
Abstract
Obesity and its comorbidities are closely related to the inflammatory environment created by expanded adipose tissue. Several mechanisms trigger inflammation in adipose tissue, including excess fatty acids, hypoxia, and activation of the inflammasome. Inflammation is characterized by the abundance of immune cells, particularly M1 macrophages and T lymphocytes, which have increased secretion of proinflammatory cytokines that act to perpetuate systemic inflammation and induce insulin resistance. The gut microbiota is also involved in obesity-induced inflammation via LPS-related endotoxemia that induces cytokine secretion and insulin resistance. Innate lymphoid type 2 cells, regulatory T cells, and interleukine (IL)-10 counteract the inflammation and insulin resistance, establishing classical or metabolically healthy obesity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obesity is a chronic disease characterized by the expansion of adipose tissue and an inflammatory component, which is highly related to the development of metabolic diseases [1]. The risk of developing obesity-related complications is proportional to the degree of obesity, and more specifically, to the accumulation of visceral fat. Increasing evidence suggests that chronic low-grade inflammation in adipose tissue affects the pathogenesis of metabolic disorders in obese patients [2].
The anatomical distribution of fat deposition has an important impact on the level of risk. Individuals with abdominal obesity, who have accumulated visceral adipose tissue (VAT), have an increased risk of developing either insulin resistance or metabolic syndrome, which precedes the development of type 2 diabetes (T2DM) and is often associated with dyslipidemia [3, 4]. Although subcutaneous adipose tissue (SAT) is more extensively studied because of easy access to clinical patients, studies have shown the crucial role of VAT on obesity inflammation [5]. Magnetic resonance imaging studies suggest that obesity-associated metabolic disorders are correlated with the expansion of VAT and are only modestly related with the expansion of SAT [6]. Alterations of adipose tissue, such as the increased volume of adipocytes, macrophage infiltration, and inflammatory cytokines production may be involved in the pathophysiology of cardiovascular disease and can modify the risk for cardiovascular disease [7]. However, the presence of metabolic disorders related to obesity varies widely among obese individuals [8].
Type 2 diabetes mellitus (T2DM) represents approximately 90 % of all cases of diabetes, and its frequency is similar to that of obesity. T2DM begins with impaired insulin signaling that is compensated for by the overproduction and secretion of pancreatic insulin (hyperinsulinemia), which characterizes the insulin resistance commonly seen in obese individuals. In adipose tissue, insulin acts by reducing lipolysis and the release of free fatty acids (FFAs), and in the liver and skeletal muscles, insulin reduces gluconeogenesis and induces glucose uptake by regulating key enzyme activities and stimulating translocation of the glucose transporter 4 (GLUT4) toward the cell membrane, respectively. As obesity is maintained or exacerbated, the pancreas fails to compensate for the insulin resistance and this eventually leads to insufficient hepatic and peripheral glucose disposal and subsequently higher circulating levels of glucose, which initiates the development of T2DM. In addition to the high levels of blood glucose, insulin resistance is characterized by high levels of circulating FFAs, which induce ectopic fat depot, primarily in the liver and muscle [9].
The Adipose Tissue
White adipose tissue comprises several cell types and includes mature lipid-filled adipocytes, macrophages, T cells and other immune cells, pre-adipocytes, which are partially committed to adipocyte differentiation, pericytes, which have the potential to become adipocytes, fibroblasts, endothelial cells and mast cells, which influence angiogenesis and remodeling. Adipose tissue is generally distinguished in subcutaneous (SAT) and visceral adipose tissue (VAT). Although SAT represents approximately 80 % of the human adipose tissue, VAT is more metabolically active, and its accumulation is more predictive of obesity-associated mortality [10].
Adipokines Orchestrate Inflammation in Obesity
Adipose tissue is dynamically involved in the regulation of cellular function, and the genesis of diseases via a complex network of endocrine, autocrine, and paracrine signals affects the response of many tissues [11, 12]. These signals are mediated in the system by various adipokines, such as leptin, adiponectin, resistin, chemokine (C-C motif) ligand 2 (CCL2), interleukin (IL) 6 and IL-1β, tumor necrosis factor (TNF), IL-10 and transforming growth factor (TGF) β [13]. The main adipokines that orchestrate inflammation in adipose tissue are discussed below.
Leptin
Leptin was first described in the Ob/Ob mouse, which has spontaneous mutation in the leptin gene, and these mice become obese, hyperphagic, intolerant to cold, and unfertile. Leptin is a non-glycosylated polypeptide of 167 amino acids and a molecular weight of 16 kDa, which can circulate freely or as a stable complex associated with alpha 2-macroglobulin [14]. It is secreted by adipose tissue, primarily SAT, in a pulsatile manner, usually 2 to 3 h after a meal. The circulating levels of leptin directly reflect the amount of energy stored in the adipose tissue; therefore, its production increases proportionally to the amount of body fat and women have greater amounts than men [15]. Additionally, leptin is expressed at other sites, such as brown adipose tissue, the gastrointestinal tract, bone, lung, intestine, kidney, liver, skin, stomach, heart, placenta, and spleen suggesting a pleiotropic effect [16]. The primary biological effect of leptin is the control of food intake and increased energy expenditure by activating its receptor, which is highly expressed in the hypothalamus [17]. The expression pattern of leptin suggests its involvement in a variety of actions in addition to the control of energy balance and the regulation of fat mass, including bone metabolism, reproductive function, and maintenance of pregnancy [18].
