
Abstract
Parkinson’s disease (PD) is a movement disorder associated with the degeneration of nigral dopaminergic (DA) neurons. One of the greatest obstacles for PD research is the lack of patient-specific nigral DA neurons for mechanistic studies and drug discovery. The advent of induced pluripotent stem cells (iPSCs) has overcome this seemingly intractable problem and changed PD research in many profound ways. In this review, we discuss recent development in the generation and analyses of patient-specific iPSC-derived midbrain DA neurons. Results from this novel platform of human cellular models of PD have offered a tantalizing glimpse of the promising future of PD research. With the development of the latest genomic modification technologies, dopaminergic neuron differentiation methodologies, and cell transplantation studies, PD research is poised to enter a new phase that utilizes the human model system to identify the unique vulnerabilities of human nigral DA neurons and disease-modifying therapies based on such mechanistic studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease. It is characterized by the progressive degeneration of dopaminergic (DA) neurons in substantia nigra pars compacta. Although the etiology of PD remains unclear, interactions between environmental and genetic factors are believed to cause the loss of nigral DA neurons and the ensuing locomotor symptoms [1–3]. Idiopathic PD accounts for the vast majority of parkinsonism. However, 2 % to 3 % of PD cases are linked to monogenic mutations. Over the past two decades, identification and mechanistic studies of these genes have shed great insights into the molecular and genetic pathogenesis of PD [4, 5].
The first gene linked to autosomal-dominant familial PD is the α-synuclein (SNCA) gene, encoding a synaptic vesicle-associated protein that shows up in high abundance in Lewy bodies [6, 7]. Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are the most common genetic causes of autosomal-dominant PD with a strong founder effect and incomplete penetrance [8, 9]. On the other hand, mutations in parkin (PRKN) [10], PTEN-induced putative kinase 1 (PINK1) [11], and DJ-1 [12] cause autosomal-recessive, early-onset PD. Many other genes are linked to familial forms of parkinsonism, often with clinical features in addition to the typical PD symptoms [13].
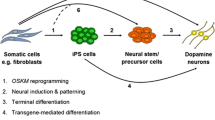
The clinical manifestations associated with the monogenic forms of PD closely resemble those of idiopathic PD, suggesting that there may be a common etiological mechanism on which genetic forms and idiopathic forms of PD converge. Thus, mechanistic studies on monogenic forms of PD may yield significant insights into the etiology of idiopathic PD. During the past decade, various rodent models of PD with genetic mutations of the disease-causing genes have been generated. Unfortunately, these animal models, along with traditional pharmacological models of the disease, do not satisfactorily mimic the salient features of PD [14]. There has been a great lack of a model system that directly reflects the genetic and physiological uniqueness of the human condition. Remarkable breakthroughs by Shinya Yamanaka and James Thomson made it possible to generate induced pluripotent stem cells (iPSCs) from human skin fibroblasts [15, 16]. During the past 4 years, iPSCs have provided a new platform to study many human diseases (eg, PD, Huntington’s disease, and amyotrophic lateral sclerosis) [17, 18]. For PD research in particular, this technology enables the generation of physiologically relevant, patient-specific midbrain DA neurons in unlimited quantities. These neurons are highly valuable for basic research on the molecular and cellular mechanisms of PD, for drug discovery research to identify disease-modifying therapies, and for cell-based therapy utilizing autologous donor materials. Thus, the iPS technology has the potential to overcome major barriers in PD research and therapy. This paper reviews recent advancements in the use of patient-specific iPSCs to model PD and discusses significant challenges that need to be met to move the field forward.
The Generation of iPSCs and Their Differentiation to Midbrain DA Neurons
In 2006, Yamanaka’s group first developed a technique that reprogrammed mouse embryonic fibroblasts into iPSCs, through virus-mediated expression of four transcription factors Oct4, Sox2, Klf4, and c-Myc [19]. This revolutionary work shows that it is possible to rewire the transcriptional control network in a cell so that the same genome can be reprogrammed to the pluripotent state, as shown in later work by the same group as well as others [20–22]. In November 2007, both Shinya Yamanaka’s group and James Thomson’s group successfully reprogrammed human skin fibroblasts to iPSCs using two different sets of transcription factors [15, 16]. The Yamanaka method used retrovirus to deliver the same reprogramming factors (Oct4, Sox2, Klf4, and c-Myc) used for mouse cells [15], while Thomson’s group used lentivirus to express Oct4, Sox2, Nanog, and Lin28 [16]. Many groups have since succeeded in reprogramming different types of human somatic cells to iPSCs [23], which demonstrates that epigenetic reprogramming is a robust process that can be applied to many, if not all, types of somatic cells.
