
Abstract
The appearance of levodopa-induced dyskinesia (LID) and ongoing degeneration of nigrostriatal dopaminergic neurons are two key features of Parkinson’s disease (PD) that current treatments fail to address. Increased glutamate transmission contributes to the motor symptoms in PD, to the striatal plasticity that underpins LID and to the progression of neurodegeneration through excitotoxic mechanisms. Glutamate receptors have therefore long been considered as potential targets for pharmacological intervention in PD, with emphasis on either blocking activation of 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA), N-methyl-d-aspartate (NMDA) or excitatory metabotropic glutamate (mGlu) 5 receptors or promoting the activation of group II/III mGlu receptors. Following a brief summary of the role of glutamate in PD and LID, this article explores the current status of pharmacological studies in pre-clinical rodent and primate models through to clinical trials, where applicable, that support the potential of glutamate-based therapeutic interventions. To date, AMPA antagonists have shown good efficacy against LID in rat and primate models, but the failure of perampanel to lessen LID in clinical trials casts doubt on the translational potential of this approach. In contrast, antagonists selective for NR2B-containing NMDA receptors were effective against LID in animal models and in small-scale clinical trials, though observed adverse cognitive effects need addressing. So far, mGlu5 antagonists or negative allosteric modulators (NAMs) look set to become the first introduced for tackling LID, with AFQ-056 reported to exhibit good efficacy in phase II clinical trials. NR2B antagonists and mGlu5 NAMs may subsequently prove to also be effective disease-modifying agents if their protective effects in rat and primate models of PD, respectively, are replicated in the next stages of investigation. Finally, group III mGlu4 agonists or positive allosteric modulators (PAMs), although in the early pre-clinical stages of investigation, are showing good efficacy against motor symptoms, neurodegeneration and LID. It is anticipated that the recent development of mGlu4 PAMs with improved systemic bioavailability will facilitate progression of these agents into the primate model of PD where their potential can be further explored.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Parkinson’s disease (PD) affects around 1 % of the general population, rising to over 3 % in those beyond 80 years of age [1]. The disease is characterized by motor disturbances such as a resting tremor, bradykinesia, rigidity and postural instability, but patients also exhibit a range of non-motor symptoms including depression and cognitive decline [2]. The key pathology associated with PD is degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc). The subsequent reduction in striatal dopamine innervation triggers a cascade of changes in firing of downstream pathways within the basal ganglia motor loop that culminate in reduced feedback to the motor cortex and the ensuing motor deficits [3, 4]. Current treatments provide relief of motor symptoms through restoring dopamine receptor stimulation. In older patients, levodopa may be introduced as first-line treatment since it appears to be fairly well tolerated. However, in younger patients, monoamine oxidase B inhibitors or dopamine agonists are usually initially recommended [5]. However, as these patients’ symptoms worsen, the introduction of levodopa becomes necessary and within as little as 2 years of commencing levodopa treatment, around one-third of patients develop disabling adverse effects in the form of motor fluctuations and levodopa-induced dyskinesia (LID) [6, 7]. Levodopa also fails to combat the ongoing degeneration, leading to progressive worsening of patients’ symptoms. Finding new drugs to treat PD that are devoid of dyskinetic adverse effects, can lessen the incidence of LID, or offer much needed neuroprotection is an ongoing challenge. Through its involvement in a number of key pathways within the basal ganglia motor loop, glutamate has long been implicated in the pathophysiology and pathogenesis of PD [8] (Fig. 1). Not surprisingly, the ionotropic and metabotropic receptors through which glutamate acts have also been extensively investigated. Of these, significant advancements have been made concerning 2-amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic acid (AMPA), N-methyl-d-aspartate (NMDA) and metabotropic glutamate (mGlu) receptors, not only in terms of identifying the role these receptors may play in the various features of PD, but also in terms of assessing their potential as targets for pharmacological intervention. In contrast, little to nothing is known about the contribution and target potential of kainate receptors in PD or LID, which is why this class is not considered further here. In this article, the role of glutamate and its receptors in the pathogenesis, development of motor symptoms and LID associated with PD is first briefly summarized. This is followed by a comprehensive, up-to-date account on the target potential of each of the three major classes of glutamate receptor (AMPA, NMDA and mGlu), highlighting findings from pharmacological studies in pre-clinical rodent and primate models right through to clinical trials, where applicable.

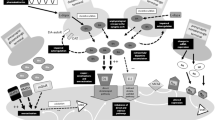
Schematic representation of the basal ganglia motor loop, showing the direct and indirect circuits that work together to control movement. Parkinson’s disease is characterized by the loss of the dopaminergic neurons in the substantia nigra pars compacta, which sets up a series of changes in firing within the basal ganglia circuitry (not shown here). A number of pathways utilize glutamate as a neurotransmitter and this figure highlights the key pathways that have been implicated in the pathogenesis, symptom generation and levodopa-induced dyskinesia associated with Parkinson’s disease. For details of how these pathways contribute, the glutamate receptors involved and relevant references, refer to the accompanying text. GPe external global pallidus, GPi internal globus pallidus, LID levodopa-induced dyskinesia, STN subthalamic nucleus, SNpc substantia nigra pars compacta, SNpr substantia nigra pars reticulata
2 Role of Glutamate in Parkinson’s Disease (PD)
2.1 In Generation of Motor Symptoms
Glutamate provides excitatory drive to both the input region (striatum) and output regions (substantia nigra pars reticulata [SNpr] and internal globus pallidus [GPi]) of the basal ganglia motor loop via the corticostriatal pathway and efferent projections from the subthalamic nucleus (STN), respectively. Increased firing of the corticostriatal pathway is thought to contribute to the pathophysiology of PD by further exciting the indirect striatopallidal pathway that is already overactive through loss of normal dopamine D2-like receptor-mediated inhibition. Increased firing of this GABAergic striatopallidal pathway inhibits GABAergic drive to the STN, leading to disinhibition of the glutamatergic STN efferents to the SNpr/GPi [9]. The increase in prevailing glutamate levels in SNpr/GPi drives inhibition of thalamocortical feedback, resulting in motor dysfunction [10]. It follows that blocking this excess glutamatergic drive may help combat the motor symptoms in PD. Our knowledge of the contribution of glutamate to PD stems largely from studies in rodent and primate models, which share many, but not all features of human PD [11]. Indeed, while increased expression of the vesicular glutamate transporters, VGLUT1 and VGLUT2, in the putamen of PD patients supports the increased glutamatergic innervation to this region [12], measurement of glutamate directly in CSF [13–15] or in the striatum or SNPr/GPi [16, 17] reveals no increases in the PD brain. Although this may reflect correction of any neurochemical imbalance by the patients’ medication, it does suggest that a degree of caution be applied when interpreting the involvement of glutamate in the generation of motor symptoms.
2.2 In Levodopa-Induced Dyskinesia (LID)
The mechanisms behind LID are still not fully understood, but appear to involve maladaptive plasticity at striatal synapses [18–21]. Since the proposed molecular mechanisms underlying glutamatergic contributions to LID have recently been extensively reviewed [22], the focus here is on summarizing points that highlight the potential of glutamate receptors as useful antidyskinetic drug targets. Within this context, a key contributory factor to LID is increased glutamate transmission from corticostriatal projections that converge with dopaminergic inputs at the affected striatal synapses [23]. Some of this increased transmission likely reflects pre-synaptic events since an increased concentration of extracellular glutamate has been measured in the striatum of 6-hydroxydopamine (6-OHDA)-lesioned rats exhibiting levodopa-induced abnormal involuntary movements (AIMs), a rodent model of LID [24]. However, post-synaptic changes in AMPA, NMDA and mGlu receptors appear to play a major role. Focusing on mGlu receptors in the first instance, mGlu5 receptor binding is increased in the putamen of 1-methyl-4-phenyl-1,2,3,6,tetrahydropyridine (MPTP)-treated primates expressing LID [25, 26] and in PD patients with motor complications [26], suggesting exaggerated mGlu5 activation may be involved in enhancing transmission at excitatory striatal synapses. Changes in mGlu2/3 receptor binding have also been measured in MPTP-treated primates with LID. For example, a reduction in mGlu2/3 binding was seen in non-dyskinetic monkeys receiving cabergoline as an adjunct to levodopa when compared with levodopa-treated dyskinetic monkeys, implying elevated mGluR2/3 levels in LID. However, given a similar reduction in binding was not seen in non-dyskinetic monkeys receiving another adjunct (the NMDA-containing NR2B antagonist, CI-1041 [besonprodil]) [27], it is likely that the changes in mGlu2/3 expression reflect crosstalk between the mGlu2/3 and D2 receptors and may not necessarily relate to the appearance or not of dyskinesia. A higher level of mGlu2/3 binding was also noted in the caudate of PD patients exhibiting wearing-off of levodopa responsiveness, compared with those with no wearing-off; however, there was again no such difference in mGlu2/3 binding between PD patients with and without dyskinesia [28]. Further studies are required to fully understand the implications of these subtle changes.
