
Abstract
Background
Copy number variants (CNVs) are increasingly recognized as an important cause of congenital malformations and likely explain over 16 % of cases of congenital anomalies of the kidney and urinary tract (CAKUT). Here, we illustrate how a molecular diagnosis of CNV can be beneficial to the clinical management of a pediatric patient presenting with CAKUT and other organ defects.
Methods
We describe a 14-year-old girl with a large de novo deletion of chromosome 3q13.31-22.1 that disrupts 101 known genes. The patient presented with CAKUT, neurodevelopmental delay, agenesis of corpus callosum (ACC), cardiac malformations, electrolyte and endocrine disorders, skeletal abnormalities and dysmorphic features. We performed extensive annotation of the deleted region to prioritize genes for specific phenotypes and to predict future disease risk.
Results
Our case defined new minimal chromosomal candidate regions for both CAKUT and ACC. The presence of the CASR gene in the deleted interval predicted a diagnosis of hypocalciuric hypercalcemia, which was confirmed by the serum and urine chemistries. Our gene annotation explained clinical hypothyroidism and predicted that the index case is at increased risk of thoracic aortic aneurysm, renal cell carcinoma and myeloproliferative disorder.
Conclusions
Extended annotation of CNV regions refines the diagnosis and uncovers previously unrecognized phenotypic features. This approach enables personalized treatment and prevention strategies in patients harboring genomic deletions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Structural genomic defects have been increasingly recognized as important causes of syndromic and isolated congenital malformations. However, because of limited resolution of standard karyotyping, large chromosomal aberrations are identified only in a small fraction of cases using current cytogenetic tools. Submicroscopic genomic structural defects that escape standard cytogenetic screening are likely to explain a substantial portion of congenital malformations [1, 2]. Such defects can be either inherited (in familial disease) or occur de novo (in sporadic disease).
In recent years, substantial technological progress has allowed for improved detection of small genomic rearrangements. Two commonly used methods include array-based comparative genomic hybridization (aCGH) and high-density single nucleotide polymorphism (SNP) arrays. Recent studies have demonstrated the ability of these methods to detect pathogenic copy number defects in a large percentage of patients with neurodevelopmental phenotypes [2, 3], schizophrenia [4, 5], autism [6], cardiac malformations [7, 8] and short stature [9, 10]. In one of the largest studies to date, diverse pathogenic copy number variants (CNVs) accounted for 14.2 % of cases among 15,767 children with intellectual disability and variable congenital defects [3]. A number of criteria are commonly used to establish the pathogenicity of CNV events, including (1) large CNV size, (2) disruption of coding gene sequence, (3) inheritance pattern (e.g. CNVs co-segregating with a phenotype in familial cases or occurring de novo in sporadic cases) and (4) absence or extremely low frequency in the general population [11]. Despite these criteria, however, some pathogenic CNVs may have incomplete penetrance and, consequently, their clinical interpretation has to be carefully considered on an individual basis.
We recently studied a large cohort of patients with congenital renal defects (renal hypodysplasia and agenesis) and found that over 16.6 % of such defects could be explained by pathogenic submicroscopic deletions or duplications [1]. The majority of these genomic variants were previously associated with variably penetrant neurodevelopmental disorders, extra-renal malformations or systemic disorders. These data indicated that disruption of genes within the CNV intervals could also explain extra-renal manifestations and that early molecular diagnosis could positively impact the clinical care of patients. For example, congenital anomalies of the kidney and urinary tract (CAKUT) patients with cystic renal dysplasia who carry a molecular diagnosis of deletions at the HNF1B locus are also at risk for developing diabetes mellitus, hyperuricemia, genital defects and infertility [12, 13]. Thus, early detection of this molecular defect is highly relevant to an individual’s reproductive health and may also alert clinicians to frequent screening for glucose intolerance, thus facilitating early detection, treatment and prevention of long-term diabetic complications.
Here, we provide detailed data on the clinical and molecular characterization of a sporadic case carrying a new de novo 15-Mb deletion of chromosome 3q13.31-22.1 that was initially missed by standard cytogenetic tools. The patient presented for re-evaluation at the age of 14 years with dysmorphic features and multiple malformations of internal organs, including right renal agenesis and grade IV left vesicoureteral reflux (VUR), at which time the CNV analysis was performed. With the results in hand, we re-evaluated the patient in the context of her genetic diagnosis.
Case
A 14-year-old Polish girl with dysmorphic features was referred to us for clinical evaluation. The proband was the first child born to healthy, non-consanguineous parents. At the time of conception, the mother was 22 and the father was 25 years old. The pregnancy was complicated by placental abruption requiring a C-section in the 35th week of pregnancy. Asphyxia was present at birth with the Apgar scores being 0, 0 and 4 at 1, 5, and 10 min after birth, respectively. Regular strong heartbeat was observed at 10 min and regular spontaneous breathing at 50 min after birth. The karyotype was 46XX, and no major chromosomal aberrations were detected by initial cytogenetic evaluation.
