
Abstract
Glutamate overactivity is well documented in Parkinson’s disease (PD) and dyskinesias induced by L-3,4-dihydroxyphenylalanine (L-DOPA), the gold-standard treatment for this disease. The contribution of metabotropic glutamate receptors type 5 (mGlu5 receptors) in PD and L-DOPA-induced dyskinesias (LID) was the topic of investigations in human PD patients and in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) monkeys.
Behaviorally, it has been shown that the prototypical mGlu5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) as well as the mGlu5 receptor antagonists 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) and mavoglurant (AFQ056) acutely attenuated LID in MPTP monkeys. Moreover, a chronic 1-month administration of MPEP to previously drug-naïve MPTP monkeys (de novo treatment) attenuated the development of LID. Acute and chronic MPEP treatments of MPTP monkeys maintained the antiparkinsonian effect of L-DOPA. Mavoglurant was also shown in some clinical studies to reduce LID in PD patients.
Using the selective mGlu5 receptors ligand [3H]ABP688, mGlu5 receptor-specific binding was measured by autoradiography in brains slices of normal and PD patients in relation to motor complications associated with an L-DOPA treatment. PD patients with motor complications (either LID or wearing-off) had higher [3H]ABP688-specific binding compared to those without motor complications and controls in putamen, external and internal globus pallidus. In monkeys with a MPTP lesion and controls, [3H]ABP688- and [3H]MPEP-specific bindings were elevated in the striatum of dyskinetic L-DOPA-treated MPTP monkeys but not in MPTP monkeys without LID compared to controls.
The brain molecular correlates of the long-term effect of a 1-month administration of MPEP with L-DOPA that attenuated the development of LID were shown to extend beyond mGlu5 receptors. In the basal ganglia, it has been showed that the L-DOPA-induced changes of NMDA and AMPA ionotropic glutamate receptors as well as mGlu2/mGlu3 receptors were prevented with the addition of MPEP. Moreover, MPEP normalized the L-DOPA-induced changes of dopamine D2 receptors, their associated signaling (ERK and Akt) and neuropeptides (preproenkephalin, preprodynorphin), as well as the serotonin receptors 5-HT2A and 5-HT1B.
In conclusion, these results have shown in humans and in a nonhuman primate model of PD reduction of LID with mGlu5 antagonism. In the basal ganglia, LID were associated with changes of various glutamatergic, dopaminergic, and serotoninergic markers that were normalized with adjunct treatment with an mGlu5 antagonist supporting the mGlu5 receptor as a good target for the treatment of LID.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Parkinson’s disease
- L-DOPA-induced dyskinesia
- Motor complications
- Glutamate receptor
- Basal ganglia
- Direct pathway
- Indirect pathway
- Receptor interaction
10.1 Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder and is likely to increase due to the aging population (Siderowf and Stern 2003). The cardinal motor manifestations of PD, tremor, bradykinesia, and rigidity are secondary to a loss of dopamine in the striatum (Olanow et al. 2009; Toulouse and Sullivan 2008). PD is principally attributed to the death of dopamine (DA) neurons in the substantia nigra, but other neurotransmitters and neuromodulators, such as glutamate, serotonin (5-HT), and adenosine, are also affected (Toulouse and Sullivan 2008). Gene mutations in familial PD are reported, but the cause for the majority of PD cases remains unknown (Olanow et al. 2009). There is currently no cure for PD. Neuroprotection or disease modification defined as an intervention that would protect or rescue vulnerable neurons, thereby slowing, stopping, or reversing disease progression, is not yet available for PD, but laboratory studies are finding promising agents (Olanow et al. 2009).
Restoring lost DA with its precursor, L-3,4-dihydroxyphenylalanine (L-DOPA), introduced 50 years ago still remains a very effective and commonly used treatment for PD (Mercuri and Bernardi 2005). However, up to 80% of PD patients will develop motor complications after 10 years of treatment with L-DOPA (Olanow and Koller 1998). These motor complications include motor fluctuations and abnormal involuntary movements, such as L-DOPA-induced dyskinesias (LID), and contribute to limit the quality of life in PD patients and can be very difficult to manage (Fabbrini et al. 2007). Motor fluctuations such as “wearing-off” are also common. Wearing-off is defined as a reduced duration of benefit from an individual L-DOPA dose and a recurrence of parkinsonian symptoms before the next normal dose of L-DOPA (Fahn et al. 2004). Hence, most PD patients initiated with DA agonist monotherapy will eventually require L-DOPA as disease progresses, and after 10 years their motor complications appear similar as they would have if started initially on L-DOPA therapy (Katzenschlager et al. 2008; Parkinson Study Group 2009). This suggests that disease progression plays the major role in the onset of dyskinesia rather than the type of dopaminergic drug treatment used. Involuntary movements such as LID are paralleled by aberrant forms of plasticity characterized by changes in various neurotransmitter systems and their intracellular signaling pathways (Jenner 2008).
Aside from some benefit of amantadine , that has anti-glutamatergic properties, no drug is yet available for LID (Meissner et al. 2011). Glutamate neurotransmission is increased in the basal ganglia in PD and LID (Klockgether and Turski 1993; Chase and Oh 2000; Calon et al. 2003a). The pathophysiology and the mechanisms involved in the development of LID are still not fully understood. However, altered dopaminergic and nondopaminergic neurotransmission in the basal ganglia is observed in LID (Blandini and Armentero 2012). Treating LID with adjunct drugs targeting nondopaminergic neurotransmitter systems is proposed such as glutamate to indirectly modulate basal ganglia DA neurotransmission (Brotchie 1998; Morin et al. 2014a). Much emphasis has therefore been placed on finding alternative nondopaminergic drugs that could circumvent some or all these problems. The design of novel agents to prevent dyskinesias requires elucidation of the adaptive changes produced in the parkinsonian brain by repeated administration of L-DOPA. The use of adjunct drugs to modulate basal ganglia DA neurotransmission is an important strategy to treat LID ( Brotchie 1998, 2003; Henry et al. 2001; Calon and Di Paolo 2002; Blanchet et al. 1999; Grondin et al. 1999).
LID are typically observed at the peak of the effect of L-DOPA in PD patients. There is also diphasic dyskinesia at the beginning and at the end of the L-DOPA dosing cycle appearing with the rise and fall of DA levels in the brain (Luquin et al. 1992) and off-dystonia (Marsden et al. 1982). LID occur in 30–80% of PD patients treated with L-DOPA (Barbeau 1980; Nutt 1990). Two conditions are necessary for their appearance: (1) the loss of DA in the nigrostriatal pathway and (2) treatment with L-DOPA or DA agonists. The development of dyskinesias in man usually requires daily treatment for 3–5 years in idiopathic PD (Klawans et al. 1977), and for parkinsonism induced in man by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), it occurs after only weeks or months of treatment (Ballard et al. 1985). The same applies to MPTP monkeys where only weeks of L-DOPA therapy are enough before dyskinesias appear (Falardeau et al. 1988; Bedard et al. 1986). MPTP primates respond to DA therapies in a similar manner than idiopathic PD patients (Jenner 2003a, b) and are currently the best model for studying LID.
Glutamate is involved in many physiological functions through its interactions with ionotropic glutamate (iGlu), ligand-gated channel, and metabotropic G-protein-coupled glutamate (mGlu) receptors. iGlu receptor drugs suppressing glutamate excitatory transmission often create undesirable side effects (Johnson et al. 2009), whereas acting on mGlu receptors could lead to a subtler and/or circuit-selective modulation of excitatory transmission (Conn et al. 2005). Pharmacologic characterization of mGlu5 receptors and its selective negative allosteric modulators (NAMs) shows therapeutic potential in animal models of PD (Grégoire et al. 2011; Johnston et al. 2010; Morin et al. 2010) and recently efficacy in human PD (Berg et al. 2011; Stocchi et al. 2013). mGlu5 receptors have been shown to play a crucial role in regulating L-DOPA-induced motor behavior, but the mechanisms involved remain unclear (Gasparini et al. 2013).
This review will cover relevant studies investigating mGlu5 receptor subtypes in the pathophysiology of PD and LID. Brain biochemical correlates of motor complications in PD patients and MPTP-lesioned monkeys will be reviewed (Morin et al. 2014a).
10.2 Nonhuman Primate Models of L-DOPA-Induced Dyskinesias
Similar etiology and functions of a particular human disease are two important characteristics to model a disease. The neurotoxin MPTP closely mimics both behavioral changes and cellular loss as seen in PD (Albanese et al. 1993). The administration of MPTP in primates promotes the development of rigidity, bradykinesia, and tremor, which are the primary motor features of PD. MPTP induces neuronal death of dopaminergic cells through a cascade of intracellular reactions by targeting specifically cells expressing the DA transporter (Smeyne and Jackson-Lewis 2005). MPTP-treated primates remain after nearly 30 years the gold standard for the study of PD and LID, as well as to evaluate the efficacy of novel compounds (Morin et al. 2014a). Acute and de novo experimental approaches are used to test new antiparkinsonian and antidyskinetic pharmacological agents. In acute approaches, animals are rendered parkinsonian with the administration of MPTP and then chronically treated with L-DOPA for several weeks, until they express well-established, constant, and stable LID. Then, the acute effects of compounds are tested with the co-administration of L-DOPA , and the motor behavior of the animals is evaluated (Grégoire et al. 2009, 2011; Bezard et al. 2004). This approach allows rapid testing of new compounds and its tolerability, and animals may be used for several studies (Morin et al. 2014a). These animals can also be used in a chronic treatment paradigm to measure possible development of tolerance to the investigated drug. In the de novo approach, two or more groups of never treated animals are rendered parkinsonian and then treated with L-DOPA alone or in combination with the new agent. This approach allows verifying if the compounds tested can reduce or prevent the development of LID and evaluating if the duration of the L-DOPA effect decreases over time, a motor complication also called “wearing-off” (Morin et al. 2012; Samadi et al. 2006; Rylander et al. 2010; Grégoire et al. 2008; Hadj Tahar et al. 2004). Moreover, measurements of biochemical changes in the brains are made possible if the animals are euthanized at the end of the protocol (Morin et al. 2012; Samadi et al. 2008a; Ouattara et al. 2010).
