
Abstract
Retrotransposable elements (RTEs) are abundant in the genomes of most species and continue to evolve and adapt to the defense mechanisms of their host cells. RTEs have contributed to the evolution of their hosts by creating germline genomic diversity, but under most circumstances retrotransposition has deleterious consequences. Our understanding of RTE activity in somatic cells and tissues has lagged, largely because we lacked effective tools to study them in these contexts. Recent evidence indicates that RTEs are more active in somatic cells than anticipated, for example in the nervous system, during the development of cancer, or in senescent cells and aging tissues. This raises the important question of whether RTEs contribute actively to these processes and the development of pathologies, and if so, how. In this review we focus on the role of RTEs in the biology of aging: the evidence for their activation, the host defense mechanisms whose failure may allow this, the consequences of the ensuing RTE activity, and the prospects that targeting RTEs may provide new avenues of treating some age-associated disorders.
Jill A. Kreiling and Brian C. Jones are co-first authors.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Aging is characterized by a failure within many cells and organs of the normal homeostatic mechanisms. It is a major risk factor for numerous disorders, including diabetes, hypertension, cardiac disease, osteoarthritis, neurodegeneration, and cancer. Slowing the rate of aging offers an opportunity to prevent, or at least delay, the onset and extent of these disorders, as well as the possibility of extending healthy human life span. Despite the biological complexity that underlies aging, it has repeatedly proven possible to extend the life span of model organisms through modifications of specific physiological systems such as chromatin maintenance, intermediary metabolism, or insulin signaling (Kenyon et al. 1993; Rogina et al. 2000; Clancy et al. 2001; Tatar et al. 2001; Giannakou et al. 2004; Hwangbo et al. 2004; Kapahi et al. 2004; Oberdoerffer and Sinclair 2007; Dang et al. 2009; Sinclair and Oberdoerffer 2009; Feser et al. 2010; Greer et al. 2010; Kenyon 2010; Feser and Tyler 2011; Maures et al. 2011; Han and Brunet 2012; Ni et al. 2012).
A critical aspect of aging is the degradation of fundamental biological structures such as chromatin (Oberdoerffer and Sinclair 2007). In somatic cells, stability of the genome and epigenome is essential for the maintenance of proper gene expression and silencing. Chromatin remodeling , including changes within regions of constitutive heterochromatin that were previously thought to retain repressive characteristics throughout the life of the cell, has emerged as an exciting area in the molecular genetics of aging. Chromatin maintenance, especially that of heterochromatin, has been shown to change with age in yeast, nematodes, flies, mice, and human cell culture, with far-reaching consequences for gene expression and cellular physiology (Kim et al. 1996; Smeal et al. 1996; Dang et al. 2009; Feser et al. 2010; Wood et al. 2010; Feser and Tyler 2011; De Cecco et al. 2013a; Jiang et al. 2013; Sedivy et al. 2013; Wood et al. 2016).
Age-related changes in chromatin states can alter gene transcription, resulting in the expression of genes that are normally silenced (or vice versa), with consequent deleterious effects on cellular physiology (Elgin and Grewal 2003; Berger 2007; Grewal and Jia 2007; Sedivy et al. 2008; Dang et al. 2009; Feser and Tyler 2011; Han and Brunet 2012). The observed loss of silencing in heterochromatic regions with age includes the increased transcription of genes native to heterochromatin, but also transcription and potential mobility of transposable elements (TEs), which make up the majority of transcripts emanating from heterochromatic regions. The ability of transposable elements not just to express themselves, but to mobilize to new genomic locations within individual somatic cells, adds an additional layer of peril to the potential consequences of the loss of heterochromatin silencing with age.
The contributions that loss of silencing of TEs in somatic cells makes toward the inevitable decline in organismal health with age are just beginning to be explored in detail. The recently discovered ability of TEs to promote aging is expected to open a new area of inquiry, with the potential of providing novel insights into the molecular mechanisms underlying the aging process, while simultaneously offering the promise of novel therapeutic interventions for the preservation of a healthier life span.
2 Remodeling of Chromatin During Aging
2.1 Yeast
The link between chromatin and aging has been well interrogated in invertebrate model systems. Early work in the budding yeast S. cerevisiae demonstrated a loss of silencing with age in heterochromatic regions of the genome, including telomeres, the mating type loci, and rDNA (Kim et al. 1996; Smeal et al. 1996; Kennedy et al. 1997). More recently, a number of studies have examined the specific chromatin changes that take place as yeasts age. Histone H4K16 acetylation levels increase with age, and Sir2 (which deacetylates H4K16ac) levels drop (Dang et al. 2009). Furthermore, subtelomeric heterochromatic regions lose both histones and silencing as cells age. Another study confirmed the observation of general histone loss with age, and also showed that increasing histone supply genetically is sufficient to extend yeast replicative life span (Feser et al. 2010). This age-related loss of histones is also associated with a breakdown in proper gene regulation, with normally silent genes becoming transcribed with age upon nucleosome loss or rearrangement (Hu et al. 2014). This is accompanied by a general increase in genomic instability, with DNA strand breaks, mitochondrial-nuclear DNA transfer, chromosomal alterations and translocations, and retrotransposition all increasing during yeast aging (Hu et al. 2014). Additionally, manipulating chromatin by deleting the ISWI family chromatin remodeling gene ISW2 also leads to an extension in life span in a manner mimicking calorie restriction (Dang et al. 2014).
2.2 Nematodes
Results observed in yeast have also been extended to metazoan invertebrate model systems. In C. elegans a number of studies have shown links between chromatin structure and life span. Disrupting the ASH-2 complex, which contains a histone H3K4 methyltransferase activity, causes an increase in life span (Greer et al. 2010, 2011). Disruption of the H3K4 demethylases has also been reported to extend life span in several studies. RNAi knockdown or null mutations of the H3K4me3 demethylase RBR-2 as well as the H3K4me1/2 demethylases LSD-1 and SPR-5 extend life span (Lee et al. 2003; McColl et al. 2008; Ni et al. 2012; Alvares et al. 2014). Manipulation of the heterochromatic H3K27me3 mark, which is associated with Polycomb group complex silencing, also showed effects on life span. Two independent studies demonstrated that disrupting the H3K27me3 demethylase UTX-1 leads to increased levels of H3K27me3 accumulation in the genome, as well as increased life span (Jin et al. 2011; Maures et al. 2011). Similarly to yeast, knocking down expression of the ISWI complex member athp-2 led to an increase in life span (Hu et al. 2014).
2.3 Fruit Flies
In addition to C. elegans, D. melanogaster has also been a useful model to investigate the association between chromatin structure and organismal life span. The characteristic enrichments of the constitutive heterochromatin mark H3K9me3 and the heterochromatin protein HP1 are lost from pericentric heterochromatin with age in flies (Wood et al. 2010). In conjunction with this observation, heterochromatic silencing of reporter genes in these same regions was lost with age in multiple tissues in the fly (Jiang et al. 2013). Overexpression of HP1 in flies is able to extend life span, suggesting the importance of maintaining proper heterochromatin structure with age (Larson et al. 2012). A study examining aging fly muscle showed an accumulation of γH2AX, a histone variant associated with DNA strand breaks, in old flies (Jeon et al. 2015). Knockdown of HP1 accelerated γH2AX accumulation and also shortened life span (Jeon et al. 2015). Aging effects are however not limited to heterochromatin. Histone acetylation levels also change with age on multiple residues, including an increase of H4K12ac, and mutation of the H4K12 acetyltransferase Chameau confers extended life span (Peleg et al. 2016).
2.4 Mammals
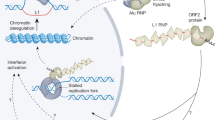
Recent studies show that large regions of the genome undergo significant reorganization in cellular senescence and in aged mammalian tissues. Cellular senescence is an irreversible cell cycle arrest that is triggered by replicative exhaustion, DNA damage, oncogene activation, or oxidative stress. Although low in numbers, senescent cells are found in aged tissues and have been shown to contribute to aging phenotypes (Baker et al. 2016). During the onset of senescence large segments of euchromatin become more closed and accumulate heterochromatic marks (Fig. 1) (Kreiling et al. 2011; De Cecco et al. 2013a; Chen et al. 2015; Criscione et al. 2016). A key feature of senescent cells is the formation of senescence-associated heterochromatin foci (SAHF) containing specific heterochromatin signatures (Narita et al. 2003; Zhang et al. 2007; Chandra et al. 2012). In contrast, regions of constitutive heterochromatin, such as lamin-associated domains (LADs) and centromeres, assume more open characteristics, as exemplified by the senescence-associated distention of satellites (SADS) (De Cecco et al. 2013a; Sadaie et al. 2013; Swanson et al. 2013). In addition, genes associated with the senescence-associated secretory phenotype (SASP) take on epigenetic signatures not found in non-senescent cells (Rai and Adams 2013; Chen et al. 2015).