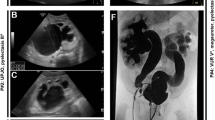
At the age of 14 years the proband was seen in the clinical genetics clinic at the Center for Medical Genetics (GENESIS, Poznan, Poland). At this time, multiple dysmorphic features were clearly noticeable (Fig. 1). Skeletal abnormalities included short stature (height 133 cm, weight 23 kg), scoliosis, contraction of the left forearm and left hand, phalangeal hypoplasia, hypoplastic thumbs, absent radial bones, left clubfoot varus and syndactyly (2nd and 3rd toes). Facial dysmorphic features included macrostomia (repaired surgically at the age of 15 months), narrow palpebral fissures and right auricular tags. Neurological evaluation revealed severe neurodevelopmental delay. A generalized hypotonia was present since birth. The patient had a reported history of epilepsy characterized by involuntary ocular movements for which she was maintained on antiepileptic medications. Brain imaging documented cerebral atrophy with agenesis of corpus callosum (ACC) (Fig. 2). In addition, the patient had a history of mild myopia, but fundoscopic exam revealed no major retinal abnormalities. Her hearing was intact.

Dysmorphic features of the proband. a Blepharophimosis, b pre-auricular tags, c macrostomia, d left foot syndactyly involving second and third toe, e left upper extremity contracture with underdeveloped phalanges and vestigial left thumb

Magnetic resonance image of the head (sagittal view) demonstrates generalized cortical atrophy and agenesis of corpus callosum
The patient also had several malformations of internal organs. Early echocardiographic evaluation revealed patent ductus arteriosus with aortic coarctation (repaired surgically at the age of 7 weeks), a small ventricular septal defect, a patent foramen ovale and an asymmetric bicuspid aortic valve. The urogenital abnormalities included complete right renal agenesis, grade IV left VUR, left-sided hydronephrosis and a prominent clitoris. A successful surgical anti-reflux procedure was performed at the age of 2 years, with follow-up voiding cystourethrogram showing only mild (grade I) residual left-sided VUR. The parameters of renal function (blood urea nitrogen and creatinine) remained within normal limits for her age, and there was no evidence of proteinuria on serial urinalyses after the procedure. Additionally the patient had left thyroid lobe hypoplasia and associated hypothyroidism. This diagnosis was established at the age of 16 months: thyroid-stimulating hormone (TSH) level was 4.2 mU/l (normal 0.4–4.0 mU/l), FT4 level was 8.1 pmol/l (normal 8.5–19.0 pmol/l), and thyroid ultrasound revealed a small hypoechoic thyroid gland with severe left lobe hypoplasia. At this time, the patient was initiated on thyroid hormone replacement therapy.
Notably, the family history was negative for neurodevelopmental disorders, hypothyroidism, heart disease or kidney failure. There was no maternal history of pre-term deliveries, miscarriages or abortions.
Results
Detection of the novel 3q13.31-22.1 deletion
Taking the extensive multi-organ involvement into consideration, we hypothesized that a submicroscopic CNV disrupting multiple genes may be responsible for the severe phenotypic features observed in this case. As outlined in our recent paper [1], we have developed a stringent analytic pipeline for identification of pathogenic CNVs in individuals with severe congenital malformations. Briefly, the first step of the protocol is a genome-wide analysis of CNVs using the 610-Quad SNP array (Illumina, San Diego, CA). The CNVs are identified using generalized genotyping methods, as implemented in the PennCNV software [14] and then mapped to the human reference genome hg18. In the case of our 14-year-old patient, we identified a large deletion of 14,960,554 bp involving chromosome 3q13.31-22.1 (Fig. 3). The breakpoints of the deletion were from rs11921574 (chr3:118,151,853 bp) to rs957919 (chr3:133,112,406 bp) and spanned over 3,088 probes on the Illumina chip. This deletion was completely novel and was absent in public databases and in our panel of 13,839 population controls genotyped with SNP chips of comparable density (Illumina’s 550 K or higher).

Detailed view of the 3q13.31-22.1 deletion region: top panel absence of heterozygous calls within the 3q13.31-22.1 locus, middle panel decreased probe intensity (Log R Ratio) in the same region, bottom panel known annotated genes within the deleted interval (NCBI v.37.2)
Deletion confirmation and annotation of the region
The deletion was confirmed by quantitative real time-PCR and SNP genotyping via traditional Sanger sequencing in the index case and the unaffected parents. The CNV was absent in both parents, indicating a de novo event. Additional genotyping of highly informative SNPs (heterozygosity approx. 0.5) in the region confirmed that the index case carried a maternal haplotype in homozygosity, thereby also confirming that the deletion occurred de novo in the paternal chromosome. We next performed comprehensive annotation of this region using public databases, including the National Center for Biotechnolgy Information (NCBI) Genome Browser (URL: http://www.ncbi.nlm.nih.gov/), Genome Reviews (URL: http://www.ebi.ac.uk/GenomeReviews/), Online Mendelian Inheritance in Man (OMIM; URL: http://www.omim.org/) and PubMed (www.ncbi.nlm.nih.gov/pubmed/). According to the NCBI Genome Browser (build 37.2), the region of the 3q13.31-22.1 deletion contains 101 known genes (Fig. 3). We cross-referenced these transcripts against the OMIM database and identified 23 annotated genes previously associated with a disease phenotype in humans (Table 1). Using the probabilistic model that integrates genomic, evolutionary, functional and network properties of haploinsufficient genes [15], we predicted that the deletion is highly deleterious with a haploinsufficiency LOD score of 49.2. We next compared the deleted region against the ECARUCA (European Cytogeneticists Association Register of Unbalanced Chromosomal Aberrations; URL: http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp) and DECIPHER (URL: http://decipher.sanger.ac.uk/) databases of rare cytogenetic abnormalities. All previously reported deletions overlapping with the 3q13.31-22.1 region are summarized in Table 2. For each of the observed phenotypes in our case, we performed detailed analysis of candidate genes. Moreover, considering the frequent occurrence of CAKUT and ACC among these cases, we performed detailed mapping of partially overlapping deletions in patients with these phenotypes (Fig. 4). The analysis was performed under the assumption of incomplete penetrance, thus only the cases with reported CAKUT or ACC phenotypes were included in the mapping.