Obesity is frequently associated with leptin resistance and hyperleptinemia, which contribute to adipocyte dysfunction, the activation of immune cells [1] and the consequences of each [19, 20]. Leptin resistance may be caused by the impaired transport of leptin across the blood–brain barrier or by alterations in neurons signaling, such as the increased expression of protein tyrosine phosphatase 1B (PTP1B), a protein that blocks leptin signaling and the suppressor of cytokine signaling 3 (SOCS3) [16]. Leptin induces the expression of proinflammatory cytokines in macrophages and T cells and activates the same receptors activated by proinflammatory cytokines, including mitogen-activated protein kinases (MAPKs), signal translator and activator of transcription 3 (JAK-STAT3), and phosphatidyl-inositol 3 kinase (PI3K). These findings demonstrate that, in addition to its role in metabolism, leptin is important during the adipose tissue inflammation [2, 19].
Adiponectin
Adiponectin is a plasma protein 30 Kd [21], that is structurally related to C1q factor of the complement system and type VIII and X collagen that circulates as multimeric forms. Adiponectin is highly expressed in the SAT, and its levels decrease with increasing body weight and adiposity [19, 22]. The adiponectin receptors AdipoR1 and R2 mediate the action of adiponectin, mainly through increasing the activity of 5′ adenosine monophosphate-activated protein kinase (AMPK) [2, 23, 24]. The metabolic effects of adiponectin occur in several organs. In the liver, adiponectin improves insulin sensitivity, inhibits free fatty acid uptake and oxidation, and reduces the secretion of glucose. In adipose tissue, adiponectin increases glucose uptake and adipogenesis and in the muscles, stimulates glucose metabolism and accelerates the oxidation of free fatty acids [11, 22]. The anti-inflammatory and antiatherogenic effects of adiponectin are mediated through the inhibition of monocyte adhesion, macrophage growth, differentiation into foam cells, and remodeling of the vascular wall muscle [25].
TNF
TNF is a proinflammatory adipokine linked to obesity, insulin resistance, and atherosclerosis. It is more highly expressed by macrophages from the vascular stroma, particularly M1 macrophages, primarily in the VAT of obese patients [11, 26–28]. TNF induces insulin resistance through the phosphorylation of a serine residue for the substrate insulin receptor 1 (IRS-1) via the c-Jun NH2-terminal kinase that has an inhibitory effect on insulin signaling [29]. Moreover, TNF inhibits lipoprotein lipase (LPL) activity and increases fatty acid mobilization from adipose tissue into the bloodstream [30, 31]. TNF influences the synthesis of proinflammatory adipokines through the activation of nuclear factor kappa B (NF-κB), increasing the expression of IL-6, chemokine (C-C motif) ligand 2 (CCL2), and TNF itself, and decreasing the expression of adiponectin [14, 32, 33].
IL-1β
IL-1β is produced by M1 macrophages, like TNF, impairs insulin signaling, and increases lipolysis. In obese patients with hyperleptinemia, circulating IL-RA levels, an IL-1β antagonist, are seven times higher than in nonobese patients, suggesting a regulatory mechanisms [25, 34].
IL-6
IL-6 is a proinflammatory cytokine that is involved in multiple physiological processes, including inflammation, tissue injury and host defense [32]. Approximately one third of circulating IL-6 originates from the adipose tissue. Increased IL-6 levels are positively correlated with obesity, glucose intolerance, insulin resistance, and T2DM and are also predictive for the development of metabolic syndrome and cardiovascular disease. Adipocyte hypertrophy and inflammatory stimuli, such as TNF, favor increased IL-6 production. This occurs primarily in the VAT, confirming the positive correlation between VAT inflammation and insulin resistance [28].
The mechanisms associated with insulin resistance and IL-6 are similar to those reported for TNF occurring via serine residue phosphorylation of IRS-1 and inhibition of lipoprotein lipase [25]. IL-6 suppresses the metabolic actions mediated by insulin in hepatocytes via a mechanism mediated by the expression of suppressor of cytokine signaling (SOCS-3) [2]. In addition, IL-6 induces the production of C-reactive protein in the liver, an important risk factor for cardiovascular disease, and has been associated with obesity-related hypertriglyceridemia by stimulating hepatic secretion of very low density lipoprotein (VLDL) [16].
Weight loss favors reduced circulating levels and IL-6 expression by adipose tissue, partially reversing the inflammatory state [2, 31].
CCL2
CCL2 is also referred to as monocyte chemotactic protein 1 (MCP1) and small inducible cytokine A2. It is mainly produced by cells of the vascular stroma and in the VAT compared with SAT, and its expression is positively regulated by leptin [28, 35]. CCL2 induces macrophage infiltration, insulin resistance, and triglyceride accumulation in the liver [35].
IL-10
IL-10 is a classic anti-inflammatory cytokine that suppresses the signal transduction of proinflammatory cytokines. IL-10 is produced primarily by M2 macrophages, Th2 T cells, and adipocytes [19, 36]. The anti-inflammatory effect of IL-10 is observed during adipocyte culture and is caused by the inhibition of the genes induced by TNF, including TNF itself, IL-6, and IL-1β, and the induction of anti-inflammatory cytokines, such as interleukin-1 receptor antagonist (IL-1ra) [29].
IL-10 also reverses the negative regulation of GLUT4 and IRS-1 stimulated by TNF, restoring appropriate insulin signaling [37]. Moreover, treatment with IL-10 improves inflammation in the adipose tissue and liver and enhances the hepatic metabolism of lipids and glucose in obese mice [38].
TGFβ
TGFβ was first discovered as a critical factor for the growth of non-immune cells, and has gradually been recognized as a critical cytokine that regulates immune responses [39]. Classically, TGFβ inhibits the growth and activation of immune cells, and also inhibits macrophages and B and T cell activation. In addition, TGFβ induces the expression of the transcription factor fork head box P3 positive (FOXP3+) in CD4+CD25− FOXP3− T cells, converting them into FOXP3+ regulatory T cells [40].
