
Abstract
Despite the enormous efforts of the scientific community over the years, effective therapeutics for many (epi)genetic brain disorders remain unidentified. The common and persistent failures to translate preclinical findings into clinical success are partially attributed to the limited efficiency of current disease models. Although animal and cellular models have substantially improved our knowledge of the pathological processes involved in these disorders, human brain research has generally been hampered by a lack of satisfactory humanized model systems. This, together with our incomplete knowledge of the multifactorial causes in the majority of these disorders, as well as a thorough understanding of associated (epi)genetic alterations, has been impeding progress in gaining more mechanistic insights from translational studies. Over the last years, however, stem cell technology has been offering an alternative approach to study and treat human brain disorders. Owing to this technology, we are now able to obtain a theoretically inexhaustible source of human neural cells and precursors in vitro that offer a platform for disease modeling and the establishment of therapeutic interventions. In addition to the potential to increase our general understanding of how (epi)genetic alterations contribute to the pathology of brain disorders, stem cells and derivatives allow for high-throughput drugs and toxicity testing, and provide a cell source for transplant therapies in regenerative medicine. In the current chapter, we will demonstrate the validity of human stem cell-based models and address the utility of other stem cell-based applications for several human brain disorders with multifactorial and (epi)genetic bases, including Parkinson’s disease (PD), Alzheimer’s disease (AD), fragile X syndrome (FXS), Angelman syndrome (AS), Prader-Willi syndrome (PWS), and Rett syndrome (RTT).
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
For decades, the scientific community has been intensively trying to translate their preclinical findings to discover and develop potent therapeutical interventions for pandemic diseases. Although (epi)genetic brain disorders, such as Alzheimer’s disease (AD), rank among the most devastating diseases and account for immeasurable socioeconomic burdens, therapeutical approaches that can prevent, stop, or even reverse them remain unidentified. This lack of effective therapeutics can be attributed to multiple factors, including the inaccessibility of human brain tissue samples, the scarcity of proper human longitudinal studies, the limitations of animal and cellular models, as well as the direct contribution of absence in understanding all brain functions together with the complex cellular heterogeneity of the brain [1, 2]. Although we have acquired an enormous body of knowledge over the years, there is an urgent and unmet demand to develop alternative model systems in order to better understand the underlying biological nature of the brain, as well as to develop new effective therapeutics to be able to reduce the suffering and costs that come along with the occurrence of these disorders.
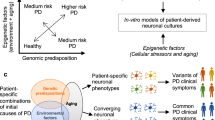
A major challenge in modeling and treating brain disorders has been the inaccessible nature of specific human neural cell types affected by the disease. Stem cell technology has contributed to overcome these challenges, and with the recent discoveries of induced pluripotency, the field has been growing at a rapid pace [3, 4]. In fact, stem cell technology has since then been offering a promising avenue to fill the gap between animal and human research. The field combines the efforts of cell biologists, (epi)genetic researchers, and clinicians to understand human biological systems and to develop effective treatment strategies for human disease. With the use of embryonic stem cells (ESCs), neural stem cells (NSCs), and induced pluripotent stem cells (iPSCs), the field of stem cell technology is dedicated to develop adequate disease model systems, preclinical platforms for (high-throughput) drugs and toxicity screenings, and strategies for transplant therapies (Fig. 23.1) [5,6,7]. Theoretically, ESCs and iPSCs allow to obtain nearly every cell type of the human body in vitro and provide an inexhaustible cell source due to their pluripotent differentiation potential and capacity of self-renewal [7]. As a consequence, these stem cells can be cultured and differentiated into NSCs and functional neural cells by using directed differentiation techniques [8, 9].

Illustration of the different stem cell-based applications currently available for human brain disorders, including disease modeling, drugs and toxicity screening, and transplant therapies in regenerative medicine. The possible origins of the stem cells are depicted and approaches to move from bench-to-bedside specified
Although there is still a lot to improve in terms of their efficiency and safety, stem cell-based models harbor high translational potential and are currently very appealing to study [10]. In fact, these model systems have proved instrumental to model in vitro molecular alterations associated with genetic mutations in disease-causing genes and allow mechanistic cellular studies of multifactorial (epi)genetic brain disorders [5, 11]. While it is well established that (epi)genetic alterations contribute to the pathophysiology of human disease, detailed epigenomic characterization of stem cell-based models and the role of epigenetic changes in the pathophysiology of these diseases remain underexplored and are currently just starting to become increasingly considered [2, 12]. The complex and interconnected network of epigenetic alterations, including DNA (hydroxy)methylation, histone modifications, and noncoding RNAs, has previously only been studied in animal models and in human postmortem brain samples. Nevertheless, with the rise and recent advances in stem cell technology, it should be accordingly expectable that stem cell-based neural models may represent valid tools to explore epigenetic changes involved in many brain disorders. The use of these models will undoubtedly contribute to a better understanding of human brain epigenetics and general physiology in the future.
In the current chapter, we will introduce the use of stem cell technology for human brain disorders with multifactorial and (epi)genetic bases, including Parkinson’s disease (PD), AD, fragile X syndrome (FXS), Angelman syndrome (AS), Prader-Willi syndrome (PWS), and Rett syndrome (RTT). We will address the recent advances of stem cell technology either with regard to disease modeling, drugs, and toxicity testing or direct clinical applications, with the aim to provide the notion of their utility in fundamental research, as well as in the field of biomedicine.
2 Parkinson’s Disease
PD is a neurodegenerative disorder affecting over ten million people worldwide [13]. It is estimated that the majority of PD cases (80–90%) are idiopathic with a multifactorial origin, while the minority of the cases (10–20%) are familial and linked to monogenic mutations in PD-related genes, including PARK2, PARK7, PINK1, ATP13A2, SCNA, LRRK2, TAU, NURR1, and GBA [6, 14]. Furthermore, there is a growing body of evidence supporting the role of epigenetics in the development and progression of PD [15, 16]. Differences in disease onset are observed, but PD pathology is common and characterized by progressive loss of dopaminergic neurons in the substantia nigra pars compacta of the mesencephalon [13, 17]. The most crucial pathological hallmarks seen in these neurons are abnormal aggregates of SNCA protein that form Lewy bodies and Lewy neurites [18, 19]. The exact molecular mechanisms underlying the initiation and progression of neurodegeneration remain elusive, but loss of dopaminergic neurons causes deficits in dopaminergic neurotransmission. These neurons are essential in the regulation of the motor functions, and their loss results in typical motor symptoms such as rest tremor, rigidity, bradykinesia, and gait abnormalities [18].
Previous therapeutic strategies have heavily relied on dopamine-enhancing drugs such as levodopa, dopamine receptor agonists, and monoamine oxidase inhibitors to compensate for the loss of dopaminergic neurotransmission [20, 21]. Alternatively, deep brain stimulation of the subthalamic nucleus represents an effective therapy in PD [22]. Even though these therapeutics are able to alleviate the symptoms, they are not able to compensate the cellular loss typical for PD. The availability of stem cell technology, however, has offered an alternative approach to treat PD and overcome this issue. Numerous studies have addressed the therapeutic potential of compensating the dopaminergic cell loss by replacing them by external cell sources, including NSCs and NSC-derived dopaminergic neurons. NSCs can be isolated from fetal brain tissue or specific regions in the adult brain, they can be obtained from differentiated ESCs and iPSCs after neural induction in vitro, or they can be derived from direct reprogramming of somatic cells [6, 23].
Although NSCs, ESCs, and iPSCs have all been studied for their therapeutic potential in PD, mesenchymal stem cells and olfactory ensheathing cells have also been considered [6]. Furthermore, the use of NSCs, ESCs, and iPSCs for disease modeling or drugs and toxicity testing in PD is another rapid-moving field of research [19]. While this area of research is also very interesting, here we will mainly focus on the direct clinical application and major problems that have been encountered when using these stem cells for the treatment of PD.
Studies using rodent and nonhuman primate models of PD have demonstrated that NSCs transplanted into animal brains differentiate into dopaminergic neurons [24, 25]. Endogenous NSCs retain their regional specificity, and therefore, fetal grafts derived from a dopaminergic- enriched region, such as the mesencephalon, are appropriate cell sources for direct transplantation [21]. When grafts of fetal brain tissue are transplanted to the midbrain of induced parkinsonism rats or nonhuman primates, they can improve many of the typical motor deficits seen in these animal models [24, 26,27,28]. Additionally, differentiation of NSCs into dopaminergic neurons can also be enhanced in vitro prior to transplantation [29, 30]. Studies that differentiated fetal grafts into enriched populations of dopaminergic neurons have also found significant cellular and motor behavior recovery in PD animal models [31, 32]. Graft-induced amelioration of motor deficits is dependent on the ability of the grafted NSCs and neurons to restore dopaminergic neurotransmission in the affected area surrounding the transplant. The mechanisms that are thought to underlie these effects can be classified into two categories: direct repair by dopaminergic neuron replacement and indirect repair trough stem cell-derived neurotropic factors [21].
Clinical trials using fetal brain mesencephalic tissue grafts were initiated in the 1990s [33,34,35,36]. Improvements have been documented in PD patients in terms of behavior, histology, and survival of the transplanted cells, and in several cases, they were even able to eliminate their dopamine-enhancing medication [37]. Interestingly, studies have demonstrated that transplanted grafts survived and remain functional up to 14 years posttransplantation, although evidence that PD pathology may propagate from host to grafts is emerging [38, 39]. On the other hand, several mild to severe side effects were also observed. One of the most troubling side effects was the occurrence of graft-induced dyskinesia [40, 41]. Furthermore, since the midbrain tissue used to treat PD patients is derived from a genetically distinct individual, i.e., allogeneic, the transplanted grafts cause immunogenic responses that need to be repressed continuously to prevent graft rejection [42]. In addition to these side effects, the use of fetal tissue grafts for PD treatment is also challenging on a large scale, given the limited accessibility and ethical concerns behind the use of primary brain tissue from aborted fetuses [43].
ESCs derived from blastocyst embryos, on the other hand, have an intrinsic capability for infinite self-renewal and are able to differentiate into nearly any cell of the human body, including NSCs or dopaminergic neurons [44]. This theoretical inexhaustible source of cells indicated ESCs as having great promise for cell transplantation therapy in PD. Likewise to fetal grafts, the functional characteristics of transplanted ESC-derived NSCs and dopaminergic neurons have been addressed in vivo by engrafting these cells into animal models of PD [45,46,47,48,49]. These experiments demonstrated that ESC-derived NSCs or dopaminergic neurons were able to integrate into the host brains and to restore dopaminergic neurotransmission, as well as to improve locomotive deficits seen in these PD models. This provided preclinical evidence of the potential of ESC-derived NSCs and dopaminergic neurons for the treatment of PD [50]. Although ESCs seemed to be a very promising cell source, the efficiency of neural cell conversion from ESCs is still limited and often results in incompletely differentiated heterogeneous cellular populations containing different neural cells [51,52,53]. Therefore, a critical issue that must be resolved and that might even increase the functional outcome after transplantation is the improvement of directed differentiation protocols, as well as cell sorting techniques.
