
Abstract
Pheochromocytomas (PHEOs) and extra-adrenal paragangliomas (PGLs) are neural crest-derived tumors associated with the sympathetic or parasympathetic nervous systems. Successful pheochromocytoma/paraganglioma management now requires an interdisciplinary team approach. Precise identification of clinical context and the patient’s genetic status enables a personalized use of genetic and biochemical tests carefully followed by the application of genotype- and biochemical-specific functional imaging modalities. Once these tumors are identified, their patient-specific management, termed personalized medicine, is applied to achieve the best treatment outcome. This chapter particularly emphasizes the current and emerging knowledge of imaging options for chromaffin tumors tailored to their unique localization, biochemical phenotype, genetic landscape, malignant predisposition, and, ultimately, metastasis.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
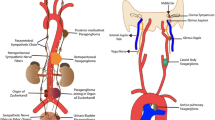

Keywords
14.1 Epidemiology and Natural History
PHEOs/PGLs are rare tumors with an annual incidence of one to eight patients per million [1]. They account for about 4 % of adrenal incidentalomas with a higher prevalence in an autopsy series [2]. Pediatric PHEOs/PGLs account for about 20 % of these tumors [3]. Approximately 75–85 % are located in the adrenal gland, and the remaining tumors are found outside of the gland, most commonly in the abdomen and thorax and less frequently in the head and neck (also termed as head and neck PGLs/HNPGLs) [4]. It is estimated that in the USA, there are about 1,000–2,000 new patients per year. Approximately 10 % of these patients present with metastatic disease upon initial diagnosis [5]. Around one third of these tumors are hereditary, caused by at least a dozen, well-characterized genes, described later on in this chapter [6, 7]. It is also estimated that about 30–50 % of these tumors are unrecognized initially, resulting in serious consequences to the patient, including death [8–10].
14.2 Tumor Origin
PHEOs and sympathetic-associated paragangliomas (symp-PGLs) develop from cells of the adrenal medulla or extra-adrenal chromaffin cells, respectively.
Chromaffin cells and sympathetic neurons derive from a common sympathoadrenal (SA) progenitor cell of neural crest origin. SA progenitor cells aggregate at the dorsal aorta, where they acquire a catecholaminergic neural fate. Subsequently, the cells migrate ventrally to invade the fetal adrenal cortex and form the adrenal medulla, as well as dorsolaterally to form sympathetic ganglia. Most extra-adrenal chromaffin cells regress via apoptosis. The organ of Zuckerkandl (OZ) constitutes the largest chromaffin paraganglia in the embryo and regresses after birth via autophagy [11]. Adrenal medulla and persistent extra-adrenal chromaffin cells located in the retroperitoneum and posterior mediastinum represent the chromaffin paraganglia system in adults. These cells possess the machineries to synthesize, store, release, and take up catecholamines, including the enzymes for noradrenaline synthesis. They have been named “chromaffin” by Kohn (1902) because of their characteristic staining property by chromium salts [12]. These embryological bases explain why PHEO and symp-PGL can be widely distributed along the sympathetic nervous system in the posterior mediastinum and retroperitoneum. In contrast, HNPGLs are derived from neural crest cells of the parasympathetic nervous system, with only 20 % producing and/or secreting catecholamines, usually dopamine or its metabolite 3-methoxytyramine [13–15]. Furthermore, these tumors are usually benign, except those caused by succinate dehydrogenase subunit B (SDHB) gene mutations, which are considered aggressive either locally or by the development of metastatic disease [5, 16–18].
14.3 Clinical Presentation and Diagnosis
PHEOs and symp-PGLs usually cause symptoms of catecholamine (norepinephrine or epinephrine) oversecretion (e.g., sustained or paroxysmal elevations in blood pressure, headache, episodic profuse sweating, palpitations, pallor, nervousness, or anxiety) [4]. These attacks can be caused with or without a trigger. The most common causes of catecholamine release from these tumors are direct manipulation, various drugs (mainly antidepressants, over-the-counter cold and allergy medicine, or antiemetics), stress, any type of local or general anesthesia, or excessive physical activity. If such an attack (sometimes called a “spell”) occurs, catecholamine concentrations can be enormous, reaching 1,000 times the normal reference limit, resulting in serious cardiovascular consequences and, in some patients, death [19–21]. Therefore, all patients with PHEO/PGL must be put on an adrenoceptor blockade that can lessen or at least partially prevent (full prevention of catecholamine effect on any organ is impossible) the deleterious effects of catecholamines on end organs [22]. Patients with a completely biochemically silent PHEO/PGL based on repeated, normal plasma and/or urine catecholamines and metanephrines are the only exception.
14.4 Biochemical Phenotypes
PHEOs/PGLs present in three well-defined biochemical phenotypes: noradrenergic, adrenergic, and dopaminergic (Table 14.1). A noradrenergic phenotype is defined by the elevation of norepinephrine or its metabolite, normetanephrine. In these tumors, the concentration of metanephrine, a metabolite of epinephrine, is usually less than 5 % of the sum of normetanephrine and metanephrine tissue concentrations [23]. These tumors are characteristic for extra-adrenal PGLs, either sporadic or those with hereditary background, mainly including mutations in succinate dehydrogenase subunits (SDHx), von Hippel-Lindau (VHL), fumarate dehydrogenase (FH), and hypoxia-inducible factor alpha (HIF2A) [7] genes. Clinically, patients with these tumors mainly present with paroxysmal or sustained hypertension and, less commonly, with tachyarrhythmia. An adrenergic phenotype is defined by the elevation of metanephrine in a PHEO/PGL tissue that is at least 10 % or more of the sum of metanephrine and normetanephrine tissue concentrations [23]. Epinephrine production in these tumors reflects the presence of the enzyme Phenylethanolamine-N-methyltransferase (PNMT) which is uniquely found in the adrenal medulla. Therefore, most of these tumors, if not all of them, are located in the adrenal medulla and as previously described called PHEOs. They can be sporadic or hereditary, including mutations in RET proto-oncogene (RET), neurofibromatosis type 1 (NF1), and transmembrane protein 127 genes. Clinically, patients with these tumors mainly present with paroxysmal or sustained tachyarrhythmia and, usually, mild hypertension. Due to the significant effect of epinephrine on beta-adrenoceptors, some of these patients may even present with hypotension due to beta-adrenoceptor-mediated vasodilation [24]. A dopaminergic phenotype is characterized by significant elevation of either dopamine or its metabolite methoxytyramine, or both. Usually, dopamine or methoxytyramine elevation is associated with an increase in norepinephrine or normetanephrine, which is commonly seen in patients with SDHx mutations [14, 15]. If only dopamine is elevated, patients do not present with any clinical signs or symptoms, unless dopamine levels are tremendously high (very rare) and hypotension may occur.
