
Abstract
Obesity is characterized by a state of chronic, low-grade inflammation. However, excessive fatty acid release may worsen adipose tissue inflammation and contributes to insulin resistance. In this case, several novel and highly active molecules are released abundantly by adipocytes like leptin, resistin, adiponectin or visfatin, as well as some more classical cytokines. Most likely cytokines that are released by inflammatory cells infiltrating obese adipose tissue are such as tumor necrosis factor-alpha (TNF-alpha), interleukin 6 (IL-6), monocyte chemoattractant protein 1 (MCP-1) (CCL-2) and IL-1. All of those molecules may act on immune cells leading to local and generalized inflammation. In this process, toll-like receptor 4 (TLR4)/phosphatidylinositol-3′-kinase (PI3K)/Protein kinase B (Akt) signaling pathway, the unfolded protein response (UPR) due to endoplasmic reticulum (ER) stress through hyperactivation of c-Jun N-terminal Kinase (JNK) -Activator Protein 1 (AP1) and inhibitor of nuclear factor kappa-B kinase beta (IKKbeta)-nuclear factor kappa B (NF-kappaB) pathways play an important role, and may also affect vascular endothelial function by modulating vascular nitric oxide and superoxide release. Additionally, systemic oxidative stress, macrophage recruitment, increase in the expression of NOD-like receptor (NLR) family protein (NLRP3) inflammasone and adipocyte death are predominant determinants in the pathogenesis of obesity-associated adipose tissue inflammation. In this chapter potential involvement of these factors that contribute to the adverse effects of obesity are reviewed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Adipose tissue macrophages (ATMs)
- Autophagy
- Ceramide
- Endoplasmic reticulum stress
- Inducible nitric oxide synthase (iNOS)
- Lipotoxicity
- M1 adipose tissue macrophages
- Macrophage migration inhibitory factor (MIF)
- Monocyte chemoattractant protein 1 (MCP-1)
- Nuclear factor kappa B (NF-kappaB)
- Obesity
- Reactive oxygen species (ROS)
- Saturated fatty acid
- Toll-like receptor 4 (TLR4)
- Tumor necrosis factor alpha (TNF-alpha)
- Vascular endothelial growth factor (VEGF)
1 Introduction
The primary event in the sequence leading to chronic inflammation in adipose tissue is metabolic dysfunction in adipocytes. These adipocytes promote inflammation via their own cytokine and chemokine synthesis machinery. Subsequently, inflammatory process is intensified by activated adipose tissue macrophages (ATMs) (Meijer et al. 2011). White adipose tissue is the primary site for the initiation and exacerbation of obesity -associated inflammation. Ten substantial molecular mechanisms have been put forward to explain the pathogenesis of obesity-associated inflammation (Ge et al. 2014); in this respect activation of toll-like receptor 4 (TLR4) by saturated fatty acids through the TLR4/phosphatidylinositol-3′-kinase (PI3K)/Protein kinase B (Akt) signaling pathway (Lee et al. 2003), increases in intracellular protein kinase C (PKC) -theta activation-associated fatty acyl-CoA and diacylglycerol (DAG) concentration with the reduction in both insulin-stimulated insulin receptor substrate-1 (IRS -1) tyrosine phosphorylation and IRS -1 associated PI3K activity (Yu et al. 2002), lipotoxicity due to increase in fatty acid release from dysfunctional and insulin-resistant adipocytes and overstimulation of hormone-sensitive lipase (HSL) (Cusi 2012), dysregulated sphingolipid biosynthesis (Summers 2006), the unfolded protein response (UPR) due to endoplasmic reticulum (ER) stress through hyperactivation of c-Jun N-terminal Kinase (JNK) -Activator Protein 1 (AP1) and inhibitor of kB (IkB) kinase (IKK) -nuclear factor kappa B (NFkappaB) pathways (Haynes et al. 2004), systemic oxidative stress through superoxide generation from nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, oxidative phosphorylation , glyceraldehyde auto-oxidation, PKC activation, and polyol and hexosamine pathways (Manna and Jain 2015), promotion of macrophage recruitment, accumulation, and persistence in white adipose tissue of obese individuals by adipocyte death (Cinti et al. 2005), increase in the expression of NOD-like receptor (NLR) family protein (NLRP3) inflammasone and caspase-1 in adipose tissue (Koenen et al. 2011), hypoxia -induced lipolysis due to activation of hypoxia Inducible factor-1alpha (HIF-1alpha) and NF-kappaB in adipocytes and macrophages (Yin et al. 2009) and dysregulation of microRNAs (miRNAs) in adipose tissue (Ge et al. 2014) are considered as currently identified predominant determinants of obesity induced-chronic inflammation.
2 Adipocyte and Inflammation
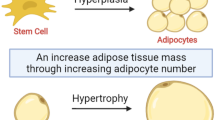
Obesity promotes inflammation in adipose tissue. Human adipocytes express many cytokines /chemokines that are biologically functional. They are able to induce inflammation and activate CD4+ T cells independent of macrophages. As mentioned above the primary event in the sequence leading to chronic inflammation in adipose tissue is the metabolic dysfunction of adipocytes (Meijer et al. 2011). Actually adipocyte hypertrophy and local hypoxia due to adipocyte expansion are two important contributing factors to the increased accumulation of macrophages in adipose tissue in the obese state. The secretion of a number of inflammation-related adipokines is upregulated by hypoxia . In this manner macrophages are phenotypically modified in response to increasing fat mass (Trayhurn 2013). Hypoxia-related adipocyte dysfunction will be discussed later.
The mRNA monocyte chemoattractant protein 1 (MCP-1) levels in human adipose tissue samples correlate with measures of adiposity (Christiansen et al. 2005). Indeed, MCP-1 expression is significantly higher in the adipocytes (Amano et al. 2014) and the MCP-1 receptor, chemokine (C-C motif) receptor 2 (CCR2), has been shown to be mostly expressed in macrophages in crown-like structures where most of proliferating ATMs are observed (Lumeng et al. 2008). These findings indicate that MCP-1 is released by adipocytes in crown-like structures, and could stimulate the proliferation of surrounding ATMs (Amano et al. 2014). In addition to MCP-1 mRNA, adiponectin, resistin, interleukin-6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) are produced by human adipocytes and are correlated with the circulating MCP-1 and body mass index (BMI) (Christiansen et al. 2005; Halberg et al. 2008). Actually subcutaneous abdominal adipose tissue MCP-1 gene overexpression is a biomarker of an inflamed adipose organ in human obesity (Christiansen et al. 2005). Expression of MCP-1 mRNA displays a 7.2-fold increase in obese mice as compared with normal ones, leading to substantially elevated MCP-1 protein levels in adipocytes (Takahashi et al. 2003). Thereby MCP-1 expression in adipose tissue attracts the monocytes bearing chemokine receptor CCR2 and contributes to the macrophage infiltration in diet- induced obese and insulin resistance animals (Kanda et al. 2006; Linton and Fazio 2003). Nearly half of the cell population in human adipose tissue consists of preadipocytes which are the undifferentiated precursors of mature adipocytes (Guo et al. 2007). Preadipocytes and pro-inflammatory macrophages share numerous functional or antigenic properties (Charrière et al. 2003). Eventually preadipocytes have a heightened inflammatory cytokine response, in particular MCP-1 expression following acute high saturated fatty acid and monounsaturated fatty acid exposure, when compared to mature adipocytes. Furthermore, preadipocytes exert a predominant role, via MCP-1, to macrophage recruitment in adipose tissue and contribute to postprandial inflammatory responses (Dordevic et al. 2014). MCP-1 over-expression in adipose tissue is under the control of the AP2 gene promoter and exhibits insulin resistance , macrophage infiltration into adipose tissue, and increased hepatic triglyceride content (Kanda et al. 2006). The abundance of macrophage infiltration in adipose tissue shows a close relationship between adipocyte size. Influence of adipocyte size on adipocyte function may be transmitted through a paracrine pathway involving ATMs (Weisberg et al. 2003). It is thought that macrophage accumulation in visceral omental and subcutaneous fat depots of obese humans positively correlated with the diameters of the fat cells in each depot. However omental macrophage infiltration is greater in comparison to the subcutaneous white adipose tissue , despite the mean diameter of omental adipose cells is significantly smaller than that of the subcutaneous cells (Cancello et al. 2006). In addition to MCP-1 , some more classical cytokines are abundantly released by adipocytes like TNF-alpha , interleukin-6 (IL-6) , IL-1 as well as several novel and highly active molecules released like adiponectin , leptin, resistin or visfatin. All may lead to local and generalized inflammation in adipose tissue (Guzik et al. 2006). The negative regulation of the nuclear hormone receptor peroxisome proliferator-activated receptor-gamma (PPAR-gamma) is important in mediating the effects of inflammatory cytokines. Furthermore, PPAR-gamma is an essential transcriptional regulator of adipogenesis and is required for maintenance of mature adipocyte function (Tamori et al. 2002). Cytokines have two dramatic effects on adipocyte function; increase in lipolysis and decrease in triglyceride synthesis. Downregulation of PPAR-gamma could strongly contribute to these effects of inflammatory cytokines on adipocytes. On the other hand, TNF-alpha can affect PPAR-gamma at multiple levels, including the transcription, translation and turnover of PPAR-gamma mRNA and protein (Guilherme et al. 2008). PPAR-gamma mRNA is rapidly degraded in adipocytes (Christianson et al. 2008). In addition to adipocytes, TNF-alpha is also produced by macrophages within adipose tissue of obese subjects. This process requires both the IKK-beta–NF-kappaB and the JNK–mitogen-activated protein kinase kinase kinase kinase-4 (MAP4K4)- AP1 signalling pathways (Guilherme et al. 2008). Indeed, free fatty acid (FFA )-mediated induction of proinflammatory cytokines in macrophages requires JNK1 activity (Solinas et al. 2007). Inflammation could also cause insulin resistance by a direct action of TNF-alpha on muscle insulin signaling , which is triggered by serine/threonine (Ser/Thr) phosphorylation of IRS proteins. On the other hand, in adipocytes, MAP4K4 contributes to the regulation of glucose metabolism as a suppressor of PPAR-gamma and adipogenesis . TNF-alpha -induced insulin-resistance on glucose uptake is rescued by MAP4K4 silencing (Bouzakri and Zierath 2007). Furthermore, the IL-6 concentration in adipose tissue is approximately 100-fold higher than that in plasma. This increase in IL-6 production after hypertrophic enlargement of the adipose cells induces adipose tissue dysfunction with impaired differentiation of the pre-adipocytes (Sopasakis et al. 2004). While IL-6 is associated with visceral adiposity, TNF-alpha rather indicates an association with total body fatness (Cartier et al. 2008). However, soluble tumour necrosis factor receptor 2 (sTNFR2) levels are more closely related to visceral adipose tissue accumulation than to the total adiposity (Cartier et al. 2010). TNF production by human adipose tissue is also regulated by weight loss in obese subjects. In addition, there is an inverse correlation between adipose TNF-alpha expression and adipose lipoprotein lipase activity (Kern et al. 1995). Hence, over-expression of TNF-alpha in subcutaneous adipose tissue of obese women is proportional to the magnitude of the fat depot. Thereby, TNF-alpha prevents further fat deposition by regulating lipoprotein lipase activity and leptin production (Bulló et al. 2002). TNF-alpha and IL-6 inhibit lipoprotein lipase, and TNF-alpha additionally stimulates HSL and induces uncoupling protein expression (Coppack 2001).
In obesity -associated adipose tissue inflammation , nuclear protein high mobility group box 1 (HMGB1) is identified as an inflammatory alarmin. Thus, HMGB1 secretion in adipose tissue is two-fold more in adipose tissue from obese compared to normo-weight individuals. Moreover, this HMGB1 release is in response to inflammatory signals of obesity rather than adipocyte death (Gunasekaran et al. 2013). Upon HMGB1 release, macrophages secrete pro-inflammatory cytokines . Thus adipose tissue HMGB1 mRNA levels correlate with the expression of inflammatory markers. Moreover, insulin resistance modifies the intracellular distribution of HMGB1 in human adipocytes. HMGB1 localizes to the cytosol in obese patients instead of nucleus (Guzmán-Ruiz et al. 2014). When it is released into the extracellular space by preadipocyte, soluble HMGB1 controls the secretion of IL-6 and MCP-1 in adipose tissues through the binding to the receptor for advanced glycation end products (RAGE) and triggers inflammatory response (Nativel et al. 2013). Excessive fatty acid release may worsen adipose tissue inflammation and contributes to insulin resistance (Morigny et al. 2016). Fat cells of omental adipose tissue obtained from morbidly obese women have at least 26-fold increase in the mRNAs for HSL, lipoprotein lipase , adipose tissue triglyceride lipase, and fatty acid translocase (FAT/CD36) when compared to the non-fat cells, whereas the mRNAs for inflammatory proteins are primarily present in the nonfat cells (Fain et al. 2008). Although the inflammatory signaling pathways primarily become activated by metabolic stresses originating from inside the adipocytes themselves, obesity also overloads the functional capacity of the ER. Obesity-related ER stress in turn leads to suppression of insulin receptor signaling through hyperactivation of JNK and subsequent Ser phosphorylation of IRS -1 (Ozcan et al. 2004).