The use of integrating viruses such as retrovirus or lentivirus poses the concern that the proviruses in the genome of the resulting iPSCs may have unforeseen consequences on the differentiation of iPSCs to cell types of interest, particularly in therapeutic settings. Many new reprogramming technologies have been developed to address this issue. For example, iPSCs can be generated using non-integrating viruses such as Sendai virus [24], synthetic modified mRNAs [25], or proteins [26]. Although integration-free iPSCs may have gene expression profiles more similar to human embryonic stem cell (hESCs) [27•], the efficiency of iPSCs to differentiate into neurons appears to have no clear relationship with different reprogramming methods [28]. Integrated proviruses do not appear to significantly affect the in vitro differentiation of iPSCs [18, 29•, 30]. A recent study showed that the efficiency of iPSC differentiation in vitro positively correlates with the number of reprogramming factors in the iPSC [31].
Like hESCs, iPSCs derived from PD patients can be efficiently differentiated into midbrain DA neurons. There are two types of protocols, depending on the use of feeder cells or not. The feeder-dependent protocols utilize mouse stromal cell lines such as PA6, which support the differentiation of pluripotent stem cells in an undefined manner [32]. The major advantage is its relative ease in adopting the technique. The disadvantages include contamination of human cells with the mouse feeders, which cannot be completely removed even with fluorescence-activated cell sorting. The other issue is that undefined differentiation of pluripotent stem cells on feeders is not synchronized, producing a variety of neurons at different developmental stages and different lineages. In contrast, the feeder-free protocols are chemically defined and seek to mimic early human embryonic development. These directed differentiation protocols generally fall into two categories, through the formation of neural tube-like “rosettes” or floor plate. The rosette-based protocol produces developmental synchronized midbrain DA neurons through multistaged enrichment for the desired populations of cells, with proper developmental restriction by various growth factors and cytokines [33]. It allows the generating of high-quality midbrain DA neurons capable of controlled dopamine release and selective dopamine reuptake, as well as many other critical features resembling the rat nigral DA neurons in vivo [29•]. The major disadvantage of the protocol is that it is technically demanding, costly, and lengthy. The more recently developed floor plate-based protocol drives the differentiation of iPSCs to floor plate, rather than neural rosette [34]. Compared with the rosette-based method, the floor plate-based method produces more tyrosine hydroxylase-positive neurons, increases dopamine levels, and enables significantly improved survival of grafted neurons [34].
iPSC Models of PD
Idiopathic PD
The majority of PD cases are idiopathic. Idiopathic PD is likely to be the result of complex interactions between genetic and environmental factors [2, 3]. One of the major obstacles to our understanding of the pathophysiology of idiopathic PD is the lack of a reliable experimental model that captures the salient features of the disease. Patient-specific iPSCs from idiopathic PD cases allow the generation of midbrain DA neurons that have the same genetic composition as the patients and share many important properties with the nigral DA neurons in the PD patients [17, 27•]. Midbrain DA neurons differentiated from idiopathic PD iPSCs can be transplanted into adult rodent striatum. These human iPSC-derived DA neurons survive at high numbers and are functionally integrated into the rodent neural circuitry, as indicated by a reduction of amphetamine- and apomorphine-induced rotational asymmetry. However, only a small number of DA neurons project into the host striatum at 16 weeks after transplantation [35•]. The long-term survival of grafts is improved with the floor plate-based differentiation protocol [34]. These transplantation studies demonstrate the feasibility of studying PD iPSC-derived DA neurons in rodent brains. The studies also lay the foundation for future autologous cell replacement therapy for PD.
Monogenic PD
Although monogenic forms of PD only account for a small percentage of PD cases [3], understanding how mutations of these genes cause the degeneration of DA neurons is critically important for the study of disease mechanism and the identification of disease-modifying drugs. The basic premise is that if PD is a deterministic process, rather than a stochastic process, no matter how PD is caused, the process converges on a common pathway that leads to the degeneration of nigral DA neurons. Research on monogenic forms of PD accounts for the vast majority of mechanistic studies of the disease since the discovery of these genes. In a recent study, iPSC lines were derived from a PD patient with triplication of the SNCA gene, which encodes α-synuclein [36•]. Mutations of SNCA cause a very rare, autosomal-dominant form of PD [6]. DA neurons derived from the PD patient produced twice the amount of α-synuclein protein, compared with normal controls. In an independent study, isogenic pairs of human iPSCs were generated that only differed in the presence of absence of the PD-causing mutation A53T in the α-synuclein gene [37•]. This is achieved by using zinc finger nuclease (ZFN)–mediated genome editing technology, which can precisely and efficiently modify genome in many types of cells including iPSCs [37•]. These iPSC lines with SNCA mutations provide a novel platform to study the cellular function of α-synuclein in human midbrain DA neurons.