14.5 Spectrum of Hereditary Syndromes and Phenotype-Genotype Correlations
Research in molecular genetics has resulted in the identification of more than 20 susceptibility genes for tumors of the entire paraganglia system [7, 25]. Most PHEOs occur sporadically, whereas the majority of symp-PGLs are associated with germline driver mutations. Depending on their location, the most commonly found gene mutations are (1) unilateral PHEO: succinate dehydrogenase complex subunit B or D (SDHB or SDHD) and VHL; (2) bilateral PHEO: SDHB, RET, VHL, NF1, MYC-associated factor X (MAX), and TMEM127; and (3) symp-PGLs with or without PHEO: SDHB, SDHD, VHL, and HIF2A. Other genes account for a small minority of cases. Recent tumor sequencing has also led to the identification of somatic events in a large number of PHEOs/PGLs (The Cancer Genome Atlas, unpublished observations) (Table 14.1). Patients presenting with metastatic disease mainly include those with SDHB and perhaps SDHD (excluding HNPGLs), FH, and MAX-related PHEOs/PGLs, although, at present, about 50 % of metastatic PHEOs/PGLs are non-hereditary [5].
14.6 Differential Diagnosis
There are several potential causes to consider for the differential diagnosis in the presence of an adrenal or extra-adrenal mass (Table 14.2). However, the masses that belong to either PHEO or PGL are usually detected by imaging-specific characteristics that include the value of Hounsfield units (HU), T2-weighted bright images, and positivity on PHEO-/PGL-specific functional imaging, as described below.
14.7 Typical PHEO/PGL Imaging Finding on CT and/or MRI
On non-contrast computed tomography (CT), PHEO/PGL can demonstrate a variety of appearances. Two thirds of PHEO/PGLs are solid, while the remainder are complex or have undergone cystic or necrotic changes [26]. Typically, the CT attenuation of PHEO/PGL is about soft tissue attenuation and thus greater than 10 HU, with most PHEOs/PGLs 20–30 HU or higher. PHEO/PGL can present with a high attenuation due to the presence of hemorrhage or calcifications. In contrast, necrotic tissue presents with a low attenuation. Typically, a PHEO/PGL demonstrates avid enhancement (often greater than 30 HU) [27]. In addition, enhancement can be heterogeneous, or there may be no enhancement due to cystic, necrotic, or degenerated regions within the lesion [28]. On magnetic resonance imaging (MRI), the classic imaging appearance of PHEO/PGL is “light bulb” bright on T2-weighted imaging. In reality, 30 % of PHEOs/PGLs demonstrate moderate or low T2-weighted signal intensity [27, 29]. PHEOs/PGLs typically demonstrate avid contrast enhancement following the administration of intravenous gadolinium-based contrast material [30, 31].
14.8 Typical Imaging Finding on Molecular Imaging
Nowadays, positron emission tomography (PET) is a cornerstone in the evaluation of hereditary as well as non-hereditary PHEOs/PGLs. The broad diversity of PET biomarkers enables assessment of different metabolic pathways and receptors. Beyond its localization value, this imaging modality provides unique opportunities for better characterizing these tumors at molecular levels (e.g., the presence of catecholamines and their metabolites, specific cell membrane receptors and transporters), mirroring ex vivo histological classification but on a whole-body, in vivo, scale (Table 14.4).
Thus, successful PHEO-/PGL-specific localization depends on the presence of molecules (imaging targets) for which PET radiopharmaceuticals are currently available. Based on several recent studies, it has been uncovered that PHEO-/PGL-specific imaging targets have various expressions based on whether these tumors belong to pseudohypoxic or kinase signaling clusters, present as metastatic, are located in or outside the adrenal gland, or are derived from the sympathetic or parasympathetic nervous system. Currently, 18F-fluorodeoxyglucose (18F-FDG) is the most accessible PET radiopharmaceutical, but lacks specificity for these tumors. 18F-fluorodopamine (18F-FDA) and 11C-hydroxyephedrine (11C-HED) are the most specific tracers for chromaffin tumors, but are available in very few centers and fail in metastatic and hereditary PHEOs/PGLs [32–34].18F-fluorodyhidroxyphenylalanine (18F-FDOPA) is available from different pharmaceutical suppliers, but its sensitivity widely depends on the genetic background and whether the PHEO/PGL is sympathetic or parasympathetic [35–37]. Metastatic behavior of these tumors can also affect the expression of amino acid transporters [38]. Newly developed 68Ga-labeled peptides, as in other neuroendocrine tumors, have shown very interesting results and, in our opinion, should be positioned first for many indications due to their exceptional affinity to somatostatin receptor type 2 found on these tumors [39–44].
