
Abstract
Autism spectrum disorder (ASD), one of the most common childhood neurodevelopmental disorders (NDDs), is diagnosed in 1 of every 68 children. ASD is incredibly heterogeneous both clinically and aetiologically. The etiopathogenesis of ASD is known to be complex, including genetic, environmental and epigenetic factors. Normal epigenetic marks modifiable by both genetics and environmental exposures can result in epigenetic alterations that disrupt the regulation of gene expression, negatively impacting biological pathways important for brain development. In this chapter we aim to summarize some of the important literature that supports a role for epigenetics in the underlying molecular mechanism of ASD. We provide evidence from work in genetics, from environmental exposures and finally from more recent studies aimed at directly determining ASD-specific epigenetic patterns, focusing mainly on DNA methylation (DNAm). Finally, we briefly discuss some of the implications of current research on potential epigenetic targets for therapeutics and novel avenues for future work.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Autism Spectrum Disorder (ASD) and Proposed Aetiologies
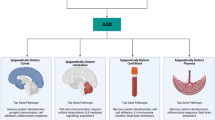
Autism spectrum disorder (ASD), one of the most common neurodevelopmental disorders (NDDs), is diagnosed in 1 of 68 children in the United States [1] with a 4:1 male-to-female sex ratio. ASD is comprised of a group of complex NDDs characterized by impaired social communication and repetitive behaviours (DSM-5). ASD also presents with a range of other features including morphological (e.g. macrocephaly), physiological (e.g. gastrointestinal, sleep problems) and psychiatric comorbidities (e.g. anxiety) [2]. Variable neuropathological features consistently described in some, but not all, cases of ASD include decreased size and number of Purkinje cells, abnormal neuronal migration, neurite outgrowth and branching and axonal guidance [3, 4]. These additional features may or may not be observed as part of the profile of syndromic cases (Sect. 4.4.1), where ASD is associated with either single-gene mutations or defined chromosomal/cytogenetic abnormalities. In May 2013, the clinical criteria for the diagnosis of ASD were redefined by the DSM-5 diagnostic manual. Several subtypes of ASD previously considered as distinct disorders (autistic disorder, childhood disintegrative disorder, pervasive developmental disorder—not otherwise specified (PDD-NOS) and Asperger syndrome) were then merged under a single umbrella diagnosis, ASD. The clinical criteria for the diagnosis of ASD are still evolving due in part to phenotypic variability and to clinical and aetiologic overlap with other NDDs (e.g. ~30% of ASD cases are comorbid with attention deficit and hyperactivity disorder [ADHD] symptoms) [5, 6]. Therefore, different NDDs are best represented not as distinct categories but as entities along a continuum with some convergence in the underlying genes and pathways. One of the greatest challenges in improving diagnosis and treatment for ASD is the degree of heterogeneity both clinically and aetiologically. It is therefore unsurprising that there are multiple proposed aetiologies and risk factors (Fig. 4.1) identified.

Diagrammatic overview of how genetic, environmental and epigenetic factors interact in the aetiology of ASD. Epigenetic load (from preconception, prenatal environment and stochastic variation) and genetic load (from familial and de novo variation) interact to alter neurodevelopmental, immune, oxidative stress and mitochondrial pathways identified through studies of ASD genetics, physiology, expression and/or DNA methylation (targeted and genome-wide). Highlighted in grey is the putative involvement of specific genes or pathways mentioned in this review. Over a certain threshold of genetic and epigenetic dysfunction, development is compromised and can lead to adverse neurodevelopmental outcomes such as ASD. Further, postnatal environments may also contribute to severity of symptoms. Abbreviations: DLGAP1 DLG-associated protein 1, OCM one carbon metabolism, OXTR oxytocin receptor, MECP2 methyl-CpG-binding protein 2, PRRT1 proline-rich transmembrane protein 1, RORA retinoic acid receptor-related orphan receptor alpha, SHANK3 SH3 and multiple ankyrin repeat domains 3, TNF-α tumour necrosis factor alpha. Adapted from Fig. 1 in [7]
Commonly proposed physiological and metabolic causes of ASD consist of immune, oxidative stress and mitochondrial dysfunction. In some ASD cases, a pro-inflammatory state [8,9,10,11,12] is suggested by alterations in immune and inflammatory markers (e.g. cells of the innate and adaptive [cytokines, interleukins] immune system and abnormalities in microglia [immune cells of the brain]). Furthermore, it has been shown that immune changes in both peripheral tissues and brain can result in anxiety and impaired social behaviour [13,14,15,16]. Studies in humans and mouse models suggest that dysbiosis of the intestinal microbiome is a novel physiological contributor to ASD risk, impacting immune function and subsequently learning, memory and behaviour [17,18,19,20]. Further, there is emerging evidence, mostly in animal models, that this gut-brain axis is, in part, regulated by epigenetic mechanisms [21,22,23]. Enhanced oxidative stress, impaired antioxidative capacity and mitochondrial dysfunction have also been extensively reviewed [12, 24]. A number of independent studies support claims that the degree of immune disruption (e.g. cytokines) and mitochondrial (e.g. phosphocreatine) dysfunction are positively correlated with ASD symptom severity [12, 24, 25]. However, it is not known whether these abnormalities existed prior to ASD diagnosis or if they play a causal role.
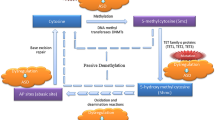
Investigations of underlying molecular mechanisms in ASD aetiology include altered genetic and epigenetic regulation. Epigenetics has emerged as a vital genome-wide regulatory layer that modulates the transcriptome, impacting transcription initiation, splicing processes and binding of transcription factors. Epigenetic regulation helps to determine the proper spatiotemporal expression of genes via a number of mechanisms including DNA methylation (DNAm), histone modifications and ATP-dependent chromatin remodelling. Epigenetics provides new avenues to investigate and refine risk estimates for NDDs beyond genetic risks alone. The failure to establish proper epigenetic marks may result in aberrant gene expression and, subsequently, various disease phenotypes. Genomic aberrations (e.g. mutations, insertions/deletions, copy number variants [CNVs]) of genes involved in epigenetic regulation (‘epigenes’) or dysregulation of epigenetically regulated genomic regions (e.g. imprinted genes/regions) can lead to epigenetic disruptions and, ultimately, NDDs. There are >600 confirmed and putative human epigenes [26], many of which are associated with NDDs such as intellectual disability (ID) and ASD (e.g. chromodomain helicase DNA-binding protein 8 [CHD8], DNA methyltransferase 3A [DNMT3A], HECT, UBA and WWE domain-containing 1, E3 ubiquitin protein [HUWE1]) [27, 28]. There is a growing body of evidence to show that there is substantial genetic overlap between risks for neuropsychiatric disorders and NDDs [29,30,31,32]. Many of these genes encode proteins involved in neuronal and synaptic pathways, while others are relevant to molecular pathways involved in epigenetic regulation [28, 32, 33] (Sects. 4.2, 4.3 and 4.4).
What is clear is that there is no single underlying cause of ASD. In order to understand the multifactorial aetiologies of ASD, we must better understand the natural history of molecular events and their regulation during critical periods of human development. ASD more likely arises as an interaction between genetic and environmental risk factors (GxE) mediated by epigenetic mechanisms. Ultimately, a better understanding of how genetics, epigenetics and environment collectively interact and contribute to ASD risk will allow us to better classify and diagnose the disorder and facilitate the application of precision-based medicine.
2 Genetics of ASD
The aetiology of ASD is known to have a strong genetic component. Early twin studies of heritability reported estimates of up to 90% heritability [34, 35]. In contrast, current estimates are closer to 10–30% according to data from more recent twin and family studies [36,37,38]. Next generation sequencing has significantly accelerated our understanding of genetic variability in individuals with ASD compared to the general population. ASD genomic risk variants are comprised of rare, de novo variants of large effect, independently of or in combination with more common and/or inherited variants of small effect [39]. Interestingly, it has recently been shown, using whole genome sequencing (WGS) of 85 ASD quartet families (parents and two affected siblings), that although ASD-relevant mutations were found in 42% of individuals with ASD, only 31% of sibling pairs carried the same variants, emphasizing the genetic heterogeneity of the disorder even within families [40]. The considerable variability of ASD necessitates genetic testing of large cohorts of patients on whole genome technologies (whole exome sequencing and WGS) which are becoming more affordable.
Despite the advantages of WGS, sequence variant classification (e.g. variants of unknown significance [VUS]) still poses a significant challenge. Such VUS are being reported at a faster rate than we are able to characterize them with respect to disease relevance. These studies underscore the aforementioned heterogeneity of the disorder; >200 ASD-risk genes have been identified [41,42,43,44,45,46]; SFARI gene: https://gene.sfari.org]. However, genomic aberrations are detected in only 25–40% of cases [40, 47, 48]. Further, no single mutation or CNV accounts for >1% of ASD cases and is variably penetrant with respect to the ASD phenotype. In addition to risk variants, six risk loci (1q21.1, 3q29, 7q11.23, 16p11.2, 15q11.2-13, 22q11.2) and several genetic syndromes (Sect. 4.4.1) are well known to be associated with ASD [47, 49, 50]. Unsurprisingly, few strong genotype-phenotype relationships have yet to be uncovered. Many genomic variants are recurrent, but rare (e.g. CHD8 mutations; Sect. 4.4.1), and therefore more patients are required to better establish such relationships.