Age-associated chromatin reorganization. In young cells chromatin is organized into regions of tightly packed heterochromatin (left) and relatively open euchromatin (right). As cells age, some regions of heterochromatin open up and other regions of euchromatin become more condensed. As a result, repressed genes (such as RTEs, indicated in blue) in heterochromatic regions become susceptible to transcription
Genome-wide changes in chromatin structure also occur in chronologically aged cells in vivo, with a closing of euchromatic regions and an accumulation of heterochromatic marks, leading to an overall reduction in mRNA expression (Sarg et al. 2002; Shumaker et al. 2006; O'Sullivan et al. 2010; Kreiling et al. 2011; De Cecco et al. 2013b). A corresponding opening of constitutive heterochromatin (De Cecco et al. 2013b) suggests an overall decompaction of the highly heterochromatic regions known to contain large numbers of retrotransposable elements (RTEs). Taken together, evidence points to large-scale changes in genome organization, with some regions becoming more closed and others more open, with the latter leading to an increase in the expression of RTEs (O'Sullivan and Karlseder 2012; Sedivy et al. 2013).
The loss of constitutive heterochromatin is correlated with a loss of DNA methylation and histone modifications associated with constitutive heterochromatin. Genome-wide methylation patterns change during cellular senescence and with age in the mammalian genome, with specific regions gaining methylation and others losing methylation (Cruickshanks et al. 2013a; Day et al. 2013; Hanzelmann et al. 2015). In young cells the repetitive regions of the genome show highest levels of DNA methylation , and these regions of hypermethylation become hypomethylated with age (Avrahami et al. 2015; Fernandez et al. 2015; Sun and Yi 2015). This global loss of methylation is coupled with a genome-wide reduction in the H3K9me3 histone modification (Scaffidi and Misteli 2006; Shumaker et al. 2006; O'Sullivan et al. 2010; Zhang et al. 2015), which is associated with repressive heterochromatin and is believed to be actively involved in the repression of RTEs (Scaffidi and Misteli 2006). As discussed below, these heterochromatic marks are involved in silencing RTEs and their loss may contribute to the derepression of these elements.
It is also important to note that results obtained in model organisms are not always completely consistent. For instance, in flies the disruption of lid, a LSD-1 H3K4 demethylase analog, shortens life span, in contrast to results observed in C. elegans (Li et al. 2010). Disruption of the H3K27 methyltransferase E(Z) in flies leads to reduced levels of H3K27me3 and increased life span (Siebold et al. 2010), in contrast to worms where higher levels of H3K27me3 were associated with long life span (Jin et al. 2011; Maures et al. 2011). Disruption of RBR-2 in worms can have differential effects depending on which allele is used (Greer et al. 2010; Alvares et al. 2014). Nevertheless, although there undoubtedly are tissue-specific and even organism-specific mechanistic details that remain to be worked out, considerable evidence has accumulated for a strong association between chromatin structure, especially that of heterochromatin, and the regulation of longevity in multiple model systems.
2.5 Changes in the 3D Structure of Chromosomes
Aging cells display dramatic alterations in chromatin accessibility, histone modifications, DNA methylation, and nuclear lamina associations. These changes in chromatin architecture were hypothesized to extend even to the 3D structure of the chromosomes. The first hint that chromosome structure may be altered in aging cells came from studies of fibroblasts from patients with the Hutchinson-Gilford progeria syndrome (HGPS) (McCord et al. 2013). HPGS is a premature aging disease that is caused by mutations in the lamin A gene (LMNA) that result in disruption of interactions between chromatin and the nuclear lamina. In cell culture HGPS patient skin fibroblasts display misshapen nuclei and a loss of the peripheral heterochromatin compartment (Goldman et al. 2004). When HGPS skin fibroblasts were examined by Hi-C, a method to investigate the three-dimensional architecture of the genome, a breakdown of the compartmentalization of active and inactive chromatin domains was observed (McCord et al. 2013). The alterations are likely caused by the disruption of nuclear lamina-chromatin interactions which normally function to restrict the inactive heterochromatin compartment to the nuclear periphery (Guelen et al. 2008).
The 3D structure of chromosomes has also been explored using Hi-C in oncogene-induced and replicative cellular senescence, which have some overlapping but also distinct features. Oncogene-induced senescence (OIS) is believed to be induced by a DNA damage response that is caused by replication stress (Hills and Diffley 2014), whereas replicative cellular senescence is caused by a DNA damage response due to the progressive shortening and deprotection of telomeres. SAHF are typically observed in OIS (Narita et al. 2003), whereas in many models of replicative senescence SAHF formation is weaker or sometimes not present (Kosar et al. 2011). In OIS regions with heterochromatic histone marks as well as LADs display loss of local interactions and gain of long-range interactions (Chandra et al. 2015). This reorganization is consistent with the presence of SAHF in OIS, since heterochromatic regions could cluster spatially over long distances to form the SAHFs (Chandra et al. 2015). The alterations in 3D chromosome structure observed in OIS are however relatively modest in comparison to the global loss of chromosome compartmentalization found in HPGS.
The alterations in chromo some structure observed during replicative senescence are more extensive than in OIS, but also not as drastic as in HPGS. In replicative senescence chromosomes displayed a global loss of long-range and increase of short-range interactions (Criscione et al. 2016). Chromosome painting experiments additionally showed that these alterations were associated with a decrease in the absolute chromosome volume in senescent cells. In replicative senescence the chromosome compartment organization remained mostly unchanged; however, a subset of compartments switched from active to repressive domains (and vice versa). Similar to the compartment switching observed during cellular differentiation (Dixon et al. 2015), compartment switching in replicative senescence also led to correlated changes in gene expression. Interestingly, similarities to cellular differentiation events were noted in studies of both OIS and replicative senescence (Chandra et al. 2015; Criscione et al. 2016). These observations highlight that cellular senescence is a programmed response to DNA damage that results in the remodeling of chromatin as well as large-scale changes in chromosome architecture, although these processes also include some distinct features that are dependent on the senescence-inducing stimuli.
3 Control of TEs and Their Activation with Aging
A significant fraction of eukaryotic genomes are comprised of repetitive sequences. Among the several types of repetitive sequences, noncoding tandem repeats (satellites, telomeres) and TEs are the most abundant. The TEs can be subdivided into two major groups, the DNA transposons and the retrotransposons (RTEs) (Huang et al. 2012). Many species, including the model organisms C. elegans and D. melanogaster discussed in this chapter, harbor active elements of both classes. The most prominent TEs in the mammalian genome are the RTEs. There are three major families of RTEs: the long terminal repeat (LTR) RTEs, which include retroviruses; the long interspersed nuclear elements (LINEs ); and the short interspersed nuclear elements (SINEs). LTR RTEs and LINEs encode a reverse transcriptase and other proteins required for retrotransposition, and hence intact elements can mobilize autonomously, whereas the SINEs are noncoding and exploit the machinery encoded by LINEs to transpose. It is believed that only the LINE L1 remains capable of autonomous retrotransposition in the human genome, whereas both LINE and LTR elements can mobilize in the mouse genome.
3.1 TEs Are Silenced by RNAi Pathways
Largely conserved across species from plants to animals, RNA interference (RNAi) pathways employ small RNAs (smRNAs) to regulate protein-coding genes as well as endogenous proviral sequences such as TEs (Shabalina and Koonin 2008). TEs are repressed by RNAi at two levels: posttranscriptional regulation by targeting mRNA, and transcriptional regulation by the recruitment of repressive heterochromatic marks to silence the target genes. smRNA pathways known to regulate TEs in animals include the microRNA (miRNA) pathway, the short interfering RNA (siRNA) pathway, and the Piwi-interacting RNA (piRNA) pathways. While these pathways are known for their roles in silencing TEs, they are largely tissue specific with the siRNA and miRNA pathways being active in all tissues while the piRNA pathway is predominantly active in the gonads (Slotkin and Martienssen 2007; Ghildiyal and Zamore 2009; Heras et al. 2013, 2014; Hamdorf et al. 2015).
Each pathway differs somewhat in its effector proteins, manner of smRNA biogenesis, and modes of silencing. The siRNA pathway employs 21 nt siRNAs derived from the cleavage of long double-stranded (dsRNA) substrates by the protein Dicer (Yang and Kazazian 2006; Brennecke et al. 2007; Czech et al. 2008; Ghildiyal et al. 2008; Kawamura et al. 2008). These siRNAs are loaded onto an argonaute (AGO) effector protein, thereby forming an RNA-induced silencing complex (RISC), which then uses its siRNA to target and cleave homologous mRNAs in the cytoplasm. The RISC can also move to the nucleus where it recruits chromatin-modifying enzymes promoting the formation of heterochromatin at the site of TE transcription (Slotkin and Martienssen 2007; Fagegaltier et al. 2009).
