
Abstract
Short interspersed elements, such as Alu elements, have propagated to more than one million copies in the human genome. They affect the genome in several ways, caused by retrotransposition, recombination between elements, gene conversion, and alterations in gene expression. These events, including novel insertions into active genes, have been associated with a number of human disorders. Hemophilia A is an X-linked severe bleeding disorder and is caused by mutations in the Factor VIII gene. The spectrum of mutations includes point mutations, rearrangements, insertions, and deletions. Recently, an Alu retrotransposition event in a coding exon has been reported in a family with a severe form of hemophilia A. This was the first report of an Alu insertion in the Factor VIII gene. Here, we report a second Alu insertion event that lies in an intron of the same gene that causes exon skipping and the complete disruption of gene expression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
During the past 65 million years, Alu elements have propagated by retrotransposition to more than one million copies in primate genomes. Despite the fact that these elements have replicated many times in the genome, under most conditions, they remain transcriptionally silent and therefore are not actively replicating (Hagan and Rudin 2002). Recombination events involving the Alu elements, including novel insertions into active genes, have been associated with a number of human disorders (Szmulewicz et al. 1998).
Hemophilia A is an X-linked severe bleeding disorder and is caused by mutations in the Factor VIII gene. The spectrum of mutations includes point mutations, rearrangements, insertions, and deletions (Antonarakis et al. 1995a, 1995b; Citron et al. 2002). Recently, an Alu retrotransposition event in a coding exon was reported in a family with a severe form of hemophilia A (Sukarova et al. 2001). This was the first report of an Alu insertion in the Factor VIII gene. Here, we report a second Alu insertion event that lies in an intron of the Factor VIII gene and that causes exon skipping and hemophilia A.
Materials and methods
Human subjects
The human subjects for this study were submitted as part of a genetic testing service. Informed consent was obtained from each individual tested. As the proband was a minor, the respective legal guardian was consented. Genetic counseling of these individuals, both prior to sampling and after the results of genetic testing became available, was provided by the respective referral centers. The consent specifically allowed research beyond routine genetic testing for hemophilia A.
Isolation of DNA from blood samples
Genomic DNA was isolated from 1~3 ml blood by using a commercial DNA isolation kit (Gentra, CA). DNA was dissolved in 500 µl 10 mM TRIS-HCl pH 8.0, 0.1 mM EDTA. Approximately 80–100 µg DNA was obtained. For the polymerase chain reaction (PCR), the DNA was diluted to 20 ng/µl, and 5 µl aliquots were used in a 50-µl PCR. A duplicate sample of DNA was isolated from a second tube of blood and stored separately from the first batch of DNA. This DNA was used for independent verification of any mutation found in the first sample.
Isolation of RNA from blood samples
Total RNA was isolated from peripheral blood lymphocytes. The lymphocytes were first separated from blood by overlaying whole blood (3 ml) collected with an anti-coagulant, on a Ficoll-Hypaque cushion (1 ml; Amersham, Pharmacia, Piscataway, NJ) in a 15-ml Falcon tube. Following centrifugation, the lymphocytes were recovered from the interface between the plasma and the Ficoll-Hypaque solution. The lymphocytes were washed twice with phosphate-buffered saline and then transferred to Trizol reagent (Invitrogen Life Technologies, Carlsbad, CA). Total RNA was isolated following instructions from the manufacturer.
DNA sequencing of PCR products
For routine clinical testing, all coding exons including exon/intron boundaries of the Factor VIII gene were amplified by PCR with genomic DNA as template and were checked on an agarose gel. The Amplitaq Gold enzyme (Perkin Elmer, ABI, CA) was used for the amplification of all exons. All coding exons were amplified for the proband and indicated the absence of deletion mutations involving any of the coding exons of the Factor VIII gene. PCR products were purified from the unincorporated nucleotides and primers by treatment with Shrimp Alkaline Phosphatase and Exonuclease (USB, Ohio) and subjected to dye-terminator cycle sequencing (ABI) by using one of the PCR primers. The sequencing reaction products were purified on a 96-well purification plate (Edge Biosystems, MD), dissolved in the standard formamide-containing gel loading buffer, and run on an ABI 377 sequencer.
Any sequence change was confirmed by repeat sequencing of a duplicate PCR product generated from a second independently isolated sample of DNA with the reverse primer and visualizing the nucleotide change in both the forward and reverse directions.