3 Adipocyte Death and Initiation of Inflammatory Response
White adipocytes are characterized by a unique morphology with unilocular lipid droplets that occupy 95% of the cell volume and thereby determine the cell size, which ranges approximately from 20 to 200 μm. Accumulation of adipose tissue depends in part on new adipocyte formation via recruitment of progenitors/preadipocytes and their differentiation into adipocytes, as well as hypertrophy of existing adipocytes (Lee et al. 2013). Adipocyte turnover is an important event for the development of hyperplasic or hypertrophic obesity . The absolute number of new hypertrophic adipocytes generation in each year is 70% less than hyperplasic adipocytes. Subcutaneous adipose hypertrophy and hyperplasia are strongly related to the total adipocyte number in adults (Arner et al. 2010). Turnover of adipocytes in adult human white adipose tissue is approximately 10% annually. Nevertheless fat cell number does not decrease in adulthood, even following long-term weight loss (Spalding et al. 2008). However chronic nutrient excess leads to visceral adipose tissue expansion and dysfunction of the adipocytes. Additionally, changing in the supporting matrix and immune cell infiltrates contribute to adipose tissue hypoxia , adipocyte stress, and ultimately adipocyte death (Revelo et al. 2014). Cell death in white adipose tissue occurs primarily by necrosis-like cell death. Further the frequency of adipocyte death is positively correlated with increased adipocyte size in obese humans (Cinti et al. 2005). Crown-like structures are formed with the accumulation of lymphocytes, macrophages, and other immune cells around dying adipocytes (Revelo et al. 2014). In severely obese patients, high B cells/T cells ratio phenotype in crown-like structures reflects long-term excess adiposity and overnutrition (McDonnell et al. 2012). Although obesity is accompanied by a substantial increase in crown-like structures in all fat depots, visceral adipose tissue contains 3.5 fold more crown-like structures when compared with the subcutaneous fat depot. Furthermore in obese mice, crown-like structures and mast cells have a similar distribution pattern in abdominal fat depots (Altintas et al. 2011). Eventually more than 90% of all macrophages in white adipose tissue of obese subjects are localized around the dead adipocytes. In contrast to adjacent viable adipocytes, perilipin immunoreactivity could not be detected on lipid droplets of adipocytes surrounded by crown like structures macrophages. Hence free lipid droplets of dead adipocytes act as persistent sites of macrophage fusion and lipid uptake (Cinti et al. 2005). Freshly isolated mature adipocytes from different adipose depots release macrophage migration inhibitory factor (MIF) approximately at a rate of 10,000 pg/ml/24 h. MIF production is positively correlated with donor BMI and most importantly it is an obesity-dependent mediator of macrophage infiltration of adipose tissue (Skurk et al. 2005). Furthermore increased expression of MIF gene in adipocytes is associated with enlarged subcutaneous abdominal adipocytes, reduced circulatory adiponectin, and impaired insulin action (Koska et al. 2009).
Recently, Wensveen et al. defined that a phenotypically distinct population of tissue-resident natural killer (NK) cells create a critical link between obesity -induced adipose stress and visceral adipose tissue inflammation . Upregulation of ligands of the NK cell-activating receptor (NCR1) on adipocytes leads to NK cell proliferation and interferon-gamma (IFN-gamma) production. Eventually macrophage polarization and insulin resistance may develop in response to obesity-induced adipocyte stress (Wensveen et al. 2015). With the development of obesity, ATMs are polarized to an M1 inflammatory phenotype . Not only the numbers of M1 ATMs and the expression of M1 marker genes, such as TNF-alpha and MCP-1 , but also the M1-to-M2 ratio were increased by high-fat diet induced obesity (Fujisaka et al. 2009; Lumeng et al. 2007). Thus, in obese subjects, when macrophages scavenge dead or dying adipocytes and free lipid droplets , they secrete IL-6 and TNF-alpha (Lumeng et al. 2007). Endocytic activities of ATMs are similar to M2 macrophages and accordingly they secrete high amounts of IL-10 and IL-1 receptor antagonist. However, basal or induced secretion of pro-inflammatory mediators, TNF-alpha , IL-6, IL-1, MCP-1 and MIP-1alpha is even higher in ATMs than in pro-inflammatory M1 macrophages (Zeyda et al. 2007). Indeed two subsets of macrophages are identified in adipose tissue of obese subjects based on their surface expression of F4/80; F4/80(high) and F4/80(low) (Guri et al. 2008). The F4/80(high) ATMs subset exhibits an enhanced ability to produce pro-inflammatory cytokines , PPAR-gamma, CD36 and TLR4 whereas the F4/80(low) subset expresses IL-4 and produces lower concentrations of cytokines (Bassaganya-Riera et al. 2009). Obese adipose tissue shows the chronic inflammation-specific macrophage infiltration with the large numbers of CD8+ effector T cells and pro-inflammatory macrophages, whereas the numbers of regulatory and CD4+ helper T cells are decreased (Xu et al. 2003). In addition to the increased levels of CD8+ T cells and macrophages, the CD11c+ dendritic cells (DCs) phenotype , F4/80(low) also increases in the adipose tissues of diet-induced obesity. In this manner there may be a link between adipose tissues derived DCs and adipose tissue inflammation which is caused by pro-inflammatory mediators of Th17 cells. Thus adipose tissues derived DCs express higher levels of IL-6, transforming growth factor-beta (TGF-beta), IL-23 that are essential cytokines for Th17 cells proliferation or differentiation (Chen et al. 2014). Indeed during obesity DCs for human CD11c+ CD1c+ and for mouse CD11c(high)F4/80(low) accumulate in adipose tissue. In patients, the presence of CD11c+ CD1c+ DCs correlates with the BMI and with an elevation in Th17 cells. Similarly, CD11c(high)F4/80(low) DCs from obese mice induce Th17 differentiation (Bertola et al. 2012).
Actually classically activated M1 macrophages create a pro-inflammatory environment that blocks adipocyte insulin action, contributing to the development of insulin resistance (Harford et al. 2011). In fact, resident ATMs uniformly express markers of alternative activation. Alternatively activated phenotype (M2a) markers are macrophage galactose N-acetyl-galactosamine specific lectin 1 (MGL1) and IL-10 (Dupasquier et al. 2006). M2a marker, MGL1+ ATMs could promote new fat cell formation. The inhibition of these processes by M1 macrophages may promote adipocyte hypertrophy and death, which has been closely linked to ATMs accumulation (Strissel et al. 2007). Obesity does not significantly alter the localization of M2a marker MGL1+ ATMs in adipose tissue but instead superimposes a new population of M1-polarized M2a marker MGL1 − CCR2+ ATMs on these resident cells. A decrease in the quantity of M2a ATMs combined with the appearance of these “recruited ATMs” ultimately shifts the balance of M1/M2a ATMs to create a more proinflammatory environment in adipose tissue. With diet-induced obesity , M2a marker, MGL1+ ATMs remain in interstitial spaces, whereas a population of M2a marker MGL1 − CCR2+ ATMs with high M1 and low M2a gene expression is recruited to clusters surrounding necrotic adipocytes. The rate of recruitment of new macrophages to M2a marker MGL1 − ATM clusters is significantly faster than that of interstitial M2a marker MGL1+ ATMs (Lumeng et al. 2008). Eventually M1-polarized macrophages are characterized by the expression of proinflammatory mediators. This phenotype forms crown-like structures in obese adipose tissue and scavenges dead adipocytes and its potentially cytotoxic remnants. Thus, ATMs synergistically increase their own absorptive capacities (Cinti et al. 2005; Lumeng et al. 2008). Crown like structures are chronic sources of TNF-alpha. Even low frequency of adipocyte death may be sufficient to cause adipose tissue inflammation and promote insulin resistance (Strissel et al. 2007).
Furthermore, macrophage markers CD68 and CD14 are closely correlated with the total number of macrophages. Consistent with this, proinflammatory markers IL-6 , MCP-1 , and TNF-alpha are significantly elevated in adipose tissue from obese subjects compared with lean subjects . A change in the macrophage phenotype may occur by obesity with a preponderance of anti-inflammatory (M2) markers and a decrement of proinflammatory (M1) markers in adipose tissue. It is suggested that this event may be a protective mechanism in counteracting with the enhanced inflammation seen in the adipose tissue in association with obesity (Fjeldborg et al. 2014).
Adipocytes-derived saturated fatty acids are endogenous ligands that induce macrophage-inducible C-type lectin (Mincle) mRNA expression in macrophages partly through the TLR4 /NF-kappaB pathway in the adipose tissue of obese subjects (Ichioka et al. 2011). At first, saturated fatty acids are released from hypertrophied adipocytes through the macrophage-induced adipocyte lipolysis, subsequently, these result in the activation of the TLR4/NF-kappaB pathway (Suganami et al. 2007). Actually, Mincle serves as a receptor for SAP130, a component of small, nuclear ribonucloprotein as a Mincle ligand that is released from damaged cells to sense cell death and induce proinflammatory cytokine production (Yamasaki et al. 2008). Scavenging of adipocyte debris is an important function of white ATMs in obese individuals. Hence, dead adipocytes associated crown-like structure or macrophage syncytia in the adipose tissue is a hallmark of chronic inflammation which is coincidental with increased TNF-alpha gene expression (Cinti et al. 2005; Strissel et al. 2007). Collectively, Mincle may play a role in sensing adipocyte death to induce proinflammatory cytokine production in the adipose tissue in obesity (Ichioka et al. 2011). In this respect, Mincle in macrophages is crucial for crown-like structure formation, expression of fibrosis -related genes and myofibroblast activation. Thereby, Mincle is localized to macrophages in crown-like structure, in a number which correlates with the extent of interstitial fibrosis (Tanaka et al. 2014). Additionally, a positive link is found between the mast cell number and fibrosis in white adipose tissue of obese nondiabetic patients (Divoux et al. 2012). The immature mast cells that infiltrate into adipose tissue at the nonobese stage, later gradually mature with the progression of obesity. Anti-mast cell protease 6 (anti-MCP-6) is secreted from mature mast cells and induces collagen 5 expression in obese adipose tissue, which may contribute to the adipose tissue fibrosis (Hirai et al. 2014). Obese subjects have more total fibrosis in omental white adipose tissue and have more pericellular fibrosis around adipocytes, when compared to lean subjects. Some adipocytes are engulfed in fibrosis and stained perilipin negative. This indicates that fibrosis is a response to dysfunctional or dying adipocytes. Thus, omental white adipose tissue fibrosis negatively correlates with omental adipocyte diameters and with triglyceride levels (Divoux et al. 2010). Adipocytes hypertrophy induces the expression of HIF-1alpha, and increased lysyl oxidase, which is one of the target genes of HIF-1alpha and mediates cross linking of collagens for adipose tissue fibrosis. Furthermore, high fat diet induces the expression of IL-13 and mediates the deposition of collagen in adipose tissue (Halberg et al. 2009). On the other hand, the extracellular matrix plays important roles in maintaining adequate adipose tissue function. Metalloelastase, MMP-12 is produced predominantly by ATMs and can be induced with both short- and long-term high fat diet challenges. MMP-12 activation leads to lipolysis (Martinez-Santibanez et al. 2015). In humans, MMP-12 expression correlated positively and significantly with insulin resistance , TNF-alpha expression, and the number of CD14+ CD206+ macrophages in adipose tissue. Despite its extensive expression in M2 macrophages and DCs, it is detected at low levels in M1 macrophages (Lee et al. 2014).
4 Inflammasome Activation
Lipid spill over from dysfunctional or necrotic adipocytes participates in dyslipidemia and lipotoxicity . Lipid metabolites serve as a source of endogenous danger-associated molecular patterns (DAMPs) that can be sensed by specific pattern recognition receptors (PRRs). Subsequently PRRs mediate activation of the cells of innate immune system (Lukens et al. 2011). In this context, obesity is associated with the increased ceramides, saturated fatty acids , reactive oxygen species (ROS) , mitochondrial dysfunction and adenosine triphosphate (ATP) release from necrotic adipocytes. All these factors have been shown to activate the NLRP3 inflammasome in macrophages (Lukens et al. 2011). The NLRP3 inflammasome is the most fully characterized inflammasome and consists of the NLRP3 scaffold, the apoptosis-associated speck-like protein containing a C-terminal – caspase recruitment domain (CARD) (ASC, encoded by PYCARD gene) adaptor, and caspase-1. Actually NLRP3 is activated upon exposure to pathogen-associated molecular patterns (PAMPs), DAMPs and by host-derived molecules (Schroder and Tschopp 2010) multiprotein complexes localize within the cytoplasm of the cell that are responsible for the maturation of proinflammatory cytokines and the activation of a highly inflammatory form of cell death, pyroptosis (Abais et al. 2015). Indeed, pyroptosis is a caspase-1-dependent programmed cell death, which features rapid plasma membrane rupture, DNA fragmentation, and release of proinflammatory intracellular contents (Xu et al. 2014). In obesity, hypertrophic adipocytes exhibit cholesterol crystals and accumulation of calcium and ROS, that might trigger the NLRP3 inflammasome pathway with subsequent massive activation of caspase-1, which ultimately results in adipocyte death from pyroptosis (Giordano et al. 2013). Dendritic cells [CD11c+CD11b+F4/80−] are exposed to high fat diet , vary toward a pro-inflammatory phenotype with increased IL-1beta secretion,Interleukin 1 receptor, type 1 (IL-1R1), TLR4 , and caspase-1 expression (Reynolds et al. 2012). Ectopic triglyceride accumulation, adipocyte size, and macrophage infiltration in adipose tissue and circulating adipokine levels are dependent on the NLRP3 inflammasome activation. Thereby inflammasomes have critical functions in obesity and insulin resistance (Stienstra et al. 2011). In this regard, NLRP3 inflammasome senses lipotoxicity -associated increases in intracellular ceramide as an obesity-associated danger signal to induce caspase-1 cleavage in macrophages and adipose tissue (Vandanmagsar et al. 2011). Actually, ceramide, a product derived from long-chain saturated fatty acids inhibits insulin-stimulated activation of Akt and translocation of GLUT4 (Chavez et al. 2003). Diet containing high amount of saturated fatty acid enhances IL-1beta-mediated adipose tissue inflammation and insulin resistance . Actually high-saturated fatty acid consumers display reduced insulin sensitivity with elevated NLRP3 inflammasome (PYCARD and caspase-1 multiprotein complex) expression in adipose tissue (Finucane et al. 2015). Eventually, activation of NLRP3 inflammasome by damaged mitochondria leads to the caspase-1-dependent secretion of the proinflammatory cytokines IL-1 beta and IL-18 (Benetti et al. 2013; Gurung et al. 2015). Processing of IL-1beta and IL-18 requires cleavage by caspase-1, a cysteine protease, which is regulated by the inflammasomes. Caspase-1 and IL-1beta activity in adipose tissue is increased both in diet-induced and genetically induced obese animals. Under these conditions caspase-1 is upregulated during adipocyte differentiation and directs adipocytes toward a more insulin-resistant phenotype . In this context, the inflammasome plays an important role in regulation of adipocyte function and insulin sensitivity through caspase-1 activity (Stienstra et al. 2010). Abdominal subcutaneous adipose tissue biopsies from obese humans revealed that NLRP3 inflammasome activation and a Th1 shift in the T cell population in adipose tissue is related to insulin resistance , which may be explained by adipose tissue inflammatory processes (Goossens et al. 2012).
