
Abstract
Renal anomalies are common birth defects that may manifest as a wide spectrum of anomalies from hydronephrosis (dilation of the renal pelvis and calyces) to renal aplasia (complete absence of the kidney(s)). Aneuploidies and mosaicisms are the most common syndromes associated with CAKUT. Syndromes with single gene and renal developmental defects are less common but have facilitated insight into the mechanism of renal and other organ development. Analysis of underlying genetic mutations with transgenic and mutant mice has also led to advances in our understanding of mechanisms of renal development.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Introduction
Renal anomalies are among the most common congenital abnormalities. Although the prevalence varies in different populations, congenital abnormalities of the kidney and urinary tract (CAKUT) complicate on average approximately 1 in 500 pregnancies (Song and Yosypiv 2011; Stoll et al. 2014). CAKUT encompass a broad spectrum of anomalies from hydronephrosis (dilation of the renal pelvis and calyces) to renal aplasia [complete absence of the kidney(s)]. CAKUT also include abnormalities of the ureters, bladder, and urethra, including ureteral obstruction at the renal pelvis (UPJ) or entry into the bladder [uretero-vesicle junction (UVJ)], vesicoureteral reflux (backup of urine in the ureters from bladder toward the kidneys), and obstruction of the urethra (e.g., posterior urethral valves). Up to 30% of children with end-stage renal disease (ESRD) in the USA have underlying CAKUT, making it the leading cause of pediatric ESRD (North American Pediatric Renal Trials and Collaborative Studies 2011).
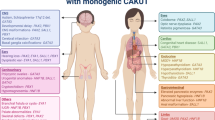
Up to one-third of children with CAKUT may have associated extrarenal manifestations (Stoll et al. 2014). Extrarenal manifestations appear to be most frequent with the severest forms of CAKUT (e.g., renal agenesis) (Stoll et al. 2014). In one large European population-based study (Stoll et al. 2014), over half of the extrarenal manifestations were part of known syndromes or chromosomal abnormalities. The three most common syndromes were VACTERL (which is comprised of vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula, renal anomalies, and limb abnormalities), Meckel–Gruber syndrome [which is comprised (Sadowski et al. 2015) of renal cystic dysplasia, occipital encephalocele/holoprosencephaly, and postaxial polydactyly], and prune belly syndrome (which is comprised of hydroureteronephrosis, defects in abdominal wall musculature, and cryptorchidism) (Stoll et al. 2014). Chromosomal aneuploidies and mosaicism, including Trisomy 18, 21, and 13, were the second most common likely to be associated with renal anomalies. Collectively, genetic syndromes associated with chromosomal microdeletions/duplications (e.g., DiGeorge) or single gene defects (e.g., Simpson–Golabie–Behmel, Pallister–Hall, Beckwith–Wiedemann) and environmental toxin-induced syndromes (e.g., fetal alcohol syndrome) accounted for the rest of the renal anomalies with identifiable syndromes (Stoll et al. 2014).
The availability of gene sequencing and microarray studies has led to significant advances in the genetics of renal anomalies (Song and Yosypiv 2011). They have rapidly expanded our knowledge of the genes that affect renal and ureter development. In addition, the ability to manipulate genes using transgenic and knockout mice has revealed that a few key signaling pathways regulate multiple stages of kidney development (Reidy and Rosenblum 2009). In particular, gene deletion with conditional knockout mice has revealed cell- and time-specific effects of developmental signaling pathways (Reidy and Rosenblum 2009). Interestingly, several of these key signaling pathways play developmental roles in other organs. Thus, genetic defects lead to both abnormal kidney development and extrarenal manifestations.
Here we will focus on a few key signaling pathways that regulate development in multiple organs and their role in renal epithelial differentiation. In particular, we will emphasize on how the identification of genetic mutations in syndromic renal malformations has advanced our insight into mechanisms of renal epithelial differentiation.
2 Manifestations of CAKUT Reflect the Impaired Stage of Renal Development
First, it is important to briefly review the stages of renal development (see Chap. x for a more detailed discussion). The mammalian kidney develops from the intermediate mesoderm. There are three primitive kidneys that form sequentially during human kidney development: the pronephros, mesonephros, and metanephros (Fig. 10.1). The pronephros consists of simple tubules that form and involute. The mesonephros forms caudal to the pronephros and consists of glomeruli and simple tubules that are able to form a filtrate. The mesonephric tubules then form the mesonephric or Wolffian duct. The final adult kidney forms when the ureteric bud grows from the mesonephric duct and invades the surrounding metanephric mesenchyme (Table 10.1).

Depiction of the three primitive kidneys
The reciprocal interactions between metanephric mesenchyme and ureteric bud are required for ureteric bud branching and conversion of the metanephric mesenchyme to renal epithelial cells. The branching ureteric bud forms the collecting ducts. The metanephric mesenchyme condenses and converts to epithelia of the renal vesicle. The renal vesicle becomes a comma and then an S-shape body. The portion of the S-shape closest to the ureteric bud tip forms the distal tubule, the mid portion forms the proximal tubule, and the portion furthest from the ureteric bud tip will form the podocytes.
Genetic defects that impair any stage of the development from the outgrowth of the ureteric bud until terminal differentiation into specialized cells of the nephron segments can manifest as developmental renal disease. For example, both defects in ureteric bud outgrowth or extra ureteric bud outgrowth are associated with CAKUT (Fig. 10.2).

Defects in the ureteric bud (UB) outgrowth lead to renal and ureteral abnormalities. Complete failure of the ureteric bud to grow or to contact the surrounding metanephric mesenchyme leads to apoptosis of the metanephric mesenchyme and renal agenesis. Defects in ureteric bud outgrowth can also result in vesicoureteral reflux (VUR). Outgrowth of multiple ureteric buds is associated with duplication of kidney and/or ureters, which are often associated with obstruction or VUR. With fully duplicated ureters, the upper ureter frequently inserts ectopically into the bladder and may be associated with an obstructing ureterocele (an out-pouching of the ureter). Obstruction at the level of the ureter entering the bladder is known as uretero-vesicular (UVJ) junction obstruction. Duplicated kidney depicted with typical clinical scenario observed (an upper pole with an ectopic, obstructed ureter, and lower pole ureter with VUR)
Defects in later stages of differentiation can result in renal dysplasia or hypoplasia. Renal dysplasia is the most severe and is classically characterized by primitive ducts with a fibromuscular collar (Kakkar et al. 2006). Non-renal tissues such as cartilage or hematopoietic cells may be present (Kakkar et al. 2006). Mouse models with defective WNT4 signaling between the ureteric bud and metanephric mesenchyme lead to disruptions in the mesenchymal-to-epithelial transition (MET) (Stark et al. 1994). Histologically this appears as primitive ducts surrounded by undifferentiated mesenchyme, analogous to human renal dysplasia (Stark et al. 1994). Supportive of this concept is the finding of dysplasia in a few families with mutations in WNT4, although the frequencies of these mutations have not been assessed in larger studies (Vivante et al. 2013; Biason-Lauber et al. 2004, 2007). Small kidneys with decreased nephron number and histologically normal nephron structures are typical findings in renal hypoplasia (Blake and Rosenblum 2014). In mouse models, defects in ureteric bud branching or premature loss of renal epithelial progenitors lead to renal hypoplasia (Zubkov et al. 2015). Dysplasia and hypoplasia may both be present in human CAKUT (known as hypo-dysplasia) (Fig. 10.3).

Defects in later differentiation result in renal dysplasia and hypoplasia. (a) Defects in the mesenchymal-to-epithelial transition lead to renal dysplasia with primitive tubules surrounded by a fibromuscular collar and metaplastic cartilage. (b) Defects in ureteric bud branching result in renal hypoplasia
3 Common Syndromes Associated with CAKUT
In genetics, syndromes are characterized by a set of associated signs and symptoms where a genetic cause is known. In some cases, there is a suspected genetic cause of associated characteristics, but the cause is unknown. These are referred to as genetic associations (e.g., VATER and VACTERL). In contrast, sequences are anomalies associated with a specific cause. For example, the Potter sequence (micrognathia, clubbed feet, and pulmonary hypoplasia) are the result of low amniotic fluid. Potter sequence may be associated with severe CAKUT, as decreased urine production for any reason leads to low amniotic fluid.