Binding to AMPA and NMDA (specifically NR1/NR2B-containing) receptors is elevated in the putamen of PD patients with LID [29] while similar increases, also encompassing NR2A subunits [30, 31], are seen in MPTP-treated primates expressing LID [32, 33] or 6-OHDA-lesioned rats exhibiting AIMs [34]. Importantly, this elevated NMDA tone is further supported by positron emission tomography (PET) studies showing increased uptake of 11C-CNS 5161, a non-competitive NMDA receptor antagonist, in the caudate-putamen of ‘on’ state PD patients expressing LID [35]. Changes in the phosphorylation state and trafficking of AMPA and NMDA receptors may add further to the functional enhancement of glutamatergic synapses in LID. Regarding AMPA receptors, increased phosphorylation of the glutamate receptor 1 subunit is seen in the mouse model of levodopa-induced AIMs [36] and accompanies the motor response alterations triggered by, and persisting after, withdrawal of chronic levodopa treatment in 6-OHDA rats [37]. Enhanced phosphorylation of glutamate receptor 1 also occurs in levodopa-treated dyskinetic primates [38]. Phosphorylation can enhance AMPA-mediated currents as well as increase trafficking to the synapse [39]. Indeed chronic levodopa treatment enhances glutamate receptor 1 levels in striatal membrane fractions from 6-OHDA-lesioned rats [40] while increased trafficking of AMPA glutamate receptor 2/3 subunits to the post-synaptic membrane has been found in the primate model of LID [41]. Concerning NMDA receptors, in 6-OHDA-lesioned rats treated chronically with levodopa and displaying either AIMs or motor response alterations, there is increased phosphorylation of NR2A and especially NR2B receptors [42] and redistribution of subunits in the post-synaptic compartment, favouring NR2A over NR2B [43, 44]. This relative increase in synaptic abundance of NR2A over NR2B subunits has also been reported in MPTP-treated primates expressing LID [30]. Whether this proposed switch in subunit composition of the NMDA receptor is key to LID remains to be established. However, it is interesting to note that depotentiation (i.e. the reversal of long-term potentiation by subsequent low-frequency stimulation), the loss of which has been implicated in the molecular mechanisms behind LID [45], requires activation of NR2A-containing NMDA receptors [46], so this switch cannot readily explain this loss of depotentiation, at least. The change in synaptic NMDA receptor composition may, however, have other implications for the pharmacological interventions required to tackle LID (described later in Sect. 4.2).
On balance, the changes in mGlu5, AMPA and NMDA receptors already described most likely contribute to an increased sensitivity of striatal neurons to glutamate, thereby supporting the view that increased glutamate signalling underpins the appearance of LID. Despite remaining uncertainties on how these receptor perturbations contribute to LID, from the nature of these changes (i.e. increased expression/sensitivity) it can be inferred that glutamate receptor antagonists may offer relief of LID.
2.3 In the Pathogenesis of PD
The mechanisms proposed to underlie cell death in PD are numerous, encompassing oxidative stress, inflammation, reduced expression of trophic factors, ubiquitin proteasome dysfunction and autophagy. However, it is the pathogenic mechanisms of mitochondrial dysfunction and excitotoxicity in which glutamate is implicated [47–49]. Glutamate-mediated excitotoxicity is the well known process whereby excess activation of NMDA receptors leads to exaggerated free intracellular calcium levels that overwhelm the cell’s buffering capacity. On the one hand, the resultant activation of calcium-dependent cysteine proteases and mitochondrial endonucleases appears to directly trigger apoptotic cell death, while on the other hand, activation of calcium-dependent neuronal nitric oxide synthase (nNOS) and subsequent peroxynitrite formation result in oxidative stress-induced damage to cellular proteins, lipids and DNA, leading subsequently to the cell’s demise [49, 50]. Glutamate-mediated excitotoxicity was considered for a long time to be involved only in progression of the ongoing neurodegeneration, rather than as an initiator. This was because while glutamatergic inputs to the SNpc from both the cortex and STN [51, 52] (Fig. 1) are certainly overactive in PD [9, 10], these changes are secondary to the reduced striatal dopamine innervation triggered by nigral cell loss so this source of elevated glutamate cannot be considered responsible for triggering nigral degeneration in the first instance. However, this idea has been challenged by findings from studies on genes associated with PD. For example, parkin, the product of the PARK2 gene, has been linked to regulation of glutamatergic synapses. Helton et al. [53] found that post-synaptic expression of parkin in cultured hippocampal neurons reduced the number and strength of excitatory synapses while, conversely, post-synaptic expression of a range of parkin mutants responsible for early-onset autosomal recessive forms of PD [54] resulted in an increase in the size and frequency of excitatory post-synaptic potentials, reflecting increased glutamatergic synapses. This proliferation of glutamatergic synapses resulted in increased vulnerability to excitotoxicity [53] and, if the same occurs in the SNpc, such a phenomenon may well contribute to the early pathogenesis of PD in these genetic cases at least. A second gene linked to early-onset PD, DJ-1 (PARK7) [55], has also been linked to excitotoxicity. Aleyasin et al. [56] discovered that cerebellar granule cells obtained from DJ-1-null mice were more sensitive to glutamate-mediated excitotoxicity while overexpression of DJ-1 provided protection against excitotoxicity. Given that DJ-1-linked PD mutations are missense or loss-of-function [55], this implies that enhanced susceptibility to excitotoxicity could also prevail in these genetic PD cases.
As reviewed by Blandini [49] in 2010, there is also mounting evidence that dopaminergic neurons in the SNpc are hypersensitive to the effects of glutamate. One early piece of evidence [57] relates to mitochondrial complex I dysfunction, which occurs in the SNpc in PD [38]. This impairment is classically associated with the production of free radicals and ensuing oxidative stress [49, 50], but the subsequently reduced adenosine triphosphate (ATP) generation also destabilizes the membrane potential, removing the Mg2+ block from the NMDA receptor, thereby potentially rendering even non-noxious levels of glutamate now more likely to trigger calcium entry [57]. This so-called weak excitotoxic hypothesis has been demonstrated in vitro, whereby pre-treatment with the mitochondrial complex I inhibitor, 1-methyl-4-phenylpyridinium (MPP+), potentiates glutamate-mediated excitotoxicity [58]. Studies using other known mitochondrial toxins, such as the pesticides rotenone and paraquat, which are classed as environmental risk factors for PD [59] and can replicate aspects of the disease in animal models [11], strengthen the link between mitochondrial dysfunction and excitotoxicity. For example, rotenone has been shown to reduce the Mg2+ block from NMDA receptors in SNpc dopaminergic neurons in slices [60], an effect that would render them more sensitive to the effects of glutamate. Similarly, paraquat-evoked dopamine neuron death in midbrain slice cultures is blocked by NMDA antagonists and nNOS inhibitors, further supporting the link between mitochondrial toxicity and excitotoxicity [61].
As far as evidence favouring excitotoxicity in the PD brain is concerned, apoptosis, which is triggered by the excessive intracellular free calcium levels [62, 63] and oxidative damage, which occurs following calcium-dependent nNOS activation and subsequent peroxynitrite formation [64], are evident in the PD brain. Given these contributions of glutamate to the pathogenesis of PD, reducing glutamate transmission in the SNpc would seem a sensible approach towards modifying, or even preventing the onset of, disease progression.
Overall, it is fair to say that substantial evidence exists to support a role for enhanced glutamate transmission in the pathogenesis, clinical symptoms and LID associated with PD. Not surprisingly, therefore, there is a wealth of published information on the potential of targeting glutamate receptors to bring about symptomatic and protective relief in PD or reduce the incidence of LID. The remainder of this article presents an up-to-date account of the current status of these therapeutic strategies from pre-clinical studies through to clinical trials, where applicable. A summary of the efficacy of targeting each receptor against PD symptoms, pathogenesis and LID is presented in Table 1.
3 Therapeutic Potential of Targeting 2-Amino-3-(5-methyl-3-oxo-1,2-oxazol-4-yl)propanoic Acid (AMPA) Receptors
Although present at relevant excitatory synapses within the SNpc, SNpr [65] and striatum [66], AMPA receptors have received relatively limited attention in the quest for improved treatments for PD.
3.1 Prospects for Ameliorating Symptoms of PD or Tackling LID
The ability of AMPA receptor antagonists to reduce parkinsonian symptoms in a range of animal models from reserpine- or 6-OHDA-treated rats to MPTP-treated primates is extremely variable [67–71], suggesting little may be gained by pursuing AMPA antagonists as monotherapy for symptomatic relief. However, AMPA antagonists do potentiate the antiparkinsonian effects of levodopa [67, 68, 70] and, more importantly, show efficacy against levodopa-induced motor response alterations. This has been shown in 6-OHDA-lesioned rats, whereby wearing off of levodopa-induced motor responses were antagonized by the competitive AMPA antagonist, LY-293558 [72]. Furthermore, in MPTP-treated primates, the non-competitive AMPA antagonist LY-300164 (telampanel) enhanced the antiparkinsonian effects of levodopa, whilst concomitantly reducing LID [70] while topiramate, which both blocks AMPA receptors and triggers their dephosphorylation, also reduced LID [71]. These beneficial outcomes, which were consistent with reversal of the aforementioned elevated AMPA receptor expression and subunit phosphorylation seen in dyskinetic patients or rat and primate models of LID [29, 32, 33, 40, 41], prompted examination of the antidyskinetic potential of the non-competitive AMPA antagonist, perampanel, in patients. Disappointingly, these randomized, double-blind, placebo-controlled trials failed to show any significant reduction in LID in patients given perampanel alongside their levodopa [73, 74]. Given the importance of calcium-dependent signalling pathways in the pathogenesis of LID [19–21], it will be interesting to see whether selective calcium-permeable AMPA receptor antagonists like IEM-1460, which are efficacious in the 6-OHDA-lesioned rat AIMs and MPTP-treated primate models of LID [75] open up a more promising line of investigation.
3.2 Prospects for Providing Disease Modification
Despite AMPA receptors being present in the SNpc [65], the AMPA receptor antagonist NBQX (NNC-079202) failed to offer protection against either 6-OHDA or MPP+ toxicity in rodent SNpc [76]. Somewhat paradoxically, it was AMPA receptor potentiators that harboured a potential neuroprotective action. LY-404187 and LY-503430 provided functional, neurochemical and histological protection in the 6-OHDA-lesioned rat and MPTP-treated mouse models of PD, even when administered up to 2 weeks post-lesion [77, 78]. These benefits were suggested to reflect a neurotrophic action, given these agents also increased brain-derived neurotrophic factor (BDNF) expression in the SNpc [77, 78], but the lack of follow-up studies suggests interest in these agents as disease modifiers in PD has waned.
