
Abstract
Cell-contacts are essential for intercellular communication and are involved in proliferation, differentiation, and homeostasis. Melanocytes establish multiple contacts with keratinocytes, which in turn control melanocyte growth and expression of cell surface receptors. Most melanoma arise within the epidermis (melanoma in situ) and then invade across the basement membrane. These melanoma cells escape from control by keratinocytes through five major mechanisms: (1) downregulation of receptors important for communication with keratinocytes such as E-cadherin, P-cadherin, desmoglein, and connexins; (2) upregulation of receptors and signaling molecules important for interactions between melanoma cells and other melanoma cells, fibroblasts, or endothelial cells, such as N-cadherin, Mel-CAM, and zonula occludens protein-1 (ZO-1); (3) deregulation of morphogens such as Notch receptors and their ligands; (4) loss of anchorage to the basement membrane due to altered expression of cell–matrix adhesion molecules; (5) increased expression of metalloproteinases.
Melanoma depends on, interacts with and reacts to its stroma, including extracellular matrix, growth factors, cytokines, fibroblasts, endothelial cells, and immune cells. In turn, melanoma is known to produce factors that influence its environment, and may force it to alter cell–cell communication.
In this chapter, we describe the alterations in cell–cell contacts in melanoma and the tumor microenvironment associated with melanoma development and progression.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
9.1 Melanoma Microenvironment
This is an update of our previous reviews on this topic (Haass et al. 2004, 2005; Kuphal and Haass 2011; Brandner and Haass 2013).
The state of a cell – quiescence, proliferation, differentiation or cell death – is under normal conditions determined by homeostasis (Bissell and Radisky 2001). A symbiotic relationship between a melanocyte and approximately 36 associated keratinocytes, which forms the epidermal melanin unit, maintains this homeostatic balance of the human epidermis (Fitzpatrick and Breathnach 1963; Jimbow et al. 1976). Within the stratum basale, the melanocytes keep a lifelong stable ratio of 1:5 with the keratinocytes (Fitzpatrick et al. 1979). This balance is maintained through regulated induction of melanocyte division coordinated through intercellular communication, which can be endocrine and paracrine via soluble factors and/or by direct contact via cell–cell and cell–matrix adhesion, or gap junctional intercellular communication (GJIC) (Haass et al. 2004, 2005). Dysregulation of this homeostasis may cause an imbalance of the epidermal melanin unit and trigger uncontrolled proliferation of the melanocytes, which may lead to the development of a nevus and/or a melanoma (Haass and Herlyn 2005).
Alterations in the interaction between neoplastic cells and their immediate microenvironment play a key role in these processes (Hanahan and Weinberg 2000, 2011; Park et al. 2000). The tumor microenvironment includes (1) the tumor stroma composed of fibroblasts, endothelial cells, immune cells, soluble molecules, and the extracellular matrix (ECM); (2) the tissue where the tumor had originated from; and (3) different sub-compartments within the tumor itself. Signals to and from the stroma via cell–cell and cell–matrix contact and/or via secretion of cytokines and growth factors may lead to a remodeling of the tumor microenvironment and consequently to promotion of melanoma development, growth, and metastasis by inducing angiogenesis, invasion, and migration (Villanueva and Herlyn 2008; Zigler et al. 2011). In addition to the interaction with the tumor stroma, primary melanoma progression as well as cutaneous melanoma metastases impact on the epidermal tumor microenvironment: the multilayered epithelium of the skin (Haass et al. 2010). Finally, different microenvironmental conditions within the tumor itself are created by differential access to nutrients and oxygen (Groebe and Mueller-Klieser 1991; Minchinton and Tannock 2006; Santiago-Walker et al. 2009; Haass et al. 2014; Haass 2015).
The microenvironment is not only important for the primary tumor, but also for colonization of a secondary organ. The “seed and soil” hypothesis implies that the metastatic process depends on the tumorigenic capacity of the cells and – again – on their interactions with the microenvironment (Fidler 2003).
9.2 Adherent Junction of Cadherins
Cross-talk between benign precursor cells, malignant cells, and surrounding host cells influences tumor development. Already in 1914, Theodor Boveri recognized the importance of changes in tumor cell adhesion for the development of cancer (Boveri 1914). Among the molecules involved in this intercellular communication are cadherins, which play a critical role for the homeostasis of normal skin and also during tumor formation and progression (Fig. 9.1). The identification of cadherins in the late 1970s and early 1980s was primarily motivated by an interest in understanding the mechanisms of cell adhesion during development (Franke 2009).

Overview of the cadherin repertoire in skin and melanoma (Illustration R.J. Bauer)
Cell–cell as well as cell–matrix adhesions are critical for cells and tissues to respond to mechanical stimuli from their environment. Both cell–cell and cell–matrix adhesions bear intrinsic mechanosensitivity, which allows them to promptly respond to stress and effectively propagate signals controlling cell shape and motility. This mechanosensitive response has been associated with pronounced changes in the size and molecular composition of specific adhesion sites and, consequently, the signals evoked by those adhesion sites. In polarized epithelia of vertebrates, the adherent junction is part of the tripartite junctional complex localized at the juxtaluminal region, which compromises the tight junction (TJ, see below), adherent junction (AJ), and desmosomes (macula adherens).
More than 80 proteins belong to the cadherin superfamily and are separated into the following “adherent junction” (AJ) subgroups in vertebrates:
-
1.
Classical adhesive cadherins of type 1 (6 members) and type 2 (13 members), e.g., E-, N-, P-, R-, and VE-cadherin. The classical cadherin family comprises 19 members that share a common domain organization of five repetitive extracellular calcium-binding subdomains (Overduin et al. 1995). Most of these classical cell–cell adhesion molecules are connected to the actin filaments and microtubules of the cellular cytoskeleton via catenins. The four known catenins, alpha-, beta-, gamma (plakoglobin)-, and delta (p120)-catenin, are important regulatory elements either for sustained cell–cell adhesion or signaling cascades into the cell.
-
2.
The “nonclassical” desmosomal cadherins, transmembrane proteins of desmosomes are, for example, desmocollin 1–3 (Dsc 1–3) and desmoglein 1–4 (Dsg 1–4). They are connected to intermediate filaments.
-
3.
Finally, there are nonclassical cadherins, like the protocadherins (e.g., protocadherin 15, cadherin 23), H-cadherin, and cadherin-like molecules (e.g., Fat, Dachsous, Flamingo, or Ret) belonging to the cadherin superfamily.
The most important classical cell–cell adhesion molecules of the skin and during melanoma development are E (epithelial)-cadherin (CDH-1), N (neuronal)-cadherin (CDH-2), and P (placental)-cadherin (CDH-3), which belong to the group of calcium-dependent glycoproteins. Certainly, this group of classical adhesion molecules can be extended with atypical VE (vascular endothelial)-cadherin (CDH-5, CD144) and the nonclassical cadherin H (heart)-cadherin (T-cadherin, CDH-13) (Fig. 9.1). In normal epidermis, melanocytes and keratinocytes are mostly connected via E-cadherin, P-cadherin, and H-cadherin (Kuphal et al. 2009; Nishimura et al. 1999; Tang et al. 1994). Whereas melanocytes in the basal layer of the epidermis seem to contain predominantly E-cadherin and H-cadherin, those residing in hair follicles are rich in P-cadherin (Nishimura et al. 1999). In contrast, N-cadherin is expressed on fibroblasts and vascular endothelial cells of normal skin (Hsu et al. 1996).
9.2.1 Loss of E-Cadherin in Tumorigenesis
E-cadherin is the major cadherin in polarized epithelial cells. Furthermore, the crosstalk between melanocytes and keratinocytes mediated by E-cadherin plays an important role in human epidermis. The normal melanocytic phenotype and controlled proliferation of melanocytes are strictly regulated by keratinocytes via E-cadherin. The E-cadherin knockout mouse is lethal in early embryonic stages (Larue et al. 1994) supporting the finding that E-cadherin has an essential role in morpho- and organogenesis. In skin development, there is evidence that E- and P-cadherin play some role in guiding melanocyte precursor cells to their final destination in the epidermis (Nishimura et al. 1999).
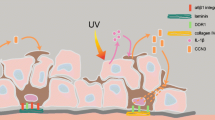
Malignant transformation of melanocytes is frequently attended by loss of E-cadherin expression and induction of N-cadherin (Hsu et al. 1996). This leads to the loss of the regulatory dominance of keratinocytes over melanocytes. The degenerated melanocytes/melanoma cells express N-cadherin to get into contact to fibroblasts and vascular endothelial cells during migration and invasion into the tumor stroma, dermis, lymph, and blood vessels (Hsu et al. 2000) (Fig. 9.2). The switch of the cadherin class is an interesting phenomenon of melanoma cells and in epithelial–mesenchymal transition (EMT) in general.

(a) Cell–cell adhesion of melanocytes and melanoma cells. Melanocytes adhere to keratinocytes via E-cadherin and desmoglein, which enables them to communicate with each other through gap junctions with cells in their environment. (b) In melanoma cells, E-cadherin is downregulated. They interact with each other through N-cadherin, Mel-CAM/Mel-CAM ligand, αvβ3 integrin/L1-CAM, ALCAM, and connexins; with fibroblasts through N-cadherin and connexins; and with endothelial cells through N-cadherin, Mel-CAM/Mel-CAM ligand, αvβ3 integrin/L1-CAM, α4β1 integrin/VCAM-1, and connexins
However, immunohistochemical examination of primary melanomas and their metastases has revealed that a proportion of melanoma cells are still E-cadherin-positive and present little, if any, N-cadherin (Danen et al. 1996; Hsu et al. 1996; Sanders et al. 1999; Silye et al. 1998). Therefore, the cadherin switch as an obligatory prerequisite of malignant behavior is still controversial and might depend on the subtype of the melanoma examined. However, immunohistochemistry data could not show whether the expressed E-cadherin is really functionally active regarding adhesion or still possesses signaling function. The general consensus is that E-cadherin is a tumor invasion suppressor.
9.2.1.1 Regulators of E-Cadherin
The mechanism by which E-cadherin expression is lost during malignancy differs between tumor entities. Loss of E-cadherin function can be caused by various genetic or epigenetic mechanisms. In patients with diffuse gastric cancer and breast cancer, the E-cadherin gene is mutated, leading to the expression of a nonfunctional protein (Strathdee 2002). The consequence is abnormal expression and abnormal subcellular localization of cadherin or the components of the cadherin-containing adhesion complex. Further, the CDH-1 gene locus can be epigenetically silenced by hypermethylation, leading to downregulation of E-cadherin expression which is known from several cancer entities, e.g., hepatocellular carcinoma (Kanai et al. 1997), squamous cell carcinoma (Saito et al. 1998), and thyroid cancer (Graff et al. 1998).
In most cases, E-cadherin expression is downregulated at the transcriptional level. The zinc-finger containing transcriptional repressor Snail1, which is a master regulator of neural crest cell specification and melanocyte migration during development in vertebrates, is mainly responsible for the loss of E-cadherin in melanoma (Batlle et al. 2000; Poser et al. 2001). The level of Snail1 expression correlates directly with the loss of E-cadherin expression, and forces overexpression of Snail in primary melanocytes downregulates E-cadherin expression (Poser et al. 2001). Slug (Hajra et al. 2002; Bolós et al. 2003), Snail2, ZEB1 and ZEB2 (Eger et al. 2005; Caramel et al. 2013), and SIP1 (Comijn et al. 2001), also members of the zinc finger transcription factor family of Snail, are further regulators of E-cadherin in melanoma, as well as basic helix–loop–helix transcription factors E12/47 (Perez-Moreno et al. 2001) and Twist (Yang et al. 2004). Additionally, the T-box transcription factor Tbx3 is overexpressed in melanoma, which enhances melanoma invasiveness through prevention of E-cadherin expression (Rodriguez et al. 2008). Furthermore, within human melanoma lesions, GLI-2, a mediator of hedgehog signaling, is associated with loss of E-cadherin (Alexaki et al. 2010).
Proteolytic degradation of E-cadherin by matrix metalloproteinases (MMPs) is another mechanism by which E-cadherin-mediated cell–cell adhesion can be ablated. In this case, cell surface E-cadherin becomes soluble by cleavage of the extracellular domain, a process known as ectodomain shedding. For melanoma, Adam-10 is responsible for E-cadherin shedding (Billion et al. 2006) (see also Chap. 8).
A family of microRNAs, such as miR-200a, miR-200b, miR-200c, and miR-205 was reported to control the expression level of E-cadherin during the epithelial–mesenchymal transition. The microRNA targets the transcriptional repressors ZEB1 and ZEB2 of E-cadherin (Gregory et al. 2008; Hurteau et al. 2007). As one example for cancer, loss of miR-200c expression is significantly correlated with early stage T1 bladder tumor progression (Wiklund et al. 2011). Another miRNA, miR-373, induces expression of genes with complementary promoter sequences. It was found that miR-373 induces E-cadherin expression by recognizing a target site in the promoter of the cdh-1 gene (Place et al. 2008). Liu et al. 2012 showed that miR-9 is downregulated in metastatic melanomas compared with primary melanomas. A tumor suppressor effect after re-expression of miR-9 in melanoma is mediated through its direct binding to sites within the NF-kB 3′-UTR, resulting in suppression of Snail1 and upregulation of E-cadherin. However, whether microRNAs are responsible for regulating cadherins directly and specifically in melanoma is still not known (see also Chap. 6).
9.2.2 Loss of P-Cadherin During Tumorigenesis
In human skin, P-cadherin is expressed mainly on cells of the epidermal basal layer (Furukawa et al. 1997) and those melanocytes residing in hair follicles (Nishimura et al. 1999). Concerning carcinogenesis, the effective role of P-cadherin remains an object of debate, since it can behave differently depending on the molecular context and tumor cell model studied. In melanoma cells, loss of full-length P-cadherin was reported (Bachmann et al. 2005; Van Marck et al. 2005; Jacobs et al. 2011). Therefore, P-cadherin has a similar tumor-suppressive behavior to E-cadherin. Additionally, a truncated 50 kDa form of the N-terminal part of P-cadherin was found, which appeared to be secreted from the melanoma cells. If this secreted form of P-cadherin is expressed from melanoma cells, it is responsible for cell migration and invasion (Bauer et al. 2005, 2006; Bauer and Bosserhoff 2006).
9.2.3 Loss of T-Cadherin During Tumorigenesis
T-cadherin (truncated-cadherin, cadherin 13, gene name CDH13) or H-cadherin, named for its strong expression in the heart, is an atypical member of the cadherin family, lacking the classical HisAlaVal recognition motif at its N-terminus, lacking the typical transmembrane and cytosolic domains and possessing a glycosylphosphatidylinositol moiety that anchors T-cadherin into the outer plasma membrane.
Immunohistochemistry of melanoma tissue samples showed positive T-cadherin staining of the endothelial cells. T-cadherin expression in endothelial cells was demonstrated to be redox sensitive (Joshi et al. 2008). The melanoma cells themselves showed loss of T-cadherin whereas healthy skin showed staining of melanocytes and keratinocytes of the basal layer of the epidermis. Loss of T-cadherin in melanoma is associated with migration and invasion of the cells (Kuphal et al. 2009). In general, the exact functional role and signaling of T-cadherin for melanoma cells itself and for the intratumoral angiogenesis are not clarified, so far. It was only shown that loss of T-cadherin in melanoma regulates AKT signaling and desensitizes for apoptosis (Bosserhoff et al. 2014). Also, a connection of loss of T-cadherin to tumor progression was speculated (Rubina et al. 2013) but not evidenced, until today.
9.2.4 N-Cadherin Expression During Tumorigenesis
N-cadherin plays a pivotal role in cell adhesion between melanoma cells and both dermal fibroblasts and vascular endothelial cells. During the cadherin class switch, loss of E-cadherin expression is accompanied by induced N-cadherin expression, which confers new adhesive properties on the cells (Fig. 9.2). The shift in cadherin profile during melanoma progression has been found not only in vitro but also in vivo (Hsu et al. 1996; Sanders et al. 1999). Experimentally, melanoma cell migration across fibroblasts is impaired upon addition of an N-cadherin neutralizing antibody (Li et al. 2001). The functional relevance of N-cadherin is to conduct migration and invasion of melanoma cells whereas N-cadherin expression correlates with progression to advanced-stage melanoma. The cell adhesion molecule N-cadherin has been suggested to represent a melanoma progression marker (Watson-Hurst and Becker 2006).
The switch of the cadherin class from E-cadherin to N-cadherin is directly connected. The transcriptional repressor Snail not only regulates E-cadherin repression but also represses the expression of the deubiquitinating enzyme CYLD. Loss of CYLD expression in melanoma in turn led to ubiquitination of Bcl-3 which is a transcriptional regulator of N-cadherin expression (Massoumi et al. 2009).
9.2.5 VE-Cadherin Expression During Tumorigenesis
The term vasculogenic mimicry describes the formation of vascular-like tubular structures and patterned networks through the connection of melanoma cells. The vascular structures are essential for the supply of the tumor. Several key molecules are responsible for the formation and maintenance of the tubular networks and these molecules are also often essential in normal blood vessels. One molecule expressed during vasculogenic mimicry of melanoma cells is VE-cadherin, previously considered to be endothelial cell specific. Analyzing VE-cadherin in detail demonstrated an interaction with EphrinA2 (EphA2), a tyrosine kinase. VE-cadherin engages the membrane-bound ligand of EphA2 and becomes phosphorylated on its tyrosines at the cytoplasmic domain. The mutual impact of VE-cadherin and EphA2 results in loosening of cell–cell adhesion and allowing for an increase in cell migration, invasion, and vasculogenic mimicry. Further studies describe the role of VE-cadherin for melanoma transendothelial migration. Here, p38 MAP kinase is necessary for increased VE-cadherin-mediated junction disassembly important for the migration processes of melanoma cells (Hendrix et al. 2001, 2003; Khanna et al. 2010).