As observed in PD patients, the macaque monkey model will display its own pattern of parkinsonian symptoms (Rajput et al. 2009). Indeed, LID involves one or more parts of the body, and each of them should therefore be quantified separately (Hadj Tahar et al. 2000). Several scales are currently available to measure and quantify dyskinesia and were recently reviewed (Fox et al. 2012). Objective measures of bradykinesia with specific motor tasks are also important to separate the antidyskinetic from antiparkinsonian activity of compounds tested (Jourdain et al. 2013). Finally, monkeys will tend to exhibit a worsening of motor symptoms before and after an acute L-DOPA challenge (Kuoppamäki et al. 2002), as seen in some PD patients (Evans et al. 2012). Thus monkey model can replicate PD conditions and underlines their usefulness in the study of new treatments (Morin et al. 2014a).
Individual titration of L-DOPA is often needed to elicit the same amount of LID among the animals (Grégoire et al. 2011; Johnston et al. 2010) even if these animals are equally denervated (Guigoni et al. 2005). However, positive correlations between L-DOPA dose and the duration and the severity of LID were observed (Kuoppamäki et al. 2007). Moreover, in dyskinetic macaques, the administration of L-DOPA after drug holiday lasting few weeks will trigger the same LID as measured before. The same observation was made in PD patients in whom L-DOPA was stopped (Goetz et al. 1982; Mayeux et al. 1985). This supports the feasibility of acute studies with the same animals therefore keeping the number needed and consequently reducing the costs.
The reappearance of LID after a withdrawal indicates that permanent or at least long-term changes are occurring in the brain basal ganglia and these changes may be studied in postmortem brain tissues. Wearing-off, described as shortening in the duration of response to L-DOPA with gradual reappearance of parkinsonian symptoms, is another important motor side effect of chronic L-DOPA administration (Fahn et al. 2004). Parkinsonian patients usually experience such end-of-dose deterioration after several months or years of treatment (Pahwa and Lyons 2009). Wearing-off can be also replicated in de novo MPTP monkeys with a shortening of the antiparkinsonian effect of L-DOPA as reported after 2 weeks of treatment (Morin et al. 2013a). Thus, the MPTP-lesioned monkey model is probably one of the best models for studying LID and PD in humans, and this model has brought significant advances for the treatment of LID. For example, docosahexaenoic acid (DHA) and cabergoline were shown to reduce the severity or delay the development of LID in MPTP-lesioned monkey in chronic and de novo treatments (Samadi et al. 2006; Belanger et al. 2003).
10.3 Glutamate Neurotransmission in PD and L-DOPA-Induced Dyskinesias
Glutamate is the brain most abundant excitatory neurotransmitter mediating as much as 70% of synaptic transmission in the central nervous system, and its overactivity is well documented in PD and LID (Klockgether and Turski 1993; Morin and Di Paolo 2014; Samadi et al. 2007). Amantadine, a noncompetitive antagonist at N-methyl-D-aspartate (NMDA) receptors, is currently the only drug used in the clinic shown to reduce the severity of LID in some PD patients without worsening parkinsonian symptoms (Meissner et al. 2011; Verhagen Metman et al. 1998a; Sawada et al. 2010). Amantadine also improves akinesia, rigidity, and tremor (Olanow et al. 2009). However, the antidyskinetic effect of amantadine may be transient and is often lost within the first year of treatment (Stocchi et al. 2008). In addition, many PD patients cannot tolerate high doses of amantadine because of cognitive impairment, which significantly limits its use (Stocchi et al. 2008).
Riluzole , a nonselective inhibitor of glutamate neurotransmission, was shown to reduce the severity of L-DOPA-induced motor complications in 6-hydroxydopamine (6-OHDA)-lesioned rat model of PD (Papa et al. 1995; Marin et al. 1996; Engber et al. 1994; Marin et al. 2000). Moreover, in the same animal model, the glutamate transporter GLT1 was reported to be increased (Oueslati et al. 2007; Robelet et al. 2004). However, riluzole was not effective in humans to reduce the expression of LID (Braz et al. 2004; Bara-Jimenez et al. 2006). Despite intensive search, no drug other than amantadine has demonstrated in the clinic an antidykinesic effect that is not associated with a worsening of parkinsonism (Olanow et al. 2009) underlying the complexity of brain changes associated with dyskinesias. mGlu receptors are topics of more recent interest in PD (Samadi et al. 2007).
Inhibiting glutamate neurotransmission in PD can also be neuroprotective since glutamate overactivity is excitotoxic (Planells-Cases et al. 2006; Gardoni and Di Luca 2006). Motor complications are in part L-DOPA dose related; protecting DA neurons could delay motor complications by using less L-DOPA. Moreover, non-motor features of PD such as depression and anxiety are common (Olanow et al. 2009) and could benefit from mGlu receptor drugs such as mGlu5 receptor antagonists that show anxiolytic and antidepressant activity (Palucha and Pilc 2007; Witkin et al. 2007).
10.4 Ionotropic Glutamate Receptors and L-DOPA-Induced Dyskinesias
iGlu receptors are classified into NMDA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate (KA) receptors (Finlay and Duty 2014; Michaelis 1998; Dingledine et al. 1999), and they mediate fast excitatory neurotransmission, whereas mGlu receptors mediate slower modulatory neurotransmission (Samadi et al. 2007). The blockade of NMDA and AMPA receptors with specific antagonists was shown to reduce the development of L-DOPA-induced motor complications in 6-OHDA-lesioned rat model (Marin et al. 2000). Moreover, in both parkinsonian patients with LID (Calon et al. 2003a) and dyskinetic MPTP monkeys (Calon et al. 2002; Huot et al. 2013), important increases in striatal NMDA and AMPA receptor -binding levels were observed. The NMDA antagonist, CI-1041, is reported to prevent the development of LID in parkinsonian monkeys (Hadj Tahar et al. 2004) and associated brain molecular changes (Morissette et al. 2006). Interestingly, in these monkeys, CI-1041 also prevented the increase of striatal mGlu5 receptor levels (Ouattara et al. 2011). Clinical trials also show the antidyskinetic profile of dextrorphan, dextromethorphan, and amantadine, known to block NMDA receptors (Meissner et al. 2011; Sawada et al. 2010; Blanchet et al. 1996, 1997; Verhagen Metman et al. 1998b, 1999; Snow et al. 2000; Rajput et al. 1998; Ruzicka et al. 2000; Luginger et al. 2000).
Kynurenic acid antagonizes glycine B site of AMPA, NMDA, and KA receptors (Schwarcz and Pellicciari 2002; Hilmas et al. 2001; Stone 1993, 2000; Moroni 1999) and inhibits glutamate release (Carpenedo et al. 2001; Nemeth et al. 2006). RO 61-8048 an inhibitor of kynurenine hydroxylase activity increases kynurenic acid levels (Stone 2001). In MPTP monkeys, an acute treatment with RO 61-8048 reduced the severity of LID (Samadi et al. 2005); chronically in de novo treated MPTP monkeys , it reduced their development (Grégoire et al. 2008).
Recent studies have explored the role and the implication of NMDA and AMPA receptor subunits in rodent and nonhuman primate models of PD in LID including the glycine site, NMDA GluN2D subunits, AMPA receptor subunit composition, and NMDA/AMPA receptor ratio (Finlay and Duty 2014; Bagetta et al. 2012; Kobylecki et al. 2010; Zhang et al. 2014; Errico et al. 2011; Heresco-Levy et al. 2013). Nevertheless, significant adverse effects such as cognitive impairment in many patients can occur which limit the use of an iGlu receptor antagonist (Stocchi et al. 2008b; Stayte and Vissel 2014). Recent efforts were devoted to pharmacologically manipulate glutamate transmission with selective mGlu receptor ligands.
10.5 Metabotropic Glutamate Receptors and L-DOPA-Induced Dyskinesias
The mGlu receptors constitute a family of G-protein-coupled receptors comprising eight subtypes that are classified into three groups based on the signal transduction pathway, homology of the amino acid sequence, and receptor pharmacology (Pisani et al. 2003; Conn and Pin 1997). Group I (mGlu1, 5) couples to Gq and stimulates polyphosphoinositide (PI) hydrolysis, while groups II (mGlu2, 3) and III (mGluR4, mGluR6, mGluR7, mGluR8) couple to Gi/Go and inhibit increase in cyclic adenosine monophosphate (cAMP) (Conn and Pin 1997). All mGlu receptor subtypes are mainly located in the brain basal ganglia, except mGlu6 receptor found primarily in the retina (Niswender and Conn 2010) (Fig. 10.1). Presynaptically localized group II and group III mGlu receptors are thought to represent the classical inhibitory autoreceptor mechanism suppressing excess glutamate release from presynaptic terminals (Schoepp 2001). The majority of group I mGlu receptor, including mGlu5, is located postsynaptically on the perisynaptic annulus of dendritic spines, which lead to enhanced neuronal excitation (Lujan et al. 1997).

