
Abstract
Antibiotic resistance can be a consequence of repeat-induced point (RIP) mutation and even by horizontal gene transfer in the pathogen genome for every chromosomal replication. On the account of a few vital antibiotic agents, point mutation of chromosomally encoded proteins is the essential instrument for resistance. Another procedure that may add to the development of resistance in the course of treatment is adaptive or induced change. Notwithstanding RIP mutation, resistance may likewise be interceded by enzymes that change the antibiotic and the target protein or lessen the intracellular concentration of the antibiotics. These systems of resistance are dispersed between microscopic organisms by horizontal gene transfer. Drug resistance grants bacterial development in the nearness of an antibiotic; in any case, it is by all account not the only variable adding to treatment failure. The resistance is also reflected in cases wherein the antibiotic fails to clear the infection regardless of the absence of resistant microbes. These microbes are tolerant, and clinical reports advocate that the level of tolerance to treatment failure and mortality in a few diseases can be as crucial as the nature of antibiotic resistance. Intelligent methodologies and awareness of potential harmful effects of drugs will expect to promise continuous worldwide access to efficient antibiotics.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Antibiotic discovery was one of the momentous advances in the field of modern medicine. Antibiotics remain as a mainstay in therapeutic regimes. Antibiotics have saved numerous lives afflicted with bacterial infections and other life-threatening infections. The successful discovery of the first β-lactam, penicillin G, prompted the exploration and subsequent development of additional and effective antibiotics. This quest eventually led to the production of countless antimicrobial compounds, which are in widespread usage today. However, the major obstacle towards this end was the rapid emergence of drug resistance amongst various microorganisms, which thwarted the efficacy of therapeutic interventions. The first known resistance was seen in Staphylococcus aureus alongside penicillin (Keeney et al. 1979).This ultimately led to reduced usefulness of the drug and consequently limited therapeutic options. Till date, almost all known drugs have counterattack by their target microorganisms (Table 1). According to various reports, steady rise of resistance in Plasmodium falciparum against artemisinin-based combination therapy, spread of methicillin-resistant Staphylococcus aureus (MRSA) amongst hospital-acquired infections, resistance of Neisseria gonorrhoeae to cephalosporins and Escherichia coli to fluoroquinolones, etc. are emerging threats to the healthcare of modern society (Espadinha et al. 2013; Johnson et al. 2013; Ashley et al. 2014; Bharara et al. 2015; Pham et al. 2015). Resistance leads to life-threatening disease conditions and prolongs infection and prognosis. It aggravates mortality and cost of treatment, too. Drug resistance refers to the phenomenon when microorganisms such as bacteria, viruses, fungi and parasites alter ways such that the medications used to cure the infections are rendered ineffective. When the microorganisms become resistant to most antimicrobial compounds, they are often referred as “superbugs” (Khan and Khan 2016). This can give rise to major health crisis because such a deadly resistance may culminate into a fatal infection and lead to spread to others imposing huge treatment costs. Thus, it can have severe impact on healthcare and wreak havoc on individuals and society.
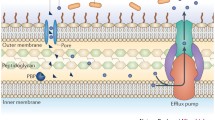
Microbes conquer antibiotic drug pressure by various biological processes (Fig. 1), which could be of two main types. First, it occurs when the microorganism has never encountered the drug against which it exhibits resistance. Second, as acquired resistance and it manifests itself following drug exposure. Antimicrobial drugs usually target a vital metabolic pathway or cell wall synthesis. To combat this threat, the microbe may either alter the target site of the drug (seen in fungi by altering cell wall composition rendering resistance to antifungal compounds) or might chemically modify it (e.g. aminoglycoside modification) (Shi et al. 2013), or plasmid-encoded degradative enzymes such as β-lactamases cleave the drug, thus hampering its action (Renneberg and Walder 1989). Genes of resistance are either plasmid-borne (Perlin and Lerner 1979) or present on mobile genetic elements (transposons) (Domingues et al. 2012) which may easily disseminate through conjugation, etc. This leads to spread of resistance in a population.

Diverse biological processes used by microbes against drug pressure
2 Aminoglycoside Resistance
Aminoglycosides represent important class of drugs. Chemically these contain an amino sugar attached to the aminocyclitol. These antibiotics are so essential attributable to their expansive range of movement against range of microscopic organisms. Amikacin, gentamicin, kanamycin, streptomycin and tobramycin are some examples and most effective in treatment of gram-negative and gram-positive bacterial infection. The drug elicits its effect by binding to the bacterial ribosome irreversibly and hindering protein synthesis though drug interactions (Chen and Murchie 2014; Dunkle et al. 2014; Song et al. 2014). Drug-modifying enzymes make the drug inactive by introducing chemical changes. Several aminoglycoside-modifying enzymes are known (Fluit and Schmitz 1999; Schmitz et al. 1999). Resistance to aminoglycoside drugs can be mediated through enzymatic chemical modifications like phosphorylations, adenylations and acetylations. Phosphorylations are catalysed by ATP-dependent O-phosphorylation (APH), nucleotidyltransferases catalysed O-adenylation (ANT) and acetyltransferases mediate N-acetylation which requires acetyl-coA-cofactor (AAC) (Marengo et al. 1974; Araoz et al. 2000; Chesneau et al. 2007). These transformations make the drug incapable of binding to the ribosome, and hence translation process remains uninhibited. In addition to enzymatic aminoglycoside-modifying enzymes, efflux pumps and ribosomal RNA mutations also contribute to reduced drug susceptibility (Kriengkauykiat et al. 2005; Corcoran et al. 2006; Takaya et al. 2013).
3 Resistance to β-Lactam
The very first β-lactamase was identified from Escherichia coli long before penicillin came in clinical use. Kirby (Kirby and Burnell 1954) extracted “penicillin inactivators” from Staphylococcus aureus. The surge in the usage of β-lactam antibiotics has put a kind of selection pressure on bacteria, which ultimately resulted in the survival of drug-resistant bacteria that have the capacity to express multiple β-lactamases. Until date, several β-lactamases have been acknowledged. These enzymes have the potential to degrade the antibiotic by hydrolysing the β-lactam ring of antimicrobial drugs like cephalosporins, penicillin, etc. (Kasik and Peacham 1968). Through this mechanism of degradation of β-lactamases, antibiotics lead to lowering down of the efficacy of these molecules, ultimately leading to the survival of the bacterial species in the presence of the drug pressure. Different β-lactamases exhibit different specificities towards the substrate and differ in host range (Hanaki et al. 2007). Various types of β-lactamases are usually secreted by gram-negative bacteria which can degrade some cephalosporins like cephalothin. In few bacterial species, these enzymes are encoded by chromosomes, for example, cephalosporinases of Pseudomonas. In other bacteria such as Enterobacteriaceae, these enzymes result due to the presence of plasmid that encodes them. As found by Dhara and Tripathi (2014), plasmid-encoded enzymes also degrade a number of penicillins, and this effect can be overcome by the presence of β-lactamase inhibitors like clavulanic acid (Song et al. 2010). β-Lactam antibiotics elicit their response by targeting and inhibiting the action of key enzymes involved in the synthesis of bacterial cell wall. The basic mechanisms linked to resistance of this class of antibiotics are bacterial synthesis of β-lactamase enzymes that has the potential to degrade antibiotic β-lactam (Song et al. 2010). This form of resistance mechanism is the most important and prevalent mode especially in gram-negative bacteria. Alteration in penicillin-binding protein (PBP) active site may be another means of attaining drug resistance that results in lower drug binding affinity (e.g. low-affinity PBP2x of Streptococcus pneumoniae) (Moisan et al. 2010). This mechanism has been reported in Neisseria spp. and Streptococcus spp. (Zapun et al. 2008). By the rigorous recombination and transformation mechanisms, these two bacterial species have developed low-affinity PBPs that are highly resistant to antibiotics. Overexpression of mecA gene that translates into penicillin-binding proteins 2a which in turn confer methicillin resistance to Staphylococcus spp. and moreover mecA overexpression desensitise bacteria against high concentrations of cephalosporins and penicillins by allowing them to synthesise new cell wall even under drug pressure (Laible et al. 1989). This antibiotic-resistant bacterial strain poses a great clinical challenge to today’s medicinal world (Neu 1984).