PHEOs and PGLs usually have highly elevated uptake values with specific radiopharmaceuticals based on their genetic background. For example, 18F-FDOPA PET/CT was replaced by 68Ga-DOTATATE PET/CT as the best available imaging modality for metastatic PHEO/PGL, especially in those with SDHB mutations and head and neck PGLs [40–42]. The Octreoscan has been suggested not to be used anymore due to its suboptimal performance in the detection of these tumors and the growing availability of 68Ga-DOTATATE PET/CT. However, the use of other 68Ga-labeled DOTA analogues (68Ga-DOTATOC and 68Ga-DOTANOC) needs to be confirmed in a large population of patients. A variety of new radiopharmaceuticals have been developed as potential competitors of 68Ga-DOTATATE (68Ga-labeled somatostatin antagonists, 64Cu-labeled SSA [45], or 18F-SiFAlin (silicon-fluoride acceptor)-modified TATE), but they need to be evaluated [46].
14.9 Relationship Between Genotype and Imaging Phenotype
Proper evaluation of PHEO/PGL is a key point for choosing the necessary treatment plan for follow-up and outcome for these patients. The presence of SDHx mutations markedly influences sensitivity of 18F-FDG, 18F-FDOPA, and 68Ga-DOTA-SSAs PET/CT. 18F-DOPA PET/CT has a sensitivity approaching 100 % for sporadic PHEO and a very high specificity (95 %), but can miss tumors in SDHx-mutated patients. 18F-FDOPA PET/CT still remains a very good modality for the detection of some metastatic PHEO/PGLs – it ranks as the second best for the detection of HNPGLs, and it can also be used for patients with non-hereditary metastatic PHEO/PGL [18, 35, 36, 38, 47, 48]. By contrast, SDHx tumors usually exhibit highly elevated 18F-FDG uptake values. However, 18F-FDG PET/CT positivity is present in about 80 % of primary PHEOs. Thus, 18F-FDG PET/CT remains a good alternative for the detection of metastatic PHEO/PGL, especially those related to SDHx mutations [49]. Several potential diagnoses should be considered in cases of 18F-FDG-avid adrenal masses (Table 14.3).
The use of 68Ga-DOTA-SSAs in the context of PHEOs/PGLs has been studied less, but has shown excellent preliminary results in localizing these tumors, especially metastatic and head and neck ones, as discussed above. A head-to-head comparison between 68Ga-DOTA-SSA and 18F-FDOPA PET has been performed in only five studies: one retrospective study from Innsbruck Medical University (68Ga-DOTATOC in 20 patients with unknown genetic background) [50], three prospective studies from the NIH (SDHB, HNPGL, and sporadic metastatic PHEOs/PGLs) (68Ga-DOTATATE in 17 and 20 patients), and one prospective study from La Timone University Hospital (68Ga-DOTATATE in 30 patients) [40–42]. In these studies, 68Ga-DOTA-SSA PET/CT detected more primary head and neck PGLs as well as SDHx-associated PGLs than 18F-FDOPA PET/CT [51]. By contrast, in the context of sporadic PHEO, 18F-FDOPA PET/CT may detect more lesions than 68Ga-DOTATATE, although larger studies are needed to confirm those results [51]. One of the main drawbacks of 68Ga-DOTA-SSA is the very high physiological uptake by healthy adrenal glands [52]. Furthermore, there are also different affinities of various DOTA-SSAs to somatostatin receptors. DOTATATE has the best affinity to somatostatin receptor type 2, mostly expressed on PHEO/PGLs, followed by DOTATOC. DOTANOC has the lowest affinity to somatostatin receptor type 2 and has some affinity to somatostatin receptor type 5, which is least abundant on PHEOs/PGLs. Therefore, the use of DOTANOC may result in suboptimal detection of PHEO/PGL, especially their metastatic lesions. Studies comparing 68Ga-DOTATATE and 68Ga-DOTATOC are currently unavailable. Excellent results with DOTA analogues in both sporadic as well as SDHx-related metastatic PHEOs/PGLs resulted in the use of 177Lu-DOTATATE (Lutathera) in radiotherapy of these tumors [53–57]. This is followed by the preparation of clinical protocols in order to properly assess the efficacy of this treatment on a large population of well-characterized patients with metastatic or inoperable PHEO/PGL (Lin, Pacak et al. NIH protocol in preparation, 2016).
14.10 Role of Radionuclide Imaging
Successful PHEO/PGL management requires an interdisciplinary team approach. Precise identification of clinical context and the genetic status of a patient enables a personalized use of functional imaging modalities. Currently, it is recommended to adopt a tailored approach using a diagnostic algorithm based on tumor location, biochemical phenotype, and any known genetic background (Table 14.5) [48, 58].
14.10.1 Diagnosis of PHEO or Symp-PGL
14.10.1.1 Adrenal Mass
Functional imaging should be used in a minority of cases, such as those with suspicion of nonfunctioning PHEO on CT/MRI, elevation of plasma or urine normetanephrine in the presence of an adrenal mass, acute cardiovascular complication in the critical care setting together with the presence of an adrenal mass, hemorrhagic adrenal masses, and either elevated plasma metanephrine or normetanephrine in renal insufficiency. Elevation of metanephrine in the plasma or urine in the presence of an adrenal mass does not call for the use of functional imaging since metanephrine is 99 % derived from the adrenal gland. Thus, its elevation highly supports the presence of PHEO, especially when plasma or urine metanephrine is 4x above the upper reference limit. Another new promising option is the use of proton single-voxel magnetic resonance spectroscopy (1H-MRS) that can detect the presence of catecholamines in PHEO and, therefore, may correctly point to the presence of this tumor [59–61].
PET imaging using 18F-FDOPA, 18F-FDA PET/CT, or 68Ga-DOTA-SSA PET/CT is highly sensitive with 18F-FDA having an excellent specificity, as described above (Fig. 14.1). The low uptake of 18F-FDOPA by normal adrenals is a potential advantage over 68Ga-DOTA-SSA for localizing a small PHEO (Figs. 14.2, 14.3, and 14.4).