The potential clinical relevance of ESCs found in animal models has opened the possibility for transplantation therapy in human PD patients, but clinical trials using ESCs have not been initiated for treating PD until very recently. Main issues that prevented these cells to move from bench-to-bedside were related to their possible phenotypic instability due to incomplete directed differentiation processes. Moreover, there is a chance of transplanting residual undifferentiated ESCs that can lead to tumor formation in vivo [54]. Furthermore, and also similar to human fetal tissue grafts, ESCs are allogeneic and harbor the problem of immune rejection [55]. Despite the methodological and potential ethical caveats, the Australian Therapeutic Goods Administration has approved a Phase I clinical trial for PD using parthenogenetic ESCs (pESCs) [56, 57]. Parthenogenetic embryos are formed by chemically activating the unfertilized human ovum, which allows induction of pESC cultures that are unable to produce a viable offspring [57]. Based on two preclinical safety studies with rats and nonhuman primates [58], the idea of the International Stem Cell Corporation is to derive NSCs from these pESCs to treat PD patients [59].
With the elegant discovery of somatic cell reprogramming and induced pluripotency by Takahashi and Yamanaka in 2006 [4], another opportunity for the treatment of PD became available. iPSCs are similar if not virtually identical to ESCs in terms of their self-renewal, differentiation potential (pluripotency), morphology, surface marker expression, and in vivo teratoma formation capacity [4, 7]. In relation to transplant therapies for PD, the major advantage of iPSCs above all the aforementioned stem cell types is that iPSCs can be generated from somatic cells of the PD patient to be treated, which allows autologous transplantation [60]. As a consequence, these cells contain the genetic background of the donor, which is speculated to minimize the risk of immune rejection [61]. Moreover, the ethical issues with regard to destruction of fetuses and embryos are circumvented with the use of iPSCs. These unique characteristics support the therapeutic potential of human iPSCs for personalized cell replacement therapy of PD.
To date, several studies have differentiated iPSCs to NSCs or dopaminergic neurons and examined their clinical potential in PD animal models. Grafted iPSC-derived NSCs survived, differentiated in vivo into dopaminergic neurons, matured, and integrated into the recipients’ brains [62]. In addition, transplanted human and non-primate iPSC-derived NSCs or dopaminergic neurons were found to have significant therapeutic effects in rat and non-primate PD models by alleviating PD phenotypes [62,63,64,65,66]. Interestingly, in one of these studies, the differentiated neural populations derived from the iPSCs were characterized and sorted based on cellular markers prior to transplantation [63]. Sorting the iPSC-derived neural populations eliminated the undifferentiated tumorigenic cells and significantly increased the number of dopaminergic neurons in the cell grafts compared to unsorted cell populations [63]. These findings demonstrate that sorted and enriched dopaminergic neuronal populations are viable, safe, and functional in vivo, as well as improve the functional impairments posttransplantation [46, 63].
Based on the animal studies, much efforts have been made to bring these potential therapeutic cells to GMP (Good Manufacturing Practice) standards so they can be translated to the clinic for treatment of PD. However, clinical trials using human iPSCs have not been reported yet. In spite of the initial positive results of iPSC-derived NSCs and dopaminergic neurons in animal models of PD, there are several hurdles that have to be elucidated to realize their full potential in regenerative medicine, including the discrepancies around the iPSCs’ epigenetic memory [67], differentiation bias [68], mitochondrial dynamics [69, 70], and the appropriate choice of reprogramming technology. In fact, current somatic cell reprogramming techniques have heavily relied on genomic integrating techniques containing factors such as c-MYC and KLF4 [3]. These techniques can affect the genome in a yet unspecified way and might alter the neurobiology of the derived cells, including their differentiation potential, as well as their survival and integration into the recipients’ brains.
Another concern related to use of patient-derived iPSCs is that these cells potentially harbor susceptibility traits to PD phenotypes because of mutations or epigenetic markers that could be present in these patients’ cells. For instance, establishing iPSC lines derived from patients that harbor PD-related genetic risk loci might make these cells more susceptible to develop PD phenotypic characteristics posttransplantation. To overcome this issue, iPSCs have been generated where the underlying mutations in the disease-causing genes were modified using genomic editing techniques [71,72,73]. Finally, whereas it has been generally assumed that autologous iPSCs should be immune-tolerated for the patient from whom the somatic cells are derived, several studies have reported immune rejection responses [61, 74].
Stem cell studies and in particular the use of iPSCs also allow for PD disease modeling in vitro. These stem cell-based studies offer a unique opportunity to unravel the (epi)genetic and environmental contributions of the disorder in patient-specific dopaminergic neurons [19]. Dopaminergic neurons from genetic PD cases have already been used to recapitulate disease phenotypes, such as impaired dopamine metabolism, SCNA accumulation, mitochondrial dysfunction, and oxidative stress vulnerability [75,76,77,78,79,80,81,82]. In addition, the study of epigenetic mechanisms using patient iPSC-derived PD models is expected to have a huge impact in understanding the pathophysiology of PD and to assist the development of therapeutic interventions [2]. Moreover, patient iPSC-derived dopaminergic cells could represent useful models to potentially recapitulate the environmental exposome through the patients’ epigenome [2]. Accordingly, iPSC-based models are expected to be helpful for investigating epigenetic changes of disorders where the environment is supposed to play a more prominent role, such as idiopathic form of PD [2]. Although the complexity of these multifactorial disorders is expected to be high, especially when taking into account possible interactions between (epi)genetic factors that could modify pathological phenotypes, iPSC-derived models provide new opportunities to investigate epigenomic alterations associated with the disorder. A pioneer study by Fernández-Santiago et al. [83] recently provided first evidence that epigenetic deregulation is associated with monogenic and idiopathic PD in an iPSC-based model system. Interestingly, their findings suggest the presence of molecular deficits in PD somatic cells that manifest only upon differentiation into dopaminergic neurons [83]. Future comparable studies will have important implications for disease modeling, as well as transplant therapies using patient-derived iPSCs.
3 Alzheimer’s Disease
AD is the most common neurodegenerative disorder and leading cause of age-related dementia [84]. In 2010 approximately 36 million people were diagnosed with AD worldwide, and the incidence is expected to double every 20 years to an estimated 115 million cases in 2050 [85]. The early onset autosomal-dominant form of AD, termed familial AD (FAD), generally occurs between 30 and 60 years of age and is estimated to represent less than 5% of all AD cases [86]. The average age of occurrence for the more common multifactorial late onset form, sporadic AD (SAD), is 65 years with an increasing likelihood of developing the disorder each subsequent year [86]. Both forms are characterized by progressive memory disorientation and cognitive disturbances, but remain clinically and neuropathologically heterogeneous [87]. Main hallmarks that are seen in AD brains include aggregation of amyloid-β (Aβ) peptides into extracellular senile plaques and accumulation of intracellular hyperphosphorylated tau protein into neurofibrillary tangles (NFTs) [88]. Furthermore, neuroinflammation, oxidative stress, and endoplasmatic reticulum (ER) stress have also been implicated in the disease [89]. Although many hypotheses have been proposed to explain the pathogenesis of AD, the interrelationships and causality of these hallmarks remain to be elucidated.
To date, AD studies have mainly relied on the use of transgenic mice models, the use of non-neural human cell cultures, and human postmortem tissue analyses [85, 90]. Although the significant impact of transgenic mice models on progress in understanding various aspect of the disorder is undeniable, they only reproduce specific AD hallmarks and do not reflect clinical phenotypes completely. Moreover, the use of non-neural cell lines omits unique neural features and therefore may fail to capture essential biological processes. The limited accessibility to postmortem tissues and an inadequate amount of cell subtype-specific samples add up to this and together hinder the study of its biological basis. Even in the case that samples are available, the use of postmortem tissue does simply not allow to differentiate between molecular hallmarks that are involved in the causes or consequences of the disease. For these reasons, there has been an ongoing demand for innovative and predictive model systems that closely resemble unique human neural features and which allow to study cause-effect relationships in a controlled setting.
The recent advances in stem cell technology make the availability of iPSCs for AD studies very relevant in this context. iPSCs derived from either FAD or SAD patients´ somatic cells contain a patient-specific pathogenic background, which offers a promising avenue for AD modeling [89]. In fact, the use of disease-relevant neural cells, by differentiating iPSCs along the neural lineage, offers an alternative approach to study the underlying neuropathological mechanisms in vitro in a humanized, personalized, and cell subtype-specific manner. iPSC-derived neuronal populations generated from AD patients with known pathogenic backgrounds can be studied, (epi)genetically probed, and treated with drug libraries to investigate their effects on molecular and cellular responses. For these reasons, there has been a growing body of research over the past years to adopt rapidly improving iPSC-derived model systems of AD for fundamental research applications, as well as for the assessment of drugs prior to the initiation of clinical trials [91, 92]. Due to this increasing interest in patient-derived iPSCs for AD research, we will here only focus on the recent progresses of iPSC studies and demonstrate their utility for disease modeling and drug discovery.
Modeling AD using patient-derived iPSCs was initiated from FAD cases with known mutations in disease-causing genes, including amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) [93]. The main goal of these preliminary studies using FAD patient-derived iPSCs has been the validation of their potential for AD modeling, in which they have been seeking to find AD-associated cellular phenotypes. It has previously been shown that mutations in APP, PSEN1, or PSEN2 may cause abnormal cleavage of APP, which results in increased levels of total Aβ or increased ratios of neurotoxic Aβ42 to Aβ40 (Aβ42/40) peptides [94].
Several studies have focused on increased copy numbers of the APP gene. The first described was a trisomy 21 Down syndrome model using both patient ESCs and iPSC-derived neurons [95]. Down syndrome individuals have an increased risk of developing AD, which has been attributed at least in part to having three copies of APP [96]. In the iPSC-derived cortical neurons, increased Aβ production and Aβ42/40 ratios were observed [95]. Furthermore, phosphorylated tau and total tau levels were seen to be upregulated and mislocalized to the neuronal dendrites [95]. In a separate study, iPSC neurons from FAD patients with a duplication of APP were analyzed [97]. Compared to non-demented control individuals, these FAD neurons exhibited significantly higher levels of Aβ40, phosphorylated tau, and glycogen synthase kinase-3β (GSK-3β) activity – a physiological kinase of tau [97]. Moreover, the neurons also accumulated large RAB5-positive endosomes, which has been seen in autopsies from SAD and some forms of FAD [97, 98].
Missense mutations in APP that are known to cause AD have also been studied in patient-derived iPSC models. A study by Kondo et al. [99] included three iPSC lines from FAD patients harboring the APP-E693Δ (Osaka) mutation. The iPSC-derived cortical neurons exhibited accumulated intracellular Aβ oligomers, leading to endoplasmic reticulum and oxidative stress [99]. Interestingly, two APP-V717 L mutant lines in the same study were tested and produced large quantities of extracellular Aβ42 that lacked intracellular accumulation, as well as the accompanying stress response hallmarks [99]. In iPSC-derived forebrain glutamatergic neurons from AD patients harboring the APP-V717I (London) mutation, significant higher levels of Aβ42 were also found [100]. The fold increase in Aβ42/40 ratio reported was highly similar to that observed in plasma from human subjects with the same mutation [100, 101]. Taken together, the pathological phenotypes found in these AD-iPSC studies mimic those that have been previously defined in mice and/or cellular models carrying the same mutations [99, 100], demonstrating the validity of iPSC-based model system.