4 Therapeutic Potential of Targeting N-methyl-d-aspartate (NMDA) Receptors
4.1 Prospects for Ameliorating Symptoms of PD
Although broad-spectrum NMDA receptor antagonists have limited clinical potential due to concerns over their propensity to induce adverse effects including psychotomimetic effects, sedation, impairments of learning and memory and dissociative anaesthesia [79, 80], these agents have been useful for proof-of-concept studies, before moving onto more subtype selective agents. That said, the inconsistency of broad-spectrum NMDA receptor antagonists like MK-801, CPP or LY-235959 to provide robust antiparkinsonian activity when administered as monotherapy to parkinsonian rodents or MPTP-treated primates [81–84] did not deter investigations into NR2B subunits on the basis that their more restricted distribution, largely within forebrain regions such as the striatum [85], may offer a more rational target. All but one study [86] revealed antiparkinsonian effects of antagonists selective for NR2B-containing NMDA receptors including ifenprodil, Ro-256981 and CP-101606 (traxoprodil) in rat [87–90] and primate models of PD [88, 91], as well as potentiation of the antiparkinsonian effects of levodopa [88, 89, 92]. However, the future use of NR2B antagonists to ameliorate motor symptoms in PD remains in doubt following a randomized, double-blind, placebo-controlled, crossover trial in which 16 patients failed to demonstrate any improvement in motor scores with single-dose administration of the NR2B-selective antagonist MK-0657 [93]. Nevertheless, NMDA receptor antagonists may yet prove helpful in alleviating some non-motor symptoms associated with PD. For example, the low-affinity non-competitive antagonist of NMDA receptors, amantadine, reduced punding (excessive repetitive non-goal-oriented activities) in four out of ten PD patients in an open-label prospective study [94] and has been linked to a delayed onset of dementia in PD patients [95]. The partial antagonist of NMDA receptors, memantine, also improves cognitive impairments for up to a year in patients with PD complicated by dementia [96, 97], opening up the possibility that this drug, already approved for use in the USA and Europe for moderate to severe Alzheimer’s disease and amantadine, which is already prescribed for LID (see below), may be used in the future against non-motor symptoms in PD patients.
4.2 Prospects for Tackling LID
Amantadine is the only drug currently licensed for the treatment of dyskinesia in PD and its effectiveness supports the involvement of enhanced NMDA signalling in LID already described [29–32, 34, 35, 42]. The clinical efficacy of amantadine against LID [98, 99] has recently been shown to last beyond 1 year [100] and memantine offers similar relief against LID [101–103]. Given the rich pharmacology of amantadine, further support for the use of NMDA antagonists per se was obtained using more selective NMDA antagonists like MK-801. While these agents reduced levodopa-induced motor fluctuations or AIMs in 6-OHDA-lesioned rats [104–106], and LID in MPTP-treated primates [83, 107], this was sometimes achieved at the expense of the antiparkinsonian action of levodopa [106, 107]. Specifically targeting NR2B-containing NMDA receptors using a seemingly ever expanding repertoire of selective antagonists (ifenprodil, traxoprodil, BZAD-01; Ro-631908, Ro-256981, Co-101244 and CI-1041 [besonprodil]) provided, with a few exceptions [90, 92], the desired alleviation of levodopa-induced motor fluctuations or AIMs in rodent models of PD [108–110] and of LID in MPTP-treated primates [86, 111, 112] without compromising the antiparkinsonian efficacy of levodopa. Alleviation of LID was accompanied by rectification of elevated striatal neuropeptide signalling (e.g. pre-proenkephalin and prodynorphin) [111, 112] that has been implicated in the pathogenesis of LID [19], supporting earlier reports of a striatal action for manifestation of these antidyskinetic effects [113].
The efficacy of the NR2B antagonists in LID has been reported in one small-scale clinical trial in which acute administration of traxoprodil suppressed LID by around 30 % [114]. However, these effects were accompanied by dose-related adverse cognitive effects (dissociation and amnesia) that require further investigation if confidence is to be strengthened in targeting NR2B receptors in the clinic [114]. Indeed not all data points to NR2B being the key subunit; some consider NR2A subunits important, especially given their increased expression in the striatum of MPTP-treated dyskinetic primates [30]. Studies with the NR2A antagonist MDL-1000453, showing a worsening of LID in MPTP-treated primates, failed to support this view [86]. However, given this drug shows only around 5-fold selectivity for NR2A versus NR2B, a recent report showing that systemic injection of a cell-permeable peptide blocking NR2A reduced the development of AIMs in 6-OHDA-lesioned levodopa-treated rats [31] suggests the future development of more selective NR2A antagonists may be worthwhile.
4.3 Prospects for Providing Disease Modification
Given the pivotal role of NMDA receptors in mediating excitotoxicity, it is not surprising that NMDA antagonists have been widely investigated as neuroprotective agents. However, their efficacy in PD remains in doubt. While some studies found the NMDA antagonists MK-801 and CPP or the strychnine-insensitive glycine site partial antagonist (R)-HA-966 provided almost complete protection against intranigral infusion of MPP+ in the rat or systemic MPTP injection in mice [76, 115], others found only partial [116, 117] or no such protection [118–120]. Protection against MPTP-induced degeneration in primates was more consistently observed [120, 121]. Block of NMDA receptors in the SNpc, or more upstream in regions of the basal ganglia such as the STN may account for any protective action seen [122, 123]. However, NMDA receptors are also expressed on human astrocytes [124]. Moreover, the protective effects of amantadine and memantine on primary midbrain dopamine neuron/glial cultures involve increased release of glial-derived neurotrophic factor (GDNF) as well as anti-inflammatory actions [125, 126]. Thus, it is possible that similar indirect actions also contribute to the protective effects of NMDA antagonists in vivo. An involvement of NR2B-containing receptors in these neuroprotective actions is supported by two recent studies. In the first, the NR2B antagonist BZAD-01 provided histological and behavioural protection against 6-OHDA-induced lesions of the nigrostriatal tract in rats when given for 11 days prior to and 3 days following 6-OHDA [127]. The second study showed that intrastriatal infusion of an NR2B-specific short interfering RNA (siRNA), commencing 7 days post-6-OHDA partial lesion produced a modest, but significant protection against nigrostriatal tract degeneration [128], again implying that actions on NMDA receptors upstream of the SNpc itself can produce the required protection. Clearly, NR2B receptors look like worthy candidates for further study in MPTP-treated primates as prospective targets for providing modification of disease progression in PD.
5 Therapeutic Potential of Targeting Metabotropic Glutamate (mGlu) Receptors
mGlu receptors are abundant in the basal ganglia: group I receptors (mGlu1 and mGlu5) are mostly post-synaptic and couple through Gq/11, leading to enhanced neuronal excitation; group II receptors (mGlu2 and mGlu3) couple through Gi/Go leading to inhibition of neuronal transmission when found on pre-synaptic terminals or to the production of neurotrophic agents when expressed on glia; group III receptors (mGlu4, mGlu7 and mGlu8) are also Gi/Go-coupled and found predominantly on pre-synaptic terminals where they regulate synaptic transmission via inhibition of transmitter release [129]. Research has therefore focused on the potential of antagonists or negative allosteric modulators (NAMs) of group I receptors while agonists or positive allosteric modulators (PAMs) of group II and III receptors are of interest.
5.1 Target Potential of Group I mGlu Receptors, Most Notably mGlu5
Of the group I receptors, it is the mGlu5 receptor that has generated most interest and has indeed progressed furthest in terms of clinical potential. However, this success is not directed towards relief of motor symptoms; indeed the efficacy seen with the mGlu5 NAMs such as MPEP and MTEP in the haloperidol and 6-OHDA rat models of PD [130–133] failed to translate into a positive outcome in MPTP-treated primates [134]. However, mGlu5 NAMs have demonstrable efficacy against a host of other symptoms that accompany PD including pain, anxiety and memory deficits [135–137]. Therefore, use of these agents for any other application in PD (such as LID) may benefit patients further in terms of tackling these non-motor signs.
Without doubt, the most successful application of mGlu5 NAMs to date is against LID. This success is consistent with suggestions that raised levels of striatal mGlu5 receptors seen in MPTP-treated primates and PD patients expressing LIDs [25, 26] may contribute to dyskinesia per se by enhancing corticostriatal glutamatergic transmission. The antidyskinetic effects of MPEP and MTEP shown first against levodopa-induced AIMs in the 6-OHDA rat were not affected by tolerance [138] and again correlated with attenuation of striatal signals causally linked to dyskinesia, such as prodynorphin [90, 139, 140] and delta Fos B [141]. An even wider range of mGlu5 NAMs (MPEP, MTEP, fenobam and AFQ-056) showed significant attenuation of LID in the MPTP-treated primate model, importantly without compromising the antiparkinsonian effects of levodopa [134, 142–144]. These antidyskinetic actions are thought to reflect blockade of abundant mGlu5 receptors on projection neurons of the direct striatal output pathway, leading to normalization of firing [134], consistent with the attenuation of striatal signals described above. Perhaps most exciting of all, AFQ-056 has produced significant antidyskinetic effects in two randomized, placebo-controlled trials of 29 patients with moderate or moderate to severe LID, again without compromising the antiparkinsonian efficacy of levodopa [145]. So here is a glutamate-based approach that shows good translation from pre-clinical through to clinical studies. Addex Therapeutics’ recent press release [146] raises further hopes that a breakthrough in the treatment of LID may be imminent. In their phase IIa, multi-centre, double-blind, placebo-controlled trial of 76 patients with moderate or severe LID, they reported the mGlu5 NAM, ADX-49621 (dipraglurant) to be safe, well tolerated and show good efficacy against LID without lessening the antiparkinsonian effects of levodopa.