The piRNA pathway operates through a mechanism whereby large genomic regions consisting of intact as well as fragmented TEs, called piRNA clusters, are transcribed into large single-stranded RNA precursors that are then processed into smaller 23–29 nt piRNAs. piRNAs are also loaded onto pathway-specific Piwi clade argonaute proteins, thus forming piRNA-RISCs (Brennecke et al. 2007; Ghildiyal and Zamore 2009). Similar to the siRNA pathway, these piRNA-RISCs are able to target TE transcripts for silencing either through catalytic cleavage or heterochromatization (Aravin et al. 2007, 2008; Carmell et al. 2007; Di Giacomo et al. 2013). These smRNA pathways have been shown to be critical in preventing the genomic damage caused by the reactivation of TEs. Evidence is also growing that the ability of these pathways to perform their vital functions of suppressing TEs, in both somatic and reproductive tissues, may be closely linked with aging phenotypes.
3.2 Disruption of RNAi Pathways Correlates with Aging Phenotypes
The role of RNAi in regulating TEs in metazoans has been well characterized in multiple model organisms. In Drosophila, mutations in genes of either the siRNA or the piRNA pathways have consistently been associated with a dramatic upregulation of TE transcript levels (Vagin et al. 2006; Rozhkov et al. 2013). This correlates with both an increase in transposition and a change in the heterochromatic marks associated with TEs (Fagegaltier et al. 2009; Gu and Elgin 2013; Perrat et al. 2013). Mutants in the siRNA genes Dcr-2 and Ago-2 have dramatically shortened life spans, and this correlates with significant reactivation of TEs (Czech et al. 2008; Ghildiyal et al. 2008; Lim et al. 2011; Li et al. 2013). Interestingly, while TEs have been shown to reactivate with age across multiple species, the transcript levels of RNAi genes that regulate TEs are not known to decline with age and in fact remain constant (Li et al. 2013; Abe et al. 2014). However, the spectrum of available smRNAs that are loaded onto RISCs is known to change with age (Abe et al. 2014). In addition, mutation of known modifiers of RNAi efficacy has been shown to modulate TE activity and life span (Savva et al. 2013). Multiple age-associated diseases are also associated with TE reactivation. For example, macular degeneration in mice and human cell culture has been shown to be dependent upon RNAi machinery where RNAi mutants exhibited increased levels of Alu RNA resulting in RNA toxicity (Kaneko et al. 2011; Tarallo et al. 2012; Gelfand et al. 2015). This suggests that while RNAi proteins themselves may remain relatively constant with age, the many dynamic components and partners of RNAi as well as the overall activity of RNAi may not be as stable. Hence, inhibition or enhancement of RNAi silencing of TEs would be expected to negatively or positively impact life span, respectively. A better understanding of the dynamics of RNAi TE silencing may allow us to control TE reactivation with age.
3.2.1 RNA Editing
In the siRNA pathway, dsRNAs serve as the substrates from which RNAi proteins produce and employ siRNAs in silencing TEs (Ghildiyal and Zamore 2009). dsRNAs in general have also long been known to be substrates for dsRNA-modifying enzymes such as ADAR proteins. These enzymes bind to dsRNAs and are able to convert adenosine bases to inosines, a base analog of guanine (Savva et al. 2012). This A-to-I editing results in a base pair mismatch between the resulting inosine and the thymine that previously paired with the edited adenosine. The capacity of ADAR proteins to edit dsRNAs has been shown to confer new properties on their substrates, including modified secondary structures, altered stability, nuclear retention, and even novel protein-coding functions (Chen et al. 2008; Jepson et al. 2011; Rieder et al. 2013).
Since dsRNAs are the substrates for siRNA formation, ADAR may also be able to edit these RNAs and thereby modulate the RNAi pathway. The Dicer proteins that catalyze the endonucleolytic cleavage of their dsRNA targets often require a high degree of base pair complementarity, a property that is impaired by RNA editing (Scadden and Smith 2001; Wang et al. 2005; Carpenter et al. 2009; Heale et al. 2009). Hence, ADAR could indirectly inhibit the ability of the siRNA pathway to silence TEs by impairing the access of Dicer to its dsRNA substrates. In fact, it was recently shown that a dsRNA trigger necessary for the silencing of a TE in Drosophila was a target of ADAR, and ADAR mutants showed reduced levels of TE transcripts, suggesting enhanced TE silencing (Savva et al. 2013). These mutants also showed altered levels of heterochromatic marks, including HP1 and H3K9me3, and dramatic changes in position effect variegation, a phenotype in Drosophila known to be dependent on heterochromatin boundaries. Finally, ADAR mutants showed a dramatic extension of life span. These results suggest that RNA editing may abrogate the TE silencing effects of RNAi and that disrupting genes that impede RNAi, such as ADAR, may enhance TE silencing and thereby extend organismal life span.
3.2.2 RISC Complex Misloading
Argonautes are the main effector proteins that perform RNAi silencing, and the siRNA, piRNA, and miRNA pathways all employ such proteins (Ghildiyal and Zamore 2009). The argonaute proteins act in concert with their respective smRNAs to mediate silencing. The miRNA pathway utilizes miRNAs (21–22 nt long) that often imperfectly base pair with their targets upon association with an argonaute protein. This miRNA-RISC then prevents translation of the target mRNA by one of the two methods: stalling or blocking ribosome access, or cleavage of the target mRNA (Ghildiyal and Zamore 2009). Mammals have four argonautes (AGO1–4), and while only AGO2 is catalytically active, all four argonautes are able to bind smRNAs and facilitate inhibition of translation (Liu 2004; Meister et al. 2004; Wilson and Doudna 2013). Interestingly, human AGO2 can accept both miRNAs and siRNAs (Hutvagner and Zamore 2002; Martinez et al. 2002).
In contrast, in Drosophila Ago1 is almost exclusively loaded with miRNAs while Ago2 is loaded mostly with siRNAs (Forstemann et al. 2007). However, recent work has shown that miRNAs and siRNAs compete for loading onto Ago2, the argonaute responsible for TE silencing in flies (Abe et al. 2014). In both flies and mammals, siRNAs are specifically 2′-O-methylated at their 3′ termini (Ghildiyal and Zamore 2009). In flies miRNAs were found to be increasingly inappropriately methylated with age, allowing them to be loaded onto Ago2, and thus reducing siRNA access (Abe et al. 2014). This study did not examine the effect of this RISC misloading on the ability of siRNAs to silence TEs. However, this is an interesting possibility, especially in mammals where siRNAs and miRNAs share AGO2 for silencing, and this competition could functionally impact TE silencing.
3.3 The Role of the piRNA Pathway in Aging
3.3.1 piRNA Deficiencies in Aging Gonads
The piRNA pathway has long been known to be a guardian of genomic integrity in the germline. These longer smRNAs (23-29 nt) associate with three Piwi clade argonaute proteins and, similar to siRNAs, are 2′-O-methylated (Brennecke et al. 2007; Ghildiyal and Zamore 2009). An exonuclease, known as Nibbler, regulates the length of miRNAs, siRNAs, and piRNAs in both somatic and gonadal tissues (Feltzin et al. 2015). Nibbler is responsible for the appropriate trimming of the 3′ termini of these diverse classes of smRNAs (Liu et al. 2011; Feltzin et al. 2015). In flies, Nibbler mutants showed age-associated accumulation of brain damage and physiological effects such as loss of climbing ability (Abe et al. 2014; Feltzin et al. 2015). Another study showed an association between increased Nibbler activity and TE reactivation (Wang et al. 2016). piRNA length was also shown to increase in aged ovaries and this correlated with lower piRNA abundance, suggesting a disruption of piRNA biogenesis. piRNA pathway mutants display increased TE reactivation and a decline or complete loss in fertility (Wang et al. 2016), and both of these phenotypes are also observed in aging animals. Aging is also known to directly affect the fertility of mammals (Ge et al. 2015) and may be related to reactivation of TEs. In support of this, studies in mice where L1 elements were transgenically overexpressed show increased embryonic lethality suggesting that TEs directly contribute to infertility (Malki et al. 2014). It is possible that reproductive output is reduced with age due to an increase in piRNA trimming, resulting in aberrant piRNA biogenesis and increased TE reactivation
3.3.2 The piRNA Pathway in Somatic Tissues
Recent evidence has begun to suggest that the piRNA pathway may also be active in tissues outside of the gonad. piRNAs and their argonautes have been found in healthy somatic tissues of flies, mice, macaques, and humans (Lee et al. 2011; Yan et al. 2011; Perrat et al. 2013; Jones et al. 2016). In addition, multiple studies have documented reactivation of piRNA pathway machinery in various types of cancer (Ross et al. 2014). However, it is not yet known why these piRNA components are expressed in these situations. It is interesting to note that multiple studies have also shown reactivation of TEs in cancer (Chenais 2013; Doucet-O’Hare et al. 2015; Ewing et al. 2015; Rodic et al. 2015) (see also chapters “Retrotransposon Contribution to Genomic Plasticity” and “LINE-1 Retrotransposons as Neoplastic Biomarkers”). One possibility is that the piRNA pathway, arguably the premier genomic defense against TEs, is activated as a compensatory response to TE derepression in cancerous or aging somatic tissues. Our knowledge of piRNA pathway activity in somatic tissues is very incomplete, and experiments determining a mechanistic cause for its presence and the role it serves in the soma have yet to be performed.