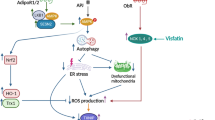
Furthermore, lipid metabolites including ceramide and diacylglycerol (DAG) can activate NAPDH oxidase and enhance ROS generation (Brookheart et al. 2009). The adenosine monophosphate-activated protein kinase (AMPK) has emerged as an essential mediator of fatty acid metabolism (Steinberg and Kemp 2009). AMPKalpha2 functions as a physiological suppressor of NADPH oxidase and ROS production (Wang et al. 2010). Thereby, FFA -enhanced ROS generation is negatively regulated by AMPK activation, which results in an increased beta oxidation of FFAs in mitochondria and decreases the lipid loading (Wang et al. 2010). Nevertheless, NAPDH oxidase-dependent generation of ROS plays an essential role in NLRP3 inflammasome activation (Schroder and Tschopp 2010). AMPK positively regulates autophagy by directly activating Unc-51 Like Autophagy Activating Kinase 1 (ULK1) (Egan et al. 2011). Saturated fatty acid-decreased AMPK activity leads to defective autophagy and the accumulation of mitochondrial ROS, by a deficiency in the clearance of dysfunctional mitochondria. Thus, excess saturated fatty acid accumulation leads to inflammasome activation and IL-1beta release through AMPK -autophagy-ROS signaling pathway (Wen et al. 2011). In fact, several autophagic genes are downregulated in obesity . These include Atg7, Becn1, Ulk1 and Atg5. Thus, activation complex 1 of the mechanistic target of rapamycin complex 1 (mammalian target of rapamycin complex 1, mTORC1) inhibits autophagy through post-translational phosphorylation of ULK1 (Sciarretta et al. 2012). Inhibition of autophagy increases the generation of ROS . In addition to ROS production and the NLRP3 inflammasome activation, FFA overload also induces ER stress, apoptosis and inflammation (Legrand-Poels et al. 2014). Furthermore, ER stress activates the NLRP3 inflammasome in human macrophages, resulting in the subsequent release of IL-1beta by human macrophages, with an activation mechanism similar to that of other known NLRP3 activators, requiring ROS generation. UPR -independent ER stress response regulates NLRP3 inflammasome activation (Menu et al. 2012).
5 Plasminogen Activator Inhibitor
Expression of enzymes involved in ceramide generation, neutral sphingomyelinase (NSMase), acid sphingomyelinase (ASMase), and serine-palmitoyl-transferase (SPT) mRNA are elevated in obese adipose tissues (Samad et al. 2006). While sphinganine ceramide significantly decreases, dihydroceramide content increases in obese humans. In these cases, Ser palmitoyltransferase and ceramidases activities (both neutral and acidic) are found elevated, whereas sphingomyelinase activities (both neutral and acidic) are reduced (Błachnio-Zabielska et al. 2012). However, high fat diet -mediated increase in ceramide is attenuated in mice lacking plasminogen activator inhibitor-1 (PAI-1). This evidence shows that PAI-1 plays a direct role in sphingolipid metabolism , which is augmented in a setting of obesity , where PAI-1 levels are elevated (Shah et al. 2008). Ceramide can induce the expression of PAI-1 and inflammatory cytokines and chemokines from adipocytes (Samad et al. 2006). The plasminogen activation system consists of urokinase plasminogen activator (uPA) and tissue plasminogen activator (tPA). Proteolitic effect of plasmin is inhibited by impeding plasminogen-plasmin cleavage. Four known protein inhibitors of uPA/tPA are defined as PAI-1, PAI-2, PAI-3 and nexin. PAI-1 is a fast-acting, highly specific inhibitor of tPA and uPA (Jankun and Skrzypczak-Jankun 2009). Obesity is characterized by elevated levels of circulating PAI-1. In fact, adipose tissue evolves into a major PAI-1 producing organ by gaining capacity during adipocyte differentiation . This is mediated by a decrease in E2F1 protein levels which is well-known transcriptional repressor of PAI-1 (Venugopal et al. 2007). However, the greater fat cell size and the adipose tissue mass has the greater contribution to circulating PAI-1 production of adipose tissue. However, visceral adipose tissue has a higher capacity to produce PAI-1 than subcutaneous adipose tissue . In addition to FFA , PAI-1 synthesis is most potently upregulated by TNF-alpha and transforming growth factor-beta (TGF-beta). Reasonably impaired fibrinolysis in obesity is probably due to an increased expression of PAI-1 in adipose tissue (Skurk and Hauner 2004). Indeed, exposure of adipocytes to TNF-alpha induces PAI-1 mRNA by increasing the rate of transcription of the PAI-1 gene. This dramatic induction of PAI-1 mRNA in response to TNF-alpha is mediated by p38, PI3K, tyrosine kinases, and the transcription factor NF-kappaB (Pandey et al. 2005). Increased FFA concentrations in blood of nondiabetic, overweight subjects markedly raise circulating PAI-1 concentrations, with a concomitant increase in the expression of the PAI-1 gene in adipose tissue. This means that adipocytes also stimulate PAI-1 expression in macrophages and potentiates the increase in PAI-1 messenger RNA expression in response to FFAs (Kishore et al. 2010). PAI-1 inhibition results in enhanced adipocyte differentiation and is also associated with significantly upregulated PPAR-gamma, CCAAT enhancer-binding protein-alpha (C/EBPalpha), and adipocyte-selective fatty acid-binding protein (aP2) expression in differentiated adipocytes. Conversely, PAI-1 overexpression in differentiated adipocytes inhibits adipocyte differentiation and is accompanied by decreases in PPARgamma, C/EBPalpha, and aP2 levels (Liang et al. 2006).
6 Activation of Toll-like Receptor 4 by Saturated Fatty Acids
Nutritional fatty acids, whose circulating levels are often increased in obesity , activate TLR4 signaling in adipocytes and macrophages and that the capacity of fatty acids to induce inflammatory signaling in adipose cells or tissue and macrophages (Shi et al. 2006). Fatty acid levels are elevated in obesity and induce inflammatory pathways by an unknown mechanism. Low grade inflammation leads to the development of insulin and leptin resistance . Recent studies show that these effects could be mediated through the activation of TLR (Fresno et al. 2011).
TLR4 -induced ER stress may be an obligatory step mediating the saturated fatty acids -mediated endothelial dysfunction (Kim et al. 2015b). TLR4-deficiency protects the mice against high-fat diet-induced ER stress. The mRNA levels of NF-kappaB regulated TNF-alpha , IL-1beta and IL-6 are not changed after hig-fat-diet. Similarly, phospho-IKB-alpha (Ser 32) is not changed after signaling downstream of TLR4. These evidences show that TLR4 is essential for the development of high-fat diet induced ER stress (Pierre et al. 2013). In fact, fatty acids derived from lipolysis of hypertrophic adipocyte are taken up by macrophages and stored as triacylglycerol droplets (Caspar-Bauguil et al. 2015). A paracrine loop involving FFA and TNF-alpha between adipocytes and macrophages establishes a vicious cycle that aggravates inflammatory changes in the adipose tissue (Suganami et al. 2005). Endogenous fatty acids, which are released from adipocytes via the beta3-adrenergic stimulation, activate the TLR4/NF-kappaB pathway. In this respect, large quantities of saturated fatty acids release from hypertrophied adipocytes via the macrophage-induced adipocyte lipolysis. These ligands, induce the inflammatory changes in both adipocytes and macrophages through TLR4 /NF-kappaB activation (Suganami et al. 2007). Actually, saturated fatty acids released via adipocyte lipolysis stimulate TLR4 signaling. Activating transcription factor 3 (ATF3) is a member of the ATF/cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) and its stimulation acts as a transcriptional repressor of saturated fatty acids/TLR4 signaling in macrophages of obese adipose tissue (Suganami et al. 2009). TNF receptor-associated factor 6 (TRAF6) is stimulated in response to fatty acid -induced inflammation . Following activation of TLR4 by saturated fatty acid, TRAF6 is upregulated. Ultimately, TRAF6 induces proinflammatory cytokine production (Tian et al. 2015). Generally, miRNAs prevent protein synthesis through degrading mRNAs and inhibit their translation. In this context, miRNAs have critical roles in TLR4 pathway (Friedman et al. 2009). During TLR4 activation by saturated fatty acids, miRNA-194 expression is downregulated. In contrast, overexpression of miRNA -194 directly decreases TRAF6 expression and attenuates the release of TNF-alpha in saturated fatty acid-activated THP-1 monocytes (Tian et al. 2015). miRNA-146 also directly decreases IL-1 receptor-associated kinase 1 (IRAK1) and TRAF6 expression and attenuates the release of proinflammatory cytokines through the inactivation of NF-kappaB P65 in hypoxia /reoxygenation-induced macrophages (Jiang et al. 2014). Adipocyte hypertrophy and local hypoxia are two of the most important factors that contribute to the accumulation of macrophages in the adipose tissue of obese subjects (Trayhurn 2013). During obesity , adipocytes release pro-inflammatory factors like chemokine (C-C) motif) ligand 2 (CCL2), TNF, or FFA that induce the recruitment and activation of ATMs (Olefsky and Glass 2010). Subsequently, ATMs release TNF-alpha , IL-6 , IL-1beta, migration inhibitory factor and resistins. All these proinflammatory factors amplify this inflammatory circuit and leads to insulin resistance by blocking insulin action in adipocytes (Biswas and Mantovani 2012). In particular, IL-6 impairs the activities of the PI3K/Akt/glycogen synthase kinase (GSK) pathway via down-regulating miRNA-200 s. This suggests that miRNA -200 s is a link between IL-6 and insulin resistance (Dou et al. 2013).
TLR4 deficiency attenuates adipose tissue inflammation in obesity but does not impede the accumulation of ATMs . Furthermore, TLR4 signaling plays a direct role in mediating adipose tissue macrophage phenotype in diet-induced obesity (Orr et al. 2012). On the other hand, the TLR and inflammasome pathways drive low-grade inflammation in obesity-related metabolic diseases. Saturated FFAs activate TLRs in macrophages and adipocytes. Thereby, an inflammatory cascade is triggered (Masters et al. 2011). Activation of TLR signaling in adipocytes causes the insulin signaling pathway inhibition, whereas upregulation of TLR4 in macrophages impairs inflammatory response to saturated FFAs and protects against insulin resistance during obesity (Nguyen et al. 2007; Olefsky and Glass 2010). Excessive fatty acid release due to increased basal lipolysis influences whole body insulin sensitivity and enhances adipose tissue inflammation. Ultimately, excessive circulating fatty acids may ectopically accumulate in insulin-sensitive tissues and impair insulin action (Morigny et al. 2016). Unfolded proteins accumulate in the ER under conditions of cellular stress induced by over-nutrition (Schröder and Kaufman 2005). The UPR functioning depends essentially on three main ER sensors involving the activation of ATF-6, protein kinase R (PKR)-like eukaryotic initiation factor 2α kinase (PERK) and inositol requiring enzyme 1 (IRE-1) . All of them together act to restrict new protein synthesis and increase the production of chaperones (Lowe et al. 2012). UPR activation links to the development of insulin resistance through JNK activation, inflammation, and oxidative stress (Brasaemle 2007). ER stress is also associated with activation of NF-kappaB and that alpha subunit of translation initiation factor 2 (eIF2alpha) phosphorylation (Deng et al. 2004) While IRS -1 tyrosine phosphorylation reduces, IRS -1 Ser phosphorylation (pS) is enhanced in obese animals compared to lean ones. In these animals IRS -1 concentration remains unchanged, IKKalpha/beta pS and JNK theronine/tyrosine phosphorylation is increased (Zolotnik et al. 2012). Thus, eIF2alpha phosphorylation-dependent NF-kappaB activation associated with decreased levels of the inhibitor IKKalpha protein (Deng et al. 2004). Increased IKKbeta Ser phosphorylation and IRS -1 Ser phosphorylation, both of which independently can have deleterious effects on insulin-stimulated PI3K activation in high-fat diet (Yaspelkis et al. 2009). Saturated fatty acid-induced NF-kappaB activation is partly mediated through the TLR4 /PI3K/Akt signaling pathway . Furthermore, saturated and polyunsaturated fatty acids reciprocally modulate the activation of TLR4 and its downstream signaling pathways involving MyD88/IRAK/TRAF6 and PI3K/Akt . Eventually, TLR4-mediated target gene expression and cellular responses are also differentially modulated by saturated and unsaturated fatty acids (Lee et al. 2003).
7 Adipose Tissue Inflammation and Insulin Resistance
Obesity-associated hyperinsulinemia drives adipose tissue inflammation which contributes to factors that suppress insulin-stimulated de novo lipogenesis and systemic insulin sensitivity (Pedersen et al. 2015). Adipokines and chemokines are key mediators that play crucial roles in crosstalk between adipocytes and macrophages in the adipose tissue inflammation (Bai and Sun 2015). TLR4 is activated with the saturated fatty acids, which are released from hypertrophied adipocytes. In this case, macrophage-induced adipocyte lipolysis stimulates NF-kappaB signaling and expression of TNF-alpha and IL-6 . Eventually, both adipocyte- and macrophage-induced inflammatory changes through NF-kappaB activation provokes insulin resistance in obesity (Song et al. 2006; Suganami et al. 2007). M1 ATMs exacerbate local inflammation by producing TNF-alpha, IL-6 and IL-1beta. These cytokines subsequently promote insulin resistance . Furthermore, macrophage migration inhibitory factor is enhanced during obesity. This chemokine is directly associated with the degree of peripheral insulin resistance (Finucane et al. 2012), whereas its deficiency partially protects from high-fat diet induced insulin resistance by attenuating macrophage infiltration (Finucane et al. 2014). Infiltration of cytotoxic T-cells into obese adipose tissue precedes macrophage accumulation. T-cell-derived cytokines promote the recruitment and activation of M1 macrophages . Activated M1 macrophages augment adipose tissue inflammation and insulin resistance (Harford et al. 2011). Moreover, tissue-resident immune cells play a major role in the regulation of obesity-induced inflammation. Besides the adipose tissue macrophages and other pro-inflammatory cells including neutrophils, Th1 CD4+ T cells, CD8+ T cells, B cells, DCs, and mast cells, however, adipose tissue also contains anti-inflammatory cells that counteract to the pro-inflammatory immune cells (Lee and Lee 2014). Compared with adipose tissue from lean subjects, adipose tissue from obese subjects contained increased areas of fibrosis , which correlated inversely with insulin sensitivity. The majority of macrophages are associated with fibrosis. Macrophages in crownlike structures are predominantly M1, but those in fibrotic areas are M2 phenotype with a lower level of IL-1 expression and a higher ratio of IL-10 to IL-12. Thus, adipose tissues of insulin-resistant obese humans demonstrate increased fibrosis with M2 macrophage abundance (Spencer et al. 2010). Human omental adipose tissue fibrosis in severe obesity is consistent with a higher degree of insulin resistance and also contains M2 macrophages. (Guglielmi et al. 2015). Particularly, protein inhibitor of activated signal transducer and activator of transcription 1 (STAT1) (PIAS1) functions in the innate immune system and is a key regulator of the inflammation cascade. PIAS1 inhibits the macrophage infiltration in adipose tissue, thus suppressing amplification of the inflammation cascade, which in turn improved insulin sensitivity (Liu et al. 2015). Adipose tissue from insulin-resistant obese have three-fold to ten-fold increases in numbers of CD4+ T cells that produce interleukin IL-22 and IL-17 compared with metabolically normal insulin-sensitive obese subjects (Fabbrini et al. 2013). Obese human adipose tissue-derived stem cells also induce Th17 promotion and monocyte activation. This proinflammatory environment inhibits insulin response of adipocyte (Eljaafari et al. 2015).