(a) Schematic representation of the localization of metabotropic glutamate receptor subtypes (groups I, II, and II) in the basal ganglia motor circuit. mGlu6 receptor subtype (group III) is absent from the figure since it is present only in the retina (Conn et al. 2005; Rascol et al. 2014; Amalric 2015). In the classical and simplified pathophysiological model (not mentioning the collateral projections from the direct to indirect pathway) of the basal ganglia in PD, reduced dopamine input by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) has a dual effect on the striatal efferents that project to the GPe (indirect pathway) and the GPi (direct pathway). Increased or facilitated neuronal activity of the indirect pathway, via decrease of the D2 receptor inhibition, increased activity of striatal GABA neurons projecting to the GPe. Over-inhibition of GABA neurons in the GPe disinhibits the STN, which, in turn, overdrives inhibitory output neurons in the GPi and SNr projecting to the thalamus, overall resulting in a decrease in thalamocortical input. (b) Postsynaptic receptors on striatal GABA efferences susceptible to be antagonized to counteract excessive corticostriatal glutamate transmission (absence of glutamate release regulation by D2 autoreceptors on corticostriatal neurons) seen in PD state in order to normalize activities of the direct and indirect pathway. Among these, mGlu5 seems to be a relevant target since this receptor acts primarily as modulator of synaptic activity. The double arrow indicates that these receptors can form complexes (heteroreceptor) providing a high degree of complexity and plasticity at different levels of basal ganglia circuitry (Fuxe et al. 2015)
While the term “antagonist ” is widely used when applied to ligands that inhibit the function of specific mGlu receptors, the more appropriate term is “negative allosteric modulator” (NAM ), since they inhibit the function of the receptor at a site distal to the actual orthosteric ligand-binding domain of the receptor and only in the presence of glutamate (Olive 2009). Group I NAM may also function as inverse agonists; in cell-based assays they can inhibit the basal (constitutive) activity of group I mGlu receptors in absence of any orthosteric agonist or even when the glutamate-binding domain is removed or mutated (Olive 2009). Moreover, mGlu receptors have a higher affinity for glutamate than iGlu receptors (Conn et al. 2005; Marino et al. 2002). On the basis of these considerations, combined with the rich distribution and diverse physiological roles of mGlu receptors within the basal ganglia, recent attention has been placed on these receptors as alternative targets to modulate glutamate hyperactivity in PD and LID (Conn et al. 2005). Studies in animal models and PD patients indicate that antagonists of group I mGlu receptor, especially mGlu5 receptor, could be considered as a suitable therapeutic approach in PD and LID.
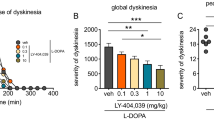
mGlu5 receptor levels were shown to be increased in the putamen of dyskinetic compared to non-dyskinetic MPTP monkeys (Samadi et al. 2008) and parkinsonian patients with motor complications (LID or wearing-off) compared to those without motor complications (Ouattara et al. 2011). Moreover, the prototypal mGlu5 receptor antagonist, 2-methyl-6-(phenylethynyl)pyridine (MPEP), and a more selective analog 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) (Carroll 2008) improved motor performance (Breysse et al. 2002) and showed antidyskinetic activity in 6-OHDA rat model (Levandis et al. 2008; Mela et al. 2007). However, the other group I mGlu receptor drugs were not effective (Dekundy et al. 2006; Rylander et al. 2009). In acute treatment in already dyskinetic MPTP-lesioned monkeys, the mGlu5 receptor antagonists MPEP, MTEP, fenobam, and AFQ056 (mavoglurant) were found effective to reduce the severity of LID (Morin et al. 2010; Rylander et al. 2010; Gregoire et al. 2011; Johnston et al. 2010). In a de novo treatment, MPEP reduced the development of L-DOPA-induced motor complications (~70%) in a chronic administration of 1 month (Morin et al. 2013a). Similarly, chronic administration of fenobam to drug-naïve monkeys attenuated the development of dyskinesia without compromising the antiparkinsonian effect of L-DOPA (Rylander et al. 2010). Moreover, the mGlu5 receptor antagonists, mavoglurant and ADX-48621 (dipraglurant), were shown to reduce LID in parkinsonian patients and were well tolerated without worsening motor symptoms (Addex Therapeutics 2012; Stocchi et al. 2013; Berg et al. 2011). For more information the reader is referred to a recent review on the use of mGlu5 antagonists for treatment of L-DOPA-induced dyskinesias including a comparison the clinical trials with mavoglurant and dipraglurant. (Rascol et al. 2014).
Group II mGlu receptor agonists have also proven effective in animal models of PD (Pisani et al. 2003). Moreover, an important decrease in mGlu2/mGlu3 receptor density in dyskinetic compared to non-dyskinetic MPTP-lesioned monkeys was observed (Morin et al. 2013b). Furthermore, changes in mGlu2/mGlu3 receptors were only observed in relation to wearing-off in postmortem brains of parkinsonian patients (Samadi et al. 2009).
More recently, agonists of group III receptors were also shown effective in rodent models of PD (Niswender and Conn 2010). In 6-OHDA-lesioned rat model, the administration of mGlu4 receptor agonist Lu AF21934 combined with L-DOPA treatment reduced the effective dose of L-DOPA and reduced the development of LID (Bennouar et al. 2013). mGlu4 receptor agonists can reduce γ-aminobutyric acid (GABA)ergic neurotransmission at striato-pallidal synapse that is overactive in PD (Macinnes and Duty 2008; Matsui and Kita 2003).
mGlu8 receptor is expressed at lower levels than mGlu4 and mGlu7 receptors but widely distributed in the brain; mGlu7 receptor has low affinity for glutamate only becoming active when glutamate levels are high thus serving as a brake for glutamate overstimulation (Niswender and Conn 2010). AMN082, an mGlu7 receptor agonist, was shown to reverse motor dysfunction associated with reduced DA activity in rodent models (Greco et al. 2010). However, the contribution of mGlu7 and mGlu8 receptors in LID is not yet reported.
10.6 The Effect of a Chronic Treatment with MPEP on L-DOPA-Induced Dyskinesias and Brain Biochemical Borrelates
The mechanisms underlying the development of LID are still unknown, but evidence suggests that LID is the result of maladaptive plasticity at striatal synapses (Cenci and Lundblad 2006; Jenner 2008; Calabresi et al. 2010; Iravani et al. 2012). Altered dopaminergic and nondopaminergic neurotransmission in the basal ganglia is observed in LID (Blandini and Armentero 2012). Glutamate neurotransmission plays a crucial role in the modulation of corticostriatal inputs and striatal output to downstream nuclei of the basal ganglia circuit (Blandini and Armentero 2012). Increased glutamate transmission involves presynaptic changes, such as increased striatal concentration of extracellular glutamate (Dupre et al. 2011) or changes in mGlu2/mGlu3 receptors (Samadi et al. 2008; Gregoire et al. 2009). Postsynaptic changes in AMPA, NMDA, and mGlu5 receptors also seem to play a major role in the development of LID (Duty 2012). mGlu5 receptors are highly expressed in the striatum and other basal ganglia nuclei including subthalamic nucleus (STN), substantia nigra, and globus pallidus (GP) (Ferraguti and Shigemoto 2006). Numerous interactions between mGlu5 receptor and NMDA, D1 DA, D2 DA, and A2A adenosine receptors suggest that these receptors may function together as closely associated signaling partners in the appearance of LID (Samadi et al. 2007). mGlu5 receptor activation of NMDA receptors (Pisani et al. 2001) as well as NMDA and mGlu5 receptor co-localization is described (Perroy et al. 2008). The blockade of mGlu5 receptors leads to antidyskinetic actions, which is associated with normalization of firing of striatal signals (Duty 2012). Moreover, chronic mGluR5 NAM treatments are reported to protect DA neurons from MPTP toxicity in mice (Battaglia et al. 2004) and monkeys (Masilamoni 2009). Moreover, Norbin, a neuron-specific protein, can physically interact in vivo with mGlu5 receptors, increases the cell surface localization of the receptor, and positively regulates mGlu5 signaling while maintaining the total amount of this receptor unchanged (Wang et al. 2009). Intracellular mGlu5 receptor was reported to activate signaling cascades distinct from cell surface counterparts (Jong et al. 2009). Thus there is still much to be learned on mGuR5 receptors and their regulation.
We reported that development of LID over a month of treatment was lower by overall ~70% with addition of MPEP to the L-DOPA treatment in de novo MPTP monkeys (Morin et al. 2013a), and this was associated with a normalization of glutamate (Morin et al. 2013b), DA (Morin et al. 2014b), and 5-HT neurotransmission (Morin et al. 2015).
More precisely, in the brain basal ganglia of these monkeys (Morin et al. 2013a), the addition of MPEP to L-DOPA treatment prevented the increase of postsynaptic mGlu5, NMDA NR1/NR2B, and AMPA glutamate receptors, while this treatment prevented the decrease of mGlu2/mGlu3 presynaptic autoreceptors (Morin et al. 2013b). The mGlu5 receptor subtype is highly expressed in striatal medium spiny neurons (Conn et al. 2005; Testa et al. 1994; Paquet and Smith 2003) and plays a key role in modulating the responses mediated by NMDA receptors and L-type calcium channels (Gubellini et al. 2004). In addition, an antagonistic interaction between the D2 DA receptor and mGlu5 receptors is reported (Fuxe et al. 2008). In rodent models of PD, striatal molecular changes relevant to LID are reported to be reversed by MPEP or MTEP, including delta FosB protein (Jimenez et al. 2009), prodynorphin mRNA (Mela et al. 2007), glutamic acid decarboxylase (GAD65 and GAD67) mRNA (Yamamoto and Soghomonian 2009), and phosphorylated extracellular signal-regulated kinases 1 and 2 (ERK1/ERK2) protein levels (Rylander et al. 2009). Hence, mGlu5 receptor antagonists reverse hyperactive GABAergic transmission in the basal ganglia in rodent models of PD and its downstream molecular changes associated with LID. Hence, mGlu5 receptor antagonist reversed hyperactive glutamate transmission in the basal ganglia of a primate model of PD thus showing the widespread normalization activity of mGlu5 receptor antagonists in the basal ganglia in PD animal models (Morin et al. 2013a).