The reported deletions in all cases with agenesis of corpus callosum (ACC, middle panel) and congenital anomalies of the kidney and urinary tract (CAKUT, bottom panel) and their relationship to the reference map of the chromosomal region 3q12.1-22.3 (top panel). Our case defines new minimal candidate regions for ACC (chr3q13.31-13.33: 118,152–123,400 kb) and CAKUT (chr3q13.31-21.3: 118,152–128,178 kb). A complete list of candidate genes in these minimal candidate regions is provided as Electronic Supplementary Material Dataset 1
Neurodevelopmental delay and ACC
Intellectual disability and neurodevelopmental delay are frequently observed in individuals harboring a variety of large genomic rearrangements, suggesting that the appropriate dosage of many different genes is critical for proper development of the central nervous system. Accordingly, neurodevelopmental delay was present in virtually all of previously reported patients carrying 3q13.31-22.1 deletions (Table 1). However, six patients (including the present case) had a specific rare finding of ACC on brain imaging (Fig. 2) [16–23]. Detailed analysis of deletion breakpoints revealed that our case effectively narrows down the minimal region of overlap for ACC to bands 3q13.32-13.33 (chr3:118,152–123,400 kb; Fig. 4). This chromosomal interval contains a total of only 34 NCBI reference genes and has a predicted haploinsufficiency LOD of 12.6. Ranking these genes based on their individual haploinsufficiency prediction scores defined GSK3B (OMIM 605004) as the top candidate gene, with 97.5 % estimated probability of having serious phenotypic manifestations [15]. The protein encoded by this gene is a serine-threonine kinase involved in neuronal cell development [24] and body pattern formation [25]. Notably, a recent aCGH study found two independent overlapping duplications of 3q13.33 as a cause of holoprosencephaly, one of the most common forms of forebrain malformations in humans [26]. While the first duplication spanned 3.8 Mb (3q13.32q21.1) and was associated with a severe alobar holoprosencephaly, the second variant was smaller (267 kb, 3q13.33) and disrupted only one gene—this gene encompassed ten of 12 exons of GSK3B and manifested with microform holoprosencephaly, including partial ACC. Therefore, our results provide further support for GSK3B involvement in the development of the central nervous system and suggest that biallelic dosage of this specific gene is critical for proper formation of the corpus callosum.
Kidney and urinary tract malformations
Among all of the deletions of 3q13.31-22.1 reported to date, six cases had a diagnosis of CAKUT. Among these cases, kidney phenotypes were highly variable, ranging from unilateral duplicated collecting system [17], unilateral hydronephrosis [20] and bilateral hydroureters with stenosis of the urethral valve [19], to grade IV left VUR with contralateral agenesis (present case). In addition to phenotypic heterogeneity, not all patients carrying the deletion of 3q13.31-21.3 had CAKUT, implying incomplete penetrance, although it is not clear if adequate screening was performed in all of the cases reported. These observations, however, are consistent with the fact that pleiotropy and incomplete penetrance are common features of the known CAKUT mutations. For example, mutations in PAX2 cause the renal-coloboma syndrome, but kidney defects vary significantly between affected individuals, ranging from relatively mild (e.g. VUR or secondary obstruction) to extremely severe (e.g. bilateral renal agenesis) [27].
As evident in Fig. 4, our case effectively defined a new minimal candidate gene region for CAKUT (3q13.31-21.3: 118,152–128,178 kb). This region contains 78 annotated transcripts and has a predicted haploinsufficiency LOD score of 25.7 (Electronic Supplementary Material Data 1). Two OMIM-annotated genes within this interval have previously been associated with a developmental renal phenotype: UPK1B (3q13.32) and IQCB1 (3q13.33). The UPK1B gene encodes uroplakin 1B, a protein strongly expressed in normal uroepithelium. UPK1B is a binding partner for UPK3, forming the UPK1B–UPK3 complex that is inserted into the apical membrane of urothelial cells and necessary for their proper differentiation [28]. Upk3-deficient mice develop VUR with evidence of overexpression and abnormal targeting of Upk1b [29]. Moreover, de novo heterozygous and potentially deleterious single nucleotide variants in UPK3A have been described in four cases of VUR and renal dysplasia [30]. However, no deleterious mutations in human UPK1B have been reported to date. The second candidate gene, IQCB1 (NPHP5), encodes a ciliary protein. Homozygous or compound heterozygous mutations in IQCB1 are responsible for Senior–Loken syndrome, a rare recessive disorder characterized by kidney disease (nephronophthisis) and retinitis pigmentosa [31]. It is not clear if haploinsufficiency for IQCB1 contributes to the observed renal defects, or if the deletion unmasks the effect of a deleterious allele in trans, or modifies the effect of other risk alleles carried by the patient. Notably, our case is also haploinsufficient for IFT122, another ciliary protein residing on 3q21. Recessive mutations in IFT122 cause cranioectodermal dysplasia-1 (Sensenbrenner syndrome) which usually involves renal failure in addition to short stature, limb defects and multiple craniofacial abnormalities [32]. Although this gene resides outside of the minimal candidate region for CAKUT, combined haploinsufficiency of both ciliary genes might modify the severity of the observed kidney defects in our case.