TA-cloning of PCR products
Fragment size analysis of exon-19-specific PCR product indicated the presence of a novel fragment. The DNA fragment was sequenced in both directions, and an insertion of ~330 bp at position −19 from the 3'-end of intron 18 was identified. There was a long poly-A tract present at the end of this insert. The exact length of this tract could not be estimated from direct sequence analysis of PCR products of exon 19 because of polymerase slippage events during the amplification process. Therefore, the PCR product was cloned by using the Topo TA cloning kit (Invitrogen Life Technologies) in pCR2.1 Topo vector and was sequenced with vector-specific primers.
Reverse transcription/PCR analysis of the Factor VIII transcripts
To analyze the transcripts specific for the Factor VIII gene, the first strand of cDNA was generated from total RNA (50 ng) by using random primers and Superscript II RNAse H− Reverse Transcriptase (RTAse, Invitrogen Life Technologies). After inactivation of the RTAse and treatment with RNAse H, the cDNA was amplified with primers specific for the Factor VIII transcript, viz., F8C.cUf, 5'-ATATCCAGATGGAAGATCCCAC-3' and F8C.cVr, 5'-CAACTCCATGCGAAGAGTGCTGC-3'. These primers are located in exons 17 and 23, respectively, and amplify a 788-bp fragment from the normal transcript. This product was further amplified by using nested primers, viz., F8C.cUf and F8C.cUr 5'-GCTCCAGGCATTGATTGATCCG-3' (located in exon 21), to generate a 479-bp fragment from the normal transcript. The PCR product was sequenced by using both forward and reverse primers.
Results
The proband in this study is a 6-year-old male with a clinical diagnosis of hemophilia A. The factor VIII level in blood at the time of diagnosis was less than 1%. Family history included a possible diagnosis of hemophilia in three maternal great-uncles, but that was never confirmed. The mother was one of two daughters with no male siblings. The affected individuals from previous generations were deceased and not available for genetic testing.
The proband and his mother were previously studied in the laboratory of Dr. D. Ginsburg, University of Michigan. The proband was negative for the common mutation, inversion of intron 22 of the Factor VIII gene. Linkage analysis was performed with three informative markers: (CA)n repeats in intron 13, the BclI restriction fragment length polymorphism (RFLP) in intron 18, and a HindIII RFLP in intron 19 (Windsor et al. 1994). The proband was shown to have an aberrant band generated during HindIII RFLP analysis of intron 18, an analysis that used PCR products amplified with primers located in introns 18 and 19.
In our assay, the amplification product for exon 19 from the genomic DNA of the proband was larger (~600 bp) than the expected size (~300 bp). The DNA fragment was sequenced in both directions and an insertion of ~330 bp at position −19 from the 3'-end of intron 18 was identified. There was a long poly-A tract present at the end of this insert. The exact length of this tract could not be estimated from direct sequence analysis of PCR products of exon 19 because of polymerase slippage events during the amplification process. The remaining exons of the Factor VIII gene were amplified from the proband, and all were sequenced as normal wild-type sequences. Therefore, the insertion at position −19 of intron 18 was the only sequence change observed in the hemophiliac male proband.
Characterization of the insert DNA
Visual inspection of the ~330 nucleotide sequence of the insert DNA showed the presence of a 37-nucleotide-long poly-A tail (with two interruptions), together with a duplication of 13 nucleotides at the site of insertion or the target site (Fig. 1). To determine the nature and origin of the insert sequence, a BLAT search against the UCSC human DNA sequence database was performed (University of California at Santa Cruz Genome Browser, http://genome.cse.ucsc.edu/). The poly-A tail was omitted from the initial BLAT search. The search came back with multiple perfect hits on a number of chromosomes indicating that the insert was likely to be a repeated sequence. When the search was repeated against the Alu database in GENBANK, the results indicated an almost perfect match with the Alu Sb2 family of subsequences (Batzer and Deininger 2002; Jurka 1993; Roy-Engel et al. 2001). There were two differences in the insert sequence with respect to the consensus sequence of the Sb2 element. Further investigation indicated that the insert sequence was a perfect match of one of the youngest Alu elements, viz., the Yb9 element (Roy-Engel et al. 2001). Thereafter, the insert was identified as an exact copy of the source sequence including the poly-A tail with its interruptions on chromosome 1q42.3 (position 231508100–231508387 on the draft human DNA sequence as frozen on November 2002 at UCSC).