The level of TGFβ in adipose tissue is highly and positively associated with the grade of obesity, and it has been suggested that TGFβ plays an important role in the biology and regulation of subcutaneous and visceral adipose tissue [41, 42]. The role of TGFβ in adipogenesis remains unclear. TGFβ induces adipogenesis in multipotent progenitor cells, but inhibits adipogenesis in populations of committed pre adipocytes [43].
Immune Cells Involved in Inflammation of Adipose Tissue
Adipose tissue inflammation is characterized by moderately increased infiltration of macrophages and immune cells, leading to high levels of circulating proinflammatory cytokines and fatty acids. Inflammation can, in turn, aggravate obesity comorbidities and induce insulin resistance and T2DM.
Adipose tissue macrophages (ATMs) represent 5 % of the stromal vascular fraction in lean adipose tissue, increasing to approximately 20-30 % during diet-induced obesity. ATMs are classified into two main types according to their surface markers and cytokine profile. The alternatively activated M2 macrophages are characterized by the presence of CD206 (mannose receptor) and macrophage galactose-type C-type lectin 1 (MGL1), which are scavenger receptors mediating phagocytosis via high levels of arginase production, which degrades induced nitric oxide synthase, and by the expression anti-inflammatory cytokines such as IL-10 and TGFβ [44]. By contrast, classically activated M1 macrophages express CD11c but not CD206 and MGL1. They are abundant in the obese adipose tissue, mainly in the VAT, and secrete a proinflammatory cytokines characterized by high levels of TNF and IL-6. These cytokines are directly involved in insulin resistance in adipocytes. The establishment of obesity induces a switch from the M2 (anti-inflammatory) to M1 (proinflammatory) phenotype, characterized by the reduced production of anti-inflammatory cytokines and increased production of proinflammatory cytokines [45]. These conditions are crucial to the development of insulin resistance, diabetes and various complications associated with obesity [36].
The death of adipocytes occurs because of hypoxia secondary to adipocyte hypertrophy. Macrophages migrate to and accumulate in regions of hypoxia, as suggested by the negative correlation between pO2 and macrophage markers in adipose tissue in humans [46]. Greater than 90 % of the adipose tissue macrophages in obese mice and humans are located around the dead adipocytes, forming crown like structures [47].
Several important hypoxia-associated genes, such as leptin and vascular endothelial growth factor (VEGF), are directly regulated by a transcription factor named hypoxia-inducible factor 1-alpha (HIF-1α). HIF-1α possesses an oxygen-sensitive subunit that has increased degradation under normoxic conditions. Therefore, hypoxia appears to be important in the accumulation of macrophages and the consequent production of various adipokines by these cells in adipose tissue [48].
Macrophages are the main cell type that infiltrates adipose tissue, although the interactions between innate and adaptive immunity involving several cell types are vital for the establishment of inflammation. Regulatory T cells, CD8+ and CD4+ (Th1 and Th2) T cells, natural killer T cells (NKT), B cells, dendritic cells, eosinophils, neutrophils, and mast cells have all been described in adipose tissue and have also been implicated in the pathogenesis of insulin resistance in obese individuals [45, 48, 49••, 50].
Dendritic cells (DCs) are professional antigen presenting leukocytes involved in macrophage activation and accumulation (M1) at the site of inflammation. As an M1 macrophage, DCs are increased in obese type 2 diabetes patients, whereas a decreased DC population in adipose tissue is associated with the improvement of insulin resistance. A novel CD11c + dendritic cell subset was described in murine adipose tissue. DCs display lower levels of CD40, CD80, CD86, major histocompatibility complex (MHC) I and MHCII expression, and higher levels of IL-6, TGF-β and IL-23 secretion than splenic DC. These cells are more frequently found in obese than in lean mice and are capable of promoting Th17 cell generation and inflammation [51••].
Neutrophils are the first immune cells that reach the site of inflammation and can exacerbate inflammation by helping to recruit macrophages and by interacting with antigen-presenting cells. Elastase is an important protease produced by neutrophils implicated in inflammation. The enzyme can degrade the IRS-1 and reduce insulin-induced serine–threonine kinase (Akt) phosphorylation in adipocytes [52••]. Deletion of neutrophil elastase leads to less intense adipose tissue inflammation in obese mice that is associated with lower adipose tissue neutrophil and macrophage contents [52••].
Eosinophils are one of the main sources of IL-4 and IL-13 in adipose tissue, and their levels are reduced with increased fat mass. These cells may improve glucose homeostasis by sustaining the levels of anti-inflammatory M2 macrophages in the white AT, possibly through IL-4 and IL-13 signaling. It has been demonstrated that the number of M2 macrophages in the adipose tissue is dependent on the eosinophil-derived IL-4 and IL-13 in the same tissue [44, 53]. Eosinophil production, secretion and recruitment are induced by IL-5, and IL-5 deficiency profoundly impairs VAT eosinophil accumulation and results in increased adiposity and insulin resistance in HFD-induced obesity [54].
Innate lymphoid type 2 cells (ILC2), also designated innate helper type 2 cells or natural helper cells, are innate cells widely distributed in mammalian tissues, including adipose tissue [54, 55]. ILC2 are resident in the VAT and are one of the major sources of IL-5 and IL-13, which promote the accumulation of eosinophils and M2 macrophages. ILC2s are resident in the VAT and promote eosinophil and M2 macrophage accumulation, which is implicated in metabolic homeostasis, and this axis is enhanced during Th2-associated immune stimulation [54].