3.4 TEs in the Mammalian Genome
Approximately 50 % of mammalian genomes are comprised of repetitive sequences (de Koning et al. 2011). Over evolutionary time most resident RTEs have acquired multiple mutations and are no longer active; however a small fraction retain the ability to transpose (Levin and Moran 2011; Sookdeo et al. 2013). In response, cells have evolved mechanisms to keep these elements tightly repressed. The front-line defense against RTEs is transcriptional silencing (Fig. 2). In mouse embryonic stem (ES) cells, LTR RTEs are silenced through multiple mechanisms including DNA methylation by the DNA methyltransferases 1 and 3L (DNMT1 and DNMT3L) (Slotkin and Martienssen 2007). In addition, ES cells use a Kruppel-associated box-associated protein 1 (KAP1)-dependent pathway that results in the tri-methylation of histone H3 on lysine 9 (H3K9me3) by the methyltransferase ESET (Matsui et al. 2010; Rowe et al. 2010). Recruitment of ESET to LTR RTEs requires the deposition of the histone variant H3.3 (Elsasser et al. 2015).

RTEs in the genome are repressed by heterochromatin. (a) Multiple pathways are involved in the establishment and maintenance of heterochromatin. In many regions of the genome, these domains of heterochromatin encompass RTEs and are instrumental in their silencing. The RB complex recruits several histone methyltransferases (HMTs) that methylate specific lysine residues on histones H3 and H4. Additional HMTs are recruited to the site of heterochromatin formation as part of a Kruppel-associated box-associated protein 1 (KAP1)-dependent pathway that requires ribosylation (Rs) by the sirtuin SIRT6. Together, these mechanisms maintain DNA methylation at cytosine residues by methyltransferases (DNMTs). (b). These processes are disrupted in aging cells resulting in the relaxation of heterochromatin that in turn allows the expression of RTEs. RB complex: a complex containing the retinoblastoma protein, elongation factor 2, histone deacetylases 1 and 2, methyl CpG-binding protein 2, and the nucleosomal and remodeling deacetylase complex; Me3: methyl group; Ac: acetyl group; MBP: methylation-binding protein
LINE RTEs are also silenced through multiple mechanisms in mammalian cells including DNA methylation and histone modification. Mouse embryonic fibroblasts (MEFs ) regulate the expression of the LINE L1 in part through a pathway involving the SIRT6-mediated ribosylation of KAP1 (Van Meter et al. 2014). In addition, MEFs and human cancer cell lines require the recruitment of the EF2/RB complex along with the histone deacetylases 1 and 2 (HDAC1 and HDAC2), the methyl CpG-binding protein 2 (MeCP2), and the nucleosomal and remodeling deacetylase (NuRD) complex to silence L1 expression (Montoya-Durango et al. 2009, 2016; Teneng et al. 2011). In human and mouse neural tissue some L1s become transiently activated during neural progenitor cell differentiation, and this process has been hypothesized to drive variation in neuronal genomes (Muotri et al. 2005; Erwin et al. 2014). Expression of L1s in neural stem cells (NSC) is repressed by SOX2, HDAC1, MeCP2, DNA methylation, and repressive histone modifications, and these factors are reduced during NSC activation (Muotri et al. 2005, 2010; Coufal et al. 2009). SINEs are repressed by DNA methylation, MeCP2, methyl-binding proteins 1 and 2 (MBP1 and MBP2), and the histone modification H3K9me3, and the removal of the latter is necessary for SINE expression (Varshney et al. 2015). The common theme among these repressive pathways is the presence of DNA methylation and the H3K9me3 histone modification. These repressive heterochromatic marks are used by the cell to silence RTEs in an effort to maintain genome integrity. However, as discussed above, these repressive pathways are altered during the aging process and can lead to the derepression of RTEs.
RTE expression increases during cellular senescence and with age in several different mouse tissues. In senescent human fibroblasts the relaxation of heterochromatic regions is correlated with increased expression of L1s and the SINEs Alu and SVA (De Cecco et al. 2013a). Since some of these elements belong to the evolutionarily youngest subfamilies and have intact sequences, they should be capable of transposition (De Cecco et al. 2013a). Indeed, increased genomic copy numbers of L1Hs were observed in senescent cells.
In mouse, members of the LINE (L1), SINE (B1 and B2), and LTR (MusD) families were found to increase expression with age in liver and skeletal muscle (De Cecco et al. 2013b). Interestingly, there appears to be variability between tissues as this increase was more pronounced in muscle than in liver. The transcription of L1s, the largest family of potentially active retrotransposons, was also analyzed in mouse liver by RNA-seq using a bioinformatic pipeline recently developed for the analysis of repetitive sequences in high-throughput DNA sequencing data (Criscione et al. 2014). Many of the L1 subfamilies in the mouse genome were found to increase their expression in liver samples from old animals (De Cecco et al. 2013b).
Expression of RTE mRNA is only the first step that eventually may lead to actual transposition, and several cellular defense mechanisms are known to be active downstream of heterochromatinization. In addition, many elements in the genome have acquired mutations rendering them incapable of transposition. However, current evidence suggests that at least a subset of the derepressed elements are capable of transposition during cellular senescence as well as aging of several mouse tissues (De Cecco et al. 2013a, b).
4 Consequences of Age-Associated TE Activation
4.1 Chimeric Transcripts
In this section we explore the links between RTE activity and changes in the transcriptome. We refer to this process as transcriptional instability, and discuss here the different forms it can take and its potential role in aging. The reader can also refer to chapters “Retrotransposon-Derived Regulatory Regions and Transcripts in Stemness” and “Retrotransposon-Driven Transcription and Cancer” for a discussion on RTE-induced transcriptome changes in the context of pluripotent cells and cancer, respectively. Transcriptional noise, defined as increased cell-to-cell variation in gene expression, has been described in the aging mouse heart (Bahar et al. 2006). Dysregulation of alternative splicing has been found in cellular senescence (Cao et al. 2011), in the aging brain (Mazin et al. 2013) and neurodegeneration (Tollervey et al. 2011), and in blood leukocytes (Harries et al. 2011). It has been argued that these changes may be of particular relevance in postmitotic cells and tissues (Warren et al. 2007). Although a direct link between transcriptional instability and RTE activity has not yet been demonstrated in aging, the ability of RTEs to affect the transcriptome is well known in other contexts. First and foremost, over the course of evolution RTEs have rewired the core regulatory network of the mammalian genome (Kunarso et al. 2010). This demonstrates their ability to influence the transcriptome by either disrupting regulatory elements or contributing new ones. For example, Alu elements harbor binding sites for nuclear hormone receptors and can compete or act as promoters for nearby genes (Polak and Domany 2006; Deininger 2011). Their presence in introns can result in alternative or aberrant splicing (Lev-Maor et al. 2008) that can lead to disease (Ganguly et al. 2003).
RTEs, including many transposition-incompetent elements, retain intact promoter sequences that are capable of driving transcription (Faulkner et al. 2009). L1s contain both sense and antisense promoters (ASPs) that can transcribe into adjacent regions to produce chimeric transcripts (Speek 2001; Cruickshanks and Tufarelli 2009). The sense promoter can promote transcription of downstream genes (Abyzov et al. 2013), and L1-ASP transcription of upstream genes has also been found (Speek 2001; Nigumann et al. 2002). Transformed cancer cell lines and prostate tumors display significant upregulation of L1 RNA expression (Criscione et al. 2014). The marked increased in L1 promoter activity in cancer cells has been linked to a variety of aberrant L1 chimeric transcripts. In colorectal cancer, hypomethylation of L1s leads to activation of the methylation-silenced MET and RAB3IP proto-oncogenes (Hur et al. 2014). A truncated isoform of the oncogene c-MET can be driven from an alternative promoter by hypomethylation of an intronic L1-ASP (Roman-Gomez et al. 2005; Weber et al. 2010; Wolff et al. 2010). Conversely, an L1-ASP-driven RNA can silence the metastasis-suppressor gene TFPI-2 (Cruickshanks et al. 2013b). This suggests that activation of L1-ASPs might lead to epigenetic silencing of tumor-suppressor genes, potentially by similar mechanisms as those described for antisense RNAs in development or several diseases (Tufarelli et al. 2003; Matzke and Birchler 2005; Yu et al. 2008; Taft et al. 2010). Hence, it is evident that RTEs are capable of interfering with the transcriptional machinery at multiple levels and could contribute a similar role to cellular dysfunction during aging.