9.2.6 FAT Expression During Tumorigenesis
FAT1, FAT2, FAT3, and FAT4 are human homologs of Drosophila Fat, which is involved in tumor suppression and planar cell polarity (PCP). FAT molecules belong to the cadherin-like protein family. FAT1 and FAT4 undergo the first proteolytic cleavage by Furin and are predicted to undergo the second cleavage by γ-secretase to release intracellular domain (ICD). Recently, it was shown using Northern blotting that human melanoma cell lines variably but universally express FAT1 and less commonly FAT2, FAT3, and FAT4. Both normal melanocytes and keratinocytes also express comparable FAT1 mRNA relative to melanoma cells. However, in melanoma cells, the non-cleaved proform of FAT1 is also expressed at the cell surface together with the furin-cleaved heterodimer. Moreover, furin-independent processing generates a potentially functional proteolytic product in melanoma cells, a persistent 65-kDa membrane-bound cytoplasmic fragment no longer in association with the extracellular fragment. In vitro localization studies of FAT1 showed that melanoma cells display high levels of cytosolic FAT1 protein. Such differences in protein distribution appear to reconcile with the different protein products generated by dual FAT1 processing. It was suggested that the uncleaved FAT1 could promote altered signaling, and the novel products of alternate processing provide a dominant negative function in melanoma (Sadeqzadeh et al. 2011). Among the human FAT gene family, FAT4 gene is recurrently mutated in several types of human cancers, such as melanoma (40 %), pancreatic cancer (8 %), HNSCC (6 %), and gastric cancer (5 %) (Nikolaev et al. 2011).
9.2.7 Signaling of Cadherins
In contrast to integrins, evidence for cadherin-induced outside–in signaling came into focus only slowly. Over the last 10 years, a number of studies have appeared to agree that signaling cascades emanating from cadherins play an important role in confluency-dependent growth arrest, migration, invasion, and differentiation. Changes in expression or function of cell adhesion molecules can therefore contribute to tumor progression both by altering the adhesion status and by affecting cell signaling. To date, no enzymatic activity has been attributed to the cytoplasmic tails of adhesion molecules like E-cadherin or N-cadherin. The signaling capability emanates from intracellularly bound kinases and phosphatases that link to the cytoplasmic tail of adhesion receptors (Fig. 9.3).

Schematic depiction of cadherin signaling in melanoma. The transcriptional repressor Snail inactivates E-cadherin expression in melanoma. With the loss of E-cadherin cytosolic beta-catenin activates the MAP kinase p38, which stimulates the transcriptional activity of NFkappaB. NFkappaB has N-cadherin as target gene. Additionally, Snail represses the expression of the tumor suppressor Cyld, which in turn leads to ubiquitination of Bcl-3 which also has N-cadherin as target gene. The overexpression of N-cadherin activates signaling cascades of SRC and PKB/Akt which leads to tumor progression (Illustration R.J. Bauer)
9.2.7.1 Signaling Cascades of E-Cadherin
Four modes of E-cadherin signaling are known:
-
1.
Modulation of receptor tyrosinase signaling (RTK) (see also Chap. 7)
-
2.
Inhibition of the Wnt signaling pathway (see also Chap. 7)
-
3.
Regulation of cytoplasmic β-catenin signaling
-
4.
Regulation of signaling through Rho GTPases
One way by which E-cadherin transmits growth-inhibiting outside–in signals appears to follow a strikingly similar scheme to that of the integrins. By using an immortalized nontumorigenic keratinocyte cell line, HaCaT, as a model system, Pece and Gutkind (2000) provide evidence that the assembly of calcium-dependent adherens junctions leads to a rapid and remarkable increase in the state of activation of MAPK and that this event is mediated by E-cadherin. Furthermore, it was found in these studies about HaCaTs that E-cadherin stimulates the MAPK pathway through ligand-independent activation of receptor tyrosine kinases, in particular EGF-receptors (Pece and Gutkind 2000). They speculated that upon adherens junction formation, signals emanating as a result of the E-cadherin-EGFR interaction might be involved in maintaining the functional and structural integrity of quiescent epithelia and, as a function of the adhesion status of the cells, possibly in promoting epithelial cell differentiation rather than proliferation. In contrast, another group detected signaling cascade inhibition through EGF-receptor/E-cadherin complex formation in melanoma and breast cancer cells (Qian et al. 2004). Unfortunately, most of the literature on E-cadherin signaling does not cover melanoma. Studies on keratinocytes and other cancer cell types revealed that the E-cadherin complex associates and cooperates with an EGF-receptor family member to activate the PI3K/Akt pathway in a Src-family kinase-dependent manner (Muller et al. 2008; Perrais et al. 2007) (see also Chap. 7).
Some studies showed that homophilic ligation of E-cadherin signals directly through Rho GTPase activity (Braga 2000; Braga et al. 1997). Loss of E-cadherin in melanoma may involve changes in the organization of the cytoskeleton which is exerted by members of the Rho family. They control not only the cytoskeletal organization but also cell motility, migration, and tumor progression to malignancy at the same time. As example, E-cadherin suppresses RhoA activity in melanoma by activating p190RhoGAP (Molina-Ortiz et al. 2009). E-cadherin overexpression led to association of p190RhoGAP and p120ctn on the plasma membrane where E-cadherin bounds p120ctn. Recently, it was shown that E-cadherin also regulates RhoC GTPase. Here, loss of E-cadherin activates the expression of the RhoC in melanoma through upregulation of the transcription factor ETS-1, which results in increased c-Jun protein stabilization and activation (Spangler et al. 2012).
In addition to its role in adhesion, nuclear β-catenin is involved in Wnt signal transduction, and it interacts with transcription factors of the leukocyte enhancer factor (LEF)/T-cell factor (TCF) family to regulate transcription of target genes implicated in cell growth control such as cyclin D1 and c-myc (van Noort and Clevers 2002). By sequestering β-catenin at the cell surface, E-cadherin has been shown to antagonize nuclear β-catenin signaling pathways and to induce growth inhibition (Gottardi et al. 2001; Shtutman et al. 1999). Furthermore, β-catenin bound to E-cadherin inhibits phosphorylation of p38 and prevents activation of NFkappaB. Unbound cytoplasmic β-catenin activates the signaling pathway ending at transcriptional activation of N-cadherin expression in melanoma cells (Kuphal et al. 2004). In general, it was shown by Onder et al. (2008) that loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. The publication presents ~84 of 617 genes differentially expressed in shE-cadherin human breast epithelial cells (HMLE). They presented, e.g., twist and TCF-8 among other 19 transcription factors as upregulated after loss of E-cadherin.
9.2.7.2 Signaling Cascades of N-Cadherin
N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells through activation of cytoplasmic signaling cascades. The Src family kinases are involved in the regulation of N-cadherin-mediated cell adhesion and signaling during, e.g., melanoma cell transendothelial migration. Src is localized at the heterotypic contacts of N-cadherin and becomes activated when melanoma cells are transmigrating across the endothelium. Activated Src has the Tyrosine-860 at the cytoplasmic domain of N-cadherin as target site for phosphorylation. The phosphorylation leads to disruption of β-catenin binding followed by nuclear translocation of this molecule to activate gene transcription of genes responsible for proliferation (Qi et al. 2006). N-cadherin mediates cell adhesion-activated antiapoptotic protein Akt/PKB and subsequently increases β-catenin and inactivates proapoptotic factor Bad (Li et al. 2001).
9.2.8 Desmosomes/Hemidesmosomes
Desmosomes, composed of desmogleins and desmocollins, are localized spot-like adhesions randomly arranged on the lateral sides of plasma membranes and are also members of the cadherin family. The extracellular domain of the desmosome is called the extracellular core domain (ECD) or the Desmoglea, and is bisected by an electron-dense midline where the desmoglein and desmocollin proteins bind to each other. On the cytoplasmic side of the plasma membrane, there are two dense structures called the outer dense plaque (ODP) and the inner dense plaque (IDP). In the ODP, the cytoplasmic domains of the cadherins desmoglein and desmocollin attach to desmoplakin via plakoglobin and plakophilin, while in the IDP, desmoplakin attaches to the intermediate filaments such as keratine filaments.
A number of melanoma cell lines synthesize, in the absence of desmosomes, the desmosomal cadherin desmoglein 2 (Dsg2) as a frequent plasma membrane glycoprotein that is not assembled into any junction but is dispersed over large parts of the cell surface. Indeed, in tissue microarrays, Dsg2 has been demonstrated in a sizable subset of nevi and primary melanomas (Rickelt et al. 2008). In contrast, Dsg1, Dsg3, and desmocollins 1–3, were absent in the analyzed melanoma cell lines but plakoglobin and plakophilin3 were also expressed in several melanoma cell lines (Schmitt et al. 2007). Future studies will have to clarify the diagnostic and prognostic significance of these different adhesion protein subtypes.
9.3 Integrins
Integrins are transmembrane adhesion receptors localized at cell–matrix contact sites where they link ECM (extracellular matrix) components, e.g., vitronectin, fibronectin, laminin, osteopontin, or collagen, to the actin cytoskeleton and interact with multiple structural and signaling molecules including talin, kindlin, paxillin, vinculin, α-actinin, FAK (focal adhesion kinase), ILK (integrin-linked kinase), Rho GTPases, and SHC (Berrier and Yamada 2007; Papusheva and Heisenberg 2010). The latter are important mediators downstream of integrins by which they interact either directly or indirectly to effect adhesion-dependent responses (Playford and Schaller 2004). The metastatic transformation of melanocytes is associated with altered expression of integrins, which transduce signals upon ligation to ECM proteins that regulate tumor growth and metastasis, apoptosis, differentiation as well as tumor angiogenesis. Integrin receptors are functional dimers of α- and β-integrin subunits, which each have a large ectodomain, a single transmembrane domain, and a generally short cytoplasmic tail (except for β4 integrin). The combination of different α- and β-subunits determines the substrate specificity of the dimer (Danen and Sonnenberg 2003). There are at least 18 known α-chains and 8 β-chains, allowing for at least 24 unique heterodimers.
The pattern of integrins on the cell surface is usually very specific, which makes the cell fit perfectly into its surrounding environment. Importantly, integrin expression patterns differ considerably in vitro versus in vivo. Thus, in vitro studies may not translate into the in vivo situation.
Several publications have shown that the expression levels mainly of αvβ3, α2β1, α3β1, α4β1, and α5β1 appear to increase from primary melanomas to metastatic melanoma tissue sections, whereas there was a significant decrease in α1β1, α2β1, and α6β1 expression levels in metastatic melanoma compared to primary melanoma (Friedl et al. 1998; Natali et al. 1993; Schadendorf et al. 1993). Although many integrins have been implicated in mediating melanoma growth and metastasis, perhaps none have been studied as much as the vitronectin receptor, αvβ3 (Danen et al. 1995; Mortarini and Anichini 1993; Seftor et al. 1999). αvβ3 integrin adheres to vitronectin, fibronectin, laminin, collagen, and osteopontin. Binding fibronectin and vitronectin induces the expression of MMP-2, which is able to degrade the collagen of the basement membrane (Felding-Habermann et al. 2002). Furthermore, osteopontin’s RGD-sequence (Arg–Gly–Asp) has high binding affinity and specificity to αvβ3. As the aggressiveness of melanoma has been associated with high osteopontin expression (Sieg et al. 2000), this interaction of αvβ3 and osteopontin is important for melanoma progression. Interaction between αvβ3 and extracellular matrix molecules serves to promote cell attachment, spreading, and migration. αvβ3 integrin also undergoes heterophilic binding with two members of the immunoglobulin superfamily of cell adhesion molecules, PECAM-1 and L1. The αv subunit is widely expressed on melanomas regardless of disease stage. This stands in contrast to the β3 subunit, which is predominantly expressed on melanoma cells in the vertical growth phase. The onset of β3 integrin expression is one of the most specific markers of the transition from radial growth phase to vertical growth phase of melanoma (Albelda et al. 1990; Danen et al. 1995; Natali et al. 1997). Although many studies on human melanoma cell lines have correlated αvβ3 integrin expression with progression and metastasis, in vivo studies are less clear.
9.3.1 Integrin Signaling in Melanoma
Apart from being involved in the attachment of cells to the ECM, integrins are also responsible for signaling between the cells and the environment. Signaling works bidirectionally: “outside–in signaling” can control behavior, proliferation, cell polarity, cell growth, and migration. “Inside–out signaling,” on the other hand, changes the integrins from a passive, weak binding state into an active, adhesive state and alters the interaction of the receptors with the extracellular environment. Integrins are receptors for cell movement in response to binding to ECM of the basement membrane or connective tissue or plasma membrane receptors expressed on endothelial cell surfaces. Additionally, integrins bind cytoplasmic adaptor proteins of the actin-myosin filaments and create a plasticity that allows the cell to move. In summary, integrins are bivalent linker proteins, binding simultaneously to extracellular ligands as well as cytoplasmic proteins including intracellular signaling molecules. They influence, for example, tyrosine kinases, serine/threonine kinases, phosphoinositides, and signaling cascades which determine the fate of a cell, letting it grow, proliferate, or die whenever it is necessary in the context of the whole organism. This paragraph introduces some of the most important and best studied proteins which are known to interact with integrins in melanoma.
There is the non-receptor protein tyrosinase kinase FAK (focal adhesion kinase) (Fig. 9.4) that co-localizes with integrins in focal adhesions. FAK becomes phosphorylated and then controls processes like cell spreading, proliferation, motility, vasculogenic mimicry, and survival (Schaller 2001). Proteins like c-SRC, SHC, CSK, PI3K, and GRB2 are known to interact with FAK to transfer the signaling into the cytoplasm and to link FAK signaling also to MAP kinases (Chakraborty et al. 2002) (see also Chap. 7). FAK expression seems to be required in melanoma cells for substrate adhesion. It has been shown that in melanoma FAK is constitutively active and that it is essential for maintaining adhesiveness in melanoma cells (Hamamura et al. 2008; Kahana et al. 2002).

Schematic depiction of the signaling pathways leading from integrins to focal adhesion kinase (FAK) and integrin-linked kinase (ILK), respectively, and further reactions of the cell (Illustration R.J. Bauer)
Furthermore, the integrin-linked kinase (ILK), a serine/threonine kinase, is implicated in connecting cell–extracellular matrix interaction and growth factor signaling to cell survival, cell migration, invasion, anchorage-independent growth, angiogenesis, and epithelial–mesenchymal transition. It has been shown that strong ILK expression was significantly associated with melanoma thickness, migration, and invasion (Wong et al. 2007). Increased expression of integrin-linked kinase is correlated with melanoma progression and poor patient survival (Dai et al. 2003). ILK directly phosphorylates PKB/Akt and glycogen synthase kinase-3 (GSK-3beta), which is inactivated upon phosphorylation (Delcommenne et al. 1998; Troussard et al. 1999). SHC is another protein which is implicated in integrin signaling. It is an adaptor protein capable of binding phosphotyrosine-containing sequences. So far, studies have demonstrated that SHC signaling is involved in pathways, which play a role in the development of malignancies like c-Myc activation (Gotoh et al. 1997), survival signaling (Friedmann et al. 1996; Sakai et al. 2000), cytoskeletal organization, and mitogenic signaling through RAS. It has been proposed that SHC is a substrate for FAK.
Also, the ERK/MAP kinase cascade is a pathway in which integrin-mediated adhesion is involved. In the ERK pathway, various stimuli of many important integrin signaling molecules like FAK or SHC converge and are able to influence nearly every profound cellular activity (Meier et al. 2005).
Epidermal growth factor receptor (EGFR) is also activated by integrins to generate cellular responses such as adhesion-dependent cell survival and proliferation in response to ECM. Subsequently, integrin-mediated EGFR activation induces ERK/MAP kinase signaling (Howe et al. 2002; Jost et al. 2001). Furthermore, Caveolin-1 (CAV1) is the main structural component of caveolae, which are plasma membrane invaginations that participate in vesicular trafficking and signal transduction events. Following integrin activation, B16F10 cells expressing CAV1 display reduced expression levels and activity of FAK and Src proteins. Furthermore, CAV1 expression markedly reduces the expression of integrin β3 in B16F10 melanoma cells. These findings provide experimental evidence that CAV1 may function as an antimetastatic gene in malignant melanoma (Trimmer et al. 2010).
9.4 Immunoglobulin Gene Superfamily of Cell Adhesion Molecules (CAMs)
Whereas normal melanocytes express few cell–cell adhesion receptors of the immunoglobulin gene superfamily of cell adhesion molecules (CAMs), melanoma cells show an increase in expression of melanoma cell adhesion molecule (MCAM, Mel-CAM, MUC18, CD146), L1 cell adhesion molecule (L1-CAM, CD171), activated leukocyte cell adhesion molecule (ALCAM, CD166), vascular cell adhesion molecule 1 (VCAM-1, CD106), intercellular cell adhesion molecule 1 (ICAM-1, CD54), and carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM-1, CD66a) (reviewed in Haass et al. 2005).