Mutations of the LRRK2 gene represent the most frequent cause of familial PD, although there is a strong founder effect and the penetrance is incomplete [8, 9]. In DA neurons differentiated from iPSCs derived from a PD patient with homozygous G2019S mutations of LRRK2, the expression levels of stress-response genes such as HSPB1, NOX1, and MAO-B are increased, as is the protein level of α-synuclein. The DA neurons with LRRK2 G2019S mutations also exhibit increased sensitivity to cell stressors such as hydrogen peroxide, MG132, and 6-hydroxydopamine [38•]. An independent study using four unrelated PD patients with LRRK2 G2019S mutation showed reduced number of neurites and their branches, as well as increased accumulation of autophagic vacuoles in iPSC-derived DA neurons with the LRRK2 mutation. These phenotypes were also observed in iPSC-derived DA neurons from seven idiopathic PD patients [39].
Mutations of the PARK2 gene, which encodes the ubiquitin-protein ligase parkin [40], represent the most frequent cause of recessively inherited PD [10]. The lack of robust phenotypes in parkin knockout mice [41] has prompted us to generate iPSCs from PD patients with parkin mutations to study the unique vulnerabilities of human midbrain DA neurons in the absence of functional parkin. We generated iPSCs from two normal subjects and two PD patients with different parkin mutations [29•]. Midbrain DA neurons differentiated from iPSCs with parkin mutations yielded significant insights on the function of parkin in the cells that are most severely affected by its mutations. Loss-of-function mutations of parkin disrupted the precision of DA transmission by increasing uncontrolled, spontaneous release of dopamine while decreasing dopamine reuptake [29•]. Since dopamine is required for the neural computational support of locomotion, the disruption of the spatial and temporal pattern of DA signals by parkin mutations would greatly affect the highly unstable bipedal movement of human, perhaps more so than parkin mutations would in other species. Aside from being a neurotransmitter, dopamine is also a toxin because it is very easily oxidized. When parkin is mutated, dopamine-induced oxidative stress is greatly increased. The mutations abrogate the ability of parkin to suppress the transcription of monoamine oxidases, which catalyze the oxidative deamination of dopamine, a reaction that produces H2O2 [29•, 42]. These dopamine-specific phenotypes, which have not been robustly observed in parkin knockout mice [41], reveal the unique functions of parkin in human midbrain DA neurons.
Mutations of the PINK1 gene, which encodes a mitochondria outer membrane protein kinase, cause a rare form of autosomal-recessive PD [11]. PINK1 is required for the mitochondrial recruitment of parkin in response to the depolarization of mitochondria inner membrane, which can be induced by damage to the respiratory chain [43, 44]. iPSC-derived DA neurons from three PD patients with different PINK1 mutations showed impaired recruitment of exogenously expressed parkin to mitochondria. The phenotype was rescued by overexpression of wild-type PINK1 [45•]. In addition, the loss of PINK1 increased mitochondria copy number and the expression level of PGC-1α, a transcription coactivator critically involved in mitochondrial biogenesis [45•]. These phenotypes suggest that PINK1 plays an important role in regulating the degradation of mitochondria through the recruitment of parkin.
Overall, the above studies using patient-specific iPSCs derived from idiopathic or monogenic forms of PD demonstrate the utility of this novel model system in analyzing human midbrain DA neurons, the type of cells significantly lost in PD. Future studies using this platform will gradually transition PD research to the human system, since now we have the right types of cells to work with.