CD4+ T cells recognize peptides presented by class II major histocompatibility complex (MHC) molecules on the surface of macrophages and dendritic cells, and secrete proinflammatory cytokines. Naïve CD4 T cells differentiate into three effector cell groups, comprising Th1, characterized by IFN-γ secretion, Th2, producing IL-4 and IL-13 cytokines, and Th17 cells, producing IL-17 cytokine. Th1 and Th17 cells mediate proinflammatory responses, whereas Th2 and Treg cells contribute to anti-inflammatory responses. IFNγ-secreting (Th1) and IL-17-secreting (Th17) T cells enhance macrophage proinflammatory functions by inducing the release of IL-1, IL-6, and TNFα. By contrast, the anti-inflammatory IL-4 and IL-13-secreting (Th2) T cells are associated with anti-inflammatory M2 macrophage activation [56]. The ratio of Th1 to Th2 cells is increased in obesity because of the increased production of IFNγ by Th1 cells, reinforcing the inflammation of obesity [56].
Regulatory CD4+FOXP3+ T cells are present in large amounts in the adipose tissue of lean mice, whereas they are significantly reduced in VAT of insulin resistant obese mice [49••]. The severity of systemic metabolic alterations is also negatively regulated and is attenuated by regulatory T cells [57]. Regulatory CD4+FOXP3+ T cells are a key population in the control of the immune response and play an important immunosuppressive role in inflammatory disorders [58, 59]. Regulatory T cells exert their effects directly by cell-cell contact or indirectly via the immunomodulatory effects of cytokines, such as IL-10 and TGFβ [40, 58, 60], which suppresses T cell proliferation, inhibits IFNγ, TNF, and IL-6 production and induces IL-10-mediated macrophage polarization to M2 cells [61]. The lack of CD4+ Foxp3+ cells in the VAT of obese mice is correlated with obesity and insulin resistance, suggesting that the modulation of Tregs in adipose tissue may be a new strategy for the treatment or control of metabolic disorders related to obesity, such as insulin resistance and type 2 diabetes mellitus [37, 62].
CD8+ T cells recognize antigens loaded by MHC class I molecules on antigen presenting cells and participate in proinflammatory cytokine secretion and cytolysis of target cells. It has been reported that the number of CD8+ T cells is elevated approximately threefold in obese adipose tissue [56, 63]. The infiltration by CD8+ T cells precedes the accumulation of macrophages, and immunological and genetic depletion of CD8+ T cells lowers macrophage infiltration and adipose tissue inflammation and ameliorates systemic insulin resistance [64].
Natural killer (NK) cells are of lymphoid origin but they do not express any antigen-specific receptors; therefore, they are considered part of the innate immunity. Although NK cells secrete a proinflammatory pattern of cytokines, their role in obesity and AT inflammation remains unclear. In models of NKT deficiency, loss of NKT cells had only a modest effect on weight gain, inflammation, and insulin resistance, suggesting a small role of these cells in obesity induced inflammation [65, 66].
B cells can also be elevated in adipose tissue after high-fat diet consumption that is followed by IgG2c concentrations in adipose tissue [63]. B cell infiltration promotes insulin resistance by inducing M1 macrophage polarization. The IgG produced by B cells induces the clearance of apoptotic and necrotic debris through antibody mediated fixation of complement proteins involved in the phagocytosis of macrophages.
The presence of regulatory B (Bregs) cells that produce interleukin-10 constitutively has been identified in adipose tissue [67]. B cell-specific IL-10 deletion enhanced adipose inflammation and insulin resistance in diet-induced obese mice, whereas adoptive transfer of adipose tissue Breg cells reversed it [67]. These findings indicate that adipose tissue Breg cells are a naturally occurring regulatory B cell subset that maintains homeostasis within the adipose tissue.
Mast cells are primarily involved in allergy and anaphylaxia. However, the presence of these cells has been described in adipose tissue. At this site, mast cells are increased several fold in obese compared with lean individuals, as suggested by the higher levels of mast cell-derived tryptase in obese patients [68]. Mast cells contain many granules loaded with various mediators, including histamine, serotonin, heparin, serine protease, eicosanoids, and cytokines. Among the mediators produced by mast cells, IL-6 and IFN-γ play major roles in the inflammation associated with obesity.
The Inflammasome
Inflammasomes are a multiproteic complex, which processes inactive pro-IL-1β and IL-18 into their active forms, regulating chronic inflammation and metabolic processes [69]. The inflammasome is primarily composed of the following three components: 1) pattern-recognition receptors (PRRs), such as toll-like receptors (TLRs) and intracellular nod-like receptors (NLR) that trigger downstream signaling cascades; 2) the adaptor molecule ASC (apoptosis associated speck-like protein containing CARD); and 3) caspase-1. Several inflammasomes have been identified, and NLRP3 (nucleotide-binding domain and leucine-rich repeat containing NLR, pyrin domain containing 3) is the most extensively studied and is linked to type 2 DM and β cell destruction, obesity, and atherosclerosis [69–71].
TLRs are activated by a variety of dietary factors and endogenous signals in response to metabolic alterations induced by obesity. Free fatty acids bind to two different toll receptors, TLR2 and TLR4. Other inflammatory stimuli, such as ceramides and oxidized LDL, bind to and activate TLR4. The downstream pathways of both TLRs activate NF-kB and MAPK to inhibit insulin signaling via serine phosphorylation of the IRS-1 and induce transcription of the proinflammatory cytokines TNF and IL-6, as well as the biologically inactive pro- IL-1β and pro-IL-18, which require posterior processing to become active [72].