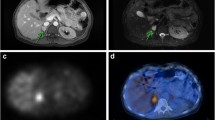
Typical imaging features of PHEO on 18F-FDOPA PET/CT and 68Ga-DOTATATE. Axial 18F-FDOPA PET (a) and PET/CT (b). Axial 68Ga-DOTATATE PET (c) and PET/CT (d). The PHEO was positive on both imaging studies (arrows). Note high 68Ga-DOTATATE uptake by the left normal adrenal gland (arrowheads)

Unilateral MEN2A-related PHEO. Multiphasic adrenal CT (a–c), 18F-FDOPA PET/CT, and (e) 18F-FDG PET/CT (f) showed a single left PHEO (arrow) with normal contralateral gland (arrowhead), while 123I-MIBG was falsely positive for the right adrenal gland. A total left adrenalectomy was performed with subsequent normalization of metanephrine. Pathological analysis found three PHEOs (20 mm for the largest tumor)

MEN2A with PHEO and persistent MTC. (a) 18F-FDOPA PET/CT (MIP image) showing persistent cervical metastatic lymph nodes from MTC (arrow), (b) unenhanced CT showing a left adrenal nodule, and (c) axial 18F-FDOPA PET and PET/CT are consistent with a small left PHEO (arrow)

18F-FDOPA PET/CT findings in hereditary PHEOs. (a, b) NF1-related right PHEO. (c, d) Bilateral VHL-related PHEO. (c, d) Bilateral MAX-related PHEO (note the presence of two foci within the left adrenal gland)
14.10.1.2 Retroperitoneal Extra-Adrenal Nonrenal Mass
In the presence of a retroperitoneal extra-adrenal, nonrenal mass, it is important to differentiate a PGL from other tumors (Table 14.2). A biopsy is not always contributory or even recommended, since it can carry a high risk of hypertensive crisis and tachyarrhythmia. Therefore, it should only be done if a PGL has been ruled out in a patient presenting with signs and symptoms of catecholamine excess. Specific functional imaging studies, which are not usually performed before biochemical results are available, are very helpful in distinguishing PGLs from other tumors (Figs. 14.5, 14.6, 14.7, and 14.8). 68Ga-DOTA-SSAs are the first-line imaging since most patients are expected to have SDHx mutations.

Sporadic extra-adrenal paraganglioma. CT scan revealed a solitary preaortic mass with a rapid and marked contrast enhancement and a slow washout pattern suspected of paraganglioma. 18F-FDG PET was slightly positive, but 18F-FDOPA PET was pathognomonic for PGL. (a) Unenhanced CT, 40 HU; (b) arterial contrast-enhanced CT, 130 HU; (c) arterial contrast-enhanced CT (reconstruction); (d) portal-phase contrast-enhanced CT, 80 HU; (e) 18F-FDG PET, moderate tumor uptake (SUVmax = 2.5); and (f) 18F-FDOPA PET, high tracer uptake (SUVmax = 8.5). Pathological analysis found a typical paraganglioma

Retroperitoneal neurofibroma. CT scan showed a retroperitoneal para-aortic solitary mass with a moderate and progressive homogeneous contrast enhancement. 18F-FDG PET showed a mildly increased 18F-FDG uptake, and 18F-FDOPA PET was negative. (a) Unenhanced CT, 28 HU; (b) arterial contrast-enhanced CT, 36 HU; (c) arterial contrast-enhanced CT (reconstruction); (d) portal-phase contrast-enhanced CT, 70 HU; (e) 18F-FDG, moderate tumor uptake (SUVmax = 3.6); and (f) 18F-FDOPA PET, absence of tumor uptake. Pathological analysis found a cellular neurofibroma with atypia (Ki-67 10–15 %)

Para-adrenal PGL in a SDHD patient. Axial 18F-FDG PET (a) and PET/CT (b) and 18F-FDOPA PET (d) and PET/CT (e). 18F-FDG PET was positive, while 18F-FDOPA PET was considered as falsely negative. Gross pathology showed that the tumor was developed from para-adrenal paraganglia (adr adrenal, pgl paraganglioma) (c)

HIF2A-related paraganglioma of the organ of Zuckerkandl. Contrast-enhanced CT arterial phase (a) showing a hypervascular and heterogeneous left para-aortic mass located at the level of the inferior mesenteric artery (arrow). 18F-FDOPA PET/CT (b) and 18F-FDG PET and PET/CT (c) showing a single tumor with heterogeneous uptake and a preferential 18F-FDOPA imaging pattern, a finding which is opposite to the classical SDHx-associated imaging phenotype (see Fig. 14.7)
14.10.2 Assessment of Locoregional Extension and Diagnosis of Malignancy
Presently, there are no reliable cytological, histological, immunohistochemical, molecular, or imaging criteria for determining malignancy [62]. The diagnosis of malignancy remains strictly based on the finding of metastases where chromaffin cells are not usually present, such as the lymph nodes, lung, bone, or liver.
Anatomical imaging appears sufficient in the staging of PHEO/PGL. Functional imaging is probably not necessary in the preoperative workup of PHEO patients meeting the following criteria: >40 years, no family history, small (less than 2.0 cm) PHEO-secreting predominantly metanephrine, and negative genetic testing. Functional imaging is strongly recommended for excluding metastatic disease in large adrenal tumors (>4–5.0 cm) and in SDHB patients, and most probably SDHD as well. It is widely accepted that tumors with an underlying SDHB mutation are associated with a higher risk of aggressive behavior, development of metastatic disease, and, ultimately, death.
To date, 18F-FDOPA PET or 68Ga-DOTA-SSA may be the imaging modality of choice in the absence of a SDHB mutation, or when genetic status is unknown. By contrast, 68Ga-DOTA-SSA or, if not available, 18F-FDG PET should be considered as the imaging modalities of reference for SDHx-related cases (for images, see Chap. 13).