4.2 Characterizing the Transposition Landscape in Aging Cells
TE sequences posed a great challenge for the initial sequencing and assembly of reference genomes. Their analysis has lagged far behind that of non-repetitive sequences, and even the most recent draft of the human genome (GRCh38) contained major updates of TE annotations. Short-read sequencing strategies, such as Illumina’s HiSeq, provide an additional challenge: it is not possible to unambiguously assign the genomic locations of many reads originating from repetitive elements. To enable the comprehensive documentation of all existing and novel RTE insertions in the genomes of human somatic cells we would ideally require long reads spanning the entire RTE and flanking sequences on both sides, sufficient coverage of the genome to make statistically significant calls, and low costs to make the profiling of many tissues and ages economically feasible. Recent advances in long-read high-throughput sequencing platforms, including Pacific Biosystems Single Molecule, Real-Time (SMRT) Sequencing, and Oxford Nanopore MinION, will likely aid in discovery of new transposition events; however, these technologies are still costly and yield low coverage.
To further complicate studying RTE mobility during cellular senescence or aging of tissues, many new insertions are likely to be “private,” i.e., occurring in an individual cell after it has ceased dividing. This is likely from theoretical considerations, because many cells in the adult organism are postmitotic. Thus characterization of the transposition landscape in aging cells is complicated by the fact that the landscape is likely to be unique for each individual cell. Two approaches have been used to overcome this obstacle: greatly enriching for RTEs before sequencing, or sequencing single-cell genomes. The principle of RTE enrichment is simply to reduce the genomic space that is sequenced in order to increase the coverage and the sensitivity of detection. The caveats of enrichment are that there is selection bias (it requires prior knowledge of active transposons) and enrichment cannot predict transposition frequency (distinguish between equivalent activity in all cells and many hits in some cells). Nevertheless, enrichment methods can provide high coverage, and have been successful in demonstrating the presence of novel events in different biological contexts by several groups (Ewing and Kazazian 2010; Huang et al. 2010; Baillie et al. 2011; Solyom et al. 2012; Shukla et al. 2013).
An attractive alternative approach is high-throughput sequencing of single-cell genomes. Single-cell sequencing was first used to identify copy number variants (CNVs) in single cells from tumors (Navin et al. 2011). Single-cell sequencing was also used to examine retrotransposition frequency in the postmortem adult brain (Evrony et al. 2012, 2015; Upton et al. 2015). While these studies clearly identified novel somatic retrotransposition events in the adult human brain, they differed on the frequency of transposition. One group (Upton et al. 2015) reported a frequency of approximately 14 new retrotranspositions per hippocampal neuron and approximately 11 per cortical neuron, while another group (Evrony et al. 2012, 2015) found that somatic retrotranspositions were relatively infrequent. The reasons for these differences, which may be technical in nature, are currently under discussion (Upton et al. 2015; Evrony et al. 2016). Hence, more work is necessary to document the retrotransposition frequency in the adult brain, and in particular to address the bioinformatic challenges of detecting novel transposition events in single-cell high-throughput sequencing data.
4.3 Transposable Elements and Autoimmunity
Studies of the negative effects of RTEs have largely focused on the damage caused by the transposition process to the genomes of their hosts. While many transposition events are abortive, they often cause DNA double-strand breaks and can promote a variety of illegitimate recombination events, such as chromosomal rearrangements (Farkash and Luning Prak 2006). Recent work has shed light on a new dimension of this host-pathogen relationship: an interesting link between RTEs and the development of autoimmune disease (Bhoj and Chen 2008).
Arguably, the primal form of infection is the parasitism of nucleic acids (Stetson 2009). Across billions of years of host-pathogen interactions many antiviral defense mechanisms have evolved and became implemented with various degrees of success (Hannon 2002; Kawai and Akira 2006; Pichlmair and Reis e Sousa 2007). Many of these response networks are centered on the detection of nucleic acids. To discriminate self from non-self, antiviral sensors must detect potentially hazardous invading nucleic acids among the copious amounts of host-derived DNA and RNA. However, overactivation or other failures of these antiviral systems can result in hyperstimulation of the immune system and autoimmune responses (Banchereau and Pascual 2006). The type I interferon (IFN-I) response is in particular important for establishing an antiviral state; however chronic IFN-I signaling can lead to hyperimmune activation and inflammation (Wilson et al. 2013).
Study of one specific human autoimmune disease, Aicardi–Goutieres syndrome (AGS ), has provided important insights into the contribution of endogenous RTEs to the development of autoimmunity (Stetson et al. 2008). The TREX1 gene encodes a 3′ exonuclease that degrades perceived invading DNA, including the cDNAs from endogenous RTEs. Unless eliminated, these DNA fragments accumulate in the cytosol and activate the IFN-stimulatory DNA (ISD) response and innate immune signaling. TREX1 was found to be mutated in AGS, and the accumulation of RTE-derived cDNAs was associated the hyperactivation of the IFN-I pathway. In addition to TREX1, mutations in the RNAseH2 enzyme also cause AGS, suggesting that accumulation of RNA-DNA hybrids derived from endogenous RTEs contributes to the chronic pro-inflammatory state (Bhoj and Chen 2008).
In a fascinating parallel, chronic inflammation was proposed many years ago to play a major role in exacerbating the aging process, referred to as the “inflammaging” theory of aging (Franceschi and Campisi 2014). Inflammaging appears to be significant risk factor for the morbidity and mortality of the elderly, as most, if not all, age-related diseases share an inflammatory component. However, the etiology of inflammaging remains largely unknown. Thus, both aging and RTEs have been independently associated with chronic IFN-I responses, and aging itself has been associated with RTE activation (De Cecco et al. 2013b). It is thus tempting to speculate a direct connection between these factors, and a causal role between the age-associated expression of RTEs and chronic IFN-I activation.
Autoimmune inflammation that may be caused by the accumulation of RTE-derived single-stranded DNA (Yang et al. 2007) can be treated with reverse transcriptase inhibitors (Beck-Engeser et al. 2011). Several different nucleoside reverse transcriptase inhibitors (NRTIs), developed to treat HIV, have been tested against the reverse transcriptase enzyme encoded by L1 elements, with varying degrees of success (Jones et al. 2008; Dai et al. 2011). Note that some of these compounds might also exert their effects indirectly by inhibiting inflammation, independently of reverse transcriptase inhibition (Fowler et al. 2014). It is thus important to consider the possibility that interventions designed specifically against RTE activities may be effective against autoimmune disorders and perhaps other age-related diseases.
5 Conclusions
In this chapter we have summarized recent discoveries documenting age-related changes in chromatin and transposable element activity. What is the significance of these changes to our understanding of aging and for the prospect of developing new interventions to ameliorate the decline of organismal function with age?
Studies in yeast, nematodes, and fruit flies have demonstrated a strong link between the loss of a “youthful” chromatin state and aging. The salient characteristic of youthfulness in this context we believe is the effective partitioning and maintenance of euchromatic and heterochromatic domains of the genome. An important (albeit not only) consequence of the loss of this chromatin homeostasis is a failure to maintain the effective repression of TE activity. The evidence linking TE activity to aging, though less abundant, is steadily growing. Studies in yeast, flies, mice, and human cell culture show that compromising the cellular TE surveillance mechanisms can result in cellular damage, age-associated diseases, and shortened life span (Czech et al. 2008; Ghildiyal et al. 2008; Wallace et al. 2008; Kaneko et al. 2011; Lim et al. 2011; Maxwell et al. 2011; Li et al. 2013; Jeon et al. 2015; Wood et al. 2016). Evidence is also emerging that augmenting the surveillance mechanisms that maintain TE repression improves cellular physiology and may extend healthy life span (Savva et al. 2013; Wood et al. 2016 ).
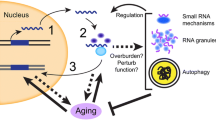
Activation of TEs in the germline has been postulated to drive evolution and create genomic diversity. We believe that the sporadic activation of RTEs in somatic cells is unlikely to be beneficial. Instead, RTE activation is more likely to result in a variety of deleterious effects, such as dysregulation of gene expression, transcriptional noise, chronic activation of an antiviral state, insertional mutagenesis, DNA damage, and genome instability (Fig. 3). The emerging understanding of the potential role of RTEs to promote these rather serious consequences has led us (and others) to envision (Li et al. 2013; Sedivy et al. 2013; Volkman and Stetson 2014) that drugs targeting RTEs, such as NRTIs, or more indirect interventions, such as improving repressive heterochromatin or bolstering some other defense mechanisms, may provide new and novel therapeutic modalities to treat diseases of aging and extend healthy life span.