Skeletal and cardiac defects
Similar to CAKUT discussed above, IFT122 haploinsufficiency may act as a modifier of some of the skeletal phenotypes, such as the short stature or distal limb malformations observed in our patient. Moreover, our patient is haploinsufficient for ARHGAP31, another gene implicated in skeletal development. ARGHAP31 encodes a GTPase-activating protein that regulates two GTPases involved in protein trafficking and cell growth, Cdc42 and Rac1. It has been shown that rare gain-of-function mutations in ARHGAP31 cause an autosomal dominant form of Adams–Oliver syndrome, a disorder characterized by skin aplasia and terminal transverse limb defects (short or absent distal phalanges with or without syndactyly) [33]. Interestingly, in mice, the expression of ARHGAP31 is strongest in the developing heart, and while there have been reports of cardiac defects in patients with other forms of Adams–Oliver syndrome, individuals harboring the gain-of-function mutations in ARHGAP31 had no cardiac involvement. However, there are no human loss-of-function mutations in this gene reported to date. Therefore, it is still possible that lower dosage of this gene contributes to the cardiac and/or skeletal defects observed in our case. Based on a literature search, we identified no other obvious known candidates for cardiac or skeletal defects in the region.
Thyroid hypoplasia and clinical hypothyroidism
Based on our gene annotations, we speculate that thyroid hypoplasia with hypothyroidism is likely due to haploinsufficiency of TRH (gene for thyrotropin-releasing hormone). The TRH gene is located in the distal portion of the 3q13.31-22.1, specifically within the band 3q22.1. Unfortunately, thyroid tests are not consistently reported for all deletion cases summarized in Table 2. Okada et al. reported a deletion of chromosome 3q12-3q21 in a patient with clinical features similar to those of our patient, including severe psychomotor retardation, progressive scoliosis, multiple joint contractures and CAKUT [17]. However, the thyroid function tests were normal in the patient described by these authors [17] , suggesting that the locus responsible for hypothyroidism resides distal to 3q21.
TRH is a hypothalamic peptide responsible for the regulation and release of TSH, a critical regulator of thyroid gland function. Congenital deficiency of TRH and loss-of-function mutations in the TRH receptor gene have previously been described as a very rare cause of central hypothyroidism [34, 35]. In central hypothyroidism, the serum free T4 level is low, but serum TSH level may vary. Normal or high serum TSH concentrations in some cases of central hypothyroidism are due, in part, to secretion of TSH that has reduced biologic activity but normal immunoreactivity [36]. Reduced bioactivity is due to abnormalities in glycosylation of the TSH subunits, which is under the control of TRH [37, 38]. A prolonged period of decreased activation of thyroid cells by TSH may result in a hypoplastic thyroid and clinical hypothyroidism, as observed in our patient.
Previously unrecognized phenotypes and disease predictions
Strikingly, the 3q13.31-22.1 deletion also encompasses the CASR gene, encoding a calcium-sensing receptor. This receptor is expressed in the chief cells of the parathyroid gland and is critical for sensing changes in serum calcium concentration and initiation of signaling pathways that modify PTH secretion. Additionally, this receptor is also expressed in regions of the kidney involved in Ca2+-regulated Ca2+ and Mg2+ reabsorption. Gain-of-function CASR mutations cause familial isolated hypoparathyroidism with hypocalcemia [39], while loss-of-function mutations cause familial hypocalciuric hypercalcemia (FHH) [40]. Importantly, the dosage of the CASR gene correlates with phenotype severity. Individuals with a single defective allele have relatively mild symptoms of FHH, while individuals carrying two deleterious alleles develop severe neonatal hyperparathyroidism [41]. Consequently, we predicted that the index case carried the diagnosis of hypocalciuric hypercalcemia. Upon subsequent review of the medical records, we noted that indeed our patient had hypercalcemia and hypermagnesemia of unclear etiology on several occasions, including serum Ca2+ levels of 12.6, 10.6, and 10.8 mg/dl (normal 8.5–10.5 mg/dl) with corresponding Mg2+ levels of 2.3, 2.6, and 2.2 mg/dl (normal 1.3–2.2 mg/dl) at the ages of 1, 8 and 12 years, respectively. As often observed in familial hypocalciuric hypercalcemia, the parathyroid hormone levels were within normal limits. Similarly, vitamin D levels were normal. Taking these findings into consideration, we performed a 24-h collection of urine and confirmed a low daily calcium clearance of 119.3 mg/24 h (normal <200 mg/24 h). Since the normal clearance threshold of 200 mg per day was established based on studies of adult subjects, we also estimated weight-normalized calcium clearance at 3.7 mg/kg/day, which was inappropriately low for a hypercalcemic patient (serum Ca2+ was 10.9 mg/dl at the time of urine collection). Finally, the calcium fractional excretion was calculated at 0.009, further supporting the diagnosis of hypocalciuric hypercalcemia.