Collectively, there are a number of potential biochemical mechanism for the contribution of proinflammatory signaling to insulin resistance (Wellen and Hotamisligil 2005). In particular, JNKs are activated within numerous metabolically important cells and mediate obesity -associated disruptions in metabolic homeostasis in obesity (Pal et al. 2016). Indeed, FFAs can activate macrophages and bone marrow-derived DCs via TLR2-, TLR4- and JNK-dependent inflammatory pathways. Metabolic pathways utilize immune signaling mechanisms to trigger the activation of CD11c+ proinflammatory cells. Consequently, FFA -induced, TLR-mediated activation of JNK proinflammatory pathways in CD11c+ immune cells promotes inflammation and subsequent cellular insulin resistance (Nguyen et al. 2007). Actually, JNK can be activated by FFAs through TNF-alpha -independent mechanisms. In obesity, activated JNK is a major contributor to FFA -induced cellular insulin resistance . Furthermore, TNF-alpha is a downstream effector of activated JNK (Nguyen et al. 2005). TNF-alpha, through activation of p38 MAPK and IKK , produces Ser phosphorylation of insulin resistance and IRS -1 . Actually, its tyrosine phosphorylation is impaired by insulin. Eventually the corresponding activation of PI3K and Akt, leads to insulin resistance (de Alvaro et al. 2004). TNF-alpha mediates insulin resistance by enhancing adipocyte lipolysis through the stimulation of JNK and IKKbeta /NF-kappaB pathway which may increase Ser/Thr phosphorylation. IL-1beta also contributes to insulin resistance by impairing insulin signaling in peripheral tissues and macrophages. Macrophages from insulin-resistant obese are associated with increased production. In this case, forkhead transcription factor-1 (FOXO1) correlates with elevated levels of IL-1beta mRNA. Furthermore, FOXO1 signaling through NF-kappaB induces insulin resistance by producing proinflammatory cytokines in obesity (Su et al. 2009). Leptin-induced signaling via STAT3 rapidly activates the negative feedback regulator suppressor of cytokine signaling-3 (SOCS3) , which inhibits leptin-induced signal transduction (Wunderlich et al. 2013). Among the most prominent cytokines that are over-represented in the bloodstream of obese individuals with obesity are TNF-alpha and IL-6 ; however only IL-6-induced signaling is mediated via JAK and STAT3 (Heinrich et al. 2003). In chronic JAK-STAT3-SOCS3 signaling in obesity, the IL-6 -bound receptor complex activates intracellular JAK2 subsequently leading to STAT3 activation, which in turn to elevating SOCS3 expression (Wunderlich et al. 2013). During the progression of obesity IL-6 may induce insulin resistance through two different signaling pathways. Firstly, IL-6 induces insulin resistance by activating STAT3 and upregulating the transcription of its target gene SOCS3 . These effects are consistent with the IL-6-dependent activation of extracellular-related kinase 1/2 (ERK1/2), a Ser-Thr protein kinase involved in Ser STAT3 phosphorylation (Serrano-Marco et al. 2012). Secondly, IL-6 induces insulin resistance by impairing the activation of the PI3K/Akt/GSK pathway and glycogenesis (Dou et al. 2013).
8 Oxidative Stress
Obesity can induce systemic oxidative stress through superoxide generation from NADPH oxidases, oxidative phosphorylation , glyceraldehyde auto-oxidation, PKC activation, and polyol and hexosamine pathways. Additional factors are relevant to the initiation of oxidative stress in obesity include hyperleptinemia, low antioxidant defense, chronic inflammation , and postprandial ROS generation (Manna and Jain 2015). Adipocytes from insulin resistant obese subject exhibit increased oxidative stress and impaired antioxidant defense. Additionally, proteasome activity is impaired in adipocytes of diet-induced obese mice. In fact, proteasomal dysfunction in adipocytes results from protein oxidation and protein misfolding. Actually, this event constitutes major pathogenic mechanism in the development of insulin resistance in obesity (Díaz-Ruiz et al. 2015). When cellular antioxidant and repair pathways fail to restore protein oxidative damage, cells activate additional defense mechanisms to prevent the accumulation of oxidized proteins. Oxidized protein residues are toxic for cells and threaten cell viability. Actually, proteasomes provide a second line of defense against the free radicals and oxidants (Grune and Davies 1997). Thereby the removal of damaged proteins is extremely important for the maintenance of normal cell function. In this context, the 20S proteasome functions primarily for removal of damaged proteins (Pickering and Davies 2012). The enhanced proteolytic activity of the proteasome is important for the preservation of cell viability. Activated proteasomes are better able to degrade oxidized proteins than the standard proteasome. Direct comparison of purified 20S proteasome and immunoproteasome demonstrated that the immunoproteasome can selectively degrade oxidized proteins (Pickering et al. 2010; Seifert et al. 2010). Proteasome dysfunction mediates obesity-induced ER stress, leading to hyperactivation of JNK and insulin resistance (Otoda et al. 2013). Inflammatory pathways diminish UPR function through inducible nitric oxide synthase (iNOS) -mediated S-nitrosylation of IRE-1alpha, which contributes defective IRE-1alpha activity, impaired ER function, and prolonged ER stress in obesity (Yang et al. 2015). Peroxisomal fatty acid metabolism , cytochrome P450 microsomal reactions and the mitochondrial respiratory chain generate large amount of free radicals, which is associated with an irregular production of adipokines . Eventually high ROS production and the decrease in antioxidant capacity lead to adipose tissue inflammation (Fernández-Sánchez et al. 2011). Formation of advanced glycation endproducts/advanced-lipoxidation endproducts and its precursors, including methylglyoxal (MGO), are increased in conditions characterized with hyperglycemia, hyperlipidemia and enhanced oxidative stress. This metabolic profile is typical for obesity (Gaens et al. 2013). Glutathione S-transferase A4 (GSTA4)-dependent production of proinflammatory glutathione metabolites, glutathionyl trans-4-hydroxy-2-nonenal (GS-HNE) and its metabolite glutathionyl-1,4-dihydroxynonene (GS-DHN) are produced by adipocyte in response to oxidative stress. In this case, increase in lipid peroxidation causes nonenzymatic formation of GS-HNE and GS-DHN. These metabolites have the potential to maintain obesity-induced inflammation, adipocyte dysfunction and insulin resistance (Frohnert et al. 2014). Obesity-induced oxidative stress in adipocytes as well as the presence of inflammatory changes in adipose tissue may lead to activation of tissue-resident macrophages (Frohnert and Bernlohr 2014). Upregulation of NADPH oxidase in multiple tissues occurs as a systemic response in obesity (Jiang et al. 2011). Intracellular ROS formation occurs upon stimulation of insulin in adipocytes. However, preadipocytes respond to insulin to a higher degree than that of adipocytes. Furthermore, NADPH oxidase 4 (NOX4)-mediated ROS could be released intracellularly and triggers adipogenesis in preadipocytes. Thus, long-term high fat diet results in massive obesity with adipocyte hypertrophy and hyperplasia. Subsequently, a significant decrease occurs in NOX4 expression. NOX4 is a hallmark for relative adipocyte mass and differentiation (Mouche et al. 2007). Additionally, NOX2 -derived ROS destroys insulin receptor and endothelial function in dietary obesity (Du et al. 2013). Mitochondria are not only a target for ROS produced by NADPH oxidase but also a significant source of ROS in obesity, which may stimulate NADPH oxidases (Dikalov 2011). On the one hand, saturated fatty acids induce carnitine palmitoyltransferase-1 expression, which may contribute to saturated fatty acid-induced ROS overproduction. In this process, accelerated beta-oxidation causes excessive electron flux in the respiratory chain. Excess electrons supply for mitochondrial oxidative phosphorylation contributes the over-production of ROS (Nakamura et al. 2009). On the other hand, protons can reenter the mitochondrial matrix through different uncoupling proteins, affecting the control of free radicals production by mitochondria. Disorders of the mitochondrial electron transport chain, over-generation of ROS and lipoperoxides or impairments in antioxidant defenses have important causal role in obesity-related inflammation (Martínez 2006). Moreover, increased expression of NADPH oxidases associated with obesity causes dysregulated production of adiponectin , PAI-1, IL-6 , and MCP-1 , and reduced expression of detoxifying enzymes (Kim et al. 2006). TNF-alpha itself stimulates endothelial NOS (eNOS) activity to generate nitric oxide (NO) through a pathway involving its lipid messenger, ceramide in obesity (Bulotta et al. 2001). In this case, TNF-alpha stimulates eNOS phosphorylation at Ser 1177 via PI3K-Akt signal transduction pathway (Kawanaka et al. 2002). Furthermore, under the conditions of nutrient excess and obesity, phosphorylation of eNOS is diminished due to saturated fatty acid-mediated induction of insulin resistance (Kim et al. 2008). Eventually elevated FFAs lower NO bioavailability (Steinberg et al. 2000) by downregulating the PI3K/Akt/eNOS-dependent insulin signaling axis (Madonna and De Caterina 2009). On the other hand elevated levels of FFAs in obesity trigger macrophage activation (Li et al. 2010). Macrophages and dendritic cells express MMP12 that restrains adipose tissue expansion while promoting the expression of inflammatory mediators. In inflammatory conditions, iNOS is expressed at high levels by DCs or macrophages. The expression of MMP12 mRNA by CD14+ CD206+ macrophages varies 100-fold in human subcutaneous adipose tissue . Moreover the MMP12 is localized in crown-like structures in adipose tissue. This indicated that MMP12 regulates NO generation by macrophages during inflammation (Lee et al. 2014). NADPH oxidases and xanthine oxidase, can be activated and produce large amounts of superoxide. Ten-fold increase in superoxide generation with the simultaneous NO formation will increase peroxynitrite formation by 100-fold. Under proinflammatory conditions, simultaneous production of superoxide and NO can be strongly activated to increase production 1000-fold, which will increase the formation of peroxynitrite by a 1,000,000-fold (Pacher et al. 2007). Hence, the expression of iNOS is an important aspect of obesity-associated chronic inflammation. As shown, simultaneously increased production of ROS could limit NO bioavailability because they convert NO to peroxynitrite that in turn leads to nitrosative stress (Codoñer-Franch et al. 2011).
Innate lymphoid type 2 cells, forkhead box P3 (FOXP3)-positive regulatory T (Treg) cells, and IL-10 counteract the low grade inflammation and insulin resistance in classical or metabolically healthy obesity (Pereira and Alvarez-Leite 2014). In fact, metabolically healthy obese individuals have effective immunoregulation to resist chronic obesity-related inflammation. These regulatory mechanisms could be important in the delayed onset of metabolic complications, even in extremely obese individuals (Pereira et al. 2014). Actually, Treg cells production is decreased in diet-induced obese mice. Meanwhile, high-fat diet also enhances apoptosis of Treg cells by diminishing their mitochondrial transmembrane potential. In this context, high-fat diet increases ROS productions and significantly decreases antioxidant enzymes expressions in the Treg cells of diet-induced obese mice (Wang et al. 2014). Basic unit of mitochondrial permeability transition pore consists of the voltage-dependent anion channel (VDAC), adenine nucleotide translocase (ANT) and the matrix-located regulatory subunit cyclophilin-D (Cyp-D). Oxidant-stimulated formation of VDAC-adenine nucleotide translocase-CypD complex can agglomerate a number of apoptotic proteins. The apoptotic pathway is triggered by the release of apoptotic proteins through the mitochondrial permeability transition pore openings. The pore complex-induced membrane rupture promotes the release of apoptosis-inducing factors during the apoptosis (Crompton 1999).
9 Hypoxia and Adipose Tissue Inflammation
In the adipose tissue, hypoxia is associated with the increased expression of inflammatory genes and decreased expression of adiponectin . In diet-induced obesity , subsequent reduction in body weight by calorie restriction is associated with an improvement of oxygenation and a reduction in inflammation (Ye et al. 2007). Adipocyte death occurs during rapid expansion of adipose tissue in hypoxic conditions. Indeed, adipose tissue hypoxia may result in disturbances in adipokine secretion, increased macrophage infiltration and inflammation in obese adipose tissue (Goossens 2008). Hypoxia develops when oxygen supply does not meet the demand of the surrounding tissues. Actually, oxygen diffusion is limited at most to 100 μm, whereas in obesity, hypertrophied adipocytes up to 140–180 μm in diameter. This excessive distance between the hypertrophic adipocytes creates a relatively hypoxic state . In brief, local tissue hypoxia and hypoperfusion of adipose tissue is common feature in obese subject. Adiponectin and PPAR-gamma mRNA expressions are reduced while PAI-1 level is increased in hypoxic adipocytes (Hosogai et al. 2007). Angiogenesis influences white adipose tissue expansion. However, the number of vessels per adipocyte and the expression level of receptor 2 of vascular endothelial growth factor (VEGF) is incompatible with the increasing number of adipocytes in obesity (Lemoine et al. 2012). In obese patients, even serum VEGF is positively associated with visceral fat content compared with lean subjects (Miyazawa-Hoshimoto et al. 2003), overweight/obese subjects have 44% lower capillary density and 58% lower VEGF. This might be due to lower PPAR-gamma 1 and higher collagen 6 mRNA expression, which correlate with adipose tissue oxygen concentration and adipose tissue inflammation in obesity (Pasarica et al. 2009). In addition, to induce the VEGF expression in adipocytes of obese subject the PI3K-Akt pathway is required for HIF-1alpha activation (He et al. 2011). Induction of HIF-1alpha in diet-induced obesity does not increase VEGF-A. Adipose tissue hypoxia serves as an early upstream initiator for adipose tissue dysfunction by inducing local fibrosis (Halberg et al. 2009). Furthermore, hypoxia stimulates the expression of the genes encoding angiopoietin-like protein 4, IL-6 , leptin, MIF and PAI-1 (Wang et al. 2007). In particular MIF is a potent macrophage migration inhibitory factor and is expressed by human adipocytes (Skurk et al. 2005). Thus, increased expression of MIF mRNA in inflamed adipose tissue indicates the activation of MIF gene transcription in response to adipose tissue inflammation (Kim et al. 2015a). Consequently, elevated MIF expression enhances angiogenesis as well as macrophage recruitment (Wang et al. 2007), whereas, MIF deficiency partially protects the organism from high-fat diet induced insulin resistance by attenuating macrophage infiltration to adipose tissue, ameliorating adipose inflammation (Finucane et al. 2014).