Studies exploring the neuroprotective potential of targeting group I mGlu receptors have again largely focused on mGlu5 NAMs like MPEP, which provided partial protection against toxin-induced lesions of the nigrostriatal tract in 6-OHDA-lesioned rats and MPTP-treated mice [123, 136, 147–149] possibly through abolishing STN hyperactivity or microglial activation in the SNpc [123, 137]. MTEP also prevented degeneration of dopaminergic neurons, as well as the development of parkinsonian symptoms, in MPTP-treated primates [150]. However, some studies failed to demonstrate protection against 6-OHDA- or rotenone-induced nigrostriatal tract degeneration in rats [133, 151] while others even suggested mGlu5 receptors play a role in enhancing degeneration, since mGlu5 knockout animals displayed less toxin-induced degeneration than their wild-type littermates [147, 152]. With such contrasting findings, further studies are needed to clarify whether mGlu5 NAMs are worthy candidates for modifying disease progression.
5.2 Target Potential of Group II mGlu Receptors, mGlu2 and mGlu3
Group II receptors have been little studied in relation to symptomatic benefit, despite early observations of reversal of akinesia in reserpine-treated rats following intracerebroventricular (i.c.v.) injection of the broad-spectrum group II agonists LY-379268 and DCG-IV [153, 154]. Moreover, in the one study against LID, LY-379268 failed to reduce AIMs in the 6-OHDA-lesioned rat when co-administered with chronic levodopa [90]. However, the disease-modifying potential of these agents looks promising. DCG-IV, APDC or LY-379268 provided partial protection against 6-OHDA- or MPTP-induced lesions in rodent models of PD [148, 154–156] and, importantly from a translational perspective, this protection was retained even when the drug was administered 48 h after the lesion was induced [157]. Whether this protection stems from activation of mGlu2 or mGlu3 remains to be established. However, studies showing that the protective effects of LY-379268 against MPTP-induced degeneration were maintained in mGlu2−/− mice, but lost in mGlu3−/− mice [158] point to the involvement of mGlu3 receptors. This is consistent with the ability of glial mGlu3 receptor activation to increase production of a range of protective growth factors including transforming growth factor-β1, BDNF and GDNF both in vitro [159, 160] and in vivo [161, 162]. Indeed, systemic injection of LY-379268 enhanced striatal GDNF expression at doses that protected against MPTP-induced degeneration in mice [161]. Given that selective activation of mGlu2 receptors alone may even be detrimental to cell viability, enhancing the toxicity of other agents such as β-amyloid for instance [163], the selective orally-available mGlu2 PAMs under development [164] are unlikely to produce favourable leads in this area. Instead, broad-spectrum agonists with better PK or selective mGlu3 PAMs (not yet available) [165] would seem a more fruitful path to explore.
5.3 Target Potential of Group III mGlu Receptors, mGlu4, mGlu7 and mGlu8
Given their pre-synaptic localization on a number of overactive pathways in the parkinsonian basal ganglia, e.g. corticostriatal, striatopallidal and subthalamonigral pathways, it is not surprising that group III receptors are attracting much attention [129, 166].
Broad-spectrum group III agonists, including LAP-4, LSOP and ACPT-I have shown good efficacy at reversing symptoms in a wide range of rodent models of PD (reserpine-induced akinesia, haloperidol-induced catalepsy or 6-OHDA-induced akinesia), following injection into various sites that receive increased transmission (striatum, globus pallidus, SNpr) [167–171] or more diffusely into the cerebral ventricles [167, 172, 173]. To date, selective targeting of mGlu4 receptors looks most promising, since a range of agonists (LSP-13081, LSP-12111) and PAMs (e.g. PHCCC) were effective in rat models following direct injection into the striatum [170], globus pallidus [174] or SNpr [175] or following i.c.v. or systemic injection [174, 176], whereas the mGlu8-selective agonist (S)-DCPG, failed to provide antiparkinsonian relief [169, 170, 174, 175]. The target potential of mGlu7 receptors requires further clarification. Although the mGlu7 preferring allosteric agonist, AMN-082, administered directly into the SNpr [175], striatum [177] or orally [177] displays antiparkinsonian actions, given the rich pharmacology of AMN-082 [178], agents with improved specificity for mGlu7 are needed for future exploratory studies. The emphasis on the target potential of mGlu4 has led to a recent rush on the development of mGlu4 PAMs with good oral bioavailability including VU-400195 [179], VU-0364770 [180] and LuAF-21934 [181]. Considering these agents consistently display good symptomatic efficacy in the rat models of PD, evidence of their effectiveness in MPTP-treated primates is now eagerly awaited. Since activation of mGlu4 also produces anxiolytic- and antipsychotic-like effects [182, 183], it follows that relief of motor and some non-motor signs may be gained by use of these agents.
Regarding the antidyskinetic potential of targeting group III mGlu receptors, recent reports of levodopa-sparing activity of the mGlu4 PAMs, VU-0364770 and LuAF-21934, in rodent models of PD [180, 181] are encouraging. Moreover, the novel mGlu4 orthosteric agonist, LSP-12111, was shown to reduce AIMs in the 6-OHDA-treated mouse when chronically co-administered with levodopa [184], while LuAF-21934 has been found to similarly reduce the incidence of AIMs in the 6-OHDA rat model of LID [181]. These actions may reflect inhibition of corticostriatal glutamate transmission, as has been demonstrated electrophysiologically [181]. Thus, it appears that a hitherto unexplored side to the target potential of mGlu4 in PD may be about to unfold. Broad-spectrum group III mGlu receptor agonists and selective mGlu4 PAMs also protect against 6-OHDA-induced lesions and ensuing motor deficits in rats [148, 171, 185] and against MPTP-induced degeneration in mice [185]. These protective actions may reflect an auto- or hetero-receptor action of mGlu4 receptors, leading to direct or indirect inhibition, respectively, of glutamate release in the SNpc [166, 171, 185, 186]. Alternatively, they may result from anti-inflammatory actions via activation of glial-located mGlu4 receptors [166, 185]. Whatever the mechanism behind this neuroprotection, it remains to be seen whether these positive outcomes in rodents can be recapitulated in MPTP-treated primates, but expectations are certainly high.
6 Conclusions
As our understanding of the role of glutamate in PD and LID has grown, so too have the opportunities for pharmacological intervention. For AMPA receptors, most hope lies with the use of AMPA antagonists in LID, but the failure of perampanel in clinical trials has dealt a blow to this approach and new approaches, perhaps targeting the calcium-permeable receptors are needed. Tackling LID is also where NMDA antagonists, especially NR2B antagonists, have progressed furthest, with reports of success in small-scale clinical trials. Avoiding adverse cognitive effects of these drugs must now be a priority. These agents also exhibit promising neuroprotective efficacy in rodent models, but MPTP-treated primate studies are now needed to challenge this potential. As far as targeting mGlu receptors is concerned, mGlu5 NAMs look most likely of all to succeed in the LID market, given their success to date in phase IIa trials. mGlu5 NAMs may also prove to be effective neuroprotective agents if the results from MPTP-treated primates translate through to patients. Targeting group II mGlu receptors, specifically mGlu3 subtypes is another promising avenue for affording neuroprotection, but the lack of selective agonists or PAMs is holding progress back. Finally, of the group III mGlu receptors, mGlu4 agonists or PAMs are generating most excitement, having displayed efficacy against symptoms, LID and neurodegeneration in rodent models of PD. With the advent of mGlu4 PAMs with improved systemic availability, we are a step closer to establishing whether these benefits translate through to the primate models so that their real potential can be exposed. Hopes are certainly high that a novel glutamate-based treatment for PD, offering improved control of dyskinesia and much needed disease modification may be in sight.
References
de Rijk MC, Tzourio C, Breteler MM, et al. Prevalence of parkinsonism and Parkinson’s disease in Europe: the EUROPARKINSON collaborative study. European community concerted action on the epidemiology of Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;62(1):10–5.
Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8(5):464–74.
Lang AE, Lozano AM. Parkinson’s disease: first of two parts. N Engl J Med. 1998;339(15):1044–53.
Blandini F, Nappi G, Tassorelli C, et al. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol. 2000;62(1):63–88.
Isaacson SH, Hauser RA. Improving symptom control in early Parkinson’s disease. Ther Adv Neurol Disord. 2009;2(6):29–41.
Marsden CD, Parkes JD. Success and problems of long-term levodopa therapy in Parkinson’s disease. Lancet. 1997;1(8007):345–9.
Hametner E, Seppi K, Poewe W. The clinical spectrum of levodopa-induced motor complications. J Neurol. 2010;257(Suppl. 2):S268–75.
Blandini F, Porter RH, Greenamayre JT. Glutamate and Parkinson’s disease. Mol Neurobiol. 1996;12(1):73–94.
Hirsch EC, Périer C, Orieux G, et al. Metabolic effects of nigrostriatal denervation in basal ganglia. Trends Neurosci. 2000;23(10 Suppl):S78–85.
Obeso JA, Rodríguez-Oroz MC, Benitez-Temino B, et al. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord. 2008;23(Suppl. 3):S548–59.
Duty S, Jenner P. Animal models of Parkinson’s disease: a source of novel treatments and clues to the cause of the disease. Br J Pharmacol. 2011;164(4):1357–91.
Kashani A, Batancur C, Giros B, et al. Altered expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in Parkinson’s disease. Neurobiol Aging. 2007;28(4):568–78.