We also noted that our patient is haploinsufficient for ADCY5, a gene encoding a membrane-bound adenylyl cyclase enzyme that mediates G protein-coupled receptor signaling. It has recently been shown that heterozygous loss-of-function mutations in this gene cause autosomal dominant familial dyskinesia with facial myokymia [42]. Individuals affected by this disorder exhibit adventitious movements associated with perioral and periorbital myokymia (brief episodes of repeated facial muscle contractions that can sometimes also involve ocular muscles). In our patient, there is a possibility that ocular myokymia was misdiagnosed as focal epilepsy early in childhood, thus the patient would benefit from additional neurological assessment and reevaluation of her antiepileptic regimen.
Finally, by identifying literature reports of disease-causing heterozygous loss-of-function mutations in the genes contained within the 3q13.31-22.1 interval, we predict that our patient may be at an increased risk of developing a thoracic aortic aneurysm and dissection (haploinsufficiency of MYLK [43]), renal cell carcinoma (DIRC2 [44]) and myelodysplasia or acute myeloid leukemia later in life (GATA2 [45, 46]). Notably, MYLK is listed as one of the 37 genes recently recommended by the American College of Medical Genetics (ACMG) for reporting to patients when confirmed to harbor pathogenic mutations detected as incidental findings in genetic studies (URL: http://www.acmg.net). Personalized screening protocols for aneurysms and malignancy should therefore be considered in this highly unusual case.
Conclusion
We report a novel 15-Mb deletion of chromosome 3q13.31-22.1 that disrupts over 101 known genes and gives rise to a variety of phenotypic features, including developmental delay, ACC, dysmorphic facial features, limb abnormalities, hypothyroidism and malformations of several internal organs, including heart and kidneys. There are several possible mechanisms through which a deletion can manifest phenotypically, including a reduced gene dosage effect of a single gene or a contiguous set of genes (a haploinsufficiency effect), unmasking of a deleterious allele(s) on the non-deleted chromosome or epistasis with deleterious allele(s) outside of the deleted locus. Some of the phenotypic features observed in our patient can be clearly explained by the gene dosage effect of individual genes within this interval, while others likely involve more complex mechanisms.
We demonstrate that genotype-driven phenotyping combined with an exhaustive analysis of positional candidate genes within the regions of structural abnormality may provide novel insights into gene function and disease pathogenesis. Our case illustrates the great utility of publicly available genomic databases in facilitating diagnoses and in establishing connections between disrupted genes and specific phenotypic features. Notably, the rate of discoveries in the field of Mendelian genetics has been greatly accelerated by the use of whole exome and genome sequencing in gene mapping studies. This technology continues to uncover new genotype–phenotype connections and, as the databases of loss-of-function phenotypes become more complete across the genome, our ability to interpret the consequences of large genomic deletions will improve. Importantly, through this case we demonstrate that detailed functional annotation of the deleted genes can uncover previously unrecognized or missed phenotypes, guide medical therapies and help to design personalized preventive approaches.
References
Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, Nees SN, Verbitsky M, Perry BJ, Sterken R, Lozanovski VJ, Materna-Kiryluk A, Barlassina C, Kini A, Corbani V, Carrea A, Somenzi D, Murtas C, Ristoska-Bojkovska N, Izzi C, Bianco B, Zaniew M, Flogelova H, Weng PL, Kacak N, Giberti S, Gigante M, Arapovic A, Drnasin K, Caridi G, Curioni S, Allegri F, Ammenti A, Ferretti S, Goj V, Bernardo L, Jobanputra V, Chung WK, Lifton RP, Sanders S, State M, Clark LN, Saraga M, Padmanabhan S, Dominiczak AF, Foroud T, Gesualdo L, Gucev Z, Allegri L, Latos-Bielenska A, Cusi D, Scolari F, Tasic V, Hakonarson H, Ghiggeri GM, Gharavi AG (2012) Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 91:987–997
Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, Nezarati MM, Asamoah A, Jackson KE, Gowans GC, Martin JA, Carmany EP, Stockton DW, Schnur RE, Penney LS, Martin DM, Raskin S, Leppig K, Thiese H, Smith R, Aberg E, Niyazov DM, Escobar LF, El-Khechen D, Johnson KD, Lebel RR, Siefkas K, Ball S, Shur N, McGuire M, Brasington CK, Spence JE, Martin LS, Clericuzio C, Ballif BC, Shaffer LG, Eichler EE (2012) Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N Engl J Med 367:1321–1331