VATER and VACTERL are genetic associations that have CAKUT as a common phenotype. They were first described in the 1970s, and the diagnosis requires at least three of the following findings: vertebral anomalies (V), anal atresia (A), cardiac defects (C), tracheoesophageal fistula and/or esophageal atresia (TE), renal anomalies (R), or limb defects (L). In some studies, up to 80% of patients with VACTERL have renal anomalies (Botto et al. 1997). Some have argued that VACTERL results from defects in midline cell fate determination and morphogen gradients, with modulation by genetic-environmental interactions (Lubinsky 2015; Gillick et al. 2003). Genetic studies of patients with VACTERL have revealed a variety of chromosomal micro-deletions and duplications, although none are consistent. Mouse models with defects in sonic hedgehog (SHH) signaling (discussed in more detail below) exhibit many VACTERL characteristics (Ngan et al. 2013; Kim et al. 2001a, b); however, human mutations of SHH signaling in patients with VACTERL have not been identified (Aguinaga et al. 2010). A rat model of teratogen (adriamycin)-induced VACTERL demonstrated ectopic expression of SHH (Gillick et al. 2003), supporting the concept that defects in SHH signaling likely contribute to the pathogenesis of VACTERL.
Rarely, monogenic autosomal recessive mutations have been identified in familial VACTERL, including mutations in TRAP1 (a heat-shock protein 90-related mitochondrial chaperone) and FGF8 (fibroblast growth factor 8) (Saisawat et al. 2014; Zeidler et al. 2014). FGFs will be discussed in further detail below.
4 Chromosomal Abnormalities in CAKUT
Chromosomal aneuploidies and mosaicism, including Trisomy 13, 18, and 21 as mentioned before, are common causes of syndromic CAKUT. Of these, Trisomy 21 is the most common (1.5 in 1000 births) (Altug-Teber et al. 2007). A study of over 700 cases of Trisomy 21 in Europe revealed 5% had CAKUT, including 2% with obstructive defects of the urinary pelvis (Stoll et al. 2015). Trisomy 13 and 18 are less common (0.05 and 0.15 per 1000 live births, respectively). The exact mechanisms by which trisomies alter renal development are unclear. An analysis of transcripts from amniocytes and chorionic villous cells from trisomy infants supports a model in which there is increased transcription (increased gene dosage) from the additional chromosome (Altug-Teber et al. 2007). The increased expression of genes may itself be deleterious, but it also appears to disrupt expression of downstream genes (not on the trisomy chromosome), known as developmental instability (Altug-Teber et al. 2007). Thus, contributions of both increased gene dosage and developmental instability likely contribute to the observed phenotypes (Altug-Teber et al. 2007).
Smaller chromosomal abnormalities are also common causes of CAKUT. Recently, it has been shown that copy number variants (CNVs) are present in 10–16% of isolated CAKUT (Caruana et al. 2015; Sanna-Cherchi et al. 2012; Xi et al. 2016). Some of these CNVs affected known developmental genes, including HNF1B. Mutations in HNF1B can manifest as syndromic or isolated CAKUT (discussed in more detail below) (Caruana et al. 2015; Verbitsky et al. 2015). CNVs also are rarely associated with VACTERL (see above) and other less common syndromes (Brosens et al. 2013; Hilger et al. 2013; Materna-Kiryluk et al. 2014; Handrigan et al. 2013). These CNVs affected multiple genes, likely explaining the multiple organ systems affected.
5 Challenges of Genotype–Phenotype Correlation
It should be emphasized that both in the setting of CNVs and single nucleotide variations (SNVs), environmental factors likely modify the phenotypic outcome. In several studies, the identified CNVs were also present in siblings and parents of affected children with CAKUT who were either asymptomatic or had mild manifestations (Xi et al. 2016; Handrigan et al. 2013). A large study of 75 patients harboring HNF1B mutations revealed a broad spectrum of manifestations, from echogenic kidneys to multi-cystic kidneys to renal hypoplasia/agenesis (Heidet et al. 2010).
6 Genetics in HNF1B are Commonly Associated with CAKUT and Extrarenal Manifestations
Genetic studies of large cohorts of children with CAKUT have revealed that the most common mutations (identified in up to 15% of European or US Caucasian patients) are in HNF1B (associated with renal cysts and diabetes syndrome) and PAX2 (associated with renal-coloboma syndrome) (Hwang et al. 2014; Madariaga et al. 2013; Weber et al. 2006). Less common but also identified in large cohorts of patients with CAKUT were mutations in SALL1 (associated with Townes–Brocks syndrome), EYA1, and SIX5 (associated with Branchio-oto-renal syndrome).
Hepatocyte nuclear factor beta (HNF1B) encodes a POU (PIT-1, OCT-1, UNC-86) homeodomain containing transcription factor. Heterozygous mutations in this gene are an important cause of developmental diseases of the kidney (Madariaga et al. 2013; Thomas et al. 2011) and renal cysts (Heidet et al. 2010). Also, patients with mutations in this gene are at an increased likelihood of developing maturity onset diabetes mellitus (MODY), secondary to pancreatic hypoplasia, which has led to the description of renal cyst and diabetes (RCAD) syndrome (Bingham et al. 2001). Since initial reporting of the early cases of HNF1B-associated disease in 1997 (Iwasaki et al. 1998; Horikawa et al. 1997), it has become evident that additional clinical features also include pancreatic hypoplasia, genital tract malformations, abnormal liver function, hypomagnesemia, hyperuricemia, and early-onset gout (Clissold et al. 2014). HNF1B-associated disease is, therefore, considered to be a multisystem disorder. It is a perfect example of a gene that is involved in the development of more than one organ, with its mutations being a cause of multiple phenotypes.
HNF1B shares >80% sequence homology with HNF1A, and they both bind to the same DNA sequence (Mendel et al. 1991). HNF1A gene mutations are the most common cause of MODY. More than 50 different HNF1B mutations have been reported, including missense, nonsense, frameshift, and splicing mutations. No correlation seems to exist between the type or position of the mutation and particular clinical features (Edghill et al. 2006), and up to 50% are spontaneous mutations not present in parents. HNF1A is most highly expressed in the liver. HNF1B is predominantly expressed in the kidneys but is also expressed in the gonads, thymus, and lungs. In situ hybridization studies using human tissue samples have shown that HNF1B mRNA is highly expressed in the fetal collecting ducts, with lower levels of expression in the metanephric mesenchyme (Kolatsi-Joannou et al. 2001).
Renal-specific inactivation of Hnf1b in mice results in polycystic kidneys. This phenotype is associated with a marked reduction in the transcriptional activation of the cystic disease genes Umod, Pkhd1, and Pkd2, which contain DNA elements to which HNF1B normally binds (Gresh et al. 2004). Hnf1b appears to act upstream of WNT9B and alters WNT signaling and thereby impairs early renal development (Carroll et al. 2005; Lokmane et al. 2010).
7 Identification of Syndromic Genes Reveals Mechanisms of Renal Epithelial Differentiation
It is challenging to categorize genes as affecting only one aspect of kidney development. However, many syndromic genes have helped to elucidate mechanisms of renal epithelial differentiation.
7.1 Genetic Syndromes Affecting Early Renal Epithelial Differentiation
Nephron progenitor cells are a self-renewing population of cells in the metanephric mesenchyme that form the nephron (O’Brien et al. 2016). The progenitor cell population is regulated by a complex network of transcription factors (Basta et al. 2014). The proper balance of these transcription factors is necessary to both induce the transcriptional program of renal differentiation and maintain the progenitor population during renal development. Studies of animal models indicate that some of the most common mutations in human CAKUT (PAX2, SALL1, EYA1, and SIX2) likely affect nephron progenitor survival and/or self-renewal.