Diagram showing the insertion of Alu Yb9 element in intron 18 of the Factor VIII gene. The underlined bases indicate the target site duplication of nucleotides −18 through –6 of intron 18. Alu-Yb9—Consensus sequence of Yb9 Alu element, Insert—sequence of the inserted DNA
To estimate the time point of retrotransposition of the Alu elements, genomic DNA samples isolated from the maternal grand mother and mother were tested. Both individuals were found to be carriers of the same Alu insertion mutation in intron 18. Therefore, the insertion event probably occurred at some time point before the last three generations.
Bioinformatic analysis of the genomic DNA sequence with the insert
The Alu insertion occurs at the −19 position of the 3'-end of intron 18 and does not affect the natural splice donor site. Moreover, the insertion is in the opposite orientation with respect to the direction of transcription of the Factor VIII gene. Therefore, the consequences of this insertion at the level of transcription are not obvious. However, the lariat branch point of intron 18 is located at the −85 position from the 3'-end of intron 18. Thus, the insert creates a longer distance between an already distant lariat and the donor site; usually, the lariats are located at −10 to −50 nucleotides from the 3'-end of the intron (Fujimaru et al. 1998). Bioinformatic analysis of the genomic sequence with and without the insert was performed. Two web-based programs, GENSCAN web server at MIT (http://genes.mit.edu/GENSCAN.html) and GeneSplicer web server at TIGR (http://www.tigr.org/tdb/GeneSplicer/gene_spl.html), were used to analyze GENBANK entry M88642 coding for exons 16–19 of the Factor VIII gene with and without the insert sequence. The output from both programs showed no difference in the predicted exons (data not shown). The size of intron 18 was predicted to be increased by ~331 nucleotides by the GENSCAN program, because of the insertion, without any consequences on the predicted position of the following exons.
RNA analysis
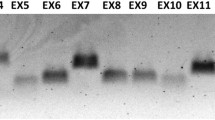
Lymphoblast cell lines were established from the proband and his mother. Total RNA was isolated, and cDNA specific for the Factor VIII gene was generated by gene-specific primers. The entire gene was amplified; the relevant fragments with altered sizes are shown in Fig. 2. The predicted size of the amplified fragment generated by using nested primers in exons 17 and 21 is 479 bp. The reverse transcription/PCR (RT-PCR) product obtained by using RNA from the proband (lane 2) for this fragment was smaller and around ~350 bp. In contrast, the RT-PCR product obtained by using the RNA isolated from the lymphoblasts of the mother showed the presence of two bands: 479 and 350 bp (lane 3). The smaller fragment from the proband was sequenced with forward and reverse primers. The results showed perfect splicing of exons 18–20, with exon 19 being spliced out. Exon 19 contains 117 nucleotides and codes for amino acids 1981–2020, which belong to the A3 domain of the Factor VIII protein. Therefore, exon skipping leads to an in-frame deletion of 39 amino acids and results in a non-functional A3 domain. There is another report of severe hemophilia A disease caused by skipping of exon 19 (Liu et al. 2002).

RT-PCR analysis of the Factor-VIII-specific cDNA amplification with the RNA isolated from the lymphoblast of the proband. Top Scheme indicating the location of primers used for nested amplification. Bottom Gel picture of the amplified RT-PCR products from the proband and his mother. Lanes 1, 4 DNA size markers (M), lanes 3, 4 the expected 479-bp fragment from the normal allele (present in the mother) and the unexpected smaller 350-bp band from the mutant allele (present in the heterozygous mother and in the proband)
Discussion
Direct sequencing of all coding exons of the Factor VIII gene has elucidated the exact nature of molecular defect in many individuals with hemophilia A (Antonarakis 1998; Citron et al. 2002). Most of the mutations affect the coding sequences such that a missense, nonsense, or frameshift mutation translates to a non-functional protein. Mutations in consensus sequences of splice sites account for a small proportion (5%~8.3%) of disease-causing mutations as indicated in the on-line databases (Hamsters Database: http://europium.csc.mrc.ac.uk/usr/WWW/WebPages/main.dir/main.htm; Human Gene Mutation Database: http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html). Beyond the changes that affect consensus sequences of the splice sites, the interpretation of any other intronic changes can be challenging.