Cooper GM, Coe BP, Girirajan S, Rosenfeld JA, Vu TH, Baker C, Williams C, Stalker H, Hamid R, Hannig V, Abdel-Hamid H, Bader P, McCracken E, Niyazov D, Leppig K, Thiese H, Hummel M, Alexander N, Gorski J, Kussmann J, Shashi V, Johnson K, Rehder C, Ballif BC, Shaffer LG, Eichler EE (2011) A copy number variation morbidity map of developmental delay. Nat Genet 43:838–846
Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J (2008) Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320:539–543
Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE, Hansen T, Jakobsen KD, Muglia P, Francks C, Matthews PM, Gylfason A, Halldorsson BV, Gudbjartsson D, Thorgeirsson TE, Sigurdsson A, Jonasdottir A, Jonasdottir A, Bjornsson A, Mattiasdottir S, Blondal T, Haraldsson M, Magnusdottir BB, Giegling I, Moller HJ, Hartmann A, Shianna KV, Ge D, Need AC, Crombie C, Fraser G, Walker N, Lonnqvist J, Suvisaari J, Tuulio-Henriksson A, Paunio T, Toulopoulou T, Bramon E, Di Forti M, Murray R, Ruggeri M, Vassos E, Tosato S, Walshe M, Li T, Vasilescu C, Muhleisen TW, Wang AG, Ullum H, Djurovic S, Melle I, Olesen J, Kiemeney LA, Franke B, GROUP, Sabatti C, Freimer NB, Gulcher JR, Thorsteinsdottir U, Kong A, Andreassen OA, Ophoff RA, Georgi A, Rietschel M, Werge T, Petursson H, Goldstein DB, Nothen MM, Peltonen L, Collier DA, St Clair D, Stefansson K (2008) Large recurrent microdeletions associated with schizophrenia. Nature 455:232–236
Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, Leotta A, Pai D, Zhang R, Lee YH, Hicks J, Spence SJ, Lee AT, Puura K, Lehtimaki T, Ledbetter D, Gregersen PK, Bregman J, Sutcliffe JS, Jobanputra V, Chung W, Warburton D, King MC, Skuse D, Geschwind DH, Gilliam TC, Ye K, Wigler M (2007) Strong association of de novo copy number mutations with autism. Science 316:445–449
Soemedi R, Wilson IJ, Bentham J, Darlay R, Topf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, Blue GM, Breckpot J, Hellens S, Zwolinkski S, Glen E, Mamasoula C, Rahman TJ, Hall D, Rauch A, Devriendt K, Gewillig M, O’Sullivan J, Winlaw DS, Bu’Lock F, Brook JD, Bhattacharya S, Lathrop M, Santibanez-Koref M, Cordell HJ, Goodship JA, Keavney BD (2012) Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 91:489–501
Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, Lionel AC, D’Amours G, Lemyre E, Cullum R, Bigras JL, Thibeault M, Chetaille P, Montpetit A, Khairy P, Overduin B, Klaassen S, Hoodless P, Nemer M, Stewart AF, Boerkoel C, Scherer SW, Richter A, Dube MP, Andelfinger G (2012) Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet 8:e1002903
Dauber A, Yu Y, Turchin MC, Chiang CW, Meng YA, Demerath EW, Patel SR, Rich SS, Rotter JI, Schreiner PJ, Wilson JG, Shen Y, Wu BL, Hirschhorn JN (2011) Genome-wide association of copy-number variation reveals an association between short stature and the presence of low-frequency genomic deletions. Am J Hum Genet 89:751–759
Zahnleiter D, Uebe S, Ekici AB, Hoyer J, Wiesener A, Wieczorek D, Kunstmann E, Reis A, Doerr HG, Rauch A, Thiel CT (2013) Rare copy number variants are a common cause of short stature. PLoS Genet 9:e1003365
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance C (2011) American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13:680–685
Mefford HC, Clauin S, Sharp AJ, Moller RS, Ullmann R, Kapur R, Pinkel D, Cooper GM, Ventura M, Ropers HH, Tommerup N, Eichler EE, Bellanne-Chantelot C (2007) Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am J Hum Genet 81:1057–1069
Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, Clauin S, Deschenes G, Bouissou F, Bensman A, Bellanne-Chantelot C (2006) Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol 17:497–503
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M (2007) PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17:1665–1674
Huang N, Lee I, Marcotte EM, Hurles ME (2010) Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6:e1001154
Mackie Ogilvie C, Rooney SC, Hodgson SV, Berry AC (1998) Deletion of chromosome 3q proximal region gives rise to a variable phenotype. Clin Genet 53:220–222
Okada N, Hasegawa T, Osawa M, Fukuyama Y (1987) A case of de novo interstitial deletion 3q. J Med Genet 24:305–308
Jenkins MB, Stang HJ, Davis E, Boyd L (1985) Deletion of the proximal long arm of chromosome 3 in an infant with features of Turner syndrome. Ann Genet 28:42–44
Genuardi M, Calvieri F, Tozzi C, Coslovi R, Neri G (1994) A new case of interstitial deletion of chromosome 3q, del(3q)(q13.12q21.3), with agenesis of the corpus callosum. Clin Dysmorphol 3:292–296