10 Conclusion
Lipid overload in obesity can indirectly stimulate the pro-inflammatory state. In addition to peripheral tissues, the hypothalamus is also similarly vulnerable to ectopic lipid accumulation and lipotoxicity (Lam et al. 2005). Excess lipids in the adipocyte increase the substrate load, but inefficient mitochondrial oxidative phosphorylation leads to generation of ROS that can damage mitochondrial constituents in obesity-associated inflammation (Goh et al. 2016). In this respect, suggested therapeutic measures in obesity should involve the mechanisms that have been described regarding the corrective effects on AMPK /IRS -1 /PI3K and TLR4 /PI3K/Akt signaling pathways , production and release of pro-inflammatory adipokines, both mitochondrial dysfunction and mTOR hyperactivation, abundantly released cytokines by adipocytes and insulin resistance .
References
Abais, J.M., M. Xia, Y. Zhang, K.M. Boini, and P.-L. Li. 2015. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxidants & Redox Signaling 22: 1111–1129. doi:10.1089/ars.2014.5994.
Altintas, M.M., A. Azad, B. Nayer, G. Contreras, J. Zaias, C. Faul, J. Reiser, and A. Nayer. 2011. Mast cells, macrophages, and crown-like structures distinguish subcutaneous from visceral fat in mice. Journal of Lipid Research 52: 480–488. doi:10.1194/jlr.M011338.
Amano, S.U., J.L. Cohen, P. Vangala, M. Tencerova, S.M. Nicoloro, J.C. Yawe, Y. Shen, M.P. Czech, and M. Aouadi. 2014. Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metabolism 19: 162–171. doi:10.1016/j.cmet.2013.11.017.
Arner, E., P.O. Westermark, K.L. Spalding, T. Britton, M. Rydén, J. Frisén, S. Bernard, and P. Arner. 2010. Adipocyte turnover: Relevance to human adipose tissue morphology. Diabetes 59: 105–109. doi:10.2337/db09-0942.
Bai, Y., and Q. Sun. 2015. Macrophage recruitment in obese adipose tissue. Obesity Reviews 16: 127–136. doi:10.1111/obr.12242.
Bassaganya-Riera, J., S. Misyak, A.J. Guri, and R. Hontecillas. 2009. PPAR gamma is highly expressed in F4/80(hi) adipose tissue macrophages and dampens adipose-tissue inflammation. Cellular Immunology 258: 138–146. doi:10.1016/j.cellimm.2009.04.003.
Benetti, E., F. Chiazza, N.S.A. Patel, and M. Collino. 2013. The NLRP3 Inflammasome as a novel player of the intercellular crosstalk in metabolic disorders. Mediators of Inflammation 2013: 678627. doi:10.1155/2013/678627.
Bertola, A., T. Ciucci, D. Rousseau, V. Bourlier, C. Duffaut, S. Bonnafous, C. Blin-Wakkach, R. Anty, A. Iannelli, J. Gugenheim, A. Tran, A. Bouloumié, P. Gual, and A. Wakkach. 2012. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes 61: 2238–2247. doi:10.2337/db11-1274.
Biswas, S.K., and A. Mantovani. 2012. Orchestration of metabolism by macrophages. Cell Metabolism 15: 432–437. doi:10.1016/j.cmet.2011.11.013.
Błachnio-Zabielska, A.U., M. Pułka, M. Baranowski, A. Nikołajuk, P. Zabielski, M. Górska, and J. Górski. 2012. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. Journal of Cellular Physiology 227: 550–557. doi:10.1002/jcp.22745.
Bouzakri, K., and J.R. Zierath. 2007. MAP4K4 gene silencing in human skeletal muscle prevents tumor necrosis factor-alpha-induced insulin resistance. The Journal of Biological Chemistry 282: 7783–7789. doi:10.1074/jbc.M608602200.
Brasaemle, D.L. 2007. Thematic review series: Adipocyte biology. The perilipin family of structural lipid droplet proteins: Stabilization of lipid droplets and control of lipolysis. Journal of Lipid Research 48: 2547–2559. doi:10.1194/jlr.R700014-JLR200.
Brookheart, R.T., C.I. Michel, and J.E. Schaffer. 2009. As a matter of fat. Cell Metabolism 10: 9–12. doi:10.1016/j.cmet.2009.03.011.
Bulló, M., P. García-Lorda, J. Peinado-Onsurbe, M. Hernández, D. Del Castillo, J.M. Argilés, and J. Salas-Salvadó. 2002. TNFalpha expression of subcutaneous adipose tissue in obese and morbid obese females: Relationship to adipocyte LPL activity and leptin synthesis. International Journal of Obesity 26: 652–658. doi:10.1038/sj.ijo.0801977.
Bulotta, S., R. Barsacchi, D. Rotiroti, N. Borgese, and E. Clementi. 2001. Activation of the endothelial nitric-oxide synthase by tumor necrosis factor-alpha. A novel feedback mechanism regulating cell death. Journal of Biological Chemistry 276: 6529–6536. doi:10.1074/jbc.M006535200.
Cancello, R., J. Tordjman, C. Poitou, G. Guilhem, J.L. Bouillot, D. Hugol, C. Coussieu, A. Basdevant, A. Bar Hen, P. Bedossa, M. Guerre-Millo, and K. Clément. 2006. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 55: 1554–1561. doi:10.2337/db06-0133.
Cartier, A., I. Lemieux, N. Alméras, A. Tremblay, J. Bergeron, and J.-P. Després. 2008. Visceral obesity and plasma glucose-insulin homeostasis: Contributions of interleukin-6 and tumor necrosis factor-alpha in men. The Journal of Clinical Endocrinology and Metabolism 93: 1931–1938. doi:10.1210/jc.2007-2191.
Cartier, A., M. Côté, J. Bergeron, N. Alméras, A. Tremblay, I. Lemieux, and J.-P. Després. 2010. Plasma soluble tumour necrosis factor-alpha receptor 2 is elevated in obesity: Specific contribution of visceral adiposity. Clinical Endocrinology 72: 349–357. doi:10.1111/j.1365-2265.2009.03671.x.
Caspar-Bauguil, S., C.-I. Kolditz, C. Lefort, I. Vila, E. Mouisel, D. Beuzelin, G. Tavernier, M.-A. Marques, A. Zakaroff-Girard, C. Pecher, M. Houssier, L. Mir, S. Nicolas, C. Moro, and D. Langin. 2015. Fatty acids from fat cell lipolysis do not activate an inflammatory response but are stored as triacylglycerols in adipose tissue macrophages. Diabetologia 58: 2627–2636. doi:10.1007/s00125-015-3719-0.
Charrière, G., B. Cousin, E. Arnaud, M. André, F. Bacou, L. Penicaud, and L. Casteilla. 2003. Preadipocyte conversion to macrophage. Evidence of plasticity. The Journal of Biological Chemistry 278: 9850–9855. doi:10.1074/jbc.M210811200.
Chavez, J.A., T.A. Knotts, L.-P. Wang, G. Li, R.T. Dobrowsky, G.L. Florant, and S.A. Summers. 2003. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. The Journal of Biological Chemistry 278: 10297–10303. doi:10.1074/jbc.M212307200.
Chen, Y., J. Tian, X. Tian, X. Tang, K. Rui, J. Tong, L. Lu, H. Xu, and S. Wang. 2014. Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS One 9: e92450. doi:10.1371/journal.pone.0092450.
Christiansen, T., B. Richelsen, and J.M. Bruun. 2005. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. International Journal of Obesity 2005(29): 146–150. doi:10.1038/sj.ijo.0802839.
Christianson, J.L., S. Nicoloro, J. Straubhaar, and M.P. Czech. 2008. Stearoyl-CoA desaturase 2 is required for peroxisome proliferator-activated receptor gamma expression and adipogenesis in cultured 3 T3-L1 cells. The Journal of Biological Chemistry 283: 2906–2916. doi:10.1074/jbc.M705656200.
Cinti, S., G. Mitchell, G. Barbatelli, I. Murano, E. Ceresi, E. Faloia, S. Wang, M. Fortier, A.S. Greenberg, and M.S. Obin. 2005. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. Journal of Lipid Research 46: 2347–2355. doi:10.1194/jlr.M500294-JLR200.
Codoñer-Franch, P., S. Tavárez-Alonso, R. Murria-Estal, J. Megías-Vericat, M. Tortajada-Girbés, and E. Alonso-Iglesias. 2011. Nitric oxide production is increased in severely obese children and related to markers of oxidative stress and inflammation. Atherosclerosis 215: 475–480. doi:10.1016/j.atherosclerosis.2010.12.035.
Coppack, S.W. 2001. Pro-inflammatory cytokines and adipose tissue. The Proceedings of the Nutrition Society 60: 349–356.
Crompton, M. 1999. The mitochondrial permeability transition pore and its role in cell death. The Biochemical Journal 341(Pt 2): 233–249.
Cusi, K. 2012. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 142: 711–725. doi:10.1053/j.gastro.2012.02.003.e6
de Alvaro, C., T. Teruel, R. Hernandez, and M. Lorenzo. 2004. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. The Journal of Biological Chemistry 279: 17070–17078. doi:10.1074/jbc.M312021200.
Deng, J., P.D. Lu, Y. Zhang, D. Scheuner, R.J. Kaufman, N. Sonenberg, H.P. Harding, and D. Ron. 2004. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Molecular and Cellular Biology 24: 10161–10168. doi:10.1128/MCB.24.23.10161-10168.2004.
Díaz-Ruiz, A., R. Guzmán-Ruiz, N.R. Moreno, A. García-Rios, N. Delgado-Casado, A. Membrives, I. Túnez, R. El Bekay, J.M. Fernández-Real, S. Tovar, C. Diéguez, F.J. Tinahones, R. Vázquez-Martínez, J. López-Miranda, and M.M. Malagón. 2015. Proteasome dysfunction associated to oxidative stress and proteotoxicity in adipocytes compromises insulin sensitivity in human obesity. Antioxidants & Redox Signaling 23: 597–612. doi:10.1089/ars.2014.5939.
Dikalov, S. 2011. Cross talk between mitochondria and NADPH oxidases. Free Radical Biology & Medicine 51: 1289–1301. doi:10.1016/j.freeradbiomed.2011.06.033.
Divoux, A., J. Tordjman, D. Lacasa, N. Veyrie, D. Hugol, A. Aissat, A. Basdevant, M. Guerre-Millo, C. Poitou, J.-D. Zucker, P. Bedossa, and K. Clément. 2010. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 59: 2817–2825. doi:10.2337/db10-0585.
Divoux, A., S. Moutel, C. Poitou, D. Lacasa, N. Veyrie, A. Aissat, M. Arock, M. Guerre-Millo, and K. Clément. 2012. Mast cells in human adipose tissue: Link with morbid obesity, inflammatory status, and diabetes. The Journal of Clinical Endocrinology and Metabolism 97: E1677–E1685. doi:10.1210/jc.2012-1532.
Dordevic, A.L., N. Konstantopoulos, and D. Cameron-Smith. 2014. 3 T3-L1 preadipocytes exhibit heightened monocyte-chemoattractant protein-1 response to acute fatty acid exposure. PLoS One 9: e99382. doi:10.1371/journal.pone.0099382.
Dou, L., T. Zhao, L. Wang, X. Huang, J. Jiao, D. Gao, H. Zhang, T. Shen, Y. Man, S. Wang, and J. Li. 2013. miR-200 s contribute to interleukin-6 (IL-6)-induced insulin resistance in hepatocytes. The Journal of Biological Chemistry 288: 22596–22606. doi:10.1074/jbc.M112.423145.
Du, J., L.M. Fan, A. Mai, and J.-M. Li. 2013. Crucial roles of Nox2-derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle-aged mice. British Journal of Pharmacology 170: 1064–1077. doi:10.1111/bph.12336.
Dupasquier, M., P. Stoitzner, H. Wan, D. Cerqueira, A. van Oudenaren, J.S.A. Voerman, K. Denda-Nagai, T. Irimura, G. Raes, N. Romani, and P.J.M. Leenen. 2006. The dermal microenvironment induces the expression of the alternative activation marker CD301/mMGL in mononuclear phagocytes, independent of IL-4/IL-13 signaling. Journal of Leukocyte Biology 80: 838–849. doi:10.1189/jlb.1005564.
Egan, D.F., D.B. Shackelford, M.M. Mihaylova, S. Gelino, R.A. Kohnz, W. Mair, D.S. Vasquez, A. Joshi, D.M. Gwinn, R. Taylor, J.M. Asara, J. Fitzpatrick, A. Dillin, B. Viollet, M. Kundu, M. Hansen, and R.J. Shaw. 2011. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461. doi:10.1126/science.1196371.
Eljaafari, A., M. Robert, M. Chehimi, S. Chanon, C. Durand, G. Vial, N. Bendridi, A.-M. Madec, E. Disse, M. Laville, J. Rieusset, E. Lefai, H. Vidal, and L. Pirola. 2015. Adipose tissue-derived stem cells from obese subjects contribute to inflammation and reduced insulin response in adipocytes through differential regulation of the Th1/Th17 balance and monocyte activation. Diabetes 64: 2477–2488. doi:10.2337/db15-0162.
Fabbrini, E., M. Cella, S.A. McCartney, A. Fuchs, N.A. Abumrad, T.A. Pietka, Z. Chen, B.N. Finck, D.H. Han, F. Magkos, C. Conte, D. Bradley, G. Fraterrigo, J.C. Eagon, B.W. Patterson, M. Colonna, and S. Klein. 2013. Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 145: 366–374 –3. doi:10.1053/j.gastro.2013.04.010.
Fain, J.N., B. Buehrer, S.W. Bahouth, D.S. Tichansky, and A.K. Madan. 2008. Comparison of messenger RNA distribution for 60 proteins in fat cells vs. the nonfat cells of human omental adipose tissue. Metabolism 57: 1005–1015. doi:10.1016/j.metabol.2008.02.019.