In addition to APP mutations, studies have also focused on patient-derived iPSCs that contain mutations in PSEN1 or PSEN2. Yagi et al. [84] demonstrated that iPSC-derived neurons harboring the PSEN1-A246E and PSEN2-N141I mutations have increased levels of Aβ42. Both mutations have also been reported to induce elevated levels of Aβ42 in human plasma, as well as in animal and other cellular models [84, 101,102,103]. Sproul et al. [104] analyzed iPSC lines with the PSEN1-A246E and PSEN1-M146L mutations, also demonstrating that mutant neural precursor cells (NPCs) presented increased Aβ42/40 ratios. Molecular profiling in this latter study identified 14 genes differently regulated in mutant PSEN1 NPCs, of which five targets were previously shown to be differentially expressed in late and intermediate AD patients [104]. While the gene expression changes identified in this study are intriguing, they also emphasize the power of AD-iPSC studies to replicate additional phenotypic features. However, future mechanistic studies in both human cells and animal models are required to determine whether they indeed play a role in AD.
APP mutants, but not PSEN1 and PSEN2 mutants’ iPSC lines, demonstrated elevated total levels of tau and tau phosphorylation in a recent study by Moore et al. [105]. They compared different iPSC-derived cortical neurons from AD patients harboring APP mutations (APP-V717I and duplication of APP) or PSEN1 mutations (PSEN1-Intron/Δ4, PSEN1-Y115C, PSEN1-M146I). While these differences might be related to unknown effects of culture conditions or timing, another testable possibility is that APP and PSEN1 or PSEN2 mutations differ in their pathological phenotypes.
Another interesting approach for studying genetic-based disorders like FAD is with the use of isogenic lines created by genome editing of ESCs or iPSCs [73, 92]. Isogenic cell lines can be generated by inducing or correcting AD mutations in the wild-type (WT) cell line or patient-derived iPSCs, respectively [106]. The use of isogenic lines with the same genetic background reduces the intrinsic variability that comes from comparing cells from different individuals and allows to determine how a single targeted mutation affects molecular and cellular mechanisms [106]. The first study to do this for FAD mutations was conducted by Woodruff et al. [107]. They were able to generate an allelic series of heterozygous and homozygous PSEN1-ΔE9 knock in iPSC lines, as well as heterozygous null mutants with WT and PSEN1-ΔE9 alleles. The authors demonstrated that PSEN1-ΔE9 mutant neurons increased the Aβ42/40 ratio in a gene dosage-dependent manner by significantly decreasing the amount of Aβ40, while moderately increasing the amount of Aβ42 [107]. The results found in this study emphasize the use of isogenic cell lines as potential promising tool for modeling AD.
iPSC-derived neurons from SAD patients have also been used to study AD, by comparing the phenotypic characteristics of these cells with iPSC-derived FAD neurons. In some of the previously mentioned reports, i.e., Israel et al. [97] and Kondo et al. [99], iPSC lines from two random SAD patients were analyzed in parallel. Interestingly, in each case only one of the two iPSC-derived neuronal populations demonstrated phenotypes consistent with the FAD lines. This heterogeneity corresponds with the complex origin of SAD and its influence on disease causation and progression. SAD is thought to be multifactorial, defined by a lack of autosomal-dominant inheritance and arises due to a complex interplay of (epi)genetic and environmental risk factors [108,109,110]. It is estimated that at least 60–80% of SAD may have a genetic underpinning [109, 111, 112]. Recent developments in genomic technologies, on the other hand, have allowed for high-throughput interrogation of the epigenome, and epigenome-wide association studies (EWAS) have identified unique epigenetic signatures that play a role in AD [15, 16, 113].
To date, only two studies have been published that directly focused on patient-derived iPSC lines that harbor known SAD genetic risk factors [114, 115]. For many years, APOE3/E4 was the only known robust genetic risk factor, but as a result of several genome-wide association studies (GWAS) collaborations, increasing sample sizes and meta-analysis, at least 20 other risk, as well as protective, loci have been reported [108, 112, 116, 117]. Duan et al. [114] analyzed iPSC lines derived from three APOE3/E4 SAD patients next to two PSEN1 FAD mutant lines (PSEN1-A246E and PSEN1-M146 L). The basal forebrain cholinergic neurons derived from AD-APOE3/E4 patients’ iPSCs showed typical AD hallmarks, including increased Aβ42/40 ratios [114]. A second study by Young et al. [115] focused on SORL1, which encodes an endocytic trafficking factor whose levels modulate the processing of APP to Aβ and other proteolytic products implicated in SAD [118]. Loss of SORL1 expression has been documented in SAD cases [119], and the SORL1 locus has been associated with SAD in both candidate gene and GWAS analyses [112]. By studying patient iPSC-derived neural cells, this latter study confirmed the importance of the SORL1/APP pathway in SAD, and their findings corroborate most previous studies in cell and animal models [115].
In another study by Hossini at el. [120], SAD iPSC-derived neurons were analyzed to assess the reflection of disease phenotype in gene expression and to examine the expression of typical AD proteins. The differentiated neuronal cells seemed to reflect the SAD phenotype by the expression of phosphorylated tau proteins and the upregulation of GSK-3β [120]. Further analysis of the neuronal cells also revealed significant changes in the expression of other genes associated with AD, including subunits of the proteasome complex [120]. Moreover, a disease-specific protein association network that models AD pathology on the transcriptome level could be generated from the AD-iPSCs [120]. Taken together, these studies have demonstrated that SAD patients’ iPSC-derived neuronal cells are able to recapitulate neuropathological processes of the disease.
Unfortunately, the contribution of epigenetic signatures, as well as environmental factors, has not been addressed yet in iPSC neurons derived from SAD, as well as FAD patients. Nonetheless, iPSC-derived neurons offer a platform to examine the casual relationships between environmental insults and the generation of molecular, cellular, and epigenomic responses in AD-relevant neuronal populations. In theory, one could test how the derived neuronal populations aggravate AD characteristic phenotypes when exposed to environmental risk factors or pathological hallmarks, such as stress hormones [121]. Furthermore, the use of iPSCs also provides the opportunity to examine the contribution of AD-associated epigenetic signatures. Based on recent advances in the field of epigenetic editing [122, 123], these AD-associated epigenetic signatures can be modified at any given locus in order to normalize the cellular phenotypes in patient iPSC-derived neurons, as well as to induce AD characteristics in control iPSC lines.
In addition to evaluating the potential of iPSC-derived neurons to model typical AD phenotypes, substantial work has been done in order to assess the possible medical relevance of AD iPSC models in terms of drug discovery and selection of appropriate therapeutics. Many of the aforementioned studies have tried to normalize the AD-associated phenotypes by previous studied therapeutics for AD. In the iPSC model of Down syndrome, for example, Shi et al. [95] speculated whether Aβ40 and Aβ42 peptide generation could be reduced by pharmacological inhibition of the γ-secretase complex. Compounds that inhibit γ-secretase, as well as β-secretase, are potential therapeutics for AD, and inhibition of these protease complexes has been shown to reduce Aβ level in mice models [124, 125]. When a γ-secretase inhibitor was administered for 4 consecutive days to the Down syndrome iPSC-derived neurons, Aβ40 and Aβ42 peptide production was reduced by almost half, whereas longer-term treatment (21 days) reduced secretion of both Aβ peptides below detectable levels [95]. Also Yagi et al. [84] found a dose-dependent reduction in Aβ42 and Aβ40 in iPSC-derived neurons form AD patients treated with γ-secretase inhibitors and modulators. A γ-secretase inhibitor was also sufficient to block Aβ production in both control and PSEN1 mutant NPCs [104], as well as in PSEN1 mutant neuronal cells [107]. Surprisingly, γ-secretase inhibitor treatment paradoxically increased Aβ40 secretion in the APOE3/E4 SAD lines [114]. The reason for this latter finding is not clear yet and will need to be addressed properly in future studies.
In other studies, FAD and SAD patient iPSC-derived neurons were treated with γ-secretase inhibitors or β-secretase inhibitors, and Aβ, GSK-3β, phosphorylated tau, and total tau levels were assessed. It was shown that β-secretase inhibitors, but not γ-secretase inhibitors, could significantly reduce the levels of phosphorylated tau and GSK-3β, while γ-secretase inhibitors only reduced the level of Aβ40 [97].These findings suggested that APP proteolytic processing had a direct relationship with GSK-3β activation and tau phosphorylation in these neuronal models [97]. In line with these findings, manipulation of APP metabolism by β-secretase and γ-secretase inhibition/modulation also affected tau protein levels in the study by Moore et al. [105]. Furthermore, inhibition of γ-secretase significantly reduced the production of extracellular Aβ38, Aβ40, and Aβ42 in neurons of all genotypes [105]. In a separate study, also significant reductions of phosphorylated tau and tau expression were found in neuronal cells differentiated from a SAD patient after treatment with a γ-secretase inhibitor [120] .
Kondo et al. [99] evaluated β-secretase inhibitors and three additional drugs that have been reported to improve ER stress or to inhibit reactive oxygen species (ROS) generation, including docosahexaenoic acid (DHA). Intracellular accumulation of Aβ oligomers disappeared and ROS formation decreased after treatment with β-secretase inhibitors in both FAD and SAD iPSC-derived neurons [99]. DHA treatment, on the other hand, decreased the generation of ROS in AD neural cells harboring the APP-E693Δ mutation, whereas the amount of Aβ oligomers in cell lysates was not altered [99]. The clinical effectiveness of DHA treatment is still controversial and, interestingly, only one of two sporadic AD neurons accumulated intracellular Aβ oligomers and showed cellular phenotypes that could respond to DHA, while the other did not [99]. This result may explain why DHA treatment is only effective for some subpopulations of SAD patients, although disease stage and timing of treatment could be other critical factors to explain this phenomenon. These patient-specific iPSCs might, therefore, provide a chance to reevaluate the effect of a drug that failed in AD clinical trials, depending on the subpopulation of patients. Finally, immunotherapy is one of the alternative strategies being studied for the treatment of AD [126]. In the study of Muratore et al. [100], Aβ-specific antibodies were able to reverse the phenotype of increased total tau in AD iPSC-derived neurons harboring the APP-V717I mutation.
To conclude, current studies represent critical first steps in assessing the potential of using iPSCs in AD research. Patient-specific iPSCs-derived neural cells have demonstrated validity on modeling AD pathological molecular alterations, such as increased Aβ42/40 ratios and tau hyperphosphorylation. Moreover, these studies have addressed the benefits of these iPSC systems for testing therapeutic intervention strategies and drug libraries. Furthermore, iPSC technology might also be a valuable tool in exploring the complex heterogeneous nature in the etiology of SAD through interrogation of functional effects of (epi)genetic variants linked to risk and protective factors. With the availability of epigenetic editing systems, we might be able to decipher how epigenetic alterations participate in AD, which may also offer opportunities for future epigenetic-based pharmacological interventions. Nevertheless, there is enormous promise in the utility of iPSC technology to predict how individual epigenetic and cellular phenotypic variation contributes to the etiology and pathophysiology of AD, as well as to pharmacological responses at clinically relevant levels.
4 Fragile X Syndrome
FXS is considered an autism spectrum disorder and the most frequent form of inherited intellectual disability with a penetrance of 1 in 2500 males and 1 in 4000 females [127]. Patients suffer from multifactorial symptoms such as learning deficits, low IQ, autism-like behavior, obesity, hypotonia in childhood, and seizures in adult life [128]. The disease is caused by the loss of fragile X mental retardation protein (FMRP), which is a cytoplasmic RNA-binding protein involved in transport and translocation of mRNA and proteins from the nucleus to neuronal dendrites [129, 130]. FMRP plays an important role in regulating synaptic development, plasticity [131], and vesicular dynamics [132]. FMRP absence is usually caused by an aberrant epigenetic silencing of fragile X mental retardation 1 (FMR1) gene due to promoter inhibition, as a consequence of CGG repeat expansion in proximity to the 5-UTR region [133, 134]. The increase in length of CGG repeats marks the region for hypermethylation. Non-affected individuals typically have 6–50 CGG repeats in the CpG island close to FMR1 locus. For reasons still unknown, some subjects are susceptible to an expansion of those repeats. Between 50 and 200 CGG repeats are found in this region in a pre-mutation state, whereas most FXS patients carry >200 repeats and characterize the full mutation state, likely leading to CpG methylation, chromatin condensation, and FMR1 transcriptional silencing [135].
