
Abstract
Nuclear factor erythroid 2-related factor (Nrf2) is a transcription factor that controls the expression of predominant antioxidant system in the central nervous system (CNS). Under normal conditions, Nrf2 is sequestered by Keap1 and degenerated by the ubiquitin system. Oxidative stress initiates Nrf2 nuclear translocation, leading to expression of antioxidant molecules and enzymes. In stroke, oxidative stress is one of the major causes of neuronal death, and Nrf2 pathway is activated in both in vitro and in vivo ischemic models. In addition to mediate self-defense in neurons, Nrf2 also actively regulates the expression of cytoprotective enzymes in other cell types within the neurovascular unit (NVU), including astrocytes and endothelial cells, and thus supports neuronal function and survival through cell–cell interaction. The roles of microglias in stroke are still controversial, but close to be clarified. In this chapter, we will briefly introduce Nrf2 pathway, followed by its key roles of nonneuronal Nrf2 in limiting ischemic injury and emerging roles in brain tissue repair after stroke.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
Ischemic stroke is one of the leading causes of disability and mortality in the world. Multiple pathological processes participate in the progression of stroke, including excitotoxicity, oxidative stress, inflammation, mitochondrial dysfunction, etc. Excessive oxidative stress is one of the most important pathogenic mechanisms in stroke. Therefore, therapeutic strategies against oxidative stress may be feasible for the treatment of stroke. Given that nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is the predominant antioxidant system, we will focus on the promising protective role of Nrf2 pathway against ischemic stroke.
1 Introduction of Nrf2 Pathway
Nrf2 is a member of the basic leucine zipper (bZIP) transcription factor family featuring a Cap ‘n’ collar structure [1, 2], and it is essential for the transcriptional induction of phase II drug-metabolizing and antioxidant enzymes, such as glutathione s-transferase (GST), heme oxygenase 1 (HO-1), and NADPH quinine oxidoreductase 1 (NQO-1) [3]. Nrf2 regulates gene transcription of these enzymes by binding the antioxidant response elements (AREs) on their promoters.
1.1 Regulation of Nrf2/ARE Pathway
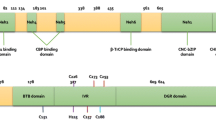
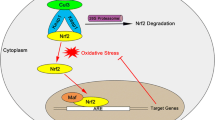
Nrf2 has six functional domains known as Nrf2-ECH homologies (Neh), designated as Neh1-6. Each Neh domain has its own function [4]. Among them, the Neh1 domain is the CNC-bZIP domain, enabling Nrf2 to form heterodimer with the ZIP domain of small musculoaponeurotic fibrosarcoma (Maf) proteins [5]; and the Neh2 domain mediates binding with the cytosolic repressor of Nrf2. Keap1 encompasses three functional domains: a bric-a-brac (BTB) domain, an intervening region (IVR), and a Kelch domain (also named DGR domain). Under non-stressed conditions, Nrf2 forms a complex with Kelch-like ECH-associated protein 1 (Keap1), which promotes rapid proteasomal degradation of Nrf2 by proteasome, giving Nrf2 a half-life time of about 20 min [3]. As a result, Keap1 functions to inhibit Nrf2.
It has been reported that alterations in the structure of Keap1 leads to dissociation of the Nrf2–Keap1 complex, and this is essential for Nrf2/ARE activation [5]. Three cysteine residues (Cys151, Cys273, and Cys288) in Keap1 are essential for Keap1 repression of Nrf2 activity under unstressed conditions [6]. The oxidation of these cysteine residues causes conformational change of Keap1 and leads to dissociation of the Nrf2-Keap1 complex. Some oxidative products can free Nrf2 from the complex and increase the expression of phase II genes [7].
Transcription factors need to translocate to the nucleus in order to transactivate. As for Nrf2, this process is quite rapid: Nrf2 can accumulate in the nucleus within 15 min after tert-Butylhydroquinone (t-BHQ) treatment [8]. The key mediators that regulate nuclear import and export of transcription factors are the nuclear localization signals (NLS) and nuclear export sequences (NES). A number of such nuclear shuttling signals have been identified on Nrf2, including three NLS motifs and two NES motifs. The direction of Nrf2 movement is determined by a homeostatic balance between import and export driving forces. Under normal condition, Nrf2 stays in the cytosol because the import force is less than the export force while NES1 is functional. Under oxidative stress, Cys183 of NES1 is adducted and NES1 loses function. Then the import force is more than the export force, resulting in Nrf2 nuclear translocation [9]. Additionally, Nrf2 nuclear translocation can also be triggered when phosphorylated by kinases. The phosphorylation sites of protein kinase C (PKC), glycogen synthase kinase-3 and Fyn have been identified. The phosphorylation site of PKC is Ser40 in Neh2 domain [10], but its role in Nrf2 nuclear shuttling is controversial. Fyn phosphorylates Tyr568 in the Neh3 domain, controlling nuclear export of Nrf2 [11, 12].