NLRP3 is then activated by long chain fatty acids, ceramides, oxidized LDL, and hyperglycemia. After activation, NLRP3 assembles with the adapter protein ASC and caspase 1, forming a multiprotein complex (inflammasome) that cleaves pro IL-1b and pro-IL-18 to the active forms IL-1b and IL-18. The islet amyloid polypeptide (IAPP or amylin) that is co-secreted with insulin, forms an amyloid structure deposited in the islet interstitium, inducing the NLRP3 inflammasome, and generating mature IL-1β in macrophages.
Both M1 macrophages and adipocytes induce inflammasome expression in the adipose tissue [73, 74]. The importance of the inflammasome in obesity and adipose tissue inflammation is confirmed by the finding that NLRP3 deletion lead to a slower weight gain, higher metabolic rate, and improved insulin sensitivity [73]. The reduction of NLRP3 adipose tissue expression is coupled with decreased inflammation and improved insulin–sensitivity in obese T2DM patients [73, 75]. The data from human studies show that caspase-1 and NLRP3 are highly expressed in human VAT and that SAT biopsies from obese–T2DM before and after one year of weight–loss intervention shows that the weight loss and enhanced insulin sensitivity are associated with a significant reduction in IL-1β and NLRP3 mRNA expression in the SAT [74, 75].
The role of Microbiota on Obesity-induced Inflammation
The role of the gut microbiota in obesity and T2DM has been studied during the last decade. Several studies have shown the association between obesity and unbalanced dominant gut phyla, with reductions in Bacteroidetes associated with a proportional increase in Firmicutes [76••, 77–81]. However, other studies have found no differences between the Firmicutes and Bacteroidetes in obese individuals [80, 82]. Two primary mechanisms have been proposed to explain this association. First, Firmicutes can increase polysaccharide fermentation and the intestinal production of the short-chain fatty acids (SCFAs) acetate, propionate, and butyrate. After absorption, SCFAs reach the liver via the portal system and could induce the synthesis of triacylglycerol, which will be stored in the adipose tissue. Second, obesity is closely related to endotoxemia because of the increased intestinal permeability [83]. In healthy lean individuals, the intestinal epithelium acts as a barrier preventing the continued translocation of LPS. It has been proposed that high fat diets increase the intestinal permeability by reducing the expression of tight junction proteins and modulating the gut microbiota, leading to the metabolic endotoxemia associated with increases in inflammatory tone, body weight gain, and insulin resistance [83]. By contrast, antibiotic administration reduces blood LPS and endotoxemia levels and is correlated with reduced glucose tolerance, body weight gain, inflammation markers, oxidative stress, and macrophage infiltration in the VAT. Studies of induced endotoxemia LPS in humans confirm that low doses of LPS induce cytokine release, low-grade inflammation in the adipose tissue, and insulin resistance [84, 85].
The importance of microbiota in obesity is reinforced by studies of microbiota transplantation from genetically obese to axenic mice [86, 87]. Microbiota transplantation from mice fed a high fat diet to lean germ-free recipients promoted greater fat deposition than transplants from lean donors. These results suggest that the phenotype of an increased capacity for energy harvesting is simply transmitted by transplantation of the obesity-associated gut microbiota to healthy and lean donors.
Metabolically Healthy but Obese Individuals
Adipose tissue is subject to a continuous process of remodeling, which normally maintains tissue health, but this process can become unregulated, leading to the death of adipocytes in association with the recruitment and activation of macrophages and systemic insulin resistance [88]. Under constant excess caloric intake, adipocytes undergo hypertrophy and hyperplasia, thereby establishing dynamic events related to the expansion of adipose tissue. Hypertrophy results in the altered secretion of products, which over time, adversely affects the remodeling of adipose tissue. In parallel, hyperplasia, although initiating increased total adiposity of obese individuals, provides valuable protection against harmful effects caused by the lipotoxic systemic effect of lipids.
An important distinction must be made between the healthy and pathological expansion of adipose tissue. The pathological expansion of adipose tissue is associated with the rapid hypertrophy of adipocytes, inducing macrophage infiltration, and activation, and the limited development of vessels, causing hypoxia and potentiating inflammation. Healthy expansion is related to a slower and more organized expansion of the fat mass, associated with the adequate recruitment of stromal cells and adipocyte precursors, which differentiate into small adipocytes in the appropriate ratios, and subsequent vascularization with a minimal induction of inflammation [89].
A subset of obese individuals appears to be protected or, at least, more resistant to developing the metabolic disorders associated with obesity. These individuals are classified as metabolically healthy but obese subjects (MHOS). MHOS is defined as the obese individual free of metabolic disorders, such as T2DM, dyslipidemia, and hypertension [90]. Despite the excess body fat, they exhibit a lower inflammatory profile, characterized by improved insulin control and lipid profiles and lower concentrations of inflammatory markers, such as IL-6, C-reactive protein and alpha-1 antitrypsin [8]. In addition, the absence of cardiometabolic risk factor clusters, such as metabolic syndrome or proinflammatory markers have also been used to categorize MHOS [91].
The mechanisms underlying the favorable metabolic profiles of these individuals are not completely understood. Characteristics of the adipose tissue, such as adipocyte size, differences related to different deposits, gene expression profiles related to metabolic pathways and the inflammatory profile, as well as muscle insulin sensitivity characteristics may be involved in the protective profile of MHO [8].
A study comparing MHOS, unhealthy obese and lean individuals highlighted the metabolic differences among these individuals. MHOS presented reduced secretion of IL-1β, reduced caspase-1 activity, reduced expression of IL-1b and NLRP3, fewer macrophages and a higher number of regulatory T cells in the visceral adipose tissue compared with unhealthy obese individuals [92].