Jiménez-Jiménez FJ, Molina JA, Vargas C, et al. Neurotransmitter amino acids in cerebrospinal fluid of patients with Parkinson’s disease. J Neurol Sci. 1996;141(1–2):39–44.
Kuiper MA, Teerlink T, Visser JJ, Bergmans PLM, et al. l-Glutamate, l-arginine and l-cirtulline levels in cerebrospinal fluid of Parkinson’s disease, multiple system atrophy, and Alzheimer’s patients. J Neural Transm. 2000;107(2):183–9.
Ondarza R, Velasco F, Velasco M, et al. Neurotransmitter levels in cerebrospinal fluid in relation to severity of symptoms and response to medical therapy in Parkinson’s disease. Stereotact Funct Neurosurg. 1994;62(1–4):90–7.
Gerlach M, Gsell W, Kornhuber J, et al. A post mortem study on neurochemical markers of dopaminergic, GABA-ergic and glutamatergic neurons in basal ganglia thalamo-cortical circuits in Parkinson syndrome. Brain Res. 1996;741(1–2):142–52.
Rinne JO, Halonen T, Riekkinen PJ, et al. Free amino acids in the brain of patients with Parkinson’s disease. Neurosci Lett. 1988;94(1–2):182–6.
Cenci MA, Lundblad M. Post- versus presynaptic plasticity in l-dopa-induced dyskinesia. J Neurochem. 2006;99(2):381–92.
Jenner P. Molecular mechanisms of l-dopa-induced dyskinesia. Nat Rev Neurosci. 2008;9(9):665–77.
Calabresi P, Di Fillippo M, Ghiglieri V, et al. Levodopa-induced dyskinesia in patients with Parkinson’s disease: filling the bench-to-bedside gap. Lancet Neurol. 2010;9(11):1106–17.
Iravani MM, McCreary AC, Jenner P. Striatal plasticity in Parkinson’s disease and l-dopa-induced dyskinesia. Parkinsonism Relat Disord. 2012;18(Suppl. 1):S123–5.
Sgambato-Faure V, Cenci MA. Glutamatergic mechanisms in the dyskinesia induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol. 2012;96(1):69–86.
Blandini F, Armentero MT. New pharmacological avenues for the treatment of l-dopa-induced dyskinesia in Parkinson’s disease: targeting glutamate and adenosine receptors. Expert Opin Investig Drugs. 2012;21(2):153–68.
Dupre KB, Ostock CY, Eskow Juanarajs KL, et al. Local modulation of striatal glutamate efflux by serotonin 1A receptor stimulation in dyskinetic, hemiparkinsonian rats. Exp Neurol. 2011;229(2):288–99.
Samadi P, Grégoire L, Morissette M, et al. mGluR5 metabotropic glutamate receptors and dyskinesia in MPTP monkeys. Neurobiol Aging. 2008;29(7):1040–51.
Outtara B, Grégoire L, Mirissette M, et al. Metabotropic glutamate receptor 5 in levodopa-induced motor complications. Neurobiol Aging. 2011;32(7):1286–95.
Samadi P, Grégoire L, Morissette M, Calon F. Hadj Tahar A, Bélanger N, Dridi M, Bédard PJ, Di Paolo T. Basal ganglia group II metabotropic glutamate receptors specific binding in non-human primate model of l-dopa-induced dyskinesias. Neuropharmacology. 2008;54(2):258–68.
Samadi P, Rajput A, Calon F, Grégoire L, Hornykiewicz O, Rajput AH, Di Paolo T. Metabotropic glutamate receptor II in the brains of Parkinsonian patients. J Neuropathol Exp Neurol. 2009;68(4):374–82.
Calon F, Rajput AH, Hornykiewicz O, et al. Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol Dis. 2003;14(3):404–16.
Hallett PJ, Dunah AW, Ravenscroft P, et al. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 2005;48(4):503–16.
Gardoni F, Sgobio C, Pendolino V, et al. Targeting NR2A-containing NMDA receptors reduces l-dopa-induced dyskinesia. Neurobiol Aging. 2012;33(9):2138–44.
Calon F, Morissette M, Ghribi O, et al. Alteration of glutamate receptors in the striatum of dyskinetic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys following dopamine agonist treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26(1):127–38.
Outtara B, Hoyer D, Grégoire L, et al. Changes of AMPA receptors in MPTP monkeys with levodopa-induced dyskinesias. Neuroscience. 2010;167(4):1160–7.
Hurley MJ, Jackson MJ, Smith LA, et al. Immunoautoradiographic analysis of NMDA receptor subunits and associated postsynaptic density proteins in the brain of dyskinetic MPTP-treated common marmosets. Eur J Neurosci. 2005;21(12):3240–50.
Ahmed I, Bose SK, Pavese N, et al. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain. 2011;134(4):979–86.
Santini E, Valjent E, Usiello A, et al. Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in l-dopa-induced dyskinesia. J Neurosci. 2007;27(26):6995–7005.
Ba M, Kong M, Yu G, et al. GluR1 phosphorylation and persistent expression of levodopa-induced motor response alterations in the hemi-parkinsonian rat. Neurochem Res. 2011;36(6):1135–44.
Santini E, Sgamboto-Faure V, Li Q, et al. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in l-dopa-induced dyskinesia. PLoS One. 2010;5(8):e12322.
Wang JQ, Arora A, Yang L, et al. Phosphorylation of AMPA receptors: mechanisms and synaptic plasticity. Mol Neurobiol. 2005;32(3):237–49.
Ba M, Kong M, Yang H, et al. Changes in subcellular distribution and phosphorylation of GluR1 in lesioned striatum of 6-hydroxydopamine-lesioned and l-dopa treated rats. Neurochem Res. 2006;31(11):1337–47.
Silverdale MA, Kobylecki C, Hallett PJ, et al. Synaptic recruitment of AMPA glutamate receptor subunits in levodopa-induced dyskinesia in the MPTP-lesioned nonhuman primate. Synapse. 2010;64(2):177–80.
Oh JD, Russell DS, Vaughan CL, et al. Enhanced tyrosine phosphorylation of striatal NMDA receptor subunits: effect of dopaminergic denervation and l-dopa administration. Brain Res. 1998;813(1):150–9.
Gardoni F, Picconi B, Ghiglieri V, et al. A critical interaction between NR2B and MAGUK in l-dopa-induced dyskinesia. J Neurosci. 2006;26(11):2914–22.
Muñoz A, Li Q, Gardoni F, et al. Combined 5-HT1A and 5-HT1B receptor agonists for the treatment of l-dopa-induced dyskinesia. Brain. 2008;131(12):3380–4.
Picconi B, Centonze D, Håkansson K, et al. Loss of bidirectional striatal synaptic plasticity in l-dopa-induced dyskinesia. Nat Neurosci. 2003;6(5):501–6.
Massey PV, Johnson BE, Moult PR, et al. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24(36):7821–8.
Olanow CW. The pathogenesis of cell death in Parkinson’s disease. Mov Disord. 2007;22(Suppl. 17):S335–42.
Schapira AH, Jenner P. Etiology and pathogenesis of Parkinson’s disease. Mov Disord. 2011;26(6):1049–55.
Blandini F. An update on the potential role of excitotoxicity in the pathogenesis of Parkinson’s disease. Funct Neurol. 2010;25(2):65–71.
Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch Eur J Physiol. 2010;460(2):525–42.
Bevan MD, Bolam JP, Crossman AR. Convergent synaptic input from the neostriatum and the subthalamus onto identified nigrothalamic neurons in the rat. Eur J Neurosci. 1994;6(3):320–34.
Kornhuber J, Kim JS, Kornhuber ME, et al. The cortico-nigral projection: reduced glutamate content in the substantia nigra following frontal cortex ablation in the rat. Brain Res. 1984;322(1):124–6.
Helton TD, Otsuka T, Lee M-C, et al. Pruning and loss of excitatory synapses by the parkin ubiquitin ligase. Proc Natl Acad Sci USA. 2008;105(49):19492–7.
Lücking CB, Dürr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med. 2000;342(21):1560–7.
Abou-Sleiman PM, Healy DG, Wood NW. Causes of Parkinson’s disease: genetics of DJ-1. Cell Tissue Res. 2004;318(1):185–8.
Aleyasin H, Rousseaux MWC, Phillips M, et al. The Parkinson’s disease gene DJ-1 is also a key regulator of stroke-induced damage. Proc Natl Acad Sci USA. 2007;104(47):18748–53.
Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42(4):733–8.
Sawada H, Shimohama S, Tamura Y, et al. Methylphenylpyridium ion (MPP+) enhances glutamate-induced cytotoxicity against dopaminergic neurons in cultures rat mesencephalon. J Neurosci Res. 1996;43(1):55–62.
Hatcher JM, Pennell KD, Miller GW. Parkinson’s disease and pesticides: a toxicological perspective. Trends Pharmacol Sci. 2008;29(6):322–9.
Wu YN, Johnson SW. Rotenone reduces Mg2+-dependent block of NMDA currents in substantia nigra dopamine neurons. Neurotoxicology. 2009;30(2):320–5.
Shimizu K, Matsubara K, Ohtaki K, et al. Paraquat leads to dopaminergic neuronal vulnerability in organotypic midbrain cultures. Neurosci Res. 2003;46(4):523–32.
Ankarcrona M, Dypbukt JM, Bonfoco E, et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15(4):961–73.
Tatton WG, Chalmers-Redman R, Brown D, et al. Apoptosis in Parkinson’s disease: signals for neuronal degradation. Ann Neurol. 2003;53(Suppl. 3):S61–70.
Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson’s disease: evidence supporting it. Ann Neurol. 1992;32(6):804–12.
Chatha BT, Bernard V, Streit P, et al. Synaptic localization of ionotropic glutamate receptors in the rat substantia nigra. Neuroscience. 2000;101(4):1037–51.