Mice models mimicking the disease phenotype were developed using FMR1 gene knockout (KO), mirroring several pathological features of FMRP impairment as FMR1 mRNA targets and FMRP functions [136,137,138]. Although very interesting, mice lack the epigenetic silencing of FMR1 in large CGG repeats, therefore, limiting the use of mice models for fully understanding the molecular mechanism of the disease. Furthermore, the mechanism of epigenetic alterations on FMR1 locus and the consequent loss of FMRP during development are still uncertain [139]. The use of human stem cells has collaborated to a better comprehension of the disease mechanisms [140], and for the purpose of this section, we will, therefore, explore the recent discoveries using iPSC-based models for FXS.
ESCs with >200 CGG repeats present unmethylated CpGs and normal levels of FMR1 gene in early stages of development and progressively become methylated during differentiation [141]. Patient-derived iPSCs, on the other hand, maintain their methylation status during the reprogramming process, which demonstrates that in iPSCs the methylation status is not reversible [140, 142]. The epigenetic silencing of FMR1 is believed to occur in a differentiation-dependent manner, although the maintenance of methylation during reprogramming of iPSC-derived neurons from FXS patients supports their use for modeling the disorder [143]. Nonetheless, iPSCs generated from patients’ fibroblasts usually show that the repeat expansion is not equally present in all the cells, thus modeling the mosaicism observed in patients [144]. This mosaicism contributes greatly to disease pathogenesis and individual phenotypic variability [127, 135, 144]. Moreover, by using iPSCs, scientists were able to illustrate the phenomenon of CGG repeat expansion. After reprogramming, fibroblasts from mutated individuals (>200 repeats) generated cells with full-length expansions and fully methylated status. However, fibroblasts with a pre-mutation genotype (between 50 and 200 repeats) generated cells with either a normal genotype (<50 repeats) or full-mutated clones [141, 145]. These data show that pre-mutation expansion length is genetically instable and that the pre-mutated state is critical for disease pathogenesis [146].
Apart from that, a growing body of evidence suggested that the epigenetic state of FMR1 locus, rather than the extension of the CGG repeats, is critical for transcriptional silencing [135, 144, 147]. In agreement, de Esch et al. [148] studied iPSC derived from unmethylated full-mutated individuals (presenting >200 repeats but no methylation on FMR1 locus). Interestingly, after fibroblast reprogramming, the obtained iPSC-derived neurons presented a full methylation status on FMR1 region. This data demonstrated that methylation is a standard mechanism in CGG repeats expansions and that individuals harboring repeats without methylation had a normal phenotype, likely attributed to the unmethylated status of FMR1.
Likewise, Park et al. [149] also illustrated the importance of (CGG)n repeats in methylation and silencing of FMR1. In their study, genetic editing with CRISPR/Cas9 technology was used to eliminate CGG repeats upstream of the promoter in FXS iPSC-derived neurons. CRISPR/Cas9 technology works as a sequence-specific nuclease inducing a double cleavage in the DNA in targeting regions [150]. After deleting a large portion of CGG repeats, the authors described that the chromatin opened and demethylation occurred, leading to transcriptional activation of FMR1. These findings do not only suggest that transcriptional silencing of FMR1 can be reverted, but also that methylation status in this region is constantly monitored.
Finally, FXS patient-derived iPSCs are also starting to be used for high-throughput drug testing and harbor high potential for drug discovery research. For instance, iPSCs have been treated with drugs that could increase FMR1 levels [151], but unfortunately no clinical relevant results were obtained yet. The possibility of reverting FXS phenotype after reestablishment of FMR1 levels is also still uncertain and might partly explain these findings. To conclude, although all the pathophysiological alterations in FXS remain to be elucidated, stem cell technology is unraveling the role of epigenetic alterations in the disorder, such as the methylation dynamics of the FMR1 locus. Research along this line will greatly facilitate the development of therapeutic interventions and will allow future studies using epigenetic editing techniques.
5 Angelman Syndrome and Prader-Willi Syndrome
AS was first described in 1965 by the pediatrician Harry Angelman who noticed autistic-like features in three different individuals [152]. AS patients present neurological problems, abnormally motor condition, severe mental retardation, epileptic seizures, and episodes of inappropriate laughter [153]. The prevalence is estimated between 1/10,000 and 1/20,000 [154]. PWS is characterized by hypotonia, hypogonadism, intellectual disability, a tendency to develop compulsive and obsessive behavior, and hyperphagia-causing obesity [155]. The incidence, on the other hand, is estimated between 1/15,000 and 1/30,000 [155]. Both disorders are caused by imprinting alterations on chromosome 15q11–13 region [156] although AS is caused by maternal inherited alterations, while PWS is caused by paternal ones.
During development, specific genes on a variety of chromosomes are subjected to silencing by a process known as genomic imprinting, which is dependent on the location of the gene on the chromosome, as well as its parent origin [157, 158]. The reason for silencing one of the parental genes is not fully understood, and estimations point to only 1% of human genes to have inherited repression markers [159]. Silencing is mediated by a set of epigenetic alterations such as methylation of promoters and histone modifications [160, 161]. An important region of chromosome 15 (15q11–13) that is critical for many cellular processes is also subjected to genomic imprinting. This region compromises a variety of genes that are exclusively expressed in a parental fashion: MKRN3, MAGEL2, NDN, C15orf2 and PWRN1 SNURF-snrpn for paternal origin, and UBE3A and ATP10A for maternal origin [162, 163]. Because many of these genes in this region are imprinted, the loss of function of one copy leads to vigorous alterations [164].
AS is caused by the reduction or loss of the maternal allele coding for UBE3A gene, while PWS results from partial deletion of the 15q11.2-q13 region of the paternal allele and affects seven genes [160]. However, not all AS or PWS diagnosed patients present the same classical alterations on 15q11.2-q13 region [153]. Both disorders and other related pathologies are also known to be caused by other genetic alterations [160], including large and small deletions and duplications of 15q11.2-q13, mutations in imprinting centers, and uniparental disomy, among others [165]. As for many epigenetic disorders, the variety of genomic alterations affects the clinical severity of the disease and the possibility of modeling it for studying.
Mice models targeting chromosome 15 have been used to study AS dynamics. The most commonly used model is the deletion of exon 2 from Ubea3, since it is the only model that induces inherited maternal loss of Ubea3 [166, 167]. Using Ubea3-null mice, researchers were able to dissect the protein function. In the brain, UBEA3 contributes to synapses formation and neuronal circuitry [168, 169] probably by downregulating the expression of other proteins [167]. As a consequence of UBEA3 loss, synapses development is impaired [169]. PWS has also been mimicked using mice models since human pattern alleles found in 15q11.2-q13 also occur in a well-conserved region in chromosome 7 in mice. Although very similar, there are differences regarding centromere distance and the absence of C15ORF2 gene and two noncoding snoRNAs in mice [170]. PWS mice targeting pattern genes in this homologous region conveniently recapitulate some PWS symptoms but still remain an incomplete model. The complexity of the human chromosomal region with genes being paternally or maternally expressed, together with the variety of genotypes/phenotypes that emerge from all the possible alterations occurring in this chromosome range, makes it difficult to model, study, and develop therapeutic approaches for both disorders [171].
To date, only a few groups have explored the potential of iPSCs in modeling pathological mechanisms in AS or PWS and addressed their potential to find therapeutic interventions for both disorders. Similar to FXS, differentiated iPSCs from AS and PWS recapitulate imprinting and methylation patterns unlike ESC, making it very suitable to study disease progression and pathological mechanisms [172]. Recently, the development of AS- and PWS iPSC-derived neurons showed that imprinting process occurs during neuronal differentiation and that this model can successfully recapitulate some of the disorder’s mechanism [173]. Although this latter study did not find neuronal differences between normal individuals and AS and PWS iPSC-derived neurons, large analyses of human iPSCs carrying chromosome 15 alterations have shown common pathological pathways for AS (deletion of 15q11–13) and other disorders harboring duplications of 15q11–13 [174].
Interestingly, most human tissues express both paternal and maternal alleles of UBE3A, whereas in neurons the paternal UBE3A is usually epigenetically silenced by the long noncoding RNA (ncRNA) UBEA3-ATS [175, 176]. Studies using iPSCs from PWS patients with microdeletion in a region critical for paternal imprinting showed that the activation of this ncRNA alone can alter imprinting patterns in UBE3A paternal allele, suggesting that in neurons UBEA3-ATS is sufficient for UBE3A paternal silencing [177, 178]. A similar model was used in an attempt to clarify the underlying mechanisms that trigger UBEA3-ATS expression in neurons [179], but the exact molecular mechanisms remain unclear. In parallel, therapeutic interventions for targeting UBEA3-ATS have been assessed in mice, with the aim of reestablishing normal levels of the protein by activating the paternal allele [180].Furthermore, the administration of selected drugs have also been used to increase paternal Ubea3 expression in a mice model of AS [181]. Both strategies successfully reduced UBEA3-ATS levels and consequently recovered Ubea3 pattern allele expression, leading to amelioration of the cognitive deficits observed in mice [180]. Although not addressed yet for AS or PWS, human stem cells could offer a valuable platform to study similar therapeutic strategies for a variety of candidate genes that are inherently repressed in the same region. Especially the use of patient-derived iPSCs for these studies could potentially help to better understand the mechanisms behind specific gene silencing, both in imprinting, as well as in disease situations, and will likely contribute to develop therapeutic interventions in AS, PWS and other related disorders.
6 Rett Syndrome
RTT is a neurodevelopment disorder affecting mostly girls, with a prevalence of 1 in 10,000 [182]. Patients first have an apparent normal intrauterus development, and symptoms only start to appear between the sixth and eightieth months of postnatal development. Typical symptoms include motor and language impairment, as well as cognitive regression. Although the symptomatology stabilizes approximately by 5 years of age, the life span of girls is severely reduced [183]. In 1999, the syndrome was correlated to de novo mutations in the methyl-CpG-binding gene (MECP2) [184], which is an important regulator of the epigenetic state highly expressed in developing neurons [185, 186]. MECP2 is involved in regulating chromatin structure and acts as a transcriptional repressor, as well as activator [187]. Although RTT is mainly associated to MECP2, several other mutations in different genes have been described in RTT-like phenotypes, such as CDKL5, TC4, JMJD1C, and FGX1, among others [188, 189].
MECP2 levels are precisely controlled during development, and studies have shown that both overexpression [190, 191] and downregulation [192] affect brain normal development. The affinity of MECP2 to target regions in the DNA is highly dependent on its sequence [193]. As a consequence, even small alterations in the protein sequence can affect the interaction of MECP2 domains with the chromatin [194,195,196], thereby altering the epigenetic state of the genome. To date, over 800 mutations have been mapped for the MECP2 locus in RTT patients [197, 198]. Several alterations, including missense and nonsense mutations, deletions, and duplications, can dramatically affect MECP2 function and contribute to a large range of clinical variability [203]. Furthermore, female embryos undergo X chromosome inactivation during development to compensate for the presence of two parental copies. Since MECP2 is located on the X chromosome (Xq28), RTT patients, therefore, can show mosaicism, which also contributes to the clinical observation of different phenotypes within the disorder [199, 200].