Moreover, a chronic treatment with MPEP prevented the decrease of D2 DA receptors, but it did not affect the D1 DA receptors (Morin et al. 2014b). MPEP also prevented the increase of striatal preproenkephalin/preprodynorphin mRNA levels and phosphorylated proteins ERK1/ERK2 as well as protein kinase B (Akt) and glycogen synthase kinase-3 (GSK3β) (Morin et al. 2014b). Denervation-induced supersensitivity of D1 and D2 receptors was initially recognized as a plausible mechanism of LID (Creese et al. 1977; Lee et al. 1978). Numerous studies measured the density of D1 and D2 receptors in the brain of human and animal models, but no general consensus emerged. Postmortem studies have shown that striatal DA receptors particularly the D2 subtype were increased in PD patients (Lee et al. 1978; Guttman et al. 1986) or unchanged (Quik et al. 1979; Rinne et al. 1981), while both D1 and D2 receptor subtypes were increased in MPTP monkeys (Falardeau et al. 1988; Bedard et al. 1986; Gagnon et al. 1990). Administration of L-DOPA was shown to reverse these increases in PD patients (Lee et al. 1978; Guttman et al. 1986) and primates in many studies (Falardeau et al. 1988; Gagnon et al. 1990; Berretta et al. 1997). No general consensus also emerged for DA receptor mRNA ( Goulet et al. 1997, 2000; Morissette et al. 1996; Aubert et al. 2005; Herrero et al. 1996). These reports support that LID are more complex than hypersensitivity due to a simple increase in the density of striatal DA receptors and its mRNA. MPEP did not affect D1 receptor levels and its mRNA but was associated with an increased in D2 receptors levels, its mRNA, and its associated signaling proteins (Morin et al. 2014b). Furthermore, mGlu receptors also have the potential to regulate the mitogen-activated protein kinase (MAPK) pathway . It has been shown that intracaudate injection of a group I mGlu receptor agonist upregulates ERK1/ERK2 phosphorylation (Choe and Wang 2001). mGlu5 receptor stimulation was also reported to lead to activation of other signaling pathways important for cell survival/proliferation, such as ERK and Akt (Mao et al. 2005).
The antidyskinetic effect of MPEP was associated with lower levels of 5-HT2A (Morin et al. 2015) and 5-HT1B (Morin et al. 2015) serotonin receptors, while 5-HT1A receptors and brain serotonin transporter (SERT) remained unaffected (Morin et al. 2015). LID are probably the result of an abnormal adaptation in the striatum due to faulty interaction between glutamate/5-HT and DA inputs in the nigrostriatal pathway. 5-HT neuron terminals in the striatum are suggested to participate in the mechanisms of action of L-DOPA (Navailles and De Deurwaerdere 2012). 5-HT axon terminals can release DA, which is considered as a false neurotransmitter and is probably one of the main presynaptic determinants of LID (Carta et al. 2007). Multiple changes in the basal ganglia glutamate and serotoninergic systems and their specific receptors have been observed (Huot et al. 2013). 5-HT receptor drugs are reported to reduce LID in PD patients (Fox et al. 2009), but changes in 5-HT activity and 5-HT receptors in the brain caused by PD and its pharmacological treatment are not completely understood, and further studies are needed.
Stimulation of 5-HT1B receptors was also shown to reduce dyskinesias induced by D1 DA receptor agonists (Jaunarajs et al. 2009). This observation suggests that in order to prevent activation of D1-expressing neurons, an increase of the inhibitory 5-HT1B heteroreceptors might represent another compensatory phenomenon to prevent activation of the direct pathway neurons, thought to be involved in the expression of LID (Greengard 2001). Moreover, an increase of 5-HT1B autoreceptors might also play a crucial role to regulate or inhibit the release of DA by 5-HT axon terminals. 5-HT axon terminals have been suggested to release DA in a nonphysiological manner following L-DOPA treatment, leading to the development of LID (Carta et al. 2007). Hence, 5-HT neurons can take up L-DOPA and convert it into DA where it is sequestered and stored for subsequent release (Maeda et al. 2005). However, 5-HT neurons lack a DA transporter and D2 DA autoreceptors, thus leading to excessive nonphysiological DA efflux that promotes LID (Navailles and De Deurwaerdere 2012). Hence, multiple changes in the basal ganglia dopaminergic, glutamate and serotoninergic systems, and their specific receptors have been observed such as modulation in the expression and the activity of subtypes of receptors, G proteins, effectors, transcription factors, and protein kinases (Samadi et al. 2007; Huot et al. 2013).
Moreover, functional interactions between 5-HT1B receptors and GABA should also be considered in the molecular mechanisms of LID. It was reported that L-DOPA administration or stimulation of D1 receptors in 6-OHDA-lesioned animals increases GABA levels in the substantia nigra pars reticulata (Aceves et al. 1992; Ochi et al. 2004; Yamamoto et al. 2006). GABA concentrations were reported to be high in the striatum of PD patients compared to controls (Kish et al. 1986), and changes in GABAA and GABAB receptor levels were also previously observed in the same PD patients, as presented in the present study, with motor complications as compared to those without and controls (Calon et al. 2003b). Hence, 5-HT1B inhibitory receptors located on GABAergic axons of striatal neurons are probably implicated in the alterations of GABA receptors and GABA concentrations measured in these brain areas (Castro et al. 1998). Hence, activation of 5-HT1B receptors could reduce GABA release (Morikawa et al. 2000; Stanford and Lacey 1996). Thus, this upregulation of 5-HT1B receptors in the basal ganglia of dyskinetic monkeys and PD patients with motor complications could be a compensatory mechanism in order to normalize GABA concentrations.
Thus, these reported results suggest that the prevention of the development of motor complications with an mGlu5 receptor antagonist was associated with the normalization of important markers and receptors of glutamate, DA, and 5-HT neurotransmission, supporting the therapeutic use of an mGlu5 receptor antagonist to treat LID.
10.7 Conclusion
Overall, these studies suggest that glutamate receptor stimulation is involved in the pathogenesis of L-DOPA-induced motor complications in PD, and some receptor subtypes, such as mGlu5 receptor subtypes, are potential selective targets for treatment of these adverse effects (Blanchet et al. 1997; Chase and Oh 2000; Verhagen Metman et al. 2000; Montastruc et al. 1997). mGlu5 receptor antagonists are very promising drugs for the management of various brain disorders, and one of its potential applications is LID therapy. Therefore, it is essential to know the long-term behavioral effects of this class of compounds, and ensuing possible biochemical adaptations of the brain, it is critical to learn as much as possible from MPTP primate models, which closely mimic human PD conditions (Morin et al. 2014a). The mGlu5 antagonist treatment in parkinsonian primates and rats affects not only glutamate receptors but also DA and 5-HT receptors. This may be indirect by restoring glutamate neurotransmission but could also involve the direct interactions of the trio mGlu5-D2-A2A receptor cross talk (Fiorentini et al. 2013; Fuxe et al. 2007). Moreover, supporting a close mGlu5-A2A receptor interaction , MTEP treatment was reported to decrease mice brain A2A receptor-specific binding and regulate the conditioned effects of cocaine (Brown et al. 2012). The implication of these receptor interaction s in mental and neurodegenerative diseases and more specifically in the development and expression of PD symptoms and LID needs further investigation to find novel targets and, ultimately, novel pharmacological treatments.