As indicated above, EYA1, SIX1, and SIX5 mutations are associated with branchio-oto-renal syndrome (Weber et al. 2008; Hoskins et al. 2007; Ruf et al. 2004). SIX2 is a gene which belongs to the same family. Although SIX2 is not found to be associated with branchio-oto-renal syndrome, its mutations are associated with hypodysplasia of the kidney (Weber et al. 2008). The EYA1 protein consists of an N-terminal transactivation domain and a C-terminal protein interaction domain. In the kidney, the EYA1 protein interaction domain binds Six proteins, facilitating EYA1 translocation from cytoplasm to the nucleus and DNA binding. Binding of EYA1 to SIX proteins converts the SIX proteins from weak to strong transcriptional activators (Xu et al. 1997; Chen et al. 1997; Pignoni et al. 1997) to specify nephron progenitor population from the intermediate mesoderm. EYA1 coordinates with SIX1 and PAX2 to induce GDNF expression in the metanephric mesenchyme during ureteric bud branching (Sajithlal et al. 2005). EYA1 is also required to induce SIX2 expression (Xu et al. 2014). EYA1 and SIX2 lead to phosphorylation of MYC to prevent apoptosis and maintain the proliferative capacity of nephron progenitors. Genetic deletion of Six2 leads to premature differentiation, depletion of the nephron progenitor population, and hypoplasia (Park et al. 2012). Genetic variants in EYA1 and SIX2 are associated with branchio-oto-renal syndrome (hearing loss, branchial fistulae, preauricular pits or tags, and renal anomalies). Similar to its actions in the nephron progenitor populations, EYA1 stimulates proliferation and inhibits apoptosis in the ear (Li et al. 2010; Zou et al. 2006; Xu et al. 1999). EYA1 regulates the growth of otic epithelium and interacts with PAX2 during the development of all sensory areas in the inner ear (Zou et al. 2006). During craniofacial development in mice, EYA1 is expressed in the cranial placodes. In these placodes, EYA1 interaction domain binds and acts as a transcriptional coactivator with two key proteins in craniofacial development, SIPL1 (Shank-interacting protein-like 1) and RBCK1 (RBCC protein interacting with PKC1) (Landgraf et al. 2010). EYA proteins are required for retinal development in Drosophila, but due to functional redundancy, individual EYA mutant mice do not exhibit a phenotype (Tadjuidje and Hegde 2013).
SALL1 autosomal dominant mutations result in Townes–Brock syndrome, which includes imperforate anus, ear malformations and hearing loss, thumb malformations (most often triphalangeal thumbs or preaxial polydactyly), and renal anomalies (Kohlhase 1993). Some patients also have congenital heart disease and genitourinary malformations. The renal phenotype includes abnormal rotation, ectopic kidneys, horseshoe kidneys, polycystic kidneys, and vesicoureteral reflux. Complete deletion of Sall1 in mice leads to failure of ureteric bud invasion, resulting in aplasia or severe dysplasia (Nishinakamura et al. 2001). Conditional knockout of Sall1 in the metanephric mesenchyme leads to ureteric bud branching defects with dysregulation of WNT signaling (Kiefer et al. 2010). SALL1 has also been shown to regulate the balance between differentiation and self-renewal in the metanephric mesenchyme by activating progenitor genes in SIX2-positive cells and repressing progenitor genes in SIX2-negative cells (Basta et al. 2014; Kanda et al. 2014). SALL1 was also recently shown to maintain cardiac progenitor cells and promote neurogenesis in mice (Harrison et al. 2008; Morita et al. 2016). Although SALL1 is expressed in the limb bud, ear, anus, heart, and kidney, Sall1 knockout mice only exhibited a renal phenotype. It is thought that SALL1 mutations may lead to truncated proteins that exert a dominant-negative effect by suppressing other SALL proteins (Kiefer et al. 2003).
The conversion of metanephric mesenchyme to renal vesicle epithelia is another key step in early nephrogenesis. Landmark work by Stark et al. identified WNT4 as a major factor regulating mesenchyme-to-epithelial transition (Stark et al. 1994). Human mutations in WNT4 are rare, but a consanguineous kindred was identified with homozygous WNT4 mutations and a phenotype of female sex reversal and dysgenesis of kidneys, adrenals, and lungs (SERKAL syndrome) (Mandel et al. 2008). Multiple WNTs are present in the developing kidney, and WNT signaling functions at multiple stages of renal development (Vainio 2003). WNT signaling is discussed in detail in Chap. X; thus, we will highlight here only its role in SERKAL syndrome. Canonical WNT signaling occurs by binding of WNTs to Frizzled receptors. This leads to an intracellular signaling cascade that ultimately results in the stabilization of beta-catenin, which translocates to the nucleus to stimulate downstream effectors. Studies suggest that both canonical and noncanonical WNT/planar cell polarity contribute to kidney development (Carroll et al. 2005; Bridgewater et al. 2008; Iglesias et al. 2007; Nishita et al. 2014). WNT9B is secreted from the collecting duct and stimulates expression of Wnt4, as well as FGF8, PAX8, and LHX1, thereby inducing nephron differentiation (Carroll et al. 2005). The gradient of WNT signaling also establishes proximal-distal patterning of the nascent nephron (Schneider et al. 2015). As in the kidney, WNT signaling plays a key role in regulation lung branching, proximal-distal patterning, and mesenchyme development (Volckaert and De Langhe 2015). Studies of conditional knockout of beta-catenin in mouse adrenal glands indicate that canonical WNT signaling is required for adrenal cortical cell renewal (Berthon et al. 2012). In the ovary, WNT4/beta-catenin activation is required for oogonial differentiation and entry into meiosis (Bernard and Harley 2007; Bernard et al. 2008; Chassot et al. 2011; Munger et al. 2013), likely explaining the sex-reversal phenotype of SERKAL.
7.2 Genetic Syndromes Affecting UB Branching
UB branching establishes the radial architecture, and the number of branches determines nephron number. This is clinically relevant because, in addition to severe defects that result in hypoplasia, the low number of nephrons (even in absence of overt renal disease in childhood) is associated with increased risk for adult-onset chronic kidney disease.
UB branching is regulated by multiple growth factors, many of which also regulate lung branching and cardiac development. Many of the growth factors that can affect branching are membrane-bound tyrosine kinase receptors. RET-GDNF is one key pathway (Schuchardt et al. 1996). RET tyrosine kinase receptors are concentrated at the tips of the ureteric buds (Riccio et al. 2016). GDNF (stimulated by the transcription factors discussed above) is secreted by the metanephric mesenchyme and binds to the RET receptor to activate intracellular signaling pathways such as PI3 kinase and MAPK/ERK proliferation signaling pathways (Shakya et al. 2005; Costantini and Shakya 2006; Costantini 2010). This leads to ureteric bud cell division and branching (Costantini and Shakya 2006; Costantini 2010). A high concentration of RET at the tip of the ureteric bud directs cell movements to regulate ureteric bud outgrowth (Riccio et al. 2016). It is important to have a single ureteric bud in the proper location to induce metanephric mesenchyme. As indicated above, extra ureteric buds or a ureteric bud in the wrong location results in duplication of ureters that may not properly drain in the bladder and become obstructed or have vesicoureteral reflux. A tyrosine kinase inhibitor, Sprouty, opposes tyrosine kinase activity to specify outgrowth of a single ureteric bud (Basson et al. 2006).
RET mutations have been associated with unilateral and bilateral renal agenesis, vesicoureteral reflux, renal hypodysplasia, and ureteropelvic junction obstruction (Hwang et al. 2014; Chatterjee et al. 2012; Jeanpierre et al. 2011). Loss-of-function GDNF and RET mutations have also been associated with Hirschsprung’s disease (congenital megacolon), which is characterized by an absence of enteric nervous system (ENS) ganglion cells in the distal intestine (Martucciello et al. 2000). In mice, GDNF and RET are required for enteric neural crest cell survival and differentiation (Natarajan et al. 2002; Taraviras et al. 1999). In addition, GDNF expressed in the developing gut acts as a chemo-attractant for ENS progenitors, and thus, mutations in GDNF or RET result in defective migration of ENS progenitors from the neural crest into the intestine (Natarajan et al. 2002). As in the kidney, PI3K and MAPK/ERK pathways appear to mediate GDNF/RET’s effects on ENS neural crest cell differentiation, survival, and migration (Natarajan et al. 2002). Ret-deficient mice exhibit Hirschspung’s disease and renal agenesis. Interestingly, studies indicate between 15 and 25% of Hirschsprung’s patients may have associated CAKUT (often hydronephrosis or hypoplasia) (Hofmann et al. 2014; Pini Prato et al. 2009).