Several studies have shown that many ASD-risk genes are involved in converging pathways relevant to the biological bases of ASD. These include development and cell proliferation, neural development, synaptic function and, of particular interest to this chapter, chromatin modifiers and transcriptional regulators [48, 49, 51]. Importantly, many of these genes are expressed in the brain during embryonic development. These functional categories relate to the neurocognitive phenotype of ASD (and other NDDs) and can help us to understand the molecular mechanisms, such as epigenetics, that are perturbed in ASD. The study of ASD-associated genetic syndromes caused by mutations in epigenes will also aid in this endeavour (Sect. 4.4.1). It is becoming increasingly apparent that some ASD-risk genes and loci confer an increased risk for other neuropsychiatric and neurological disorders including ID, ADHD, schizophrenia, epilepsy, motor impairment and sleep disturbance [30, 47, 52].
The genomic architecture of ASD is further elucidated through examinations of large numbers of families and individuals with ASD [40, 43,44,45,46, 49]. Better genotype-phenotype relationships are being defined; for example, the presence of de novo loss-of-function (LOF) mutations or CNVs is associated with lower IQ [43, 49, 53, 54]. Higher mutational burden has also been correlated with certain ASD features such as seizures and head circumference, observed in subsets of individuals [49, 55]. Genomic studies of ASD have identified genomic features relevant to ASD aetiology [49]. For example, de novo variants are distributed in a non-random fashion, enriched in epigenetically relevant regions (e.g. simple repeats and DNase I hypersensitivity sites, marks of open chromatin) [56].
Epigenetic mechanisms can help fill the aetiologic knowledge gap where genetic information alone is insufficient to explain the aetiology of all ASD cases. Epigenetic outcomes, much like genetic outcomes, are also expected to be heterogeneous (Sect. 4.4.2). Combining genetic and epigenetic data is likely to provide a more comprehensive understanding of the molecular landscape of the aetiology of ASD. The discovery of consistent molecular (genotype, epigenotype) and biochemical associations with ASD or ASD subtype-specific phenotypes will allow clinicians to better classify individuals, facilitate earlier diagnosis, and improve prognosis. In parallel, gaining molecular insights into the disorder will also help us to identify more homogeneous subgroups of individuals, which will allow for better patient stratification for behavioural and pharmacological treatments.
3 Environmental Exposures and ASD Risk
Twin, adoption and sibling studies have defined the heterogeneous and complex etiopathogenic nature of ASD and have supported potential contributions of environmental factors to ASD risk. Results from such studies suggest that ASD aetiology can be attributed to ~50% genetic contribution and ~50% influenced by non-shared environmental factors [37, 57,58,59,60]. The epigenome acts as an interface between the genome and the environment, transforming the genome into a regulator of cell type and developmental time-specific transcription. The epigenome is programmed during embryonic/foetal development by multiple genetic factors including genes that encode DNA methyl transferases (DNMTs), histone deacetylases (HDACs) and chromatin remodelling factors. Epigenetic errors can arise as primary stochastic events or in response to genetic mutations and/or environmental exposures. At critical times during development, typical foetal programming can be dysregulated by gene mutations, environmental exposures or epigenetic errors potentially leading to adverse long-term health outcomes [61,62,63]. There have been extensive investigations during the critical period of maternal gestation to examine the effects of exposures to both exogenous and endogenous environmental factors, which will be summarized below.
3.1 Exogenous Environment
Smoking, alcohol, medications (e.g. valproic acid [VPA], selective serotonin reuptake inhibitors) and environmental chemicals (e.g. pesticides, metals, bisphenol A [BPA]) are the most commonly studied exogenous exposures in relation to adverse foetal neurodevelopmental outcomes. Gestational smoking and alcohol exposure studies are inconsistent with respect to ASD risk [64,65,66,67], likely due to differences in study cohorts and methodologies, but also because it is extremely difficult to accurately estimate levels and timing of exposure in the mothers and more critically in the foetus. These challenges are compounded with the fact that the mechanisms of action and effects of maternal vs. foetal metabolism are not well understood enough to directly infer causation. In general, the impact of complex GxE interactions requires further investigation. Integrating genetic, environmental and epigenetic datasets will enable us to better understand the synergistic effects of these interactions. Animal models are critical for such studies, allowing for more precise and quantitative manipulations of environmental exposures. Further, we need to be able to distinguish between the direct effects of the exposure itself (e.g. direct perturbation of neurodevelopmentally important genes by gestational exposure to maternal smoking) and the downstream effects that may result (e.g. the fact that maternal smoking has been linked to decreased birth weight and reduced in utero brain growth). Interestingly, several studies demonstrate that environmental exposures affect epigenetic marks. Altered DNAm or DNMT expression/activity has been reported in various tissues of both human and animal models following exposure to a variety of toxicants including alcohol, cigarette smoking, BPA and VPA [68,69,70,71,72,73,74,75,76]. Some of these DNAm alterations have been further associated with ASD-relevant endpoints (e.g. behavioural outcomes, neurite outgrowth, axon formation and in ASD-relevant brain regions) (Table 2 in [65]).
3.2 Endogenous Environment
Preconception environmental risks include maternal [37, 77] and paternal [77, 78] age, which have been positively associated with an increase in ASD risk (relative risk [RR] of 1.16 to >1.5), both independently and with a joint effect. Interestingly, genetics may partially explain this finding; a greater number of de novo mutations in ASD probands have been found as a function of paternal age [43, 46]. Age-related epigenetic changes such as altered DNAm observed in both sperm and oocytes [79,80,81] may also contribute to this association. The mechanisms by which these factors introduce enhanced ASD risk still need to be further explored.
Epidemiologic studies show that preterm birth, due to various causes, significantly increases (3–14-fold) the risk of developing ASD [82,83,84]. A recent study [85] tested for DNAm differences between preterm and term foetal placental tissues at preselected ASD candidate genes. These include OXTR, SHANK3, BCL2, apoptosis regulator (BCL-2) and RORA that are known to have altered DNAm in some ASD cases. A significant gain of methylation (GOM) was found only in OXTR. More studies are needed to understand whether this DNAm mark at OXTR has a functional impact on ASD risk.
There have been inconsistent reports regarding the risk of ASD following the use of assisted reproductive technologies (ART; e.g. in vitro fertilization, intracytoplasmic sperm injection) [86,87,88]. ART currently account for ~1.6% of live births in the United States and rates of use are increasing [89, 90]. Although results are controversial, epidemiological studies have shown a possible increase in the incidence of ASD in offspring conceived with ART [86, 87]. There are several reasons why ART should be carefully considered. First, there are inherent risks of ART including preterm labour, multiple births and low birth weight that, independently of ART, already confer an increased risk for ASD [91,92,93]. Second, ART are used during critical windows of gametogenesis and early embryogenesis, when epigenetic reprogramming is occurring [92, 94]. Previous studies in the aetiologically heterogeneous paediatric imprinting disorders (Beckwith-Wiedemann [BWS] and Angelman syndromes [AS]) demonstrate that ART have a significant impact on epigenetic outcomes. These disorders are caused by loss of methylation (LOM) at critical imprinted sites on chromosomes 11p15 and 15q11 in individuals with BWS or AS, respectively. Most molecular alterations identified in these subjects following ART arose from epigenetic rather than genetic alterations; LOM is increased in frequency following the use of ART [95,96,97,98,99,100]. Currently, it is unclear if a parallel effect of ART occurs in the context of the development of ASD.
The endogenous maternal gestational environment has been studied extensively with respect to ASD risk [101,102,103]. Modest increases in ASD risk have been found to be associated with certain perinatal complications. Two of the main hypotheses relate back to two proposed underlying aetiologies of ASD (Sect. 4.2)—oxidative stress and immune function. Epidemiological data have shown that hypoxia-related obstetric complications pose a significant increase in ASD risk (effect estimate >1.4) [91, 101]. The maternal immune system is also central to several associations, although they are yet to be well established or replicated. Infection during pregnancy [82, 104,105,106] (OR: 1.24–1.37) and autoimmune disorders [107] (OR: 1.34) are linked to an increased risk for ASD. Recent studies report the maternal production of antibodies against circulating foetal brain proteins, detected in ~20% of mothers of children with ASD, but only in 1% of mothers of neurotypical children [108, 109]. Replication and further studies are required to solidify the potential role of these antibodies as biomarkers and their functional effect on foetal outcome. Maternal metabolic factors such as obesity and gestational diabetes mellitus (GDM) have both been associated with an increased risk for ASD [110,111,112,113]. There are convincing data demonstrating a role for epigenetic regulation of these metabolic processes. One study of women of South Asian origin, who have a high risk of GDM, showed statistically significant DNAm differences in cord blood and placental tissue, identifying differentially methylated genes involved in embryonic development and intracellular metabolic processes [114]. Other studies have reported DNAm changes in general immune, metabolic and endocrine pathways in placenta and cord blood of infants exposed to GDM [115,116,117]. There are no existing data available yet to validate these individual studies in cohorts of ASD patients exposed to GDM or maternal obesity.