Retrotransposition theory of aging. RTEs are epigenetically silenced in young somatic cells by their incorporation into constitutive heterochromatin, and additionally targeted by RNAi pathways and a variety of antiviral surveillance systems. Due to an accumulation of macromolecular damage and loss of homeostatic capacity, caused by a variety of extrinsic as well as intrinsic stresses, these cellular defense mechanisms become weakened with age. One consequence of this decline is the activation of dormant RTEs. The age-related increase in RTE expression and mobilization in turn causes further damage, and thus promotes the dysregulation of cellular physiology, loss of tissue function, and ultimately many of the deleterious aspects of aging
References
Abe M, Naqvi A, Hendriks GJ et al (2014) Impact of age-associated increase in 2′-O-methylation of miRNAs on aging and neurodegeneration in Drosophila. Genes Dev 28:44–57
Abyzov A, Iskow R, Gokcumen O et al (2013) Analysis of variable retroduplications in human populations suggests coupling of retrotransposition to cell division. Genome Res 23:2042–2052
Alvares SM, Mayberry GA, Joyner EY et al (2014) H3K4 demethylase activities repress proliferative and postmitotic aging. Aging Cell 13:245–253
Aravin AA, Sachidanandam R, Bourc’his D et al (2008) A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31:785–799
Aravin AA, Sachidanandam R, Girard A et al (2007) Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 316:744–747
Avrahami D, Li C, Zhang J et al (2015) Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved beta cell function. Cell Metab 22:619–632
Bahar R, Hartmann CH, Rodriguez KA et al (2006) Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature 441:1011–1014
Baillie JK, Barnett MW, Upton KR et al (2011) Somatic retrotransposition alters the genetic landscape of the human brain. Nature 479:534–537
Baker DJ, Childs BG, Durik M et al (2016) Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530:184–189
Banchereau J, Pascual V (2006) Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25:383–392
Beck-Engeser GB, Eilat D, Wabl M (2011) An autoimmune disease prevented by anti-retroviral drugs. Retrovirology 8:91
Berger SL (2007) The complex language of chromatin regulation during transcription. Nature 447:407–412
Bhoj VG, Chen ZJ (2008) Linking retroelements to autoimmunity. Cell 134:569–571
Brennecke J, Aravin AA, Stark A et al (2007) Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128:1089–1103
Cao K, Blair CD, Faddah DA et al (2011) Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J Clin Invest 121:2833–2844
Carmell MA, Girard A, van de Kant HJ et al (2007) MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12:503–514
Carpenter JA, Keegan LP, Wilfert L et al (2009) Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae). BMC Genet 10:75
Chandra T, Ewels PA, Schoenfelder S et al (2015) Global reorganization of the nuclear landscape in senescent cells. Cell Rep 10:471–483
Chandra T, Kirschner K, Thuret JY et al (2012) Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47:203–214
Chen H, Ruiz PD, McKimpson WM et al (2015) MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated Secretory phenotype. Mol Cell 59:719–731
Chen LL, DeCerbo JN, Carmichael GG (2008) Alu element-mediated gene silencing. EMBO J 27:1694–1705
Chenais B (2013) Transposable elements and human cancer: a causal relationship? Biochim Biophys Acta 1835:28–35
Clancy DJ, Gems D, Harshman LG et al (2001) Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292:104–106
Coufal NG, Garcia-Perez JL, Peng GE et al (2009) L1 retrotransposition in human neural progenitor cells. Nature 460:1127–1131
Criscione SW, De Cecco M, Siranosian B et al (2016) Reorganization of chromosome architecture in replicative cellular senescence. Sci Adv 2:e1500882
Criscione SW, Zhang Y, Thompson W et al (2014) Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15:583
Cruickshanks HA, McBryan T, Nelson DM et al (2013a) Senescent cells harbour features of the cancer epigenome. Nat Cell Biol 15:1495–1506
Cruickshanks HA, Tufarelli C (2009) Isolation of cancer-specific chimeric transcripts induced by hypomethylation of the LINE-1 antisense promoter. Genomics 94:397–406
Cruickshanks HA, Vafadar-Isfahani N, Dunican DS et al (2013b) Expression of a large LINE-1-driven antisense RNA is linked to epigenetic silencing of the metastasis suppressor gene TFPI-2 in cancer. Nucleic Acids Res 41:6857–6869
Czech B, Malone CD, Zhou R et al (2008) An endogenous small interfering RNA pathway in Drosophila. Nature 453:798–802
Dai L, Huang Q, Boeke JD (2011) Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC Biochem 12:18
Dang W, Steffen KK, Perry R et al (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459:802–807
Dang W, Sutphin GL, Dorsey JA et al (2014) Inactivation of yeast Isw2 chromatin remodeling enzyme mimics longevity effect of calorie restriction via induction of genotoxic stress response. Cell Metab 19:952–966
Day K, Waite LL, Thalacker-Mercer A et al (2013) Differential DNA methylation with age displays both common and dynamic features across human tissues that are influenced by CpG landscape. Genome Biol 14:R102
De Cecco M, Criscione SW, Peckham EJ et al (2013a) Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12:247–256
De Cecco M, Criscione SW, Peterson AL et al (2013b) Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging (Albany NY) 5:867–883
de Koning AP, Gu W, Castoe TA et al (2011) Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet 7:e1002384
Deininger P (2011) Alu elements: know the SINEs. Genome Biol 12:236
Di Giacomo M, Comazzetto S, Saini H et al (2013) Multiple epigenetic mechanisms and the piRNA pathway enforce LINE1 silencing during adult spermatogenesis. Mol Cell 50:601–608
Dixon JR, Jung I, Selvaraj S et al (2015) Chromatin architecture reorganization during stem cell differentiation. Nature 518:331–336
Doucet-O’Hare TT, Rodic N, Sharma R et al (2015) LINE-1 expression and retrotransposition in Barrett's esophagus and esophageal carcinoma. Proc Natl Acad Sci U S A 112:E4894–E4900
Elgin SC, Grewal SI (2003) Heterochromatin: silence is golden. Curr Biol 13:R895–R898
Elsasser SJ, Noh KM, Diaz N et al (2015) Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522:240–244
Erwin JA, Marchetto MC, Gage FH (2014) Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci 15:497–506
Evrony GD, Cai X, Lee E et al (2012) Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151:483–496
Evrony GD, Lee E, Mehta BK et al (2015) Cell lineage analysis in human brain using endogenous retroelements. Neuron 85:49–59
Evrony GD, Lee E, Park PJ et al (2016) Resolving rates of mutation in the brain using single-neuron genomics. Elife 5:e12966
Ewing AD, Gacita A, Wood LD et al (2015) Widespread somatic L1 retrotransposition occurs early during gastrointestinal cancer evolution. Genome Res 25:1536–1545
Ewing AD, Kazazian HH Jr (2010) High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res 20:1262–1270
Fagegaltier D, Bouge AL, Berry B et al (2009) The endogenous siRNA pathway is involved in heterochromatin formation in Drosophila. Proc Natl Acad Sci U S A 106:21258–21263
Farkash EA, Luning Prak ET (2006) DNA damage and L1 retrotransposition. J Biomed Biotechnol 2006:37285
Faulkner GJ, Kimura Y, Daub CO et al (2009) The regulated retrotransposon transcriptome of mammalian cells. Nat Genet 41:563–571
Feltzin VL, Khaladkar M, Abe M et al (2015) The exonuclease Nibbler regulates age-associated traits and modulates piRNA length in Drosophila. Aging Cell 14:443–452
Fernandez AF, Bayon GF, Urdinguio RG et al (2015) H3K4me1 marks DNA regions hypomethylated during aging in human stem and differentiated cells. Genome Res 25:27–40
Feser J, Truong D, Das C et al (2010) Elevated histone expression promotes life span extension. Mol Cell 39:724–735
Feser J, Tyler J (2011) Chromatin structure as a mediator of aging. FEBS Lett 585:2041–2048
Forstemann K, Horwich MD, Wee L et al (2007) Drosophila microRNAs are sorted into functionally distinct argonaute complexes after production by dicer-1. Cell 130:287–297
Fowler BJ, Gelfand BD, Kim Y et al (2014) Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 346:1000–1003
Franceschi C, Campisi J (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69(Suppl 1):S4–S9
Ganguly A, Dunbar T, Chen P et al (2003) Exon skipping caused by an intronic insertion of a young Alu Yb9 element leads to severe hemophilia A. Hum Genet 113:348–352
Ge ZJ, Schatten H, Zhang CL et al (2015) Oocyte ageing and epigenetics. Reproduction 149:R103–R114
Gelfand BD, Wright CB, Kim Y et al (2015) Iron toxicity in the retina requires Alu RNA and the NLRP3 inflammasome. Cell Rep 11:1686–1693