The present report describes the first case of an intronic Alu insertion in the complimentary strand of the coding sequence of the Factor VIII gene. This insertion leads to exon skipping and elimination of 39 critical amino acids of the A3 domain of the Factor VIII protein. There are reports of exonic Alu insertions causing disease in hemophilia. An Alu insertion in exon 14 of the Factor VIII gene gives rise to a premature stop codon within the insert and causes hemophilia A (Sukarova et al. 2001). Three separate Alu inserts in different exons of the Factor IX gene caused hemophilia B (Li et al. 2001; Vidaud et al. 1993; Wulff et al. 2000). In addition, a few reports of intronic Alu insertions causing aberrant splicing have been published. The insertion of an Alu-like element 2.4 kb downstream from the 5' end of intron 11 of the dystrophin gene (Ferlini and Muntoni 1998) causes activation of a cryptic splice site in intron 11, resulting in an alternate transcript with parts of the Alu-like sequence and intron 11 included. This transcript is prematurely truncated upon translation. However, this transcript is present together with the normal transcript in a tissue-specific manner and modulates the disease manifestation. Similarly, the Alu insert in an intron of NF1 gene causes exon skipping and aberrant mRNA product (Wallace et al. 1991) and the Alu-mediated intronic deletion causes Hunter's disease (Ricci et al. 2003).
It is interesting to note that intron 18 (total length: 1736 nucleotides) of the Factor VIII gene naturally harbors Alu-like sequences between nucleotides 427 and 950. This repeat element has partial homology to the J subfamily of Alu sequences and to subfamily Sb2 sequences (data not shown). Thus, there are two Alu elements residing head to head in close proximity within intron 18 in this proband and his relatives and this can lead to instability in the genome. In addition, the normal splice donor sequence of intron 18 in the Factor VIII gene is TATAAG/GT as opposed to the consensus sequence of YYNYAG/GT (Senapathy et al. 1990; Shapiro and Senapathy 1987). Therefore, the possibility of a weak splice donor site resulting from differences in the bold bases from the consensus sequence, together with an increased separation from the putative lariat junction, is the probable cause of the downstream exon being skipped.
Bioinformatic analysis of the consequences of the Alu insertion in the intron by using either the Gene or splice junction prediction programs did not predict exon skipping. This indicates the limited usefulness of these programs in a clinical genetic testing laboratory.
The Alu insert characterized in this study is a perfect copy of the source site on chromosome 1q42.3 (according to the November 2002 freeze of the Human Genome database at http://genome.cse.ucsc.edu/). The length of the poly-A tail at the end of an Alu element has been suggested to determine the retrotransposition capability of the element (Roy-Engel et al. 2002). The length of the poly-A tail at the source Alu element on chromosome 1 is 37 nucleotides with two interruptions. The same length is retained at the site of insertion on chromosome X in our proband.
The target site duplication of 13 nucleotides at the site of Alu insertion (bold and underlined in Fig. 1) is a signature of Alu retrotransposition (Batzer and Deininger 2002). Alu retrotransposition has also been reported to take place with the help of L1 endonuclease (Cost and Boeke 1998; Jurka and Klonowski 1996). The target site for this endonuclease activity is specific and has a consensus sequence of TTTT-NN-AA. In the present case, the insertion took place at the position of the gap within the TTTT-AT-AA sequence.
In conclusion, this report describes the characterization of a recent retrotransposition event of an Alu Yb9 element in an intron of the Factor VIII gene. The molecular consequence of this event is exon skipping that leads to hemophilia A. The Alu Yb9 elements are human-specific and belong to a sub-family of very recently transposed sequences (Batzer and Deininger 2002; Roy-Engel et al. 2001). These retrotransposition events, although rare, seem to be ongoing and continue to shape the mammalian genome.