Other tyrosine kinases may contribute to ureteric bud outgrowth. In particular, in mice, fibroblast growth factors (FGF) can stimulate ureteric bud formation in the absence of GDNF and Sprouty (Michos et al. 2010). Elegant studies of ureteric bud and metanephric mesenchyme-specific knockout mice of FGF receptors 1 and 2 have revealed time and spatial specific roles for the FGF receptors (Bates 2011). Knockout of both Fgfr1 and Fgfr2 in the metanephric mesenchyme led to renal dysgenesis due to the failure of the ureteric bud to elongate and branch (Poladia et al. 2006). In the ureteric bud, FGFR2 is the key receptor, and deletion in the ureteric bud leads to branching defects and hypoplasia (Bates 2011; Zhao et al. 2004). FGFs are also key factors in bone growth and remodeling (Chen and Deng 2005; Su et al. 2014). Mutations in FGFR1 and FGFR2 are associated with craniosynostosis syndromes such as Apert’s syndrome, Antley–Bixler, Pfeiffer, Beare–Stevenson syndromes (Marie et al. 2005; Passos-Bueno et al. 1999). FGFR 1 or 2 mutations may cause craniosynostosis via effects of FGFs on osteoblast differentiation and apoptosis, although differences between humans and mouse models have made understanding the pathophysiology challenging. CAKUT has been described in association with craniosynostosis syndromes, including solitary kidneys, hydroureter, and vesicoureteral reflux (Seyedzadeh et al. 2008; Hains et al. 2010).
Closely related to GDNF/RET and the FGFs are Fraser syndrome FRAS1 and FRAS1-related extracellular matrix (ECM) gene 2 (FREM2) (Pavlakis et al. 2011). Fraser syndrome is characterized by CAKUT (often uni- or bilateral renal agenesis), eye anomalies (Cryptophthalmos), embryonic skin blistering, and cutaneous syndactyly and results from autosomal recessive mutations in FRAS1, FREM2, or GRIP1 (a scaffolding protein that links to FRAS1 and FREM2). Autosomal recessive mutations in Fraser syndrome genes are also associated with isolated CAKUT (Kohl et al. 2014). FRAS1 and FREM2 are extracellular matrix proteins that line the ureteric bud epithelia, and mouse models of mutant FRAS1 develop renal agenesis due to the failure of ureteric bud outgrowth (Pitera et al. 2008). Impaired GDNF expression likely contributes to the failure of ureteric bud outgrowth. Fras1 mutant mice can be rescued by decreased Sprouty activity, likely via FGF-mediated ureteric bud induction (Pitera et al. 2012). Fras1/Frem contribute to epithelial integrity and mutations lead to disrupted dermal-epidermal attachment in the plane of sublamina densa likely leading to the embryonic skin blistering (Pavlakis et al. 2011). Syndactyly likely results from downregulation of Msx2, a transcription factor that stimulates apoptosis and interdigital cell death (Hines et al. 2016).
7.3 Genetic Defects Affecting Renal Patterning
Several developmental patterning signaling pathways including Hox and Sonic Hedgehog (SHH) contribute to renal development (Reidy and Rosenblum 2009). As indicated above, defects in Sonic Hedgehog (SHH) signaling may contribute to VACTERL associated with CAKUT. Genetic mutations in SHH signaling component GLI3 are associated with Pallister–Hall syndrome, which is characterized by polydactyly, imperforate anus, hypothalamic hamartoma, and CAKUT (Hill et al. 2007). SHH is a secreted morphogen that binds the Patched receptor. The Patched receptor constitutively inhibits Smoothened, and binding of SHH allows Smoothened to stimulate translocation of GLI1 and 2 effectors to the nucleus. In the kidney, GLI1 and 2 induce transcription of cell cycle proliferative proteins MYC and CYCLIND1 as well as nephron differentiation factors PAX2 and SALL1. The GLI3 mutations associated with Pallister–Hall syndrome lead to the expression of a truncated GLI3 that inhibits GLI1 and GLI2. In the hand, GLI3 restricts regulators of cell cycle entry and also promotes differentiation into chondrocytes (Lopez-Rios et al. 2012). A gradient of SHH also establishes the anteroposterior limb axis and digit identities, and defects in GLI3 lead to polydactyly in mice (Hill et al. 2007).
7.4 Genetic Defects Affecting Nephron Segmentation
NOTCH signaling is a highly conserved evolutionary pathway that is important for cell–cell signaling and determines cell fate and differentiation during renal development. To date, five NOTCH ligands (delta-like protein [DLL]-1, DLL-3, DLL-4, Jagged1, and Jagged2) and four NOTCH receptors (NOTCH 1–4) have been identified in mammals. Both the receptors and their ligands are single-pass type I membrane proteins. NOTCH ligands consist of an extracellular N-terminal region called as the delta/serrate/LAG2 (DSL) domain which is required for their binding to the receptors (Kamath et al. 2013). NOTCH receptors consist of an extracellular segment, formed of multiple EGF-like repeats, a transmembrane domain, and an intracellular domain. Once the receptor–ligand interaction has occurred, the intracellular domain of the NOTCH receptor undergoes a series of cleavages that allows the intracellular domain to translocate to the nucleus, where it regulates the transcription of downstream genes, such as HES1 and HEY2 (transcription factors that are NOTCH effectors) (Kopan 2012).
In developing kidneys, receptors NOTCH2 and NOTCH1, along with ligands, Jagged1 and DLL-1, are expressed. Both receptors and ligands are expressed in pretubular aggregates. In later stages of glomerular development, the NOTCH receptors are expressed in podocytes and Bowman’s capsule, whereas the ligands are expressed in endothelial cells (McCright 2003). This change in expression pattern over development suggests that NOTCH signaling is operating via inductive signaling and lateral inhibition to control cell-fate determination. In inductive signaling, the ligand-expressing cell induces differentiation in receptor-expressing cell. In lateral inhibition, the ligand-expressing cell differentiates and interacts with receptor expressing cell and inhibits it from adopting a differentiated state. The developing kidney may use both types of NOTCH signaling (McCright 2003). This process is comparable to the commitment of hepatoblasts to a biliary cell fate that occurs within the ductal plate of the developing liver, which is also dependent on NOTCH signaling (Si-Tayeb et al. 2010).
Various models of Jag1 and Notch2 mutations in mice have been developed to study the role of NOTCH signaling during renal development. Mice homozygous for a hypomorphic allele of Notch2 die shortly after birth owing to a lack of functional nephrons (McCright et al. 2001). The early stages of kidney development (ureteric bud migration and mesenchymal aggregate formation) occur normally in these mice; however, podocytes and proximal tubules do not form, and the developing glomerulus has vascularization defects. This phenotype is consistent with the known role of NOTCH signaling in tissue specification and the formation of major arteries and veins (Krebs et al. 2000; Uyttendaele et al. 2001; Xue et al. 1999). Mouse metanephrons cultured in the presence of an inhibitor of NOTCH intracellular signaling have a severe deficiency of proximal tubules and glomerular podocytes (Cheng et al. 2003). In the collecting duct, NOTCH acts by lateral inhibition to determine cell fate (Guo et al. 2015). Collecting duct-specific deletion of the ADAM10 metalloproteinase that cleaves the intracellular domain of NOTCH led to a reduction of principal cells and increase of intercalated cells in the collecting duct (Guo et al. 2015).
In humans, abnormalities in NOTCH signaling present as Alagille syndrome, an autosomal dominant disorder with variable multisystem organ involvement that is caused by mutations in one of two genes in the NOTCH signaling pathway, JAG1 or NOTCH2. The diagnosis is based on the presence of intrahepatic bile duct paucity in liver biopsy samples in association with at least three of the following major clinical features: chronic cholestasis, cardiac disease (most often peripheral pulmonary stenosis), skeletal abnormalities (typically butterfly vertebrae), ocular abnormalities (primarily posterior embryotoxon), and characteristic facial features. JAG1 mutations have been identified in 94% of patients with clinically defined Alagille syndrome (Warthen et al. 2006). Renal anomalies, such as impaired function and structural abnormalities, have been documented in almost 40% of patients with Alagille syndrome who carry JAG1 mutations. Renal dysplasia (58.9%) is the most common renal abnormality. Other common renal anomalies include renal tubular acidosis (9.5%), ureteropelvic or vesicoureteral urinary obstruction (8.2%), and VUR (also 8.2%). Since renal involvement is common in Alagille syndrome, it has been suggested to be included as the sixth diagnostic criteria for this syndrome (Kamath et al. 2012, 2013).