Lowering down the expression of outer membrane proteins (OMPs) is yet another vital mechanism of resistance. OMPs facilitate the drug to traverse through them and interact with PBPs present on the inner side of the plasma membrane in gram-negative bacteria. For example, resistance against carbapenems in some Enterobacteriaceae (Klebsiella pneumoniae and Enterobacter spp.) has been developed due to downregulation of these OMPs (Doumith et al. 2009); lowering down the expression of OprD gene is also linked with resistance towards imipenem and decrease in efficacy of meropenem in non-fermenter Pseudomonas aeruginosa (Moghoofei et al. 2015; Rodriguez-Beltran et al. 2015). Resistance against meropenem and imipenem has been reported due to downregulation of the CarO outer membrane protein (OMP) in multidrug-resistant clinical isolates of Acinetobacter baumannii (Fernandez-Cuenca et al. 2011). Various point mutations or insertion sequences in the genes coding for these porins proteins can produce altered OMPs that have loss in function or retarded function and permeability. Activation of efflux pumps provides intrinsic or acquired resistance phenotype. These efflux pumps are main determinants of multidrug resistance in various gram-negative pathogens, above all in Acinetobacter spp. and Pseudomonas aeruginosa (Morita et al. 2012). In P. aeruginosa, upregulation of the MexA-MexB-OprD framework and organism’s low outer membrane permeability have been reported in several reports (Tamber et al. 2006), which have been attributed to formidable drug resistance such as decreased susceptibility against antibiotics like tetracycline (tet), penicillins, cephalosporins, chloramphenicol and quinolones. Moreover, upregulation of efflux pumps (e.g. AdeABC which is resistance-nodulation-division (RND) family sort efflux pump ordinarily found in A. baumannii) has been reported to confer carbapenem resistance by synthesising catalytically poor form of β-lactamase (del Mar et al. 2005).
3.1 Penicillin Resistance
The most potent mechanism of evading the action of penicillin by pneumococci is elicited through alteration in the penicillin-binding proteins (PBPs). These proteins are absolutely essential for the cell wall synthesis and serve to enforce the efficacy of β-lactam antibiotics by binding to them; therefore these alterations substantially decrease the affinity of PBPS to the drug and related classes of drugs, hence effectively hampering drug action and effect (Dowson et al. 1990). “Mosaics” comprising of mixed regions of native and acquired foreign DNA segments are responsible for encoding these altered PBPs. More often than not, the DNA from foreign source belongs to the more resistant strains like viridians streptococci. There are evidences about the transfer of such mixed and hybrid genetic elements between pneumococci and gram-positive bacteria like Streptococcus oralis (Sibold et al. 1994).
4 Quinolone Resistance
Nalidixic acid was the first discovered quinolone. Many derivatives have been made available since then, fluoroquinolones being the most important ones (Emmerson and Jones 2003). These compounds possess a fluorine substitution at sixth position on the quinolone moiety, making it highly efficient against gram-positive to gram-negative bacteria and anaerobes. Quinolone drug action is brought about by inhibiting an important class of enzymes called as bacterial topoisomerases (DNA gyrase and topoisomerase IV) (Chen et al. 1996). These enzymes play an important role in bacterial DNA replication and are central to the maintenance of bacterial replication fork by modifying the topology of double-stranded DNA. Enzyme structure comprises of two subunits, namely, A and B that are heterotetrameric in nature making it highly efficient against gram-positive to gram-negative bacteria and anaerobes (Chen et al. 1996; Higgins et al. 2003).
Two principal mechanisms can very well explain the resistance seen against quinolones. Firstly by modifying the target enzyme and second by limiting the permeability of the drug (Nikaido 1998; Hernandez et al. 2011). Quite plausibly, most changes are centred at the active domains of the enzyme, which drastically reduce drug binding. DNA gyrase activity is mostly inhibited in gram-negative bacteria, but in gram-positive either DNA gyrase or topoisomerase IV can be inhibited depending on the choice of fluoroquinolones used (Jacoby 2005). In majority of the cases, there is an amino acid substitution in quinolone-resistance-determining region, which introduces a bulky hydrophobic residue instead of a polar hydroxyl group (Mehla and Ramana 2016). Mutations in gyrA gene modify the enzyme-binding site or alter a charge that leads to conformational changes essential in maintaining drug enzyme interaction. Alterations in the outer membrane structure culminate into resistance as displayed by most gram-positive bacteria (Ferrero et al. 1994). Consequently, there is reduced drug influx and uptake. More unexpectedly, resistance mechanism spread by transmission leading to fluoroquinolone inactivation has also surfaced. This mechanism has cropped up because of the ability of aminoglycoside N-acetyltransferases to modify a secondary amine on the fluoroquinolones thus leading to lowered activity. The latter mechanism confers a low-level tolerance favouring the selection of resistance mutants (Robicsek et al. 2006). Mycobacterium smegmatis and Mycobacterium bovis elicit a basal and low-level resistance to several fluoroquinolones (Montero et al. 2001). The chromosomal gene called MfpA expression is plasmid encoded, the plasmid being present in multiple copies within the bacterium. Conversely, MfpA gene disruptions enhance drug efficacy and making wild type M. smegmatis more prone to drug action. Hence, drug susceptibility is directly linked to MfpA expression level.
A very fascinating mechanism of resistance against fluoroquinolones has been investigated in Mycobacterium tuberculosis. Studies elucidating M. tuberculosis MfpA structure have unfolded a unique three-dimensional structure of MfpA that shows similarity to bacterial DNA double helix. It is speculated that MfpA could serve to sequester the entire drug and free the bacterial DNA from drug effect. Therefore, target mimicry seen in mycobacterium affords protection against fluoroquinolones (Hegde et al. 2005). In addition, point mutations in genes like cytochrome b, or dihydrofolate reductase, are known to cause atovaquone resistance or pyrimethamine resistance, respectively (Meneceur et al. 2008).