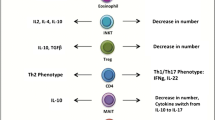
Another clinical study compared the characteristics of the SAT and VAT of 16 morbidly obese but metabolically healthy individuals and non-obese individuals [93•]. The results suggested that the favorable metabolic state of MHOS is associated with the higher production of anti-inflammatory cytokines, particularly IL-10, TGFβ, FOXP3+ Treg cells, and MMPs in the adipose tissue, suggesting greater anti-inflammatory control. The increased expression of MMP2, MMP8, and MMP9 in the VAT of obese individuals favors adipogenesis and adipose tissue remodeling. In adipose tissue, adipogenesis functions as a beneficial event by increasing the number of adipocytes and the fat storage capacity, thereby preventing ectopic deposition. Furthermore, because they induce angiogenesis, MMPs reduce hypertrophic adipocytes, hypoxia, preventing necrosis, M1 macrophage infiltration and the induction of inflammation. These regulatory mechanisms may be important in the delayed onset of metabolic complications, even in individuals with extreme obesity. The main events linked to the development of unhealthy or healthy obesity are shown in Fig. 1.

The expansion of adipose tissue and alterations of gut microbiota induce increased free fatty acid and LPS concentrations in blood that. Those factors, associated with the hipertrophy induced hypoxia, trigger the low-grade inflammatory response characterized by the infiltration and activation of M1 macrophage and other immune cells that will secrete pro-inflammatory cytokines and activate NLRP2 inflammasome. These events lead to the establishment of chronic obesity-related comorbidities such as type 2 diabetes mellitus (T2DM) and atherosclerosis. Alternatively, adipose tissue expansion will induce preadipocytes differentiation, generating new adipocytes (hyperplasy) related to a lower pro-inflammatory stimulus and a predominance of regulatory/anti-inflammatory immune cells and cytokines. This scenario is suitable to a more benign obesity evolution defined as metabolically healthy obesity
Conclusion
Several factors determine the establishment and severity of low-grade inflammation in obese individuals. An adequate diet may reduce several unfavorable factors, such as the adipocyte hypertrophy that leads to adipocyte hypoxia; and the increase of intestinal permeability and unbalanced gut microbiota that trigger metabolic endotoxemia. After initiation, the inflammatory stimuli activates a cascade of inflammatory events in adipocytes and infiltrated immune cells, which determines the presence and severity of obesity comorbidities.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
de Heredia FP, Gomez-Martinez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. 2012;71:332–8.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85–97.
Fain, J.N. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: a review. Mediators Inflamm 2010, 513948.
van Greevenbroek MM, Schalkwijk CG, Stehouwer CD. Obesity-associated low-grade inflammation in type 2 diabetes mellitus: causes and consequences. Neth J Med. 2013;71:174–87.
Bigornia SJ, Farb MG, Mott MM, Hess DT, Carmine B, Fiscale A, et al. Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes. 2012;2:e30.
Despres JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28:1039–49.
Marinou K, Tousoulis D, Antonopoulos AS, Stefanadi E, Stefanadis C. Obesity and cardiovascular disease: from pathophysiology to risk stratification. Int J Cardiol. 2010;138:3–8.
Primeau V, Coderre L, Karelis AD, Brochu M, Lavoie ME, Messier V, et al. Characterizing the profile of obese patients who are metabolically healthy. Int J Obes (Lond). 2011;35:971–81.
Zeyda M, Stulnig TM. Obesity, inflammation, and insulin resistance–a mini-review. Gerontology. 2009;55:379–86.
Girard J, Lafontan M. Impact of visceral adipose tissue on liver metabolism and insulin resistance. Part II: Visceral adipose tissue production and liver metabolism. Diabetes Metab. 2008;34:439–45.
Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;9:191–200.
Li ZY, Wang P, Miao CY. Adipokines in inflammation, insulin resistance and cardiovascular disease. Clin Exp Pharmacol Physiol. 2011;38:888–96.
Lemoine AY, Ledoux S, Larger E. Adipose tissue angiogenesis in obesity. Thromb Haemost. 2013;110.
Gualillo O, Gonzalez-Juanatey JR, Lago F. The emerging role of adipokines as mediators of cardiovascular function: physiologic and clinical perspectives. Trends Cardiovasc Med. 2007;17:275–83.
Pujanek M, Bronisz A, Malecki P, Junik R. Pathomechanisms of the development of obesity in some endocrinopathies - an overview. Endokrynol Pol. 2013;64:150–5.
Leal Vde O, Mafra D. Adipokines in obesity. Clin Chim Acta. 2013;419:87–94.
Jung CH, Kim MS. Molecular mechanisms of central leptin resistance in obesity. Arch Pharm Res. 2013;36:201–7.
D’Ippolito S, Tersigni C, Scambia G, Di Simone N. Adipokines, an adipose tissue and placental product with biological functions during pregnancy. Biofactors. 2012;38:14–23.
Ye J, McGuinness OP. Inflammation during obesity is not all bad: evidence from animal and human studies. Am J Physiol Endocrinol Metab. 2013;304:E466–77.
Vazquez-Vela ME, Torres N, Tovar AR. White adipose tissue as endocrine organ and its role in obesity. Arch Med Res. 2008;39:715–28.
Yadav A, Kataria MA, Saini V. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta. 2013;417:80–4.
Lenz A, Diamond Jr FB. Obesity: the hormonal milieu. Curr Opin Endocrinol Diabetes Obes. 2008;15:9–20.
Yamauchi T, Kadowaki T. Physiological and pathophysiological roles of adiponectin and adiponectin receptors in the integrated regulation of metabolic and cardiovascular diseases. Int J Obes (Lond). 2008;32 Suppl 7:S13–8.