Stem Cell Replacement Therapies for PD
The clinical symptoms of PD are generally not observed until the degeneration of nigral DA neurons reaches 60 % to 80 % [46]. This suggests that the system is very robust and small percentages of increase in nigral DA neurons may significantly restore locomotor functions. Although results from transplantation studies using fetal brain tissues are unclear due to a variety of issues [47], there have been intense studies on this topic since the discovery of hESCs. However, hESC-based therapy is currently limited by a variety of issues, such as ethical concerns regarding embryos, the potential problem for graft rejection, effectiveness of the grafted cells, etc. The discovery of iPSCs significantly alleviates some of the concerns. Since iPSCs are derived from somatic cells such as skin cells, most people do not view iPSCs as a potential concern for ethical issues. Patient-specific iPSCs offer the possibility for autologous transplant, which would significantly reduce, if not eliminate, graft rejection. However, a recent study showed that iPSCs derived from C57BL/6 mouse using a non-integrating method were rejected by the same strain of mice [48]. In the same setting, mouse ESCs from C56BL/6 were not rejected by C57BL/6 mice. Gene expression profiling experiments show that certain genes involved in immune rejection are activated in iPSCs, compared with ESCs derived from the same genome [48]. The study casts a cautious note on the use of patient-specific autologous transplant of iPSC-derived cells.
Another significant issue of cell replacement therapy for PD is how to differentiate human pluripotent stem cells (hESCs or iPSCs) to the right kind of cells so that they can be effectively integrated into the patient’s neural network. This is perhaps the most difficult issue, despite many studies. The recent floor plate-based differentiation protocol showed that grafted DA neurons differentiated from hESCs or iPSCs have long-term survival and exhibit electrophysiological properties in rodent and rhesus monkey brains [34]. While it is very encouraging, the DA neurons grafted to striatum reside in a very artificial environment. Normally, a nigral DA neuron has its cell body in substantia nigra and extends a single axon with massive axon arborization to innervate striatum [49]. For a single rat nigral DA neuron, the average length of its axon arbors is 45 cm. If we use the cube root of brain weight (1350 g for human vs 2 g for rat) to estimate linear dimension, a human nigral DA neuron would have axon arbors of 400 cm. This extraordinarily elaborate structure suggests a function that would be impossible to achieve otherwise. With current technology, it is impossible to generate midbrain DA neurons with such a massive axon arbor in vivo. Many transplantation studies show that the grafted DA neurons only make limited projections in striatum [35•]. To restore locomotor control, which is impaired in PD patients, it seems necessary to have a large number of DA neurons transplanted into the brain to cover a significant portion of striatum. The current neurosurgical techniques do not have a reliably safe way to deliver cells to many locations.
A perplexing issue for cell replacement therapy is the observation that grafted neurons from healthy fetal brain tissues develop Lewy bodies in some PD patients [50, 51]. This striking feature suggests that components of Lewy bodies (eg, α-synuclein) in the brain of the PD patients may induce changes in the graft in a prion-like mechanism. Many studies show that misfolded proteins such as α-synuclein can be transmitted from one cell to another cell [52]. Thus, stem cell–based replacement therapies for PD need to overcome many obstacles to become useful for patients.
Conclusions and Future Directions
It is perhaps useful to first consider the essence of the iPS technology to appreciate its advantages and limitations. Fundamentally, the technology enables us to capture the unique genome of an individual and reprogram that genome to cell types of interest. The strength of the technology is that it allows us to recreate the intrinsic, mostly cell autonomous functions of an individual genome. The weakness is that during in vitro differentiation, most of the developmental queues cannot be recreated to mimic human embryonic development, on which we have very little information at the molecular and cellular levels. This becomes a chicken and egg issue in that we cannot study human embryonic development effectively without hESCs or human iPSCs, and we cannot find a good differentiation protocol for these human pluripotent stem cells without a comprehensive knowledge of human embryonic development. Studying the embryonic development of rodents provides a crucial starting point, but is not sufficient, because a human brain is quite different from a rodent brain. It will become necessary to study the embryonic development of non-human primates such as rhesus monkeys. In conjunction with study of aborted human fetus, we can hope to gain a more complete and accurate understanding of human embryonic development. The human iPSCs and hESCs are very valuable for this purpose as they provide a test bed to examine and validate an idea on human embryonic development in an in vitro setting.
Once we have a significantly improved differentiation protocol, we can perhaps generate midbrain DA neurons with properties highly similar to nigral DA neurons in vivo, including proper synaptic connections with other types of neurons in an environment like a miniorgan. The miniorgan should preserve the basic functional modules of the nigrostriatal pathway in that it contains cells resembling the nigral DA neurons and the striatal medium spiny neurons. These neurons should form synaptic connections with characteristics seen in vivo. It might be quite difficult to recreate the three-dimensional environment and developmental cues in vitro to allow the differentiation of iPSCs or hESCs to a miniorgan that recapitulate key characteristics of the nigrostriatal pathway. One possible route is to transplant iPSC-derived neuroepithelial cells, which are the precursors for neurons, to the brain of a developing rhesus monkey or rat, and thus provide the in vivo environment for the human neuroepithelial cells to develop functional neural networks.