5 Tetracycline Resistance
The ease of availability, broad range of activity and cost-effectiveness make tetracyclines as the most favourite and widely used antibiotics. Since their discovery in the 1940s, they have been readily used for therapeutic interventions (Nguyen et al. 2014). The drug elicits its inhibitory effects by impeding bacterial translation through the prevention of aminoacyl-tRNA attachment to the ribosomes (Connell et al. 2013). These antibiotics successfully combat pathogenic challenges from a wide array of microorganisms including gram-positive and gram-negative microbes, atypical life forms, for example, protozoan, Chlamydiae, Rickettsiae and Mycoplasma parasites. Tetracyclines include agents like tetracycline, minocycline, oxytetracycline and doxycycline (Rasmussen et al. 1997). The phenomenon of resistance against this class of drugs can be due to drug efflux, protection of bacterial ribosomes or chemical modification of the drug. Export proteins can contribute to the resistance by mediating drug efflux (Piddock et al. 2000). These gatherings of proteins have a place with the real facilitator superfamily. Tetracycline (tet) efflux pumps encode these fare proteins and subsequently encourage drug efflux (Stavropoulos and Strathdee 2000; Tuckman et al. 2000). The expulsion of drug ultimately lowers the drug concentration, and the inhibitory effects on the ribosomes are diminished. Ribosome protection proteins that are cytoplasmic in nature aid ribosomal protection. This mode of resistance is mostly prevalent in case of doxycycline and minocycline, whereas drug efflux is the major mechanism imparting resistance against most other classes of tetracyclines (Kobayashi et al. 2007). These efflux proteins share a marked homology with other class of efflux proteins that confer multidrug resistance to various other classes of antibiotics (Wang et al. 2004). Large plasmids, encoding for such efflux genes, are transmitted through conjugation and are believed to confer resistance to gram-negative bacteria (Roberts 1997). Another important means of resistance involves enzymatic inactivation of the drug. The role of tet (X) gene has been implicated in altering tetracycline function. The tet (X) gene encodes a 44 kDa product that is capable of modifying tetracycline chemically in the presence of oxygen and NADPH. This resistance gene is present on transposons and found in anaerobic Bacteroides species (Speer et al. 1991). More recently tetracycline destructases have been discovered. These are a novel class of inactivating enzymes belonging to flavoenzyme family and catalyse oxidation of the drug. Consequently, there is modification in the structure and function of the drug (Forsberg et al. 2015).
6 Peptide-Based Drug Resistance
The intrinsic and widespread resistance to most common and rampantly used antibiotics has led to the emergence of enterococci garnering the ability to survive in a hospital-borne environment (Canton et al. 1999). Their extensive survival and resilience to the most front-line drugs used in trauma care and hospital can be attributed to wide arrays of genomic changes such as mutations, acquisition of foreign genetic element harbouring resistance genes, plasmid transfer, transposons, etc. (Rossi et al. 2014; Hu et al. 2015a). The commonest resistance ensues against drugs of classes β-lactam and glycopeptides. Synergising antibiotics such as glycopeptides with an aminoglycoside can prove to be extremely fruitful in circumventing the deleterious emergence of hospital-borne antibiotic resistance (Hu et al. 2015b). A highly regulated clustered gene unit termed as operon is believed to mediate the acquisition of glycopeptide resistance in enterococci (James et al. 2012). This operon encodes an alternative pathway responsible for the production of a transformed cell wall component. Consequently, vancomycin binds to this modified precursor peptidoglycan more readily as the normal substrate remains unaffected and available for cell wall synthesis. Thus, the progression of the normal biosynthetic pathway remains unhindered (Fraise et al. 1997). Innate resistance to vancomycin can be attributed to two types of gene cluster designated as vanA and vanB gene clusters (Grissom-Arnold et al. 1997; Baptista et al. 1997). These confer resistance by modifying target from d-alanine-d-alanine to d-alanine-d-lactate (Marshall et al. 1997).
7 Resistance to Echinocandins and Azoles
Reduced susceptibility to echinocandins can be linked to genetic events such as mutations or instigation of an adaptive stress response (Astruey-Izquierdo et al. 2011). Mutations are mostly centred around regions known as “hot spots”. These are much conserved gene clusters and are hubs of intrinsic point mutations. Mutation in FKS gene encoding, fungal FKS subunits of β(1,3)D-glucan synthase lead to cross resistance and decreased drug efficacy (Marti-Carrizosa et al. 2015; Dichtl et al. 2015). The drug-induced threat is bypassed by fungal cells through commencement of a stress response. This compensates for the drug-induced loss of a cell wall component by overproducing one or more other constituents.
The cell wall synthesis is highly regulated. In response to the drug, chitin levels are unregulated to balance the inhibition by echinocandins (Prasad et al. 2016). These elaborate metabolic changes are believed to be mediated via high-osmolarity glycerol, protein kinase C responses and Ca2+-calcineurin signalling pathways. This helps in negating the fatal effects of echinocandins (Walker et al. 2010). A pivotal role is also played by genomic plasticity in aggravating resistance. This is achieved by loss of heterozygosity and is acquired by genetic rearrangements and amplifications majorly at genetic regions that are linked with resistance (Niimi et al. 2010). Frequent and rampant use of antifungal compounds, particularly fluconazole, has prompted the rise of resistance amongst different types of Candida species. These organisms display varied levels of susceptibility depending on the amount of selection pressure and the prevalence of infections (Mane et al. 2016). The activity of an important enzyme catalyst, i.e. lanosterol 14-α-sterol demethylase involved in cell wall biosynthesis, is inhibited by azoles (Warrilow et al. 2012). Additionally, accrual of a toxic by-product, namely, 14-α-methyl-3,6-diol, further contributes to the inhibitory effects of the antifungal agent (Warrilow et al. 2012). As a result, cell wall structure, with regard to ergosterol content, is altered leading to disruptions in membrane integrity and functioning.
Candida species are adept in manifesting resistance and can successfully evade prophylactic and therapeutic regimes. Three major mechanisms dictate this first mechanism involves upregulation and overexpression of efflux pumps. This leads to significantly lowered drug levels inside the cells (Niimi 2004). Efflux pumps of Candida spp. are CDR gene encoded belonging to ATP-binding superfamily or are products of MDR1 locus which encode major facilitator superfamily (MFS) proteins (Shao et al. 2016). Enhanced expression of CDR gene product culminating into expanded no. of efflux pumps presents imperviousness to all known azoles. Nevertheless, MDR-encoded efflux pumps bolster just fluconazole resistance. Examples of Candida spp. evoking azole resistance are as per the following: Candida glabrata (CgCDR1, CgCDR2) and C. albicans (CDR1, CDR2, MDR1) (Shao et al. 2016). One more resistance mechanism comprises the alteration or overproduction of the target molecule. A key player determining this mode of resistance is ERG11 gene that codes for the enzyme lanosterol 14-a-demethylase. Mutations pertaining to this gene lead to subtle modifications in the target enzyme that change the binding affinity of the enzyme to the drug (Martel et al. 2010). Lowered susceptibility of ERG11p to fluconazole as seen in C. krusei is due to this kind of altered binding (Martel et al. 2010). Overproduction of ERG11p makes the antifungal agent ineffective at its normal dosage, which is due to the outnumbering of target molecules with reference to drug molecules. Consequently, drug is present in insufficient amounts as compared to its target to successfully exhibit the inhibitory effects (Wang et al. 2009). ERG3 gene mutations circumvent the accumulation of toxic metabolic products and lead to formation of proper and functional cell membrane. Devising an alternate/bypass biosynthetic pathway paves way for another mechanism of resistance in fungi (Wang et al. 2009; Lo et al. 2015).