Bernard V, Somogyi P, Bolam JP. Cellular, subcellular, and subsynaptic distribution of AMPA-type glutamate receptor subunits in the neostriatum of the rat. J Neurosci. 1997;17(2):819–33.
Klockgether T, Turski L, Honoré T, et al. The AMPA receptor antagonist NBQX has antiparkinsonian effects in monoamine-depleted rats and MPTP-treated monkeys. Ann Neurol. 1991;39(5):717–23.
Löschmann PA, Lange KW, Kunow M, et al. Synergism of the AMPA-antagonist NBQX and the NMDA-antagonist CPP with l-dopa in models of Parkinson’s disease. J Neural Transm Park Dis Dement Sect. 1991;3(3):203–13.
Luquin MR, Obeso JA, Laguna J, et al. The AMPA receptor antagonist NBQX does not alter the motor response induced by selective dopamine agonists in MPTP-treated monkeys. Eur J Pharmacol. 1993;235(2–3):297–300.
Konitsiotis S, Blanchet PJ, Verhagen L, et al. AMPA receptor blockade improves levodopa-induced dyskinesia in MPTP monkeys. Neurology. 2000;54(8):1589–95.
Silverdale MA, Nicholson SL, Crossman AR, et al. Topiramate reduces levodopa-induced dyskinesia in the MPTP-lesioned marmoset model of Parkinson’s disease. Mov Disord. 2005;20(4):403–9.
Marin C, Jimenez A, Bonastre M, et al. LY293558, an AMPA glutamate receptor antagonist, prevents and reverses levodopa-induced motor alterations in Parkinsonian rats. Synapse. 2001;42(1):40–7.
Eggert K, Squillacote D, Barone P, et al. Safety and efficacy of perampanel in advanced Parkinson’s disease: a randomized, placebo-controlled study. Mov Disord. 2010;25(7):896–905.
Lees A, Fahn S, Eggert KM, et al. Perampanel, an AMPA antagonist, found to have no benefit in reducing “off” time in Parkinson’s disease. Mov Disord. 2012;27(2):284–8.
Kobylecki C, Cenci MA, Crossman AR, et al. Calcium-permeable AMPA receptors are involved in the induction and expression of l-dopa-induced dyskinesia in Parkinson’s disease. J Neurochem. 2010;114(2):499–511.
Turski L, Bressler K, Rettig KJ, et al. Protection of substantia nigra from MPP+ neurotoxicity by N-methyl-d-aspartate antagonists. Nature. 1991;349(6308):414–8.
Murray TK, Whalley K, Robinson CS, et al. LY503430, a novel alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor potentiator with functional, neuroprotective and neurotrophic effects in rodent models of Parkinson’s disease. J Pharmacol Exp Ther. 2003;306(2):752–62.
O’Neill MJ, Murray TK, Whalley K, et al. Neurotrophic actions of the novel AMPA receptor potentiator, LY404187, in rodent models of Parkinson’s disease. Eur J Pharmacol. 2004;486(2):163–74.
Muir KW, Grosset DG, Gamzu E, et al. Pharmacological effects of the non-competitive NMDA antagonist CNS 1102 in normal volunteers. Br J Clin Pharmacol. 1994;38(1):33–8.
Low SJ, Roland CL. Review of NMDA antagonist-induced neurotoxicity and implications for clinical development. Int J Clin Pharmacol Ther. 2004;42(1):1–14.
Carlsson M, Svensson A. The non-competitive NMDA antagonists MK-801 and PCP, as well as the competitive NMDA antagonist SDZEAA494 (D-CPPene), interact synergistically with clonidine to promote locomotion in monoamine-depleted mice. Life Sci. 1990;47(19):1729–36.
Crossman AR, Peggs D, Boyce S, et al. Effect of the NMDA antagonist MK-801 on MPTP-induced parkinsonism in the monkey. Neuropharmacology. 1989;28(11):1271–3.
Papa SM, Chase TN. Levodopa-induced dyskinesias improved by a glutamate antagonist in Parkinsonian monkeys. Ann Neurol. 1996;39(5):574–8.
Kelsey JE, Mague SD, Pijanowski RS, et al. NMDA receptor antagonists ameliorate the stepping deficits produce by unilateral medial forebrain bundle injections of 6-OHDA in rats. Psychopharmacology (Berl). 2004;175(2):179–88.
Kosinski CM, Standaert DG, Counihan TJ, et al. Expression of N-methyl-d-aspartate receptor subunit mRNAs in the human brain: striatum and globus pallidus. J Comp Neurol. 1998;390(1):63–74.
Blanchet PJ, Konitsiotis S, Whittemore ER, et al. Differing effects of N-methyl-d-aspartate receptor subtype selective antagonists on dyskinesia in levodopa-treated 1-methyl-4-phenyl-tetrahydropyridine monkeys. J Pharmacol Exp Ther. 1999;290(3):1034–40.
Nash JE, Hill MP, Brotchie JM. Anti-parkinsonian actions of blockade of NR2B-containing NMDA receptors in the reserpine-treated rat. Exp Neurol. 1999;155(1):42–8.
Steece-Collier K, Chambers LK, Jaw-Tsai SS, et al. Antiparkinsonian actions of CP-101,606, an antagonist of NR2B subunit-containing N-methyl-d-aspartate receptors. Exp Neurol. 2000;163(1):239–43.
Löschmann PA, De Groote C, Smith L, et al. Antiparkinsonian activity of Ro 25-6981, a NR2B subunit specific NMDA receptor antagonist, in animal models of Parkinson’s disease. Exp Neurol. 2004;187(1):86–93.
Rylander D, Recchia A, Mela F, et al. Pharmacological modulation of glutamate transmission in a rat model of l-dopa-induced dyskinesia: effects on motor behaviour and striatal nuclear signaling. J Pharmacol Exp Ther. 2009;330(1):227–35.
Nash JE, Fox SH, Henry B, et al. Antiparkinsonian actions of ifenprodil in the MPTP-lesoned marmoset model of Parkinson’s disease. Exp Neurol. 2000;165(1):136–42.
Nash JE, Ravenscroft P, McGuire S, et al. The NR2B-selective NMDA receptor antagonist CP-101,606 exacerbates l-dopa-induced dyskinesia and provides mild potentiation of anti-parkinsonian effects of l-dopa in the MPTP-treated marmoset model of Parkinson’s disease. Exp Neurol. 2004;188(2):471–9.
Addy C, Assaid C, Hreniuk D, et al. Single-dose administration of MK-0657, an NR2B-selective NMDA antagonist, does not result in clinically meaningful improvement in motor function in patients with moderate Parkinson’s disease. J Clin Pharmacol. 2009;49(7):856–64.
Fasano A, Ricciardi L, Pettorusso M, et al. Management of punding in Parkinson’s disease: an open-label prospective study. J Neurol. 2011;258(4):656–60.
Inzelberg R, Bonuccelli U, Schechtman E, et al. Association between amantadine and the onset of dementia in Parkinson’s disease. Mov Disord. 2006;21(9):1375–9.
Emre M, Tsolaki M, Bonucccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomized, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969–77.
Litvinenko IV, Odinak MM, Mogli’naya VI, et al. Use of memantine (akatinol) for the correction of cognitive impairments in Parkinson’s disease complicated by dementia. Neurosci Behav Physiol. 2010;40(2):149–55.
Metman VL, Del Dotto P, LePoole K, et al. Amantadine for levodopa-induced dyskinesia: a 1-year follow-up study. Arch Neurol. 1999;56(11):1383–6.
Luginger E, Wenning GK, Bösch S, et al. Beneficial effects of amantadine on l-dopa-induced dyskinesias in Parkinson’s disease. Mov Disord. 2000;15(5):873–8.
Wolf E, Seppi K, Katzenschlager R, et al. Long-term antidyskinetic efficacy of amantadine in Parkinson’s disease. Mov Disord. 2010;25(10):1357–63.
Hanağasi HA, Kaptanoglu G, Sahin HA, et al. The use of NMDA antagonist memantine in drug-resistant dyskinesia resulting from l-dopa. Mov Disord. 2000;15(5):1016–7.
Varanese S, Howard J, Di Rocco A. NMDA antagonist memantine improves levodopa-induced dyskinesia and “on–off” phenomena in Parkinson’s disease. Mov Disord. 2010;24(4):508–10.
Merello M, Nouzeilles MI, Cammarota A, et al. Effect of memantine (NMDA antagonist) on Parkinson’s disease: a double-blind crossover randomized study. Clin Neuropharmacol. 1999;22(5):273–6.
Engber TM, Papa SM, Boldry RC, et al. NMDA receptor blockade reverses motor response alterations induced by levodopa. Neuroreport. 1994;5(18):2586–8.
Dupre KB, Eskow KL, Steiniger A, et al. Effects of coincident 5-HT1A receptor stimulation and NMDA receptor antagonism on l-dopa-induced dyskinesia and rotational behaviours in the hemi-parkinsonian rat. Psychopharmacology. 2008;199(1):99–108.
Paquette MA, Anderson AM, Lewis JR, et al. MK-801 inhibits l-dopa-induced abnormal involuntary movements only at doses that worsen parkinsonism. Neuropharmacology. 2010;58(7):1002–8.
Gomez-Mancilla B, Bédard PJ. Effect of nondopaminergic drugs on l-dopa-induced dyskinesia in MPTP-treated monkeys. Clin Neuropharmacol. 1993;16(5):418–27.
Wessell RH, Ahmed SM, Meniti FS, et al. NR2B selective NMDA receptor antagonist CP-101,606 prevents levodopa-induced motor response alterations in hemi-parkinsonian rats. Neuropharmacology. 2004;47(2):184–94.