7.5 Genetic Syndromes Affecting Terminal Epithelial Differentiation of Podocytes
Over 40 genes have now been associated with nephrotic syndrome (Sadowski et al. 2015). Some of these gene defects affect podocyte differentiation and are also associated with extrarenal manifestations (Sadowski et al. 2015). Genetic defects in the nuclear transcription factor WT1 associated with Denys–Drash and Frasier syndrome are perhaps some of the best-understood examples (Morrison et al. 2008). Denys–Drash is associated with autosomal dominant missense mutations in the zinc finger region of exons 8 or 9 of the WT1 gene (Patek et al. 2003). These mutations lead to decreased nuclear expression of WT1, which allows for abnormal persistent expression of PAX2, likely leading to diffuse mesangial sclerotic lesion. The WT1 zinc finger mutations alter DNA binding and may act as a dominant-negative fashion during genitourinary development. In contrast, mutations in a WT1 splice site lead to Frasier syndrome (Lefebvre et al. 2015). The splice site mutations lead to loss of the most common WT1 isoform that includes three amino acids (known as +KTS). The WT1 isoform that lacks those three amino acids (-KTS) has a different DNA binding and nuclear localization patterns (Lefebvre et al. 2015). Thus, loss of this +KTS isoform likely leads to gonadal dysgenesis, male–female sex reversal, progressive podocyte damage, and focal glomerulosclerosis (Lahiri et al. 2007).
7.6 Genetic Syndromes with Progressive Defects in Tubular Epithelial Differentiation
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by the growth of dysgenic lesions in multiple organs including the brain, skin, kidney, heart, lungs, and retina. TSC arises from inactivating mutations of either TSC1 (chromosome locus 9q34.3) or TSC2 (16p13.3), which encode hamartin and tuberin, respectively. These proteins are believed to function as tumor suppressors by forming a complex that regulates cellular proliferation (Plank et al. 1998; van Slegtenhorst et al. 1998).
Hamartin and tuberin, together with a third protein, TBC1D7 (Dibble et al. 2012), form the TSC protein complex, which regulates multiple cellular processes and importantly acts as a critical negative regulator of the mechanistic target of rapamycin (mTOR) complex 1 (mTORC1) (DiMario et al. 2015; Inoki et al. 2002; Tee et al. 2002), a serine/threonine kinase that is central to many cell functions including cell growth and proliferation. RHEB (RAS homolog enriched in brain) is a specific GTPase downstream of the TSC protein complex that functionally links TSC1/TSC2 to mTORC1. The TSC1/TSC2 complex functions as a GTPase-activating protein (GAP) for Rheb and stimulates the conversion of RHEB-GTP to a GDP-bound state, thereby inactivating RHEB signaling and thus removing its stimulatory effect on mTORC1. Conversely, loss-of-function mutations in either TSC1 or TSC2 lead to enhanced RHEB-GTP signaling and mTORC1 activation. Constitutively, active mTORC1 signaling thus constitutes the molecular basis of TSC and responsible for the dysgenic lesions. When in its active state, mTORC1 phosphorylates the translational regulators 4E-BP1 (eukaryotic translation initiation factor 4E-binding protein 1) and S6K1 (S6 kinase 1), which, in turn, stimulate protein synthesis. Active mTORC1 stimulates the biosynthesis of ribosomes, lipid biogenesis, glucose metabolism, nucleotide synthesis, mitochondrial and lysosomal biogenesis, ATP, and amino acid production. mTORC1 also functions as a negative regulator of autophagy, another key cellular pathway that has been implicated in TSC. mTORC1 inhibitors have proven efficacy in TSC and are now the first line of therapy in growing angiomyolipomas of the kidney and in surgically unresectable symptomatic giant cell tumors of the brain (Kingswood et al. 2016; Krueger et al. 2010).
TSC most often presents with neurologic symptoms, and approximately 90% of affected individuals experience seizures and about half of patients experience cognitive impairment, autism, or other behavioral disorders. Renal manifestations are the second most common findings associated with TSC, with angiomyolipomas occurring in up to 55–75% and renal cystic disease in 30–45% of patients (Crino et al. 2006; Rakowski et al. 2006).
Angiomyolipomas are the prototype of a family of lesions called perivascular epithelioid cell tumors or PEComas which exhibit immunoreactivity for both melanocytic markers (as detected by the HMB-45 and melanin-A antibodies) and smooth-muscle markers (actin and desmin). All components of angiomyolipomas, including the vascular cells, immature smooth-muscle-like spindle cells, epithelioid cells, and fat cells, contain somatic mutations that, combined with their germline mutation, render the cells deficient in either tuberin or hamartin. Presumably, this deficiency disrupts the integrated control of cell growth leading to the angiomyolipoma (Siroky et al. 2011; Dixon et al. 2011).
Renal cysts are also a common complication of TSC. Fifty percent of the patients with TSC develop renal cysts. New insight has been gained into the different mechanisms by which TSC pathway interacts with polycystin-1 (PC-1). PC-1 is a protein encoded by PKD1 which is the gene involved in adult-onset polycystic kidney disease (ADPKD). Two percent of patients with TSC have a contiguous germline deletion of TSC2 and PKD1 (chromosome 16p13.3) and develop severe infantile polycystic kidney disease (Brook-Carter et al. 1994), suggesting a functional cooperation between their gene products. ADPKD is discussed in more detail in another chapter and is caused secondary to defects in PKD1 (81% of the cases) and PKD2 (19% of the cases) (Mao et al. 2016). It has been recently shown that mTORC-1 upregulation in TSC-1 mutant mice causes decreased protein neo-synthesis of PC-1 which cystogenesis in TSC (Pema et al. 2016). There are other studies in Tsc1+/- Tsc2+/- mice that have shown the presence of early cysts with no evidence of mTOR activation measured by phosphorylated S6K staining, suggesting that many TSC-associated renal lesions initially develop via mTOR-independent pathways. Primary cilia are anchored to the cell via the basal body. Hamartin has been localized to the basal body (Hartman et al. 2009), and tuberin interacts with PC-1 (Shillingford et al. 2006), which has been localized to the primary cilium (Yoder et al. 2002). Similarities between TSC and polycystic kidney disease have been demonstrated by showing how hamartin, tuberin, and polycystin-1 defects affect primary cilia length (Bonnet et al. 2009). Primary cilia is also an organelle important for cell polarity and is thought to underlie numerous disorders associated with cystic kidney disease, as discussed in detail elsewhere in the volume (Hartman et al. 2009). Further defects in apicobasal cell polarity and misoriented dividing tubular cells have been seen, pointing out another possible mechanism for cystogenesis (Bonnet et al. 2009).
8 Conclusions
CAKUT is a common feature of many syndromes. The clinical manifestations of CAKUT depend on the stage at which renal development is impaired. Aneuploidies and mosaicisms are the most common syndromes associated with CAKUT. Single gene defects are less common but have facilitated insight into the mechanism of renal and other organ development. Analysis of underlying genetic mutations with transgenic and mutant mice has also led to advances in our understanding of mechanisms of renal development.