Abbreviations
- 5-HT:
-
serotonin
- 6-OHDA:
-
6-hydroxydopamine
- Akt:
-
protein kinase B
- AMPA:
-
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- cAMP:
-
cyclic adenosine monophosphate
- DA:
-
dopamine
- DHA:
-
docosahexaenoic acid
- dipraglurant:
-
ADX-48621
- ERK:
-
extracellular signal-regulated kinase
- GABA:
-
γ-aminobutyric acid
- GAD:
-
glutamic acid decarboxylase
- GSK3β:
-
glycogen synthase kinase-3 β
- GP:
-
globus pallidus;
- iGlu:
-
ionotropic glutamate
- KA:
-
kainate
- L-DOPA:
-
levodopa (L-3,4-dihydroxyphenylalanine)
- LID:
-
L-DOPA-induced dyskinesias
- MAPK:
-
mitogen-activated protein kinase
- mavoglurant:
-
AFQ056
- mGlu:
-
metabotropic glutamate
- MPEP:
-
2-methyl-6-(phenylethynyl)pyridine
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MTEP:
-
3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine
- NAM:
-
negative allosteric modulator
- NMDA:
-
N-methyl-D-aspartate
- PD:
-
Parkinson’s disease
- PI:
-
polyphosphoinositide
- SERT:
-
serotonin transporter
- STN:
-
subthalamic nucleus
References
Aceves J, Floran B, Martinez-Fong D, Benitez J, Sierra A, Flores G (1992) Activation of D1 receptors stimulates accumulation of gamma-aminobutyric acid in slices of the pars reticulata of 6-hydroxydopamine-lesioned rats. Neurosci Lett 145(1):40–42
Addex Therapeutics (2012) Geneva, Switzerland [media release]. 2012 Mar 21 [online]. Available from URL: http://www.addextherapeutics.com/investors/press-releases/news-details/article/addex-reports-positive-top-line-phase-iia-data-for-dipraglurant-in-parkinsons-disease-levodopa-indu/. Accessed 11 Feb 2013
Albanese A, Granata R, Gregori B, Piccardi MP, Colosimo C, Tonali P (1993) Chronic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine to monkeys: behavioural, morphological and biochemical correlates. Neuroscience 55(3):823–832
Amalric M (2015) Targeting metabotropic glutamate receptors (mGluRs) in Parkinson’s disease. Curr Opin Pharmacol 20:29–34
Aubert I, Guigoni C, Hakansson K, Li Q, Dovero S, Barthe N et al (2005) Increased D1 dopamine receptor signaling in levodopa-induced dyskinesia. Ann Neurol 57(1):17–26
Bagetta V, Sgobio C, Pendolino V, Del Papa G, Tozzi A, Ghiglieri V et al (2012) Rebalance of striatal NMDA/AMPA receptor ratio underlies the reduced emergence of dyskinesia during D2-like dopamine agonist treatment in experimental Parkinson’s disease. J Neurosci 32(49):17921–17931
Ballard PA, Tetrud JW, Langston JW (1985) Permanent human parkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): seven cases. Neurology 35(7):949–956
Bara-Jimenez W, Dimitrova TD, Sherzai A, Aksu M, Chase TN (2006) Glutamate release inhibition ineffective in levodopa-induced motor complications. Mov Disord Off J Mov Disord Soc 21(9):1380–1383
Barbeau A (1980) High level levodopa therapy in several akinetic parkinsonian patients: twelve years later. In: Rinne JK, Klinger H, Stamm G (eds) Parkinson’s disease: current progress, problems and management. Elsevier, Amsterdam, pp 229–239
Battaglia G, Busceti CL, Molinaro G, Biagioni F, Storto M, Fornai F et al (2004) Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J Neurosci Off J Soc Neurosci 24(4):828–835
Bedard PJ, Di Paolo T, Falardeau P, Boucher R (1986) Chronic treatment with L-DOPA, but not bromocriptine induces dyskinesia in MPTP-parkinsonian monkeys. Correlation with [3H]spiperone binding. Brain Res 379(2):294–299
Belanger N, Gregoire L, Hadj Tahar A, Bedard PJ (2003) Chronic treatment with small doses of cabergoline prevents dopa-induced dyskinesias in parkinsonian monkeys. Mov Disord Off J Mov Disord Soc 18(12):1436–1441
Bennouar KE, Uberti MA, Melon C, Bacolod MD, Jimenez HN, Cajina M et al (2013) Synergy between L-DOPA and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson’s disease treatment and dyskinesia. Neuropharmacology 66:158–169
Berg D, Godau J, Trenkwalder C, Eggert K, Csoti I, Storch A et al (2011) AFQ056 treatment of levodopa-induced dyskinesias: results of 2 randomized controlled trials. Mov Disord 26(7):1243–1250
Berretta S, Parthasarathy HB, Graybiel AM (1997) Local release of GABAergic inhibition in the motor cortex induces immediate-early gene expression in indirect pathway neurons of the striatum. J Neurosci Off J Soc Neurosci 17(12):4752–4763
Bezard E, Hill MP, Crossman AR, Brotchie JM, Michel A, Grimée R et al (2004) Levetiracetam improves choreic levodopa-induced dyskinesia in the MPTP-treated macaque. Eur J Pharmacol 485(1-3):159–164
Blanchet PJ, Metman LV, Mouradian MM, Chase TN (1996) Acute pharmacologic blockade of dyskinesias in Parkinson’s disease. Mov Disord Off J Mov Disord Socy 11(5):580–581
Blanchet PJ, Papa SM, Metman LV, Mouradian MM, Chase TN (1997) Modulation of levodopa-induced motor response complications by NMDA antagonists in Parkinson’s disease. Neurosci Biobehav Rev 21(4):447–453
Blanchet PJ, Konitsiotis S, Whittemore ER, Zhou ZL, Woodward RM, Chase TN (1999) Differing effects of N-methyl-D-aspartate receptor subtype selective antagonists on dyskinesias in levodopa-treated 1-methyl-4-phenyl- tetrahydropyridine monkeys. J Pharmacol Exp Ther 290(3):1034–1040
Blandini F, Armentero MT (2012) New pharmacological avenues for the treatment of L-DOPA-induced dyskinesias in Parkinson’s disease: targeting glutamate and adenosine receptors. Expert Opin Investig Drugs 21(2):153–168
Braz CA, Borges V, Ferraz HB (2004) Effect of riluzole on dyskinesia and duration of the on state in Parkinson disease patients: a double-blind, placebo-controlled pilot study. Clin Neuropharmacol 27(1):25–29
Breysse N, Baunez C, Spooren W, Gasparini F, Amalric M (2002) Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of parkinsonism. J Neurosci Off J Soc Neurosci 22(13):5669–5678
Brotchie JM (1998) Adjuncts to dopamine replacement: a pragmatic approach to reducing the problem of dyskinesia in Parkinson’s disease. Mov Disord 13(6):871–876
Brotchie JM (2003) CB1 cannabinoid receptor signalling in Parkinson’s disease. Curr Opin Pharmacol 3(1):54–61
Brown RM, Duncan JR, Stagnitti MR, Ledent C, Lawrence AJ (2012) mGlu5 and adenosine A2A receptor interactions regulate the conditioned effects of cocaine. Int J Neuropsychopharmacol/Off Sci J Coll Int Neuropsychopharmacol 15(7):995–1001
Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B (2010) Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol 9(11):1106–1117
Calon F, Di Paolo T (2002) Levodopa response motor complications -GABA receptors and preproenkephalin expression in human brain. Parkinsonism Relat Disord 8(6):449–454
Calon F, Morissette M, Ghribi O, Goulet M, Grondin R, Blanchet PJ et al (2002) Alteration of glutamate receptors in the striatum of dyskinetic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys following dopamine agonist treatment. Prog Neuro-Psychopharmacol Biol Psychiatry 26(1):127–138
Calon F, Rajput AH, Hornykiewicz O, Bedard PJ, Di Paolo T (2003a) Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol Dis 14(3):404–416
Calon F, Morissette M, Rajput AH, Hornykiewicz O, Bedard PJ, Di Paolo T (2003b) Changes of GABA receptors and dopamine turnover in the postmortem brains of parkinsonians with levodopa-induced motor complications. Mov Disord Off J Mov Disord Soc 18(3):241–253
Carpenedo R, Pittaluga A, Cozzi A, Attucci S, Galli A, Raiteri M et al (2001) Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur J Neurosci 13(11):2141–2147
Carroll FI (2008) Antagonists at metabotropic glutamate receptor subtype 5: structure activity relationships and therapeutic potential for addiction. Ann N Y Acad Sci 1141:221–232
Carta M, Carlsson T, Kirik D, Bjorklund A (2007) Dopamine released from 5-HT terminals is the cause of L-DOPA-induced dyskinesia in parkinsonian rats. Brain J Neurol 130(Pt 7):1819–1833
Castro ME, Pascual J, Romon T, Berciano J, Figols J, Pazos A (1998) 5-HT1B receptor binding in degenerative movement disorders. Brain Res 790(1-2):323–328
Cenci MA, Lundblad M (2006) Post- versus presynaptic plasticity in L-DOPA-induced dyskinesia. J Neurochem 99(2):381–392
Chase TN, Oh JD (2000) Striatal mechanisms and pathogenesis of parkinsonian signs and motor complications. Ann Neurol 47(4 Suppl 1):S122–S129. discussion S9-30
Choe ES, Wang JQ (2001) Group I metabotropic glutamate receptor activation increases phosphorylation of cAMP response element-binding protein, Elk-1, and extracellular signal-regulated kinases in rat dorsal striatum. Brain Res Mol Brain Res 94(1-2):75–84
Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37:205–237
Conn PJ, Battaglia G, Marino MJ, Nicoletti F (2005) Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci 6(10):787–798
Creese I, Burt DR, Snyder SH (1977) Dopamine receptor binding enhancement accompanies lesion-induced behavioral supersensitivity. Science 197(4303):596–598
Dekundy A, Pietraszek M, Schaefer D, Cenci MA, Danysz W (2006) Effects of group I metabotropic glutamate receptors blockade in experimental models of Parkinson’s disease. Brain Res Bull 69(3):318–326
Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51(1):7–61
Dupre KB, Ostock CY, Eskow Jaunarajs KL, Button T, Savage LM, Wolf W et al (2011) Local modulation of striatal glutamate efflux by serotonin 1A receptor stimulation in dyskinetic, hemiparkinsonian rats. Exp Neurol 229(2):288–299