Prenatal maternal stress has been identified as a small but robust risk factor for ADHD and ASD [118]. Maternal stress has been correlated with offspring autistic traits [119]. General and social communication scores were associated with altered DNAm of OXTR, a recurring ASD-risk gene of interest (Sect. 4.4.2). Further, specific single-nucleotide polymorphism (SNP) genotypes of OXTR were found to be predictive of methylation outcomes. However, this study was unable to find a GxE interaction, where associations between maternal stress and autistic traits were not related to OXTR methylation or genotype. Maternal prenatal nutrition, specifically folate supplementation, has been shown to have a protective effect, showing a reduced risk of ASD (OR: 0.61) [120,121,122]. Folate is well known for its role in preventing neural tube defects. Its protective effect for ASD may derive from the role of folate in OCM. OCM recycles homocysteine to generate cysteine and methionine for the process of methylation and antioxidative capacity through the formation of S-adenosyl methionine (SAM), supporting epigenetic processes important for typical neurodevelopment.
In summary, it will be important to more thoroughly explore whether the associations between environment and ASD outcome may be reflected in stable epigenetic marks detectable in the foetus or neonate. This would require large sample sizes to achieve sufficient power and well-annotated exposure data. Ultimately, the goal is to discover replicable GxE effects associated with ASD, perhaps in parallel with prenatal genetic testing, that are not only statistically but also biologically significant.
4 The Direct Role of Epigenetics in ASD
4.1 Genetic Syndromes Involving Epigenes and ASD
Syndromic ASD accounts for ~10–15% of cases [123,124,125]. A significant number of such genetic syndromes involve mutations in epigenes and are associated with increased risk for ASD (Table 4.1). Some of these epigenes function as epigenetic writers (DNMTs, histone methyltransferases and acetyltransferases), erasers (HDACs, lysine demethylases), readers (proteins containing bromo-, chromo- or Tudor domains), chromatin remodelling factors (e.g. CHD8) and epigenetic regulators of imprinted regions (ZFP57 zinc finger protein). Others exert more indirect effects through OCM, noncoding RNA processing or recruitment of methyl-CpG-binding proteins (MBDs) to modify histones and regulate transcription. Genetic syndromes caused by mutations in epigenes are also highly comorbid with ID, sometimes making it difficult to estimate ASD risk. As data for WGS of larger numbers of well-phenotyped ASD cases become available, additional genetic syndromes involving epigenes may be identified.
Rett syndrome (RTT; OMIM 312750) has been described in detail in Chaps. 1 and 2 and therefore will not be discussed in detail here. ASD symptoms can appear in early infancy, but the clinical phenotype becomes more distinct as RTT features (e.g. loss of hand skills, deceleration of head growth) develop with age. Interestingly, a large proportion of patients (>70%) with milder RTT variants exhibit ASD-like features [126]. Rare MECP2 mutations associated with ASD but not RTT have also been identified [127,128,129,130]. Typically, these mutations are found to be intronic and located in the 3′ untranslated region of the gene as opposed to LOF mutations which lead to RTT. The functional role that MECP2 may play in ASD pathogenesis has yet to be identified.
Mutations (single-nucleotide variants and small indels) in the chromatin modifier gene CHD8 have recently been described as a novel genetic syndrome with a strong association (>87%) with an ASD phenotype, amongst other common features such as macrocephaly (>80%), tall stature (86%) and gastrointestinal problems (80%) [131, 132]. Individuals with mutations in a related gene, chromodomain helicase DNA-binding protein 7 (CHD7; CHARGE syndrome, OMIM 214800), have a lesser but still significant risk (40%) for ASD [133,134,135]. The two genes have different interacting proteins and target binding sites [136], explaining at least in part the differences in phenotype.
Sotos syndrome (SS; OMIM 117550) is a congenital overgrowth disorder caused primarily (90%) by mutations in the nuclear receptor SET (su(var)3–9, enhancer-of-zeste, trithorax) domain-containing protein-1 gene (NSD1), a developmentally important histone methyltransferase. SS presents an example of a genetic syndrome with robust functionally relevant genome-wide epigenetic alterations [137]. Notably, reports show that >55% of SS patients display ASD symptomatology above clinical cutoffs [138,139,140]. It has recently been shown that individuals with SS have a specific blood DNAm signature that distinguishes individuals with pathogenic NDS1 mutations from controls [137]. Further, this DNAm signature is able to classify NSD1 VUS, which holds great potential for clinical application and molecular diagnostics. Examining a genetically homogeneous group of individuals as an approach for the study of ASD may eliminate some of the resultant epigenetic heterogeneity. The ability to refine and make more consistent molecular and/or phenotypic observations within subsets of individuals with ASD will help to establish causal roles for aetiologic factors.
4.2 Direct Assessment of Epigenetic Marks in ASD
Specific genomic alterations (e.g. mutations, CNVs) are known to confer increased risks for ASD, but these risks often are fairly broad ranging. Given the anticipated role of epigenetic dysregulation in ASD aetiology, multiple studies of different epigenetic marks in ASD cases have investigated stable epigenetic biomarkers either with or without an underlying genomic change. Stable biomarkers found in easily accessible, peripheral tissues such as blood would have a profound impact in the clinical diagnostic arena, especially if blood biomarkers were confirmed to reflect biomarkers in the brain. Examining cross-tissue markers, specifically brain vs. peripheral tissues, would help to further elucidate the underlying biological pathways involved in the aetiology of ASD. This section will focus mainly on assessments of the most stable and commonly studied epigenetic mark, DNAm (Table 4.2). We will also review data for epigenetic marks that are less frequently examined that will complement DNAm data in the future.
Although there is a solid rationale supporting a role for epigenetics in ASD molecular aetiology, there are relatively few studies that have directly measured epigenetic marks in ASD patients, especially when compared with the number of genetic studies available. Differentially methylated variants (DMVs) at specific CpG sites or differentially methylated regions (DMRs) spanning multiple CpGs have been measured in a variety of tissue types: lymphoblastoid cell lines [155], whole blood [156], buccal [157], sperm [158] and post-mortem brain [159,160,161]. The epigenome is characterized by cell-, tissue- and brain region-specific methylation patterns [161,162,163,164,165,166,167], making it impossible to directly compare data across these studies. However, as previously mentioned, from a biomarker and pathophysiological standpoint, it will be important to define intersecting ASD-specific DMVs/DMRs and pathways across cell types and tissues.
Genome-wide studies performed primarily using DNAm microarrays have yielded variable results for several reasons: differences in tissue type, ASD cohorts, methods (single site vs. region specific, i.e. DMVs vs. DMRs) and limited sample size (<50 cases) will affect the epigenetic output. These results emphasize the need for increased power in genome-wide DNAm studies focused on discovery of ASD-specific DNAm alterations across heterogeneous ASD groups. Most findings in ASD are reported with overwhelmingly modest effect sizes (<10% absolute difference), and some are statistically unreliable (e.g. without correction for multiple testing). Other possible confounding variables have yet to be addressed for their potential impact on DNAm outcome, including sex, age, post-mortem interval and cause of death (for post-mortem brain samples) and brain cell type-specific DNAm patterns, to name a few. Replication of these results in larger cohorts of ASD patients will strengthen the support for a role for dysregulation of DNAm in ASD neuropathology. Only two studies [160, 161] have shown replication of differentially methylated sites that were hypomethylated in the 3′ untranslated region of PRRT1, tetraspanin 32 (TSPAN32) and C11orf21 in brains of ASD patients when compared with controls. For many of the other identified differentially methylated genes across all studies, there is no known function in the context of ASD. Others appear to be functionally relevant, with potential roles in brain electrophysiological function (e.g. PRRT1), immunity (e.g. C1Q, TNF-α) and/or involving known ASD-risk genes (e.g. AT-rich interaction domain 1B [ARID1B], glutamate ionotropic receptor NMDA type subunit 2B [GRIN2B], neurexin 1 [NRXN1], phosphatase and tensin homolog [PTEN]).