Ghildiyal M, Seitz H, Horwich MD et al (2008) Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science 320:1077–1081
Ghildiyal M, Zamore PD (2009) Small silencing RNAs: an expanding universe. Nat Rev Genet 10:94–108
Giannakou ME, Goss M, Junger MA et al (2004) Long-lived Drosophila with overexpressed dFOXO in adult fat body. Science 305:361
Goldman RD, Shumaker DK, Erdos MR et al (2004) Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 101:8963–8968
Greer EL, Maures TJ, Hauswirth AG et al (2010) Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466:383–387
Greer EL, Maures TJ, Ucar D et al (2011) Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479:365–371
Grewal SI, Jia S (2007) Heterochromatin revisited. Nat Rev Genet 8:35–46
Gu T, Elgin SC (2013) Maternal depletion of Piwi, a component of the RNAi system, impacts heterochromatin formation in Drosophila. PLoS Genet 9:e1003780
Guelen L, Pagie L, Brasset E et al (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453:948–951
Hamdorf M, Idica A, Zisoulis DG et al (2015) miR-128 represses L1 retrotransposition by binding directly to L1 RNA. Nat Struct Mol Biol 22:824–831
Han S, Brunet A (2012) Histone methylation makes its mark on longevity. Trends Cell Biol 22:42–49
Hannon GJ (2002) RNA interference. Nature 418:244–251
Hanzelmann S, Beier F, Gusmao EG et al (2015) Replicative senescence is associated with nuclear reorganization and with DNA methylation at specific transcription factor binding sites. Clin Epigenetics 7:19
Harries LW, Hernandez D, Henley W et al (2011) Human aging is characterized by focused changes in gene expression and deregulation of alternative splicing. Aging Cell 10:868–878
Heale BS, Keegan LP, McGurk L et al (2009) Editing independent effects of ADARs on the miRNA/siRNA pathways. EMBO J 28:3145–3156
Heras SR, Macias S, Caceres JF et al (2014) Control of mammalian retrotransposons by cellular RNA processing activities. Mob Genet Elements 4:e28439
Heras SR, Macias S, Plass M et al (2013) The Microprocessor controls the activity of mammalian retrotransposons. Nat Struct Mol Biol 20:1173–1181
Hills SA, Diffley JF (2014) DNA replication and oncogene-induced replicative stress. Curr Biol 24:R435–R444
Hu Z, Chen K, Xia Z et al (2014) Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev 28:396–408
Huang CR, Burns KH, Boeke JD (2012) Active transposition in genomes. Annu Rev Genet 46:651–675
Huang CR, Schneider AM, Lu Y et al (2010) Mobile interspersed repeats are major structural variants in the human genome. Cell 141:1171–1182
Hur K, Cejas P, Feliu J et al (2014) Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 63:635–646
Hutvagner G, Zamore PD (2002) A microRNA in a multiple-turnover RNAi enzyme complex. Science 297:2056–2060
Hwangbo DS, Gershman B, Tu MP et al (2004) Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429:562–566
Jeon HJ, Kim YS, Park JS et al (2015) Age-related change in gammaH2AX of Drosophila muscle: its significance as a marker for muscle damage and longevity. Biogerontology 16:503–516
Jepson JE, Savva YA, Yokose C et al (2011) Engineered alterations in RNA editing modulate complex behavior in Drosophila: regulatory diversity of adenosine deaminase acting on RNA (ADAR) targets. J Biol Chem 286:8325–8337
Jiang N, Du G, Tobias E et al (2013) Dietary and genetic effects on age-related loss of gene silencing reveal epigenetic plasticity of chromatin repression during aging. Aging 5:813–824
Jin C, Li J, Green CD et al (2011) Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab 14:161–172
Jones RB, Garrison KE, Wong JC et al (2008) Nucleoside analogue reverse transcriptase inhibitors differentially inhibit human LINE-1 retrotransposition. PLoS One 3:e1547
Jones BC, Wood JG, Chang C et al (2016) A somatic piRNA pathway in the Drosophila fat body ensures metabolic homeostasis and normal lifespan. Nat Com (in press)
Kaneko H, Dridi S, Tarallo V et al (2011) DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 471:325–330
Kapahi P, Zid BM, Harper T et al (2004) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 14:885–890
Kawai T, Akira S (2006) Innate immune recognition of viral infection. Nat Immunol 7:131–137
Kawamura Y, Saito K, Kin T et al (2008) Drosophila endogenous small RNAs bind to Argonaute 2 in somatic cells. Nature 453:793–797
Kennedy BK, Gotta M, Sinclair DA et al (1997) Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89:381–391
Kenyon C, Chang J, Gensch E et al (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366:461–464
Kenyon CJ (2010) The genetics of ageing. Nature 464:504–512
Kim S, Villeponteau B, Jazwinski SM (1996) Effect of replicative age on transcriptional silencing near telomeres in Saccharomyces cerevisiae. Biochem Biophys Res Commun 219:370–376
Kosar M, Bartkova J, Hubackova S et al (2011) Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 10:457–468
Kreiling JA, Tamamori-Adachi M, Sexton AN et al (2011) Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 10:292–304
Kunarso G, Chia NY, Jeyakani J et al (2010) Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet 42:631–634
Larson K, Yan SJ, Tsurumi A et al (2012) Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet 8:e1002473
Lee EJ, Banerjee S, Zhou H et al (2011) Identification of piRNAs in the central nervous system. RNA 17:1090–1099
Lee SS, Kennedy S, Tolonen AC et al (2003) DAF-16 target genes that control C. elegans life-span and metabolism. Science 300:644–647
Lev-Maor G, Ram O, Kim E et al (2008) Intronic Alus influence alternative splicing. PLoS Genet 4:e1000204
Levin HL, Moran JV (2011) Dynamic interactions between transposable elements and their hosts. Nat Rev Genet 12:615–627
Li L, Greer C, Eisenman RN et al (2010) Essential functions of the histone demethylase lid. PLoS Genet 6:e1001221
Li W, Prazak L, Chatterjee N et al (2013) Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16:529–531
Lim DH, Oh CT, Lee L et al (2011) The endogenous siRNA pathway in Drosophila impacts stress resistance and lifespan by regulating metabolic homeostasis. FEBS Lett 585:3079–3085
Liu J (2004) Argonaute2 is the catalytic engine of mammalian RNAi. Science 305:1437–1441
Liu N, Abe M, Sabin LR et al (2011) The exoribonuclease Nibbler controls 3′ end processing of microRNAs in Drosophila. Curr Biol 21:1888–1893
Malki S, van der Heijden GW, O’Donnell KA et al (2014) A role for retrotransposon LINE-1 in fetal oocyte attrition in mice. Dev Cell 29:521–533
Martinez J, Patkaniowska A, Urlaub H et al (2002) Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 110:563–574
Matsui T, Leung D, Miyashita H et al (2010) Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 464:927–931
Matzke MA, Birchler JA (2005) RNAi-mediated pathways in the nucleus. Nat Rev Genet 6:24–35
Maures TJ, Greer EL, Hauswirth AG et al (2011) The H3K27 demethylase UTX‐1 regulates C. elegans lifespan in a germline‐independent, insulin‐dependent manner. Aging Cell 10:980–990
Maxwell PH, Burhans WC, Curcio MJ (2011) Retrotransposition is associated with genome instability during chronological aging. Proc Natl Acad Sci U S A 108:20376–20381
Mazin P, Xiong J, Liu X et al (2013) Widespread splicing changes in human brain development and aging. Mol Syst Biol 9:633
McColl G, Killilea DW, Hubbard AE et al (2008) Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem 283:350–357
McCord RP, Nazario-Toole A, Zhang H et al (2013) Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res 23:260–269
Meister G, Landthaler M, Patkaniowska A et al (2004) Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell 15:185–197
Montoya-Durango DE, Liu Y, Teneng I et al (2009) Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat Res 665:20–28
Montoya-Durango DE, Ramos KA, Bojang P et al (2016) LINE-1 silencing by retinoblastoma proteins is effected through the nucleosomal and remodeling deacetylase multiprotein complex. BMC Cancer 16:38
Muotri AR, Chu VT, Marchetto MC et al (2005) Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435:903–910
Muotri AR, Marchetto MC, Coufal NG et al (2010) L1 retrotransposition in neurons is modulated by MeCP2. Nature 468:443–446
Narita M, Nunez S, Heard E et al (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113:703–716
Navin N, Kendall J, Troge J et al (2011) Tumour evolution inferred by single-cell sequencing. Nature 472:90–94
Ni Z, Ebata A, Alipanahiramandi E et al (2012) Two SET domain containing genes link epigenetic changes and aging in Caenorhabditis elegans. Aging Cell 11:315–325
Nigumann P, Redik K, Matlik K et al (2002) Many human genes are transcribed from the antisense promoter of L1 retrotransposon. Genomics 79:628–634