9.4.1 Melanoma Cell Adhesion Molecule (MCAM, Mel-CAM, MUC18, CD146)
Mel-CAM mediates homologous and heterologous interactions between melanoma cells and endothelial cells, respectively, via a heterophilic Ca2+-independent adhesion to its ligand (Shih et al. 1997a, b; Johnson et al. 1997). Recently, Laminin-411 (α4β1γ1 integrin) and Galectin-1 have been identified as Mel-CAM ligands (Flanagan et al. 2012; Jouve et al. 2013; Yazawa et al. 2015). In melanocytic cells, expression of Mel-CAM is first found in nevi, when the cells have separated from the epidermal keratinocytes and have migrated into the dermis (Shih et al. 1994; Kraus et al. 1997). With progression to malignancy, Mel-CAM expression gradually increases and is highest in metastatic melanoma cells (Xie et al. 1997; Johnson et al. 1996; Shih et al. 1994; Lehmann et al. 1987, 1989). In vitro and in vivo data supporting an important role of Mel-CAM in melanoma progression was demonstrated in several experimental studies (reviewed in Haass et al. 2005; Lei et al. 2015). Recently, the zinc finger transcription factor ZBTB7A was found to repress melanoma metastasis by directly binding to the promoter and transcriptionally repressing Mel-CAM (Liu et al. 2015).
An evaluation of tissue arrays of primary and metastatic melanomas revealed that in patients meeting the current criteria for sentinel lymph node dissection, both Mel-CAM expression positivity and intensity were independently predictive of survival and development of lymph node disease in primary melanoma over and above established markers of prognosis, such as Breslow thickness. Mel-CAM-negative patients had a 5-year survival of 92 % compared with 40 % for Mel-CAM-positive patients (Pearl et al. 2008). Recently, a study on 175 patients revealed that sequential molecular detection of Mel-CAM mRNA in the peripheral blood correlated with poor prognosis. The authors suggested to utilize Mel-CAM expression as a “molecular warning of progression” even in early stage patients in otherwise disease-free conditions (Rapanotti et al. 2014). However, larger trials to confirm this finding as a biomarker are still pending.
9.4.2 L1-Cell Adhesion Molecule (L1-CAM, CD171)
L1-CAM, originally described as a neuronal cell adhesion molecule, has also been detected in a number of other non-neuroendocrine tissues and in several malignant tumors, including melanoma (Nolte et al. 1999; Thies et al. 2002b). L1-CAM mediates adhesion both via homophilic (L1-CAM-L1-CAM) and heterophilic (L1-CAM-αvβ3 integrin) mechanisms (Hortsch 1996). In melanoma/melanoma cell and in melanoma/endothelial cell interactions, L1-CAM binds to αvβ3 integrin (Montgomery et al. 1996). The interaction of L1-CAM and αvβ3 integrin plays an important role in transendothelial migration of melanoma cells (Voura et al. 2001) whereas overexpression of L1-CAM promotes conversion from radial to vertical growth phase melanoma without upregulation of αvβ3 integrin expression (Meier et al. 2006). There is an increase in L1-CAM immunoreactivity in melanomas and metastases of melanoma compared to acquired melanocytic nevi (Fogel et al. 2003). A study that systematically identified novel melanoma-specific genes confirmed that L1-CAM is not expressed in normal skin and melanocytic nevi, but is highly and differentially expressed in primary melanoma tissues and melanoma lymph node metastases (Talantov et al. 2005). Evaluation of specimens of nevi, primary melanomas, sentinel lymph nodes, and distant metastases showed that L1-CAM can serve as a highly sensitive and specific diagnostic marker for melanoma (Thies et al. 2007). A 10-year retrospective biomarker study, evaluating 100 melanoma specimens, showed that the expression of L1-CAM in human primary cutaneous melanoma is significantly associated with metastatic spread and that L1-CAM expression is an independent predictor for the risk of metastasis (Thies et al. 2002b). A recent study revealed that the CE7 epitope of L1-CAM on a variety of tumors (however, melanoma was not included in the study) may be amenable to targeting by CE7R+ T cells, making it a promising target for adoptive immunotherapy (Hong et al. 2014).
9.4.3 Activated Leukocyte Cell Adhesion Molecule (ALCAM, CD166)
ALCAM is involved in homophilic (ALCAM-ALCAM) (Degen et al. 1998) and heterophilic (ALCAM-CD6) (Patel et al. 1995) cell–cell adhesion interactions. ALCAM is expressed in metastatic human melanoma cells, whereas it is absent in non-metastatic cells (Degen et al. 1998). Immunohistochemistry on a series of common nevi, primary melanomas, and melanoma metastases revealed that ALCAM expression correlates with melanoma progression (van Kempen et al. 2000). ALCAM is therefore proposed to be a molecular melanoma progression marker. Intact cell adhesion function of ALCAM favored primary tumor growth and represented a rate-limiting step for tissue invasion, which supported the view that dynamic control of ALCAM plays an important role in progression (van Kempen et al. 2004). An immunohistochemical biomarker study, evaluating tissue microarrays showed that a significantly greater percentage of melanomas (combined primary and metastatic) than nevi contained cells that expressed ALCAM (Klein et al. 2007). Interestingly, a recent study evaluating ALCAM expression and long-term survival in melanoma patients suggested that, in primary melanomas, high ALCAM expression was a marker of negative outcome, but in regional lymph node melanoma metastases low expression of ALCAM was a feature associated with unfavorable prognosis (Donizy et al. 2015). ALCAM upregulation in metastatic melanoma cells is driven by miR-214 and depends on transcriptional mechanisms mediated by TFAP2 and posttranscriptional mechanisms mediated by miR-148b, which itself is controlled by TFAP2. Therefore, miR-214 and miR-148b have opposite effects on melanoma cell dissemination and are part of a regulatory loop (Penna et al. 2013).
9.4.4 Intercellular Adhesion Molecule-1 (ICAM-1, CD54)
ICAM-1 can be induced in a cell-specific manner by several cytokines, e.g., TNF-α (tumor necrosis factor-alpha), IL-1 (interleukin-1), and IFN-γ (interferon-gamma). The ligands of ICAM-1 are αLβ2 (lymphocyte function-associated antigen 1, LFA-1) and Mac1 on lymphocytes (van de Stolpe and van der Saag 1996). ICAM-1 correlates with melanoma progression and increased risk of metastasis (Johnson et al. 1989). Its expression in melanoma is stronger than in common nevi and increases with the Breslow index in primary melanomas (Natali et al. 1990, 1997; Schadendorf et al. 1993, 1995). The observation that stage I patients with ICAM-1-positive melanomas had a significantly shorter disease-free interval and overall survival than those with ICAM-1-negative tumors (Natali et al. 1997) and that the suppression of ICAM-1 in an animal model reduced the metastatic capacity (Miele et al. 1994), supported the role of ICAM-1 in melanoma progression and metastasis. However, the specific role of ICAM-1 in melanoma progression remains to be determined. Expression of ICAM-1 may promote aggregate formation with leucocytes, which can enhance survival in the vascular system and encourage extravasation (Aeed et al. 1988). On the other hand, ICAM-1 is shed from melanoma cells (Giavazzi et al. 1992) – possibly in a form that inhibits lymphocyte–tumor cell interaction and thus contributes to tumor survival (Becker et al. 1993). A recent study has unraveled a mechanism by which shear flow-regulated melanoma cell adhesion to the endothelium can upregulate endothelial ICAM-1 expression (Zhang et al. 2014). Elevated ICAM-1 levels may serve as receptors to recruit neutrophils and bind fibrin, which assists melanoma cell adhesion and migration. An increase of ICAM-1 expression on endothelial cells could be a result of direct ligation of tumor CD44 and endothelial E-selectin, through the PKCα-p38-SP-1 pathway. This suggests a new mechano-signaling cascade triggered by stretching E-selectin to induce ICAM-1 expression (Zhang et al. 2014).
9.4.5 Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 (CEACAM-1, CD66a)
CEACAM1 is involved in intercellular adhesion and subsequent signal transduction events in a number of epithelia. In epithelial cells, CEACAM1 is believed to act as a growth suppressor, since its expression was shown to be lost or significantly down- or dysregulated in carcinomas of liver, prostate, endometrium, breast, and colon (reviewed in Haass et al. 2005). On the other hand, CEACAM1 is upregulated in non-small cell lung cancer (Sienel et al. 2003). CEACAM1 interacts with the β3 integrin subunit via the CEACAM1 cytoplasmic domain. CEACAM1 and the β3 integrin subunit co-localize at the tumor–stroma interface of invading melanoma masses, suggesting that CEACAM1–integrin β3 interaction plays a role in melanoma cell migration and invasion (Brummer et al. 2001). The expression of CEACAM1 in primary melanomas is associated with the subsequent development of metastatic disease (Thies et al. 2002a). Furthermore, the overexpression of CEACAM1 in CEACAM1-negative melanocytic cells and melanoma cell lines increases the migratory and invasive growth potentials in vitro (Ebrahimnejad et al. 2004) supporting the role of CEACAM1 in melanoma progression and metastasis. Evaluation of specimens of nevi, primary melanomas, sentinel lymph nodes, and distant metastases showed that CEACAM1 can serve as a highly sensitive and specific diagnostic marker for melanoma (Thies et al. 2007). Indeed, CEACAM1 was shown to be one of the seven plasma markers best able to identify metastatic melanoma patients (Kluger et al. 2011).
9.5 Gap Junctions/Connexins
Connexins belong to a family of transmembrane proteins that form gap junctions (GJs), cell–cell junctions that are essential for intercellular communication. Gap junctional intercellular communication (GJIC) in the skin is involved in maintenance of homeostasis, regulation of proliferation, differentiation, barrier function, and recruitment of inflammatory cells. GJIC is thus a critical factor in the life and death balance of cells (Djalilian et al. 2006; Langlois et al. 2007; Maass et al. 2004; Man et al. 2007) (reviewed in Kretz et al. 2004; Mese et al. 2007). Furthermore, GJIC is critical in keratinocyte–melanocyte interaction (Hsu et al. 2000; Satyamoorthy et al. 2001). Alternatively, connexins can form hemichannels, which allow release (e.g., ATP, NAD+) or putative uptake of molecules and ions to and from the cellular environment (Barr et al. 2013; Chandrasekhar and Bera 2012). Finally, connexins, especially Cx43, interact with structural and signaling molecules, which may add further functions to these molecules (Herve et al. 2007).
GJs form channels between adjacent cells allowing the intercellular transport of small metabolites, second messengers, and ions (Loewenstein 1981; Spray 1994). In addition to molecular weight and size, the ability of a solute to transverse these channels depends on its net charge, shape, and interactions with specific connexins that constitute gap junctions in particular cells (Goldberg et al. 2004). Each GJ channel consists of two hemichannels called connexons, each formed by six connexins (reviewed in Richard 2000). Twenty-one connexins have been identified, 11 of which are in the skin (Di et al. 2001; Willecke et al. 2002; Zucker et al. 2013). GJs can be homotypic, heterotypic, homomeric, and heteromeric (reviewed in Richard 2000). A connexon is homomeric if it is composed of six identical connexin subunits (e.g., Cx32 only), or heteromeric if it is composed of more than one connexin species (e.g., Cx32 and Cx43 and/or others). Channels are homotypic if both connexons are homomeric of the same type, heterotypic if homomeric connexons are of different types, and heteromeric if both connexons are heteromeric. Not all connexins are equally compatible at forming a connexon – even though they may co-exist in the same cell (reviewed in Haass et al. 2004). The type of connexin-forming GJ channels influences their selectivity and thereby controls the specificity of GJIC. For example, channels formed by Cx26 prefer cations, while those formed by Cx32 prefer anions (Brissette et al. 1994; Elfgang et al. 1995; Veenstra 1996). Thus, the up- or downregulation of a certain connexin in a tissue may change its GJIC considerably. In addition, connexins can also form hemichannels, which have been shown to be able to exchange molecules with the extracellular microenvironment. These hemichannels are relevant for signal propagation and especially for calcium homeostasis (reviewed in Evans et al. 2006).
9.5.1 Connexins Are Conditional Tumor Suppressors
Loss of gap junctional activity and/or downregulation of connexins have been reported both in cell lines as well as in tissues of many tumor types, such as hepatocellular carcinoma, gastric carcinoma, prostate cancer, lung cancer, glioma, mammary carcinoma, basal cell carcinoma, squamous cell carcinoma, and melanoma. This phenomenon was first observed half a century ago (Loewenstein and Kanno 1966) and summarized in a number of review articles (Cronier et al. 2009; Mesnil et al. 2005; Naus and Laird 2010). The type of connexins lost during tumor progression varies according to tumor type. In the 1980s and 1990s, a series of studies were published showing that reagents and/or oncogenes that promote tumor onset or progression frequently inhibit GJIC or downregulate connexin expression (Lampe 1994; Trosko et al. 1990; Atkinson et al. 1981). The role of connexins as potential tumor suppressors was also shown in gene knockdown studies (Shao et al. 2005). Correspondingly, ectopic expression of connexins in tumors restored functional communication and reduced tumor proliferation and growth both in vitro and in vivo (reviewed in Naus and Laird 2010). Importantly, ectopic expression of connexins partially differentiated transformed cells (Zhu et al. 1991; McLachlan et al. 2006; Hellmann et al. 1999; Hirschi et al. 1996). Moreover, functional abrogation of connexins, using antisense or dominant-negative mutant approaches, have demonstrated an enhancement of the malignant phenotype in several tumor types, such as Cx26 in HeLa cells (Duflot-Dancer et al. 1997), Cx32 in hepatocellular carcinoma (Dagli et al. 2004), Cx43 in lung cancer (Avanzo et al. 2004), Cx43 in glioma (Omori and Yamasaki 1998), and Cx43 in bladder carcinoma (Krutovskikh et al. 1998). Finally, Cx32 knock-out mice have an increased incidence of tumor onset when challenged with carcinogens (Temme et al. 1997; King and Lampe 2004a, b; Moennikes et al. 2000).
This may lead to the assumption that connexins are general tumor suppressors, but it appears that this is only the case in the earlier steps of cancerogenesis. In fact, the role of connexins in invasion and metastasis is very complex, and connexins might facilitate invasion, intravasation, extravasation, and metastasis (Krutovskikh et al. 1994; el-Sabban and Pauli 1991, 1994; Ito et al. 2000; Saunders et al. 2001; Lin et al. 2002; Miekus et al. 2005; Pollmann et al. 2005; Kanczuga-Koda et al. 2006; Bates et al. 2007; Li et al. 2007; Dobrowolski et al. 2008; Cotrina et al. 2008; Elzarrad et al. 2008; Ezumi et al. 2008). The following model supports both the tumor suppressor and the tumor driver theories (Cronier et al. 2009): for the step from primary to invasive tumors, there is a need for disruption of intercellular junctions including GJs, consistent with the model that connexins are tumor suppressors. In contrast, for the tumor cell dissemination and metastasis steps, increased cell contacts and communication are needed in order to enable interaction with the tumor stroma – especially between cancer cells and endothelial cells. Therefore, connexins might be better classified as conditional tumor suppressors that modulate cell proliferation as well as adhesion and migration (Naus and Laird 2010).
9.5.2 Cx43 in Cancer
Cx43 is decreased in prostate cancer (Tsai et al. 1996), mammary cancer (Hirschi et al. 1996), glioma (Huang et al. 1999), lung cancer (Jinn et al. 1998; Zhang et al. 1998), bladder carcinoma (Krutovskikh et al. 2000), cervical carcinoma (King et al. 2000), and various skin cancers including melanoma (Haass et al. 2006; Tada and Hashimoto 1997; Wilgenbus et al. 1992). Electron microscopy investigations have shown that basal and squamous cell carcinomas do not have fully developed GJs, and that Cx43 is not restricted to these poorly developed GJs but is present in the cytoplasm (Tada and Hashimoto 1997). In several cancers, Cx43 acts as a tumor suppressor gene with loss of Cx43 contributing to metastasis (Czyz 2008; Gershon et al. 2008; Shen et al. 2007). Functional abrogation of Cx43 enhances the malignant phenotype in lung cancer (Avanzo et al. 2004), glioma (Omori and Yamasaki 1998), and bladder carcinoma (Krutovskikh et al. 1998).
In contrast to other cancers, hepatocellular carcinoma is associated with an induction of Cx43, which is, however, localized in the cytoplasm, and thus is not involved in GJIC (Krutovskikh et al. 1994). The loss of GJIC might help the tumor cells to survive, as GJIC has been shown to spread cell-killing signals, most likely Ca2+ ions (Krutovskikh et al. 2002). In addition, downregulation of Cx43 expression or function resulted in increased proliferation and migration in primary keratinocytes, implying a contribution of Cx43 to controlling early stages of tumorigenesis (Mori et al. 2006; Wright et al. 2009; Pollok et al. 2011). Finally, increased opening of hemichannels formed by connexins resulted in cell death in cochlear supporting cells of the ear and in keratinocytes of the epidermis (Xu and Nicholson 2013).
Conversely, expression of Cx43 has also been shown to increase tumor metastasis in breast cancer, glioma as well as in melanoma through increased attachment and communication with the vascular endothelium (Bates et al. 2007; Kanczuga-Koda et al. 2006; Cotrina et al. 2008; Lin et al. 2002; el-Sabban and Pauli 1991, 1994; Pollmann et al. 2005; Elzarrad et al. 2008).
9.5.3 Cx32 in Cancer
Cx32 is downregulated in gastric carcinoma (Uchida et al. 1995), lung cancer (Jinn et al. 1998), and hepatocellular carcinoma (Eghbali et al. 1991; Loewenstein and Rose 1992; Krutovskikh et al. 1994; Yamaoka et al. 1995). In the latter case, the remaining Cx32 is localized in the cytoplasm or in the plasma membrane free from contact with other cells. In addition, it was found that there was no mutation in the coding sequence of Cx32 in hepatocellular carcinoma; instead, it appears that the aberrant localization of Cx32 is a consequence of the disruption of Cx32 gap junction plaque formation (Krutovskikh et al. 1994). Functional abrogation of Cx32 enhances the malignant phenotype in hepatocellular carcinoma (Dagli et al. 2004). Cx32 knock-out mice have an increased incidence of tumor onset when challenged with carcinogens (Temme et al. 1997; King and Lampe 2004a, b; Moennikes et al. 2000). In contrast to most other tumors, Cx32 is upregulated in some breast cancer cells (Saunders et al. 2001).