References
Aguinaga M, Zenteno JC, Perez-Cano H, Moran V (2010) Sonic hedgehog mutation analysis in patients with VACTERL association. Am J Med Genet Part A 152a:781–783. doi:10.1002/ajmg.a.33293
Altug-Teber O et al (2007) Specific transcriptional changes in human fetuses with autosomal trisomies. Cytogenet Genome Res 119:171–184. doi:10.1159/000112058
Basson MA et al (2006) Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Dev Biol 299:466–477. doi:10.1016/j.ydbio.2006.08.051
Basta JM, Robbins L, Kiefer SM, Dorsett D, Rauchman M (2014) Sall1 balances self-renewal and differentiation of renal progenitor cells. Development 141:1047–1058. doi:10.1242/dev.095851
Bates CM (2011) Role of fibroblast growth factor receptor signaling in kidney development. Am J Physiol Renal Physiol 301:F245–F251. doi:10.1152/ajprenal.00186.2011
Bernard P, Harley VR (2007) Wnt4 action in gonadal development and sex determination. Int J Biochem Cell Biol 39:31–43. doi:10.1016/j.biocel.2006.06.007
Bernard P, Sim H, Knower K, Vilain E, Harley V (2008) Human SRY inhibits beta-catenin-mediated transcription. Int J Biochem Cell Biol 40:2889–2900. doi:10.1016/j.biocel.2008.06.006
Berthon A, Martinez A, Bertherat J, Val P (2012) Wnt/beta-catenin signalling in adrenal physiology and tumour development. Mol Cell Endocrinol 351:87–95. doi:10.1016/j.mce.2011.09.009
Biason-Lauber A, Konrad D, Navratil F, Schoenle EJ (2004) A WNT4 mutation associated with Mullerian-duct regression and virilization in a 46,XX woman. N Engl J Med 351:792–798. doi:10.1056/NEJMoa040533
Biason-Lauber A et al (2007) WNT4 deficiency—a clinical phenotype distinct from the classic Mayer–Rokitansky–Kuster–Hauser syndrome: a case report. Hum Reprod 22:224–229. doi:10.1093/humrep/del360
Bingham C et al (2001) Mutations in the hepatocyte nuclear factor-1beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet 68:219–224. doi:10.1086/316945
Blake J, Rosenblum ND (2014) Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin Cell Dev Biol 36:2–12. doi:10.1016/j.semcdb.2014.07.011
Bonnet CS et al (2009) Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum Mol Genet 18:2166–2176. doi:10.1093/hmg/ddp149
Botto LD et al (1997) The spectrum of congenital anomalies of the VATER association: an international study. Am J Med Genet 71:8–15
Bridgewater D et al (2008) Canonical WNT/beta-catenin signaling is required for ureteric branching. Dev Biol 317:83–94. doi:10.1016/j.ydbio.2008.02.010
Brook-Carter PT et al (1994) Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease—a contiguous gene syndrome. Nat Genet 8:328–332. doi:10.1038/ng1294-328
Brosens E et al (2013) VACTERL association etiology: the impact of de novo and rare copy number variations. Mol Syndromol 4:20–26. doi:10.1159/000345577
Carroll TJ, Park JS, Hayashi S, Majumdar A, McMahon AP (2005) Wnt9b plays a central role in the regulation of mesenchymal to epithelial transitions underlying organogenesis of the mammalian urogenital system. Dev Cell 9:283–292. doi:10.1016/j.devcel.2005.05.016
Caruana G et al (2015) Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 30:487–495. doi:10.1007/s00467-014-2962-9
Chassot AA et al (2011) RSPO1/β-catenin signaling pathway regulates oogonia differentiation and entry into meiosis in the mouse fetal ovary. PLoS One 6. doi:10.1371/journal.pone.0025641
Chatterjee R et al (2012) Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum Genet 131:1725–1738. doi:10.1007/s00439-012-1181-3
Chen L, Deng CX (2005) Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci J Virtual Libr 10:1961–1976
Chen R, Amoui M, Zhang Z, Mardon G (1997) Dachshund and eyes absent proteins form a complex and function synergistically to induce ectopic eye development in Drosophila. Cell 91:893–903
Cheng HT et al (2003) Gamma-secretase activity is dispensable for mesenchyme-to-epithelium transition but required for podocyte and proximal tubule formation in developing mouse kidney. Development 130:5031–5042. doi:10.1242/dev.00697
Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C (2014) HNF1B-associated renal and extra-renal disease—an expanding clinical spectrum. Nat Rev Nephrol 11:102–112. doi:10.1038/nrneph.2014.232
Costantini F (2010) GDNF/Ret signaling and renal branching morphogenesis: from mesenchymal signals to epithelial cell behaviors. Organogenesis 6:252–262. doi:10.4161/org.6.4.12680
Costantini F, Shakya R (2006) GDNF/Ret signaling and the development of the kidney. Bioessays 28:117–127. doi:10.1002/bies.20357
Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355:1345–1356. doi:10.1056/NEJMra055323
Dibble CC et al (2012) TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell 47:535–546. doi:10.1016/j.molcel.2012.06.009
DiMario FJ, Sahin M, Ebrahimi-Fakhari D (2015) Tuberous sclerosis complex. Pediatr Clin N Am 62:633–648. doi:10.1016/j.pcl.2015.03.005
Dixon BP, Hulbert JC, Bissler JJ (2011) Tuberous sclerosis complex renal disease. Nephron Exp Nephrol 118:e15–e20. doi:10.1159/000320891
Edghill EL, Bingham C, Ellard S, Hattersley AT (2006) Mutations in hepatocyte nuclear factor-1beta and their related phenotypes. J Med Genet 43:84–90. doi:10.1136/jmg.2005.032854
Gillick J, Mooney E, Giles S, Bannigan J, Puri P (2003) Notochord anomalies in the adriamycin rat model: a morphologic and molecular basis for the VACTERL association. J Pediatr Surg 38, 469–473; discussion 469–473. doi:10.1053/jpsu.2003.50081
Gresh L et al (2004) A transcriptional network in polycystic kidney disease. EMBO J 23:1657–1668. doi:10.1038/sj.emboj.7600160
Guo Q et al (2015) Adam10 mediates the choice between principal cells and intercalated cells in the kidney. J Am Soc Nephrol JASN 26:149–159. doi:10.1681/asn.2013070764
Hains DS et al (2010) High incidence of vesicoureteral reflux in mice with Fgfr2 deletion in kidney mesenchyma. J Urol 183:2077–2084. doi:10.1016/j.juro.2009.12.095
Handrigan GR et al (2013) Deletions in 16q24.2 are associated with autism spectrum disorder, intellectual disability and congenital renal malformation. J Med Genet 50:163–173. doi:10.1136/jmedgenet-2012-101288
Harrison SJ, Nishinakamura R, Monaghan AP (2008) Sall1 regulates mitral cell development and olfactory nerve extension in the developing olfactory bulb. Cereb Cortex (New York, N.Y.: 1991) 18:1604–1617. doi:10.1093/cercor/bhm191
Hartman TR et al (2009) The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet 18:151–163. doi:10.1093/hmg/ddn325
Heidet L et al (2010) Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol CJASN 5:1079–1090. doi:10.2215/cjn.06810909
Hilger A et al (2013) De novo microduplications at 1q41, 2q37.3, and 8q24.3 in patients with VATER/VACTERL association. Eur J Hum Genet EJHG 21:1377–1382. doi:10.1038/ejhg.2013.58
Hill P, Wang B, Ruther U (2007) The molecular basis of Pallister Hall associated polydactyly. Hum Mol Genet 16:2089–2096. doi:10.1093/hmg/ddm156
Hines EA et al (2016) Syndactyly in a novel Fras1(rdf) mutant results from interruption of signals for interdigital apoptosis. Dev Dyn Off Publ Am Assoc Anat 245:497–507. doi:10.1002/dvdy.24389
Hofmann AD, Duess JW, Puri P (2014) Congenital anomalies of the kidney and urinary tract (CAKUT) associated with Hirschsprung’s disease: a systematic review. Pediatr Surg Int 30:757–761. doi:10.1007/s00383-014-3529-3
Horikawa Y et al (1997) Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet 17:384–385. doi:10.1038/ng1297-384
Hoskins BE et al (2007) Transcription factor SIX5 is mutated in patients with branchio-oto-renal syndrome. Am J Hum Genet 80:800–804. doi:10.1086/513322
Hwang DY et al (2014) Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int 85:1429–1433. doi:10.1038/ki.2013.508
Iglesias DM et al (2007) Canonical WNT signaling during kidney development. Am J Physiol Renal Physiol 293:F494–F500. doi:10.1152/ajprenal.00416.2006
Inoki K, Li Y, Zhu T, Wu J, Guan K-L (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4:648–657. doi:10.1038/ncb839