Mice models of RTT are widely used for modeling the disorder [201,202,203,204] and recapitulate several pathological hallmarks of the disease, such as abnormalities in dendritic morphology and neuronal connectivity [205, 206]. Interestingly, mice models have also shown that distinct neuronal populations are differently altered due to MECP2 loss [207,208,209]. Furthermore, those models have demonstrated that RTT phenotypes can be rescued by different treatment strategies [210,211,212]. One of the disadvantages of these RTT mice models, however, is that they are usually generated by MECP2 knockouts, while RTT patients likely present missense mutations in MECP2. Another caveat in these models is that the defects only appear very late during development in contrast to the early onset seen in humans [213]. Moreover, there are many biological differences in neurodevelopment and brain functions among mice and humans, which makes the use of humanized models more appealing for understanding RTT [11, 214].
The use of stem cells and specifically iPSC has shed a new light in the comprehension of RTT mechanisms [215]. The first in vitro neuronal model derived from RTT iPSCs with MECP2 mutations demonstrated most of the cellular abnormalities found in human and mice brains, such as lack of complex synapses (e.g., defects in synaptic outgrowth) and reduced number of dendrites [216]. Also, this study showed abnormalities in glutamatergic synapses and impaired electrophysiological properties [216]. Neurons obtained from iPSC from RTT patients also demonstrated defects in synaptic transmission [217] and connectivity [218], a phenomenon that is likely related to a reduced number of dendrites and which could be ameliorated through the administration of choline [217]. It has also been shown that iPSCs from RTT patients have maturation defects [219] and harbor a possible early deviation into the astrocytic lineage during differentiation [220], compromising neuronal commitment and proliferation. Furthermore, it is important to point out that cells derived from patients likely recapitulate the random X chromosome inactivation during development and differentiation [218, 221], providing construct validity to the model.
The possible contribution of glial cells in RTT pathology has also been addressed by studying iPSC-derived astrocytes [222, 223]. Mutated astrocytes presented impairment in vesicular transport [223] and interacted with normal neurons affecting neuronal morphology and maturation [222]. By using a 3D culture model of iPSCs from RTT patients, Zhang et al. [224] assessed the consequences of MECP2 impairment in neuronal migration and in the interaction of neurons and astrocytes [224]. In this 3D RTT model, normal neurons and astrocytes were combined with mutated cells in a set of experiments for cellular migration. In the control group, normal astrocytes and neurons migrated toward each other, establishing cohesion. Interestingly, when normal astrocytes and RTT-derived neurons were combined, the migration impairment was similar to the impairment noticed in RTT-derived astrocytes and normal neurons. This shows that MECP2 can be important to cellular migration during developing of the brain and that the involvement of glia cells in the process shouldn’t be overlooked. One interesting conclusion from the abovementioned studies, however, is that the implication of MECP2 in migration process contradicts the general assumption that the lack of complex connections on MECP2-impaired brain occurs due to poor synapses development, rather than migration problems.
Besides MECP2 mutations as a cause of RTT, several other mutations have been identified in RTT-like phenotypes [186, 188, 189]. For example, CDKL5-mutated iPSC were recently studied [225, 226], not only in the context of RTT but also for modeling other CDKL5-related disorders, such as epileptic encephalopathy or West syndrome. Cellular abnormalities caused by disruption of CDKL5 are similar to MECP2 mutations, but underlying mechanisms are different and CDLK5 protein appears to have a direct role in synaptic control [225, 226].
Most studies modeling MECP2 loss are not only interested in the basic pathological pathways of RTT but also in the function of MECP2 itself as a key component of epigenetic regulatory mechanisms in the brain [227], affecting gene expression [228, 229] and chromatin structure [230]. The analysis of in vitro phenotypes of RTT-derived iPSCs will collaborate to understanding the function of each MECP2 protein domains in RTT and potentially enable the development of mutation specific therapy [182]. By using iPSC-based approaches or allogenic stem cell [231] populations in combination with CRISPR/Cas9-induced mutations, we can nowadays assess specific consequences of selected mutations in neuronal function. In this perspective, iPSC-derived neurons from RTT patients will greatly contribute to unveil molecular targets and cellular process within neuronal development.
7 Discussion and Future Perspectives
In the current chapter, we have addressed the growing body of scientific interest in stem cells and their utility in terms of their potential to increase our general knowledge of several (epi)genetic brain disorders. A universal challenge in the field of translational neuroscience has been the development of animal and cellular models that effectively recapitulate the biology of the human brain. Stem cells, including ESCs, NSCs, and iPSCs, have entered the field as a potential breakthrough to overcome this hurdle and presented an alternative model for human brain studies. The field has been developing at a rapid pace, and methods to obtain, maintain, and differentiate these cells are in continuous adaptation and optimization. Stem cell technology and directed neural differentiation techniques allow to examine the broad repertoire of neural cells found in the human brain, with the goal to elucidate developmental, cellular, and molecular features that were previously inaccessible in animal models or clinical studies. Aside from contributing to the understanding of underlying neurobiology and consequences of personal molecular variations on healthy brain functioning and disease, stem cell-based studies have the potential to enhance the development of new and effective therapeutic interventions. The use of stem cells and derivatives for high-throughput screening of compound libraries and toxicological analysis before the initiation of clinical trials becomes increasingly favored for the aim of drug discovery. Moreover, fully functional neural cells or neural precursors that are differentiated from human stem cells have direct therapeutic potential in the field of regenerative medicine and might be employed as a cell replacement therapy for multiple neurodegenerative disorders such as Parkinson’s disease. In summary, stem cell technology has been offering unprecedented possibilities to investigate unique human biological features in a cell subtype-specific, as well as personalized matter, and is expected to greatly contribute to the development of therapeutic interventions [232].
Especially relevant in the light of current chapter, these stem cell-based studies offer a unique opportunity to further reveal the (epi)genetic and environmental contributions of brain disorders. In fact, stem cell-based studies of (epi)genetic influences in combination with the environmental exposome are expected to have a huge impact in enhancing our understanding of brain disorders and will also assist the development of (epi)genetic-based therapeutic intervention. Moreover, with the current advances in genetic [233] and epigenetic [234] editing techniques, human stem cells and derivatives can be (epi)genetically altered in a controlled setting to induce, aggravate, or recover cellular disease phenotypes in vitro. This directly allows to study the role of individual (epi)genetic signatures on disease causation and progression. In fact, a unique opportunity of human stem cell-based systems is the ability to probe how (epi)genomic architectures predispose individuals and how they influence the behavior of various, participating cell types. A consequent translational contribution, therefore, is the possibility of early detection of brain disorders that will improve timeliness and efficacy of diagnostic and therapeutic interventions. Furthermore, there is enormous promise in the utility of human neural culture systems to predict how individual (epi)genetic and cellular phenotypic variations contribute to the response of pharmacological interventions at clinically relevant levels. Finally, the (epi)genetic technologies available may also allow the deciphering of how (epi)genomic architectures found in individual humans act together to generate susceptibility and variation in response to the environmental insults that may also contribute to, or pharmacologically modify, disease phenotypes in patients. Taken together, these study strategies provide an elegant and dynamic tool for modeling, following, and understanding various essential pathological mechanisms of multiple brain disorders.
Although considerable progress has been made in validating these stem cell-based applications, further research is necessary to realize their full potential [235]. To date, there remains widespread lab-to-lab variability in culture methods and a lack in differentiation techniques to obtain homogeneous neural populations, as well as a lack of data regarding the desired cell types for modeling or treating certain brain disorders. Cellular heterogeneity, in combination with unidentified effects of reprogramming processes, can act as a potential confounder in epigenetic research and may lead to an under- or overestimation of the observed epigenetic differences. Furthermore, the simplicity of an in vitro model system is both an advantage and a significant disadvantage when culture purity in this case becomes a liability. Such models lack the complex mixture of neural cells found in the human brain, the complex extracellular matrices, and their integrated 3D organizations, which might also cause confounding effects on experimental outcomes. Although the latter might be circumvented by combined cultures, 3D culture systems, or the use of organoids [236], the future development of stem cell-based approaches will require extensive analysis and standardization, and any translation from bench to bedside must be undertaken gradually, with great caution and based on profound experimental data [1]. Altogether, stem cell technology is hypothesized to revolutionize the field of neuroscience the next 10 years, contributing substantially to improve our knowledge of brain functions and epigenetic dysregulation in disease and to identify new druggable targets.
References
Horvath P, Aulner N, Bickle M, Davies AM, Del Nery E, Ebner D, et al. Screening out irrelevant cell-based models of disease. Nat Rev Drug Discov. 2016;15:751–69.
Fernández-Santiago R, Ezquerra M. Epigenetic research of neurodegenerative disorders using patient iPSC-based models. Stem Cells Int. 2016;2016:1–16.
Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol. 2016;17:183–93.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76.
Rubin LL. Stem cells and drug discovery: the beginning of a new era? Cell. 2008;132:549–52.
Han F, Baremberg D, Gao J, Duan J, Lu X, Zhang N, et al. Development of stem cell-based therapy for Parkinson’s disease. Transl Neurodegener. 2015;4:16.
Yap MS, Nathan KR, Yeo Y, Lim LW, Poh CL, Richards M, et al. Neural differentiation of human pluripotent stem cells for nontherapeutic applications: toxicology, pharmacology, and in vitro disease modeling. Stem Cells Int. 2015;2015:105172.
Ma L, Liu Y, Zhang SC. Directed differentiation of dopamine neurons from human pluripotent stem cells. Methods Mol Biol. 2011;767:411–8.
Santos DP, Kiskinis E. Generation of spinal motor neurons from human pluripotent stem cells. Synap Dev Methods Protoc. 2017;53–66
Payne NL, Sylvain A, O’Brien C, Herszfeld D, Sun G, Bernard CCA. Application of human induced pluripotent stem cells for modeling and treating neurodegenerative diseases. New Biotechnol. 2015;32:212–28.
Wen Z, Christian KM, Song H, Ming G. Modeling psychiatric disorders with patient-derived iPSCs. Curr Opin Neurobiol. 2016;36:118–27.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68.
Badger JL, Cordero-Llana O, Hartfield EM, Wade-Martins R. Parkinson’s disease in a dish - Using stem cells as a molecular tool. Neuropharmacology. 2014;76(Pt A):88–96.
Pu J, Jiang H, Zhang B, Feng J. Redefining Parkinson’s disease research using induced pluripotent stem cells. Curr Neurol Neurosci Rep. 2012;12:392–8.
Sanchez-Mut JV, Heyn H, Vidal E, Moran S, Sayols S, Delgado-Morales R, et al. Human DNA methylomes of neurodegenerative diseases show common epigenomic patterns. Transl Psychiatry. 2016;6:e718.
Lardenoije R, Iatrou A, Kenis G, Kompotis K, Steinbusch HW, Mastroeni D, et al. The epigenetics of aging and neurodegeneration. Prog Neurobiol. 2015;131:21–64.
Damier P, Hirsch EC, Agid Y, Graybiel AM. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain. 1999;8:1437–48.
Beitz JM. Parkinson’s disease: a review. Front Biosci (Schol Ed). 2014;6:65–74.