After translocating into the nucleus, Nrf2 can form heterodimer with Maf proteins [13], which further enhances the specificity of Nrf2 to bind to a cis-acting enhancer ARE [14] located at the promoter of phase II genes [1, 15]. Subsequently, the transcription of phase II gene is initiated.
Recent studies have also proposed a Keap1-independent ubiquitination model of Nrf2 degradation [16]; in which, GSK3β is a key player because it can phosphorylate Nrf2 at Ser342 and Ser347 located at Neh6. Phosphorylated Neh6 can bind with an ubiquitin ligase adaptor beta-transducin repeat-containing protein (β-TrCP). β-TrCP is a scaffolding protein that directly links Nrf2 to the Cullin1/Rbx1 ubiquitination complex. Therefore, GSK-3β-mediated phosphorylation of Neh6 causes the ubiquitination and degradation of Nrf2 via β-TrCP. This model is supported by the stabilization of Nrf2 by GSK-3β inhibitors in Keap1−/− mouse embryo fibroblasts (MEFs) [12]. Additionally, cancer-chemo preventive agent nordihydroguaiaretic acid can activate Nrf2 and increase HO-1 protein levels through inhibiting GSK-3β phosphorylation in Keap1−/− MEFs [17].
Additionally, there are some other regulatory mechanisms to prevent excessive activation of Nrf2. For example, AREs are located in the promoter region of Cul3, Rbx1, and Keap1 genes; while activation of Nrf2/ARE pathway boots the expression of phase II genes, it also upregulates these inhibitory proteins at the same time. This negative feedback loop is known as an autoregulatory arm of the Nrf2/ARE pathway [18]. In addition, the Keap1-Cul3-Rbx1 complex can enter the nucleus, mediated by prothymosin α (ProTα), a Keap1-binding protein with a NLS. As a result, 10–15 % of Keap1-Cul3-Rbx1 complex is localized in the nucleus. Once entering the nucleus, the Keap1-Cul3-Rbx1 complex drops ProTα and binds Nrf2, leading to the ubiquitination and degradation of nuclear Nrf2 [19].
1.2 Inducers and Effectors of Nrf2 Pathway
It is important to identify inducers and effectors of Nrf2 pathway, because they could be promising therapeutic targets. As discussed above, Keap1 is the major inhibitor of Nrf2 activation; changes in its structure and/or its dissociation from Nrf2 results in the release and activation of Nrf2, which is recognized as the target mechanisms of nearly all Nrf2 inducers. There are several ways to classify Nrf2 inducers. Based on their origin, inducers are divided into two classes, the exogenous and the endogenous; based on their chemical structure, inducers are divided into ten groups [20]; based on the Keap1 domains that the inducers react with, they can be divided into four categories [21].
Currently, over 200 Nrf2/ARE-driven proteins are described for detoxification and antioxidant defense. Among which, phase 2 enzymes initially caught the interest of scientists for their preventative action against carcinogens [22]. Later, their function has been expanded to neuroprotection [23, 24]. Table 1 lists the major cytoprotective enzymes that participate in protection against neuronal injury in common neurological diseases.
2 Nrf2 Provides Neuroprotection Against Ischemic Stroke Via Self-Defense and Cell–Cell Interaction
In ischemic stroke, several pathological mechanisms known as the ischemic cascade are triggered, and consequently cause rapid and irreversible neuronal death within the ischemic core due to severe and rapid loss of energy. In the surrounding hypoperfused brain tissues, known as the penumbral areas, the neurons may be potentially salvageable due to relatively moderate or mild energy loss. Therefore, salvaging the neurons in penumbra is of great translational significance. To do so, multiple protection and supports may be needed, because neuronal death in penumbra is not a stand-along event, instead, it is the consequence of neuronal injury itself plus dysfunction of all kinds of cells within the neurovascular unit (NVU), including endothelial cells, astrocytes, and others [25–27].