Duty S (2012) Targeting glutamate receptors to tackle the pathogenesis, clinical symptoms and levodopa-induced dyskinesia associated with Parkinson’s disease. CNS drugs 26(12):1017–1032
Engber TM, Papa SM, Boldry RC, Chase TN (1994) NMDA receptor blockade reverses motor response alterations induced by levodopa. Neuroreport 5(18):2586–2588
Errico F, Bonito-Oliva A, Bagetta V, Vitucci D, Romano R, Zianni E et al (2011) Higher free D-aspartate and N-methyl-D-aspartate levels prevent striatal depotentiation and anticipate L-DOPA-induced dyskinesia. Exp Neurol 232(2):240–250
Evans AH, Farrell MJ, Gibson SJ, Helme RD, Lim SY (2012) Dyskinetic patients show rebound worsening of affect after an acute L-dopa challenge. Parkinsonism Relat Disord 18(5):514–519
Fabbrini G, Brotchie JM, Grandas F, Nomoto M, Goetz CG (2007) Levodopa-induced dyskinesias. Mov Disord 22(10):1379–1389. quiz 523
Fahn S, Oakes D, Shoulson I, Kieburtz K, Rudolph A, Lang A et al (2004) Levodopa and the progression of Parkinson’s disease. N Engl J Med 351(24):2498–2508
Falardeau P, Bouchard S, Bedard PJ, Boucher R, Di Paolo T (1988) Behavioral and biochemical effect of chronic treatment with D-1 and/or D-2 dopamine agonists in MPTP monkeys. Eur J Pharmacol 150(1-2):59–66
Ferraguti F, Shigemoto R (2006) Metabotropic glutamate receptors. Cell Tissue Res 326(2):483–504
Finlay C, Duty S (2014) Therapeutic potential of targeting glutamate receptors in Parkinson’s disease. J Neural Transm 121(8):861–880
Fiorentini C, Savoia P, Savoldi D, Missale C (2013) Receptor heteromers in Parkinson’s disease and L-DOPA-induced dyskinesia. CNS Neurol Disord Drug Targets 12(8):1101–1113
Fox SH, Chuang R, Brotchie JM (2009) Serotonin and Parkinson’s disease: on movement, mood, and madness. Mov Disord Off J Mov Disord Soc 24(9):1255–1266
Fox SH, Johnston TH, Li Q, Brotchie J, Bezard E (2012) A critique of available scales and presentation of the non-human primate dyskinesia rating scale. Mov Disord Off J Mov Disord Soc 27(11):1373–1378
Fuxe K, Ferre S, Genedani S, Franco R, Agnati LF (2007) Adenosine receptor-dopamine receptor interactions in the basal ganglia and their relevance for brain function. Physiol Behav 92(1-2):210–217
Fuxe K, Marcellino D, Rivera A, Diaz-Cabiale Z, Filip M, Gago B et al (2008) Receptor-receptor interactions within receptor mosaics. Impact on neuropsychopharmacology. Brain Res Rev 58(2):415–452
Fuxe K, Guidolin D, Agnati LF, Borroto-Escuela DO (2015) Dopamine heteroreceptor complexes as therapeutic targets in Parkinson’s disease. Expert Opin Ther Targets 19(3):377–398
Gagnon C, Bedard PJ, Di Paolo T (1990) Effect of chronic treatment of MPTP monkeys with dopamine D-1 and/or D-2 receptor agonists. Eur J Pharmacol 178(1):115–120
Gardoni F, Di Luca M (2006) New targets for pharmacological intervention in the glutamatergic synapse. Eur J Pharmacol 545(1):2–10
Gasparini F, Di Paolo T, Gomez-Mancilla B (2013) Metabotropic glutamate receptors for Parkinson’s disease therapy. Park Dis 2013:196028
Goetz CG, Tanner CM, Klawans HL (1982) Drug holiday in the management of Parkinson disease. Clin Neuropharmacol 5(4):351–364
Goulet M, Morissette M, Calon F, Blanchet PJ, Falardeau P, Bedard PJ et al (1997) Continuous or pulsatile chronic D2 dopamine receptor agonist (U91356A) treatment of drug-naive 4-phenyl-1,2,3,6-tetrahydropyridine monkeys differentially regulates brain D1 and D2 receptor expression: in situ hybridization histochemical analysis. Neuroscience 79(2):497–507
Goulet M, Grondin R, Morissette M, Maltais S, Falardeau P, Bedard PJ et al (2000) Regulation by chronic treatment with cabergoline of dopamine D1 and D2 receptor levels and their expression in the striatum of Parkinsonian-monkeys. Prog Neuro-Psychopharmacol Biol Psychiatry 24(4):607–617
Greco B, Lopez S, van der Putten H, Flor PJ, Amalric M (2010) Metabotropic glutamate 7 receptor subtype modulates motor symptoms in rodent models of Parkinson’s disease. J Pharmacol Exp Ther 332(3):1064–1071
Greengard P (2001) The neurobiology of slow synaptic transmission. Science 294(5544):1024–1030
Grégoire L, Rassoulpour A, Guidetti P, Samadi P, Bédard PJ, Izzo E et al (2008) Prolonged kynurenine 3-hydroxylase inhibition reduces development of levodopa-induced dyskinesias in parkinsonian monkeys. Behav Brain Res 186(2):161–167
Grégoire L, Samadi P, Graham J, Bédard PJ, Bartoszyk GD, Di Paolo T (2009) Low doses of sarizotan reduce dyskinesias and maintain antiparkinsonian efficacy of L-Dopa in parkinsonian monkeys. Parkinsonism Relat Disord 15(6):445–452
Gregoire L, Morin N, Ouattara B, Gasparini F, Bilbe G, Johns D et al (2011) The acute antiparkinsonian and antidyskinetic effect of AFQ056, a novel metabotropic glutamate receptor type 5 antagonist, in L-Dopa-treated parkinsonian monkeys. Parkinsonism Relat Disord 17(4):270–276
Grondin R, Bedard PJ, Hadj Tahar A, Gregoire L, Mori A, Kase H (1999) Antiparkinsonian effect of a new selective adenosine A2A receptor antagonist in MPTP-treated monkeys. Neurology 52(8):1673–1677
Gubellini P, Pisani A, Centonze D, Bernardi G, Calabresi P (2004) Metabotropic glutamate receptors and striatal synaptic plasticity: implications for neurological diseases. Prog Neurobiol 74(5):271–300
Guigoni C, Dovero S, Aubert I, Li Q, Bioulac BH, Bloch B et al (2005) Levodopa-induced dyskinesia in MPTP-treated macaques is not dependent on the extent and pattern of nigrostrial lesioning. Eur J Neurosci 22(1):283–287
Guttman M, Seeman P, Reynolds GP, Riederer P, Jellinger K, Tourtellotte WW (1986) Dopamine D2 receptor density remains constant in treated Parkinson’s disease. Ann Neurol 19(5):487–492
Hadj Tahar A, Grégoire L, Bangassoro E, Bédard PJ (2000) Sustained cabergoline treatment reverses levodopa-induced dyskinesias in parkinsonian monkeys. Clin Neuropharmacol 23(4):195–202
Hadj Tahar A, Grégoire L, Darré A, Bélanger N, Meltzer L, Bédard PJ (2004) Effect of a selective glutamate antagonist on L-dopa-induced dyskinesias in drug-naive parkinsonian monkeys. Neurobiol Dis 15(2):171–176
Henry B, Fox SH, Crossman AR, Brotchie JM (2001) Mu- and delta-opioid receptor antagonists reduce levodopa-induced dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Exp Neurol 171(1):139–146
Heresco-Levy U, Shoham S, Javitt DC (2013) Glycine site agonists of the N-methyl-D-aspartate receptor and Parkinson’s disease: a hypothesis. Mov Disord 28(4):419–424
Herrero MT, Augood SJ, Asensi H, Hirsch EC, Agid Y, Obeso JA et al (1996) Effects of L-DOPA-therapy on dopamine D2 receptor mRNA expression in the striatum of MPTP-intoxicated parkinsonian monkeys. Brain Res Mol Brain Res 42(1):149–155
Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX (2001) The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci Off J Soc Neurosci 21(19):7463–7473
Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM (2013) The pharmacology of L-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol Rev 65(1):171–222
Iravani MM, McCreary AC, Jenner P (2012) Striatal plasticity in Parkinson’s disease and L-dopa induced dyskinesia. Parkinsonism Relat Disord 18(Suppl 1):S123–S125
Jaunarajs KL, Dupre KB, Steiniger A, Klioueva A, Moore A, Kelly C et al (2009) Serotonin 1B receptor stimulation reduces D1 receptor agonist-induced dyskinesia. Neuroreport 20(14):1265–1269
Jenner P (2003a) The contribution of the MPTP-treated primate model to the development of new treatment strategies for Parkinson’s disease. Parkinsonism Relat Disord 9(3):131–137
Jenner P (2003b) The MPTP-treated primate as a model of motor complications in PD: primate model of motor complications. Neurology 61(6 Suppl 3):S4–11
Jenner P (2008) Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci 9(9):665–677
Jimenez A, Bonastre M, Aguilar E, Marin C (2009) Effect of the metabotropic glutamate antagonist MPEP on striatal expression of the Homer family proteins in levodopa-treated hemiparkinsonian rats. Psychopharmacology 206(2):233–242
Johnson KA, Conn PJ, Niswender CM (2009) Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol Disord Drug Targets 8(6):475–491
Johnston TH, Fox SH, McIldowie MJ, Piggott MJ, Brotchie JM (2010) Reduction of L-DOPA-induced dyskinesia by the selective metabotropic glutamate receptor 5 antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J Pharmacol Exp Ther 333(3):865–873
Jong YJ, Kumar V, O’Malley KL (2009) Intracellular metabotropic glutamate receptor 5 (mGluR5) activates signaling cascades distinct from cell surface counterparts. J Biol Chem 284(51):35827–35838
Jourdain VA, Gregoire L, Morissette M, Morin N, Parent M, Di Paolo T (2013) Potentiation of response to low doses of levodopa in MPTP-injected monkeys by chemical unilateral subthalamotomy. J Neurosurg 118(1):180–191
Katzenschlager R, Head J, Schrag A, Ben-Shlomo Y, Evans A, Lees AJ et al (2008) Fourteen-year final report of the randomized PDRG-UK trial comparing three initial treatments in PD. Neurology 71(7):474–480
Kish SJ, Rajput A, Gilbert J, Rozdilsky B, Chang LJ, Shannak K et al (1986) Elevated gamma-aminobutyric acid level in striatal but not extrastriatal brain regions in Parkinson’s disease: correlation with striatal dopamine loss. Ann Neurol 20(1):26–31