Truong L, Allbutt HN, Coster MJ, et al. Behavioural effects of a selective NMDA NR1A/2B receptor antagonist in rats with unilateral 6-OHDA + parafasicular lesions. Brain Res Bull. 2009;78(2–3):91–6.
Warraich ST, Allbutt HN, Billing R, et al. Evaluation of behavioural effects of a selective NMDA NR1A/2B receptor antagonist in the unilateral 6-OHDA lesion rat model. Brain Res Bull. 2009;78(2–3):85–90.
Morissette M, Dridi M, Calon F, et al. Prevention of levodopa-induced dyskinesias by a selective NR1/NR2B N-methyl-d-aspartate receptor antagonist in parkinsonian monkeys: implication of preproenkephalin. Mov Disord. 2006;21(1):9–17.
Tamim MK, Samadi P, Morissette M, et al. Effect of non-dopaminergic drug treatment on levodopa induced dyskinesia in MPTP monkeys: common implications of striatal peptides. Neuropharmacology. 2010;58(1):286–96.
Papa SM, Boldry RC, Enger TM, et al. Reversal of levodopa-induced motor fluctuations in experimental parkinsonism by NMDA receptor blockade. Brain Res. 1995;701(1–2):13–8.
Nutt JG, Gunzler SA, Kirchnoff T, et al. Effects of a NR2B selective NMDA glutamate antagonist, CP-101,606, on dyskinesia and Parkinsonism. Mov Disord. 2008;23(13):1860–6.
Kanthasamy AG, Kanthasamy A, Matsumoto RR, et al. Neuroprotective effects of the strychnine-insensitive glycine site NMDA antagonist (R)-HA-966 in an experimental model of Parkinson’s disease. Brain Res. 1997;759(1):1–8.
Brouillet E, Beal MF. NMDA antagonists partially protect against MPTP induced neurotoxicity in mice. Neuroreport. 1993;4(4):387–90.
Chan P, Di Monte DA, Langston JW, et al. (+)MK-801 does not prevent MPTP-induced loss of nigral neurons in mice. J Pharmacol Exp Ther. 1997;280(1):439–46.
Sonsalla PK, Zeevalk GD, Manzino L, et al. MK-801 fails to protect against the dopaminergic neuropathology produced by systemic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice or intranigral 1-methyl-4-phenylpyridinium in rats. J Neurochem. 1992;58(5):1979–82.
Kupsch A, Löschmann PA, Suer H, et al. Do NMDA receptor antagonists protect against MPTP-toxicity? Biochemical and immunocytochemical analyses in black mice. Brain Res. 1992;592(1–2):74–83.
Zuddas A, Vaglini F, Fornai F, et al. Pharmacologic modulation of MPTP toxicity: MK-801: prevention of dopaminergic cell death in monkeys and mice. Ann N Y Acad Sci. 1992;648:268–71.
Lange KW, Löschmann PA, Sofic E, et al. The competitive NMDA antagonist CPP protects substantia nigra neurons from MPTP-induced degeneration in primates. Naunyn Schmeidbergs Arch Pharmacol. 1993;348(6):586–92.
Blandini F, Nappi G, Greenamyre JT. Subthalamic infusion of an NMDA antagonist prevents basal ganglia metabolic changes and nigral degeneration in a rodent model of Parkinson’s disease. Ann Neurol. 2001;49(4):525–9.
Armentero M-T, Fancekku R, Nappi G, et al. Prolonged blockade of NMDA or mGluR5 glutamate receptors reduces nigrostriatal degeneration while inducing selective metabolic changes in the basal ganglia circuitry in a rodent model of Parkinson’s disease. Neurobiol Dis. 2006;22(1):1–9.
Lee MC, Ting KK, Adams S, et al. Characterisation of the expression of NMDA receptors in human astrocytes. PLoS One. 2010;5(11):e14123.
Wu HM, Tzeng NS, Qian L, et al. Novel neuroprotective mechanisms of memantine: increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology. 2009;34(10):2344–57.
Ossola B, Schendzielorz N, Chen SH, et al. Amantadine protects dopamine neurons by a dual action: reducing activation of microglia and inducing expression of GDNF in astroglia. Neuropharmacology. 2011;61(4):574–82.
Leaver KR, Allbutt HN, Creber NJ, et al. Neuroprotective effects of a selective N-methyl-d-aspartate NR2B receptor antagonist in the 6-hydroxydopamine rat model of Parkinson’s disease. Clin Exp Pharmacol Physiol. 2008;35(11):1388–94.
Ng OT, Chen LW, Chan YS, et al. Small interfering RNA specific for N-methyl-d-aspartate receptor 2B offers neuroprotection to dopamine neurons through activation of MAP kinase. Neurosignals (Epub 2012 Feb 23).
Conn PJ, Battaglia G, Marino MJ, et al. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci. 2005;6(10):787–98.
Ossowska K, Konieczny J, Wolfarth S, et al. Blockade of the metabotropic glutamate receptor subtype 5 (mGluR5) produces antiparkinsonian-like effects in rats. Neuropharmacology. 2001;41(4):413–20.
Breysse N, Baunez C, Spooren W, et al. Chronic but not acute treatment with a metabotropic glutamate 5 receptor antagonist reverses the akinetic deficits in a rat model of Parkinson’s disease. J Neurosci. 2002;22(13):5669–78.
Dekundy A, Pietraszek M, Schefer D, et al. Effects of group I metabotropic glutamate receptors blockade in experimental models of Parkinson’s disease. Brain Res Bull. 2006;69(3):318–26.
Ambrosi G, Arementero MT, Levandis G, et al. Effects of early and delayed treatment with an mGluR5 antagonist on motor impairment, nigrostriatal damage and neuroinflammation in a rodent model of Parkinson’s disease. Brain Res Bull. 2010;82(1–2):29–38.
Johnston TH, Fox SH, McIldowie MJ, et al. Reduction of l-dopa-induced dyskinesia by the selective metabotropic glutamate receptor 5 antagonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J Pharmacol Exp Ther. 2010;333(3):865–73.
De Leonibus E, Managò F, Giordani F, et al. Metabotropic glutamate receptors 5 blockade reverses spatial memory deficits in a mouse model of Parkinson’s disease. Neuropsychopharmacology. 2009;34(3):729–38.
Chen L, Liu J, Ali U, et al. Chronic, systemic treatment with a metabotropic glutamate receptor 5 antagonist produces anxiolytic-like effects and reverses abnormal firing activity of projection neurons in the basolateral amygdala in rats with bilateral 6-OHDA lesions. Brain Res Bull. 2011;84(3):215–23.
Hsieh MH, Ho SC, Yeh KY, et al. Blockade of metabotropic glutamate receptors inhibits cognition and neurodegeneration in an MPTP-induced Parkinson’s disease rat model. Pharmacol Biochem Behav. 2012;102(1):64–71.
Gravius A, Dekundy A, Nagel J, et al. Investigation on tolerance development to subchronic blockade of mGluR5 in models of learning, anxiety, and levodopa-induced dyskinesia in rats. J Neural Transm. 2008;115(12):1609–19.
Mela F, Marti M, Dekundy A, et al. Antagonism of metabotropic glutamate receptor type 5 attenuates l-dopa-induced dyskinesia and its molecular and neurochemical correlates in a rat model of Parkinson’s disease. J Neurochem. 2007;101(2):483–97.
Yamamoto N, Soghomonian JJ. Metabotropic glutamate mGluR5 receptor blockade opposes abnormal involuntary movement and the increases in glutamic acid decarboxylase mRNA levels in striatal neurons of 6-hydroxydopamine-lesioned rats. Neuroscience. 2009;163(4):1171–80.
Levandis G, Bazzini E, Armentero MT, et al. Systemic administration of an mGluR5 antagonist, but not unilateral subthalamic lesion, counteracts l-dopa-induced dyskinesias in a rodent model of Parkinson’s disease. Neurobiol Dis. 2008;29(1):161–8.
Rylander D, Iderberg H, Li Q, et al. A mGluR5 antagonist under clinical development improves l-dopa-induced dyskinesia in parkinsonian rats and monkeys. Neurobiol Dis. 2010;39(3):352–61.
Morin N, Grégoire L, Gomez-Mancilla B, et al. Effect of the metabotropic glutamate receptor type 5 antagonists MPEP and MTEP in parkinsonian monkeys. Neuropharmacology. 2010;58(7):981–6.
Grégoire L, Morin N, Ouattara B, et al. The acute antiparkinsonian and antidyskinetic effect of AFQ056, a novel metabotropic glutamate receptor 5 antagonist. l-dopa-treated parkinsonian monkeys. Parkinsonism Relat Disord. 2011;17(4):270–6.
Berg D, Godau J, Trenkwalder C, et al. AFQ056 treatment of levodopa-induced dyskinesia: results of 2 randomized controlled trials. Mov Disord. 2011;26(7):1243–50.
Addex Therapeutics. Geneva, Switzerland [media release]. 2012 Mar 21 [online]. Available from URL: http://www.addextherapeutics.com/investors/press-releases/news-details/article/addex-reports-positive-top-line-phase-iia-data-for-dipraglurant-in-parkinsons-disease-levodopa-indu/ (Accessed 2012 Oct 3).
Battaglia G, Busceti CL, Molinaro G, et al. Endogenous activation of mGlu5 metabotropic glutamate receptors contributes to the development of nigro-striatal damage induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. J Neurosci. 2004;24(4):828–35.
Vernon AC, Palmer S, Datla K, et al. Neuroprotective effects of metabotropic glutamate receptor ligands in a 6-hydroxydopamine rodent model of Parkinson’s disease. Eur J Neurosci. 2005;22(7):1799–806.