9.5.4 Cx26 in Cancer
Whereas in mammary carcinoma cells, there is a downregulation of both Cx43 and Cx26 (Hirschi et al. 1996); in human basal cell carcinoma, Cx43 is downregulated but there is an induction of Cx26 (Haass et al. 2006; Wilgenbus et al. 1992). Cx26 is also highly expressed in HeLa cells, where its functional abrogation enhances the malignant phenotype (Duflot-Dancer et al. 1997).
9.5.5 Connexins in Melanoma
Reflecting the situation in many other cancer types as discussed above, the role of connexins and GJIC is still highly controversial also in melanoma and its tumor microenvironment.
Cx43 is the most-studied connexin in melanoma. Western blotting revealed Cx43 protein expression in foreskin-derived melanocytes and several melanoma cell lines (Hsu et al. 2000). This was confirmed by immunofluorescence detecting Cx43 expression in human melanoma cell lines (Lin et al. 2010). While neither study quantified the Cx43 protein expression levels, a qRT-PCR and immunofluorescence study demonstrated lower Cx43 expression levels in human melanoma cell lines compared to human melanocytes (Schiffner et al. 2011). Also, a microarray study revealed that Cx43 was expressed at low levels in human melanoma cell lines and, importantly, that its overexpression suppressed anchorage-independent growth in colony-forming efficiency assays, suggesting a tumor-suppressor role of Cx43 in melanoma (Su et al. 2000). By qRT-PCR, no expression for Cx26, Cx30, Cx31.1, Cx36, and Cx37; low expression for Cx30.3 and Cx31; and higher expression levels for Cx32, Cx40, Cx43, and Cx45 were detected in human melanoma cell lines (Zucker et al. 2013). Surprisingly, Western blotting showed much higher Cx43 expression levels in migrating than in non-migrating cells (Zucker et al. 2013). Consistently, high levels of Cx43 protein expression were found in human metastatic melanoma cell lines (Villares et al. 2009). Loss of protease-activated receptor-1 (PAR-1) expression resulted in the loss of Cx43 and, correspondingly, overexpression of PAR-1 contributed to melanoma metastasis via upregulation of Cx43 (Villares et al. 2009, 2011). Interestingly, while initial levels of Cx43 were low in B16 mouse melanoma cells, Cx43 protein levels increased after infection with bacteria or treatment with interferon-γ (Saccheri et al. 2010). This was followed by the transfer of preprocessed antigenic peptides from melanoma cells to dendritic cells, which then presented those peptides on their surface and activated cytotoxic T cells against the tumor antigen. Correspondingly, melanoma cells in which Cx43 had been silenced, failed to elicit a cytotoxic antitumor response after infection with bacteria (Saccheri et al. 2010).
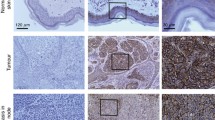
In addition to the discussed in vitro data, there are also a number of studies on human melanoma tissue. Using immunofluorescence on human tissue samples, we did not detect Cx43 (nor Cx26 and Cx30) in nevi, primary melanomas, or cutaneous melanoma metastases, while the internal controls (adjacent epidermis) were positive in the expected layers (Haass et al. 2006, 2010). In contrast, using immunohistochemistry, other groups reported Cx43 expression in human melanoma tissue, higher than in human nevi (Rezze et al. 2011; Sargen et al. 2013). However, neither of these studies provided high magnification images to confirm the subcellular localization nor did they show appropriate positive and negative controls. Indeed, in both studies, Cx43 expression in melanoma cells appeared to be cytoplasmic and hence would argue for a cell–cell or cell–matrix communication-independent role of these connexins. This would not support the mechanism for melanoma survival in brain metastasis proposed by Lin and colleagues, who showed that reactive astrocytes protect metastatic melanoma cells in the brain from chemotherapy by sequestering intracellular calcium through direct cell–cell communication (Lin et al. 2010). Moreover, in the Rezze and Sargen studies, the expression pattern of Cx43 in nevi and different melanoma stages appeared very variable and the typical Cx43 staining in the epidermis was missing (Rezze et al. 2011; Sargen et al. 2013). An Oncomine analysis of human tissue showed that increased Cx43 (and Cx26) gene expression in primary lesions correlated with metastasis and poor patient survival (Stoletov et al. 2013).
Cx26 and Cx30 are much less studied. Cx26 was found to be upregulated in the highly aggressive BL6 sub-line of B16 mouse melanoma cells compared to the less aggressive F10 sub-line (Ito et al. 2000). F10 cells transfected with wild-type Cx26 exhibited similar metastatic behavior to the BL6 cells. Correspondingly, BL6 cells transfected with a dominant-negative Cx26 mutant showed the less aggressive behavior characteristic of F10 cells. Cx26 was not found to be expressed in human melanoma in situ but was upregulated in invasive melanomas (Ito et al. 2000). However, in this study, Cx26 staining in both melanoma cells and epidermal keratinocytes was cytoplasmic. Moreover, the study did not distinguish between Cx26 and Cx30. In contrast, we showed in immunofluorescence studies on human melanoma tissue samples, that all areas of melanocytic nevi, primary melanomas, and cutaneous melanoma metastases lacked Cx26 and Cx30 expression (Haass et al. 2006, 2010) – similar to our findings in Merkel cell carcinoma (Haass et al. 2003a). This was confirmed by other groups who did not detect Cx26 in melanoma using immunohistochemistry on human tissue samples (Sargen et al. 2013) or did not find Cx26 and Cx30 expression in human melanoma cell lines using qRT-PCR (Zucker et al. 2013). Contrastingly, a positive correlation between Cx26 expression and metastatic potential was reported using Cx26 shRNA in B16 mouse melanoma cells (Stoletov et al. 2013). This was supported by an Oncomine analysis of human tissue, which showed that increased Cx26 expression in primary lesions correlated with metastasis and poor patient survival (Stoletov et al. 2013).
Interestingly, loss of Pannexin 1, a channel-forming glycoprotein remotely related to connexins, attenuated melanoma progression by reversion to a melanocyte-like phenotype (Penuela et al. 2013).
The Oncomine data (Stoletov et al. 2013) do not seem to match the data on primary melanomas in other studies; however, it would be interesting to re-analyze these data more in detail. As there appears to be a correlation to tumor thickness, is there no or little expression on thin tumors and a differential expression pattern in different areas of thick melanomas?
The discrepancies between the different studies in Cx43, Cx26, and Cx30 in melanoma may be due to the following reasons:
-
1.
Several studies investigated the molecules on mRNA level only. The presence of mRNA does not necessarily mean that the respective protein is present.
-
2.
In tissues, it is difficult to separate between connexins present in melanoma cells and those present in epidermal, mesenchymal, or endothelial tissues enclosed by the tumor.
-
3.
Immunohistochemistry is often dependent on staining conditions and can result in false-positive and false-negative results. Appropriate positive and negative controls showing the sensitivity and specificity of the antibody are indispensable for the interpretation of these results. For example, the Cx26 antibody used in some of the discussed studies shows cross-reactivity with Cx30.
Importantly, most of the apparent discrepancies in this paragraph can be explained by a model, which implies that connexins are tumor suppressors during early melanomagenesis but tumor drivers during metastasis (Cronier et al. 2009). During early melanomagenesis, the respective connexins are typically located in the cell membranes indicating that they are functioning through GJIC. In contrast, in advanced stages, connexins are typically located in the cytoplasm indicating a different function – possibly through interaction with signaling molecules.
9.5.6 Connexins in the Epidermal Tumor Environment of Melanoma
Keratinocytes communicate with melanocytes but not with melanoma cells via GJIC; instead, melanoma cells communicate among themselves and with fibroblasts and endothelial cells (Hsu et al. 2000). This switch in communication partners coincides with the E- to N-cadherin switch, suggesting that the gain of N-cadherin with the concurrent loss of E-cadherin facilitates GJ formation with fibroblasts and endothelial cells (Hsu et al. 2000). Additionally, GJ formation in human melanoma cell lines appears to require MCAM (Satyamoorthy et al. 2001). This switch will allow melanoma cells to de-couple from the epidermal microenvironment and to communicate with cell types important for their metastatic spread. Several studies have suggested that connexins may promote metastasis in melanoma and other tumors by forming intercellular connections between cancer cells and vascular endothelium that are able to initiate tumor cell diapedesis (Hsu et al. 2000; Villares et al. 2009; el-Sabban and Pauli 1991, 1994; Saito-Katsuragi et al. 2007; Pollmann et al. 2005). Melanoma cells expressing higher levels of Cx43 show increased coupling to vascular endothelial cells (el-Sabban and Pauli 1991) and the ability of tumor cells to metastasize appears to correlate with the ability of tumor cells to communicate with endothelial cells (Pollmann et al. 2005). Also, Cx26 may contribute to the metastasis of melanoma by facilitating communication between melanoma cells and their surrounding endothelial cells (Saito-Katsuragi et al. 2007). Cx26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer (Naoi et al. 2007).
Melanoma brain metastases are surrounded and infiltrated by astrocytes, and these astrocytes can play a role similar to their established ability to protect neurons from apoptosis (Lin et al. 2010). In co-culture experiments, astrocytes reduced apoptosis in human melanoma cells treated with various chemotherapeutic drugs. This chemoprotective effect was dependent on physical contact and GJIC between astrocytes, which express high levels of Cx43, and tumor cells. Moreover, the protective effect of astrocytes resulted from their sequestering calcium from the cytoplasm of tumor cells. These data suggest that brain metastases can harness the neuroprotective effects of reactive astrocytes for their own survival (Lin et al. 2010). In a chick embryo model, B16 mouse melanoma cells, which express Cx26 but not Cx43, colonized the chicken brain forming numerous microtumors invading along the preexisting vasculature (Stoletov et al. 2013). In contrast, Cx26 knockdown B16 cells formed significantly fewer and less invasive tumors, suggesting that in metastatic melanoma cells Cx26 expression enhances microtumor formation in the brain in association with the existing vasculature (Stoletov et al. 2013).
While these studies demonstrate the interaction of melanoma cells with the stroma and the role of connexins and/or GJIC in the early and late steps of melanomagenesis, interactions between melanoma and the epidermal tumor microenvironment (ETM) – the multilayered epithelium of the skin – are poorly understood. In this regard, we have demonstrated the induction of Cx26 and Cx30 in the epidermis adjacent to malignant tumors (e.g., melanoma and Merkel cell carcinoma), but not in the epidermis adjacent to benign tumors (e.g., melanocytic nevi and angiomas) (Haass et al. 2003a, 2006). Subsequently, we found correlation between (a) tumor thickness (Breslow index) and vertical Cx26 and Cx30 expression in the ETM, (b) tumor thickness and horizontal Cx26 dissemination in the ETM, (c) metastasis and horizontal Cx26 expression in the ETM, and (d) vertical epidermal expression patterns of Cx26 and Cx30 and the proliferative index in the ETM. We thus provided evidence for the association of ETM alteration with tumor malignancy and progression (Haass et al. 2010). The results of this study, which included dysplastic nevi as well as thin melanomas which are often difficult to distinguish (reviewed in Haass and Smalley 2009), suggest that membrane expression of Cx26 and Cx30 in the epidermal tumor microenvironment may be a useful diagnostic aid for the distinction of melanomas and melanocytic nevi (Haass et al. 2010). As neither Cx26 nor Cx30 are expressed in the melanoma itself, but both are induced in its tumor microenvironment, they may be useful complementary melanoma markers.
Cx26 and Cx30 upregulation in the epidermal tumor microenvironment did not correlate with the proliferative index of the melanoma cells, but correlated significantly with the proliferative index in the epidermis. In transgenic mice expressing Cx26 ectopically, proliferation was increased in the epidermis (Djalilian et al. 2006), suggesting that Cx26 influences keratinocyte proliferation and not vice versa. Interestingly, Cx26 overexpressing mice showed a delay in wound healing, which needs to be explored with regards to ulceration, a biomarker associated with very poor prognosis for melanoma patients (Balch et al. 2001). In our study, all melanomas with ulceration showed Cx26 (and Cx30) expression in all layers of the epidermal tumor microenvironment (Haass et al. 2010). Induction of angiogenesis by the hyperplastic epithelium could stimulate growth and progression of melanoma (McCarty et al. 2003). This suggests a positive feedback mechanism: tumor cells induce alterations in keratinocytes, which results in the production of growth factors which, in turn, stimulate tumor survival via endothelial cells. The induction of Cx26 and Cx30 in the epidermis adjacent to melanoma putatively leading to GJIC or signaling via hemichannels may play a role in this feedback mechanism by inducing proliferation and other functions. An interruption of this vicious circle may provide a novel therapeutic approach.
9.6 Tight Junctions
In simple epithelia and endothelia, tight junctions (TJs) are responsible for the formation and maintenance of the tissue barrier between distinct compartments by controlling the paracellular pathway (“barrier function”) (reviewed in Stevenson and Keon 1998; Tsukita et al. 2001). Subsequently, the involvement of TJs in the barrier function of a complex epithelium, the epidermis, was shown (Pummi et al. 2001; Brandner et al. 2002, 2003; Furuse et al. 2002; Langbein et al. 2002). In addition, TJs prevent the diffusion of membrane proteins and lipids from the apical to the basolateral side of an epithelial cell sheet, helping to maintain cell polarity (“fence function”) (reviewed in Mitic and Anderson 1998; Tsukita et al. 2001). Therefore, TJs are crucial for the epithelium to generate chemical and electrical gradients that is necessary for vectorial transport processes such as absorption and secretion (reviewed in Martin and Jiang 2009). Moreover, TJ molecules act as intermediates and transducers in cell signaling, thus playing a role in the processes of polarity, cell differentiation, cell growth, and differentiation. Finally, TJs act as cell–cell adhesion molecules and as a barrier to cell migration (reviewed in Martin and Jiang 2009).
TJs are composed of integral transmembrane proteins (claudin 1–24, occludin, and junctional adhesion molecules A-C, 4 (JAMs)), peripheral plaque proteins (zonula occludens (ZO) proteins 1–3, MAGI 1–3, MUPP-1, PAR-3, PAR-6, AF-6, CASK, and CAROM), and associated proteins (symplekin, ZONAB, cingulin, Rab-13, Rab-3B, c-src, α-catenin, PKA, ZAK, and Rho GTPases). The molecular composition of TJs is highly complex and varies according to the cell type and degree of differentiation. TJ molecules from neighboring cells associate and form paired strands which seal the paracellular pathway and which contain aqueous pores or paracellular channels, explaining the ion and size selectivity for passaging molecules of TJ (Tsukita and Furuse 2000).
In cancer, disruption of TJs should occur in three critical steps: (1) detachment of the tumor cell from the primary tumor, (2) intravasation of the tumor through the endothelium, and (3) extravastion of the circulating tumor cell (reviewed in Martin and Jiang 2009). Early studies have shown a correlation between lack of TJs and tumor differentiation and there is evidence that TJs need to be overcome by cancer cells in order to metastasize (reviewed in Martin and Jiang 2001, 2009). Cancer cells frequently exhibit deficiencies in TJ function, as well as decreased differentiation and cell polarity (Weinstein et al. 1976; Soler et al. 1999). Loss of TJ integrity may be particularly important in allowing the diffusion of nutrients and other factors necessary for the survival and growth of the tumor cells (Mullin et al. 1997). In addition, decreased polarity and differentiation may be important for the metastatic phenotype, where individual cells must leave the primary site and enter the blood vessels to reach distant sites (Ren et al. 1990).
Electron microscopy studies in human thyroid tumors showed that TJs decrease in number and are attenuated during carcinogenesis, which is associated with loss of tumor differentiation (Kerjaschki et al. 1979). Expression of TJ proteins is decreased in some cancer types, e.g., ZO-1 and occludin in gastrointestinal adenocarcinoma (Kimura et al. 1997), occludin in epithelial-derived tumors (Li and Mrsny 2000), claudin 3 in glioblastoma multiforme (Wolburg et al. 2003), claudin 1 in sporadic and hereditary breast cancer (Kramer et al. 2000), and claudin 7 in ductal carcinoma of the breast (Kominsky et al. 2003). On the other hand, some TJ molecules appear to be upregulated in some cancers. We found protein expression of claudins 3, 4, and 5, occludin, and ZO-1 in Merkel cell carcinoma cells (Haass et al. 2003b). Strikingly, expression of some claudin family members is highly elevated in various human cancers, e.g., claudin 7 in two breast cancer cell lines (Nacht et al. 1999), claudin 1 in colorectal cancer (Miwa et al. 2000), and claudins 3 and 4 in ovarian (Hough et al. 2001; Rangel et al. 2003) and prostate cancer (Long et al. 2001).
The expression of TJ proteins in melanoma tissues and cultured melanoma cells was described on RNA and on protein level (Cohn et al. 2005; Smalley et al. 2005; Leotlela et al. 2007; Schmitt et al. 2007; Morita et al. 2008). In a tissue array study, Claudin-1 was found to be significantly reduced in metastatic melanoma (Cohn et al. 2005). These data were, however, directly contradicted by another study (Leotlela et al. 2007). In this study Claudin-1 appeared to contribute to melanoma cell invasion, as transient transfection of melanoma cells with Claudin-1 increased metalloproteinase 2 (MMP-2) secretion and activation, and subsequently, motility of melanoma cells as demonstrated by wound-healing assays. Conversely, knockdown of CLDN1 by siRNA resulted in the inhibition of motility, as well as decreases in MMP-2 secretion and activation (Leotlela et al. 2007).