Klawans HL, Goetz C, Nausieda PA, Weiner WJ (1977) Levodopa-induced dopamine receptor hypersensitivity. Trans Am Neurol Assoc 102:80–83
Klockgether T, Turski L (1993) Toward an understanding of the role of glutamate in experimental parkinsonism: agonist-sensitive sites in the basal ganglia. Ann Neurol 34(4):585–593
Kobylecki C, Cenci MA, Crossman AR, Ravenscroft P (2010) Calcium-permeable AMPA receptors are involved in the induction and expression of l-DOPA-induced dyskinesia in Parkinson’s disease. J Neurochem 114(2):499–511
Kuoppamäki M, Al-Barghouthy G, Jackson M, Smith L, Zeng B-Y, Quinn N et al (2002) Beginning-of-dose and rebound worsening in MPTP-treated common marmosets treated with levodopa. Mov Disord Off J Mov Disord Soc 17(6):1312–1317
Kuoppamäki M, Al-Barghouthy G, Jackson MJ, Smith LA, Quinn N, Jenner P (2007) L-dopa dose and the duration and severity of dyskinesia in primed MPTP-treated primates. J Neural Transm 114(9):1147–1153
Lee T, Seeman P, Rajput A, Farley IJ, Hornykiewicz O (1978) Receptor basis for dopaminergic supersensitivity in Parkinson’s disease. Nature 273(5657):59–61
Levandis G, Bazzini E, Armentero MT, Nappi G, Blandini F (2008) Systemic administration of an mGluR5 antagonist, but not unilateral subthalamic lesion, counteracts l-DOPA-induced dyskinesias in a rodent model of Parkinson’s disease. Neurobiol Dis 29(1):161–168
Luginger E, Wenning GK, Bosch S, Poewe W (2000) Beneficial effects of amantadine on L-dopa-induced dyskinesias in Parkinson’s disease. Mov Disord Off J Mov Disord Soc 15(5):873–878
Lujan R, Roberts JD, Shigemoto R, Ohishi H, Somogyi P (1997) Differential plasma membrane distribution of metabotropic glutamate receptors mGluR1 alpha, mGluR2 and mGluR5, relative to neurotransmitter release sites. J Chem Neuroanat 13(4):219–241
Luquin MR, Scipioni O, Vaamonde J, Gershanik O, Obeso JA (1992) Levodopa-induced dyskinesias in Parkinson’s disease: clinical and pharmacological classification. Mov Disord Off J Mov Disord Soc 7(2):117–124
Macinnes N, Duty S (2008) Group III metabotropic glutamate receptors act as hetero-receptors modulating evoked GABA release in the globus pallidus in vivo. Eur J Pharmacol 580(1-2):95–99
Maeda T, Nagata K, Yoshida Y, Kannari K (2005) Serotonergic hyperinnervation into the dopaminergic denervated striatum compensates for dopamine conversion from exogenously administered l-DOPA. Brain Res 1046(1-2):230–233
Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ (2005) The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci Off J Soc Neurosci 25(10):2741–2752
Marin C, Papa S, Engber TM, Bonastre M, Tolosa E, Chase TN (1996) MK-801 prevents levodopa-induced motor response alterations in parkinsonian rats. Brain Res 736(1-2):202–205
Marin C, Jimenez A, Bonastre M, Chase TN, Tolosa E (2000) Non-NMDA receptor-mediated mechanisms are involved in levodopa-induced motor response alterations in parkinsonian rats. Synapse 36(4):267–274
Marino MJ, Awad H, Poisik O, Wittmann M, Conn PJ (2002) Localization and physiological roles of metabotropic glutamate receptors in the direct and indirect pathways of the basal ganglia. Amino Acids 23(1-3):185–191
Marsden C, Parkes J, Quinn N (1982) Fluctuations of disability in Parkinson’s disease – clinical aspects. In: Marsden CD, Fahn S (eds) Movement disorders. Butterworths Scientific, London, pp 96–122
Masilamoni GJ, Bogenpohl JW, Alagille D, Delevich K, Tamagnan G, Votaw JR, Wichmann T, Smith Y (2011) Metabotropic glutamate receptor 5 antagonist protects dopaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain 134:2057–2073
Matsui T, Kita H (2003) Activation of group III metabotropic glutamate receptors presynaptically reduces both GABAergic and glutamatergic transmission in the rat globus pallidus. Neuroscience 122(3):727–737
Mayeux R, Stern Y, Mulvey K, Cote L (1985) Reappraisal of temporary levodopa withdrawal ("drug holiday") in Parkinson’s disease. N Engl J Med 313(12):724–728
Meissner WG, Frasier M, Gasser T, Goetz CG, Lozano A, Piccini P et al (2011) Priorities in Parkinson’s disease research. Nat Rev Drug Discov 10(5):377–393
Mela F, Marti M, Dekundy A, Danysz W, Morari M, Cenci MA (2007) Antagonism of metabotropic glutamate receptor type 5 attenuates l-DOPA-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J Neurochem 101(2):483–497
Mercuri NB, Bernardi G (2005) The ‘magic’ of L-dopa: why is it the gold standard Parkinson’s disease therapy? Trends Pharmacol Sci 26(7):341–344
Michaelis EK (1998) Molecular biology of glutamate receptors in the central nervous system and their role in excitotoxicity, oxidative stress and aging. Prog Neurobiol 54(4):369–415
Montastruc JL, Rascol O, Senard JM (1997) Glutamate antagonists and Parkinson’s disease: a review of clinical data. Neurosci Biobehav Rev 21(4):477–480
Morikawa H, Manzoni OJ, Crabbe JC, Williams JT (2000) Regulation of central synaptic transmission by 5-HT(1B) auto- and heteroreceptors. Mol Pharmacol 58(6):1271–1278
Morin N, Di Paolo T (2014) Pharmacological treatments inhibiting levodopa-induced dyskinesias in MPTP-lesioned monkeys: brain glutamate biochemical correlates. Front Neurol 5:144
Morin N, Gregoire L, Gomez-Mancilla B, Gasparini F, Di Paolo T (2010) Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in parkinsonian monkeys. Neuropharmacology 58(7):981–986
Morin N, Grégoire L, Morissette M, Desrayaud S, Gomez-Mancilla B, Gasparini F et al (2012) MPEP, an mGlu5 receptor antagonist, reduces the development of l-DOPA-induced motor complications in de novo parkinsonian monkeys: biochemical correlates. Neuropharmacology 66:355–364
Morin N, Gregoire L, Morissette M, Desrayaud S, Gomez-Mancilla B, Gasparini F et al (2013a) MPEP, an mGlu5 receptor antagonist, reduces the development of L-DOPA-induced motor complications in de novo parkinsonian monkeys: biochemical correlates. Neuropharmacology 66:355–364
Morin N, Morissette M, Gregoire L, Gomez-Mancilla B, Gasparini F, Di Paolo T (2013b) Chronic treatment with MPEP, an mGlu5 receptor antagonist, normalizes basal ganglia glutamate neurotransmission in L-DOPA-treated parkinsonian monkeys. Neuropharmacology 73:216–231
Morin N, Jourdain VA, Di Paolo T (2014a) Modeling dyskinesia in animal models of Parkinson disease. Exp Neurol 256:105–116
Morin N, Jourdain VA, Morissette M, Gregoire L, Di Paolo T (2014b) Long-term treatment with l-DOPA and an mGlu5 receptor antagonist prevents changes in brain basal ganglia dopamine receptors, their associated signaling proteins and neuropeptides in parkinsonian monkeys. Neuropharmacology 79:688–706
Morin N, Morissette M, Gregoire L, Di Paolo T (2015) Effect of a chronic treatment with an mGlu5 receptor antagonist on brain serotonin markers in parkinsonian monkeys. Prog Neuropsychopharmacol Biol Psychiatry 56:27–38
Morissette M, Goulet M, Calon F, Falardeau P, Blanchet PJ, Bedard PJ et al (1996) Changes of D1 and D2 dopamine receptor mRNA in the brains of monkeys lesioned with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine: correction with chronic administration of L-3,4-dihydroxyphenylalanine. Mol Pharmacol 50(5):1073–1079
Morissette M, Dridi M, Calon F, Hadj Tahar A, Meltzer LT, Bedard PJ et al (2006) Prevention of levodopa-induced dyskinesias by a selective NR1A/2B N-methyl-D-aspartate receptor antagonist in parkinsonian monkeys: implication of preproenkephalin. Mov Disord 21(1):9–17
Moroni F (1999) Tryptophan metabolism and brain function: focus on kynurenine and other indole metabolites. Eur J Pharmacol 375(1-3):87–100
Navailles S, De Deurwaerdere P (2012) Imbalanced Dopaminergic Transmission Mediated by Serotonergic Neurons in L-DOPA-Induced Dyskinesia. Park Dis 2012:1–16
Nemeth H, Toldi J, Vecsei L (2006) Kynurenines, Parkinson’s disease and other neurodegenerative disorders: preclinical and clinical studies. J Neural Transm Suppl 70:285–304
Niswender CM, Conn PJ (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322
Nutt JG (1990) Levodopa-induced dyskinesia: review, observations, and speculations. Neurology 40(2):340–345
Ochi M, Shiozaki S, Kase H (2004) L-DOPA-induced modulation of GABA and glutamate release in substantia nigra pars reticulata in a rodent model of Parkinson’s disease. Synapse 52(2):163–165
Olanow CW, Koller WC (1998) An algorithm (decision tree) for the management of Parkinson’s disease: treatment guidelines. American Academy of Neurology. Neurology 50(3 Suppl 3):S1–57
Olanow CW, Stern MB, Sethi K (2009) The scientific and clinical basis for the treatment of Parkinson disease (2009). Neurology 72(21 Suppl 4):S1–136
Olive MF (2009) Metabotropic glutamate receptor ligands as potential therapeutics for addiction. Curr Drug Abuse Rev 2(1):83–989
Ouattara B, Gasparini F, Morissette M, Grégoire L, Samadi P, Gomez-Mancilla B et al (2010) Effect of L-Dopa on metabotropic glutamate receptor 5 in the brain of parkinsonian monkeys. J Neurochem 113(3):715–724
Ouattara B, Gregoire L, Morissette M, Gasparini F, Vranesic I, Bilbe G et al (2011) Metabotropic glutamate receptor type 5 in levodopa-induced motor complications. Neurobiol Aging 32(7):1286–1295