Lago F, Gomez R, Gomez-Reino JJ, Dieguez C, Gualillo O. Adipokines as novel modulators of lipid metabolism. Trends Biochem Sci. 2009;34:500–10.
Pereira SS, Alvarez-Leite JI. Adipokines: biological functions and metabolically healthy obese profile. J Recept Ligand Channel Res. 2014;7:1–11.
Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010;316:129–39.
Zou C, Shao J. Role of adipocytokines in obesity-associated insulin resistance. J Nutr Biochem. 2008;19:277–86.
Maury E, Brichard SM. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol Cell Endocrinol. 2010;314:1–16.
Balistreri CR, Caruso C, Candore G. The role of adipose tissue and adipokines in obesity-related inflammatory diseases. Mediat Inflamm. 2010;2010:802078.
Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–77.
Hermsdorff HH, Angeles Zulet M, Bressan J, Alfredo Martinez J. Effect of diet on the low-grade and chronic inflammation associated with obesity and metabolic syndrome. Endocrinol Nutr. 2008;55:409–19.
Bray GA, Clearfield MB, Fintel DJ, Nelinson DS. Overweight and obesity: the pathogenesis of cardiometabolic risk. Clin Cornerstone. 2009;9:30–40. discussion 41–32.
Lee J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 Diabetes. Arch Pharm Res. 2013;36:208–22.
Sun S, Ji Y, Kersten S, Qi L. Mechanisms of inflammatory responses in obese adipose tissue. Annu Rev Nutr. 2012;32:261–86.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest. 2006;116:1494–505.
Itoh M, Suganami T, Hachiya R, Ogawa Y. Adipose tissue remodeling as homeostatic inflammation. Int J Inflamm. 2011;2011:720926.
Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15:930–9.
Gotoh K, Inoue M, Masaki T, Chiba S, Shimasaki T, Ando H, et al. A novel anti-inflammatory role for spleen-derived interleukin-10 in obesity-induced inflammation in white adipose tissue and liver. Diabetes. 2012;61:1994–2003.
Chen W, Konkel JE. TGF-beta and ‘adaptive’ Foxp3(+) regulatory T cells. J Mol Cell Biol. 2010;2:30–6.
Chen X, Oppenheim JJ. Resolving the identity myth: key markers of functional CD4 + FoxP3+ regulatory T cells. Int Immunopharmacol. 2011;11:1489–96.
Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011;14:67–79.
Tan CK, Chong HC, Tan EH, Tan NS. Getting ‘Smad’ about obesity and diabetes. Nutr Diabetes. 2012;2:e29.
Cristancho AG, Lazar MA. Forming functional fat: a growing understanding of adipocyte differentiation. Nat Rev Mol Cell Biol. 2011;12:722–34.
Chmelar J, Chung KJ, Chavakis T. The role of innate immune cells in obese adipose tissue inflammation and development of insulin resistance. Thromb Haemost. 2013;109:399–406.
Kalupahana NS, Moustaid-Moussa N, Claycombe KJ. Immunity as a link between obesity and insulin resistance. Mol Aspects Med. 2012;33:26–34.
Harford KA, Reynolds CM, McGillicuddy FC, Roche HM. Fats, inflammation and insulin resistance: insights to the role of macrophage and T-cell accumulation in adipose tissue. Proc Nutr Soc. 2011;70:408–17.
Cinti S, Mitchell G, Barbatelli G, Murano I, Ceresi E, Faloia E, et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J Lipid Res. 2005;46:2347–55.
Lolmede K, Duffaut C, Zakaroff-Girard A, Bouloumie A. Immune cells in adipose tissue: key players in metabolic disorders. Diabetes Metab. 2011;37:283–90.
Bertola A, Ciucci T, Rousseau D, Bourlier V, Duffaut C, Bonnafous S, et al. Identification of adipose tissue dendritic cells correlated with obesity-associated insulin-resistance and inducing Th17 responses in mice and patients. Diabetes. 2012;61:2238–47. This paper shows for the first time the presence of specific DCs in adipose tissue in mouse and human obesity.
Patel PS, Buras ED, Balasubramanyam A. The role of the immune system in obesity and insulin resistance. J Obes. 2013;2013:616193.
Chen Y, Tian J, Tian X, Tang X, Rui K, Tong J, et al. Adipose tissue dendritic cells enhances inflammation by prompting the generation of Th17 cells. PLoS ONE. 2014;9:e92450. The results of this study indicates the existence of CD11c + DCs in adipose tissue, which displays an immature phenotype but possessing pro-inflammatory function.
Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–12. The authors show that treatment of hepatocytes with neutrophil elastase causes insulin resistance and that deletion of neutrophil elastase in obese mice reduce inflammation.
Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science. 2011;332:243–7.
Molofsky AB, Nussbaum JC, Liang HE, Van Dyken SJ, Cheng LE, Mohapatra A, et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J Exp Med. 2013;210:535–49.
Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature. 2010;463:540–4.
Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, et al. Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med. 2009;15:921–9.
Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. 2012;486:549–53.
Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory T cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes. 2011;60:2954–62.
Cipolletta D, Kolodin D, Benoist C, Mathis D. Tissular T(regs): a unique population of adipose-tissue-resident Foxp3 + CD4+ T cells that impacts organismal metabolism. Semin Immunol. 2011;23:431–7.
Deiuliis J, Shah Z, Shah N, Needleman B, Mikami D, Narula V, et al. Visceral adipose inflammation in obesity is associated with critical alterations in tregulatory cell numbers. PLoS ONE. 2011;6:e16376.
Tiemessen MM, Jagger AL, Evans HG, van Herwijnen MJ, John S, Taams LS. CD4 + CD25 + Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc Natl Acad Sci U S A. 2007;104:19446–51.