Several studies have focused on the targeted quantification of DNAm in promoters of ASD candidate genes (glutamic acid decarboxylase 65 [GAD65], OXTR, SHANK3, reelin (RELN), UBE3A and MECP2) [168,169,170,171,172,173] in different tissues (blood, specific regions in post-mortem brain). No differences between ASD and controls were found for DNAm of GAD65 or RELN [173] nor for one of the OXTR studies [168]. The latter result did not agree with an earlier study covering an overlapping region of OXTR [169], where significant GOM was found at specific CpG sites overall and in a sex-specific manner. In summary, significant ASD-specific DMVs at OXTR, SHANK3, UBE3A and MECP2 were identified at specific promoter CpG sites in each gene (i.e. not across all sites analysed) [168,169,170,171,172]. Absolute differences were found to be modest (~twofold) on average and did not affect all ASD cases equally. Overall, the variability in results observed in the genome-wide DNAm studies is also reflected in these targeted studies, for many of the same reasons (tissue type, unselected ASD cases examined, methods, sample size).
Only two studies to date have looked at differences in histone marks between individuals with ASD and neurotypical controls [174, 175]. The two studies are difficult to compare since each study differed in the histone marks examined (H3K4me3 vs. H3K27ac), brain regions (prefrontal cortex vs. prefrontal cortex, temporal cortex and cerebellum), patient cohorts and methodology. However, each study independently found ASD-specific patterns of histone mark methylation to varying degrees. Shulha et al. (2012) report that there were no global alterations of H3K4me3, but rather an expansion in the presence of H3K4me3 at specific genomic regions in ASD. Sun et al. (2016) describe brain region- and ASD-specific differentially acetylated regions. Differences were found to correspond to functionally relevant genes involved in synaptic transmission, neuronal connectivity, immunity and behaviour. Researchers are also just beginning to look at differential miRNA and long noncoding RNA (lncRNA) expression in ASD [176,177,178,179]. Thus far, there is a lack of consistent findings across these studies.
In spite of current limitations, the studies cited above will act as a catalyst for the study of ASD to identify epigenetic biomarkers for prediction or classification of individuals with ASD. Additionally, they have brought to light the many critical variables that need to be considered to improve study design and interpretation of data going forward.
5 Therapeutics
Identifying epigenetic targets with therapeutic potential exploits the dynamic and modifiable nature of epigenetic pathways, allowing for new approaches to ameliorate ASD symptoms. Pharmacologic agents could be used to target direct epigenetic regulators such as histone acetylation (HDACs) or to target indirect and/or downstream pathways (e.g. OCM). Some of these targets already have existing therapies/drugs for testing in clinical trials.
There are several significant challenges to this endeavour. Will general inhibition/activation of epigenetic processes be too disruptive of other mechanisms? Conversely, how can we develop more targeted (e.g. tissue and cell type, enzyme isoform-specific) epigenetic drugs? One of the most critical obstacles to overcome for the treatment of ASD neurobehavioural deficits is ensuring that a drug is able to pass the blood–brain barrier (BBB). Currently, HDAC and DNAm inhibitors have poor brain penetrance and potency, although some recent work is showing improvement in this area. Improved delivery systems are being tested with a novel HDAC inhibitor analogue [180] and image-guided (positron emission tomography) radiolabelled drug delivery [181]. However, the BBB issue may be bypassed by harnessing the therapeutic potential of the microbiome to effect downstream neurobehavioural outcomes.
Indirect targets and pathways that affect epigenetic mechanisms may also have pharmacologic potential. The importance of folate in maintaining proper SAM levels and therefore methyl donors (Sect. 4.4) is demonstrated in its apparent protective effect on ASD risk. The neuropeptide hormone oxytocin, which binds to OXTR, has been highlighted as a promising pharmacological agent in several clinical trials [182,183,184,185] for the treatment of certain neuropsychiatric disorders including ASD. Although results have been mixed, some positive results [186,187,188] support the use of oxytocin for improving specific deficits seen in ASD such as emotion recognition and eye gaze, as well as for its prosocial and anxiolytic properties.
The exploration of epigenetic therapeutic targets for ASD is in its infancy since researchers are still uncovering the molecular conundrum by which epigenetic mechanisms are perturbed in ASD. However, genomic and epigenomic insights are uncovering potential biological pathways that may be targeted for therapeutics. With more comprehensive classifications of ASD patients, we may identify subgroups of individuals that will be candidates for more precision-based therapies (e.g. pathways susceptible to environmental influences, immune, metabolic, chromatin modifiers, etc.).
6 Future Directions/Summary
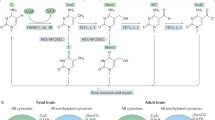
There is still much to learn about ASD in the context of epigenetics. As technology advances, we may interrogate the genome and epigenome with higher resolution. This will allow researchers to refine DNAm studies with increased genomic coverage, to expand on our knowledge of ASD-specific histone marks and to explore the role of noncoding regions (e.g. enhancers, intergenic regions, noncoding RNAs). Beyond CpG methylation, non-CpG methylation (CpH where H = A, C, or T) and 5-hydroxymethylcytosine (5-hmC) should not be overlooked. Both of these alternative types of methylation have been found to be important during neurogenesis [163, 189, 190] and thus could play a role in the pathogenesis of ASD. 5-hmC, an intermediate in the process of oxidative demethylation, is highly abundant in the brain relative to 5-methylcytosine (5mC) [191] and could reveal important regulatory brain region-specific epigenetic patterns.
One emerging method of better defining these mechanisms is to tackle the issue of heterogeneity in ASD by examining more homogeneous subsets of ASD patients based on various factors. Presented in this chapter were examples of environmental (preterm labour) and genetic (NSD1 mutations in SS) stratification, which demonstrate the strength of this approach and are paving the road for the future of research into the aetiologies of ASD. It is clear from the current literature that many roads lead back to epigenetics; from genetics to structural, physiological and biochemical hallmarks of the disorder to environment, epigenetic mechanisms are intimately involved in interfacing with and regulating these aetiologic factors. The complexity of epigenetic mechanisms, its intermediary role bridging multifactorial risk factors through GxE interactions and its malleable nature underscore both the challenges of studying ASD in the context of epigenetics and the exciting potential for this area of research.
Although we were unable to touch upon these areas of research and knowledge in this chapter, there are several important additional topics to consider in the context of ASD aetiology and epigenetics.
Additional Reading
Abbreviations
- 5-hmC:
-
5-Hydroxymethylcytosine
- 5-mC:
-
5-Methylcytosine
- Δβ:
-
Difference in DNA methylation
- ADHD:
-
Attention deficit and hyperactivity disorder
- ARID1B:
-
AT-rich interaction domain 1B
- ART:
-
Assisted reproductive technologies
- AS:
-
Angelman syndrome
- ASD:
-
Autism spectrum disorder
- ATP:
-
Adenosine triphosphate
- BA10:
-
Brodmann area 10
- BA19:
-
Brodmann area 19
- BA24:
-
Brodmann area 24
- BBB:
-
Blood–brain barrier
- BCL-2:
-
BCL2, apoptosis regulator
- BPA:
-
Bisphenol A
- BWS:
-
Beckwith-Wiedemann syndrome
- C11orf21:
-
Chromosome 11 open reading frame 21
- C1Q:
-
Complement subcomponent C1q
- CBL:
-
Cerebellum
- CDKL5:
-
Cyclin-dependent kinase-like 5
- CHARGE:
-
Coloboma of the eye, heart defects, atresia of the nasal choanae, retardation of growth and/or development, genital and/or urinary abnormalities and ear abnormalities/deafness
- CHD7:
-
Chromodomain helicase DNA-binding protein 7
- CHD8:
-
Chromodomain helicase DNA-binding protein 8
- CNV:
-
Copy number variant
- DGCR6:
-
DiGeorge critical region 6
- DGCR8:
-
DiGeorge critical region 8
- DLGAP1:
-
DLG associated protein 1
- DLGAP2:
-
DLG associated protein 2
- DNAm:
-
DNA methylation
- DNMT:
-
DNA methyl transferases
- FDR:
-
False discovery rate
- FMR1:
-
Fragile X mental retardation 1
- GAD65:
-
Glutamic acid decarboxylase 65
- GDM:
-
Gestational diabetes mellitus
- GOM:
-
Gain of methylation
- GRIN2B:
-
Glutamate ionotropic receptor NMDA type subunit 2B
- GxE:
-
Gene by environment interactions
- H3K27ac:
-
Histone 3 lysine 27 acetylation
- H3K4me3:
-
Histone 3 lysine 4 trimethylation
- HDAC:
-
Histone deacetylase
- HUWE1:
-
HECT, UBA and WWE domain-containing 1, E3 ubiquitin protein
- IC:
-
Imprinting centre
- ID:
-
Intellectual disability
- KDM6A:
-
Lysine demethylase 6A
- KMT2D:
-
Lysine methyltransferase 2D
- lncRNA:
-
Long noncoding RNA
- LOF:
-
Loss of function
- LOM:
-
Loss of methylation
- MBDs:
-
Methyl-CpG-binding proteins
- MECP2:
-
Methyl-CpG-binding protein 2
- miRNA:
-
Micro-RNA
- MZ:
-
Monozygotic
- NDD:
-
Neurodevelopmental disorder
- NRXN1:
-
Neurexin 1
- NSD1:
-
Nuclear receptor SET (su(var)3–9, enhancer-of-zeste, trithorax) domain-containing protein-1 gene
- OCM:
-
One carbon metabolism
- OR:
-
Odds ratio
- OXTR:
-
Oxytocin receptor
- PDD-NOS:
-
Pervasive developmental disorder – not otherwise specified
- PFC:
-
Prefrontal cortex
- PRRT1:
-
Proline-rich transmembrane protein 1
- PTEN:
-
Phosphatase and tensin homolog
- RELN:
-
Reelin
- RORA:
-
Retinoic acid-related orphan receptor
- RTT:
-
Rett syndrome
- SAM:
-
S-Adenosyl methionine
- SHANK3:
-
SH3 and multiple ankyrin repeat domains 3
- SNP:
-
Single-nucleotide polymorphism
- SNRPN:
-
Small nuclear ribonucleoprotein polypeptide N
- TC:
-
Temporal cortex
- TNF-α:
-
Tumour necrosis factor alpha
- TSPAN32:
-
Tetraspanin 32
- UBE3A:
-
Ubiquitin protein ligase
- UBE3A:
-
Ubiquitin protein ligase E3A
- UPD:
-
Uniparental disomy
- VPA:
-
Valproic acid
- VUS:
-
Variant of unknown significance
- WGS:
-
Whole genome sequencing
- ZFP57:
-
ZFP57 zinc finger protein
References
Christensen DL, Baio J, Braun KV, Bilder D, Charles J, Constantino JN, et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years — Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill Summ. 2016;65(No. SS-3)(No. SS-3):1–23.