Oueslati A, Sgambato-Faure V, Melon C, Kachidian P, Gubellini P, Amri M et al (2007) High-frequency stimulation of the subthalamic nucleus potentiates L-DOPA-induced neurochemical changes in the striatum in a rat model of Parkinson’s disease. J Neurosci Off J Soc Neurosci 27(9):2377–2386
Pahwa R, Lyons KE (2009) Levodopa-related wearing-off in Parkinson’s disease: identification and management. Curr Med Res Opin 25(4):841–849
Palucha A, Pilc A (2007) Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol Ther 115(1):116–147
Papa SM, Boldry RC, Engber TM, Kask AM, Chase TN (1995) Reversal of levodopa-induced motor fluctuations in experimental parkinsonism by NMDA receptor blockade. Brain Res 701(1-2):13–18
Paquet M, Smith Y (2003) Group I metabotropic glutamate receptors in the monkey striatum: subsynaptic association with glutamatergic and dopaminergic afferents. J Neurosci Off J Soc Neurosci 23(20):7659–7669
Parkinson Study Group (2009) Long-term effect of initiating pramipexole vs levodopa in early Parkinson disease. Arch Neurol 66(5):563–570
Perroy J, Raynaud F, Homburger V, Rousset MC, Telley L, Bockaert J et al (2008) Direct interaction enables cross-talk between ionotropic and group I metabotropic glutamate receptors. J Biol Chem 283(11):6799–6805
Pisani A, Gubellini P, Bonsi P, Conquet F, Picconi B, Centonze D et al (2001) Metabotropic glutamate receptor 5 mediates the potentiation of N-methyl-D-aspartate responses in medium spiny striatal neurons. Neuroscience 106(3):579–587
Pisani A, Bonsi P, Centonze D, Gubellini P, Bernardi G, Calabresi P (2003) Targeting striatal cholinergic interneurons in Parkinson’s disease: focus on metabotropic glutamate receptors. Neuropharmacology 45(1):45–56
Planells-Cases R, Lerma J, Ferrer-Montiel A (2006) Pharmacological intervention at ionotropic glutamate receptor complexes. Curr Pharm Des 12(28):3583–3596
Quik M, Spokes EG, Mackay AV, Bannister R (1979) Alterations in [3H]spiperone binding in human caudate nucleus, substantia nigra and frontal cortex in the Shy-Drager syndrome and Parkinson’s disease. J Neurol Sci 43(3):429–437
Rajput AH, Rajput A, Lang AE, Kumar R, Uitti RJ, Galvez-Jimenez N (1998) New use for an old drug: amantadine benefits levodopa-induced dyskinesia. Mov Disord Off J Mov Disord Soc 13(5):851
Rajput AH, Voll A, Rajput ML, Robinson CA, Rajput A (2009) Course in Parkinson disease subtypes: a 39-year clinicopathologic study. Neurology 73(3):206–212
Rascol O, Fox S, Gasparini F, Kenney C, Di Paolo T, Gomez-Mancilla B (2014) Use of metabotropic glutamate 5-receptor antagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat Disord 20(9):947–956
Rinne UK, Lonnberg P, Koskinen V (1981) Dopamine receptors in the Parkinsonian brain. J Neural Transm 51(1-2):97–106
Robelet S, Melon C, Guillet B, Salin P, Kerkerian-Le GL (2004) Chronic L-DOPA treatment increases extracellular glutamate levels and GLT1 expression in the basal ganglia in a rat model of Parkinson’s disease. Eur J Neurosci 20(5):1255–1266
Ruzicka E, Streitova H, Jech R, Kanovsky P, Roth J, Rektorova I et al (2000) Amantadine infusion in treatment of motor fluctuations and dyskinesias in Parkinson’s disease. J Neural Transm 107(11):1297–1306
Rylander D, Recchia A, Mela F, Dekundy A, Danysz W, Cenci MA (2009) Pharmacological modulation of glutamate transmission in a rat model of L-DOPA-induced dyskinesia: effects on motor behavior and striatal nuclear signaling. J Pharmacol Exp Ther 330(1):227–235
Rylander D, Iderberg H, Li Q, Dekundy A, Zhang J, Li H et al (2010) A mGluR5 antagonist under clinical development improves L-DOPA-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis 39(3):352–361
Samadi P, Gregoire L, Rassoulpour A, Guidetti P, Izzo E, Schwarcz R et al (2005) Effect of kynurenine 3-hydroxylase inhibition on the dyskinetic and antiparkinsonian responses to levodopa in Parkinsonian monkeys. Mov Disord 20(7):792–802
Samadi P, Grégoire L, Rouillard C, Bédard PJ, Di Paolo T, Lévesque D (2006) Docosahexaenoic acid reduces levodopa-induced dyskinesias in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine monkeys. Ann Neurol 59(2):282–288
Samadi P, Rouillard C, Bedard PJ, Di Paolo T (2007) Functional neurochemistry of the basal ganglia. Handb Clin Neurol 83:19–66
Samadi P, Grégoire L, Morissette M, Calon F, Hadj Tahar A, Dridi M et al (2008a) mGluR5 metabotropic glutamate receptors and dyskinesias in MPTP monkeys. Neurobiol Aging 29(7):1040–1051
Samadi P, Gregoire L, Morissette M, Calon F, Hadj Tahar A, Belanger N et al (2008b) Basal ganglia group II metabotropic glutamate receptors specific binding in non-human primate model of L-Dopa-induced dyskinesias. Neuropharmacology 54(2):258–268
Samadi P, Rajput A, Calon F, Gregoire L, Hornykiewicz O, Rajput AH et al (2009) Metabotropic glutamate receptor II in the brains of Parkinsonian patients. J Neuropathol Exp Neurol 68(4):374–382
Sawada H, Oeda T, Kuno S, Nomoto M, Yamamoto K, Yamamoto M et al (2010) Amantadine for dyskinesias in Parkinson’s disease: a randomized controlled trial. PLoS One 5(12):e15298
Schoepp DD (2001) Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther 299(1):12–20
Schwarcz R, Pellicciari R (2002) Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther 303(1):1–10
Siderowf A, Stern M (2003) Update on Parkinson disease. Ann Intern Med 138(8):651–658
Smeyne RJ, Jackson-Lewis V (2005) The MPTP model of Parkinson’s disease. Brain Res Mol Brain Res 134(1):57–66
Snow BJ, Macdonald L, McAuley D, Wallis W (2000) The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 23(2):82–85
Stanford IM, Lacey MG (1996) Differential actions of serotonin, mediated by 5-HT1B and 5-HT2C receptors, on GABA-mediated synaptic input to rat substantia nigra pars reticulata neurons in vitro. J Neurosci Off J Soc Neurosci 16(23):7566–7573
Stayte S, Vissel B (2014) Advances in non-dopaminergic treatments for Parkinson’s disease. Front Neurosci 8:113
Stocchi F, Tagliati M, Olanow CW (2008) Treatment of levodopa-induced motor complications. Mov Disord Off J Mov Disord Soc 23(Suppl 3):S599–S612
Stocchi F, Rascol O, Destee A, Hattori N, Hauser RA, Lang AE et al (2013) AFQ056 in Parkinson patients with levodopa-induced dyskinesia: 13-week, randomized, dose-finding study. Mov Disord 28(13):1838–1846
Stone TW (1993) Neuropharmacology of quinolinic and kynurenic acids. Pharmacol Rev 45(3):309–379
Stone TW (2000) Development and therapeutic potential of kynurenic acid and kynurenine derivatives for neuroprotection. Trends Pharmacol Sci 21(4):149–154
Stone TW (2001) Kynurenines in the CNS: from endogenous obscurity to therapeutic importance. Prog Neurobiol 64(2):185–218
Testa CM, Standaert DG, Young AB, Penney JB Jr (1994) Metabotropic glutamate receptor mRNA expression in the basal ganglia of the rat. J Neurosci Off J Soc Neurosci 14(5 Pt 2):3005–3018
Toulouse A, Sullivan AM (2008) Progress in Parkinson’s disease-where do we stand? Prog Neurobiol 85(4):376–392
Verhagen Metman L, Del Dotto P, van den Munckhof P, Fang J, Mouradian MM, Chase TN (1998a) Amantadine as treatment for dyskinesias and motor fluctuations in Parkinson’s disease. Neurology 50(5):1323–1326
Verhagen Metman L, Del Dotto P, Natte R, van den Munckhof P, Chase TN (1998b) Dextromethorphan improves levodopa-induced dyskinesias in Parkinson’s disease. Neurology 51(1):203–206
Verhagen Metman L, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN (1999) Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 56(11):1383–1386
Verhagen Metman L, Konitsiotis S, Chase TN (2000) Pathophysiology of motor response complications in Parkinson’s disease: hypotheses on the why, where, and what. Mov Disord Off J Mov Disord Soc 15(1):3–8
Wang H, Westin L, Nong Y, Birnbaum S, Bendor J, Brismar H et al (2009) Norbin is an endogenous regulator of metabotropic glutamate receptor 5 signaling. Science 326(5959):1554–1557
Witkin JM, Marek GJ, Johnson BG, Schoepp DD (2007) Metabotropic glutamate receptors in the control of mood disorders. CNS Neurol Disord Drug Targets 6(2):87–100
Yamamoto N, Soghomonian JJ (2009) Metabotropic glutamate mGluR5 receptor blockade opposes abnormal involuntary movements and the increases in glutamic acid decarboxylase mRNA levels induced by l-DOPA in striatal neurons of 6-hydroxydopamine-lesioned rats. Neuroscience 163(4):1171–1180
Yamamoto N, Pierce RC, Soghomonian JJ (2006) Subchronic administration of L-DOPA to adult rats with a unilateral 6-hydroxydopamine lesion of dopamine neurons results in a sensitization of enhanced GABA release in the substantia nigra, pars reticulata. Brain Res 1123(1):196–200
Zhang X, Feng ZJ, Chergui K (2014) GluN2D-containing NMDA receptors inhibit neurotransmission in the mouse striatum through a cholinergic mechanism: implication for Parkinson’s disease. J Neurochem 129(4):581–590
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Morin, N., Di Paolo, T. (2017). mGlu5 Receptors in Parkinson’s Disease and MPTP-Lesioned Monkeys: Behavior and Brain Molecular Correlates. In: Ngomba, R., Di Giovanni, G., Battaglia, G., Nicoletti, F. (eds) mGLU Receptors. The Receptors, vol 31. Humana Press, Cham. https://doi.org/10.1007/978-3-319-56170-7_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-56170-7_10
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-56168-4
Online ISBN: 978-3-319-56170-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)