Iwasaki N et al (1998) Liver and kidney function in Japanese patients with maturity-onset diabetes of the young. Diabetes Care 21:2144–2148
Jeanpierre C et al (2011) RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J Med Genet 48:497–504. doi:10.1136/jmg.2010.088526
Kakkar N, Menon S, Radotra BD (2006) Histomorphology of renal dysplasia—an autopsy study. Fetal Pediatr Pathol 25:73–86. doi:10.1080/15513810600788764
Kamath BM, Spinner NB, Rosenblum ND (2013) Renal involvement and the role of Notch signalling in Alagille syndrome. Nat Rev Nephrol 9:409–418. doi:10.1038/nrneph.2013.102
Kamath BM et al (2012) Renal anomalies in Alagille syndrome: a disease-defining feature. Am J Med Genet A 158A:85–89. doi:10.1002/ajmg.a.34369
Kanda S et al (2014) Sall1 maintains nephron progenitors and nascent nephrons by acting as both an activator and a repressor. J Am Soc Nephrol JASN 25:2584–2595. doi:10.1681/asn.2013080896
Kiefer SM et al (2003) Expression of a truncated Sall1 transcriptional repressor is responsible for Townes–Brocks syndrome birth defects. Hum Mol Genet 12:2221–2227. doi:10.1093/hmg/ddg233
Kiefer SM et al (2010) Sall1-dependent signals affect Wnt signaling and ureter tip fate to initiate kidney development. Development 137:3099–3106. doi:10.1242/dev.037812
Kim J, Kim P, Hui CC (2001a) The VACTERL association: lessons from the Sonic hedgehog pathway. Clin Genet 59:306–315
Kim PC, Mo R, Hui CC (2001b) Murine models of VACTERL syndrome: role of sonic hedgehog signaling pathway. J Pediatr Surg 36:381–384
Kingswood JC et al (2016) Review of the tuberous sclerosis renal guidelines from the 2012 consensus conference: current data and future study. Nephron 133. doi:10.1159/000448293
Kohl S et al (2014) Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol JASN 25:1917–1922. doi:10.1681/asn.2013101103
Kohlhase J (1993) In: Pagon RA et al (eds) GeneReviews(R). University of Washington, Seattle University of Washington, Seattle
Kolatsi-Joannou M et al (2001) Hepatocyte nuclear factor-1beta: a new kindred with renal cysts and diabetes and gene expression in normal human development. J Am Soc Nephrol JASN 12:2175–2180
Kopan R (2012) Notch signaling. Cold Spring Harb Perspect Biol 4:a011213. doi:10.1101/cshperspect.a011213
Krebs LT et al (2000) Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14:1343–1352
Krueger DA et al (2010) Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363:1801–1811. doi:10.1056/NEJMoa1001671
Lahiri D et al (2007) Nephropathy and defective spermatogenesis in mice transgenic for a single isoform of the Wilms’ tumour suppressor protein, WT1-KTS, together with one disrupted Wt1 allele. Mol Reprod Dev 74:300–311. doi:10.1002/mrd.20491
Landgraf K et al (2010) Sipl1 and Rbck1 are novel Eya1-binding proteins with a role in craniofacial development. Mol Cell Biol 30:5764–5775. doi:10.1128/mcb.01645-09
Lefebvre J et al (2015) Alternatively spliced isoforms of WT1 control podocyte-specific gene expression. Kidney Int 88:321–331. doi:10.1038/ki.2015.140
Li Y, Manaligod J, Weeks D (2010) EYA1 mutations associated with the branchio-oto-renal syndrome result in defective otic development in Xenopus laevis. Biol Cell 102:277–292. doi:10.1042/bc20090098
Lokmane L, Heliot C, Garcia-Villalba P, Fabre M, Cereghini S (2010) vHNF1 functions in distinct regulatory circuits to control ureteric bud branching and early nephrogenesis. Development 137:347–357. doi:10.1242/dev.042226
Lopez-Rios J et al (2012) GLI3 constrains digit number by controlling both progenitor proliferation and BMP-dependent exit to chondrogenesis. Dev Cell 22:837–848. doi:10.1016/j.devcel.2012.01.006
Lubinsky M (2015) The VACTERL Association as a disturbance of cell fate determination. Am J Med Genet Part A 167a:2582–2588. doi:10.1002/ajmg.a.37238
Madariaga L et al (2013) Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol CJASN 8:1179–1187. doi:10.2215/cjn.10221012
Mandel H et al (2008) SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet 82:39–47. doi:10.1016/j.ajhg.2007.08.005
Mao Z, Chong J, Ong ACM (2016) Autosomal dominant polycystic kidney disease: recent advances in clinical management. F1000Research 5. doi:10.12688/f1000research.9045.1
Marie PJ, Coffin JD, Hurley MM (2005) FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J Cell Biochem 96:888–896. doi:10.1002/jcb.20582
Martucciello G, Ceccherini I, Lerone M, Jasonni V (2000) Special basic science review: pathogenesis of Hirschsprung’s disease. J Pediatr Surg 35:1017–1025. doi:10.1053/jpsu.2000.7763
Materna-Kiryluk A et al (2014) The emerging role of genomics in the diagnosis and workup of congenital urinary tract defects: a novel deletion syndrome on chromosome 3q13.31-22.1. Pediatr Nephrol 29:257–267. doi:10.1007/s00467-013-2625-2
McCright B (2003) Notch signaling in kidney development. Curr Opin Nephrol Hypertens 12:5–10. doi:10.1097/01.mnh.0000049802.69874.c0
McCright B et al (2001) Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development 128:491–502
Mendel DB, Hansen LP, Graves MK, Conley PB, Crabtree GR (1991) HNF-1 alpha and HNF-1 beta (vHNF-1) share dimerization and homeo domains, but not activation domains, and form heterodimers in vitro. Genes Dev 5:1042–1056
Michos O et al (2010) Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS Genet 6:e1000809. doi:10.1371/journal.pgen.1000809
Morita Y et al (2016) Sall1 transiently marks undifferentiated heart precursors and regulates their fate. J Mol Cell Cardiol 92:158–162. doi:10.1016/j.yjmcc.2016.02.008
Morrison AA, Viney RL, Saleem MA, Ladomery MR (2008) New insights into the function of the Wilms tumor suppressor gene WT1 in podocytes. Am J Physiol Renal Physiol 295:F12–F17. doi:10.1152/ajprenal.00597.2007
Munger SC, Natarajan A, Looger LL, Ohler U, Capel B (2013) Fine time course expression analysis identifies cascades of activation and repression and maps a putative regulator of mammalian sex determination. PLoS Genet 9. doi:10.1371/journal.pgen.1003630
North American Pediatric Renal Trials and Collaborative Studies (2011) NAPRTCS 2011 annual dialysis report. The National Institute of Diabetes and Digestive and Kidney Diseases, pp 1–26
Natarajan D, Marcos-Gutierrez C, Pachnis V, de Graaff E (2002) Requirement of signalling by receptor tyrosine kinase RET for the directed migration of enteric nervous system progenitor cells during mammalian embryogenesis. Development 129:5151–5160
Ngan ES, Kim KH, Hui CC (2013) Sonic hedgehog signaling and VACTERL association. Mol Syndromol 4:32–45. doi:10.1159/000345725
Nishinakamura R et al (2001) Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development 128:3105–3115
Nishita M et al (2014) Role of Wnt5a-Ror2 signaling in morphogenesis of the metanephric mesenchyme during ureteric budding. Mol Cell Biol 34:3096–3105. doi:10.1128/mcb.00491-14
O’Brien LL et al (2016) Differential regulation of mouse and human nephron progenitors by the Six family of transcriptional regulators. Development 143:595–608. doi:10.1242/dev.127175
Park JS et al (2012) Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev Cell 23:637–651. doi:10.1016/j.devcel.2012.07.008
Passos-Bueno MR et al (1999) Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat 14:115–125. doi:10.1002/(sici)1098-1004(1999)14:2<115::aid-humu3>3.0.co;2-2
Patek CE et al (2003) Murine Denys–Drash syndrome: evidence of podocyte de-differentiation and systemic mediation of glomerulosclerosis. Hum Mol Genet 12:2379–2394. doi:10.1093/hmg/ddg240
Pavlakis E, Chiotaki R, Chalepakis G (2011) The role of Fras1/Frem proteins in the structure and function of basement membrane. Int J Biochem Cell Biol 43:487–495. doi:10.1016/j.biocel.2010.12.016
Pema M et al (2016) mTORC1-mediated inhibition of polycystin-1 expression drives renal cyst formation in tuberous sclerosis complex. Nat Commun 7:10786. doi:10.1038/ncomms10786
Pignoni F et al (1997) The eye-specification proteins So and Eya form a complex and regulate multiple steps in Drosophila eye development. Cell 91:881–891