8 Methicillin-Resistant Staphylococcus aureus
The presence of MRSA has been detected in both the community-acquired and hospital settings. These are found to express mecA gene, which confers them resistance against methicillin and other β-lactams (Neu 1984). However, mecA gene’s genetic environment is found to be different for the hospital-acquired and community-acquired isolates. Nosocomial MRSA is an example of multidrug resistance (Panda et al. 2016). Imperviousness to methicillin and a few other β-lactams has been connected to the ability of mecA to encode low-affinity penicillin-binding protein PBP2a (Roychoudhury et al. 1994). PBP2a-encoding mecA gene is located on a genetically mobile element that is termed as staphylococcal chromosomal cassette (SCC-mec) (Hososaka et al. 2007). In particular, resistance towards fluoroquinolone is regarded as a hallmark for nosocomial MRSA. Expression of EMRSA-17 lead to development of resistance towards a wide range of antibiotics like methicillin, macrolides (erythromycin), fluoroquinolones (ciprofloxacin), tetracycline, fusidic acid, aminoglycosides (kanamycin, streptomycin, gentamicin and neomycin) and rifampicin (Aucken et al. 2002).
9 Prospective
Augmentation in a number of diabetic patients and burn patients increases the susceptibility to acquired infections and spread of resistance. The resistant strains may evolve naturally when microorganisms replicate themselves in an erroneous fashion or by the exchange of resistant traits between them. Our abuse of antibiotics in people incomprehensibly quickened the procedure of drug resistance; however drug administration in intensive care units of hospitals and treatment of immunocompromised patients further leads to expansion of multidrug resistance and prevalence of nosocomial infections. To contain antibiotic resistance, motivating forces for drug organisation, clinics, specialist and patients have to be devised to act in ways that may restrain the exhaustion of antimicrobial efficacy.
References
Araoz R, Anhalt E, Rene L, Badet-Denisot MA, Courvalin P, Badet B (2000) Mechanism-based inactivation of VanX, a D-alanyl-D-alanine dipeptidase necessary for vancomycin resistance. Biochemistry 39:15971–15979. doi:10.1021/bi001408b
Arthur M, Courvalin P (1993) Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob Agents Chemother 37:1563–1571. doi:10.1128/AAC.37.8.1563
Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, Sreng S, Anderson JM, Mao S, Sam B, Sopha C, Chuor CM, Nguon C, Sovannaroth S, Pukrittayakamee S, Jittamala P, Chotivanich K, Chutasmit K, Suchatsoonthorn C, Runcharoen R, Hien TT, Thuy-Nhien NT, Thanh NV, Phu NH, Htut Y, Han KT, Aye KH, Mokuolu OA, Olaosebikan RR, Folaranmi OO, Mayxay M, Khanthavong M, Hongvanthong B, Newton PN, Onyamboko MA, Fanello CI, Tshefu AK, Mishra N, Valecha N, Phyo AP, Nosten F, Yi P, Tripura R, Borrmann S, Bashraheil M, Peshu J, Faiz MA, Ghose A, Hossain MA, Samad R, Rahman MR, Hasan MM, Islam A, Miotto O, Amato R, MacInnis B, Stalker J, Kwiatkowski DP, Bozdech Z, Jeeyapant A, Cheah PY, Sakulthaew T, Chalk J, Intharabut B, Silamut K, Lee SJ, Vihokhern B, Kunasol C, Imwong M, Tarning J, Taylor WJ, Yeung S, Woodrow CJ, Flegg JA, Das D, Smith J, Venkatesan M, Plowe CV, Stepniewska K, Guerin PJ, Dondorp AM, Day NP, White NJ (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 371:411–423. doi:10.1056/NEJMoa1314981
Astruey-Izquierdo A, Cuesta I, Ros L, Mellado E, Rodriguez-Tudela JL (2011) Antifungal susceptibility profile of clinical Alternaria spp. identified by molecular methods. J Antimicrob Chemother 66:2585–2587. doi:10.1093/jac/dkr365
Aucken HM, Ganner M, Murchan S, Cookson BD, Johnson AP (2002) A new UK strain of epidemic methicillin-resistant Staphylococcus aureus (EMRSA-17) resistant to multiple antibiotics. J Antimicrob Chemother 50:171–175. doi:10.1093/jac/dkf117
Baptista M, Depardieu F, Reynolds P, Courvalin P, Arthur M (1997) Mutations leading to increased levels of resistance to glycopeptide antibiotics in VanB-type enterococci. Mol Microbiol 25:93–105. doi:10.1046/j.1365-2958.1997.4401812.x
Bharara T, Bhalla P, Rawat D, Garg VK, Sardana K, Chakravarti A (2015) Rising trend of antimicrobial resistance among Neisseria gonorrhoeae isolates and the emergence of N. gonorrhoeae isolate with decreased susceptibility to ceftriaxone. Indian J Med Microbiol 33:39–42. doi:10.4103/0255-0857.148374
Boucher HW, Sakoulas G (2007) Perspectives on Daptomycin resistance, with emphasis on resistance in Staphylococcus aureus. Clin Infect Dis 45:601–608. doi:10.1086/520655
Bryan LE, Kwan S (1983) Roles of ribosomal binding, membrane potential, and electron transport in bacterial uptake of streptomycin and gentamicin. Antimicrob Agents Chemother 23:835–845. doi:10.1128/AAC.23.6.835
Busse HJ, Wostmann C, Bakker EP (1992) The bactericidal action of streptomycin: membrane permeabilization caused by the insertion of mistranslated proteins into the cytoplasmic membrane of Escherichia coli and subsequent caging of the antibiotic inside the cells due to degradation of these proteins. J Gen Microbiol 138:551–561. doi:10.1099/00221287-138-3-551
Cai Y, Chai D, Wang R, Liang B, Bai N (2012) Colistin resistance of Acinetobacter baumannii: clinical reports, mechanisms and antimicrobial strategies. J Antimicrob Chemother 67:1607–1615. doi:10.1093/jac/dks084
Canton R, Mir N, Sanchez M, Baquero F (1999) MIC distribution and inoculum effect of LY333328: a study of vancomycin-susceptible and VanA-type and VanC-type enterococci obtained from intensive care unit patient surveillance cultures. Clin Microbiol Infect 5:554–559. doi:10.1111/j.1469-0691.1999.tb00434.x
Chen D, Murchie AI (2014) An aminoglycoside sensing riboswitch controls the expression of aminoglycoside resistance acetyltransferase and adenyltransferases. Biochim Biophys Acta 1839:951–958. doi:10.1016/j.bbagrm.2014.02.019