Doshi-Velez F, Ge Y, Kohane I. Comorbidity clusters in autism spectrum disorders: an electronic health record time-series analysis. Pediatrics. 2014;133(1):e54–63.
Bailey A, Luthert P, Dean A, Harding B, Janota I, Montgomery M, et al. A clinicopathological study of autism. Brain. 1998;121(Pt 5):889–905.
Donovan AP, Basson MA. The neuroanatomy of autism - a developmental perspective. J Anat. 2016;230(1):4–15.
Rao PA, Landa RJ. Association between severity of behavioral phenotype and comorbid attention deficit hyperactivity disorder symptoms in children with autism spectrum disorders. Autism. 2014;18(3):272–80.
Ronald A, Simonoff E, Kuntsi J, Asherson P, Plomin R. Evidence for overlapping genetic influences on autistic and ADHD behaviours in a community twin sample. J Child Psychol Psychiatry. 2008;49(5):535–42.
Loke YJ, Hannan AJ, Craig JM. The Role of Epigenetic Change in Autism Spectrum Disorders. Front Neurol. 2015;6:107.
Gesundheit B, Rosenzweig JP, Naor D, Lerer B, Zachor DA, Prochazka V, et al. Immunological and autoimmune considerations of Autism Spectrum Disorders. J Autoimmun. 2013;44:1–7.
McDougle CJ, Landino SM, Vahabzadeh A, O'Rourke J, Zurcher NR, Finger BC, et al. Toward an immune-mediated subtype of autism spectrum disorder. Brain Res. 1617;2015:72–92.
Nardone S, Elliott E. The interaction between the immune system and epigenetics in the etiology of Autism Spectrum disorders. Front Neurosci. 2016;10:329.
Noriega DB, Savelkoul HF. Immune dysregulation in autism spectrum disorder. Eur J Pediatr. 2014;173(1):33–43.
Rossignol DA, Frye RE. A review of research trends in physiological abnormalities in autism spectrum disorders: immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol Psychiatry. 2012;17(4):389–401.
Filiano AJ, Gadani SP, Kipnis J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 1617;2015:18–27.
Onore C, Careaga M, Ashwood P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav Immun. 2012;26(3):383–92.
Rilett KC, Friedel M, Ellegood J, MacKenzie RN, Lerch JP, Foster JA. Loss of T cells influences sex differences in behavior and brain structure. Brain Behav Immun. 2015;46:249–60.
Sidor MM, Halgren CR, Foster JA. The impact of early life immune challenge in behavior and microglia during postnatal development. Inflamm Cell Signal. 2014;1:51–60.
Bilbo SD, Nevison CD, Parker W. A model for the induction of autism in the ecosystem of the human body: the anatomy of a modern pandemic? Microb Ecol Health Dis. 2015;26:26253.
Cao X, Lin P, Jiang P, Li C. Characteristics of the gastrointestinal microbiome in children with autism spectrum disorder: a systematic review. Shanghai Arch Psychiatry. 2013;25(6):342–53.
Stilling RM, Dinan TG, Cryan JF. Microbial genes, brain & behaviour—epigenetic regulation of the gut-brain axis. Genes Brain Behav. 2014;13(1):69–86.
Zhang YJ, Li S, Gan RY, Zhou T, Xu DP, Li HB. Impacts of gut bacteria on human health and diseases. Int J Mol Sci. 2015;16(4):7493–519.
Alenghat T, Osborne LC, Saenz SA, Kobuley D, Ziegler CG, Mullican SE, et al. Histone deacetylase 3 coordinates commensal-bacteria-dependent intestinal homeostasis. Nature. 2013;504(7478):153–7.
Espallergues J, Teegarden SL, Veerakumar A, Boulden J, Challis C, Jochems J, et al. HDAC6 regulates glucocorticoid receptor signaling in serotonin pathways with critical impact on stress resilience. J Neurosci. 2012;32(13):4400–16.
Jochems J, Boulden J, Lee BG, Blendy JA, Jarpe M, Mazitschek R, et al. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology. 2014;39(2):389–400.
Rossignol DA, Frye RE. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front Physiol. 2014;5:150.
Rossignol DA, Frye RE. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol Psychiatry. 2012;17(3):290–314.
Turinsky AL, Turner B, Borja RC, Gleeson JA, Heath M, Pu S, et al. DAnCER: disease-annotated chromatin epigenetics resource. Nucleic Acids Res. 2010;39(Database issue):D889–94.
van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104.
McCarthy SE, Gillis J, Kramer M, Lihm J, Yoon S, Berstein Y, et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol Psychiatry. 2014;19(6):652–8.
Carroll LS, Owen MJ. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009;1(10):102.
Cukier HN, Dueker ND, Slifer SH, Lee JM, Whitehead PL, Lalanne E, et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol Autism. 2014;5(1):1.
Doherty JL, Owen MJ. Genomic insights into the overlap between psychiatric disorders: implications for research and clinical practice. Genome Med. 2014;6(4):29.
Network, Pathway Analysis Subgroup of Psychiatric Genomics C. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015;18(2):199–209.
Kiser DP, Rivero O, Lesch KP. Annual research review: the (epi)genetics of neurodevelopmental disorders in the era of whole-genome sequencing—unveiling the dark matter. J Child Psychol Psychiatry. 2015;56(3):278–95.
Freitag CM. The genetics of autistic disorders and its clinical relevance: a review of the literature. Mol Psychiatry. 2007;12(1):2–22.
Smalley SL, Asarnow RF, Spence MA. Autism and genetics. A decade of research. Arch Gen Psychiatry. 1988;45(10):953–61.
Ozonoff S, Young GS, Carter A, Messinger D, Yirmiya N, Zwaigenbaum L, et al. Recurrence risk for autism spectrum disorders: a Baby Siblings Research Consortium study. Pediatrics. 2011;128(3):e488–95.
Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311(17):1770–7.
Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68(11):1095–102.
Gratten J, Wray NR, Keller MC, Visscher PM. Large-scale genomics unveils the genetic architecture of psychiatric disorders. Nat Neurosci. 2014;17(6):782–90.
Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med. 2015;21(2):185–91.
Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77.
Devlin B, Scherer SW. Genetic architecture in autism spectrum disorder. Curr Opin Genet Dev. 2012;22(3):229–37.
Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515(7526):216–21.
Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74(2):285–99.
O'Roak BJ, Stessman HA, Boyle EA, Witherspoon KT, Martin B, Lee C, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nat Commun. 2014;5:5595.
Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485(7397):237–41.
Jeste SS, Geschwind DH. Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat Rev Neurol. 2014;10(2):74–81.
Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet. 2014;94(5):677–94.
Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, et al. Insights into Autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87(6):1215–33.
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–88.
Lasalle JM. Autism genes keep turning up chromatin. OA Autism. 2013;1(2):14.
Li J, Cai T, Jiang Y, Chen H, He X, Chen C, et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol Psychiatry. 2016;21(2):298.
Robinson EB, Samocha KE, Kosmicki JA, McGrath L, Neale BM, Perlis RH, et al. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc Natl Acad Sci U S A. 2014;111(42):15161–5.
Samocha KE, Robinson EB, Sanders SJ, Stevens C, Sabo A, McGrath LM, et al. A framework for the interpretation of de novo mutation in human disease. Nat Genet. 2014;46(9):944–50.