Aguirre JA, Kehr J, Yoshitake T, et al. Protection but maintained dysfunction of nigral dopaminergic nerve cell bodies and striatal dopaminergic terminals in MPTP-lesioned mice after acute treatment with the mGluR5 antagonist MPEP. Brain Res. 2005;1033(2):216–20.
Masilamoni GJ, Bogenpohl JW, Alagille D, et al. Metabotropic glutamate receptor 5 antagonist protects domaminergic and noradrenergic neurons from degeneration in MPTP-treated monkeys. Brain. 2011;134(7):2057–73.
Alam M, Danysz W, Schmidt WJ, et al. Effects of glutamate and alpha-2-noradrenergic receptor antagonists on the development of neurotoxicity produced by chronic rotenone in rats. Toxicol Appl Pharmacol. 2009;240(2):198–207.
Black YD, Xiao D, Pellegrino D, et al. Protective effect of metabotropic glutamate mGluR5 receptor elimination in a 6-hydroxydopamine model of Parkinson’s disease. Neurosci Lett. 2010;486(3):161–5.
Dawson L, Chadha A, Megalou M, et al. The group II metabotropic glutamate receptor agonist, DCG-IV, alleviates akinesia following intranigral or intracerebroventricular administration in the reserpine-treated rat. Br J Pharmacol. 2000;129(3):541–6.
Murray TK, Messenger MJ, Ward MA, et al. Evaluation of the mGluR2/3 agonist LY379268 in rodent models of Parkinson’s disease. Pharmacol Biochem Behav. 2002;73(2):455–66.
Battaglia G, Busceti CL, Pontarelli F, et al. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology. 2003;45(2):155–66.
Matarredona ER, Santiago M, Venero JL, et al. Group II metabotropic glutamate receptor activation protects striatal dopaminergic nerve terminals against MPP+-induced neurotoxicity along with brain-derived neurotrophic factor induction. J Neurochem. 2001;76(2):351–60.
Chan H, Paur H, Vernon AC, et al. Neuroprotection and functional recovery associated with decreased microglial activation following selective activation of mGluR2/3 receptors in a rodent model of Parkinson’s disease. Parkinsons Dis. 2010;2010. pii: 190450.
Corti C, Battaglia G, Molinaro G, et al. The use of knock-out mice unravels distinct roles for mGlu2 and mGlu3 metabotropic glutamate receptors in mechanisms of neurodegeneration/neuroprotection. J Neurosci. 2007;27(31):8297–308.
Bruno V, Sureda FX, Storto M, et al. The neuroprotective activity of group II metabotropic glutamate receptors requires new protein synthesis and involves a glial-neuronal signaling. J Neurosci. 1997;17(6):1891–7.
Ciccarelli R, Di Iorio P, Bruno V, et al. Activation of A(1) adenosine or mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100beta protein from cultured astrocytes. Glia. 1999;27(3):275–81.
Battaglia G, Molinaro G, Riozzi B, et al. Activation of mGlu3 receptors stimulates the production of GDNF in striatal neurons. PLoS ONE. 2009;4(8):e6591.
Di Liberto V, Bonomo A, Frinchi M, et al. Group II metabotropic glutamate receptor activation by agonist LY379268 treatment increases the expression of brain derived neurotrophic factor in the mouse brain. Neuroscience. 2010;165(3):863–73.
Caraci F, Molinaro G, Battaglia G, et al. Targeting group II metabotropic glutamate (mGlu) receptors for the treatment of psychosis associated with Alzheimer’s disease: selective activation of mGlu2 receptors amplifies beta-amyloid toxicity in cultures neurons, whereas dual activation of mGlu2 and mGlu3 is neuroprotective. Mol Pharmacol. 2011;79(3):618–26.
D’Alessandro PL, Corti C, Roth A, et al. The identification of structurally novel, selective, orally bioavailable positive modulators of mGluR2. Bioorg Med Chem Lett. 2010;20(2):759–62.
Caraci F, Battaglia G, Sortino MA, et al. Metabotropic glutamate receptors in neurodegeneration/neuroprotection: still a hot topic? Neurochem Int. 2012;61(4):559565.
Duty S. Therapeutic potential of targeting group III metabotropic glutamate receptors in the treatment of Parkinson’s disease. Br J Pharmacol. 2010;161(2):271–87.
MacInnes N, Messenger MJ, Duty S. Activation of group III metabotropic glutamate receptors in selected regions of the basal ganglia alleviates akinesia in the reserpine-treated rat. Br J Pharmacol. 2004;141(1):15–22.
Konieczny J, Wardas J, Kuter K, et al. The influence of group III metabotropic glutamate receptor stimulation by (1S,3R,4S)-1-aminocyclopentane-1,3,4-tricarboxylic acid on the parkinsonian like akinesia and striatal proenkephalin and prodynorphin expression in rats. Neuroscience. 2007;145(2):611–20.
Lopez S, Turle-Lorenzo N, Acher F, et al. Targeting group III metabotropic glutamate receptors produces complex behavioural effects in rodent models of Parkinson’s disease. J Neurosci. 2007;27(25):6701–11.
Cuomo D, Martella G, Barabino E, et al. Metabotropic glutamate receptor subtype 4 selectively modulates both glutamate and GABA transmission in the striatum: implications for Parkinson’s disease treatment. J Neurochem. 2009;109(4):1096–105.
Austin PJ, Betts MJ, Broadstock M, et al. Symptomatic and neuroprotective effects following activation of nigral group III metabotropic glutamate receptors in rodent models of Parkinson’s disease. Br J Pharmacol. 2010;160(7):1741–53.
Valenti O, Marino MJ, Wittmann M, et al. Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J Neurosci. 2003;23(18):7218–26.
Lopez S, Turle-Lorenzo N, Johnston TH, et al. Functional interaction between adenosine A2A and group III metabotropic glutamate receptors to reduce parkinsonian symptoms in rats. Neuropharmacology. 2008;55(4):483–90.
Beurrier C, Lopez S, Révy D, et al. Electrophysiological and behavioural evidence that modulation of metabotropic glutamate receptor 4 with a new agonist reverses experimental parkinsonism. FASEB J. 2009;23(10):3619–28.
Broadstock M, Ausin PJ, Betts MJ, et al. Antiparkinsonian potential of targeting group III metabotropic glutamate receptor subtypes in the rodent substantia nigra pars reticulata. Br J Pharmacol. 2012;165(4b):1034–45.
Marino MJ, Williams DL Jr, O’Brien JA, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc Natl Acad Sci USA. 2003;100(23):13668–73.
Greco B, Lopez S, van der Putten H, et al. Metabotropic glutamate 7 receptor subtype modulates motor symptoms in rodent models of Parkinson’s disease. J Pharmacol Exp Ther. 2010;332(3):1064–71.
Sukoff-Rizzo S, Leonard SK, Gilbert A, et al. The mGluR7 allosteric agonist AMN082 is a monoaminergic agent in disguise? J Pharmacol Exp Ther. 2011;338(1):345–52.
Jones CK, Engers DW, Thompson AD, et al. Discovery, synthesis and structure-activity relationship development of a series of N-4-(2,5-dioxopyrrolidin-1-yl)phenylpicolinamides (VU0400195, ML182): characterization of novel positive allosteric modulator of the metabotropic glutamate receptor 4 (mGlu(4)) with oral efficacy in an antiparkinsonian animal model. J Med Chem. 2011;54(21):7639–47.
Jones CK, Bubser M, Thompson AD, et al. The metabotropic glutamate receptor 4-positive allosteric modulator VU0364770 produces efficacy alone and in combination with l-dopa or an adenosine 2A antagonist in preclinical rodent models of Parkinson’s disease. J Pharmacol Exp Ther. 2012;340(2):404–21.
Bennouar KE, Uberti MA, Melon C, et al. Synergy between l-dopa and a novel positive allosteric modulator of metabotropic glutamate receptor 4: implications for Parkinson’s disease treatment and dyskinesia. Neuropharmacology (Epub 2012 Apr 3).
Wierońska JM, Stachowicz K, Palucha-Ponciewiera A, et al. Metabotropic glutamate receptor 4 novel agonist LSP1-2111 with anxiolytic, but not antidepressant activity, mediated by serotonergic and GABAergic systems. Neuropharmacology. 2010;59(7–8):627–34.
Wierońska JM, Stachowicz K, Acher F, et al. Opposing efficacy of group III mGlu receptor activators, LSP1-2111 and AMN082, in animal models of positive symptoms of schizophrenia. Psychopharmacology (Berl). 2012;220(3):481–94.
Lopez S, Bonito-Oliva A, Pallottino S, et al. Activation of metabotropic glutamate 4 receptors decreases l-dopa-induced dyskinesia in a mouse model of Parkinson’s disease. J Parkinsons Dis. 2011;1(4):339–46.
Betts MJ, O’Neill MJ, Duty S. Allosteric modulation of the group III mGlu receptor 4 provides functional neuroprotection in the 6-OHDA rat model of Parkinson’s disease. Br J Pharmacol. 2012;166(8):2317–30.
Battaglia G, Busceti CL, Molinaro G, et al. Pharmacological activation of mGlu4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurosci. 2006;26(27):7222–9.
Acknowledgments
SD has received grant funding/research support on projects relating to glutamate in Parkinson’s disease from the Wellcome Trust, UK Medical Research Council, Guy’s King’s and St Thomas’ Charitable trustees, Parkinson’s UK and Eli Lilly and Co Ltd. However, no sources of funding have been used to prepare this review. There are no conflicts of interest to note.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Duty, S. Targeting Glutamate Receptors to Tackle the Pathogenesis, Clinical Symptoms and Levodopa-Induced Dyskinesia Associated with Parkinson’s Disease. CNS Drugs 26, 1017–1032 (2012). https://doi.org/10.1007/s40263-012-0016-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-012-0016-z