Fernández-Sánchez, A., E. Madrigal-Santillán, M. Bautista, J. Esquivel-Soto, A. Morales-González, C. Esquivel-Chirino, I. Durante-Montiel, G. Sánchez-Rivera, C. Valadez-Vega, and J.A. Morales-González. 2011. Inflammation, oxidative stress, and obesity. International Journal of Molecular Sciences 12: 3117–3132. doi:10.3390/ijms12053117.
Finucane, O.M., C.M. Reynolds, F.C. McGillicuddy, and H.M. Roche. 2012. Insights into the role of macrophage migration inhibitory factor in obesity and insulin resistance. The Proceedings of the Nutrition Society 71: 622–633. doi:10.1017/S0029665112000730.
Finucane, O.M., C.M. Reynolds, F.C. McGillicuddy, K.A. Harford, M. Morrison, J. Baugh, and H.M. Roche. 2014. Macrophage migration inhibitory factor deficiency ameliorates high-fat diet induced insulin resistance in mice with reduced adipose inflammation and hepatic steatosis. PLoS One 9: e113369. doi:10.1371/journal.pone.0113369.
Finucane, O.M., C.L. Lyons, A.M. Murphy, C.M. Reynolds, R. Klinger, N.P. Healy, A.A. Cooke, R.C. Coll, L. McAllan, K.N. Nilaweera, M.E. O’Reilly, A.C. Tierney, M.J. Morine, J.F. Alcala-Diaz, J. Lopez-Miranda, D.P. O’Connor, L.A. O’Neill, F.C. McGillicuddy, and H.M. Roche. 2015. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes 64: 2116–2128. doi:10.2337/db14-1098.
Fjeldborg, K., S.B. Pedersen, H.J. Møller, T. Christiansen, M. Bennetzen, and B. Richelsen. 2014. Human adipose tissue macrophages are enhanced but changed to an anti-inflammatory profile in obesity. Journal of Immunology Research 2014: 309548. doi:10.1155/2014/309548.
Fresno, M., R. Alvarez, and N. Cuesta. 2011. Toll-like receptors, inflammation, metabolism and obesity. Archives of Physiology and Biochemistry 117: 151–164. doi:10.3109/13813455.2011.562514.
Friedman, R.C., K.K.-H. Farh, C.B. Burge, and D.P. Bartel. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Research 19: 92–105. doi:10.1101/gr.082701.108.
Frohnert, B.I., and D.A. Bernlohr. 2014. Glutathionylated products of lipid peroxidation: A novel mechanism of adipocyte to macrophage signaling. Adipocytes 3: 224–229. doi:10.4161/adip.28851.
Frohnert, B.I., E.K. Long, W.S. Hahn, and D.A. Bernlohr. 2014. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 63: 89–100. doi:10.2337/db13-0777.
Fujisaka, S., I. Usui, A. Bukhari, M. Ikutani, T. Oya, Y. Kanatani, K. Tsuneyama, Y. Nagai, K. Takatsu, M. Urakaze, M. Kobayashi, and K. Tobe. 2009. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes 58: 2574–2582. doi:10.2337/db08-1475.
Gaens, K.H.J., C.D.A. Stehouwer, and C.G. Schalkwijk. 2013. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Current Opinion in Lipidology 24: 4–11. doi:10.1097/MOL.0b013e32835aea13.
Ge, Q., S. Brichard, X. Yi, and Q. Li. 2014. microRNAs as a new mechanism regulating adipose tissue inflammation in obesity and as a novel therapeutic strategy in the metabolic syndrome. Journal of Immunology Research 2014: 987285. doi:10.1155/2014/987285.
Giordano, A., I. Murano, E. Mondini, J. Perugini, A. Smorlesi, I. Severi, R. Barazzoni, P.E. Scherer, and S. Cinti. 2013. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. Journal of Lipid Research 54: 2423–2436. doi:10.1194/jlr.M038638.
Goh, J., K.P. Goh, and A. Abbasi. 2016. Exercise and adipose tissue macrophages: New frontiers in obesity research? Frontiers in Endocrinology 7: 65. doi:10.3389/fendo.2016.00065.
Goossens, G.H. 2008. The role of adipose tissue dysfunction in the pathogenesis of obesity-related insulin resistance. Physiology & Behavior 94: 206–218. doi:10.1016/j.physbeh.2007.10.010.
Goossens, G.H., E.E. Blaak, R. Theunissen, A.M. Duijvestijn, K. Clément, J.-W.C. Tervaert, and M.M. Thewissen. 2012. Expression of NLRP3 inflammasome and T cell population markers in adipose tissue are associated with insulin resistance and impaired glucose metabolism in humans. Molecular Immunology 50: 142–149. doi:10.1016/j.molimm.2012.01.005.
Grune, T., and K.J. Davies. 1997. Breakdown of oxidized proteins as a part of secondary antioxidant defenses in mammalian cells. BioFactors 6: 165–172.
Guglielmi, V., M. Cardellini, F. Cinti, F. Corgosinho, I. Cardolini, M. D’Adamo, M.C. Zingaretti, A. Bellia, D. Lauro, P. Gentileschi, M. Federici, S. Cinti, and P. Sbraccia. 2015. Omental adipose tissue fibrosis and insulin resistance in severe obesity. Nutrition and Diabetes 5: e175. doi:10.1038/nutd.2015.22.
Guilherme, A., J.V. Virbasius, V. Puri, and M.P. Czech. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nature Reviews. Molecular Cell Biology 9: 367–377. doi:10.1038/nrm2391.
Gunasekaran, M.K., W. Viranaicken, A.-C. Girard, F. Festy, M. Cesari, R. Roche, and L. Hoareau. 2013. Inflammation triggers high mobility group box 1 (HMGB1) secretion in adipose tissue, a potential link to obesity. Cytokine 64: 103–111. doi:10.1016/j.cyto.2013.07.017.
Guo, W., S. Wong, W. Xie, T. Lei, and Z. Luo. 2007. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3 T3-L1 and rat primary preadipocytes. American Journal of Physiology. Endocrinology and Metabolism 293: E576–E586. doi:10.1152/ajpendo.00523.2006.
Guri, A.J., R. Hontecillas, G. Ferrer, O. Casagran, U. Wankhade, A.M. Noble, D.L. Eizirik, F. Ortis, M. Cnop, D. Liu, H. Si, and J. Bassaganya-Riera. 2008. Loss of PPAR gamma in immune cells impairs the ability of abscisic acid to improve insulin sensitivity by suppressing monocyte chemoattractant protein-1 expression and macrophage infiltration into white adipose tissue. The Journal of Nutritional Biochemistry 19: 216–228. doi:10.1016/j.jnutbio.2007.02.010.
Gurung, P., J.R. Lukens, and T.-D. Kanneganti. 2015. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends in Molecular Medicine 21: 193–201. doi:10.1016/j.molmed.2014.11.008.
Guzik, T.J., D. Mangalat, and R. Korbut. 2006. Adipocytokines - novel link between inflammation and vascular function? Journal of Physiology and Pharmacology 57: 505–528.
Guzmán-Ruiz, R., F. Ortega, A. Rodríguez, R. Vázquez-Martínez, A. Díaz-Ruiz, S. Garcia-Navarro, M. Giralt, A. Garcia-Rios, D. Cobo-Padilla, F.J. Tinahones, J. López-Miranda, F. Villarroya, G. Frühbeck, J.M. Fernández-Real, and M.M. Malagón. 2014. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. International Journal of Obesity 2005(38): 1545–1554. doi:10.1038/ijo.2014.36.
Halberg, N., I. Wernstedt-Asterholm, and P.E. Scherer. 2008. The adipocyte as an endocrine cell. Endocrinology and Metabolism Clinics of North America 37: 753–768. doi:10.1016/j.ecl.2008.07.002.x–xi
Halberg, N., T. Khan, M.E. Trujillo, I. Wernstedt-Asterholm, A.D. Attie, S. Sherwani, Z.V. Wang, S. Landskroner-Eiger, S. Dineen, U.J. Magalang, R.A. Brekken, and P.E. Scherer. 2009. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Molecular and Cellular Biology 29: 4467–4483. doi:10.1128/MCB.00192-09.
Harford, K.A., C.M. Reynolds, F.C. McGillicuddy, and H.M. Roche. 2011. Fats, inflammation and insulin resistance: Insights to the role of macrophage and T-cell accumulation in adipose tissue. The Proceedings of the Nutrition Society 70: 408–417. doi:10.1017/S0029665111000565.
Haynes, C.M., E.A. Titus, and A.A. Cooper. 2004. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Molecular Cell 15: 767–776. doi:10.1016/j.molcel.2004.08.025.
He, Q., Z. Gao, J. Yin, J. Zhang, Z. Yun, and J. Ye. 2011. Regulation of HIF-1{alpha} activity in adipose tissue by obesity-associated factors: Adipogenesis, insulin, and hypoxia. American Journal of Physiology. Endocrinology and Metabolism 300: E877–E885. doi:10.1152/ajpendo.00626.2010.
Heinrich, P.C., I. Behrmann, S. Haan, H.M. Hermanns, G. Müller-Newen, and F. Schaper. 2003. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. The Biochemical Journal 374: 1–20. doi:10.1042/BJ20030407.
Hirai, S., C. Ohyane, Y.-I. Kim, S. Lin, T. Goto, N. Takahashi, C.-S. Kim, J. Kang, R. Yu, and T. Kawada. 2014. Involvement of mast cells in adipose tissue fibrosis. American Journal of Physiology. Endocrinology and Metabolism 306: E247–E255. doi:10.1152/ajpendo.00056.2013.
Hosogai, N., A. Fukuhara, K. Oshima, Y. Miyata, S. Tanaka, K. Segawa, S. Furukawa, Y. Tochino, R. Komuro, M. Matsuda, and I. Shimomura. 2007. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56: 901–911. doi:10.2337/db06-0911.
Ichioka, M., T. Suganami, N. Tsuda, I. Shirakawa, Y. Hirata, N. Satoh-Asahara, Y. Shimoda, M. Tanaka, M. Kim-Saijo, Y. Miyamoto, Y. Kamei, M. Sata, and Y. Ogawa. 2011. Increased expression of macrophage-inducible C-type lectin in adipose tissue of obese mice and humans. Diabetes 60: 819–826. doi:10.2337/db10-0864.
Jankun, J., and E. Skrzypczak-Jankun. 2009. Yin and yang of the plasminogen activator inhibitor. Polskie Archiwum Medycyny Wewnętrznej 119: 410–417.
Jiang, F., H.K. Lim, M.J. Morris, L. Prior, E. Velkoska, X. Wu, and G.J. Dusting. 2011. Systemic upregulation of NADPH oxidase in diet-induced obesity in rats. Redox Report 16: 223–229. doi:10.1179/174329211X13049558293713.
Jiang, W., L. Kong, Q. Ni, Y. Lu, W. Ding, G. Liu, L. Pu, W. Tang, and L. Kong. 2014. miR-146a ameliorates liver ischemia/reperfusion injury by suppressing IRAK1 and TRAF6. PLoS One 9: e101530. doi:10.1371/journal.pone.0101530.
Kanda, H., S. Tateya, Y. Tamori, K. Kotani, K. Hiasa, R. Kitazawa, S. Kitazawa, H. Miyachi, S. Maeda, K. Egashira, and M. Kasuga. 2006. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. The Journal of Clinical Investigation 116: 1494–1505. doi:10.1172/JCI26498.
Kawanaka, H., M.K. Jones, I.L. Szabo, D. Baatar, R. Pai, K. Tsugawa, K. Sugimachi, I.J. Sarfeh, and A.S. Tarnawski. 2002. Activation of eNOS in rat portal hypertensive gastric mucosa is mediated by TNF-alpha via the PI 3-kinase-Akt signaling pathway. Hepatology 35: 393–402. doi:10.1053/jhep.2002.30958.
Kern, P.A., M. Saghizadeh, J.M. Ong, R.J. Bosch, R. Deem, and R.B. Simsolo. 1995. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. The Journal of Clinical Investigation 95: 2111–2119. doi:10.1172/JCI117899.
Kim, J., M. Montagnani, K.K. Koh, and M.J. Quon. 2006. Reciprocal relationships between insulin resistance and endothelial dysfunction: Molecular and pathophysiological mechanisms. Circulation 113: 1888–1904. doi:10.1161/CIRCULATIONAHA.105.563213.
Kim, F., M. Pham, E. Maloney, N.O. Rizzo, G.J. Morton, B.E. Wisse, E.A. Kirk, A. Chait, and M.W. Schwartz. 2008. Vascular inflammation, insulin resistance, and reduced nitric oxide production precede the onset of peripheral insulin resistance. Arteriosclerosis, Thrombosis, and Vascular Biology 28: 1982–1988. doi:10.1161/ATVBAHA.108.169722.
Kim, B.-S., R. Rongisch, S. Hager, G. Grieb, M. Nourbakhsh, H.-O. Rennekampff, R. Bucala, J. Bernhagen, and N. Pallua. 2015a. Macrophage Migration Inhibitory Factor in Acute Adipose Tissue Inflammation. PLoS One 10: e0137366. doi:10.1371/journal.pone.0137366.
Kim, J.-A., H.-J. Jang, and D.H. Hwang. 2015b. Toll-like receptor 4-induced endoplasmic reticulum stress contributes to impairment of vasodilator action of insulin. American Journal of Physiology. Endocrinology and Metabolism 309: E767–E776. doi:10.1152/ajpendo.00369.2015.
Kishore, P., W. Li, J. Tonelli, D.-E. Lee, S. Koppaka, K. Zhang, Y. Lin, S. Kehlenbrink, P.E. Scherer, and M. Hawkins. 2010. Adipocyte-derived factors potentiate nutrient-induced production of plasminogen activator inhibitor-1 by macrophages. Science Translational Medicine 2: 20ra15. doi:10.1126/scitranslmed.3000292.
Koenen, T.B., R. Stienstra, L.J. van Tits, L.A.B. Joosten, J.F. van Velzen, A. Hijmans, J.A. Pol, J.A. van der Vliet, M.G. Netea, C.J. Tack, A.F.H. Stalenhoef, and J. de Graaf. 2011. The inflammasome and caspase-1 activation: A new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology 152: 3769–3778. doi:10.1210/en.2010-1480.