Chen CR, Malik M, Snyder M, Drlica K (1996) DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J Mol Biol 258:627–637. doi:10.1006/jmbi.1996.0274
Chesneau O, Tsvetkova K, Courvalin P (2007) Resistance phenotypes conferred by macrolide phosphotransferases. FEMS Microbiol Lett 269:317–322. doi:10.1111/j.1574-6968.2007.00643
Connell SR, Tracz DM, Nierhaus KH, Taylor DE (2013) Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Chemother 47:3675–3681. doi:10.1128/AAC.47.12.3675-3681.2003
Corcoran D, Quinn T, Cotter L, Fanning S (2006) An investigation of the molecular mechanisms contributing to high-level erythromycin resistance in Campylobacter. Int J Antimicrob Agents 27:40–45. doi:10.1016/j.ijantimicag.2005.08.019
del Mar TM, Beceiro A, Perez A, Velasco D, Moure R, Villanueva R, Martinez-Beltran J, Bou G (2005) Cloning and functional analysis of the gene encoding the 33- to 36-kilodalton outer membrane protein associated with carbapenem resistance in Acinetobacter baumannii. Antimicrob Agents Chemother 49:5172–5175. doi:10.1128/AAC.49.12.5172-5175.2005
Dhara L, Tripathi A (2014) Genetic and structural insights into plasmid-mediated extended-spectrum beta-lactamase activity of CTX-M and SHV variants among pathogenic Enterobacteriaceae infecting Indian patients. Int J Antimicrob Agents 43:518–526. doi:10.1016/j.ijantimicag.2014.03.002
Dichtl K, Samantaray S, Aimanianda V, Zhu Z, Prevost MC, Latge JP, Ebel F, Wagener J (2015) Aspergillus fumigatus devoid of cell wall beta-1,3-glucan is viable, massively sheds galactomannan and is killed by septum formation inhibitors. Mol Microbiol 95:458–471. doi:10.1111/mmi.12877
Domingues S, da Silva GJ, Nielsen KM (2012) Integrons: vehicles and pathways for horizontal dissemination in bacteria. Mob Genet Elements 2:211–223. doi:10.4161/mge.22967
Doumith M, Ellington MJ, Livermore DM, Woodford N (2009) Molecular mechanisms disrupting porin expression in ertapenem-resistant Klebsiella and Enterobacter spp. clinical isolates from the UK. J Antimicrob Chemother 63:659–667. doi:10.1093/jac/dkp029
Dowson CG, Hutchison A, Woodford N, Johnson AP, George RC, Spratt BG (1990) Penicillin-resistant viridans streptococci have obtained altered penicillin-binding protein genes from penicillin-resistant strains of Streptococcus pneumoniae. Proc Natl Acad Sci U S A 87:5858–5862. doi:10.1073/pnas.87.15.5858
Dunkle JA, Vinal K, Desai PM, Zelinskaya N, Savic M, West DM, Conn GL, Dunham CM (2014) Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc Natl Acad Sci U S A 111:6275–6280. doi:10.1073/pnas.1402789111
Emmerson AM, Jones AM (2003) The quinolones: decades of development and use. J Antimicrob Chemother 51:13–20. doi:10.1093/jac/dkg208
Espadinha D, Faria NA, Miragaia M, Lito LM, Melo-Cristino J, de Lencastre H (2013) Extensive dissemination of methicillin-resistant Staphylococcus aureus (MRSA) between the hospital and the community in a country with a high prevalence of nosocomial MRSA. PLoS One 8, e59960. doi:10.1371/journal.pone.0059960
Fernandez-Cuenca F, Smani Y, Gomez-Sanchez MC, Docobo-Perez F, Caballero-Moyano FJ, Dominguez-Herrera J, Pascual A, Pachon J (2011) Attenuated virulence of a slow-growing pandrug-resistant Acinetobacter baumannii is associated with decreased expression of genes encoding the porins CarO and OprD-like. Int J Antimicrob Agents 38:548–549. doi:10.1016/j.ijantimicag.2011.08.002
Ferrero L, Cameron B, Manse B, Lagneaux D, Crouzet J, Famechon A, Blanche F (1994) Cloning and primary structure of Staphylococcus aureus DNA topoisomerase IV: a primary target of fluoroquinolones. Mol Microbiol 13:641–653. doi:10.1111/j.1365-2958.1994.tb00458.x
Ferrero L, Cameron B, Crouzet J (1995) Analysis of gyrA and grlA mutations in stepwise-selected ciprofloxacin-resistant mutants of Staphylococcus aureus. Antimicrob Agents Chemother 39:554–1558. doi:10.1128/AAC.39.7.1554
Fluit AC, Schmitz FJ (1999) Class 1 integrons, gene cassettes, mobility, and epidemiology. Eur J Clin Microbiol Infect Dis 18:761–770. doi:10.1007/s100960050398
Fluit AC, Florijn A, Verhoef J, Milatovic D (2005) Presence of tetracycline resistance determinants and susceptibility to tigecycline and minocycline. Antimicrob Agents Chemother 49:1636–1638. doi:10.1128/AAC.49.4.1636-1638.2005
Forsberg KJ, Patel S, Wencewicz TA, Dantas G (2015) The tetracycline destructases: a novel family of tetracycline-inactivating enzymes. Chem Biol 22:888–897. doi:10.1016/j.chembiol.2015.05.017
Fraise AP, Andrews J, Wise R (1997) Activity of a new glycopeptide antibiotic (LY333328) against enterococci and other resistant gram-positive organisms. J Antimicrob Chemother 40:423–425. doi:10.1093/jac/40.3.423
Grissom-Arnold J, Alborn WE Jr, Nicas TI, Jaskunas SR (1997) Induction of VanA vancomycin resistance genes in Enterococcus faecalis: use of a promoter fusion to evaluate glycopeptide and nonglycopeptide induction signals. Microb Drug Resist 3:53–64. doi:10.1089/mdr.1997.3.53
Hanaki H, Koide Y, Yamazaki H, Kubo R, Nakano T, Atsuda K, Sunakawa K (2007) Substrate specificity of HMRZ-86 for beta-lactamases, including extended-spectrum beta-lactamases (ESBLs). J Infect Chemother 13:390–395. doi:10.1007/s10156-007-0563-2
Hegde SS, Vetting MW, Roderick SL, Mitchenall LA, Maxwell A, Takiff HE, Blanchard JS (2005) A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 308:1480–1483. doi:10.1126/science.1110699
Hernandez A, Sanchez MB, Martinez JL (2011) Quinolone resistance: much more than predicted. Front Microbiol 2:22. doi:10.3389/fmicb.2011.00022
Higgins PG, Fluit AC, Schmitz FJ (2003) Fluoroquinolones: structure and target sites. Curr Drug Targets 4:181–190. doi:10.2174/1389450033346920
Holmes AH, Moore LS, Sundsfjord A, Steinbakk M, Regmi S, Karkey A, Guerin PJ, Piddock LJ (2016) Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 387:176–187. doi:10.1016/S0140-6736(15)00473-0