McMorrow LE, Reid CS, Coleman J, Medeiros A, D’Andrea M, Santucci T, McCormack MK (1986) A new interstitial deletion of the long arm of chromosome 3. Am J Hum Genet 39:A124
Fujita H, Meng J, Kawamura M, Tozuka N, Ishii F, Tanaka N (1992) Boy with a chromosome del (3)(q12q23) and blepharophimosis syndrome. Am J Med Genet 44:434–436
Lawson-Yuen A, Berend SA, Soul JS, Irons M (2006) Patient with novel interstitial deletion of chromosome 3q13.1q13.3 and agenesis of the corpus callosum. Clin Dysmorphol 15:217–220
Molin AM, Andrieux J, Koolen DA, Malan V, Carella M, Colleaux L, Cormier-Daire V, David A, de Leeuw N, Delobel B, Duban-Bedu B, Fischetto R, Flinter F, Kjaergaard S, Kok F, Krepischi AC, Le Caignec C, Ogilvie CM, Maia S, Mathieu-Dramard M, Munnich A, Palumbo O, Papadia F, Pfundt R, Reardon W, Receveur A, Rio M, Ronsbro Darling L, Rosenberg C, Sa J, Vallee L, Vincent-Delorme C, Zelante L, Bondeson ML, Anneren G (2012) A novel microdeletion syndrome at 3q13.31 characterised by developmental delay, postnatal overgrowth, hypoplastic male genitals, and characteristic facial features. J Med Genet 49:104–109
Jiang H, Guo W, Liang X, Rao Y (2005) Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3beta and its upstream regulators. Cell 120:123–135
Liu KJ, Arron JR, Stankunas K, Crabtree GR, Longaker MT (2007) Chemical rescue of cleft palate and midline defects in conditional GSK-3beta mice. Nature 446:79–82
Rosenfeld JA, Ballif BC, Martin DM, Aylsworth AS, Bejjani BA, Torchia BS, Shaffer LG (2010) Clinical characterization of individuals with deletions of genes in holoprosencephaly pathways by aCGH refines the phenotypic spectrum of HPE. Hum Genet 127:421–440
Eccles MR, Schimmenti LA (1999) Renal-coloboma syndrome: a multi-system developmental disorder caused by PAX2 mutations. Clin Genet 56:1–9
Deng FM, Liang FX, Tu L, Resing KA, Hu P, Supino M, Hu CC, Zhou G, Ding M, Kreibich G, Sun TT (2002) Uroplakin IIIb, a urothelial differentiation marker, dimerizes with uroplakin Ib as an early step of urothelial plaque assembly. J Cell Biol 159:685–694
Hu P, Deng FM, Liang FX, Hu CM, Auerbach AB, Shapiro E, Wu XR, Kachar B, Sun TT (2000) Ablation of uroplakin III gene results in small urothelial plaques, urothelial leakage, and vesicoureteral reflux. J Cell Biol 151:961–972
Jenkins D, Bitner-Glindzicz M, Malcolm S, Hu CC, Allison J, Winyard PJ, Gullett AM, Thomas DF, Belk RA, Feather SA, Sun TT, Woolf AS (2005) De novo Uroplakin IIIa heterozygous mutations cause human renal adysplasia leading to severe kidney failure. J Am Soc Nephrol 16:2141–2149
Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, Utsch B, Sayer JA, Lillo C, Jimeno D, Coucke P, De Paepe A, Reinhardt R, Klages S, Tsuda M, Kawakami I, Kusakabe T, Omran H, Imm A, Tippens M, Raymond PA, Hill J, Beales P, He S, Kispert A, Margolis B, Williams DS, Swaroop A, Hildebrandt F (2005) Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat Genet 37:282–288
Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW (2010) Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet 86:949–956
Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, Branney PA, Fisher M, Lee GJ, Simpson MA, He Y, Bradshaw TY, Blaumeiser B, Winship WS, Reardon W, Maher ER, FitzPatrick DR, Wuyts W, Zenker M, Lamarche-Vane N, Trembath RC (2011) Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet 88:574–585
Niimi H, Inomata H, Sasaki N, Nakajima H (1982) Congenital isolated thyrotrophin releasing hormone deficiency. Arch Dis Child 57:877–878
Collu R, Tang J, Castagne J, Lagace G, Masson N, Huot C, Deal C, Delvin E, Faccenda E, Eidne KA, Van Vliet G (1997) A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab 82:1561–1565
Persani L, Ferretti E, Borgato S, Faglia G, Beck-Peccoz P (2000) Circulating thyrotropin bioactivity in sporadic central hypothyroidism. J Clin Endocrinol Metab 85:3631–3635
Beck-Peccoz P, Amr S, Menezes-Ferreira MM, Faglia G, Weintraub BD (1985) Decreased receptor binding of biologically inactive thyrotropin in central hypothyroidism. Effect of treatment with thyrotropin-releasing hormone. N Engl J Med 312:1085–1090
Miura Y, Perkel VS, Papenberg KA, Johnson MJ, Magner JA (1989) Concanavalin-A, lentil, and ricin lectin affinity binding characteristics of human thyrotropin: differences in the sialylation of thyrotropin in sera of euthyroid, primary, and central hypothyroid patients. J Clin Endocrinol Metab 69:985–995
Pollak MR, Brown EM, Estep HL, McLaine PN, Kifor O, Park J, Hebert SC, Seidman CE, Seidman JG (1994) Autosomal dominant hypocalcaemia caused by a Ca(2+)-sensing receptor gene mutation. Nat Genet 8:303–307
Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG (1993) Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75:1297–1303
Pollak MR, Chou YH, Marx SJ, Steinmann B, Cole DE, Brandi ML, Papapoulos SE, Menko FH, Hendy GN, Brown EM (1994) Familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Effects of mutant gene dosage on phenotype. J Clin Invest 93:1108–1112
Chen YZ, Matsushita MM, Robertson P, Rieder M, Girirajan S, Antonacci F, Lipe H, Eichler EE, Nickerson DA, Bird TD, Raskind WH (2012) Autosomal dominant familial dyskinesia and facial myokymia: single exome sequencing identifies a mutation in adenylyl cyclase 5. Arch Neurol 69:630–635
Wang L, Guo DC, Cao J, Gong L, Kamm KE, Regalado E, Li L, Shete S, He WQ, Zhu MS, Offermanns S, Gilchrist D, Elefteriades J, Stull JT, Milewicz DM (2010) Mutations in myosin light chain kinase cause familial aortic dissections. Am J Hum Genet 87:701–707
Bodmer D, Eleveld M, Kater-Baats E, Janssen I, Janssen B, Weterman M, Schoenmakers E, Nickerson M, Linehan M, Zbar B, van Kessel AG (2002) Disruption of a novel MFS transporter gene, DIRC2, by a familial renal cell carcinoma-associated t(2;3)(q35;q21). Hum Mol Genet 11:641–649
Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, Li XC, Babic M, Lin M, Carmagnac A, Lee YK, Kok CH, Gagliardi L, Friend KL, Ekert PG, Butcher CM, Brown AL, Lewis ID, To LB, Timms AE, Storek J, Moore S, Altree M, Escher R, Bardy PG, Suthers GK, D’Andrea RJ, Horwitz MS, Scott HS (2011) Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet 43:1012–1017
Ostergaard P, Simpson MA, Connell FC, Steward CG, Brice G, Woollard WJ, Dafou D, Kilo T, Smithson S, Lunt P, Murday VA, Hodgson S, Keenan R, Pilz DT, Martinez-Corral I, Makinen T, Mortimer PS, Jeffery S, Trembath RC, Mansour S (2011) Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet 43:929–931
Malan V, Chevallier S, Soler G, Coubes C, Lacombe D, Pasquier L, Soulier J, Morichon-Delvallez N, Turleau C, Munnich A, Romana S, Vekemans M, Cormier-Daire V, Colleaux L (2010) Array-based comparative genomic hybridization identifies a high frequency of copy number variations in patients with syndromic overgrowth. Eur J Hum Genet 18:227–232
Lawson CT, Toomes C, Fryer A, Carette MJ, Taylor GM, Fukushima Y, Dixon MJ (1995) Definition of the blepharophimosis, ptosis, epicanthus inversus syndrome critical region at chromosome 3q23 based on the analysis of chromosomal anomalies. Hum Mol Genet 4:963–967
Wolstenholme J, Brown J, Masters KG, Wright C, English CJ (1994) Blepharophimosis sequence and diaphragmatic hernia associated with interstitial deletion of chromosome 3 (46, XY, del(3)(q21q23)). J Med Genet 31:647–648
Jewett T, Rao PN, Weaver RG, Stewart W, Thomas IT, Pettenati MJ (1993) Blepharophimosis, ptosis, and epicanthus inversus syndrome (BPES) associated with interstitial deletion of band 3q22: review and gene assignment to the interface of band 3q22.3 and 3q23. Am J Med Genet 47:1147–1150
Acknowledgments
We are grateful to the devoted family of the index case for their ongoing participation. This study is the result of scientific collaboration between the Department of Medical Genetics, Poznan University of Medical Sciences in Poland and the Division of Nephrology at Columbia University in New York, USA. Our funding sources include: A.M.K., A.L.B., and The Polish Registry of Congenital Malformations (PRCM) are supported by the Polish Ministry of Health; A.G.G. and K.K. are supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grants R01DK080099 and K23-DK090207, respectively. S.S.C. is supported by the American Heart Association Grant in Aid 13GRNT14680075 and by the American Society of Nephrology Carl W Gottschalk Research Scholar Grant.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Anna Materna-Kiryluk and Krzysztof Kiryluk contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic Supplementary Material Dataset 1
Reference genes in the minimal candidate regions for CAKUT and ACC. Column legend: chromosome, transcript start (bp, hg-18), transcript stop (bp, hg-18), gene name, predicted probability of haploinsufficiency according to the probabilistic model of Huang et al. [15], membership in the minimal candidate region for CAKUT and ACC (TXT 3 kb)
Rights and permissions
About this article
Cite this article
Materna-Kiryluk, A., Kiryluk, K., Burgess, K.E. et al. The emerging role of genomics in the diagnosis and workup of congenital urinary tract defects: a novel deletion syndrome on chromosome 3q13.31-22.1. Pediatr Nephrol 29, 257–267 (2014). https://doi.org/10.1007/s00467-013-2625-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-013-2625-2