In contrast to most cancers, where levels of ZO-1 are typically downregulated, leading to increased motility, we found that ZO-1 expression is upregulated in melanoma cells and is located at adherens junctions between melanoma cells and fibroblasts (Smalley et al. 2005). Immunofluorescence and co-immunoprecipitation studies showed co-localization of ZO-1 with N-cadherin. Downregulation of ZO-1 in melanoma cells through RNA interference produced marked changes in cell morphology – leading to a less dendritic, more rounded phenotype. Consistent with a role in N-cadherin-based adhesion, RNAi-treated melanoma cells were less adherent and invasive when grown in a collagen gel. These data provided the first evidence that increased ZO-1 expression in melanoma contributes to the oncogenic behavior of this tumor and further illustrated that protein products of genes, such as ZO-1, can function in either a pro- or anti-oncogenic manner when expressed in different cellular contexts (Smalley et al. 2005).
In summary, while it appears that functional TJs may be tumor suppressors, the upregulation of certain TJ proteins can contribute to oncogenic behavior. The relationship between TJ protein overexpression and cancer initiation or progression is thus unclear at present, but may be explained by the lack of functional TJs and that the upregulated TJ proteins therefore likely function through TJ-independent mechanisms.
Abbreviations
- CDH:
-
Cadherin
- Cx:
-
Connexin(s)
- Dsc 1–3:
-
Desmocollin
- Dsg:
-
Desmoglein
- GJ:
-
Gap junction
- GJIC:
-
Gap junctional intercellular communication
References
Aeed PA, Nakajima M, Welch DR (1988) The role of polymorphonuclear leukocytes (PMN) on the growth and metastatic potential of 13762NF mammary adenocarcinoma cells. Int J Cancer 42(5):748–759
Albelda SM, Mette SA, Elder DE, Stewart R, Damjanovich L, Herlyn M, Buck CA (1990) Integrin distribution in malignant melanoma: association of the beta 3 subunit with tumor progression. Cancer Res 50:6757–6764
Alexaki VI, Javelaud D, Van Kempen LC, Mohammad KS, Dennler S, Luciani F, Hoek KS, Juarez P, Goydos JS, Fournier PJ, Sibon C, Bertolotto C, Verrecchia F, Saule S, Delmas V, Ballotti R, Larue L, Saiag P, Guise TA, Mauviel A (2010) GLI2-mediated melanoma invasion and metastasis. J Natl Cancer Inst 102:1148–1159
Atkinson MM, Menko AS, Johnson RG, Sheppard JR, Sheridan JD (1981) Rapid and reversible reduction of junctional permeability in cells infected with a temperature-sensitive mutant of avian sarcoma virus. J Cell Biol 91(2 Pt 1):573–578
Avanzo JL, Mesnil M, Hernandez-Blazquez FJ, Mackowiak II, Mori CM, da Silva TC, Oloris SC, Garate AP, Massironi SM, Yamasaki H, Dagli ML (2004) Increased susceptibility to urethane-induced lung tumors in mice with decreased expression of connexin43. Carcinogenesis 25(10):1973–1982. doi:10.1093/carcin/bgh193bgh193 [pii]
Bachmann IM, Straume O, Puntervoll HE, Kalvenes MB, Akslen LA (2005) Importance of P-cadherin, beta-catenin, and Wnt5a/frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res 11:8606–8614
Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, Urist M, McMasters KM, Ross MI, Kirkwood JM, Atkins MB, Thompson JA, Coit DG, Byrd D, Desmond R, Zhang Y, Liu PY, Lyman GH, Morabito A (2001) Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 19(16):3622–3634
Barr TP, Albrecht PJ, Hou Q, Mongin AA, Strichartz GR, Rice FL (2013) Air-stimulated ATP release from keratinocytes occurs through connexin hemichannels. PLoS One 8(2):e56744. doi:10.1371/journal.pone.0056744
Bates DC, Sin WC, Aftab Q, Naus CC (2007) Connexin43 enhances glioma invasion by a mechanism involving the carboxy terminus. Glia 55(15):1554–1564. doi:10.1002/glia.20569
Batlle E, Sancho E, Francí C, Domínguez D, Monfar M, Baulida J, García De Herreros A (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2(2):84–89
Bauer R, Bosserhoff AK (2006) Functional implication of truncated P-cadherin expression in malignant melanoma. Exp Mol Pathol 81:224–230
Bauer R, Hein R, Bosserhoff AK (2005) A secreted form of P-cadherin is expressed in malignant melanoma. Exp Cell Res 305:418–426
Bauer R, Wild PJ, Meyer S, Bataille F, Pauer A, Klinkhammer-Schalke M, Hofstaedter F, Bosserhoff AK (2006) Prognostic relevance of P-cadherin expression in melanocytic skin tumours analysed by high-throughput tissue microarrays. J Clin Pathol 59:699–705
Becker JC, Termeer C, Schmidt RE, Brocker EB (1993) Soluble intercellular adhesion molecule-1 inhibits MHC-restricted specific T cell/tumor interaction. J Immunol 151(12):7224–7232
Berrier AL, Yamada KM (2007) Cell-matrix adhesion. J Cell Physiol 213:565–573
Bissell MJ, Radisky D (2001) Putting tumours in context. Nat Rev Cancer 1(1):46–54
Billion K, Ibrahim H, Mauch C, Niessen CM (2006) Increased soluble E-cadherin in melanoma patients. Skin Pharmacol Physiol 19:65–70
Bolós V, Peinado H, Pérez-Moreno MA, Fraga MF, Esteller M, Cano A (2003) The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 116(Pt 3):499–511
Boveri T (1914) Zur frage der entstehung maligner tumoren. Gustav Fischer Verlag, Jena
Brandner JM, Haass NK (2013) Melanoma’s connections to the tumour microenvironment. Pathology 45(5):443–452. doi:10.1097/PAT.0b013e328363b3bd
Brandner JM, Kief S, Grund C, Rendl M, Houdek P, Kuhn C, Tschachler E, Franke WW, Moll I (2002) Organization and formation of the tight junction system in human epidermis and cultured keratinocytes. Eur J Cell Biol 81(5):253–263
Brandner JM, McIntyre M, Kief S, Wladykowski E, Moll I (2003) Expression and localization of tight junction-associated proteins in human hair follicles. Arch Dermatol Res 295(5):211–221
Braga V (2000) Epithelial cell shape: cadherins and small GTPases. Exp Cell Res 261:83–90
Braga VM, Machesky LM, Hall A, Hotchin NA (1997) The small GTPases Rho and Rac are required for the establishment of cadherin-dependent cell-cell contacts. J Cell Biol 137:1421–1431
Brissette JL, Kumar NM, Gilula NB, Hall JE, Dotto GP (1994) Switch in gap junction protein expression is associated with selective changes in junctional permeability during keratinocyte differentiation. Proc Natl Acad Sci U S A 91(14):6453–6457
Brummer J, Ebrahimnejad A, Flayeh R, Schumacher U, Loning T, Bamberger AM, Wagener C (2001) cis Interaction of the cell adhesion molecule CEACAM1 with integrin beta(3). Am J Pathol 159(2):537–546
Caramel J, Papadogeorgakis E, Hill L, Browne GJ, Richard G, Wierinckx A, Saldanha G, Osborne J, Hutchinson P, Tse G, Lachuer J, Puisieux A, Pringle JH, Ansieau S, Tulchinsky E (2013) A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24:466–480
Chandrasekhar A, Bera AK (2012) Hemichannels: permeants and their effect on development, physiology and death. Cell Biochem Funct 30(2):89–100. doi:10.1002/cbf.2794
Chakraborty C, Gleeson LM, McKinnon T, Lala PK (2002) Regulation of human trophoblast migration and invasiveness. Can J Physiol Pharmacol 80:116–124
Cohn ML, Goncharuk VN, Diwan AH, Zhang PS, Shen SS, Prieto VG (2005) Loss of claudin-1 expression in tumor-associated vessels correlates with acquisition of metastatic phenotype in melanocytic neoplasms. J Cutan Pathol 32(8):533–536, CUP324 [pii]10.1111/j.0303-6987.2005.00324.x
Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F (2001) The two-handed E box binding zinc fingerprotein SIP1 downregulates E-cadherin and induces invasion. Mol Cell 7(6):1267–1278
Cotrina ML, Lin JH, Nedergaard M (2008) Adhesive properties of connexin hemichannels. Glia 56(16):1791–1798. doi:10.1002/glia.20728
Cronier L, Crespin S, Strale PO, Defamie N, Mesnil M (2009) Gap junctions and cancer: new functions for an old story. Antioxid Redox Signal 11(2):323–338. doi:10.1089/ars.2008.2153
Czyz J (2008) The stage-specific function of gap junctions during tumourigenesis. Cell Mol Biol Lett 13(1):92–102. doi:10.2478/s11658-007-0039-5
Dai DL, Makretsov N, Campos EI, Huang C, Zhou Y, Huntsman D, Martinka M, Li G (2003) Increased expression of integrin-linked kinase is correlated with melanoma progression and poor patient survival. Clin Cancer Res 9:4409–4414
Danen EH, Sonnenberg A (2003) Integrins in regulation of tissue development and function. J Pathol 201:632–641
Danen EH, Jansen KF, Van Kraats AA, Cornelissen IM, Ruiter DJ, van Muijen GN (1995) Alpha v-integrins in human melanoma: gain of alpha v beta 3 and loss of alpha v beta 5 are related to tumor progression in situ but not to metastatic capacity of cell lines in nude mice. Int J Cancer 61:491–496
Danen EH, de Vries TJ, Morandini R, Ghanem GG, Ruiter DJ, van Muijen GN (1996) E-cadherin expression in human melanoma. Melanoma Res 6:127–131
Dagli ML, Yamasaki H, Krutovskikh V, Omori Y (2004) Delayed liver regeneration and increased susceptibility to chemical hepatocarcinogenesis in transgenic mice expressing a dominant-negative mutant of connexin32 only in the liver. Carcinogenesis 25(4):483–492. doi:10.1093/carcin/bgh050bgh050 [pii]
Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A 95:11211–11216
Degen WG, van Kempen LC, Gijzen EG, van Groningen JJ, van Kooyk Y, Bloemers HP, Swart GW (1998) MEMD, a new cell adhesion molecule in metastasizing human melanoma cell lines, is identical to ALCAM (activated leukocyte cell adhesion molecule). Am J Pathol 152(3):805–813
Di WL, Rugg EL, Leigh IM, Kelsell DP (2001) Multiple epidermal connexins are expressed in different keratinocyte subpopulations including connexin 31. J Invest Dermatol 117(4):958–964
Djalilian AR, McGaughey D, Patel S, Seo EY, Yang C, Cheng J, Tomic M, Sinha S, Ishida-Yamamoto A, Segre JA (2006) Connexin 26 regulates epidermal barrier and wound remodeling and promotes psoriasiform response. J Clin Invest 116(5):1243–1253
Dobrowolski R, Sasse P, Schrickel JW, Watkins M, Kim JS, Rackauskas M, Troatz C, Ghanem A, Tiemann K, Degen J, Bukauskas FF, Civitelli R, Lewalter T, Fleischmann BK, Willecke K (2008) The conditional connexin43G138R mouse mutant represents a new model of hereditary oculodentodigital dysplasia in humans. Hum Mol Genet 17(4):539–554. doi:10.1093/hmg/ddm329
Donizy P, Zietek M, Halon A, Leskiewicz M, Kozyra C, Matkowski R (2015) Prognostic significance of ALCAM (CD166/MEMD) expression in cutaneous melanoma patients. Diagn Pathol 10:86. doi:10.1186/s13000-015-0331-z
Duflot-Dancer A, Mesnil M, Yamasaki H (1997) Dominant-negative abrogation of connexin-mediated cell growth control by mutant connexin genes. Oncogene 15(18):2151–2158. doi:10.1038/sj.onc.1201393
Ebrahimnejad A, Streichert T, Nollau P, Horst AK, Wagener C, Bamberger AM, Brummer J (2004) CEACAM1 enhances invasion and migration of melanocytic and melanoma cells. Am J Pathol 165(5):1781–1787
Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R (2005) DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 24(14):2375–2385
Eghbali B, Kessler JA, Reid LM, Roy C, Spray DC (1991) Involvement of gap junctions in tumorigenesis: transfection of tumor cells with connexin 32 cDNA retards growth in vivo. Proc Natl Acad Sci U S A 88(23):10701–10705
el-Sabban ME, Pauli BU (1991) Cytoplasmic dye transfer between metastatic tumor cells and vascular endothelium. J Cell Biol 115(5):1375–1382
el-Sabban ME, Pauli BU (1994) Adhesion-mediated gap junctional communication between lung-metastatatic cancer cells and endothelium. Invasion Metastasis 14(1–6):164–176
Elfgang C, Eckert R, Lichtenberg-Frate H, Butterweck A, Traub O, Klein RA, Hulser DF, Willecke K (1995) Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J Cell Biol 129(3):805–817
Elzarrad MK, Haroon A, Willecke K, Dobrowolski R, Gillespie MN, Al-Mehdi AB (2008) Connexin-43 upregulation in micrometastases and tumor vasculature and its role in tumor cell attachment to pulmonary endothelium. BMC Med 6:20. doi:10.1186/1741-7015-6-20, 1741-7015-6-20 [pii]
Evans WH, De Vuyst E, Leybaert L (2006) The gap junction cellular internet: connexin hemichannels enter the signalling limelight. Biochem J 397(1):1–14
Ezumi K, Yamamoto H, Murata K, Higashiyama M, Damdinsuren B, Nakamura Y, Kyo N, Okami J, Ngan CY, Takemasa I, Ikeda M, Sekimoto M, Matsuura N, Nojima H, Monden M (2008) Aberrant expression of connexin 26 is associated with lung metastasis of colorectal cancer. Clin Cancer Res 14(3):677–684. doi:10.1158/1078-0432.CCR-07-1184
Felding-Habermann B, Fransvea E, O’Toole TE, Manzuk L, Faha B, Hensler M (2002) Involvement of tumor cell integrin alpha v beta 3 in hematogenous metastasis of human melanoma cells. Clin Exp Metastasis 19:427–436
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3(6):453–458. doi:10.1038/nrc1098
Fitzpatrick TB, Breathnach AS (1963) The epidermal melanin unit system. Dermatol Wochenschr 147:481–489
Fitzpatrick TB, Szabo G, Seiji M, Quevedo WC Jr (1979) Biology of the melanin pigmentary system. In: Fitzpatrick TB, Eisen A, Wolff K, Freedberg I, Austen K (eds) Dermatology in general medicine. McGraw-Hill, New York, pp 131–145
Flanagan K, Fitzgerald K, Baker J, Regnstrom K, Gardai S, Bard F, Mocci S, Seto P, You M, Larochelle C, Prat A, Chow S, Li L, Vandevert C, Zago W, Lorenzana C, Nishioka C, Hoffman J, Botelho R, Willits C, Tanaka K, Johnston J, Yednock T (2012) Laminin-411 is a vascular ligand for MCAM and facilitates TH17 cell entry into the CNS. PLoS One 7(7):e40443. doi:10.1371/journal.pone.0040443
Fogel M, Mechtersheimer S, Huszar M, Smirnov A, Abu-Dahi A, Tilgen W, Reichrath J, Georg T, Altevogt P, Gutwein P (2003) L1 adhesion molecule (CD 171) in development and progression of human malignant melanoma. Cancer Lett 189(2):237–247
Franke WW (2009) Discovering the molecular components of intercellular junctions – a historical view. Cold Spring Harb Perspect Biol 1:a003061
Friedl P, Zanker KS, Brocker EB (1998) Cell migration strategies in 3-D extracellular matrix: differences in morphology, cell matrix interactions, and integrin function. Microsc Res Tech 43:369–378
Friedmann MC, Migone TS, Russell SM, Leonard WJ (1996) Different interleukin 2 receptor beta-chain tyrosines couple to at least two signaling pathways and synergistically mediate interleukin 2-induced proliferation. Proc Natl Acad Sci U S A 93:2077–2082
Furuse M, Hata M, Furuse K, Yoshida Y, Haratake A, Sugitani Y, Noda T, Kubo A, Tsukita S (2002) Claudin-based tight junctions are crucial for the mammalian epidermal barrier: a lesson from claudin-1-deficient mice. J Cell Biol 156(6):1099–1111
Furukawa F, Fujii K, Horiguchi Y, Matsuyoshi N, Fujita M, Toda K, Imamura S, Wakita H, Shirahama S, Takigawa M (1997) Roles of E- and P-cadherin in the human skin. Microsc Res Tech 38:343–352
Gershon E, Plaks V, Dekel N (2008) Gap junctions in the ovary: expression, localization and function. Mol Cell Endocrinol 282(1–2):18–25. doi:10.1016/j.mce.2007.11.001, S0303-7207(07)00411-X [pii]
Giavazzi R, Chirivi RG, Garofalo A, Rambaldi A, Hemingway I, Pigott R, Gearing AJ (1992) Soluble intercellular adhesion molecule 1 is released by human melanoma cells and is associated with tumor growth in nude mice. Cancer Res 52(9):2628–2630
Goldberg GS, Valiunas V, Brink PR (2004) Selective permeability of gap junction channels. Biochim Biophys Acta 1662(1–2):96–101. doi:10.1016/j.bbamem.2003.11.022
Gottardi CJ, Wong E, Gumbiner BM (2001) E-cadherin suppresses cellular transformation by inhibiting beta-catenin signaling in an adhesion-independent manner. J Cell Biol 153:1049–1060