Hososaka Y, Hanaki H, Endo H, Suzuki Y, Nagasawa Z, Otsuka Y, Nakae T, Sunakawa K (2007) Characterization of oxacillin-susceptible mecA-positive Staphylococcus aureus: a new type of MRSA. J Infect Chemother 13:79–86. doi:10.1007/s10156-006-0502-7
Hu J, Zhang X, Liu X, Chen C, Sun B (2015a) Mechanism of reduced vancomycin susceptibility conferred by walK mutation in community-acquired methicillin-resistant Staphylococcus aureus strain MW2. Antimicrob Agents Chemother 59:1352–1355. doi:10.1128/AAC.04290-14
Hu Y, Liu A, Vaudrey J, Vaiciunaite B, Moigboi C, McTavish SM, Kearns A, Coates A (2015b) Combinations of beta-lactam or aminoglycoside antibiotics with plectasin are synergistic against methicillin-sensitive and methicillin-resistant Staphylococcus aureus. PLoS One 10, e0117664. doi:10.1371/journal.pone.0117664
Huovinen P (2001) Resistance to trimethoprim-sulfamethoxazole. Clin Infect Dis 32:1608–1614. doi:10.1086/320532
Jacoby GA (2005) Mechanisms of resistance to quinolones. Clin Infect Dis 41(Suppl 2):S120–S126. doi:10.1086/428052
James RC, Pierce JG, Okano A, Xie J, Boger DL (2012) Redesign of glycopeptide antibiotics: back to the future. ACS Chem Biol 7:797–804. doi:10.1021/cb300007j
Jensen LB, Hammerum AM, Aarestrup FM (2000) Linkage of vat(E) and erm(B) in streptogamin-resistant Enterococcus faecium isolates from Europe. Antimicrob Agents Chemother 44:2231–2232. doi:10.1128/AAC.44.8.2231-2232.2000
Johnson R, Tchesnokova V, Johnston B, Clabots C, Roberts PL, Billig M, Riddell K, Rogers P, Qin X, Butler-Wu S, Price LB, Aziz M, Nicolas-Chanoine MH, Debroy C, Robicsek A, Hansen G, Urban C, Platell J, Trott DJ, Zhanel G, Weissman SJ, Cookson BT, Fang FC, Limaye AP, Scholes D, Chattopadhyay S, Hooper DC, Sokurenko EV (2013) Abrupt emergence of a single dominant multidrug-resistant strain of Escherichia coli. J Infect Dis 207:919–928. doi:10.1093/infdis/jis933
Kasik JE, Peacham L (1968) Properties of beta-lactamases produced by three species of mycobacteria. Biochem J 107:675–682. doi:10.1042/bj1070675
Keeney RE, Seamans ML, Russo RM, Gururaj VJ, Allen JE (1979) The comparative efficacy of minocycline and penicillin-V in Staphylococcus aureus skin and soft tissue infections. Cutis 23:711–718
Khan SN, Khan AU (2016) Breaking the spell: combating multidrug resistant ‘superbugs’. Front Microbiol 7:174. doi:10.3389/fmicb.2015.01574
Kirby WM, Burnell JM (1954) Effect of combinations of antibiotics on lysis of Staphylococcus aureus by penicillin. J Bacteriol 67:50–52
Kobayashi T, Nonaka L, Maruyama F, Suzuki S (2007) Molecular evidence for the ancient origin of the ribosomal protection protein that mediates tetracycline resistance in bacteria. J Mol Evol 65:228–235. doi:10.1007/s00239-007-9006-z
Kriengkauykiat J, Porter E, Lomovskaya O, Wong-Beringer A (2005) Use of an efflux pump inhibitor to determine the prevalence of efflux pump-mediated fluoroquinolone resistance and multidrug resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 49:565–570. doi:10.1128/AAC.49.2.565-570.2005
Laible G, Hakenbeck R, Sicard MA, Joris B, Ghuysen JM (1989) Nucleotide sequences of the pbpX genes encoding the penicillin-binding proteins 2x from Streptococcus pneumoniae R6 and a cefotaxime-resistant mutant, C506. Mol Microbiol 3:1337–1348. doi:10.1111/j.1365-2958.1989.tb00115.x
Leclercq R (2002) Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin Infect Dis 34:482–492. doi:10.1086/324626
Leclercq R, Courvalin P (1991) Bacterial resistance to macrolide, lincosamide, and streptogramin antibiotics by target modification. Antimicrob Agents Chemother 35:1267–1272. doi:10.1128/AAC.35.7.1267
Linkevicius M, Sandegren L, Andersson DI (2015) Potential of tetracycline resistance proteins to evolve tigecycline resistance. Antimicrob Agents Chemother 60:789–796. doi:10.1128/AAC.02465-15
Lo HJ, Tseng KY, Kao YY, Tsao MY, Lo HL, Yang YL (2015) Cph1p negatively regulates MDR1 involved in drug resistance in Candida albicans. Int J Antimicrob Agents 45:617–621. doi:10.1016/j.ijantimicag.2015.01.017
Mahbub AM, Kobayashi N, Ishino M, Sumi A, Kobayashi K, Uehara N, Watanabe N (2005) Detection of a novel aph(2″) allele (aph[2″]-Ie) conferring high-level gentamicin resistance and a spectinomycin resistance gene ant(9)-Ia (aad 9) in clinical isolates of enterococci. Microb Drug Resist 11:239–247. doi:10.1089/mdr.2005.11.239
Mane A, Vidhate P, Kusro C, Waman V, Saxena V, Kulkarni-Kale U, Risbud A (2016) Molecular mechanisms associated with Fluconazole resistance in clinical Candida albicans isolates from India. Mycoses 59:93–100. doi:10.1111/myc.12439
Marengo PB, Chenoweth ME, Overturf GD, Wilkins J (1974) Phosphorylation of kanamycin, lividomycin A, and butirosin B by Providencia stuartii. Antimicrob Agents Chemother 6:821–824. doi:10.1128/AAC.6.6.821
Marshall CG, Broadhead G, Leskiw BK, Wright GD (1997) D-Ala-D-Ala ligases from glycopeptide antibiotic-producing organisms are highly homologous to the enterococcal vancomycin-resistance ligases VanA and VanB. Proc Natl Acad Sci U S A 94:6480–6483. doi:10.1073/pnas.94.12.6480
Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AG, Kelly DE, Kelly SL (2010) A clinical isolate of Candida albicans with mutations in ERG11 (encoding sterol 14alpha-demethylase) and ERG5 (encoding C22 desaturase) is cross resistant to azoles and amphotericin B. Antimicrob Agents Chemother 54:3578–3583. doi:10.1128/AAC.00303-10
Marti-Carrizosa M, Sanchez-Reus F, March F, Canton E, Coll P (2015) Implication of Candida parapsilosis FKS1 and FKS2 mutations in reduced echinocandin susceptibility. Antimicrob Agents Chemother 59:3570–3573. doi:10.1128/AAC.04922-14
Mehla K, Ramana J (2016) Structural signature of Ser83Leu and Asp87Asn mutations in DNA gyrase from enterotoxigenic Escherichia coli and impact on quinolone resistance. Gene 576:28–35. doi:10.1016/j.gene.2015.09.063
Meka VG, Gold HS (2004) Antimicrobial resistance to linezolid. Clin Infect Dis 39:1010–1015. doi:10.1086/423841