References
Antonarakis SE (1998) Molecular genetics of coagulation factor VIII gene and haemophilia A. Haemophilia 4 (Suppl 2):1-11
Antonarakis SE, Kazazian HH, Gitschier J, Hutter P, Moerloose P de, Morris MA (1995a) Molecular etiology of factor VIII deficiency in hemophilia A. Adv Exp Med Biol 386:19–34
Antonarakis SE, Rossiter JP, Young M, Horst J, Moerloose P de, Sommer SS, Ketterling RP, Kazazian HH Jr, Negrier C, Vinciguerra C (1995b) Factor VIII gene inversions in severe hemophilia A: results of an international consortium study. Blood 86:2206–2212
Batzer MA, Deininger PL (2002) Alu repeats and human genomic diversity. Nat Rev Genet 3:370–379
Citron M, Godmilow L, Ganguly T, Ganguly A (2002) High throughput mutation screening of the factor VIII gene (F8C) in hemophilia A: 37 novel mutations and genotype-phenotype correlation. Hum Mutat 20:267–274
Cost GJ, Boeke JD (1998) Targeting of human retrotransposon integration is directed by the specificity of the L1 endonuclease for regions of unusual DNA structure. Biochemistry 37:18081–18093
Ferlini A, Muntoni F (1998) The 5' region of intron 11 of the dystrophin gene contains target sequences for mobile elements and three overlapping ORFs. Biochem Biophys Res Commun 242:401–406
Fujimaru M, Tanaka A, Choeh K, Wakamatsu N, Sakuraba H, Isshiki G (1998) Two mutations remote from an exon/intron junction in the beta-hexosaminidase beta-subunit gene affect 3'-splice site selection and cause Sandhoff disease. Hum Genet 103:462–469
Hagan CR, Rudin CM (2002) Mobile genetic element activation and genotoxic cancer therapy: potential clinical implications. Am J Pharmacogenomics 2:25–35
Jurka J (1993) A new subfamily of recently retroposed human Alu repeats. Nucleic Acids Res 21:2252
Jurka J, Klonowski P (1996) Integration of retroposable elements in mammals: selection of target sites. J Mol Evol 43:685–689
Li X, Scaringe WA, Hill KA, Roberts S, Mengos A, Careri D, Pinto MT, Kasper CK, Sommer SS (2001) Frequency of recent retrotransposition events in the human factor IX gene. Hum Mutat 17:511–519
Liu ML, Nakaya S, Thompson AR (2002) Non-inversion factor VIII mutations in 80 hemophilia A families including 24 with alloimmune responses. Thromb Haemost 87:273–276
Ricci V, Regis S, Di Duca M, Filocamo M (2003) An Alu-mediated rearrangement as cause of exon skipping in Hunter disease. Hum Genet 112:419–425
Roy-Engel AM, Carroll ML, Vogel E, Garber RK, Nguyen SV, Salem AH, Batzer MA, Deininger PL (2001) Alu insertion polymorphisms for the study of human genomic diversity. Genetics 159:279–290
Roy-Engel AM, Salem AH, Oyeniran OO, Deininger L, Hedges DJ, Kilroy GE, Batzer MA, Deininger PL (2002) Active Alu element "A-tails": size does matter. Genome Res 12:1333–1344
Senapathy P, Shapiro MB, Harris NL (1990) Splice junctions, branch point sites, and exons: sequence statistics, identification, and applications to genome project. Methods Enzymol 183:252–278
Shapiro MB, Senapathy P (1987) RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15:7155–7174
Sukarova E, Dimovski AJ, Tchacarova P, Petkov GH, Efremov GD (2001) An Alu insert as the cause of a severe form of hemophilia A. Acta Haematol 106:126–129
Szmulewicz MN, Novick GE, Herrera RJ (1998) Effects of Alu insertions on gene function. Electrophoresis 19:1260–1264
Vidaud D, Vidaud M, Bahnak BR, Siguret V, Gispert Sanchez S, Laurian Y, Meyer D, Goossens M, Lavergne JM (1993) Haemophilia B due to a de novo insertion of a human-specific Alu subfamily member within the coding region of the factor IX gene. Eur J Hum Genet 1:30–36
Wallace MR, Andersen LB, Saulino AM, Gregory PE, Glover TW, Collins FS (1991) A de novo Alu insertion results in neurofibromatosis type 1. Nature 353:864–866
Windsor S, Taylor SA, Lillicrap D (1994) Multiplex analysis of two intragenic microsatellite repeat polymorphisms in the genetic diagnosis of haemophilia A. Br J Haematol 86:810–815
Wulff K, Gazda H, Schroder W, Robicka-Milewska R, Herrmann FH (2000) Identification of a novel large F9 gene mutation—an insertion of an Alu repeated DNA element in exon e of the factor 9 gene. Hum Mutat 15:299
Acknowledgements
The authors thank the family members of the affected individual for participation in this study. We also acknowledge the generous contribution from Dr. K. Shiomi, Osaka, Japan, which has made this study possible. A special note of thanks is extended to Dr. Tamim Shaikh, Department of Human Genetics, Children's Hospital of Philadelphia for careful reading of the manuscript and valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ganguly, A., Dunbar, T., Chen, P. et al. Exon skipping caused by an intronic insertion of a young Alu Yb9 element leads to severe hemophilia A. Hum Genet 113, 348–352 (2003). https://doi.org/10.1007/s00439-003-0986-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-003-0986-5