys D, Twell D (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol 5:1465–6906
Huijser P, Schmid M (2011) The control of developmental phase transitions in plants. Development 138:4117–4129
Huntzinger E, Izaurralde E (2011) Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 12:99–110
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4:249–264
Jiang P, Wu H, Wang W, Ma W, Sun X, Lu Z (2007) MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res 35:W339–W344
Johnson C, Kasprzewska A, Tennessen K, Fernandes J, Nan GL, Walbot V, Sundaresan V, Vance V, Bowman LH (2009) Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res 19:1429–1440
Jover-Gil S, Candela H, Ponce MR (2005) Plant microRNAs and development. Int J Dev Biol 49:733–744
Kapoor M, Arora R, Lama T, Nijhawan A, Khurana JP, Tyagi AK, Kapoor S (2008) Genome-wide identification, organization and phylogenetic analysis of Dicer-like, Argonaute and RNA-dependent RNA polymerase gene families and their expression analysis during reproductive development and stress in rice. Bmc Genomics 9:1471–2164
Kruger J, Rehmsmeier M (2006) RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res 34:W451–W454
Lacombe S, Nagasaki H, Santi C, Duval D, Piegu B, Bangratz M, Breitler JC, Guiderdoni E, Brugidou C, Hirsch J, Cao X, Brice C, Panaud O, Karlowski WM, Sato Y, Echeverria M (2008) Identification of precursor transcripts for 6 novel miRNAs expands the diversity on the genomic organisation and expression of miRNA genes in rice. BMC Plant Biol 8:123
Lanet E, Delannoy E, Sormani R, Floris M, Brodersen P, Crete P, Voinnet O, Robaglia C (2009) Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant Cell 21:1762–1768
Laubinger S, Sachsenberg T, Zeller G, Busch W, Lohmann JU, Ratsch G, Weigel D (2008) Dual roles of the nuclear cap-binding complex and SERRATE in pre-mRNA splicing and microRNA processing in Arabidopsis thaliana. Proc Natl Acad Sci USA 105:8795–8800
Le Trionnaire G, Grant-Downton RT, Kourmpetli S, Dickinson HG, Twell D (2010) Small RNA activity and function in angiosperm gametophytes. J Exp Bot 62:1601–1610
Li C (2008) Automating dChip: toward reproducible sharing of microarray data analysis. BMC Bioinform 9:1471–2105
Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang WX, Sunkar R (2010) Transcriptome-wide identification of microRNA targets in rice. Plant J 62:742–759
Liu B, Li P, Li X, Liu C, Cao S, Chu C, Cao X (2005) Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol 139:296–305
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(−Delta Delta C) method. Methods 25:402–408
Llave C, Xie Z, Kasschau KD, Carrington JC (2002) Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 297:2053–2056
Lu C, Jeong DH, Kulkarni K, Pillay M, Nobuta K, German R, Thatcher SR, Maher C, Zhang L, Ware D, Liu B, Cao X, Meyers BC, Green PJ (2008) Genome-wide analysis for discovery of rice microRNAs reveals natural antisense microRNAs (nat-miRNAs). Proc Nat Acad Sci USA 105:4951–4956
Lutter D, Marr C, Krumsiek J, Lang EW, Theis FJ (2010) Intronic microRNAs support their host genes by mediating synergistic and antagonistic regulatory effects. Bmc Genomics 11:1471–2164
Mallory A, Vaucheret H (2010) Form, function, and regulation of ARGONAUTE proteins. Plant Cell 22:3879–3889
McCormick S (1993) Male gametophyte development. Plant Cell 5:1265–1275
Meyersa C, Axtellb MJ, Bartelc B, Barteld DP, Baulcombee D, Bowmanf JL, Caog X (2008) Criteria for annotation of plant microRNAs. Plant Cell 20:3186–3190
Mi SJ, Cai T, Hu YG, Chen Y, Hodges E, Ni FR, Wu L, Li S, Zhou H, Long CZ, Chen S, Hannon GJ, Qi YJ (2008) Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 133:116–127
Montgomery TA, Howell MD, Cuperus JT, Li D, Hansen JE, Alexander AL, Chapman EJ, Fahlgren N, Allen E, Carrington JC (2008) Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 133:128–141
Moxon S, Schwach F, Dalmay T, MacLean D, Studholme DJ, Moulton V (2008) A toolkit for analysing large-scale plant small RNA datasets. Bioinformatics 24:2252–2253
Nonomura K, Morohoshi A, Nakano M, Eiguchi M, Miyao A, Hirochika H, Kurata N (2007) A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 19:2583–2594
Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud-Nissen F, Malek RL, Lee Y, Zheng L, Orvis J, Haas B, Wortman J, Buell CR (2007) The TIGR rice genome annotation resource: improvements and new features. Nucleic Acids Res 35:D883–D887