Meneceur P, Bouldouyre MA, Aubert D, Villena I, Menotti J, Sauvage V, Garin JF, Derouin F (2008) In vitro susceptibility of various genotypic strains of Toxoplasma gondii to pyrimethamine, sulfadiazine and atovaquone. Antimicrob Agents Chemother 52:1269–1277. doi:10.1128/AAC.01203-07
Mingoia M, Vecchi M, Cochetti I, Tili E, Vitali LA, Manzin A, Varaldo PE, Montanari MP (2007) Composite structure of Streptococcus pneumoniae containing the erythromycin efflux resistance gene mefI and the chloramphenicol resistance gene catQ. Antimicrob Agents Chemother 51:3983–3987. doi:10.1128/AAC.00790-07
Moghoofei M, Fazeli H, Poursina F, Nasr EB, Moghim S, Vaez H, Hadifar S, Ghasemian SH (2015) Morphological and bactericidal effects of amikacin, meropenem and imipenem on Pseudomonas aeruginosa. Jundishapur J Microbiol 8, e25250. doi:10.5812/jjm.25250
Moisan H, Pruneau M, Malouin F (2010) Binding of ceftaroline to penicillin-binding proteins of Staphylococcus aureus and Streptococcus pneumoniae. J Antimicrob Chemother 65:713–716. doi:10.1093/jac/dkp503
Montero C, Mateu G, Rodriguez R, Takiff H (2001) Intrinsic resistance of Mycobacterium smegmatis to fluoroquinolones may be influenced by new pentapeptide protein MfpA. Antimicrob Agents Chemother 45:3387–3392. doi:10.1128/AAC.45.12.3387-3392.2001
Morita Y, Tomida J, Kawamura Y (2012) MexXY multidrug efflux system of Pseudomonas aeruginosa. Front Microbiol 3:408. doi:10.3389/fmicb.2012.00408
Neu HC (1984) Changing patterns of hospital infections: implications for therapy. Changing mechanisms of bacterial resistance. Am J Med 77:11–23. doi:10.1016/j.eimc.2008.11.001
Nguyen F, Starosta AL, Arenz S, Sohmen D, Donhofer A, Wilson DN (2014) Tetracycline antibiotics and resistance mechanisms. Biol Chem 395:559–575. doi:10.1515/hsz-2013-0292
Niimi M (2004) An efficient system for functional hyper-expression of multidrug efflux pumps in Saccharomyces cerevisiae. Nihon Ishinkin Gakkai Zasshi 45:63–69. doi:10.3314/jjmm.45.63
Niimi K, Monk BC, Hirai A, Hatakenaka K, Umeyama T, Lamping E, Maki K, Tanabe K, Kamimura T, Ikeda F, Uehara Y, Kano R, Hasegawa A, Cannon RD, Niimi M (2010) Clinically significant micafungin resistance in Candida albicans involves modification of a glucan synthase catalytic subunit GSC1 (FKS1) allele followed by loss of heterozygosity. J Antimicrob Chemother 65:842–852. doi:10.1093/jac/dkq073
Nikaido H (1998) Antibiotic resistance caused by gram-negative multidrug efflux pumps. Clin Infect Dis 27:S32–S41. doi:10.1086/514920
Palomino JC, Martin A (2014) Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics 3:317–340. doi:10.3390/antibiotics3030317
Panda RK, Mahapatra A, Mallick B, Chayani N (2016) Evaluation of genotypic and phenotypic methods for detection of methicillin resistant Staphylococcus aureus in a tertiary care hospital of Eastern Odisha. J Clin Diagn Res 10:DC19–DC21. doi:10.7860/JCDR/2016/17476.7278
Perlin MH, Lerner SA (1979) Amikacin resistance associated with a plasmid-borne aminoglycoside phosphotransferase in Escherichia coli. Antimicrob Agents Chemother 16:598–604. doi:10.1128/AAC.16.5.598
Pham QD, Do NT, Le YN, Nguyen TV, Nguyen DB, Huynh TK, Bui DD, Van KN, Nguyen PD, Luong AQ, Bui HT, Nguyen HH, McConnell M, Nguyen LT, Zhang L, Truong LX (2015) Pretreatment HIV-1 drug resistance to first-line drugs: results from a baseline assessment of a large cohort initiating ART in Vietnam, 2009-10. J Antimicrob Chemother 70:941–947. doi:10.1093/jac/dku473
Philippon A, Labia R, Jacoby G (1989) Extended-spectrum beta-lactamases. Antimicrob Agents Chemother 33:1131–1136. doi:10.1128/AAC.33.8.1131
Piddock LJ, White DG, Gensberg K, Pumbwe L, Griggs DJ (2000) Evidence for an efflux pump mediating multiple antibiotic resistance in Salmonella enterica serovar Typhimurium. Antimicrob Agents Chemother 44:3118–3121. doi:10.1128/AAC.44.11.3118-3121.2000
Pournaras S, Maniati M, Spanakis N, Ikonomidis A, Tassios PT, Tsakris A, Legakis NJ, Maniatis AN (2005) Spread of efflux pump-overexpressing, non-metallo-beta-lactamase-producing, meropenem-resistant but ceftazidime-susceptible Pseudomonas aeruginosa in a region with blaVIM endemicity. J Antimicrob Chemother 56:761–764. doi:10.1093/jac/dki296
Prasad R, Shah AH, Rawal MK (2016) Antifungals: mechanism of action and drug resistance. Adv Exp Med Biol 892:327–349. doi:10.1007/978-3-319-25304-6_14
Prystowsky J, Siddiqui F, Chosay J, Shinabarger DL, Millichap J, Peterson LR, Noskin GA (2001) Resistance to linezolid: characterization of mutations in rRNA and comparison of their occurrences in vancomycin-resistant enterococci. Antimicrob Agents Chemother 45:2154–2156. doi:10.1128/AAC.45.7.2154-2156.2001
Rasmussen BA, Bush K, Tally FP (1997) Antimicrobial resistance in anaerobes. Clin Infect Dis 24:S110–S120. doi:10.1093/clinids/24.Supplement_1.S110
Renneberg J, Walder M (1989) The role of beta-lactamase in mixed infections in mice in relation to treatment with ampicillin. J Infect Dis 160:337–341. doi:10.1093/infdis/160.2.337
Roberts MC (1997) Genetic mobility and distribution of tetracycline resistance determinants. Ciba Found Symp 207:206–218. doi:10.1002/9780470515358.ch13
Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Bush K, Hooper DC (2006) Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat Med 12:83–88. doi:10.1038/nm1347
Rodriguez-Beltran J, Cabot G, Valencia EY, Costas C, Bou G, Oliver A, Blazquez J (2015) N-acetylcysteine selectively antagonizes the activity of imipenem in Pseudomonas aeruginosa by an OprD-mediated mechanism. Antimicrob Agents Chemother 59:3246–3251. doi:10.1128/AAC.00017-15
Ross JI, Eady EA, Cove JH, Cunliffe WJ, Baumberg S, Wootton JC (1990) Inducible erythromycin resistance in staphylococci is encoded by a member of the ATP-binding transport super-gene family. Mol Microbiol 4:1207–1214. doi:10.1111/j.1365-2958.1990.tb00696.x