For mechanistic studies on the transplanted human neurons, we need to label them to enable experiments that require live neurons. Recent development in genetic modifications of human pluripotent stem cells provides many timely tools. It is now possible to genetically label iPSCs or hESCs with fluorescent markers such as green fluorescent protein (GFP), under the control of endogenous genes such as Pitx3, a transcription factor critically involved in determining the differentiation of midbrain DA neurons. Using ZFN or transcription activator-like effector nuclease (TALEN), homologous recombination in iPSCs or hESCs can take place efficiently to allow the precise modification of the human genome [53•, 54•]. The modified human pluripotent stem cells will turn on the expression of GFP only when they are differentiated to midbrain DA neurons. To improve the efficiency of gene targeting in iPSCs, one can also push iPSCs to a developmentally earlier stage of naïve pluripotency, a state of the cells in the inner cell mass [55]. The naïve-state human iPSCs or hESCs exhibit properties very similar to the mouse ESCs including a dome-shaped colony morphology, resistance to trypsinization, and high clonal efficiency [56, 57]. Homologous recombination is much more efficient in naïve-state iPSCs than in the regular (ie, primed-state) iPSCs [56, 57].
With these new technologies, PD research is poised to enter a new phase, in which mechanistic studies, biomarker discoveries, and drug development will become more dependent on using iPSC-derived human midbrain DA neurons, both in vitro and in the brain of an animal model. These will open up unprecedented opportunities to change PD research significantly by redefining the disease in molecular and cellular terms, by predicting PD and tracking its progression, and by identifying new therapeutic strategies that can prevent or slow down the degeneration of nigral DA neurons. With rapid progress in iPSC-based technology, one can envision a bright future for PD research. Now that we have direct access to the neurons that are affected in PD, the pace of discovery should speed up and the cure for PD should be an attainable goal.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Langston JW. Parkinson’s disease: current and future challenges. Neurotoxicology. 2002;23:443–50.
Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–54.
Dawson TM, Dawson VL. Rare genetic mutations shed light on the pathogenesis of Parkinson disease. J Clin Invest. 2003;111:145–51.
Klein C, Schlossmacher MG. Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology. 2007;69:2093–104.
Gasser T. Update on the genetics of Parkinson’s disease. Mov Disord. 2007;22:S343–50.
Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7.
Spillantini MG, Schmidt ML, Lee VM, et al. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40.
Nichols WC, Pankratz N, Hernandez D, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet. 2005;365:410–2.
Paisan-Ruiz C, Lang AE, Kawarai T, et al. LRRK2 gene in Parkinson disease: Mutation analysis and case control association study. Neurology. 2005;65:696–700.
Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–8.
Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60.
Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9.
Hardy J. Genetic analysis of pathways to Parkinson disease. Neuron. 2010;68:201–6.
Dawson TM, Ko HS, Dawson VL. Genetic animal models of Parkinson’s disease. Neuron. 2010;66:646–61.
Takahashi K, Tanabe K, Ohnukl M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. This is the first paper to report the reprogramming of human somatic cells to iPSCs, a landmark breakthrough in biomedical research.
Yu JY, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. This independent study shows that derivation of iPSCs from human somatic cells is possible using a different set of transcription factors.
Park IH, Arora N, Huo H, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–86. In this study, iPSCs are generated for the first time from patients with a variety of diseases, including idiopathic PD.
Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–21.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. This is the first report on the generation of iPSCs from mouse somatic cells. It lays the foundation for the whole field.
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–7.
Maherali N, Sridharan R, Xie W, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70.
Meissner A, Wernig M, Jaenisch R. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol. 2007;25:1177–81.
Hanna JH, Saha K, Jaenisch R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell. 2010;143:508–25.
Fusaki N, Ban H, Nishiyama A, et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–62.
Warren L, Manos PD, Ahfeldt T, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–30.
Rhee YH, Ko JY, Chang MY, et al. Protein-based human iPS cells efficiently generate functional dopamine neurons and can treat a rat model of Parkinson disease. J Clin Invest. 2011;121:2326–35.
• Soldner F, Hockemeyer D, Beard C, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–77. The study describes the generation of virus-free iPSCs from fibroblasts of five idiopathic PD patients, using cre recombinase-mediated excision of integrated lentiviruses.
Hu BY, Weick JP, Yu J, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A. 2010;107:4335–40.