14.10.3 Detection of Multifocality
Beyond malignancy risk, inherited (especially SDHx, VHL, and MEN2) or symp-PGL raises the problem of multifocality. Based on recent published data, 68Ga-DOTA-SSA has gained an increasing role and should get a leading position in this setting. In absence of available 68Ga-DOTA-SSA, 18F-FDG should be preferred to 18F-FDOPA in SDHx patients, whereas 18F-FDOPA appears to be a very good imaging tool in other genotypes and sporadic cases.
14.10.4 Imaging Follow-Up of Sporadic PHEO/PGL Patients
For sporadic PHEO/PGL patients, imaging follow-up is necessary, especially in those patients presenting with primary tumors larger than 4–5 cm and those with an extra-adrenal location [63]. Currently, there is no guideline regarding the frequency of imaging follow-up, but it is recommended to be done at least every 2 years, with either CT or MRI, preferably alternatively, to reduce the amount of radiation delivered to the patient.
14.10.5 Imaging Follow-Up of Mutation Carriers
An optimal follow-up algorithm has not yet been validated in mutation carriers for predisposing genes for PHEO/PGL. MRI offers several physical advantages over CT and does not expose patients to ionizing radiation, which is critical in a patient population submitted to lifelong imaging surveillance. However, MRI can be less sensitive than radionuclide imaging for detecting small lesions. Follow-up should include annual biochemical screening, and CT or MRI can be delayed on 2-year intervals. Indications for PET imaging studies should be discussed on an individual basis.
14.11 Current Proposed Imaging Algorithm in the Diagnosis and Localization of PHEO and Symp-PGL
Successful PPGL management requires an interdisciplinary team approach. Precise identification of clinical context and genetic status of patients enables a personalized use of functional imaging modalities [64–67]. Although the extra cost and availability of new PET tracers can prove problematic, the option of not employing them might lead to inappropriate management with health-related consequences that should not be underestimated.
Based on the currently available imaging techniques, we propose the following approach to investigate a patient with HNPGL:
-
1.
For diagnosis, the specificity provided by functional imaging techniques using 18F-FDOPA PET/CT or 68Ga-SSTa is superior to anatomical imaging.
-
2.
For detecting additional tumor sites (multifocality, metastases), functional imaging techniques are superior to anatomical imaging. Based on the most recent studies and personal experience, PPGLs are well detected with 18F-FDOPA PET/CT. 68Ga-DOTA-SSA is also very sensitive for staging, regardless of the genotype. In the absence of available 68Ga-DOTA-SSA, 18F-FDG PET/CT should be used as the first-line radionuclide imaging tool in SDHx cases.
-
3.
For determining the locoregional extension of PHEO/symp-PGL, anatomic imaging remains the first-line modality.
This algorithm should be adapted to the practical situation within each institution and should evolve with time as new techniques become available (Table 14.5). 18F-FDOPA and 68Ga-DOTA-SSA are currently available in many clinical and research centers around the world.
14.11.1 Image-Based Treatment of PHEOs/PGLs
14.11.1.1 Adrenal-Sparing Surgery
A subtotal (cortical-sparing) adrenalectomy is a valid option in patients with MEN2, NF1, or VHL. In cases with bilateral PHEOs, this strategy offers the advantage of potentially avoiding steroid supplementation. Therefore, it is crucial to perform regular imaging follow-ups of known PHEOs in addition to biochemical testing for determining the optimal time to schedule a cortical-sparing surgery. CT is preferable over MRI due to its excellent resolution, which provides detailed anatomical locations of tumor extension within the adrenal gland and, for MEN2 patients, the number of tumors within the adrenal medulla. On the other hand, the advantage of using MRI over CT is the lack of exposure to ionizing radiation, which is an important factor in hereditary cases undergoing continuous follow-up. In selected cases, functional imaging may be used in addition to anatomical imaging. There is a clear advantage of 18F-FDOPA PET/CT over MIBG and other specific PET tracers due to the lack of significant uptake in normal adrenal glands [68]. 18F-FDOPA PET may also identify metastases from medullary thyroid carcinoma with persistent hypercalcitoninemia [69].
14.11.1.2 Theranostics
123I-MIBG scintigraphy is used as a companion imaging agent to assist in radionuclide therapy selection. A special advantage of labeled SSAs is that, unlike 18F-FDOPA, they can be used in the radioactive treatment of these tumors (as theranostic agents). To date, peptide receptor radionuclide therapy (PRRT) using 90Y-/177Lu-labeled somatostatin agonists has been evaluated in a limited number of PHEO/PGL cases [53–57, 70]. On average, response rates (mainly partial responses) have been 30–60 %. Disease stabilization is frequent but more difficult to interpret, since these tumors often exhibit a slow growing pattern. Larger studies, including various hereditary and non-hereditary PHEOs/PGLs, are needed in order to conclude which PHEOs/PGLs can best be treated using this therapy and whether PRRT should be used together or as a “replacement” to other treatment modalities.