Rossi F, Diaz L, Wollam A, Panesso D, Zhou Y, Rincon S, Narechania A, Xing G, Di Gioia TS, Doi A, Tran TT, Reyes J, Munita JM, Carvajal LP, Hernandez-Roldan A, Brandao D, van der Heijden I, Murray BE, Planet PJ, Weinstock GM, Arias CA (2014) Transferable vancomycin resistance in a community-associated MRSA lineage. N Engl J Med 370:1524–1531. doi:10.1056/NEJMoa1303359
Roychoudhury S, Dotzlaf JE, Ghag S, Yeh WK (1994) Purification, properties, and kinetics of enzymatic acylation with beta-lactams of soluble penicillin-binding protein 2a. A major factor in methicillin-resistant Staphylococcus aureus. J Biol Chem 269:12067–12073
Sanchez MB, Martinez JL (2015) The efflux pump SmeDEF contributes to trimethoprim-sulfamethoxazole resistance in Stenotrophomonas maltophilia. Antimicrob Agents Chemother 59:4347–4348. doi:10.1128/AAC.00714-15
Schmitz FJ, Fluit AC, Gondolf M, Beyrau R, Lindenlauf E, Verhoef J, Heinz HP, Jones ME (1999) The prevalence of aminoglycoside resistance and corresponding resistance genes in clinical isolates of staphylococci from 19 European hospitals. J Antimicrob Chemother 43:253–259. doi:10.1093/jac/43.2.253
Schwarz S, Kehrenberg C, Doublet B, Cloeckaert A (2004) Molecular basis of bacterial resistance to chloramphenicol and florfenicol. FEMS Microbiol Rev 28:519–542. doi:10.1016/j.femsre.2004.04.001
Shao J, Zhang M, Wang T, Li Y, Wang C (2016) The roles of CDR1, CDR2, and MDR1 in kaempferol-induced suppression with fluconazole-resistant Candida albicans. Pharm Biol 54:984–992. doi:10.3109/13880209.2015.1091483
Shi K, Caldwell SJ, Fong DH, Berghuis AM (2013) Prospects for circumventing aminoglycoside kinase mediated antibiotic resistance. Front Cell Infect Microbiol 3:22. doi:10.3389/fcimb.2013.00022
Sibold C, Henrichsen J, Konig A, Martin C, Chalkley L, Hakenbeck R (1994) Mosaic pbpX genes of major clones of penicillin-resistant Streptococcus pneumoniae have evolved from pbpX genes of a penicillin-sensitive Streptococcus oralis. Mol Microbiol 12:1013–1023. doi:10.1111/j.1365-2958.1994.tb01089.x
Song JY, Jensen SE, Lee KJ (2010) Clavulanic acid biosynthesis and genetic manipulation for its overproduction. Appl Microbiol Biotechnol 88:659–669. doi:10.1007/s00253-010-2801-2
Song W, Kim YH, Sim SH, Hwang S, Lee JH, Lee Y, Bae J, Hwang J, Lee K (2014) Antibiotic stress-induced modulation of the endoribonucleolytic activity of RNase III and RNase G confers resistance to aminoglycoside antibiotics in Escherichia coli. Nucleic Acids Res 42:4669–4681. doi:10.1093/nar/gku093
Speer BS, Bedzyk L, Salyers AA (1991) Evidence that a novel tetracycline resistance gene found on two Bacteroides transposons encodes an NADP-requiring oxidoreductase. J Bacteriol 173:176–183
Spratt BG, Cromie KD (1988) Penicillin-binding proteins of gram-negative bacteria. Rev Infect Dis 10:699–711. doi:10.1093/clinids/10.4.699
Stavropoulos TA, Strathdee CA (2000) Expression of the tetA(C) tetracycline efflux pump in Escherichia coli confers osmotic sensitivity. FEMS Microbiol Lett 190:147–150
Takaya A, Sato Y, Shoji T, Yamamoto T (2013) Methylation of 23S rRNA nucleotide G748 by RlmAII methyltransferase renders Streptococcus pneumoniae telithromycin susceptible. Antimicrob Agents Chemother 57:3789–3796. doi:10.1128/AAC.00164-13
Tamber S, Ochs MM, Hancock RE (2006) Role of the novel OprD family of porins in nutrient uptake in Pseudomonas aeruginosa. J Bacteriol 188:45–54. doi:10.1128/JB.188.1.45-54.2006
Tenover FC (2006) Mechanisms of antimicrobial resistance in bacteria. Am J Med 119:S3–S10. doi:10.1016/j.amjmed.2006.03.011
Tuckman M, Petersen PJ, Projan SJ (2000) Mutations in the interdomain loop region of the tetA(A) tetracycline resistance gene increase efflux of minocycline and glycylcyclines. Microb Drug Resist 6:277–282. doi:10.1089/mdr.2000.6.277
Walker LA, Gow NA, Munro CA (2010) Fungal echinocandin resistance. Fungal Genet Biol 47:117–126. doi:10.1016/j.fgb.2009.09.003
Wang Y, Shoemaker NB, Salyers AA (2004) Regulation of a Bacteroides operon that controls excision and transfer of the conjugative transposon CTnDOT. J Bacteriol 186:2548–2557. doi:10.1128/JB.186.9.2548-2557.2004
Wang H, Kong F, Sorrell TC, Wang B, McNicholas P, Pantarat N, Ellis D, Xiao M, Widmer F, Chen SC (2009) Rapid detection of ERG11 gene mutations in clinical Candida albicans isolates with reduced susceptibility to fluconazole by rolling circle amplification and DNA sequencing. BMC Microbiol 9:167. doi:10.1186/1471-2180-9-167
Warrilow AG, Mullins JG, Hull CM, Parker JE, Lamb DC, Kelly DE, Kelly SL (2012) S279 point mutations in Candida albicans Sterol 14-alpha demethylase (CYP51) reduce in vitro inhibition by fluconazole. Antimicrob Agents Chemother 56:2099–2107. doi:10.1128/AAC.05389-11
Webber MA, Piddock LJ (2001) Absence of mutations in marRAB or soxRS in acrB-overexpressing fluoroquinolone-resistant clinical and veterinary isolates of Escherichia coli. Antimicrob Agents Chemother 45:1550–1552. doi:10.1128/AAC.45.5.1550-1552.2001
Zapun A, Contreras-Martel C, Vernet T (2008) Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol Rev 32:361–385. doi:10.1111/j.1574-6976.2007.00095.x
Acknowledgements
The authors wish to thank the Director of Dr. B.R. Ambedkar Center for Biomedical Research, University of Delhi (North Campus), Delhi, India, for providing necessary facilities. Brijendra K. Tiwari is thankful to UGC for granting Dr. D.S. Kothari Postdoctoral Fellowship.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Tiwari, B.K., Kak, G., Sharma, D., Natarajan, K. (2017). How Pathogens Survive Drug Pressure?. In: Arora, G., Sajid, A., Kalia, V. (eds) Drug Resistance in Bacteria, Fungi, Malaria, and Cancer. Springer, Cham. https://doi.org/10.1007/978-3-319-48683-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-48683-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48682-6
Online ISBN: 978-3-319-48683-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)