Byers B, Lee HL, Reijo PR. Modeling Parkinson’s disease using induced pluripotent stem cells. Curr Neurol Neurosci Rep. 2012;12:237–42.
Nishimura K, Takahashi J. Therapeutic application of stem cell technology toward the treatment of Parkinson’s disease. Biol Pharm Bull. 2013;36:171–5.
Fu MH, Li CL, Lin HL, Chen PC, Calkins MJ, Chang YF, et al. Stem cell transplantation therapy in Parkinson’s disease. Springerplus. 2015;4:597.
Kocabicak E, Tan SK, Temel Y. Deep brain stimulation of the subthalamic nucleus in Parkinson’s disease: why so successful? Surg Neurol Int. 2012;3:S312–4.
Kornblum HI. Introduction to neural stem cells. Stroke. 2007;38:810–6.
Nishino H, Hida H, Takei N, Kumazaki M, Nakajima K, Baba H. Mesencephalic neural stem (progenitor) cells develop to dopaminergic neurons more strongly in dopamine-depleted striatum than in intact striatum. Exp Neurol. 2000;164:209–14.
Redmond DE, Bjugstad KB, Teng YD, Ourednik V, Ourednik J, Wakeman DR, et al. Behavioral improvement in a primate Parkinson’s model is associated with multiple homeostatic effects of human neural stem cells. Proc Natl Acad Sci U S A. 2007;104:12175–80.
Lee CS, Cenci MA, Schulzer M, Björklund A. Embryonic ventral mesencephalic grafts improve levodopa-induced dyskinesia in a rat model of Parkinson’s disease. Brain. 2000;123:1365–79.
Kondoh T, Pundt LL, Low WC. Development of human fetal ventral mesencephalic grafts in rats with 6-OHDA lesions of the nigrostriatal pathway. Neurosci Res. 1995;21:223–33.
Redmond Jr DE, Vinuela A, Kordower JH, Isacson O. Influence of cell preparation and target location on the behavioral recovery after striatal transplantation of fetal dopaminergic neurons in a primate model of Parkinson’s disease. Neurobiol Dis. 2008;29:103–16.
Yang H, Wang J, Wang F, Liu X, Chen H, Duan W, et al. Dopaminergic neuronal differentiation from the forebrain-derived human neural stem cells induced in cultures by using a combination of BMP-7 and Pramipexole with growth factors. Front Neural Circuits. 2016;10:1172.
Park CH, Kang JS, Shin YH, Chang MY, Chung S, Koh HC, et al. Acquisition of in vitro and in vivo functionality of Nurr1-induced dopamine neurons. FASEB J. 2006;20:2553–5.
Parish CL, Castelo-Branco G, Rawal N, Tonnesen J, Sorensen AT, Salto C, et al. Wnt5a-treated midbrain neural stem cells improve dopamine cell replacement therapy in parkinsonian mice. J Clin Invest. 2008;118:149–60.
Studer L, Tabar V, McKay RD. Transplantation of expanded mesencephalic precursors leads to recovery in parkinsonian rats. Nat Neurosci. 1998;1:290–5.
Hagell P, Schrag A, Piccini P, Jahanshahi M, Brown R, Rehncrona S, et al. Sequential bilateral transplantation in Parkinson’s disease. Brain. 1999;122:1121–32.
Levivier M, Dethy S, Rodesch F, Peschanski M, Vandesteene A, David P, et al. Intracerebral transplantation of fetal ventral mesencephalon for patients with advanced Parkinson’s disease. Stereotact Funct Neurosurg. 1998;69:99–111.
Freed CR, Breeze RE, Rosenberg NL, Schneck SA, Kriek E, Qi J, et al. Survival of implanted fetal dopamine cells and neurologic improvement 12 to 46 months after transplantation for Parkinson’s disease. N Engl J Med. 1992;327:1549–55.
Freeman TB, Olanow CW, Hauser RA, Nauert GM, Smith DA, Borlongan CV, et al. Bilateral fetal nigral transplantation into the postcommissural putamen in Parkinson’s disease. Ann Neurol. 1995;38:379–88.
Lindvall O, Sawle G, Widner H, Rothwell JC, Björklund A, Brooks D, et al. Evidence for long-term survival and function of dopaminergic grafts in progressive Parkinson’s disease. Ann Neurol. 1994;35:172–80.
Mendez I, Viñuela A, Astradsson A, Mukhida K, Hallett P, Robertson H, et al. Dopamine neurons implanted into people with Parkinson’s disease survive without pathology for 14 years. Nat Med. 2008;14:507–9.
Chu Y, Kordower JH. Lewy body pathology in fetal grafts. Ann N Y Acad Sci. 2010;1184:55–67.
Hagell P, Piccini P, Björklund A, Brundin P, Rehncrona S, Widner H, et al. Dyskinesias following neural transplantation in Parkinson’s disease. Nat Neurosci. 2002;5:627–8.
Olanow CW, Goetz CG, Kordower JH, Stoessl AJ, Sossi V, Brin MF, et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol. 2003;54:403–14.
Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EGA, Willemze R, Fibbe WE. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood. 2006;108:2114–20.
Turner DA, Kearney W. Scientific and ethical concerns in neural fetal tissue transplantation. Neurosurgery. 1993;33:1031–7.
Kirkeby A, Grealish S, Wolf DA, Nelander J, Wood J, Lundblad M, et al. Generation of regionally specified neural progenitors and functional neurons from human embryonic stem cells under defined conditions. Cell Rep. 2012;1:703–14.
Muramatsu S, Okuno T, Suzuki Y, Nakayama T, Kakiuchi T, Takino N, et al. Multitracer assessment of dopamine function after transplantation of embryonic stem cell-derived neural stem cells in a primate model of Parkinson’s disease. Synapse. 2009;63:541–8.
Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–51.
Takagi Y, Takahashi J, Saiki H, Morizane A, Hayashi T, Kishi Y, et al. Dopaminergic neurons generated from monkey embryonic stem cells function in a Parkinson primate model. J Clin Invest. 2005;115:102–9.
Kikuchi T, Morizane A, Doi D, Onoe H, Hayashi T, Kawasaki T, et al. Survival of human induced pluripotent stem cell-derived midbrain dopaminergic neurons in the brain of a primate model of Parkinson’s disease. J Parkinsons Dis. 2011;1:395–412.
Falkner S, Grade S, Dimou L, Conzelmann KK, Bonhoeffer T, Götz M, et al. Transplanted embryonic neurons integrate into adult neocortical circuits. Nature. 2016;539:248–53.
Grealish S, Diguet E, Kirkeby A, Mattsson B, Heuer A, Bramoulle Y, et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell. 2014;15:653–65.
Arenas E, Denham M, Villaescusa JC. How to make a midbrain dopaminergic neuron. Development. 2015;142:1918–36.
Brederlau A, Correia AS, Anisimov SV, Elmi M, Paul G, Roybon L, et al. Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson’s disease: effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells. 2006;24:1433–40.
Ben-Hur T, Idelson M, Khaner H, Pera M, Reinhartz E, Itzik A, et al. Transplantation of human embryonic stem cell–derived neural progenitors improves behavioral deficit in parkinsonian rats. Stem Cells. 2004;22:1246–55.
Blum B, Benvenisty N. The Tumorigenicity of human embryonic stem cell. Adv Cancer Res. 2008;100:133–58.
Grinnemo KH, Kumagai-Braesch M, Mânsson-Broberg A, Skottman H, Hao X, Siddiqui A, et al. Human embryonic stem cells are immunogenic in allogeneic and xenogeneic settings. Reprod Biomed Online. 2006;13:712–24.
Trounson A, McDonald C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell. 2015;17:11–22.
Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17:194–200.
Gonzalez R, Garitaonandia I, Crain A, Poustovoitov M, Abramihina T, Noskov A, et al. Proof of concept studies exploring the safety and functional activity of human parthenogenetic-derived neural stem cells for the treatment of Parkinson’s disease. Cell Transplant. 2015;24:681–90.
Fikes BJ. Parkinson’s stem cell therapy OK’d for testing. 2015. http://www.sandiegouniontribune.com/business/biotech/sdut-international-stem-cell-parkinsons-australia-2015dec14-story.html. Accessed 20 Nov 2016.
Hallett PJ, Deleidi M, Astradsson A, Smith GA, Cooper O, Osborn TM, et al. Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell. 2015;16:269–74.
Zhao T, Zhang ZN, Rong Z, Xu Y. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–5.
Han F, Wang W, Chen B, Chen C, Li S, Lu X, et al. Human induced pluripotent stem cell-derived neurons improve motor asymmetry in a 6-hydroxydopamine-induced rat model of Parkinson’s disease. Cytotherapy. 2015;17:665–79.
Sundberg M, Bogetofte H, Lawson T, Jansson J, Smith G, Astradsson A, et al. Improved cell therapy protocols for Parkinson’s disease based on differentiation efficiency and safety of hESC-, hiPSC-, and non-human primate iPSC-derived dopaminergic neurons. Stem Cells. 2013;31:1548–62.
Rhee YH, Ko JY, Chang MY, Yi SH, Kim D, Kim CH, et al. Protein-based human iPS cells efficiently generate functional dopamine neurons and can treat a rat model of Parkinson disease. J Clin Invest. 2011;121:2326–35.
Hargus G, Cooper O, Deleidi M, Levy A, Lee K, Marlow E, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in parkinsonian rats. Proc Natl Acad Sci U S A. 2010;107:15921–6.
Emborg ME, Liu Y, Xi J, Zhang X, Yin Y, Lu J, et al. Induced pluripotent stem cell-derived neural cells survive and mature in the nonhuman primate brain. Cell Rep. 2013;3:646–50.
Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–90.
Kim DS, Ross PJ, Zaslavsky K, Ellis J. Optimizing neuronal differentiation from induced pluripotent stem cells to model ASD. Front Cell Neurosci. 2014;8:109.
Parker GC, Acsadi G, Brenner CA. Mitochondria: determinants of stem cell fate? Stem Cells Dev. 2009;18:803–6.
Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell. 2016;19:232–47.
Soldner F, Laganiere J, Cheng AW, Hockemeyer D, Gao Q, Alagappan R, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–31.
Ross CA, Akimov SS. Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum Mol Genet. 2014;23:R17–26.
Maeder ML, Gersbach CA. Genome-editing technologies for gene and cell therapy. Mol Ther. 2016;24:430–46.
Wang L, Cao J, Wang Y, Lan T, Liu L, Wang W, et al. Immunogenicity and functional evaluation of iPSC-derived organs for transplantation. Cell Discov. 2015;1:15015.
Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra90–0.
Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–80.
Jiang H, Ren Y, Yuen EY, Zhong P, Ghaedi M, Hu Z, et al. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun. 2012;3:668.
Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52.
Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun. 2011;2:440.
Reinhardt P, Schmid B, Burbulla LF, Schöndorf DC, Wagner L, Glatza M, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–67.
Woodard CM, Campos BA, Kuo SH, Nirenberg MJ, Nestor MW, Zimmer M, et al. iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep. 2014;9:1173–82.
Sánchez-Danés A, Richaud-Patin Y, Carballo-Carbajal I, Jiménez-Delgado S, Caig C, Mora S, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4:380–95.
Fernández-Santiago R, Carballo-Carbajal I, Castellano G, Torrent R, Richaud Y, Sánchez-Danés A, et al. Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med. 2015;7:1529–46.
Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–9.