We believe that data from PRRT in midgut NETs will provide a powerful impetus for wider application of the 68Ga-/177Lu-DOTATATE strategy in the management of inoperable (including metastatic ones) PHEOs/PGLs. The toxicity profile (kidney, bone marrow) of 177Lu-DOTATATE is acceptable due to the low penetration range of 177Lu (Table 14.6). 90Y has a pure beta emission with long-range particles that, besides the direct action, also lead to irradiation of non-receptor-expressing tumor cell (cross-fire effect). For these reasons, 90Y-peptides could be preferable for the treatment of larger and inhomogeneous lesions. However, toxicity is more frequent than with 177Lu, and clinical implementation of dosimetry is much more complex than for 177Lu. 68Ga can be used as a surrogate isotope for dosimetry purposes, but it provides only limited information due to its very short physical half-life (68 min) compared to 90Y and 177Lu (Table 14.6). 64Cu-DOTATATE was also found to provide excellent imaging quality for NET imaging [45] and can be used for dosimetry purposes. Beyond 90Y and 177Lu, the terbium radioisotopes 155Tb (SPECT), 152Tb (PET), and 161Tb (therapeutic isotope) can also be considered as theranostic pairs. 161Tb has medium-energy beta particles similar to 177Lu, but has the advantage over 177Lu to emit a high level of internal conversion and Auger electrons that may act synergistically to beta particles. Modeling studies have shown that 161Tb and 177Lu can deliver equivalent absorbed doses in 10 mm spheres. However, 161Tb would be more effective than 177Lu at a smaller scale [71]. Therefore, the use of 161Tb would enable destruction of macroscopic residual disease but also small cell clusters (micrometastases, isolated cells). Therapeutic nuclear medicine may also include the administration of bone-seeking radiopharmaceuticals. Beyond the use of beta particles and Auger emitters for therapeutic applications, there are significant advantages to use alpha emitters. However, until present, only a few studies have targeted NETs.
Recent reports have shown that cellular internalization might shorten the residual time of 177Lu within tumor cells compared to radiolabeled SST antagonists. SST antagonists also have higher affinities for SST receptors than agonists, and lower internalization rates, resulting in a longer retention time on cell membrane. According to these observations, in the future, somatostatin antagonists might be considered as an alternative to agonists for PRRT.
References
Stenstrom G, Svardsudd K. Pheochromocytoma in Sweden 1958–1981. An analysis of the National Cancer Registry Data. Acta Med Scand. 1986;220(3):225–32.
Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85(2):637–44.
Barontini M, Levin G, Sanso G. Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann N Y Acad Sci. 2006;1073:30–7.
Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366(9486):665–75.
Turkova H, Prodanov T, Maly M, Martucci V, Adams K, Widimsky Jr J, et al. Characteristics and outcomes of metastatic Sdhb and sporadic pheochromocytoma/paraganglioma: an National Institutes of Health Study. Endocr Pract. 2016;22(3):302–14.
Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44(5):328–33.
Pacak K, Wimalawansa SJ. Pheochromocytoma and paraganglioma. Endocr Pract. 2015;21(4):406–12.
McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30(6):648–52.
Platts JK, Drew PJ, Harvey JN. Death from phaeochromocytoma: lessons from a post-mortem survey. J R Coll Physicians Lond. 1995;29(4):299–306.
Sutton MG, Sheps SG, Lie JT. Prevalence of clinically unsuspected pheochromocytoma. Review of a 50-year autopsy series. Mayo Clin Proc. 1981;56(6):354–60.
Schober A, Parlato R, Huber K, Kinscherf R, Hartleben B, Huber TB, et al. Cell loss and autophagy in the extra-adrenal chromaffin organ of Zuckerkandl are regulated by glucocorticoid signalling. J Neuroendocrinol. 2013;25(1):34–47.
Kohn A. Die Paraganglien. Arch Mikrosk Anat. 1903;52:262–365.
van Duinen N, Steenvoorden D, Kema IP, Jansen JC, Vriends AH, Bayley JP, et al. Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomas. J Clin Endocrinol Metab. 2010;95(1):209–14.
Eisenhofer G, Lenders JW, Siegert G, Bornstein SR, Friberg P, Milosevic D, et al. Plasma methoxytyramine: a novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur J Cancer. 2012;48(11):1739–49.
Eisenhofer G, Lenders JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57(3):411–20.
Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92(10):3822–8.
Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91(3):827–36.
Taïeb D, Kaliski A, Boedeker CC, Martucci V, Fojo T, Adler Jr JR, et al. Current approaches and recent developments in the management of head and neck paragangliomas. Endocr Rev. 2014;35(5):795–819.
Schuttler J, Westhofen P, Kania U, Ihmsen H, Kammerecker S, Hirner A. Quantitative assessment of catecholamine secretion as a rational principle of anesthesia management in pheochromocytoma surgery. Anasthesiol Intensivmed Notfallmed Schmerzther. 1995;30(6):341–9.
Pacak K. Preoperative management of the pheochromocytoma patient. J Clin Endocrinol Metab. 2007;92(11):4069–79.
Lenders JW, Eisenhofer G. Pathophysiology and diagnosis of disorders of the adrenal medulla: focus on pheochromocytoma. Compr Physiol. 2014;4(2):691–713.
Luiz HV, Tanchee MJ, Pavlatou MG, Yu R, Nambuba J, Wolf K, et al. Are patients with hormonally functional phaeochromocytoma and paraganglioma initially receiving a proper adrenoceptor blockade? A retrospective cohort study. Clin Endocrinol (Oxf). 2016;85(1):62–9.
Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, et al. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem. 2005;51:735–44.
Kantorovich V, Pacak K. A new concept of unopposed beta-adrenergic overstimulation in a patient with pheochromocytoma. Ann Intern Med. 2005;142(12 Pt 1):1026–8.
Jochmanova I, Yang C, Zhuang Z, Pacak K. Hypoxia-inducible factor signaling in pheochromocytoma: turning the rudder in the right direction. J Natl Cancer Inst. 2013;105(17):1270–83.
Park BK, Kim CK, Kwon GY, Kim JH. Re-evaluation of pheochromocytomas on delayed contrast-enhanced CT: washout enhancement and other imaging features. Eur Radiol. 2007;17(11):2804–9.
Blake MA, Kalra MK, Maher MM, Sahani DV, Sweeney AT, Mueller PR, et al. Pheochromocytoma: an imaging chameleon. Radiographics. 2004;24 Suppl 1:S87–99.
Park BK, Kim B, Ko K, Jeong SY, Kwon GY. Adrenal masses falsely diagnosed as adenomas on unenhanced and delayed contrast-enhanced computed tomography: pathological correlation. Eur Radiol. 2006;16(3):642–7.