Gotoh N, Toyoda M, Shibuya M (1997) Tyrosine phosphorylation sites at amino acids 239 and 240 of Shc are involved in epidermal growth factor-induced mitogenic signaling that is distinct from Ras/mitogen-activated protein kinase activation. Mol Cell Biol 17:1824–1831
Graff JR, Greenberg VE, Herman JG, Westra WH, Boghaert ER, Ain KB, Saji M, Zeiger MA, Zimmer SG, Baylin SB (1998) Distinct patterns of E-cadherin CpG island methylation in papillary, follicular, Hurthle’s cell, and poorly differentiated human thyroid carcinoma. Cancer Res 58:2063–2066
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ (2008) The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10:593–601
Groebe K, Mueller-Klieser W (1991) Distributions of oxygen, nutrient, and metabolic waste concentrations in multicellular spheroids and their dependence on spheroid parameters. Eur Biophys J 19(4):169–181
Haass NK (2015) Dynamic tumor heterogeneity in melanoma therapy: how do we address this in a novel model system? Melanoma Manag 2(2):93–95
Haass NK, Beaumont KA, Hill DS, Anfosso A, Mrass P, Munoz MA, Kinjyo I, Weninger W (2014) Real-time cell cycle imaging during melanoma growth, invasion, and drug response. Pigment Cell Melanoma Res 27(5):764–776. doi:10.1111/pcmr.12274
Haass NK, Herlyn M (2005) Normal human melanocyte homeostasis as a paradigm for understanding melanoma. J Investig Dermatol Symp Proc 10(2):153–163
Haass NK, Houdek P, Brandner JM, Moll I (2003a) Expression patterns of connexins in Merkel cell carcinoma and adjacent epidermis. In: Baumann KI, Moll I, Halata Z (eds) The Merkel cell – structure – development – function – and cancerogenesis. Springer, Berlin/Heidelberg/New York/Tokyo, pp 219–222
Haass NK, Houdek P, Wladykowski E, Moll I, Brandner JM (2003b) Expression patterns of tight junction proteins in Merkel cell carcinoma. In: Baumann KI, Moll I, Halata Z (eds) The Merkel cell – structure – development – function – and cancerogenesis. Springer, Berlin/Heidelberg/New York/Tokyo, pp 223–226
Haass NK, Ripperger D, Wladykowski E, Dawson P, Gimotty PA, Blome C, Fischer F, Schmage P, Moll I, Brandner JM (2010) Melanoma progression exhibits a significant impact on connexin expression patterns in the epidermal tumor microenvironment. Histochem Cell Biol 133(1):113–124. doi:10.1007/s00418-009-0654-5
Haass NK, Smalley KS (2009) Melanoma biomarkers: current status and utility in diagnosis, prognosis, and response to therapy. Mol Diagn Ther 13(5):283–296. doi:10.2165/11317270-000000000-00000, 2 [pii]
Haass NK, Smalley KS, Herlyn M (2004) The role of altered cell-cell communication in melanoma progression. J Mol Histol 35(3):309–318
Haass NK, Smalley KS, Li L, Herlyn M (2005) Adhesion, migration and communication in melanocytes and melanoma. Pigment Cell Res 18(3):150–159
Haass NK, Wladykowski E, Kief S, Moll I, Brandner JM (2006) Differential induction of connexins 26 and 30 in skin tumors and their adjacent epidermis. J Histochem Cytochem 54(2):171–182
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hamamura K, Tsuji M, Ohkawa Y, Nakashima H, Miyazaki S, Urano T, Yamamoto N, Ueda M, Furukawa K, Furukawa K (2008) Focal adhesion kinase as well as p130Cas and paxillin is crucially involved in the enhanced malignant properties under expression of ganglioside GD3 in melanoma cells. Biochim Biophys Acta 1780:513–519
Hajra KM, Chen DY, Fearon ER (2002) The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res 62(6):1613–1618
Hendrix MJ, Seftor EA, Meltzer PS, Gardner LM, Hess AR, Kirschmann DA, Schatteman GC, Seftor RE (2001) Expression and functional significance of VE-cadherin in aggressive human melanoma cells: role in vasculogenic mimicry. Proc Natl Acad Sci U S A 98:8018–8023
Hendrix MJ, Seftor EA, Hess AR, Seftor RE (2003) Molecular plasticity of human melanoma cells. Oncogene 22:3070–3075
Hellmann P, Grummer R, Schirrmacher K, Rook M, Traub O, Winterhager E (1999) Transfection with different connexin genes alters growth and differentiation of human choriocarcinoma cells. Exp Cell Res 246(2):480–490. doi:10.1006/excr.1998.4332, S0014-4827(98)94332-4 [pii]
Herve JC, Bourmeyster N, Sarrouilhe D, Duffy HS (2007) Gap junctional complexes: from partners to functions. Prog Biophys Mol Biol 94(1–2):29–65. doi:10.1016/j.pbiomolbio.2007.03.010
Hirschi KK, Xu CE, Tsukamoto T, Sager R (1996) Gap junction genes Cx26 and Cx43 individually suppress the cancer phenotype of human mammary carcinoma cells and restore differentiation potential. Cell Growth Differ 7(7):861–870
Hong H, Stastny M, Brown C, Chang WC, Ostberg JR, Forman SJ, Jensen MC (2014) Diverse solid tumors expressing a restricted epitope of L1-CAM can be targeted by chimeric antigen receptor redirected T lymphocytes. J Immunother 37(2):93–104. doi:10.1097/CJI.0000000000000018
Hortsch M (1996) The L1 family of neural cell adhesion molecules: old proteins performing new tricks. Neuron 17(4):587–593
Hough CD, Cho KR, Zonderman AB, Schwartz DR, Morin PJ (2001) Coordinately up-regulated genes in ovarian cancer. Cancer Res 61(10):3869–3876
Howe AK, Aplin AE, Juliano RL (2002) Anchorage-dependent ERK signaling – mechanisms and consequences. Curr Opin Genet Dev 12:30–35
Hsu MY, Wheelock MJ, Johnson KR, Herlyn M (1996) Shifts in cadherin profiles between human normal melanocytes and melanomas. J Investig Dermatol Symp Proc 1:188–194
Hsu M, Andl T, Li G, Meinkoth JL, Herlyn M (2000) Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J Cell Sci 113(Pt 9):1535–1542
Huang RP, Hossain MZ, Sehgal A, Boynton AL (1999) Reduced connexin43 expression in high-grade human brain glioma cells. J Surg Oncol 70(1):21–24. doi:10.1002/(SICI)1096-9098(199901)70:1<21::AID-JSO4>3.0.CO;2–0, [pii]#
Hurteau GJ, Carlson JA, Spivack SD, Brock GJ (2007) Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 67:7972–7976
Ito A, Katoh F, Kataoka TR, Okada M, Tsubota N, Asada H, Yoshikawa K, Maeda S, Kitamura Y, Yamasaki H, Nojima H (2000) A role for heterologous gap junctions between melanoma and endothelial cells in metastasis. J Clin Invest 105(9):1189–1197
Jacobs K, Feys L, Vanhoecke B, Van Marck V, Bracke M (2011) P-cadherin expression reduces melanoma growth, invasion, and responsiveness to growth factors in nude mice. Eur J Cancer Prev 20(3):207–216
Jimbow K, Quevedo WC Jr, Fitzpatrick TB, Szabo G (1976) Some aspects of melanin biology: 1950–1975. J Invest Dermatol 67(1):72–89
Jinn Y, Ichioka M, Marumo F (1998) Expression of connexin32 and connexin43 gap junction proteins and E-cadherin in human lung cancer. Cancer Lett 127(1–2):161–169
Johnson JP, Bar-Eli M, Jansen B, Markhof E (1997) Melanoma progression-associated glycoprotein MUC18/MCAM mediates homotypic cell adhesion through interaction with a heterophilic ligand. Int J Cancer 73(5):769–774
Johnson JP, Rummel MM, Rothbacher U, Sers C (1996) MUC18: a cell adhesion molecule with a potential role in tumor growth and tumor cell dissemination. Curr Top Microbiol Immunol 213(Pt 1):95–105
Johnson JP, Stade BG, Holzmann B, Schwable W, Riethmuller G (1989) De novo expression of intercellular-adhesion molecule 1 in melanoma correlates with increased risk of metastasis. Proc Natl Acad Sci U S A 86(2):641–644
Joshi MB, Ivanov D, Philippova M, Kyriakakis E, Erne P, Resink TJ (2008) A requirement for thioredoxin in redox-sensitive modulation of T-cadherin expression in endothelial cells. Biochem J 416:271–280
Jost M, Huggett TM, Kari C, Boise LH, Rodeck U (2001) Epidermal growth factor receptor-dependent control of keratinocyte survival and Bcl-xL expression through a MEK-dependent pathway. J Biol Chem 276:6320–6326
Jouve N, Despoix N, Espeli M, Gauthier L, Cypowyj S, Fallague K, Schiff C, Dignat-George F, Vely F, Leroyer AS (2013) The involvement of CD146 and its novel ligand Galectin-1 in apoptotic regulation of endothelial cells. J Biol Chem 288(4):2571–2579. doi:10.1074/jbc.M112.418848
Kanai Y, Ushijima S, Hui AM, Ochiai A, Tsuda H, Sakamoto M, Hirohashi S (1997) The E-cadherin gene is silenced by CpG methylation in human hepatocellular carcinomas. Int J Cancer 71:355–359
Kanczuga-Koda L, Sulkowski S, Lenczewski A, Koda M, Wincewicz A, Baltaziak M, Sulkowska M (2006) Increased expression of connexins 26 and 43 in lymph node metastases of breast cancer. J Clin Pathol 59(4):429–433. doi:10.1136/jcp.2005.029272, 59/4/429 [pii]
Kahana O, Micksche M, Witz IP, Yron I (2002) The focal adhesion kinase (P125FAK) is constitutively active in human malignant melanoma. Oncogene 21:3969–3977
Kerjaschki D, Krisch K, Sleyter UB, Umrath W, Jakesz R, Depisch D, Kokoschka R, Horandner H (1979) The structure of tight junctions in human thyroid tumors. A systematic freeze-fracture study. Am J Pathol 96(1):207–225
Khanna P, Yunkunis T, Muddana HS, Peng HH, August A, Dong C (2010) p38 MAP kinase is necessary for melanoma-mediated regulation of VE-cadherin disassembly. Am J Physiol Cell Physiol 298:C1140–C1150
Kimura Y, Shiozaki H, Hirao M, Maeno Y, Doki Y, Inoue M, Monden T, Ando-Akatsuka Y, Furuse M, Tsukita S, Monden M (1997) Expression of occludin, tight-junction-associated protein, in human digestive tract. Am J Pathol 151(1):45–54
King TJ, Fukushima LH, Hieber AD, Shimabukuro KA, Sakr WA, Bertram JS (2000) Reduced levels of connexin43 in cervical dysplasia: inducible expression in a cervical carcinoma cell line decreases neoplastic potential with implications for tumor progression. Carcinogenesis 21(6):1097–1109
King TJ, Lampe PD (2004a) The gap junction protein connexin32 is a mouse lung tumor suppressor. Cancer Res 64(20):7191–7196. doi:10.1158/0008-5472.CAN-04-0624, 64/20/7191 [pii]
King TJ, Lampe PD (2004b) Mice deficient for the gap junction protein Connexin32 exhibit increased radiation-induced tumorigenesis associated with elevated mitogen-activated protein kinase (p44/Erk1, p42/Erk2) activation. Carcinogenesis 25(5):669–680. doi:10.1093/carcin/bgh071bgh071 [pii]
Klein WM, Wu BP, Zhao S, Wu H, Klein-Szanto AJ, Tahan SR (2007) Increased expression of stem cell markers in malignant melanoma. Mod Pathol 20(1):102–107
Kluger HM, Hoyt K, Bacchiocchi A, Mayer T, Kirsch J, Kluger Y, Sznol M, Ariyan S, Molinaro A, Halaban R (2011) Plasma markers for identifying patients with metastatic melanoma. Clin Cancer Res 17(8):2417–2425. doi:10.1158/1078-0432.CCR-10-2402
Kominsky SL, Argani P, Korz D, Evron E, Raman V, Garrett E, Rein A, Sauter G, Kallioniemi OP, Sukumar S (2003) Loss of the tight junction protein claudin-7 correlates with histological grade in both ductal carcinoma in situ and invasive ductal carcinoma of the breast. Oncogene 22(13):2021–2033
Kramer F, White K, Kubbies M, Swisshelm K, Weber BH (2000) Genomic organization of claudin-1 and its assessment in hereditary and sporadic breast cancer. Hum Genet 107(3):249–256
Kraus A, Masat L, Johnson JP (1997) Analysis of the expression of intercellular adhesion molecule-1 and MUC18 on benign and malignant melanocytic lesions using monoclonal antibodies directed against distinct epitopes and recognizing denatured, non-glycosylated antigen. Melanoma Res 7(Suppl 2):S75–S81
Kretz M, Maass K, Willecke K (2004) Expression and function of connexins in the epidermis, analyzed with transgenic mouse mutants. Eur J Cell Biol 83(11–12):647–654
Krutovskikh V, Mazzoleni G, Mironov N, Omori Y, Aguelon AM, Mesnil M, Berger F, Partensky C, Yamasaki H (1994) Altered homologous and heterologous gap-junctional intercellular communication in primary human liver tumors associated with aberrant protein localization but not gene mutation of connexin 32. Int J Cancer 56(1):87–94
Krutovskikh VA, Piccoli C, Yamasaki H (2002) Gap junction intercellular communication propagates cell death in cancerous cells. Oncogene 21(13):1989–1999. doi:10.1038/sj.onc.1205187
Krutovskikh VA, Troyanovsky SM, Piccoli C, Tsuda H, Asamoto M, Yamasaki H (2000) Differential effect of subcellular localization of communication impairing gap junction protein connexin43 on tumor cell growth in vivo. Oncogene 19(4):505–513. doi:10.1038/sj.onc.1203340
Krutovskikh VA, Yamasaki H, Tsuda H, Asamoto M (1998) Inhibition of intrinsic gap-junction intercellular communication and enhancement of tumorigenicity of the rat bladder carcinoma cell line BC31 by a dominant-negative connexin 43 mutant. Mol Carcinog 23(4):254–261. doi:10.1002/(SICI)1098-2744(199812)23:4<254::AID-MC9>3.0.CO;2-4 [pii]
Kuphal S, Poser I, Jobin C, Hellerbrand C, Bosserhoff AK (2004) Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene 23:8509–8519
Kuphal S, Martyn AC, Pedley J, Crowther LM, Bonazzi VF, Parsons PG, Bosserhoff AK, Hayward NK, Boyle GM (2009) H-cadherin expression reduces invasion of malignant melanoma. Pigment Cell Melanoma Res 22:296–306
Bosserhoff AK, Ellmann L, Quast AS, Eberle J, Boyle GM, Kuphal S (2014) Loss of T-cadherin (CDH-13) regulates AKT signaling and desensitizes cells to apoptosis in melanoma. Mol Carcinog 53(8):635–647
Kuphal S, Haass NK (2011) Cell–cell and cell–matrix contacts in melanoma and the tumor microenvironment. In: Bosserhoff AK (ed) Melanoma development – molecular biology, genetics and clinical application. Springer, Wien, pp 181–215
Lampe PD (1994) Analyzing phorbol ester effects on gap junctional communication: a dramatic inhibition of assembly. J Cell Biol 127(6 Pt 2):1895–1905
Langbein L, Grund C, Kuhn C, Praetzel S, Kartenbeck J, Brandner JM, Moll I, Franke WW (2002) Tight junctions and compositionally related junctional structures in mammalian stratified epithelia and cell cultures derived therefrom. Eur J Cell Biol 81(8):419–435
Langlois S, Maher AC, Manias JL, Shao Q, Kidder GM, Laird DW (2007) Connexin levels regulate keratinocyte differentiation in the epidermis. J Biol Chem 282(41):30171–30180
Larue L, Ohsugi M, Hirchenhain J, Kemler R (1994) E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc Natl Acad Sci U S A 91:8263–8267
Lehmann JM, Holzmann B, Breitbart EW, Schmiegelow P, Riethmuller G, Johnson JP (1987) Discrimination between benign and malignant cells of melanocytic lineage by two novel antigens, a glycoprotein with a molecular weight of 113,000 and a protein with a molecular weight of 76,000. Cancer Res 47(3):841–845
Lehmann JM, Riethmuller G, Johnson JP (1989) MUC18, a marker of tumor progression in human melanoma, shows sequence similarity to the neural cell adhesion molecules of the immunoglobulin superfamily. Proc Natl Acad Sci U S A 86(24):9891–9895
Lei X, Guan CW, Song Y, Wang H (2015) The multifaceted role of CD146/MCAM in the promotion of melanoma progression. Cancer Cell Int 15(1):3. doi:10.1186/s12935-014-0147-z
Leotlela PD, Wade MS, Duray PH, Rhode MJ, Brown HF, Rosenthal DT, Dissanayake SK, Earley R, Indig FE, Nickoloff BJ, Taub DD, Kallioniemi OP, Meltzer P, Morin PJ, Weeraratna AT (2007) Claudin-1 overexpression in melanoma is regulated by PKC and contributes to melanoma cell motility. Oncogene 26(26):3846–3856. doi:10.1038/sj.onc.1210155, 1210155 [pii]
Li D, Mrsny RJ (2000) Oncogenic Raf-1 disrupts epithelial tight junctions via downregulation of occludin. J Cell Biol 148(4):791–800
Li G, Satyamoorthy K, Herlyn M (2001) N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res 61:3819–3825