Wojda U, Kuznicki J. Alzheimer’s disease modeling: ups, downs, and perspectives for human induced pluripotent stem cells. J Alzheimers Dis. 2013;34:563–88.
Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23:213–27.
Lam B, Masellis M, Freedman M, Stuss DT, Black SE. Clinical, imaging, and pathological heterogeneity of the Alzheimer’s disease syndrome. Alzheimers Res Ther. 2013;5:1.
Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1:a006189.
Yang J, Li S, He XB, Cheng C, Le W. Induced pluripotent stem cells in Alzheimer’s disease: applications for disease modeling and cell-replacement therapy. Mol Neurodegener. 2016;11:39.
Sullivan SE, Young-Pearse TL. Induced pluripotent stem cells as a discovery tool for Alzheimers disease. Brain Res. 2015; doi:10.2016/j.brainres.2015.10.005.
Inoue H, Nagata N, Kurokawa H, Yamanaka S. iPS cells: a game changer for future medicine. EMBO J. 2014;33:409–17.
Sproul AA. Being human: the role of pluripotent stem cells in regenerative medicine and humanizing Alzheimer’s disease models. Mol Asp Med. 2015;43–44:54–65.
Mohamet L, Miazga NJ, Ward CM. Familial Alzheimer’s disease modelling using induced pluripotent stem cell technology. World J Stem Cells. 2014;6:239–47.
Weggen S, Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res Ther. 2012;4:9.
Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4:124ra9.
Prasher VP. Down syndrome and Alzheimer’s disease: biological correlates: Radcliffe Publishing; 2006.
Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–20.
Cataldo A, Rebeck GW, Ghetri B, Hulette C, Lippa C, Van Broeckhoven C, et al. Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann Neurol. 2001;50:661–5.
Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell. 2013;12:487–96.
Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LN, et al. The familial Alzheimer’s disease APPV717I mutation alters APP processing and tau expression in iPSC-derived neurons. Hum Mol Genet. 2014;23:3523–36.
Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med. 1996;2:864–70.
Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, et al. Familial Alzheimer’s disease–linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–13.
Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, et al. The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci U S A. 1997;94:2025–30.
Sproul AA, Jacob S, Pre D, Kim SH, Nestor MW, Navarro-Sobrino M, et al. Characterization and molecular profiling of PSEN1 familial Alzheimer’s disease iPSC-derived neural progenitors. PLoS One. 2014;9:e84547.
Moore S, Evans LD, Andersson T, Portelius E, Smith J, Dias TB, et al. APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 2015;11:689–96.
Young JE, Goldstein LSB. Alzheimer’s disease in a dish: promises and challenges of human stem cell models. Hum Mol Genet. 2012;21:R82–9.
Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, et al. The presenilin-1 ΔE9 mutation results in reduced γ-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013;5:974–85.
Tanzi RE. The genetics of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006296.
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74.
Delgado-Morales R, Esteller M. Opening up the DNA methylome of dementia. Mol Psychiatry. 2017; doi:10.1038/mp.2016.242.
Lord J, Lu AJ, Cruchaga C. Identification of rare variants in Alzheimer’s disease. Front Genet. 2014;5:369.
Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421–30.
Iatrou A, Kenis G, Rutten BPF, Lunnon K, van den Hove DLA. Epigenetic dysregulation of brainstem nuclei in the pathogenesis of Alzheimer’s disease: looking in the correct place at the right time? Cell Mol Life Sci. 2016;1–15
Duan L, Bhattacharyya BJ, Belmadani A, Pan L, Miller RJ, Kessler JA. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol Neurodegener. 2014;9:3.
Young JE, Boulanger-Weill J, Williams DA, Woodruff G, Buen F, Revilla AC, et al. Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell. 2015;16:373–85.
Alagiakrishnan K, Gill SS, Fagarasanu A. Genetics and epigenetics of Alzheimer’s disease. Postgrad Med J. 2012;88:522–9.
Medway C, Morgan K. The genetics of Alzheimer’s disease; putting flesh on the bones. Neuropathol Appl Neurobiol. 2014;40:97–105.
Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–6.
Dodson SE, Gearing M, Lippa CF, Montine TJ, Levey AI, Lah JJ. LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:866–72.
Hossini AM, Megges M, Prigione A, Lichtner B, Toliat MR, Wruck W, et al. Induced pluripotent stem cell-derived neuronal cells from a sporadic Alzheimer’s disease donor as a model for investigating AD-associated gene regulatory networks. BMC Genomics. 2015;16:84.
Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, et al. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2015;86:1299–306.
Kungulovski G, Jeltsch A. Epigenome editing: state of the art, concepts, and perspectives. Trends Genet. 2016;32:101–13.
Thakore PI, Black JB, Hilton IB, Gersbach CA. Editing the epigenome: technologies for programmable transcription and epigenetic modulation. Nat Methods. 2016;13:127–37.
Barten DM, Meredith Jr JE, Zaczek R, Houston JG, Albright CF. Gamma-secretase inhibitors for Alzheimer’s disease: balancing efficacy and toxicity. Drugs R D. 2006;7:87–97.
Ghosh AK, Osswald HL. BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem Soc Rev. 2014;43:6765–813.
Yiannopoulou KG, Papageorgiou SG. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2013;6:19–33.
Lozano R, Rosero CA, Hagerman RJ. Fragile X spectrum disorders. Intractable Rare Dis Res. 2014;3:134–46.
Saldarriaga W, Tassone F, González-Teshima LY, Forero-Forero JV, Ayala-Zapata S, Hagerman R. Fragile X syndrome. Colomb Med. 2014;45:190–8.
Eberhart DE, Malter HE, Feng Y, Warren ST. The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet. 1996;5:1083–91.
Ashley CT, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993;262:563–6.
Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile X mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–55.
Broek JAC. Lin Z, de Gruiter HM, van ‘t Spijker H, Haasdijk ED, cox D, et al. Synaptic vesicle dynamic changes in a model of fragile X Mol Autism. 2016;7:17.
Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, et al. Mapping of DNA instability at the fragile-X to a Trinucleotide repeat sequence P(Ccg)N. Science. 1991;252:1711–4.
Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1:397–400.
Mailick MR, Hong J, Rathouz P, Baker MW, Greenberg JS, Smith L, et al. Low-normal FMR1 CGG repeat length: phenotypic associations. Front Genet. 2014;5:309.
Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, et al. RNA cargoes associating with in cellular functioning in Fmrl Fmr1 reveal deficits null mice. Neuron. 2003;37:417–31.
Primerano B, Tassone F, Hagerman RJ, Hagerman P, Amaldi F, Bagni C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. RNA. 2002;8:1482–8.
Suhl JA, Warren ST. Single-nucleotide mutations in FMR1 reveal novel functions and regulatory mechanisms of the fragile X syndrome protein FMRP. J Exp Neurosci. 2015;9:35–41.
Brouwer JR, Mientjes EJ, Bakker CE, Nieuwenhuizen IM, Severijnen LA, Van der Linde HC, et al. Elevated Fmr1 mRNA levels and reduced protein expression in a mouse model with an unmethylated fragile X full mutation. Exp Cell Res. 2007;313:244–53.
Mor-Shaked H, Eiges R. Modeling fragile X syndrome using human pluripotent stem cells. Genes (Basel). 2016;7:77.
Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–11.
Khalfallah O, Jarjat M, Davidovic L, Nottet N, Cestèle S, Mantegazza M, et al. Depletion of the fragile X mental retardation protein in embryonic stem cells alters the kinetics of neurogenesis. Stem Cells. 2016; doi:10.1002/stem.2505.
Ben-Reuven L, Reiner O. Modeling the autistic cell: iPSCs recapitulate developmental principles of syndromic and nonsyndromic ASD. Develop Growth Differ. 2016;58:481–91.
Sheridan SD, Theriault KM, Reis SA, Zhou F, Madison JM, Daheron L, et al. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203.
Doers ME, Musser MT, Nichol R, Berndt ER, Baker M, Gomez TM, et al. iPSC-derived forebrain neurons from FXS individuals show defects in initial Neurite outgrowth. Stem Cells Dev. 2014;23:1777–87.
Usdin K, Kumari D. Repeat-mediated epigenetic dysregulation of the FMR1 gene in the fragile X-related disorders. Front Genet. 2015;6:192.
Liu J, Kościelska KA, Cao Z, Hulsizer S, Grace N, Mitchell G, et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum Mol Genet. 2012;21:3795–805.
de Esch CEF, Ghazvini M, Loos F, Schelling-Kazaryan N, Widagdo W, Munshi ST, et al. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 2014;3:548–55.
Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, et al. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13:234–41.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Kumari D, Swaroop M, Southall N, Huang W, Zheng W, Usdin K. High-throughput screening to identify compounds that increase fragile X mental retardation protein expression in neural stem cells differentiated from fragile X syndrome patient-derived induced pluripotent stem cells. Stem Cells Transl Med. 2015;4:800–8.
Angelman H. “Puppet” children a report on three cases. Dev Med Child Neurol. 1965;7:681–8.
Van Buggenhout G, Fryns JP. Angelman syndrome (AS, MIM 105830). Eur J Hum Genet. 2009;17:1367–73.
Trillingsgaard A, ØStergaard JR. Autism in Angelman syndrome: an exploration of comorbidity. Autism. 2004;8:163–74.
Cassidy SB, Driscoll DJ. Prader-Willi syndrome. Eur J Hum Genet. 2009;17:3–13.
Kalsner L, Chamberlain SJ. Prader-Willi, Angelman, and 15q11-q13 duplication syndromes. Pediatr Clin N Am. 2015;62:587–606.
Koerner MV, Barlow DP. Genomic imprinting - an epigenetic gene-regulatory model. Curr Opin Genet Dev. 2010;20:164–70.
Sanchez-Delgado M, Court F, Vidal E, Medrano J, Monteagudo-Sánchez A, Martin-Trujillo A, et al. Human oocyte-derived methylation differences persist in the placenta revealing widespread transient imprinting. PLoS Genet. 2016;12:e1006427.
Wilkinson LS, Davies W, Isles AR. Genomic imprinting effects on brain development and function. Nat Rev Neurosci. 2007;8:832–43.
Horsthemke B, Wagstaff J. Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet. 2008;146A:2041–52.
Constância M, Pickard B, Kelsey G, Reik W. Imprinting mechanisms. Genome Res. 1998;8:881–900.
Butler MG. Prader-Willi syndrome: obesity due to genomic imprinting. Curr Genomics. 2011;12(3):204–15.
Cavaillé J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci U S A. 2000;97:14311–6.
Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D, et al. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 2016;32:444–55.
LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics. 2015;7:1213–28.
Hulbert SW, Jiang YH. Monogenic mouse models of autism spectrum disorders: common mechanisms and missing links. Neuroscience. 2016;321:3–23.
Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, et al. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811.
Scheiffele P, Beg AA. Neuroscience: Angelman syndrome connections. Nature. 2010;468:907–8.
Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, et al. The Angelman syndrome protein Ube3A regulates synapse development by Ubiquitinating arc. Cell. 2010;140:704–16.
Bervini S, Herzog H. Mouse models of Prader–Willi syndrome: a systematic review. Front Neuroendocrinol. 2013;34:107–19.
Tan WH, Bird LM, Thibert RL, Williams CA. If not Angelman, what is it? A review of Angelman-like syndromes. Am J Med Genet A. 2014;164:975–92.