Chaste P, Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, et al. Adjusting head circumference for covariates in autism: clinical correlates of a highly heritable continuous trait. Biol Psychiatry. 2013;74(8):576–84.
Michaelson JJ, Shi Y, Gujral M, Zheng H, Malhotra D, Jin X, et al. Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151(7):1431–42.
Colvert E, Tick B, McEwen F, Stewart C, Curran SR, Woodhouse E, et al. Heritability of Autism spectrum disorder in a UK population-based twin sample. JAMA Psychiat. 2015;72(5):415–23.
Frazier TW, Thompson L, Youngstrom EA, Law P, Hardan AY, Eng C, et al. A twin study of heritable and shared environmental contributions to autism. J Autism Dev Disord. 2014;44(8):2013–25.
Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. Most genetic risk for autism resides with common variation. Nat Genet. 2014;46(8):881–5.
Ronald A, Hoekstra RA. Autism spectrum disorders and autistic traits: a decade of new twin studies. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(3):255–74.
El Hajj N, Schneider E, Lehnen H, Haaf T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction. 2014;148(6):R111–20.
Roseboom TJ, Painter RC, van Abeelen AF, Veenendaal MV, de Rooij SR. Hungry in the womb: what are the consequences? Lessons from the Dutch famine. Maturitas. 2011;70(2):141–5.
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592.
Dufour-Rainfray D, Vourc'h P, Tourlet S, Guilloteau D, Chalon S, Andres CR. Fetal exposure to teratogens: evidence of genes involved in autism. Neurosci Biobehav Rev. 2011;35(5):1254–65.
Keil KP, Lein PJ. DNA methylation: a mechanism linking environmental chemical exposures to risk of autism spectrum disorders? Environ Epigenet. 2016;2(1).
Kim YS, Leventhal BL. Genetic epidemiology and insights into interactive genetic and environmental effects in autism spectrum disorders. Biol Psychiatry. 2015;77(1):66–74.
Lyall K, Schmidt RJ, Hertz-Picciotto I. Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int J Epidemiol. 2014;43(2):443–64.
Laufer BI, Diehl EJ, Singh SM. Neurodevelopmental epigenetic etiologies: insights from studies on mouse models of fetal alcohol spectrum disorders. Epigenomics. 2013;5(5):465–8.
Laufer BI, Kapalanga J, Castellani CA, Diehl EJ, Yan L, Singh SM. Associative DNA methylation changes in children with prenatal alcohol exposure. Epigenomics. 2015;7(8):1259–74.
Laufer BI, Mantha K, Kleiber ML, Diehl EJ, Addison SM, Singh SM. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis Model Mech. 2013;6(4):977–92.
Lee KW, Richmond R, Hu P, French L, Shin J, Bourdon C, et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ Health Perspect. 2015;123(2):193–9.
Portales-Casamar E, Lussier AA, Jones MJ, MacIsaac JL, Edgar RD, Mah SM, et al. DNA methylation signature of human fetal alcohol spectrum disorder. Epigenetics Chromatin. 2016;9:25.
Roullet FI, Lai JK, Foster JA. In utero exposure to valproic acid and autism—a current review of clinical and animal studies. Neurotoxicol Teratol. 2013;36:47–56.
Smith EG. Additional effect size measures helpful in understanding lithium and valproate trial results. Am J Psychiatry. 2012;169(1):97–8.
O'Brien E, Dolinoy DC, Mancuso P. Perinatal bisphenol A exposures increase production of pro-inflammatory mediators in bone marrow-derived mast cells of adult mice. J Immunotoxicol. 2014;11(3):205–12.
Yaoi T, Itoh K, Nakamura K, Ogi H, Fujiwara Y, Fushiki S. Genome-wide analysis of epigenomic alterations in fetal mouse forebrain after exposure to low doses of bisphenol A. Biochem Biophys Res Commun. 2008;376(3):563–7.
Sandin S, Schendel D, Magnusson P, Hultman C, Suren P, Susser E, et al. Autism risk associated with parental age and with increasing difference in age between the parents. Mol Psychiatry. 2016;21(5):693–700.
Frans EM, Lichtenstein P, Hultman CM, Kuja-Halkola R. Age at fatherhood: heritability and associations with psychiatric disorders. Psychol Med. 2016;46(14):2981–8.
Ge ZJ, Schatten H, Zhang CL, Sun QY. Oocyte ageing and epigenetics. Reproduction. 2015;149(3):R103–14.
Jenkins TG, Aston KI, Pflueger C, Cairns BR, Carrell DT. Age-associated sperm DNA methylation alterations: possible implications in offspring disease susceptibility. PLoS Genet. 2014;10(7):e1004458.
Milekic MH, Xin Y, O'Donnell A, Kumar KK, Bradley-Moore M, Malaspina D, et al. Age-related sperm DNA methylation changes are transmitted to offspring and associated with abnormal behavior and dysregulated gene expression. Mol Psychiatry. 2015;20(8):995–1001.
Atladottir HO, Schendel DE, Henriksen TB, Hjort L, Parner ET. Gestational age and autism spectrum disorder: trends in risk over time. Autism Res. 2016;9(2):224–31.
Kuzniewicz MW, Wi S, Qian Y, Walsh EM, Armstrong MA, Croen LA. Prevalence and neonatal factors associated with autism spectrum disorders in preterm infants. J Pediatr. 2014;164(1):20–5.
Leavey A, Zwaigenbaum L, Heavner K, Burstyn I. Gestational age at birth and risk of autism spectrum disorders in Alberta. Can J Pediatr. 2013;162(2):361–8.
Behnia F, Parets SE, Kechichian T, Yin H, Dutta EH, Saade GR, et al. Fetal DNA methylation of autism spectrum disorders candidate genes: association with spontaneous preterm birth. Am J Obstet Gynecol. 2015;212(4):533 e1–9.
Conti E, Mazzotti S, Calderoni S, Saviozzi I, Guzzetta A. Are children born after assisted reproductive technology at increased risk of autism spectrum disorders? A systematic review. Hum Reprod. 2013;28(12):3316–27.
Fountain C, Zhang Y, Kissin DM, Schieve LA, Jamieson DJ, Rice C, et al. Association between assisted reproductive technology conception and autism in California, 1997–2007. Am J Public Health. 2015;105(5):963–71.
Schieve LA, Fountain C, Boulet SL, Yeargin-Allsopp M, Kissin DM, Jamieson DJ, et al. Does Autism diagnosis age or symptom severity differ among children according to whether assisted reproductive technology was used to achieve pregnancy? J Autism Dev Disord. 2015;45(9):2991–3003.
Sunderam S, Kissin DM, Crawford SB, Folger SG, Jamieson DJ, Warner L, et al. Assisted Reproductive Technology Surveillance — United States, 2014. MMWR Surveill Summ. 2017;66(No. SS-6):1–24.
Schieve LA, Devine O, Boyle CA, Petrini JR, Warner L. Estimation of the contribution of non-assisted reproductive technology ovulation stimulation fertility treatments to US singleton and multiple births. Am J Epidemiol. 2009;170(11):1396–407.
Gardener H, Spiegelman D, Buka SL. Perinatal and neonatal risk factors for autism: a comprehensive meta-analysis. Pediatrics. 2011;128(2):344–55.
Grafodatskaya D, Cytrynbaum C, Weksberg R. The health risks of ART. EMBO Rep. 2013;14(2):129–35.
Savage T, Peek J, Hofman PL, Cutfield WS. Childhood outcomes of assisted reproductive technology. Hum Reprod. 2011;26(9):2392–400.
Iliadou AN, Janson PC, Cnattingius S. Epigenetics and assisted reproductive technology. J Intern Med. 2011;270(5):414–20.
DeBaun MR, Niemitz EL, Feinberg AP. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am J Hum Genet. 2003;72(1):156–60.
Doornbos ME, Maas SM, McDonnell J, Vermeiden JP, Hennekam RC. Infertility, assisted reproduction technologies and imprinting disturbances: a Dutch study. Hum Reprod. 2007;22(9):2476–80.
Gicquel C, Gaston V, Mandelbaum J, Siffroi JP, Flahault A, Le Bouc Y. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am J Hum Genet. 2003;72(5):1338–41.
Ludwig M, Katalinic A, Gross S, Sutcliffe A, Varon R, Horsthemke B. Increased prevalence of imprinting defects in patients with Angelman syndrome born to subfertile couples. J Med Genet. 2005;42(4):289–91.
Maher ER, Brueton LA, Bowdin SC, Luharia A, Cooper W, Cole TR, et al. Beckwith-Wiedemann syndrome and assisted reproduction technology (ART). J Med Genet. 2003;40(1):62–4.
Sutcliffe AG, Peters CJ, Bowdin S, Temple K, Reardon W, Wilson L, et al. Assisted reproductive therapies and imprinting disorders—a preliminary British survey. Hum Reprod. 2006;21(4):1009–11.