Li Q, Omori Y, Nishikawa Y, Yoshioka T, Yamamoto Y, Enomoto K (2007) Cytoplasmic accumulation of connexin32 protein enhances motility and metastatic ability of human hepatoma cells in vitro and in vivo. Int J Cancer 121(3):536–546. doi:10.1002/ijc.22696
Liu S, Kumar SM, Lu H, Liu A, Yang R, Pushparajan A, Guo W, Xu X (2012) MicroRNA-9 up-regulates E-cadherin through inhibition of NF-kB1-Snail1 pathway in melanoma. J Pathol 226:61–72
Lin JH, Takano T, Cotrina ML, Arcuino G, Kang J, Liu S, Gao Q, Jiang L, Li F, Lichtenberg-Frate H, Haubrich S, Willecke K, Goldman SA, Nedergaard M (2002) Connexin 43 enhances the adhesivity and mediates the invasion of malignant glioma cells. J Neurosci 22(11):4302–4311, 2002645022/11/4302 [pii]
Lin Q, Balasubramanian K, Fan D, Kim SJ, Guo L, Wang H, Bar-Eli M, Aldape KD, Fidler IJ (2010) Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia 12(9):748–754
Liu XS, Genet MD, Haines JE, Mehanna EK, Wu S, Chen HI, Chen Y, Qureshi AA, Han J, Chen X, Fisher DE, Pandolfi PP, Yuan ZM (2015) ZBTB7A suppresses melanoma metastasis by transcriptionally repressing MCAM. Mol Cancer Res 13(8):1206–1217. doi:10.1158/1541-7786.MCR-15-0169
Loewenstein WR (1981) Junctional intercellular communication: the cell-to-cell membrane channel. Physiol Rev 61(4):829–913
Loewenstein WR, Kanno Y (1966) Intercellular communication and the control of tissue growth: lack of communication between cancer cells. Nature 209(5029):1248–1249
Loewenstein WR, Rose B (1992) The cell-cell channel in the control of growth. Semin Cell Biol 3(1):59–79
Long H, Crean CD, Lee WH, Cummings OW, Gabig TG (2001) Expression of Clostridium perfringens enterotoxin receptors claudin-3 and claudin-4 in prostate cancer epithelium. Cancer Res 61(21):7878–7881
Maass K, Ghanem A, Kim JS, Saathoff M, Urschel S, Kirfel G, Grummer R, Kretz M, Lewalter T, Tiemann K, Winterhager E, Herzog V, Willecke K (2004) Defective epidermal barrier in neonatal mice lacking the C-terminal region of connexin43. Mol Biol Cell 15(10):4597–4608
Man YK, Trolove C, Tattersall D, Thomas AC, Papakonstantinopoulou A, Patel D, Scott C, Chong J, Jagger DJ, O’Toole EA, Navsaria H, Curtis MA, Kelsell DP (2007) A deafness-associated mutant human connexin 26 improves the epithelial barrier in vitro. J Membr Biol 218(1–3):29–37
Martin TA, Jiang WG (2001) Tight junctions and their role in cancer metastasis. Histol Histopathol 16(4):1183–1195
Martin TA, Jiang WG (2009) Loss of tight junction barrier function and its role in cancer metastasis. Biochim Biophys Acta 1788(4):872–891. doi:10.1016/j.bbamem.2008.11.005, S0005-2736(08)00373-8 [pii]
Massoumi R, Kuphal S, Hellerbrand C, Haas B, Wild P, Spruss T, Pfeifer A, Fassler R, Bosserhoff AK (2009) Down-regulation of CYLD expression by snail promotes tumor progression in malignant melanoma. J Exp Med 206:221–232
McCarty MF, Bielenberg DR, Nilsson MB, Gershenwald JE, Barnhill RL, Ahearne P, Bucana CD, Fidler IJ (2003) Epidermal hyperplasia overlying human melanoma correlates with tumour depth and angiogenesis. Melanoma Res 13(4):379–387
McLachlan E, Shao Q, Wang HL, Langlois S, Laird DW (2006) Connexins act as tumor suppressors in three-dimensional mammary cell organoids by regulating differentiation and angiogenesis. Cancer Res 66(20):9886–9894. doi:10.1158/0008-5472.CAN-05-4302, 66/20/9886 [pii]
Meier F, Busch S, Gast D, Goppert A, Altevogt P, Maczey E, Riedle S, Garbe C, Schittek B (2006) The adhesion molecule L1 (CD171) promotes melanoma progression. Int J Cancer 119(3):549–555
Meier F, Schittek B, Busch S, Garbe C, Smalley K, Satyamoorthy K, Li G, Herlyn M (2005) The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci 10(2986–3001):2986–3001
Mese G, Richard G, White TW (2007) Gap junctions: basic structure and function. J Invest Dermatol 127(11):2516–2524
Mesnil M, Crespin S, Avanzo JL, Zaidan-Dagli ML (2005) Defective gap junctional intercellular communication in the carcinogenic process. Biochim Biophys Acta 1719(1–2):125–145. doi:10.1016/j.bbamem.2005.11.004, S0005-2736(05)00361-5 [pii]
Miekus K, Czernik M, Sroka J, Czyz J, Madeja Z (2005) Contact stimulation of prostate cancer cell migration: the role of gap junctional coupling and migration stimulated by heterotypic cell-to-cell contacts in determination of the metastatic phenotype of dunning rat prostate cancer cells. Biol Cell 97(12):893–903. doi:10.1042/BC20040129
Miele ME, Bennett CF, Miller BE, Welch DR (1994) Enhanced metastatic ability of TNF-alpha-treated malignant melanoma cells is reduced by intercellular adhesion molecule-1 (ICAM-1, CD54) antisense oligonucleotides. Exp Cell Res 214(1):231–241
Minchinton AI, Tannock IF (2006) Drug penetration in solid tumours. Nat Rev Cancer 6(8):583–592
Mitic LL, Anderson JM (1998) Molecular architecture of tight junctions. Annu Rev Physiol 60:121–142
Miwa N, Furuse M, Tsukita S, Niikawa N, Nakamura Y, Furukawa Y (2000) Involvement of claudin-1 in the beta-catenin/Tcf signaling pathway and its frequent upregulation in human colorectal cancers. Oncol Res 12(11–12):469–476
Moennikes O, Buchmann A, Willecke K, Traub O, Schwarz M (2000) Hepatocarcinogenesis in female mice with mosaic expression of connexin32. Hepatology 32(3):501–506. doi:10.1053/jhep.2000.16598, S0270913900790100
Molina-Ortiz I, Bartolome RA, Hernandez-Varas P, Colo GP, Teixido J (2009) Overexpression of E-cadherin on melanoma cells inhibits chemokine-promoted invasion involving p190RhoGAP/p120ctn-dependent inactivation of RhoA. J Biol Chem 284:15147–15157
Montgomery AM, Becker JC, Siu CH, Lemmon VP, Cheresh DA, Pancook JD, Zhao X, Reisfeld RA (1996) Human neural cell adhesion molecule L1 and rat homologue NILE are ligands for integrin alpha v beta 3. J Cell Biol 132(3):475–485
Mori R, Power KT, Wang CM, Martin P, Becker DL (2006) Acute downregulation of connexin43 at wound sites leads to a reduced inflammatory response, enhanced keratinocyte proliferation and wound fibroblast migration. J Cell Sci 119(Pt 24):5193–5203
Morita K, Morita NI, Nemoto K, Nakamura Y, Miyachi Y, Muto M (2008) Expression of claudin in melanoma cells. J Dermatol 35(1):36–38. doi:10.1111/j.1346-8138.2007.00409.x, JDE409 [pii]
Mortarini R, Anichini A (1993) From adhesion to signalling: roles of integrins in the biology of human melanoma. Melanoma Res 3:87–97
Mullin JM, Kampherstein JA, Laughlin KV, Saladik DT, Soler AP (1997) Transepithelial paracellular leakiness induced by chronic phorbol ester exposure correlates with polyp-like foci and redistribution of protein kinase C-alpha. Carcinogenesis 18(12):2339–2345
Muller EJ, Williamson L, Kolly C, Suter MM (2008) Outside-in signaling through integrins and cadherins: a central mechanism to control epidermal growth and differentiation? J Invest Dermatol 128:501–516
Nacht M, Ferguson AT, Zhang W, Petroziello JM, Cook BP, Gao YH, Maguire S, Riley D, Coppola G, Landes GM, Madden SL, Sukumar S (1999) Combining serial analysis of gene expression and array technologies to identify genes differentially expressed in breast cancer. Cancer Res 59(21):5464–5470
Naoi Y, Miyoshi Y, Taguchi T, Kim SJ, Arai T, Tamaki Y, Noguchi S (2007) Connexin26 expression is associated with lymphatic vessel invasion and poor prognosis in human breast cancer. Breast Cancer Res Treat 106(1):11–17. doi:10.1007/s10549-006-9465-8
Natali P, Nicotra MR, Cavaliere R, Bigotti A, Romano G, Temponi M, Ferrone S (1990) Differential expression of intercellular adhesion molecule 1 in primary and metastatic melanoma lesions. Cancer Res 50(4):1271–1278
Natali PG, Hamby CV, Felding-Habermann B, Liang B, Nicotra MR, Di Filippo F, Giannarelli D, Temponi M, Ferrone S (1997) Clinical significance of alpha(v)beta3 integrin and intercellular adhesion molecule-1 expression in cutaneous malignant melanoma lesions. Cancer Res 57(8):1554–1560
Natali PG, Nicotra MR, Bartolazzi A, Cavaliere R, Bigotti A (1993) Integrin expression in cutaneous malignant melanoma: association of the alpha 3/beta 1 heterodimer with tumor progression. Int J Cancer 54:68–72
Naus CC, Laird DW (2010) Implications and challenges of connexin connections to cancer. Nat Rev Cancer 10(6):435–441. doi:10.1038/nrc2841, nrc2841 [pii]
Nikolaev SI, Rimoldi D, Iseli C, Valsesia A, Robyr D, Gehrig C, Harshman K, Guipponi M, Bukach O, Zoete V, Michielin O, Muehlethaler K, Speiser D, Beckmann JS, Xenarios I, Halazonetis TD, Jongeneel CV, Stevenson BJ, Antonarakis SE (2011) Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet 44:133–139
Nishimura EK, Yoshida H, Kunisada T, Nishikawa SI (1999) Regulation of E- and P-cadherin expression correlated with melanocyte migration and diversification. Dev Biol 215:155–166
Nolte C, Moos M, Schachner M (1999) Immunolocalization of the neural cell adhesion molecule L1 in epithelia of rodents. Cell Tissue Res 298(2):261–273
Omori Y, Yamasaki H (1998) Mutated connexin43 proteins inhibit rat glioma cell growth suppression mediated by wild-type connexin43 in a dominant-negative manner. Int J Cancer 78(4):446–453. doi:10.1002/(SICI)1097-0215(19981109)78:4<446::AID-IJC10>3.0.CO;2-4 [pii]
Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA (2008) Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res 68:3645–3654
Overduin M, Harvey TS, Bagby S, Tong KI, Yau P, Takeichi M, Ikura M (1995) Solution structure of the epithelial cadherin domain responsible for selective cell adhesion. Science 267:386–389
Papusheva E, Heisenberg CP (2010) Spatial organization of adhesion: force-dependent regulation and function in tissue morphogenesis. EMBO J 29:2753–2768
Park CC, Bissell MJ, Barcellos-Hoff MH (2000) The influence of the microenvironment on the malignant phenotype. Mol Med Today 6(8):324–329
Patel DD, Wee SF, Whichard LP, Bowen MA, Pesando JM, Aruffo A, Haynes BF (1995) Identification and characterization of a 100-kD ligand for CD6 on human thymic epithelial cells. J Exp Med 181(4):1563–1568
Pearl RA, Pacifico MD, Richman PI, Wilson GD, Grover R (2008) Stratification of patients by melanoma cell adhesion molecule (MCAM) expression on the basis of risk: implications for sentinel lymph node biopsy. J Plast Reconstr Aesthet Surg 61(3):265–271
Pece S, Gutkind JS (2000) Signaling from E-cadherins to the MAPK pathway by the recruitment and activation of epidermal growth factor receptors upon cell-cell contact formation. J Biol Chem 275:41227–41233
Penna E, Orso F, Cimino D, Vercellino I, Grassi E, Quaglino E, Turco E, Taverna D (2013) miR-214 coordinates melanoma progression by upregulating ALCAM through TFAP2 and miR-148b downmodulation. Cancer Res 73(13):4098–4111. doi:10.1158/0008-5472.CAN-12-3686
Penuela S, Gehi R, Laird DW (2013) The biochemistry and function of pannexin channels. Biochim Biophys Acta 1828(1):15–22. doi:10.1016/j.bbamem.2012.01.017
Perrais M, Chen X, Perez-Moreno M, Gumbiner BM (2007) E-cadherin homophilic ligation inhibits cell growth and epidermal growth factor receptor signaling independently of other cell interactions. Mol Biol Cell 18:2013–2025
Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A (2001) A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem 276(29):27424–27431
Pollmann MA, Shao Q, Laird DW, Sandig M (2005) Connexin 43 mediated gap junctional communication enhances breast tumor cell diapedesis in culture. Breast Cancer Res 7(4):R522–R534. doi:10.1186/bcr1042, bcr1042 [pii]
Pollok S, Pfeiffer AC, Lobmann R, Wright CS, Moll I, Martin PE, Brandner JM (2011) Connexin 43 mimetic peptide Gap27 reveals potential differences in the role of Cx43 in wound repair between diabetic and non-diabetic cells. J Cell Mol Med 15(4):861–873. doi:10.1111/j.1582-4934.2010.01057.x
Poser I, Dominguez D, de Herreros AG, Varnai A, Buettner R, Bosserhoff AK (2001) Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. J Biol Chem 276:24661–24666
Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R (2008) MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A 105:1608–1613
Playford MP, Schaller MD (2004) The interplay between Src and integrins in normal and tumor biology. Oncogene 23:7928–7946
Pummi K, Malminen M, Aho H, Karvonen SL, Peltonen J, Peltonen S (2001) Epidermal tight junctions: ZO-1 and occludin are expressed in mature, developing, and affected skin and in vitro differentiating keratinocytes. J Invest Dermatol 117(5):1050–1058
Qi J, Wang J, Romanyuk O, Siu CH (2006) Involvement of Src family kinases in N-cadherin phosphorylation and beta-catenin dissociation during transendothelial migration of melanoma cells. Mol Biol Cell 17:1261–1272
Qian X, Karpova T, Sheppard AM, McNally J, Lowy DR (2004) E-cadherin-mediated adhesion inhibits ligand-dependent activation of diverse receptor tyrosine kinases. EMBO J 23:1739–1748
Rangel LB, Agarwal R, D’Souza T, Pizer ES, Alo PL, Lancaster WD, Gregoire L, Schwartz DR, Cho KR, Morin PJ (2003) Tight junction proteins claudin-3 and claudin-4 are frequently overexpressed in ovarian cancer but not in ovarian cystadenomas. Clin Cancer Res 9(7):2567–2575
Rapanotti MC, Suarez Viguria TM, Costanza G, Ricozzi I, Pierantozzi A, Di Stefani A, Campione E, Bernardini S, Chimenti S, Orlandi A, Bianchi L (2014) Sequential molecular analysis of circulating MCAM/MUC18 expression: a promising disease biomarker related to clinical outcome in melanoma. Arch Dermatol Res 306(6):527–537. doi:10.1007/s00403-014-1473-7
Ren J, Hamada J, Takeichi N, Fujikawa S, Kobayashi H (1990) Ultrastructural differences in junctional intercellular communication between highly and weakly metastatic clones derived from rat mammary carcinoma. Cancer Res 50(2):358–362
Rezze GG, Fregnani JH, Duprat J, Landman G (2011) Cell adhesion and communication proteins are differentially expressed in melanoma progression model. Hum Pathol 42(3):409–418. doi:10.1016/j.humpath.2010.09.004
Richard G (2000) Connexins: a connection with the skin. Exp Dermatol 9(2):77–96
Rickelt S, Franke WW, Doerflinger Y, Goerdt S, Brandner JM, Peitsch WK (2008) Subtypes of melanocytes and melanoma cells distinguished by their intercellular contacts: heterotypic adherens junctions, adhesive associations, and dispersed desmoglein 2 glycoproteins. Cell Tissue Res 334:401–422
Rodriguez M, Aladowicz E, Lanfrancone L, Goding CR (2008) Tbx3 represses E-cadherin expression and enhances melanoma invasiveness. Cancer Res 68(19):7872–7881
Rubina KA, Yurlova EI, Sysoeva VY, Semina EV, Kalinina NI, Poliakov AA, Mikhaylova IN, Andronova NV, Treshalina HM (2013) T-cadherin stimulates melanoma cell proliferation and mesenchymal stromal cell recruitment, but inhibits angiogenesis in a mouse melanoma model. In: Jianyuan Chai (ed) Cardiology and Cardiovascular Medicine. “Research Directions in Tumor Angiogenesis”, pp 143–174
Sadeqzadeh E, de Bock CE, Zhang XD, Shipman KL, Scott NM, Song C, Yeadon T, Oliveira CS, Jin B, Hersey P, Boyd AW, Burns GF, Thorne RF (2011) Dual processing of FAT1 cadherin protein by human melanoma cells generates distinct protein products. J Biol Chem 286:28181–28191
Saccheri F, Pozzi C, Avogadri F, Barozzi S, Faretta M, Fusi P, Rescigno M (2010) Bacteria-induced gap junctions in tumors favor antigen cross-presentation and antitumor immunity. Sci Transl Med 2(44):44ra57. doi:10.1126/scitranslmed.3000739
Saito-Katsuragi M, Asada H, Niizeki H, Katoh F, Masuzawa M, Tsutsumi M, Kuniyasu H, Ito A, Nojima H, Miyagawa S (2007) Role for connexin 26 in metastasis of human malignant melanoma: communication between melanoma and endothelial cells via connexin 26. Cancer 110(5):1162–1172