Koska, J., N. Stefan, S. Dubois, C. Trinidad, R.V. Considine, T. Funahashi, J.C. Bunt, E. Ravussin, and P.A. Permana. 2009. mRNA concentrations of MIF in subcutaneous abdominal adipose cells are associated with adipocyte size and insulin action. International Journal of Obesity 2005(33): 842–850. doi:10.1038/ijo.2009.106.
Lam, T.K.T., G.J. Schwartz, and L. Rossetti. 2005. Hypothalamic sensing of fatty acids. Nature Neuroscience 8: 579–584. doi:10.1038/nn1456.
Lee, B.-C., and J. Lee. 2014. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochimica et Biophysica Acta 1842: 446–462. doi:10.1016/j.bbadis.2013.05.017.
Lee, J.Y., J. Ye, Z. Gao, H.S. Youn, W.H. Lee, L. Zhao, N. Sizemore, and D.H. Hwang. 2003. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. The Journal of Biological Chemistry 278: 37041–37051. doi:10.1074/jbc.M305213200.
Lee, M.-J., Y. Wu, and S.K. Fried. 2013. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Molecular Aspects of Medicine 34: 1–11. doi:10.1016/j.mam.2012.10.001.
Lee, J.-T., N. Pamir, N.-C. Liu, E.A. Kirk, M.M. Averill, L. Becker, I. Larson, D.K. Hagman, K.E. Foster-Schubert, B. van Yserloo, K.E. Bornfeldt, R.C. LeBoeuf, M. Kratz, and J.W. Heinecke. 2014. Macrophage metalloelastase (MMP12) regulates adipose tissue expansion, insulin sensitivity, and expression of inducible nitric oxide synthase. Endocrinology 155: 3409–3420. doi:10.1210/en.2014-1037.
Legrand-Poels, S., N. Esser, L. L’homme, A. Scheen, N. Paquot, and J. Piette. 2014. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochemical Pharmacology 92: 131–141. doi:10.1016/j.bcp.2014.08.013.
Lemoine, A.Y., S. Ledoux, I. Quéguiner, S. Caldérari, C. Mechler, S. Msika, P. Corvol, and E. Larger. 2012. Link between adipose tissue angiogenesis and fat accumulation in severely obese subjects. The Journal of Clinical Endocrinology and Metabolism 97: E775–E780. doi:10.1210/jc.2011-2649.
Li, P., M. Lu, M.T.A. Nguyen, E.J. Bae, J. Chapman, D. Feng, M. Hawkins, J.E. Pessin, D.D. Sears, A.-K. Nguyen, A. Amidi, S.M. Watkins, U. Nguyen, and J.M. Olefsky. 2010. Functional heterogeneity of CD11c-positive adipose tissue macrophages in diet-induced obese mice. The Journal of Biological Chemistry 285: 15333–15345. doi:10.1074/jbc.M110.100263.
Liang, X., T. Kanjanabuch, S.-L. Mao, C.-M. Hao, Y.-W. Tang, P.J. Declerck, A.H. Hasty, D.H. Wasserman, A.B. Fogo, and L.-J. Ma. 2006. Plasminogen activator inhibitor-1 modulates adipocyte differentiation. American Journal of Physiology. Endocrinology and Metabolism 290: E103–E113. doi:10.1152/ajpendo.00605.2004.
Linton, M.F., and S. Fazio. 2003. Macrophages, inflammation, and atherosclerosis. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 27(Suppl 3): S35–S40. doi:10.1038/sj.ijo.0802498.
Liu, Y., X. Ge, X. Dou, L. Guo, Y. Liu, S.-R. Zhou, X.-B. Wei, S.-W. Qian, H.-Y. Huang, C.-J. Xu, W.-P. Jia, Y.-J. Dang, X. Li, and Q.-Q. Tang. 2015. Protein inhibitor of activated STAT 1 (PIAS1) protects against obesity-induced insulin resistance by inhibiting inflammation cascade in adipose tissue. Diabetes 64: 4061–4074. doi:10.2337/db15-0278.
Lowe, C.E., R.J. Dennis, U. Obi, S. O’Rahilly, and J.J. Rochford. 2012. Investigating the involvement of the ATF6α pathway of the unfolded protein response in adipogenesis. International Journal of Obesity 2005(36): 1248–1251. doi:10.1038/ijo.2011.233.
Lukens, J.R., V.D. Dixit, and T.-D. Kanneganti. 2011. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discovery Medicine 12: 65–74.
Lumeng, C.N., J.L. Bodzin, and A.R. Saltiel. 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of Clinical Investigation 117: 175–184. doi:10.1172/JCI29881.
Lumeng, C.N., J.B. DelProposto, D.J. Westcott, and A.R. Saltiel. 2008. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 57: 3239–3246. doi:10.2337/db08-0872.
Madonna, R., and R. De Caterina. 2009. Prolonged exposure to high insulin impairs the endothelial PI3-kinase/Akt/nitric oxide signalling. Thrombosis and Haemostasis 101: 345–350.
Manna, P., and S.K. Jain. 2015. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: Causes and therapeutic strategies. Metabolic Syndrome and Related Disorders 13: 423–444. doi:10.1089/met.2015.0095.
Martínez, J.A. 2006. Mitochondrial oxidative stress and inflammation: An slalom to obesity and insulin resistance. Journal of Physiology and Biochemistry 62: 303–306.
Martinez-Santibanez, G., K. Singer, K.W. Cho, J.L. DelProposto, T. Mergian, and C.N. Lumeng. 2015. Obesity-induced remodeling of the adipose tissue elastin network is independent of the metalloelastase MMP-12. Adipocytes 4: 264–272. doi:10.1080/21623945.2015.1027848.
Masters, S.L., E. Latz, and L.A.J. O’Neill. 2011. The inflammasome in atherosclerosis and type 2 diabetes. Science Translational Medicine 3: 81ps17. doi:10.1126/scitranslmed.3001902.
McDonnell, M.E., L.M. Ganley-Leal, A. Mehta, S.J. Bigornia, M. Mott, Q. Rehman, M.G. Farb, D.T. Hess, L. Joseph, N. Gokce, and C.M. Apovian. 2012. B lymphocytes in human subcutaneous adipose crown-like structures. Obesity (Silver Spring) 20: 1372–1378. doi:10.1038/oby.2012.54.
Meijer, K., M. de Vries, S. Al-Lahham, M. Bruinenberg, D. Weening, M. Dijkstra, N. Kloosterhuis, R.J. van der Leij, H. van der Want, B.-J. Kroesen, R. Vonk, and F. Rezaee. 2011. Human primary adipocytes exhibit immune cell function: Adipocytes prime inflammation independent of macrophages. PLoS One 6: e17154. doi:10.1371/journal.pone.0017154.
Menu, P., A. Mayor, R. Zhou, A. Tardivel, H. Ichijo, K. Mori, and J. Tschopp. 2012. ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death & Disease 3: e261. doi:10.1038/cddis.2011.132.
Miyazawa-Hoshimoto, S., K. Takahashi, H. Bujo, N. Hashimoto, and Y. Saito. 2003. Elevated serum vascular endothelial growth factor is associated with visceral fat accumulation in human obese subjects. Diabetologia 46: 1483–1488. doi:10.1007/s00125-003-1221-6.
Morigny, P., M. Houssier, E. Mouisel, and D. Langin. 2016. Adipocyte lipolysis and insulin resistance. Biochimie 125: 259–266. doi:10.1016/j.biochi.2015.10.024.
Mouche, S., S.B. Mkaddem, W. Wang, M. Katic, Y.-H. Tseng, S. Carnesecchi, K. Steger, M. Foti, C.A. Meier, P. Muzzin, C.R. Kahn, E. Ogier-Denis, and I. Szanto. 2007. Reduced expression of the NADPH oxidase NOX4 is a hallmark of adipocyte differentiation. Biochimica et Biophysica Acta 1773: 1015–1027. doi:10.1016/j.bbamcr.2007.03.003.
Nakamura, S., T. Takamura, N. Matsuzawa-Nagata, H. Takayama, H. Misu, H. Noda, S. Nabemoto, S. Kurita, T. Ota, H. Ando, K.-I. Miyamoto, and S. Kaneko. 2009. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. The Journal of Biological Chemistry 284: 14809–14818. doi:10.1074/jbc.M901488200.
Nativel, B., M. Marimoutou, V.G. Thon-Hon, M.K. Gunasekaran, J. Andries, G. Stanislas, C. Planesse, C.R. Da Silva, M. Césari, T. Iwema, P. Gasque, and W. Viranaicken. 2013. Soluble HMGB1 is a novel adipokine stimulating IL-6 secretion through RAGE receptor in SW872 preadipocyte cell line: Contribution to chronic inflammation in fat tissue. PLoS One 8: e76039. doi:10.1371/journal.pone.0076039.
Nguyen, M.T.A., H. Satoh, S. Favelyukis, J.L. Babendure, T. Imamura, J.I. Sbodio, J. Zalevsky, B.I. Dahiyat, N.-W. Chi, and J.M. Olefsky. 2005. JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3 T3-L1 adipocytes. The Journal of Biological Chemistry 280: 35361–35371. doi:10.1074/jbc.M504611200.
Nguyen, M.T.A., S. Favelyukis, A.-K. Nguyen, D. Reichart, P.A. Scott, A. Jenn, R. Liu-Bryan, C.K. Glass, J.G. Neels, and J.M. Olefsky. 2007. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. The Journal of Biological Chemistry 282: 35279–35292. doi:10.1074/jbc.M706762200.
Olefsky, J.M., and C.K. Glass. 2010. Macrophages, inflammation, and insulin resistance. Annual Review of Physiology 72: 219–246. doi:10.1146/annurev-physiol-021909-135846.
Orr, J.S., M.J. Puglisi, K.L.J. Ellacott, C.N. Lumeng, D.H. Wasserman, and A.H. Hasty. 2012. Toll-like receptor 4 deficiency promotes the alternative activation of adipose tissue macrophages. Diabetes 61: 2718–2727. doi:10.2337/db11-1595.
Otoda, T., T. Takamura, H. Misu, T. Ota, S. Murata, H. Hayashi, H. Takayama, A. Kikuchi, T. Kanamori, K.R. Shima, F. Lan, T. Takeda, S. Kurita, K. Ishikura, Y. Kita, K. Iwayama, K. Kato, M. Uno, Y. Takeshita, M. Yamamoto, K. Tokuyama, S. Iseki, K. Tanaka, and S. Kaneko. 2013. Proteasome dysfunction mediates obesity-induced endoplasmic reticulum stress and insulin resistance in the liver. Diabetes 62: 811–824. doi:10.2337/db11-1652.
Ozcan, U., Q. Cao, E. Yilmaz, A.-H. Lee, N.N. Iwakoshi, E. Ozdelen, G. Tuncman, C. Görgün, L.H. Glimcher, and G.S. Hotamisligil. 2004. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461. doi:10.1126/science.1103160.
Pacher, P., J.S. Beckman, and L. Liaudet. 2007. Nitric oxide and peroxynitrite in health and disease. Physiological Reviews 87: 315–424. doi:10.1152/physrev.00029.2006.
Pal, M., M.A. Febbraio, and G.I. Lancaster. 2016. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. The Journal of Physiology 594: 267–279. doi:10.1113/JP271457.
Pandey, M., D.J. Loskutoff, and F. Samad. 2005. Molecular mechanisms of tumor necrosis factor-alpha-mediated plasminogen activator inhibitor-1 expression in adipocytes. The FASEB Journal 19: 1317–1319. doi:10.1096/fj.04-3459fje.
Pasarica, M., O.R. Sereda, L.M. Redman, D.C. Albarado, D.T. Hymel, L.E. Roan, J.C. Rood, D.H. Burk, and S.R. Smith. 2009. Reduced adipose tissue oxygenation in human obesity: Evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58: 718–725. doi:10.2337/db08-1098.
Pedersen, D.J., A. Guilherme, L.V. Danai, L. Heyda, A. Matevossian, J. Cohen, S.M. Nicoloro, J. Straubhaar, H.L. Noh, D. Jung, J.K. Kim, and M.P. Czech. 2015. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Molecular Metabolism 4: 507–518. doi:10.1016/j.molmet.2015.04.003.
Pereira, S.S., and J.I. Alvarez-Leite. 2014. Low-Grade Inflammation, Obesity, and Diabetes. Current Obesity Reports 3: 422–431. doi:10.1007/s13679-014-0124-9.
Pereira, S., L. Teixeira, E. Aguilar, M. Oliveira, A. Savassi-Rocha, J.N. Pelaez, L. Capettini, M.T. Diniz, A. Ferreira, and J. Alvarez-Leite. 2014. Modulation of adipose tissue inflammation by FOXP3+ Treg cells, IL-10, and TGF-β in metabolically healthy class III obese individuals. Nutrition 30: 784–790. doi:10.1016/j.nut.2013.11.023.
Pickering, A.M., and K.J.A. Davies. 2012. Degradation of damaged proteins: The main function of the 20S proteasome. Progress in Molecular Biology and Translational Science 109: 227–248. doi:10.1016/B978-0-12-397863-9.00006-7.
Pickering, A.M., A.L. Koop, C.Y. Teoh, G. Ermak, T. Grune, and K.J.A. Davies. 2010. The immunoproteasome, the 20S proteasome and the PA28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. The Biochemical Journal 432: 585–594. doi:10.1042/BJ20100878.
Pierre, N., L. Deldicque, C. Barbé, D. Naslain, P.D. Cani, and M. Francaux. 2013. Toll-like receptor 4 knockout mice are protected against endoplasmic reticulum stress induced by a high-fat diet. PLoS One 8: e65061. doi:10.1371/journal.pone.0065061.
Revelo, X.S., H. Luck, S. Winer, and D.A. Winer. 2014. Morphological and inflammatory changes in visceral adipose tissue during obesity. Endocrine Pathology 25: 93–101. doi:10.1007/s12022-013-9288-1.
Reynolds, C.M., F.C. McGillicuddy, K.A. Harford, O.M. Finucane, K.H.G. Mills, and H.M. Roche. 2012. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Molecular Nutrition & Food Research 56: 1212–1222. doi:10.1002/mnfr.201200058.
Samad, F., K.D. Hester, G. Yang, Y.A. Hannun, and J. Bielawski. 2006. Altered adipose and plasma sphingolipid metabolism in obesity: A potential mechanism for cardiovascular and metabolic risk. Diabetes 55: 2579–2587. doi:10.2337/db06-0330.
Schröder, M., and R.J. Kaufman. 2005. The mammalian unfolded protein response. Annual Review of Biochemistry 74: 739–789. doi:10.1146/annurev.biochem.73.011303.074134.