Froehlich-Santino W, Londono Tobon A, Cleveland S, Torres A, Phillips J, Cohen B, et al. Prenatal and perinatal risk factors in a twin study of autism spectrum disorders. J Psychiatr Res. 2014;54:100–8.
Guinchat V, Thorsen P, Laurent C, Cans C, Bodeau N, Cohen D. Pre-, peri- and neonatal risk factors for autism. Acta Obstet Gynecol Scand. 2012;91(3):287–300.
Schieve LA, Clayton HB, Durkin MS, Wingate MS, Drews-Botsch C. Comparison of perinatal risk factors associated with Autism Spectrum Disorder (ASD), Intellectual Disability (ID), and co-occurring ASD and ID. J Autism Dev Disord. 2015;45(8):2361–72.
Brown AS. Epidemiologic studies of exposure to prenatal infection and risk of schizophrenia and autism. Dev Neurobiol. 2012;72(10):1272–6.
Fang SY, Wang S, Huang N, Yeh HH, Chen CY. Prenatal infection and Autism Spectrum disorders in childhood: a population-based case-control study in Taiwan. Paediatr Perinat Epidemiol. 2015;29(4):307–16.
Lee BK, Magnusson C, Gardner RM, Blomstrom A, Newschaffer CJ, Burstyn I, et al. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav Immun. 2015;44:100–5.
Chen SW, Zhong XS, Jiang LN, Zheng XY, Xiong YQ, Ma SJ, et al. Maternal autoimmune diseases and the risk of autism spectrum disorders in offspring: a systematic review and meta-analysis. Behav Brain Res. 2016;296:61–9.
Braunschweig D, Krakowiak P, Duncanson P, Boyce R, Hansen RL, Ashwood P, et al. Autism-specific maternal autoantibodies recognize critical proteins in developing brain. Transl Psychiatry. 2013;3:e277.
Braunschweig D, Van de Water J. Maternal autoantibodies in autism. Arch Neurol. 2012;69(6):693–9.
Krakowiak P, Walker CK, Tancredi D, Hertz-Picciotto I, Van de Water J. Autism-specific maternal anti-fetal brain autoantibodies are associated with metabolic conditions. Autism Res. 2016;10(1):89–98.
Li M, Fallin MD, Riley A, Landa R, Walker SO, Silverstein M, et al. The association of maternal obesity and diabetes with autism and other developmental disabilities. Pediatrics. 2016;137(2):e20152206.
Li YM, Ou JJ, Liu L, Zhang D, Zhao JP, Tang SY. Association between maternal obesity and autism spectrum disorder in offspring: a meta-analysis. J Autism Dev Disord. 2016;46(1):95–102.
Xiang AH, Wang X, Martinez MP, Walthall JC, Curry ES, Page K, et al. Association of maternal diabetes with autism in offspring. JAMA. 2015;313(14):1425–34.
Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, et al. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. 2015;24(11):3021–9.
Binder AM, LaRocca J, Lesseur C, Marsit CJ, Michels KB. Epigenome-wide and transcriptome-wide analyses reveal gestational diabetes is associated with alterations in the human leukocyte antigen complex. Clin Epigenetics. 2015;7:79.
Petropoulos S, Guillemin C, Ergaz Z, Dimov S, Suderman M, Weinstein-Fudim L, et al. Gestational diabetes alters offspring DNA methylation profiles in human and rat: identification of key pathways involved in endocrine system disorders, insulin signaling, diabetes signaling, and ILK signaling. Endocrinology. 2015;156(6):2222–38.
Ruchat SM, Hivert MF, Bouchard L. Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr Rev. 2013;71(Suppl 1):S88–94.
Ronald A, Pennell CE, Whitehouse AJ. Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front Psychol. 2010;1:223.
Rijlaarsdam J, Pappa I, Walton E, Bakermans-Kranenburg MJ, Mileva-Seitz VR, Rippe RC, et al. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: a model approach for replication. Epigenetics. 2016;11(2):140–9.
Schmidt RJ. Maternal folic acid supplements associated with reduced autism risk in the child. Evid Based Med. 2013;18(6):e53.
Schmidt RJ, Tancredi DJ, Ozonoff S, Hansen RL, Hartiala J, Allayee H, et al. Maternal periconceptional folic acid intake and risk of autism spectrum disorders and developmental delay in the CHARGE (CHildhood Autism Risks from Genetics and Environment) case-control study. Am J Clin Nutr. 2012;96(1):80–9.
Suren P, Roth C, Bresnahan M, Haugen M, Hornig M, Hirtz D, et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA. 2013;309(6):570–7.
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, et al. Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008;113(6):427–38.
Moretti P, Zoghbi HY. MeCP2 dysfunction in Rett syndrome and related disorders. Curr Opin Genet Dev. 2006;16(3):276–81.
Zafeiriou DI, Ververi A, Dafoulis V, Kalyva E, Vargiami E. Autism spectrum disorders: the quest for genetic syndromes. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(4):327–66.
Renieri A, Mari F, Mencarelli MA, Scala E, Ariani F, Longo I, et al. Diagnostic criteria for the Zappella variant of Rett syndrome (the preserved speech variant). Brain Dev. 2009;31(3):208–16.
Beyer KS, Blasi F, Bacchelli E, Klauck SM, Maestrini E, Poustka A, et al. Mutation analysis of the coding sequence of the MECP2 gene in infantile autism. Hum Genet. 2002;111(4–5):305–9.
Carney RM, Wolpert CM, Ravan SA, Shahbazian M, Ashley-Koch A, Cuccaro ML, et al. Identification of MeCP2 mutations in a series of females with autistic disorder. Pediatr Neurol. 2003;28(3):205–11.
Coutinho AM, Oliveira G, Katz C, Feng J, Yan J, Yang C, et al. MECP2 coding sequence and 3'UTR variation in 172 unrelated autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(4):475–83.
Suter B, Treadwell-Deering D, Zoghbi HY, Glaze DG, Neul JL. Brief report: MECP2 mutations in people without Rett syndrome. J Autism Dev Disord. 2014;44(3):703–11.
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–76.
Merner N, Forgeot d'Arc B, Bell SC, Maussion G, Peng H, Gauthier J, et al. A de novo frameshift mutation in chromodomain helicase DNA-binding domain 8 (CHD8): a case report and literature review. Am J Med Genet A. 2016;170A(5):1225–35.
Hartshorne TS, Grialou TL, Parker KR. Autistic-like behavior in CHARGE syndrome. Am J Med Genet A. 2005;133A(3):257–61.
Johansson M, Rastam M, Billstedt E, Danielsson S, Stromland K, Miller M, et al. Autism spectrum disorders and underlying brain pathology in CHARGE association. Dev Med Child Neurol. 2006;48(1):40–50.
Smith IM, Nichols SL, Issekutz K, Blake K, Canadian Paediatric Surveillance P. Behavioral profiles and symptoms of autism in CHARGE syndrome: preliminary Canadian epidemiological data. Am J Med Genet A. 2005;133A(3):248–56.
Micucci JA, Sperry ED, Martin DM. Chromodomain helicase DNA-binding proteins in stem cells and human developmental diseases. Stem Cells Dev. 2015;24(8):917–26.
Choufani S, Cytrynbaum C, Chung BH, Turinsky AL, Grafodatskaya D, Chen YA, et al. NSD1 mutations generate a genome-wide DNA methylation signature. Nat Commun. 2015;6:10207.
Lane C, Milne E, Freeth M. Cognition and behaviour in Sotos syndrome: a systematic review. PLoS One. 2016;11(2):e0149189.
Lane C, Milne E, Freeth M. Characteristics of autism spectrum disorder in Sotos syndrome. J Autism Dev Disord. 2016;47(1):135–43.
Sheth K, Moss J, Hyland S, Stinton C, Cole T, Oliver C. The behavioral characteristics of Sotos syndrome. Am J Med Genet A. 2015;167A(12):2945–56.
Richards C, Jones C, Groves L, Moss J, Oliver C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: a systematic review and meta-analysis. Lancet Psychiatry. 2015;2(10):909–16.
Lam CW, Yeung WL, Ko CH, Poon PM, Tong SF, Chan KY, et al. Spectrum of mutations in the MECP2 gene in patients with infantile autism and Rett syndrome. J Med Genet. 2000;37(12):E41.
Shibayama A, Cook Jr EH, Feng J, Glanzmann C, Yan J, Craddock N, et al. MECP2 structural and 3'-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet B Neuropsychiatr Genet. 2004;128B(1):50–3.
Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: human genome epidemiology review. Genet Med. 2001;3(5):359–71.
Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Neuropsychiatric disorders in the 22q11 deletion syndrome. Genet Med. 2001;3(1):79–84.
Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil. 2009;30(4):763–73.
Angkustsiri K, Goodlin-Jones B, Deprey L, Brahmbhatt K, Harris S, Simon TJ. Social impairments in chromosome 22q11.2 deletion syndrome (22q11.2DS): autism spectrum disorder or a different endophenotype? J Autism Dev Disord. 2014;44(4):739–46.