Mayo-Smith WW, Boland GW, Noto RB, Lee MJ. State-of-the-art adrenal imaging. Radiographics. 2001;21(4):995–1012.
Mitchell DG. Chemical shift magnetic resonance imaging: applications in the abdomen and pelvis. Top Magn Reson Imaging. 1992;4(3):46–63.
Varghese JC, Hahn PF, Papanicolaou N, Mayo-Smith WW, Gaa JA, Lee MJ. MR differentiation of phaeochromocytoma from other adrenal lesions based on qualitative analysis of T2 relaxation times. Clin Radiol. 1997;52(8):603–6.
Mamede M, Carrasquillo JA, Chen CC, Del Corral P, Whatley M, Ilias I, et al. Discordant localization of 2-[18F]-fluoro-2-deoxy-D-glucose in 6-[18F]-fluorodopamine- and [123I]-metaiodobenzylguanidine-negative metastatic pheochromocytoma sites. Nucl Med Commun. 2006;27(1):31–6.
Ilias I, Yu J, Carrasquillo JA, Chen CC, Eisenhofer G, Whatley M, et al. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab. 2003;88(9):4083–7.
Shulkin BL, Wieland DM, Schwaiger M, Thompson NW, Francis IR, Haka MS, et al. PET scanning with hydroxyephedrine: an approach to the localization of pheochromocytoma. J Nucl Med. 1992;33:1125–31.
Imani F, Agopian VG, Auerbach MS, Walter MA, Imani F, Benz MR, et al. 18F-FDOPA PET and PET/CT accurately localize pheochromocytomas. J Nucl Med. 2009;50(4):513–9.
Fottner C, Helisch A, Anlauf M, Rossmann H, Musholt TJ, Kreft A, et al. 6-18F-fluoro-L-dihydroxyphenylalanine positron emission tomography is superior to 123I-metaiodobenzyl-guanidine scintigraphy in the detection of extraadrenal and hereditary pheochromocytomas and paragangliomas: correlation with vesicular monoamine transporter expression. J Clin Endocrinol Metab. 2010;95(6):2800–10.
King KS, Chen CC, Alexopoulos DK, Whatley MA, Reynolds JC, Patronas N, et al. Functional imaging of SDHx-related head and neck paragangliomas: comparison of 18F-fluorodihydroxyphenylalanine, 18F-fluorodopamine, 18F-fluoro-2-deoxy-D-glucose PET, 123I-metaiodobenzylguanidine scintigraphy, and 111In-pentetreotide scintigraphy. J Clin Endocrinol Metab. 2011;96(9):2779–85.
Taïeb D, Tessonnier L, Sebag F, Niccoli-Sire P, Morange I, Colavolpe C, et al. The role of 18F-FDOPA and 18F-FDG-PET in the management of malignant and multifocal phaeochromocytomas. Clin Endocrinol (Oxf). 2008;69(4):580–6.
Maurice JB, Troke R, Win Z, Ramachandran R, Al-Nahhas A, Naji M, et al. A comparison of the performance of (6)(8)Ga-DOTATATE PET/CT and (1)(2)(3)I-MIBG SPECT in the diagnosis and follow-up of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2012;39(8):1266–70.
Janssen I, Blanchet EM, Adams K, Chen CC, Millo CM, Herscovitch P, et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2015;21(17):3888–95.
Janssen I, Chen CC, Millo CM, Ling A, Taïeb D, Lin FI, et al. PET/CT comparing Ga-DOTATATE and other radiopharmaceuticals and in comparison with CT/MRI for the localization of sporadic metastatic pheochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2016;43(10):1784–91.
Janssen I, Chen CC, Taïeb D, Patronas NJ, Millo CM, Adams KT, et al. 68Ga-DOTATATE PET/CT in the localization of head and neck paragangliomas compared with other functional imaging modalities and CT/MRI. J Nucl Med. 2016;57(2):186–91.
Reubi JC, Schar JC, Waser B, Wenger S, Heppeler A, Schmitt JS, et al. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med. 2000;27(3):273–82.
Reubi JC, Waser B, Khosla S, Kvols L, Goellner JR, Krenning E, et al. In vitro and in vivo detection of somatostatin receptors in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 1992;74(5):1082–9.
Pfeifer A, Knigge U, Binderup T, Mortensen J, Oturai P, Loft A, et al. 64Cu-DOTATATE PET for neuroendocrine tumors: a prospective head-to-head comparison with 111In-DTPA-octreotide in 112 patients. J Nucl Med. 2015;56(6):847–54.
Niedermoser S, Chin J, Wangler C, Kostikov A, Bernard-Gauthier V, Vogler N, et al. In vivo Evaluation of (1)(8)F-SiFAlin-modified TATE: a potential challenge for (6)(8)Ga-DOTATATE, the clinical gold standard for somatostatin receptor imaging with PET. J Nucl Med. 2015;56(7):1100–5.
Charrier N, Deveze A, Fakhry N, Sebag F, Morange I, Gaborit B, et al. Comparison of [(1)(1)(1)In]pentetreotide-SPECT and [(1)(8)F]FDOPA-PET in the localization of extra-adrenal paragangliomas: the case for a patient-tailored use of nuclear imaging modalities. Clin Endocrinol (Oxf). 2011;74(1):21–9.
Taïeb D, Timmers HJ, Hindie E, Guillet BA, Neumann HP, Walz MK, et al. EANM 2012 guidelines for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2012;39(12):1977–95.
Timmers HJ, Chen CC, Carrasquillo JA, Whatley M, Ling A, Eisenhofer G, et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J Natl Cancer Inst. 2012;104(9):700–8.
Kroiss A, Putzer D, Frech A, Decristoforo C, Uprimny C, Gasser RW, et al. A retrospective comparison between (68)Ga-DOTA-TOC PET/CT and (18)F-DOPA PET/CT in patients with extra-adrenal paraganglioma. Eur J Nucl Med Mol Imaging. 2013;40(12):1800–8.