Park W, Li J, Song R, Messing J, Chen X (2002) CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12:1484–1495
Phizicky EM, Hopper AK (2010) tRNA biology charges to the front. Genes Dev 24:1832–1860
Pua EC, Davey MR, Twell D (2010) Male gametophyte development. In: Plant developmental biology—biotechnological perspectives. Springer, Berlin, pp 225–244
Qiu CX, Wang JA, Yao PY, Wang E, Cui QH (2010) microRNA evolution in a human transcription factor and microRNA regulatory network. BMC Syst Biol 4:90
Romier C, Cocchiarella F, Mantovani R, Moras D (2003) The NF-YB/NF-YC structure gives insight into DNA binding and transcription regulation by CCAAT factor NF-Y. J Biol Chem 278:1336–1345
Ron M, Alandete Saez M, Eshed Williams L, Fletcher JC, McCormick S (2010) Proper regulation of a sperm-specific cis-nat-siRNA is essential for double fertilization in Arabidopsis. Genes Dev 24:1010–1021
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34:374–378
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK (2008) Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol 8:25
Suzuki G (2009) Recent progress in plant reproduction research: the story of the male gametophyte through to successful fertilization. Plant Cell Physiol 50:1857–1864
Thirumurugan T, Ito Y, Kubo T, Serizawa A, Kurata N (2008) Identification, characterization and interaction of HAP family genes in rice. Mol Genet Genomics 279:279–289
Van Ex F, Jacob Y, Martienssen RA (2011) Multiple roles for small RNAs during plant reproduction. Curr Opin Plant Biol 14:588–593
Vazquez F, Legrand S, Windels D (2010) The biosynthetic pathways and biological scopes of plant small RNAs. Trends Plant Sci 15:337–345
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wang YL, Juranek S, Li HT, Sheng G, Wardle GS, Tuschl T, Patel DJ (2009) Nucleation, propagation and cleavage of target RNAs in ago silencing complexes. Nature 461:754–761
Wang XS, Tong YG, Wang SH (2010) Rapid and accurate detection of plant miRNAs by liquid northern hybridization. Int J Mol Sci 11:3138–3148
Wei LQ, Xu WY, Deng ZY, Su Z, Xue Y, Wang T (2010) Genome-scale analysis and comparison of gene expression profiles in developing and germinated pollen in Oryza sativa. Bmc Genomics 11:338
Wei LQ, Yan LF, Wang T (2011) Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol 12:R53
Weijland A, Harmark K, Cool RH, Anborgh PH, Parmeggiani A (1992) Elongation factor Tu: a molecular switch in protein biosynthesis. Mol Microbiol 6:683–688
Wettenhall JM, Simpson KM, Satterley K, Smyth GK (2006) affylmGUI: A graphical user interface for linear modeling of single channel microarray data. Bioinformatics 22:897–899
Wilson ZA, Zhang DB (2009) From Arabidopsis to rice: pathways in pollen development. J Exp Bot 60:1479–1492
Wu L, Zhang QQ, Zhou HY, Ni FR, Wu XY, Qi YJ (2009) Rice MicroRNA effector complexes and targets. Plant Cell 21:3421–3435
Wu L, Zhou HY, Zhang QQ, Zhang JG, Ni FR, Liu C, Qi YJ (2010) DNA methylation mediated by a MicroRNA pathway. Mol Cell 38:465–475
Xie F, Zhang B (2010) Target-align: a tool for plant microRNA target identification. Bioinformatics 26:3002–3003
Zhang Y (2005) miRU: An automated plant miRNA target prediction server. Nucleic Acids Res 33:W701–W704
Zhou M, Gu L, Li P, Song X, Wei L, Chen Z, Cao X (2010) Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica). Front Biol 5:67–90
Zhu QH, Helliwell CA (2011) Regulation of flowering time and floral patterning by miR172. J Exp Bot 62:487–495
Zhu QH, Spriggs A, Matthew L, Fan L, Kennedy G, Gubler F, Helliwell C (2008) A diverse set of microRNAs and microRNA-like small RNAs in developing rice grains. Genome Res 18:1456–1465
Acknowledgments
This paper is supported by National Natural Science Foundation of China (No. 31070276).
Author information
Authors and Affiliations
Corresponding author
Additional information
Hua Peng, Jun Chun, Tao-bo Ai, and Yong-ao Tong contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Peng, H., Chun, J., Ai, Tb. et al. MicroRNA profiles and their control of male gametophyte development in rice. Plant Mol Biol 80, 85–102 (2012). https://doi.org/10.1007/s11103-012-9898-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11103-012-9898-x