Descheemaeker MJ, Govers V, Vermeulen P, Fryns JP. Pervasive developmental disorders in Prader-Willi syndrome: the Leuven experience in 59 subjects and controls. Am J Med Genet A. 2006;140(11):1136–42.
Veltman MW, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF. Prader-Willi syndrome—a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. Eur Child Adolesc Psychiatry. 2004;13(1):42–50.
Bennett JA, Germani T, Haqq AM, Zwaigenbaum L. Autism spectrum disorder in Prader-Willi syndrome: a systematic review. Am J Med Genet A. 2015;167A(12):2936–44.
Hogart A, Wu D, Lasalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2008;38(2):181–91.
Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463(7280):474–84.
Parisi L, Di Filippo T, Roccella M. Autism spectrum disorder in Kabuki syndrome: clinical, diagnostic and rehabilitative aspects assessed through the presentation of three cases. Minerva Pediatr. 2015;67(4):369–75.
Creswell C, Skuse DH. Autism in association with Turner syndrome: genetic implications for male vulnerability to pervasive developmental disorders. Neurocase. 1999;5(6):511–8.
Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010;24(8):3036–51.
Wong CC, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, et al. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19(4):495–503.
Berko ER, Suzuki M, Beren F, Lemetre C, Alaimo CM, Calder RB, et al. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. PLoS Genet. 2014;10(5):e1004402.
Feinberg JI, Bakulski KM, Jaffe AE, Tryggvadottir R, Brown SC, Goldman LR, et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int J Epidemiol. 2015;44(4):1199–210.
Ginsberg MR, Rubin RA, Falcone T, Ting AH, Natowicz MR. Brain transcriptional and epigenetic associations with autism. PLoS One. 2012;7(9):e44736.
Ladd-Acosta C, Hansen KD, Briem E, Fallin MD, Kaufmann WE, Feinberg AP. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry. 2014;19(8):862–71.
Nardone S, Sams DS, Reuveni E, Getselter D, Oron O, Karpuj M, et al. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl Psychiatry. 2014;4:e433.
Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20(6):1164–72.
Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905.
Lokk K, Modhukur V, Rajashekar B, Martens K, Magi R, Kolde R, et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 2014;15(4):r54.
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30.
Slieker RC, Bos SD, Goeman JJ, Bovee JV, Talens RP, van der Breggen R, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450 k array. Epigenetics Chromatin. 2013;6(1):26.
Lowe R, Slodkowicz G, Goldman N, Rakyan VK. The human blood DNA methylome displays a highly distinctive profile compared with other somatic tissues. Epigenetics. 2015;10(4):274–81.
Elagoz Yuksel M, Yuceturk B, Faruk Karatas O, Ozen M, Dogangun B. The altered promoter methylation of oxytocin receptor gene in autism. J Neurogenet. 2016;30:280–4.
Gregory SG, Connelly JJ, Towers AJ, Johnson J, Biscocho D, Markunas CA, et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009;7:62.
Jiang YH, Sahoo T, Michaelis RC, Bercovich D, Bressler J, Kashork CD, et al. A mixed epigenetic/genetic model for oligogenic inheritance of autism with a limited role for UBE3A. Am J Med Genet A. 2004;131(1):1–10.
Nagarajan RP, Hogart AR, Gwye Y, Martin MR, LaSalle JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics. 2006;1(4):e1–11.
Zhu L, Wang X, Li XL, Towers A, Cao X, Wang P, et al. Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum Mol Genet. 2014;23(6):1563–78.
Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A, Grayson DR. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl Psychiatry. 2014;4:e349.
Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, et al. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry. 2012;69(3):314–24.
Sun W, Poschmann J, Cruz-Herrera Del Rosario R, Parikshak NN, Hajan HS, Kumar V, et al. Histone acetylome-wide association study of autism spectrum disorder. Cell. 2016;167(5):1385–97. e11
Fregeac J, Colleaux L, Nguyen LS. The emerging roles of MicroRNAs in autism spectrum disorders. Neurosci Biobehav Rev. 2016;71:729–38.
Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 2016;540:423–7.
Ziats MN, Rennert OM. Aberrant expression of long noncoding RNAs in autistic brain. J Mol Neurosci. 2013;49(3):589–93.
Wang Y, Zhao X, Ju W, Flory M, Zhong J, Jiang S, et al. Genome-wide differential expression of synaptic long noncoding RNAs in autism spectrum disorder. Transl Psychiatry. 2015;5:e660.
Grinshtein N, Rioseco CC, Marcellus R, Uehling D, Aman A, Lun X, et al. Small molecule epigenetic screen identifies novel EZH2 and HDAC inhibitors that target glioblastoma brain tumor-initiating cells. Oncotarget. 2016;7:59360–76.
Seo YJ, Kang Y, Muench L, Reid A, Caesar S, Jean L, et al. Image-guided synthesis reveals potent blood-brain barrier permeable histone deacetylase inhibitors. ACS Chem Nerosci. 2014;5(7):588–96.
Bakermans-Kranenburg MJ, van I Jzendoorn MH. Sniffing around oxytocin: review and meta-analyses of trials in healthy and clinical groups with implications for pharmacotherapy. Transl Psychiatry. 2013;3:e258.
Guastella AJ, Hickie IB. Oxytocin treatment, circuitry, and autism: a critical review of the literature placing oxytocin into the autism context. Biol Psychiatry. 2016;79(3):234–42.
Preti A, Melis M, Siddi S, Vellante M, Doneddu G, Fadda R. Oxytocin and autism: a systematic review of randomized controlled trials. J Child Adolesc Psychopharmacol. 2014;24(2):54–68.
Young LJ, Barrett CE. Neuroscience. Can oxytocin treat autism? Science. 2015;347(6224):825–6.
Anagnostou E, Soorya L, Brian J, Dupuis A, Mankad D, Smile S, et al. Intranasal oxytocin in the treatment of autism spectrum disorders: a review of literature and early safety and efficacy data in youth. Brain Res. 2014;1580:188–98.
Anagnostou E, Soorya L, Chaplin W, Bartz J, Halpern D, Wasserman S, et al. Intranasal oxytocin versus placebo in the treatment of adults with autism spectrum disorders: a randomized controlled trial. Mol Autism. 2012;3(1):16.
Auyeung B, Lombardo MV, Heinrichs M, Chakrabarti B, Sule A, Deakin JB, et al. Oxytocin increases eye contact during a real-time, naturalistic social interaction in males with and without autism. Transl Psychiatry. 2015;5:e507.
Guo JU, Szulwach KE, Su Y, Li Y, Yao B, Xu Z, et al. Genome-wide antagonism between 5-hydroxymethylcytosine and DNA methylation in the adult mouse brain. Front Biol (Beijing). 2014;9(1):66–74.
Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14(12):1607–16.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–30.
Lai MC, Baron-Cohen S, Buxbaum JD. Understanding autism in the light of sex/gender. Mol Autism. 2015;6:24.
Lai MC, Lombardo MV, Auyeung B, Chakrabarti B, Baron-Cohen S. Sex/gender differences and autism: setting the scene for future research. J Am Acad Child Adolesc Psychiatry. 2015;54(1):11–24.
Werling DM, Geschwind DH. Understanding sex bias in autism spectrum disorder. Proc Natl Acad Sci U S A. 2013;110(13):4868–9.
Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M, et al. Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry. 2015;20(1):118–25.
Ey E, Leblond CS, Bourgeron T. Behavioral profiles of mouse models for autism spectrum disorders. Autism Res. 2011;4(1):5–16.
Moy SS, Nadler JJ. Advances in behavioral genetics: mouse models of autism. Mol Psychiatry. 2008;13(1):4–26.
Nakai N, Otsuka S, Myung J, Takumi T. Autism spectrum disorder model mice: focus on copy number variation and epigenetics. Sci China Life Sci. 2015;58(10):976–84.
Petrinovic MM, Kunnecke B. Neuroimaging endophenotypes in animal models of autism spectrum disorders: lost or found in translation? Psychopharmacology (Berl). 2014;231(6):1167–89.
Barry G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol Psychiatry. 2014;19(4):410–6.
Mellios N, Sur M. The emerging role of micrornas in schizophrenia and autism spectrum disorders. Front Psychiatry. 2012;3:39.
Roberts TC, Morris KV, Wood MJ. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos Trans R Soc Lond B Biol Sci. 2014;369(1652).
Wilkinson B, Campbell DB. Contribution of long noncoding RNAs to autism spectrum disorder risk. Int Rev Neurobiol. 2013;113:35–59.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Siu, M.T., Weksberg, R. (2017). Epigenetics of Autism Spectrum Disorder. In: Delgado-Morales, R. (eds) Neuroepigenomics in Aging and Disease. Advances in Experimental Medicine and Biology(), vol 978. Springer, Cham. https://doi.org/10.1007/978-3-319-53889-1_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-53889-1_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53888-4
Online ISBN: 978-3-319-53889-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)