Pini Prato A et al (2009) Hirschsprung disease and congenital anomalies of the kidney and urinary tract (CAKUT): a novel syndromic association. Medicine 88:83–90. doi:10.1097/MD.0b013e31819cf5da
Pitera JE, Scambler PJ, Woolf AS (2008) Fras1, a basement membrane-associated protein mutated in Fraser syndrome, mediates both the initiation of the mammalian kidney and the integrity of renal glomeruli. Hum Mol Genet 17:3953–3964. doi:10.1093/hmg/ddn297
Pitera JE, Woolf AS, Basson MA, Scambler PJ (2012) Sprouty1 haploinsufficiency prevents renal agenesis in a model of Fraser syndrome. J Am Soc Nephrol JASN 23:1790–1796. doi:10.1681/asn.2012020146
Plank TL, Yeung RS, Henske EP (1998) Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res 58:4766–4770
Poladia DP et al (2006) Role of fibroblast growth factor receptors 1 and 2 in the metanephric mesenchyme. Dev Biol 291:325–339. doi:10.1016/j.ydbio.2005.12.034
Rakowski SK et al (2006) Renal manifestations of tuberous sclerosis complex: incidence, prognosis, and predictive factors. Kidney Int 70:1777–1782. doi:10.1038/sj.ki.5001853
Reidy KJ, Rosenblum ND (2009) Cell and molecular biology of kidney development. Semin Nephrol 29:321–337. doi:10.1016/j.semnephrol.2009.03.009
Riccio P, Cebrian C, Zong H, Hippenmeyer S, Costantini F (2016) Ret and Etv4 promote directed movements of progenitor cells during renal branching morphogenesis. PLoS Biol 14:e1002382. doi:10.1371/journal.pbio.1002382
Ruf RG et al (2004) SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1–SIX1–DNA complexes. Proc Natl Acad Sci USA 101:8090–8095. doi:10.1073/pnas.0308475101
Sadowski CE et al (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol JASN 26:1279–1289. doi:10.1681/asn.2014050489
Saisawat P et al (2014) Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int 85:1310–1317. doi:10.1038/ki.2013.417
Sajithlal G, Zou D, Silvius D, Xu PX (2005) Eya 1 acts as a critical regulator for specifying the metanephric mesenchyme. Dev Biol 284:323–336. doi:10.1016/j.ydbio.2005.05.029
Sanna-Cherchi S et al (2012) Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91:987–997. doi:10.1016/j.ajhg.2012.10.007
Schneider J, Arraf AA, Grinstein M, Yelin R, Schultheiss TM (2015) Wnt signaling orients the proximal-distal axis of chick kidney nephrons. Development 142:2686–2695. doi:10.1242/dev.123968
Schuchardt A, D’Agati V, Pachnis V, Costantini F (1996) Renal agenesis and hypodysplasia in ret-k-mutant mice result from defects in ureteric bud development. Development 122:1919–1929
Seyedzadeh A, Kompani F, Esmailie E, Samadzadeh S, Farshchi B (2008) High-grade vesicoureteral reflux in Pfeiffer syndrome. Urol J 5:200–202
Shakya R, Watanabe T, Costantini F (2005) The role of GDNF/Ret signaling in ureteric bud cell fate and branching morphogenesis. Dev Cell 8:65–74. doi:10.1016/j.devcel.2004.11.008
Shillingford JM et al (2006) The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA 103:5466–5471. doi:10.1073/pnas.0509694103
Siroky BJ, Yin H, Bissler JJ (2011) Clinical and molecular insights into tuberous sclerosis complex renal disease. Pediatr Nephrol 26:839–852. doi:10.1007/s00467-010-1689-5
Si-Tayeb K, Lemaigre FP, Duncan SA (2010) Organogenesis and development of the liver. Dev Cell 18:175–189. doi:10.1016/j.devcel.2010.01.011
Song R, Yosypiv IV (2011) Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 26:353–364. doi:10.1007/s00467-010-1629-4
Stark K, Vainio S, Vassileva G, McMahon AP (1994) Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372:679–683. doi:10.1038/372679a0
Stoll C, Dott B, Alembik Y, Roth MP (2014) Associated nonurinary congenital anomalies among infants with congenital anomalies of kidney and urinary tract (CAKUT). Eur J Med Genet 57:322–328. doi:10.1016/j.ejmg.2014.04.014
Stoll C, Dott B, Alembik Y, Roth MP (2015) Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet 58:674–680. doi:10.1016/j.ejmg.2015.11.003
Su N, Jin M, Chen L (2014) Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res 2:14003. doi:10.1038/boneres.2014.3
Tadjuidje E, Hegde RS (2013) The eyes absent proteins in development and disease. Cell Mol Life Sci 70:1897–1913. doi:10.1007/s00018-012-1144-9
Taraviras S et al (1999) Signalling by the RET receptor tyrosine kinase and its role in the development of the mammalian enteric nervous system. Development 126:2785–2797
Tee AR et al (2002) Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA 99:13571–13576. doi:10.1073/pnas.202476899
Thomas R et al (2011) HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 26:897–903. doi:10.1007/s00467-011-1826-9
Uyttendaele H, Ho J, Rossant J, Kitajewski J (2001) Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc Natl Acad Sci USA 98:5643–5648. doi:10.1073/pnas.091584598
Vainio SJ (2003) Nephrogenesis regulated by Wnt signaling. J Nephrol 16:279–285
van Slegtenhorst M et al (1998) Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 7:1053–1057
Verbitsky M et al (2015) Genomic imbalances in pediatric patients with chronic kidney disease. J Clin Invest 125:2171–2178. doi:10.1172/jci80877
Vivante A et al (2013) Renal hypodysplasia associates with a WNT4 variant that causes aberrant canonical WNT signaling. J Am Soc Nephrol JASN 24:550–558. doi:10.1681/asn.2012010097
Volckaert T, De Langhe SP (2015) Wnt and FGF mediated epithelial-mesenchymal crosstalk during lung development. Dev Dyn Off Publ Am Assoc Anat 244:342–366. doi:10.1002/dvdy.24234
Warthen DM et al (2006) Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Hum Mutat 27:436–443. doi:10.1002/humu.20310
Weber S et al (2006) Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol JASN 17:2864–2870. doi:10.1681/asn.2006030277
Weber S et al (2008) SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol JASN 19:891–903. doi:10.1681/asn.2006111282
Xi Q et al (2016) Copy number variations in multicystic dysplastic kidney: update for prenatal diagnosis and genetic counseling. Prenat Diagn. doi:10.1002/pd.4807
Xu PX, Cheng J, Epstein JA, Maas RL (1997) Mouse Eya genes are expressed during limb tendon development and encode a transcriptional activation function. Proc Natl Acad Sci USA 94:11974–11979
Xu PX et al (1999) Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet 23:113–117. doi:10.1038/12722
Xu J et al (2014) Eya1 interacts with Six2 and Myc to regulate expansion of the nephron progenitor pool during nephrogenesis. Dev Cell 31:434–447. doi:10.1016/j.devcel.2014.10.015
Xue Y et al (1999) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8:723–730
Yoder BK, Hou X, Guay-Woodford LM (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol JASN 13:2508–2516
Zeidler C et al (2014) Heterozygous FGF8 mutations in patients presenting cryptorchidism and multiple VATER/VACTERL features without limb anomalies. Birth Defects Res A Clin Mol Teratol 100:750–759. doi:10.1002/bdra.23278
Zhao H et al (2004) Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol 276:403–415. doi:10.1016/j.ydbio.2004.09.002
Zou D, Silvius D, Rodrigo-Blomqvist S, Enerbäck S, Xu PX (2006) Eya1 regulates the growth of otic epithelium and interacts with Pax2 during the development of all sensory areas in the inner ear. Dev Biol 298. doi:10.1016/j.ydbio.2006.06.049
Zubkov VS et al (2015) A spatially-averaged mathematical model of kidney branching morphogenesis. J Theor Biol 379:24–37. doi:10.1016/j.jtbi.2015.04.015
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Pal, A., Reidy, K.J. (2017). Genetic Syndromes Affecting Kidney Development. In: Miller, R. (eds) Kidney Development and Disease. Results and Problems in Cell Differentiation, vol 60. Springer, Cham. https://doi.org/10.1007/978-3-319-51436-9_10
Download citation
DOI: https://doi.org/10.1007/978-3-319-51436-9_10
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-51435-2
Online ISBN: 978-3-319-51436-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)