Yang J, Cai J, Zhang Y, Wang X, Li W, Xu J, et al. Induced pluripotent stem cells can be used to model the genomic imprinting disorder Prader-Willi syndrome. J Biol Chem. 2010;285:40303–11.
Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, et al. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader – Willi syndromes. Proc Natl Acad Sci U S A. 2010;107:17668–73.
Germain ND, Chen PF, Plocik AM, Glatt-Deeley H, Brown J, Fink JJ, et al. Gene expression analysis of human induced pluripotent stem cell-derived neurons carrying copy number variants of chromosome 15q11-q13.1. Mol Autism. 2014;5:44.
Rougeulle C, Cardoso C, Fontés M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19:15–6.
Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17:12–3.
Martins-Taylor K, Hsiao JS, Chen PF, Glatt-Deeley H, De Smith AJ, Blakemore AIF, et al. Imprinted expression of UBE3A in non-neuronal cells from a Prader-willi syndrome patient with an atypical deletion. Hum Mol Genet. 2014;23:2364–73.
Chamberlain SJ, Germain ND, Chen PF, Hsiao JS, Glatt-Deeley H. Modeling genomic imprinting disorders using induced pluripotent stem cells. Methods Mol Biol. 2016;1353:45–64.
Chen PF, Hsiao JS, Sirois CL, Chamberlain SJ. RBFOX1 and RBFOX2 are dispensable in iPSCs and iPSC-derived neurons and do not contribute to neural-specific paternal UBE3A silencing. Sci Rep. 2016;6:25368.
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F. Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature. 2015;518:409–12.
Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011;481:185–9.
Katz DM, Bird A, Coenraads M, Gray SJ, Menon DU, Philpot BD, et al. Rett syndrome: crossing the threshold to clinical translation. Trends Neurosci. 2016;39:100–13.
Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–37.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8.
Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. AJP Cell Physiol. 2011;300:C723–42.
Dragich JM, Kim YH, Arnold AP, Schanen NC. Differential distribution of the MeCP2 splice variants in the postnatal mouse brain. J Comp Neurol. 2007;501:526–42.
Della Ragione F, Vacca M, Fioriniello S, Pepe G, D'Esposito M. MECP2, a multi-talented modulator of chromatin architecture. Brief Funct Genomics. 2016;15:420–31.
Sáez MA, Fernández-Rodríguez J, Moutinho C, Sanchez-Mut JV, Gomez A, Vidal E, et al. Mutations in JMJD1C are involved in Rett syndrome and intellectual disability. Genet Med. 2016;18:378–85.
Lucariello M, Vidal E, Vidal S, Saez M, Roa L, Huertas D, et al. Whole exome sequencing of Rett syndrome-like patients reveals the mutational diversity of the clinical phenotype. Hum Genet. 2016;135:1343–54.
Petazzi P, Akizu N, García A, Estarás C. Martínez de Paz a, Rodríguez-Paredes M, et al. an increase in MECP2 dosage impairs neural tube formation. Neurobiol Dis. 2014;67:49–56.
Meins M, Lehmann J, Gerresheim F, Herchenbach J, Hagedorn M, Hameister K, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42:e12–2.
Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol Psychiatry. 2006;59:468–76.
Ausió J. Martinez de Paz a, Esteller M. MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol Med. 2014;20:487–98.
Kucukkal TG, Yang Y, Uvarov O, Cao W, Alexov E. Impact of Rett syndrome mutations on MeCP2 MBD stability. Biochemistry. 2015;54:6357–68.
Brown K, Selfridge J, Lagger S, Connelly J, De Sousa D, Kerr A, et al. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum Mol Genet. 2016;25:558–70.
Stuss DP, Cheema M, Ng MK, Martinez de Paz A, Williamson B, Missiaen K, et al. Impaired in vivo binding of MeCP2 to chromatin in the absence of its DNA methyl-binding domain. Nucleic Acids Res. 2013;41:4888–900.
Christodoulou J, Grimm A, Maher T, Bennetts B. RettBASE: the IRSA MECP2 variation database-a new mutation database in evolution. Hum Mutat. 2003;21:466–72.
Gold WA, Christodoulou J. The utility of next-generation sequencing in Gene discovery for mutation-negative patients with Rett syndrome. Front Cell Neurosci. 2015;9:266.
Naidu S, Bibat G, Kratz L, Kelley RI, Pevsner J, Hoffman E, et al. Clinical variability in Rett syndrome. J Child Neurol. 2003;18:662–8.
Bao X, Jiang S, Song F, Pan H, Meirong Li WX-R. X chromosome inactivation in Rett syndrome and its correlations with MeCP2 mutations and phenotype. J Child Neurol. 2008;23:22–5.
Huang TW, Kochukov MY, Ward CS, Merritt J, Thomas K, Nguyen T, et al. Progressive changes in a distributed neural circuit underlie breathing abnormalities in mice lacking MeCP2. J Neurosci. 2016;36:5572–86.
Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, et al. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–54.
Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger-Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 2007;146:907–21.
Pelka GJ, Watson CM, Radziewic T, Hayward M, Lahooti H, Christodoulou J, et al. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006;129:887–98.
Calfa G, Percy AK, Pozzo-Miller L. Experimental models of Rett syndrome based on Mecp2 dysfunction. Exp Biol Med. 2011;236:3–19.
Rietveld L, Stuss DP, McPhee D, Delaney KR. Genotype-specific effects of Mecp2 loss-of-function on morphology of layer V pyramidal neurons in heterozygous female Rett syndrome model mice. Front Cell Neurosci. 2015;9:145.
Medrihan L, Tantalaki E, Aramuni G, Sargsyan V, Dudanova I, Missler M, et al. Early defects of GABAergic synapses in the brain stem of a MeCP2 mouse model of Rett syndrome. J Neurophysiol. 2008;99:112–21.
Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65.
Viemari JC, Roux JC, Tryba AK, Saywell V, Burnet H, Peña F, et al. Mecp2 deficiency disrupts norepinephrine and respiratory systems in mice. J Neurosci. 2005;25:11521–30.
Szczesna K, de la Caridad O, Petazzi P, Soler M, Roa L, Saez MA, et al. Improvement of the Rett syndrome phenotype in a MeCP2 mouse model upon treatment with levodopa and a dopa-decarboxylase inhibitor. Neuropsychopharmacology. 2014;39:2846–56.
Ward CS, Arvide EM, Huang TW, Yoo J, Noebels JL, Neul JL. MeCP2 is critical within HoxB1 derived tissues of mice for normal lifespan. J Neurosci. 2011;31:10359–70.
Kim S, Broströmer E, Xing D, Jin J, Chong S, Ge H, et al. Probing allostery through DNA. Science. 2013;339:816–9.
Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet. 2015;16:261–75.
Beltrão-Braga PCB, Muotri AR. Modeling autism spectrum disorders with human neurons. Brain Res. 2016;1–8
Dajani R, Koo SE, Sullivan GJ, Park IH. Investigation of Rett syndrome using pluripotent stem cells. J Cell Biochem. 2013;114:2446–53.
Marchetto MCN, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, et al. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell. 2010;143:527–39.
Chin EWM, Marcy G, Yoon SI, Ma D, Rosales FJ, Augustine GJ, et al. Choline ameliorates disease phenotypes in human iPSC models of Rett syndrome. NeuroMolecular Med. 2016;18:364–77.
Djuric U, Cheung AYL, Zhang W, Mok RS, Lai W, Piekna A, et al. MECP2e1 isoform mutation affects the form and function of neurons derived from Rett syndrome patient iPS cells. Neurobiol Dis. 2015;76:37–45.
Kim KY, Hysolli E, Park IH. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 2011;108:14169–74.
Andoh-Noda T, Akamatsu W, Miyake K, Matsumoto T, Yamaguchi R, Sanosaka T, et al. Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol Brain. 2015;8:31.
Cheung AYL, Horvath LM, Carrel L, Ellis J. X-chromosome inactivation in Rett syndrome human induced pluripotent stem cells. Front Psychiatry. 2012;3:24.
Williams EC, Zhong X, Mohamed A, Li R, Liu Y, Dong Q, et al. Mutant astrocytes differentiated from Rett syndrome patients-specific iPSCs have adverse effects on wild-type neurons. Hum Mol Genet. 2014;23:2968–80.
Delépine C, Meziane H, Nectoux J, Opitz M, Smith AB, Ballatore C, et al. Altered microtubule dynamics and vesicular transport in mouse and human MeCP2-deficient astrocytes. Hum Mol Genet. 2016;25:146–57.
Zhang ZN, Freitas BC, Qian H, Lux J, Acab A, Trujillo CA, et al. Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc Natl Acad Sci U S A. 2016;113:3185–90.
Amenduni M, De Filippis R, Cheung AYL, Disciglio V, Epistolato MC, Ariani F, et al. iPS cells to model CDKL5-related disorders. Eur J Hum Genet. 2011;19:1246–55.
Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol. 2012;14:911–23.
Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–26.
Aldinger KA, Plummer JT, Levitt P. Comparative DNA methylation among females with neurodevelopmental disorders and seizures identifies TAC1 as a MeCP2 target gene. J Neurodev Disord. 2013;5:15.
Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13:446–58.
Akbarian S, Huang HS. Epigenetic regulation in human brain-focus on histone lysine methylation. Biol Psychiatry. 2009;65:198–203.
Ananiev G, Williams EC, Li H, Chang Q. Isogenic pairs of wild type and mutant induced pluripotent stem cell (iPSC) lines from Rett syndrome patients as in vitro disease model. PLoS One. 2011;6:e25255.
Kimbrel EA, Lanza R. Current status of pluripotent stem cells: moving the first therapies to the clinic. Nat Rev Drug Discov. 2015;14:681–92.
Hendriks WT, Warren CR, Cowan CA. Genome editing in human pluripotent stem cells: approaches, pitfalls, and solutions. Cell Stem Cell. 2016;18:53–65.
Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, et al. Editing DNA methylation in the mammalian genome. Cell. 2016;167:233–47.e17.
Tapia N, Schöler HR. Molecular obstacles to clinical translation of iPSCs. Cell Stem Cell. 2016;19:298–309.
Passier R, Orlova V, Mummery C. Complex tissue and disease modeling using hiPSCs. Cell Stem Cell. 2016;18:309–21.
Acknowledgments
Renzo J. M. Riemens is supported by Maastricht University (Maastricht, The Netherlands) and Julius Maximilians University (Würzburg, Germany). Edilene Siqueira Soares is supported by the National Council for Scientific and Technological Development (CNPQ; grant n. 202074/2015-3), Ministry of Science, Brazil. The work of laboratory is supported by, among other institutions, the EU Joint Programme – Neurodegenerative Disease Research (JPND), Cellex Foundation, the Health and Science Departments of the Catalan Government (Generalitat de Catalunya), the E-Rare (ERA-Net for research programs on rare diseases), and EuroRETT (a European network on Rett syndrome, funded by the European Commission under its 6th Framework Program since 2006). Dr. Manel Esteller is an ICREA Research Professor.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Riemens, R.J.M., Soares, E.S., Esteller, M., Delgado-Morales, R. (2017). Stem Cell Technology for (Epi)genetic Brain Disorders. In: Delgado-Morales, R. (eds) Neuroepigenomics in Aging and Disease. Advances in Experimental Medicine and Biology(), vol 978. Springer, Cham. https://doi.org/10.1007/978-3-319-53889-1_23
Download citation
DOI: https://doi.org/10.1007/978-3-319-53889-1_23
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53888-4
Online ISBN: 978-3-319-53889-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)