Chen, X., Wu, Y., and Wang, L. Fat-resident Tregs: an emerging guard protecting from obesity-associated metabolic disorders. Obes Rev. 2013;14:568–78.
Huh JY, Park YJ, Ham M, Kim JB. Crosstalk between Adipocytes and Immune Cells in Adipose Tissue Inflammation and Metabolic Dysregulation in Obesity. Mol Cells. 2014;37(5):365–71.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20.
Kotas ME, Lee HY, Gillum MP, Annicelli C, Guigni BA, Shulman GI, et al. Impact of CD1d deficiency on metabolism. PLoS ONE. 2011;6:e25478.
Mantell BS, Stefanovic-Racic M, Yang X, Dedousis N, Sipula IJ, O’Doherty RM. Mice lacking NKT cells but with a complete complement of CD8+ T-cells are not protected against the metabolic abnormalities of diet-induced obesity. PLoS ONE. 2011;6:e19831.
Nishimura, S., Manabe, I., Takaki, S., Nagasaki, M., Otsu, M., Yamashita, H., Sugita, J., Yoshimura, K., Eto, K., Komuro, I., et al. (2013). Adipose Natural Regulatory B Cells Negatively Control Adipose Tissue Inflammation. Cell Metab.
Liu J, Divoux A, Sun J, Zhang J, Clément K, Glickman JN, et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat Med. 2009;15:940–5.
Robbins GR, Wen H, Ting JP. Inflammasomes and Metabolic Disorders: Old Genes in Modern Diseases. Mol Cell. 2014;54:297–308.
Wieser V, Moschen AR, Tilg H. Inflammation, cytokines and insulin resistance: a clinical perspective. Arch Immunol Ther Exp (Warsz). 2013;61:119–25.
Skeldon AM, Faraj M, Saleh M. Caspases and inflammasomes in metabolic inflammation. Immunol Cell Biol. 2014;92:304–13.
Jin C, Flavell RA. Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol. 2013;132:287–94.
Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108:15324–9.
Koenen TB, Stienstra R, van Tits LJ, Joosten LA, van Velzen JF, Hijmans A, et al. The inflammasome and caspase-1 activation: a new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology. 2011;152:3769–78.
Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17:179–88.
Tilg, H., and Moschen, AR.Microbiota and diabetes: an evolving relationship. Gut. 2014. The paper reviews the role of the microbiota in diabetes and provide new aspects regarding its pathophysiological relevance.
Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef Microbes. 2014;5:33–43.
Moreno-Indias I, Cardona F, Tinahones FJ, Queipo-Ortuño MI. Impact of the gut microbiota on the development of obesity and type 2 diabetes mellitus. Front Microbiol. 2014;5:190.
Kemp DM. Does chronic low-grade endotoxemia define susceptibility of obese humans to insulin resistance via dietary effects on gut microbiota? Adipocyte. 2013;2:188–90.
Jumpertz R, Le DS, Turnbaugh PJ, Trinidad C, Bogardus C, Gordon JI, et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am J Clin Nutr. 2011;94:58–65.
Furet JP, Kong LC, Tap J, Poitou C, Basdevant A, Bouillot JL, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes. 2010;59:3049–57.
Mai V, McCrary QM, Sinha R, Glei M. Associations between dietary habits and body mass index with gut microbiota composition and fecal water genotoxicity: an observational study in African American and Caucasian American volunteers. Nutr J. 2009;8:49.
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57:1470–81.
Mehta NN, Heffron SP, Patel PN, Ferguson J, Shah RD, Hinkle CC, et al. A human model of inflammatory cardio-metabolic dysfunction; a double blind placebo-controlled crossover trial. J Transl Med. 2012;10:124.
Mehta NN, McGillicuddy FC, Anderson PD, Hinkle CC, Shah R, Pruscino L, et al. Experimental endotoxemia induces adipose inflammation and insulin resistance in humans. Diabetes. 2010;59:172–81.
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–23.
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31.
Lee MJ, Wu Y, Fried SK. Adipose tissue remodeling in pathophysiology of obesity. Curr Opin Clin Nutr Metab Care. 2010;13:371–6.
Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Investig. 2011;121:2094–101.
Bluher M. The distinction of metabolically ‘healthy’ from ‘unhealthy’ obese individuals. Curr Opin Lipidol. 2010;21:38–43.
Pataky Z, Bobbioni-Harsch E, Golay A. Open questions about metabolically normal obesity. Int J Obes (Lond). 2010;34 Suppl 2:S18–23.
Esser N, L’homme L, De Roover A, Kohnen L, Scheen AJ, Moutschen M, et al. Obesity phenotype is related to NLRP3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia. 2013;56:2487–97.
Pereira SS, Teixeira LG, Aguilar EC, Chaves OM, Savassi-Rocha AL, Pelaez JMN, et al. Modulation of Adipose Tissue Inflammation by Foxp3+ Treg Cells, Il-10 and Tgfβ In Metabolically Healthy Class Iii Obese Individuals. Nutrition. 2014. doi:10.1016/j.nut.2013.11.023. This paper studies the visceral and subcutaneous adipose tissue from grade III obese individuals showing that Treg and IL10 are the important to control inflammation in those individuals.
Compliance with Ethics Guidelines
Conflicts of Interest
Solange S. Pereira and Jacqueline I Alvarez-Leite declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pereira, S.S., Alvarez-Leite, J.I. Low-Grade Inflammation, Obesity, and Diabetes. Curr Obes Rep 3, 422–431 (2014). https://doi.org/10.1007/s13679-014-0124-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-014-0124-9