• Jiang H, Ren Y, Yuen EY, et al. Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun. 2012;3:668. This is the first paper on iPSCs from PD patients with parkin mutations. The study reveals the unique functions of parkin in human midbrain DA neurons.
Major T, Menon J, Auyeung G, et al. Transgene excision has no impact on in vivo integration of human iPS derived neural precursors. PLoS One. 2011;6:e24687.
Lohle M, Hermann A, Glass H, et al. Differentiation efficiency of induced pluripotent stem cells depends on the number of reprogramming factors. Stem Cells. 2012;30:570–9.
Kawasaki H, Mizuseki K, Nishikawa S, et al. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000;28:31–40.
Yan Y, Yang D, Zarnowska ED, et al. Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells. 2005;23:781–90.
Kriks S, Shim JW, Piao J, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–51.
• Hargus G, Cooper O, Deleidi M, et al. Differentiated Parkinson patient-derived induced pluripotent stem cells grow in the adult rodent brain and reduce motor asymmetry in Parkinsonian rats. Proc Natl Acad Sci U S A. 2010;107:15921–6. This study transplants iPSC-derived cells from PD patients to striatum of rat PD models. The grafts can differentiate into DA neurons and reduce motor asymmetry in the PD rat models.
• Devine MJ, Ryten M, Vodicka P, et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun. 2011;2:440. This is the first paper on iPSCs from PD patients with α-synuclein triplication.
• Soldner F, Laganière J, Cheng AW, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146:318–31. This paper used ZFN to generate isogenic pairs of iPSCs with or without PD-linked α-synuclein mutations.
• Nguyen HN, Byers B, Cord B, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–80. This is the first paper on iPSCs from PD patients with LRRK2 mutations.
Sanchez-Danes A, Richaud-Patin Y, Carballo-Carbajal I, et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med. 2012;4:380–95.
Shimura H, Hattori N, Kubo S, et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–5.
Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A. 2005;102:2174–9.
Ren Y, Jiang H, Ma D, et al. Parkin degrades estrogen-related receptors to limit the expression of monoamine oxidases. Hum Mol Genet. 2011;20:1074–83.
Rakovic A, Grünewald A, Seibler P, et al. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum Mol Genet. 2010;19:3124–37.
Vives-Bauza C, Zhou C, Huang Y, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–83.
• Seibler P, Graziotto J, Jeong H, et al. Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci. 2011;31(16):5970–6. This is the first paper on iPSCs from PD patients with PINK1 mutations.
Barbeau A. Parkinson’s disease: clinical features and etiopathology. In: Viken PJ, Bruyn GW, Klawans HL, editors. Handbook of clinical neurology. Amsterdam: Elsevier Science Publishers; 1986. p. 87–108.
Braak H, Del Tredici K. Assessing fetal nerve cell grafts in Parkinson’s disease. Nat Med. 2008;14:483–5.
Zhao T, Zhang ZN, Rong Z, et al. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474(7350):212–5.
Matsuda W, Furuta T, Nakamura KC, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29(2):444–53.
Li JY, Englund E, Holton JL, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14(5):501–3.
Kordower JH, Chu Y, Hauser RA, et al. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14(5):504–6.
Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci USA. 2009;106(31):13010–5.
• Hockemeyer D, Soldner F, Beard C, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–7. This is the first study to use ZFN to target GFP to the Pitx3 locus for the genetic labeling of midbrain DA neurons differentiated from iPSCs.
• Hockemeyer D, Wang H, Kiani S, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29:731–4. This is the first study to use TALEN to target GFP to the Pitx3 locus for the genetic labeling of midbrain DA neurons differentiated from iPSCs.
Nichols J, Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4(6):487–92.
Hanna J, Cheng AW, Saha K, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci U S A. 2010;107(20):9222–7.
Buecker C, Chen HH, Polo JM, et al. A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell. 2010;6(6):535–46.
Disclosure
Conflicts of interest: J. Pu: none; H. Jiang: none; B. Zhang: has received grant support from Zhejiang Province S&T Office; J. Feng: has received grant support from NIH/NINDS (grants NS061856 and NS070276), NYSTEM (contracts C024406 and C026714), Michael J. Fox Foundation, and State University of New York; and has received support for travel to meetings for the study or otherwise from NSFC.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pu, J., Jiang, H., Zhang, B. et al. Redefining Parkinson’s Disease Research Using Induced Pluripotent Stem Cells. Curr Neurol Neurosci Rep 12, 392–398 (2012). https://doi.org/10.1007/s11910-012-0288-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-012-0288-1