Saito Y, Takazawa H, Uzawa K, Tanzawa H, Sato K (1998) Reduced expression of E-cadherin in oral squamous cell carcinoma: relationship with DNA methylation of 5’ CpG island. Int J Oncol 12:293–298
Sakai R, Henderson JT, O’Bryan JP, Elia AJ, Saxton TM, Pawson T (2000) The mammalian ShcB and ShcC phosphotyrosine docking proteins function in the maturation of sensory and sympathetic neurons. Neuron 28:819–833
Sanders DS, Blessing K, Hassan GA, Bruton R, Marsden JR, Jankowski J (1999) Alterations in cadherin and catenin expression during the biological progression of melanocytic tumours. Mol Pathol 52:151–157
Santiago-Walker A, Li L, Haass NK, Herlyn M (2009) Melanocytes: from morphology to application. Skin Pharmacol Physiol 22(2):114–121
Sargen MR, Gormley RH, Pasha TL, Yum S, Acs G, Xu X, Zhang PJ (2013) Melanocytic tumors express connexin 43 but not 26: immunohistochemical analysis with potential significance in melanocytic oncogenesis. Am J Dermatopathol 35(8):813–7. doi:10.1097/DAD.0b013e318278d401
Satyamoorthy K, Muyrers J, Meier F, Patel D, Herlyn M (2001) Mel-CAM-specific genetic suppressor elements inhibit melanoma growth and invasion through loss of gap junctional communication. Oncogene 20(34):4676–4684
Saunders MM, Seraj MJ, Li Z, Zhou Z, Winter CR, Welch DR, Donahue HJ (2001) Breast cancer metastatic potential correlates with a breakdown in homospecific and heterospecific gap junctional intercellular communication. Cancer Res 61(5):1765–1767
Schadendorf D, Gawlik C, Haney U, Ostmeier H, Suter L, Czarnetzki BM (1993) Tumour progression and metastatic behaviour in vivo correlates with integrin expression on melanocytic tumours. J Pathol 170(4):429–434
Schadendorf D, Heidel J, Gawlik C, Suter L, Czarnetzki BM (1995) Association with clinical outcome of expression of VLA-4 in primary cutaneous malignant melanoma as well as P-selectin and E-selectin on intratumoral vessels. J Natl Cancer Inst 87(5):366–371
Schaller MD (2001) Biochemical signals and biological responses elicited by the focal adhesion kinase. Biochim Biophys Acta 1540:1–21
Schiffner S, Zimara N, Schmid R, Bosserhoff AK (2011) p54nrb is a new regulator of progression of malignant melanoma. Carcinogenesis 32(8):1176–1182. doi:10.1093/carcin/bgr103
Schmitt CJ, Franke WW, Goerdt S, Falkowska-Hansen B, Rickelt S, Peitsch WK (2007) Homo- and heterotypic cell contacts in malignant melanoma cells and desmoglein 2 as a novel solitary surface glycoprotein. J Invest Dermatol 127(9):2191–2206. doi:10.1038/sj.jid.5700849, 5700849 [pii]
Seftor RE, Seftor EA, Hendrix MJ (1999) Molecular role(s) for integrins in human melanoma invasion. Cancer Metastasis Rev 18:359–375
Shao Q, Wang H, McLachlan E, Veitch GI, Laird DW (2005) Down-regulation of Cx43 by retroviral delivery of small interfering RNA promotes an aggressive breast cancer cell phenotype. Cancer Res 65(7):2705–2711. doi:10.1158/0008-5472.CAN-04-2367, 65/7/2705 [pii]
Shen Y, Khusial PR, Li X, Ichikawa H, Moreno AP, Goldberg GS (2007) SRC utilizes Cas to block gap junctional communication mediated by connexin43. J Biol Chem 282(26):18914–18921. doi:10.1074/jbc.M608980200, M608980200 [pii]
Shih IM, Elder DE, Speicher D, Johnson JP, Herlyn M (1994) Isolation and functional characterization of the A32 melanoma-associated antigen. Cancer Res 54(9):2514–2520
Shih IM, Speicher D, Hsu MY, Levine E, Herlyn M (1997a) Melanoma cell-cell interactions are mediated through heterophilic Mel-CAM/ligand adhesion. Cancer Res 57(17):3835–3840
Shih LM, Hsu MY, Palazzo JP, Herlyn M (1997b) The cell-cell adhesion receptor Mel-CAM acts as a tumor suppressor in breast carcinoma. Am J Pathol 151(3):745–751
Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, Ben Ze’ev A (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A 96:5522–5527
Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD (2000) FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol 2:249–256
Sienel W, Dango S, Woelfle U, Morresi-Hauf A, Wagener C, Brummer J, Mutschler W, Passlick B, Pantel K (2003) Elevated expression of carcinoembryonic antigen-related cell adhesion molecule 1 promotes progression of non-small cell lung cancer. Clin Cancer Res 9(6):2260–2266
Silye R, Karayiannakis AJ, Syrigos KN, Poole S, van Noorden S, Batchelor W, Regele H, Sega W, Boesmueller H, Krausz T, Pignatelli M (1998) E-cadherin/catenin complex in benign and malignant melanocytic lesions. J Pathol 186:350–355
Smalley KS, Brafford P, Haass NK, Brandner JM, Brown E, Herlyn M (2005) Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am J Pathol 166(5):1541–1554
Soler AP, Marano CW, Bryans M, Miller RD, Garulacan LA, Mauldin SK, Stamato TD, Mullin JM (1999) Activation of NF-kappaB is necessary for the restoration of the barrier function of an epithelium undergoing TNF-alpha-induced apoptosis. Eur J Cell Biol 78(1):56–66
Spangler B, Kappelmann M, Schittek B, Meierjohann S, Vardimon L, Bosserhoff AK, Kuphal S (2012) ETS1/RhoC signaling regulates the transcription factor c-Jun in melanoma. Int J Cancer 130:2801–2811
Spray DC (1994) Physiological and pharmacological regulation of gap junction channels. In: Citi S (ed) Molecular mechanisms of epithelial cell junctions: from development to disease. R. G. Landes Company, Austin, pp 195–215
Stevenson BR, Keon BH (1998) The tight junction: morphology to molecules. Annu Rev Cell Dev Biol 14:89–109
Stoletov K, Strnadel J, Zardouzian E, Momiyama M, Park FD, Kelber JA, Pizzo DP, Hoffman R, Vandenberg SR, Klemke RL (2013) Role of connexins in metastatic breast cancer and melanoma brain colonization. J Cell Sci 126(Pt 4):904–913. doi:10.1242/jcs.112748
Strathdee G (2002) Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol 12:373–379
Su YA, Bittner ML, Chen Y, Tao L, Jiang Y, Zhang Y, Stephan DA, Trent JM (2000) Identification of tumor-suppressor genes using human melanoma cell lines UACC903, UACC903(+6), and SRS3 by comparison of expression profiles. Mol Carcinog 28(2):119–127
Tada J, Hashimoto K (1997) Ultrastructural localization of gap junction protein connexin 43 in normal human skin, basal cell carcinoma, and squamous cell carcinoma. J Cutan Pathol 24(10):628–635
Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y (2005) Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res 11(20):7234–7242
Tang A, Eller MS, Hara M, Yaar M, Hirohashi S, Gilchrest BA (1994) E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J Cell Sci 107:983–992
Temme A, Buchmann A, Gabriel HD, Nelles E, Schwarz M, Willecke K (1997) High incidence of spontaneous and chemically induced liver tumors in mice deficient for connexin32. Curr Biol 7(9):713–716
Thies A, Berlin A, Brunner G, Schulze HJ, Moll I, Pfuller U, Wagener C, Schachner M, Altevogt P, Schumacher U (2007) Glycoconjugate profiling of primary melanoma and its sentinel node and distant metastases: implications for diagnosis and pathophysiology of metastases. Cancer Lett 248(1):68–80
Thies A, Moll I, Berger J, Wagener C, Brummer J, Schulze HJ, Brunner G, Schumacher U (2002a) CEACAM1 expression in cutaneous malignant melanoma predicts the development of metastatic disease. J Clin Oncol 20(10):2530–2536
Thies A, Schachner M, Moll I, Berger J, Schulze HJ, Brunner G, Schumacher U (2002b) Overexpression of the cell adhesion molecule L1 is associated with metastasis in cutaneous malignant melanoma. Eur J Cancer 38(13):1708–1716
Trimmer C, Whitaker-Menezes D, Bonuccelli G, Milliman JN, Daumer KM, Aplin AE, Pestell RG, Sotgia F, Lisanti MP, Capozza F (2010) CAV1 inhibits metastatic potential in melanomas through suppression of the integrin/Src/FAK signaling pathway. Cancer Res 70:7489–7499
Trosko JE, Chang CC, Madhukar BV, Klaunig JE (1990) Chemical, oncogene and growth factor inhibition gap junctional intercellular communication: an integrative hypothesis of carcinogenesis. Pathobiology 58(5):265–278
Troussard AA, Tan C, Yoganathan TN, Dedhar S (1999) Cell-extracellular matrix interactions stimulate the AP-1 transcription factor in an integrin-linked kinase- and glycogen synthase kinase 3-dependent manner. Mol Cell Biol 19:7420–7427
Tsai H, Werber J, Davia MO, Edelman M, Tanaka KE, Melman A, Christ GJ, Geliebter J (1996) Reduced connexin 43 expression in high grade, human prostatic adenocarcinoma cells. Biochem Biophys Res Commun 227(1):64–69
Tsukita S, Furuse M (2000) Pores in the wall: claudins constitute tight junction strands containing aqueous pores. J Cell Biol 149(1):13–16
Tsukita S, Furuse M, Itoh M (2001) Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol 2(4):285–293
Uchida Y, Matsuda K, Sasahara K, Kawabata H, Nishioka M (1995) Immunohistochemistry of gap junctions in normal and diseased gastric mucosa of humans. Gastroenterology 109(5):1492–1496
van Marck V, Stove C, Van Den Bossche K, Stove V, Paredes J, Vander Haeghen Y (2005) P-cadherin promotes cell-cell adhesion and counteracts invasion in human melanoma. Cancer Res 65(19):8774–8783
van de Stolpe A, van der Saag PT (1996) Intercellular adhesion molecule-1. J Mol Med 74(1):13–33
van Kempen LC, Meier F, Egeblad M, Kersten-Niessen MJ, Garbe C, Weidle UH, Van Muijen GN, Herlyn M, Bloemers HP, Swart GW (2004) Truncation of activated leukocyte cell adhesion molecule: a gateway to melanoma metastasis. J Invest Dermatol 122(5):1293–1301
van Kempen LC, van den Oord JJ, van Muijen GN, Weidle UH, Bloemers HP, Swart GW (2000) Activated leukocyte cell adhesion molecule/CD166, a marker of tumor progression in primary malignant melanoma of the skin. Am J Pathol 156(3):769–774
van Noort M, Clevers H (2002) TCF transcription factors, mediators of Wnt-signaling in development and cancer. Dev Biol 244:1–8
Veenstra RD (1996) Size and selectivity of gap junction channels formed from different connexins. J Bioenerg Biomembr 28(4):327–337
Villanueva J, Herlyn M (2008) Melanoma and the tumor microenvironment. Curr Oncol Rep 10(5):439–446
Villares GJ, Dobroff AS, Wang H, Zigler M, Melnikova VO, Huang L, Bar-Eli M (2009) Overexpression of protease-activated receptor-1 contributes to melanoma metastasis via regulation of connexin 43. Cancer Res 69(16):6730–6737. doi:10.1158/0008-5472.CAN-09-0300, 69/16/6730 [pii]
Villares GJ, Zigler M, Bar-Eli M (2011) The emerging role of the thrombin receptor (PAR-1) in melanoma metastasis – a possible therapeutic target. Oncotarget 2(1–2):8–17
Voura EB, Ramjeesingh RA, Montgomery AM, Siu CH (2001) Involvement of integrin alpha(v)beta(3) and cell adhesion molecule L1 in transendothelial migration of melanoma cells. Mol Biol Cell 12(9):2699–2710
Watson-Hurst K, Becker D (2006) The role of N-cadherin, MCAM and beta3 integrin in melanoma progression, proliferation, migration and invasion. Cancer Biol Ther 5:1375–1382
Weinstein RS, Merk FB, Alroy J (1976) The structure and function of intercellular junctions in cancer. Adv Cancer Res 23:23–89
Wilgenbus KK, Kirkpatrick CJ, Knuechel R, Willecke K, Traub O (1992) Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int J Cancer 51(4):522–529
Wiklund ED, Bramsen JB, Hulf T, Dyrskjot L, Ramanathan R, Hansen TB, Villadsen SB, Gao S, Ostenfeld MS, Borre M, Peter ME, Orntoft TF, Kjems J, Clark SJ (2011) Coordinated epigenetic repression of the miR-200 family and miR-205 in invasive bladder cancer. Int J Cancer 128:1327–1334
Willecke K, Eiberger J, Degen J, Eckardt D, Romualdi A, Guldenagel M, Deutsch U, Sohl G (2002) Structural and functional diversity of connexin genes in the mouse and human genome. Biol Chem 383(5):725–737
Wolburg H, Wolburg-Buchholz K, Kraus J, Rascher-Eggstein G, Liebner S, Hamm S, Duffner F, Grote EH, Risau W, Engelhardt B (2003) Localization of claudin-3 in tight junctions of the blood–brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol (Berl) 105(6):586–592
Wong RP, Ng P, Dedhar S, Li G (2007) The role of integrin-linked kinase in melanoma cell migration, invasion, and tumor growth. Mol Cancer Ther 6:1692–1700
Wright CS, van Steensel MA, Hodgins MB, Martin PE (2009) Connexin mimetic peptides improve cell migration rates of human epidermal keratinocytes and dermal fibroblasts in vitro. Wound Repair Regen 17(2):240–249. doi:10.1111/j.1524-475X.2009.00471.x
Xie S, Luca M, Huang S, Gutman M, Reich R, Johnson JP, Bar-Eli M (1997) Expression of MCAM/MUC18 by human melanoma cells leads to increased tumor growth and metastasis. Cancer Res 57(11):2295–2303
Xu J, Nicholson BJ (2013) The role of connexins in ear and skin physiology – functional insights from disease-associated mutations. Biochim Biophys Acta 1828(1):167–178. doi:10.1016/j.bbamem.2012.06.024
Yamaoka K, Nouchi T, Tazawa J, Hiranuma S, Marumo F, Sato C (1995) Expression of gap junction protein connexin 32 and E-cadherin in human hepatocellular carcinoma. J Hepatol 22(5):536–539, 0168-8278(95)80447-1 [pii]
Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117(7):927–939
Yazawa EM, Geddes-Sweeney JE, Cedeno-Laurent F, Walley KC, Barthel SR, Opperman MJ, Liang J, Lin JY, Schatton T, Laga AC, Mihm MC, Qureshi AA, Widlund HR, Murphy GF, Dimitroff CJ (2015) Melanoma cell galectin-1 ligands functionally correlate with malignant potential. J Invest Dermatol 135(7):1849–1862. doi:10.1038/jid.2015.95
Zhang P, Goodrich C, Fu C, Dong C (2014) Melanoma upregulates ICAM-1 expression on endothelial cells through engagement of tumor CD44 with endothelial E-selectin and activation of a PKCalpha-p38-SP-1 pathway. FASEB J 28(11):4591–4609. doi:10.1096/fj.11-202747
Zhang ZQ, Zhang W, Wang NQ, Bani-Yaghoub M, Lin ZX, Naus CC (1998) Suppression of tumorigenicity of human lung carcinoma cells after transfection with connexin43. Carcinogenesis 19(11):1889–1894
Zhu D, Caveney S, Kidder GM, Naus CC (1991) Transfection of C6 glioma cells with connexin 43 cDNA: analysis of expression, intercellular coupling, and cell proliferation. Proc Natl Acad Sci U S A 88(5):1883–1887
Zigler M, Kamiya T, Brantley EC, Villares GJ, Bar-Eli M (2011) PAR-1 and thrombin: the ties that bind the microenvironment to melanoma metastasis. Cancer Res 71(21):6561–6566. doi:10.1158/0008-5472.CAN-11-1432
Zucker SN, Bancroft TA, Place DE, Des Soye B, Bagati A, Berezney R (2013) A dominant negative Cx43 mutant differentially affects tumorigenic and invasive properties in human metastatic melanoma cells. J Cell Physiol 228(4):853–859. doi:10.1002/jcp.24235
Acknowledgment
NKH is a Cameron Fellow of the Melanoma and Skin Cancer Research Institute, Australia, and a Sydney Medical School Foundation Fellow. NKH also thanks the German Research Foundation (DFG, HA26801), Cancer Council NSW (RG 09–08, RG 13–06), Cancer Australia/Cure Cancer Australia Foundation (570778), Cancer Institute New South Wales (08/RFG/1-27), and the National Health and Medical Research Council Australia (1003637, 1084893) for contributing grant support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Kuphal, S., Haass, N.K. (2017). Cell–Cell Contacts in Melanoma and the Tumor Microenvironment. In: Bosserhoff, A. (eds) Melanoma Development. Springer, Cham. https://doi.org/10.1007/978-3-319-41319-8_9
Download citation
DOI: https://doi.org/10.1007/978-3-319-41319-8_9
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-41317-4
Online ISBN: 978-3-319-41319-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)