Archier A, Varoquaux A, Garrigue P, Montava M, Guerin C, Gabriel S, et al. Prospective comparison of (68)Ga-DOTATATE and (18)F-FDOPA PET/CT in patients with various pheochromocytomas and paragangliomas with emphasis on sporadic cases. Eur J Nucl Med Mol Imaging. 2016;43(7):1248–57.
Kroiss A, Putzer D, Decristoforo C, Uprimny C, Warwitz B, Nilica B, et al. 68Ga-DOTA-TOC uptake in neuroendocrine tumour and healthy tissue: differentiation of physiological uptake and pathological processes in PET/CT. Eur J Nucl Med Mol Imaging. 2013;40(4):514–23.
Delpassand ES, Samarghandi A, Zamanian S, Wolin EM, Hamiditabar M, Espenan GD, et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE for patients with somatostatin receptor-expressing neuroendocrine tumors: the first US phase 2 experience. Pancreas. 2014;43(4):518–25.
Strosberg JR, Wolin EM, Chasen B, Kulke MH, Bushnell DL, Caplin ME, et al. NETTER-1 phase III: Progression-free survival, radiographic response, and preliminary overall survival results in patients with midgut neuroendocrine tumors treated with 177-Lu-Dotatate. J Clin Oncol. 2016;34 (suppl 4S; abstr 194).
van Essen M, Krenning EP, Kooij PP, Bakker WH, Feelders RA, de Herder WW, et al. Effects of therapy with [177Lu-DOTA0, Tyr3]octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J Nucl Med. 2006;47(10):1599–606.
Vinik AI. Advances in diagnosis and treatment of pancreatic neuroendocrine tumors. Endocr Pract. 2014;20(11):1222–30.
Zovato S, Kumanova A, Dematte S, Sansovini M, Bodei L, Di Sarra D, et al. Peptide receptor radionuclide therapy (PRRT) with 177Lu-DOTATATE in individuals with neck or mediastinal paraganglioma (PGL). Horm Metab Res. 2012;44(5):411–4.
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99(6):1915–42.
Imperiale A, Battini S, Averous G, Mutter D, Goichot B, Bachellier P, et al. In vivo detection of catecholamines by magnetic resonance spectroscopy: A potential specific biomarker for the diagnosis of pheochromocytoma. Surgery. 2016;159(4):1231–3.
Imperiale A, Moussallieh FM, Roche P, Battini S, Cicek AE, Sebag F, et al. Metabolome profiling by HRMAS NMR spectroscopy of pheochromocytomas and paragangliomas detects SDH deficiency: clinical and pathophysiological implications. Neoplasia. 2015;17(1):55–65.
Varoquaux A, le Fur Y, Imperiale A, Reyre A, Montava M, Fakhry N, et al. Magnetic resonance spectroscopy of paragangliomas: new insights into in vivo metabolomics. Endocr Relat Cancer. 2015;22(4):M1–8.
Gimm O, DeMicco C, Perren A, Giammarile F, Walz MK, Brunaud L. Malignant pheochromocytomas and paragangliomas: a diagnostic challenge. Langenbecks Arch Surg. 2012;397(2):155–77.
Schovanek J, Martucci V, Wesley R, Fojo T, Del Rivero J, Huynh T, et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer. 2014;14:523.
Havekes B, King K, Lai EW, Romijn JA, Corssmit EP, Pacak K. New imaging approaches to phaeochromocytomas and paragangliomas. Clin Endocrinol (Oxf). 2010;72(2):137–45.
Taïeb D, Neumann H, Rubello D, Al-Nahhas A, Guillet B, Hindie E. Modern nuclear imaging for paragangliomas: beyond SPECT. J Nucl Med. 2012;53(2):264–74.
Timmers HJ, Taïeb D, Pacak K. Current and future anatomical and functional imaging approaches to pheochromocytoma and paraganglioma. Horm Metab Res. 2012;44(5):367–72.
Taïeb D, Rubello D, Al-Nahhas A, Calzada M, Marzola MC, Hindie E. Modern PET imaging for paragangliomas: relation to genetic mutations. Eur J Surg Oncol. 2011;37(8):662–8.
Luster M, Karges W, Zeich K, Pauls S, Verburg FA, Dralle H, et al. Clinical value of 18F-fluorodihydroxyphenylalanine positron emission tomography/computed tomography (18F-DOPA PET/CT) for detecting pheochromocytoma. Eur J Nucl Med Mol Imaging. 2010;37(3):484–93.
Treglia G, Rufini V, Salvatori M, Giordano A, Giovanella L. PET imaging in recurrent medullary thyroid carcinoma. Int J Mol Imaging. 2012;2012:324686.
Puranik AD, Kulkarni HR, Singh A, Baum RP. Peptide receptor radionuclide therapy with (90)Y/(177)Lu-labelled peptides for inoperable head and neck paragangliomas (glomus tumours). Eur J Nucl Med Mol Imaging. 2015;42(8):1223–30.
Hindie E, Zanotti-Fregonara P, Quinto MA, Morgat C, Champion C. Dose deposits from 90Y, 177Lu, 111In, and 161Tb in micrometastases of various sizes: implications for radiopharmaceutical therapy. J Nucl Med. 2016;57(5):759–64.
Funding
This research did not receive a specific grant from any funding agency in the public, commercial, or not-for-profit sector.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Taïeb, D., Pacak, K. (2017). Radionuclide Imaging of Chromaffin Cell Tumors. In: Pacak, K., Taïeb, D. (eds) Diagnostic and Therapeutic Nuclear Medicine for Neuroendocrine Tumors. Contemporary Endocrinology. Humana Press, Cham. https://doi.org/10.1007/978-3-319-46038-3_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-46038-3_14
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-46036-9
Online ISBN: 978-3-319-46038-3
eBook Packages: MedicineMedicine (R0)