O'Sullivan RJ, Karlseder J (2012) The great unravelling: chromatin as a modulator of the aging process. Trends Biochem Sci 37:466–476
O'Sullivan RJ, Kubicek S, Schreiber SL et al (2010) Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17:1218–1225
Oberdoerffer P, Sinclair DA (2007) The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol 8:692–702
Peleg S, Feller C, Forne I et al (2016) Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep 17:455–469
Perrat PN, DasGupta S, Wang J et al (2013) Transposition-driven genomic heterogeneity in the Drosophila brain. Science 340:91–95
Pichlmair A, Reis e Sousa C (2007) Innate recognition of viruses. Immunity 27:370–383
Polak P, Domany E (2006) Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics 7:133
Rai TS, Adams PD (2013) Lessons from senescence: chromatin maintenance in non-proliferating cells. Biochim Biophys Acta 1819:322–331
Rieder LE, Staber CJ, Hoopengardner B et al (2013) Tertiary structural elements determine the extent and specificity of messenger RNA editing. Nat Commun 4:2232
Rodic N, Steranka JP, Makohon-Moore A et al (2015) Retrotransposon insertions in the clonal evolution of pancreatic ductal adenocarcinoma. Nat Med 21:1060–1064
Rogina B, Reenan RA, Nilsen SP et al (2000) Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 290:2137–2140
Roman-Gomez J, Jimenez-Velasco A, Agirre X et al (2005) Promoter hypomethylation of the LINE-1 retrotransposable elements activates sense/antisense transcription and marks the progression of chronic myeloid leukemia. Oncogene 24:7213–7223
Ross RJ, Weiner MM, Lin H (2014) PIWI proteins and PIWI-interacting RNAs in the soma. Nature 505:353–359
Rowe HM, Jakobsson J, Mesnard D et al (2010) KAP1 controls endogenous retroviruses in embryonic stem cells. Nature 463:237–240
Rozhkov NV, Hammell M, Hannon GJ (2013) Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev 27:400–412
Sadaie M, Salama R, Carroll T et al (2013) Redistribution of the Lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev 27:1800–1808
Sarg B, Koutzamani E, Helliger W et al (2002) Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem 277:39195–39201
Savva YA, Jepson JE, Chang YJ et al (2013) RNA editing regulates transposon-mediated heterochromatic gene silencing. Nat Commun 4:2745
Savva YA, Rieder LE, Reenan RA (2012) The ADAR protein family. Genome Biol 13:252
Scadden AD, Smith CW (2001) RNAi is antagonized by A→I hyper-editing. EMBO Rep 2:1107–1111
Scaffidi P, Misteli T (2006) Lamin A-dependent nuclear defects in human aging. Science 312:1059–1063
Sedivy JM, Banumathy G, Adams PD (2008) Aging by epigenetics—a consequence of chromatin damage? Exp Cell Res 314:1909–1917
Sedivy JM, Kreiling JA, Neretti N et al (2013) Death by transposition—the enemy within? Bioessays 35:1035–1043
Shabalina SA, Koonin EV (2008) Origins and evolution of eukaryotic RNA interference. Trends Ecol Evol 23:578–587
Shukla R, Upton KR, Munoz-Lopez M et al (2013) Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 153:101–111
Shumaker DK, Dechat T, Kohlmaier A et al (2006) Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A 103:8703–8708
Siebold AP, Banerjee R, Tie F et al (2010) Polycomb repressive complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc Natl Acad Sci U S A 107:169–174
Sinclair DA, Oberdoerffer P (2009) The ageing epigenome: damaged beyond repair? Ageing Res Rev 8:189–198
Slotkin RK, Martienssen R (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8:272–285
Smeal T, Claus J, Kennedy B et al (1996) Loss of transcriptional silencing causes sterility in old mother cells of S. cerevisiae. Cell 84:633–642
Solyom S, Ewing AD, Rahrmann EP et al (2012) Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res 22:2328–2338
Sookdeo A, Hepp CM, McClure MA et al (2013) Revisiting the evolution of mouse LINE-1 in the genomic era. Mob DNA 4:3
Speek M (2001) Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Mol Cell Biol 21:1973–1985
Stetson DB (2009) Connections between antiviral defense and autoimmunity. Curr Opin Immunol 21:244–250
Stetson DB, Ko JS, Heidmann T et al (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134:587–598
Sun D, Yi SV (2015) Impacts of chromatin states and long-range genomic segments on aging and DNA methylation. PLoS One 10:e0128517
Swanson EC, Manning B, Zhang H et al (2013) Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J Cell Biol 203:929–942
Taft RJ, Pang KC, Mercer TR et al (2010) Non-coding RNAs: regulators of disease. J Pathol 220:126–139
Tarallo V, Hirano Y, Gelfand BD et al (2012) DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 149:847–859
Tatar M, Kopelman A, Epstein D et al (2001) A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292:107–110
Teneng I, Montoya-Durango DE, Quertermous JL et al (2011) Reactivation of L1 retrotransposon by benzo(a)pyrene involves complex genetic and epigenetic regulation. Epigenetics 6:355–367
Tollervey JR, Wang Z, Hortobagyi T et al (2011) Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res 21:1572–1582
Tufarelli C, Stanley JA, Garrick D et al (2003) Transcription of antisense RNA leading to gene silencing and methylation as a novel cause of human genetic disease. Nat Genet 34:157–165
Upton KR, Gerhardt DJ, Jesuadian JS et al (2015) Ubiquitous L1 mosaicism in hippocampal neurons. Cell 161:228–239
Vagin VV, Sigova A, Li C et al (2006) A distinct small RNA pathway silences selfish genetic elements in the germline. Science 313:320–324
Van Meter M, Kashyap M, Rezazadeh S et al (2014) SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun 5:5011
Varshney D, Vavrova-Anderson J, Oler AJ et al (2015) SINE transcription by RNA polymerase III is suppressed by histone methylation but not by DNA methylation. Nat Commun 6:6569
Volkman HE, Stetson DB (2014) The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol 15:415–422
Wallace NA, Belancio VP, Deininger PL (2008) L1 mobile element expression causes multiple types of toxicity. Gene 419:75–81
Wang H, Ma Z, Niu K et al (2016) Antagonistic roles of Nibbler and Hen1 in modulating piRNA 3′ ends in Drosophila. Development 143:530–539
Wang Q, Zhang Z, Blackwell K et al (2005) Vigilins bind to promiscuously A-to-I-edited RNAs and are involved in the formation of heterochromatin. Curr Biol 15:384–391
Warren LA, Rossi DJ, Schiebinger GR et al (2007) Transcriptional instability is not a universal attribute of aging. Aging Cell 6:775–782
Weber B, Kimhi S, Howard G et al (2010) Demethylation of a LINE-1 antisense promoter in the cMet locus impairs Met signalling through induction of illegitimate transcription. Oncogene 29:5775–5784
Wilson EB, Yamada DH, Elsaesser H et al (2013) Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207
Wilson RC, Doudna JA (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42:217–239
Wolff EM, Byun HM, Han HF et al (2010) Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet 6:e1000917
Wood JG, Hillenmeyer S, Lawrence C et al (2010) Chromatin remodeling in the aging genome of Drosophila. Aging Cell 9:971–978
Wood JG, Jones BC, Jiang N et al. (2016) Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc Natl Acad Sci USA, 113:11277-11282
Yan Z, Hu HY, Jiang X et al (2011) Widespread expression of piRNA-like molecules in somatic tissues. Nucleic Acids Res 39:6596–6607
Yang N, Kazazian HH Jr (2006) L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat Struct Mol Biol 13:763–771
Yang YG, Lindahl T, Barnes DE (2007) Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131:873–886
Yu W, Gius D, Onyango P et al (2008) Epigenetic silencing of tumour suppressor gene p15 by its antisense RNA. Nature 451:202–206
Zhang R, Chen W, Adams PD (2007) Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27:2343–2358
Zhang W, Li J, Suzuki K et al (2015) Aging stem cells. A Werner syndrome stem cell model unveils heterochromatin alterations as a driver of human aging. Science 348:1160–1163
Acknowledgments
The authors gratefully acknowledge the following sources of financial support: J.A.K., NIH K01 AG039410; B.C.J., NIH T32 AG041688, F31 AG047736; M.D.C., AFAR postdoctoral fellowship; S.W.C., NIH T32 GM007601, F31 AG050365; N.N., NIH R56 AG050582; S.L.H., NIH R01 AG024353, NIH P01 AG051449 and J.M.S., NIH R37 AG016694, NIH P01 AG051449.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Kreiling, J.A. et al. (2017). Contribution of Retrotransposable Elements to Aging. In: Cristofari, G. (eds) Human Retrotransposons in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-319-48344-3_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-48344-3_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-48343-6
Online ISBN: 978-3-319-48344-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)