Schroder, K., and J. Tschopp. 2010. The inflammasomes. Cell 140: 821–832. doi:10.1016/j.cell.2010.01.040.
Sciarretta, S., M. Volpe, and J. Sadoshima. 2012. Is reactivation of autophagy a possible therapeutic solution for obesity and metabolic syndrome? Autophagy 8: 1252–1254. doi:10.4161/auto.20670.
Seifert, U., L.P. Bialy, F. Ebstein, D. Bech-Otschir, A. Voigt, F. Schröter, T. Prozorovski, N. Lange, J. Steffen, M. Rieger, U. Kuckelkorn, O. Aktas, P.-M. Kloetzel, and E. Krüger. 2010. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell 142: 613–624. doi:10.1016/j.cell.2010.07.036.
Serrano-Marco, L., E. Barroso, I. El Kochairi, X. Palomer, L. Michalik, W. Wahli, and M. Vázquez-Carrera. 2012. The peroxisome proliferator-activated receptor (PPAR) β/δ agonist GW501516 inhibits IL-6-induced signal transducer and activator of transcription 3 (STAT3) activation and insulin resistance in human liver cells. Diabetologia 55: 743–751. doi:10.1007/s00125-011-2401-4.
Shah, C., G. Yang, I. Lee, J. Bielawski, Y.A. Hannun, and F. Samad. 2008. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. The Journal of Biological Chemistry 283: 13538–13548. doi:10.1074/jbc.M709950200.
Shi, H., M.V. Kokoeva, K. Inouye, I. Tzameli, H. Yin, and J.S. Flier. 2006. TLR4 links innate immunity and fatty acid-induced insulin resistance. The Journal of Clinical Investigation 116: 3015–3025. doi:10.1172/JCI28898.
Skurk, T., and H. Hauner. 2004. Obesity and impaired fibrinolysis: Role of adipose production of plasminogen activator inhibitor-1. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 28: 1357–1364. doi:10.1038/sj.ijo.0802778.
Skurk, T., C. Herder, I. Kräft, S. Müller-Scholze, H. Hauner, and H. Kolb. 2005. Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinology 146: 1006–1011. doi:10.1210/en.2004-0924.
Solinas, G., C. Vilcu, J.G. Neels, G.K. Bandyopadhyay, J.-L. Luo, W. Naugler, S. Grivennikov, A. Wynshaw-Boris, M. Scadeng, J.M. Olefsky, and M. Karin. 2007. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metabolism 6: 386–397. doi:10.1016/j.cmet.2007.09.011.
Song, M.J., K.H. Kim, J.M. Yoon, and J.B. Kim. 2006. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochemical and Biophysical Research Communications 346: 739–745. doi:10.1016/j.bbrc.2006.05.170.
Sopasakis, V.R., M. Sandqvist, B. Gustafson, A. Hammarstedt, M. Schmelz, X. Yang, P.-A. Jansson, and U. Smith. 2004. High local concentrations and effects on differentiation implicate interleukin-6 as a paracrine regulator. Obesity Research 12: 454–460. doi:10.1038/oby.2004.51.
Spalding, K.L., E. Arner, P.O. Westermark, S. Bernard, B.A. Buchholz, O. Bergmann, L. Blomqvist, J. Hoffstedt, E. Näslund, T. Britton, H. Concha, M. Hassan, M. Rydén, J. Frisén, and P. Arner. 2008. Dynamics of fat cell turnover in humans. Nature 453: 783–787. doi:10.1038/nature06902.
Spencer, M., A. Yao-Borengasser, R. Unal, N. Rasouli, C.M. Gurley, B. Zhu, C.A. Peterson, and P.A. Kern. 2010. Adipose tissue macrophages in insulin-resistant subjects are associated with collagen VI and fibrosis and demonstrate alternative activation. American Journal of Physiology. Endocrinology and Metabolism 299: E1016–E1027. doi:10.1152/ajpendo.00329.2010.
Steinberg, G.R., and B.E. Kemp. 2009. AMPK in Health and Disease. Physiological Reviews 89: 1025–1078. doi:10.1152/physrev.00011.2008.
Steinberg, H.O., G. Paradisi, G. Hook, K. Crowder, J. Cronin, and A.D. Baron. 2000. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes 49: 1231–1238.
Stienstra, R., L.A.B. Joosten, T. Koenen, B. van Tits, J.A. van Diepen, S.A.A. van den Berg, P.C.N. Rensen, P.J. Voshol, G. Fantuzzi, A. Hijmans, S. Kersten, M. Müller, W.B. van den Berg, N. van Rooijen, M. Wabitsch, B.-J. Kullberg, J.W.M. van der Meer, T. Kanneganti, C.J. Tack, and M.G. Netea. 2010. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metabolism 12: 593–605. doi:10.1016/j.cmet.2010.11.011.
Stienstra, R., J.A. van Diepen, C.J. Tack, M.H. Zaki, F.L. van de Veerdonk, D. Perera, G.A. Neale, G.J. Hooiveld, A. Hijmans, I. Vroegrijk, S. van den Berg, J. Romijn, P.C.N. Rensen, L.A.B. Joosten, M.G. Netea, and T.-D. Kanneganti. 2011. Inflammasome is a central player in the induction of obesity and insulin resistance. Proceedings of the National Academy of Sciences of the United States of America 108: 15324–15329. doi:10.1073/pnas.1100255108.
Strissel, K.J., Z. Stancheva, H. Miyoshi, J.W. Perfield, J. DeFuria, Z. Jick, A.S. Greenberg, and M.S. Obin. 2007. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56: 2910–2918. doi:10.2337/db07-0767.
Su, D., G.M. Coudriet, D. Hyun Kim, Y. Lu, G. Perdomo, S. Qu, S. Slusher, H.M. Tse, J. Piganelli, N. Giannoukakis, J. Zhang, and H.H. Dong. 2009. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes 58: 2624–2633. doi:10.2337/db09-0232.
Suganami, T., J. Nishida, and Y. Ogawa. 2005. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arteriosclerosis, Thrombosis, and Vascular Biology 25: 2062–2068. doi:10.1161/01.ATV.0000183883.72263.13.
Suganami, T., K. Tanimoto-Koyama, J. Nishida, M. Itoh, X. Yuan, S. Mizuarai, H. Kotani, S. Yamaoka, K. Miyake, S. Aoe, Y. Kamei, and Y. Ogawa. 2007. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology 27: 84–91. doi:10.1161/01.ATV.0000251608.09329.9a.
Suganami, T., X. Yuan, Y. Shimoda, K. Uchio-Yamada, N. Nakagawa, I. Shirakawa, T. Usami, T. Tsukahara, K. Nakayama, Y. Miyamoto, K. Yasuda, J. Matsuda, Y. Kamei, S. Kitajima, and Y. Ogawa. 2009. Activating transcription factor 3 constitutes a negative feedback mechanism that attenuates saturated Fatty acid/toll-like receptor 4 signaling and macrophage activation in obese adipose tissue. Circulation Research 105: 25–32. doi:10.1161/CIRCRESAHA.109.196261.
Summers, S.A. 2006. Ceramides in insulin resistance and lipotoxicity. Progress in Lipid Research 45: 42–72. doi:10.1016/j.plipres.2005.11.002.
Takahashi, K., S. Mizuarai, H. Araki, S. Mashiko, A. Ishihara, A. Kanatani, H. Itadani, and H. Kotani. 2003. Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. The Journal of Biological Chemistry 278: 46654–46660. doi:10.1074/jbc.M309895200.
Tamori, Y., J. Masugi, N. Nishino, and M. Kasuga. 2002. Role of peroxisome proliferator-activated receptor-gamma in maintenance of the characteristics of mature 3 T3-L1 adipocytes. Diabetes 51: 2045–2055.
Tanaka, M., K. Ikeda, T. Suganami, C. Komiya, K. Ochi, I. Shirakawa, M. Hamaguchi, S. Nishimura, I. Manabe, T. Matsuda, K. Kimura, H. Inoue, Y. Inagaki, S. Aoe, S. Yamasaki, and Y. Ogawa. 2014. Macrophage-inducible C-type lectin underlies obesity-induced adipose tissue fibrosis. Nature Communications 5: 4982. doi:10.1038/ncomms5982.
Tian, H., C. Liu, X. Zou, W. Wu, C. Zhang, and D. Yuan. 2015. MiRNA-194 regulates palmitic acid-induced toll-like receptor 4 inflammatory responses in THP-1 cells. Nutrients 7: 3483–3496. doi:10.3390/nu7053483.
Trayhurn, P. 2013. Hypoxia and adipose tissue function and dysfunction in obesity. Physiological Reviews 93: 1–21. doi:10.1152/physrev.00017.2012.
Vandanmagsar, B., Y.-H. Youm, A. Ravussin, J.E. Galgani, K. Stadler, R.L. Mynatt, E. Ravussin, J.M. Stephens, and V.D. Dixit. 2011. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature Medicine 17: 179–188. doi:10.1038/nm.2279.
Venugopal, J., K. Hanashiro, and Y. Nagamine. 2007. Regulation of PAI-1 gene expression during adipogenesis. Journal of Cellular Biochemistry 101: 369–380. doi:10.1002/jcb.21173.
Wang, B., I.S. Wood, and P. Trayhurn. 2007. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflügers Archiv 455: 479–492. doi:10.1007/s00424-007-0301-8.
Wang, S., M. Zhang, B. Liang, J. Xu, Z. Xie, C. Liu, B. Viollet, D. Yan, and M.-H. Zou. 2010. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: Role of 26S proteasomes. Circulation Research 106: 1117–1128. doi:10.1161/CIRCRESAHA.109.212530.
Wang, B., J. Sun, Y. Ma, G. Wu, Y. Shi, and G. Le. 2014. Increased oxidative stress and the apoptosis of regulatory T cells in obese mice but not resistant mice in response to a high-fat diet. Cellular Immunology 288: 39–46. doi:10.1016/j.cellimm.2014.02.003.
Weisberg, S.P., D. McCann, M. Desai, M. Rosenbaum, R.L. Leibel, and A.W. Ferrante. 2003. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of Clinical Investigation 112: 1796–1808. doi:10.1172/JCI19246.
Wellen, K.E., and G.S. Hotamisligil. 2005. Inflammation, stress, and diabetes. The Journal of Clinical Investigation 115: 1111–1119. doi:10.1172/JCI25102.
Wen, H., D. Gris, Y. Lei, S. Jha, L. Zhang, M.T.-H. Huang, W.J. Brickey, and J.P.-Y. Ting. 2011. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nature Immunology 12: 408–415. doi:10.1038/ni.2022.
Wensveen, F.M., V. Jelenčić, S. Valentić, M. Šestan, T.T. Wensveen, S. Theurich, A. Glasner, D. Mendrila, D. Štimac, F.T. Wunderlich, J.C. Brüning, O. Mandelboim, and B. Polić. 2015. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nature Immunology 16: 376–385. doi:10.1038/ni.3120.
Wunderlich, C.M., N. Hövelmeyer, and F.T. Wunderlich. 2013. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAK-STAT 2: e23878. doi:10.4161/jkst.23878.
Xu, H., G.T. Barnes, Q. Yang, G. Tan, D. Yang, C.J. Chou, J. Sole, A. Nichols, J.S. Ross, L.A. Tartaglia, and H. Chen. 2003. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of Clinical Investigation 112: 1821–1830. doi:10.1172/JCI19451.
Xu, J., Y. Jiang, J. Wang, X. Shi, Q. Liu, Z. Liu, Y. Li, M.J. Scott, G. Xiao, S. Li, L. Fan, T.R. Billiar, M.A. Wilson, and J. Fan. 2014. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death and Differentiation 21: 1229–1239. doi:10.1038/cdd.2014.40.
Yamasaki, S., E. Ishikawa, M. Sakuma, H. Hara, K. Ogata, and T. Saito. 2008. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nature Immunology 9: 1179–1188. doi:10.1038/ni.1651.
Yang, L., E.S. Calay, J. Fan, A. Arduini, R.C. Kunz, S.P. Gygi, A. Yalcin, S. Fu, and G.S. Hotamisligil. 2015. METABOLISM. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science 349: 500–506. doi:10.1126/science.aaa0079.
Yaspelkis, B.B., I.A. Kvasha, and T.Y. Figueroa. 2009. High-fat feeding increases insulin receptor and IRS-1 coimmunoprecipitation with SOCS-3, IKKalpha/beta phosphorylation and decreases PI-3 kinase activity in muscle. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology 296: R1709–R1715. doi:10.1152/ajpregu.00117.2009.
Ye, J., Z. Gao, J. Yin, and Q. He. 2007. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. American Journal of Physiology. Endocrinology and Metabolism 293: E1118–E1128. doi:10.1152/ajpendo.00435.2007.
Yin, J., Z. Gao, Q. He, D. Zhou, Z. Guo, and J. Ye. 2009. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. American Journal of Physiology. Endocrinology and Metabolism 296: E333–E342. doi:10.1152/ajpendo.90760.2008.
Yu, C., Y. Chen, G.W. Cline, D. Zhang, H. Zong, Y. Wang, R. Bergeron, J.K. Kim, S.W. Cushman, G.J. Cooney, B. Atcheson, M.F. White, E.W. Kraegen, and G.I. Shulman. 2002. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. The Journal of Biological Chemistry 277: 50230–50236. doi:10.1074/jbc.M200958200.
Zeyda, M., D. Farmer, J. Todoric, O. Aszmann, M. Speiser, G. Györi, G.J. Zlabinger, and T.M. Stulnig. 2007. Human adipose tissue macrophages are of an anti-inflammatory phenotype but capable of excessive pro-inflammatory mediator production. International Journal of Obesity 2005(31): 1420–1428. doi:10.1038/sj.ijo.0803632.
Zolotnik, I.A., T.Y. Figueroa, and B.B. Yaspelkis. 2012. Insulin receptor and IRS-1 co-immunoprecipitation with SOCS-3, and IKKα/β phosphorylation are increased in obese Zucker rat skeletal muscle. Life Sciences 91: 816–822. doi:10.1016/j.lfs.2012.08.038.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Engin, A. (2017). The Pathogenesis of Obesity-Associated Adipose Tissue Inflammation. In: Engin, A., Engin, A. (eds) Obesity and Lipotoxicity. Advances in Experimental Medicine and Biology, vol 960. Springer, Cham. https://doi.org/10.1007/978-3-319-48382-5_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-48382-5_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48380-1
Online ISBN: 978-3-319-48382-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)