2.1 Oxidative Stress and Nrf2 Activation in Neurons After Brain Ischemia
Following ischemia and reperfusion, the cascade of events that leads to neuronal deaths can be summarized as below [28–30]. Energy failure is the first event of the cascade caused by decreased ATP production, the latter then disrupts Na+/K+-ATPase, Ca2+/H+-ATPase, and Na+/Ca2+-transporter, leading to depolarization of cellular membrane. After depolarization, excitotoxicity amino acids, especially glutamate, are released into the synaptic cleft, which consequently activates N-methyl-d-aspartic acid (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), and metabotropic glutamate receptors increasing intracellular Ca2+ load. A variety of Ca2+ dependent enzymes are then activated and hence induce protein phosphorylation, proteolysis, mitochondrial damage, and the generation of free radical, i.e., reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS and RNS eventually lead to oxidative stress, which cause lipid peroxidation, membrane injury, DNA damage and moreover, allowing the infiltration of leukocytes and activating inflammation [31]. Multiple neuroinflammatory cascades are also activated, inducing secondary brain injury that leads to cell death [32].
During the procedure of neuronal death in the penumbra, oxidative stress triggered by ROS and RNS generation is considered the “key” event. The predominant ROS and RNS produced are superoxide anion (O2 −•), hydrogen peroxide (H2O2), hydroxyl radical (•OH), nitric oxide (•NO), peroxynitrite anion (ONOO−), and nitrogen dioxide (•NO2) [28]. Ischemic insults produce an excessive amount of free radicals especially in the reperfused regions, and the imbalance in the formation and clearance of ROS and RNS leads to oxidative stress, and subsequently alters the cell dynamics [33] and impacts major cellular components [26]. Complex signaling network involving numerous survival/death pathways have been reported to be redox mediated [34]. ROS and perhaps RNS, may act directly as executioners of cell death [31]. ROS leads to increase of mitochondrial permeability transition pore which subsequently causes mitochondrial swelling and neuronal death [35]. ROS also initiates apoptosis through activating p53 and p38 MAPK [36]. Another damage effect acted by ROS to ischemic brain is that ROS can directly increase the permeability of the blood–brain barrier (BBB) [37]. Focal ischemia induces a potent inflammatory response within a few hours after onset of ischemia, and BBB disruption allows various blood immune cells infiltrate into ischemic area [38–40]. These migrated immune cells, along with injured brain cells, produce inflammatory mediators, enhancing neuronal death [41].
The brain is sensitive to oxidative stress. Constituting only 2 % of the total body weight, the brain uses 20 % of the total oxygen consumed by the whole body, making the brain the major oxygen user and probably a major source of ROS [28]. In addition, a high level of unsaturated lipid content, multiple chemical reactions in dopamine oxidation and high concentrations of iron also contribute ROS generation in the brain [42, 43]. To defense against oxidative stress, both direct and indirect antioxidants are needed [44–46]. Direct antioxidants denote a group of low molecular weight compounds that can directly undergo redox reactions and scavenge reactive oxidation products, ROS and RNS. This group includes glutathione (GSH), ascorbate, tocopherols, lipoid acid, carotenes, and ubiquinones. Direct antioxidants are consumed in the process, therefore need replenishment or regeneration. Indirect antioxidants, however, may not present the redox activity themselves. They exert their antioxidant effects through the upregulation phase 2 and antioxidant enzymes (Table 1), or so-called “phase 2 response” [5, 47, 48], which display a wide variety of antioxidant activities.
Several studies suggest that ROS is generated in the brain after ischemia. Peters et al. reported that rats subjected to middle cerebral artery occlusion (MCAO) first exhibited a short period (10–30 min) of decreased ROS level after the occlusion, followed by a significant increase (to 162 ± 51 %; baseline = 100 %) from 100-min ischemia to permanent MCAO onward [49]. Moreover, reperfusion after 1 h of MCAO led to a burst-like pattern of ROS production (about fivefold), and lasted at least to 3 h post MCAO. Liu et al. studied the spatial distribution of ROS in ischemic rat brain and found that ROS was significantly increased in the infarct core during both ischemia and reperfusion, but in penumbra ROS was only increased during reperfusion [50, 51].
Nrf2 pathway is activated in post-ischemic stroke. Tanaka et al. [52] explored the expression of Nrf2 and Keap1 in mice subjected to MCAO using immunohistochemistry and immunofluorescence. Their results showed that, Keap1 was significantly decreased from 24 to 72 h post MCAO, paralleled with increased expression of Nrf2. In consistency with previous results, rats subjected to MCAO also exhibited significantly upregulated Nrf2 expression 24 h later, predominantly located in the penumbra compared to the core [53]. Li et al. [54] detected Nrf2 levels in rat brain by immunohistochemistry and western blotting analyses after MCAO and reperfusion, and found that, Nrf2 and hemeoxygenase-1 (HO-1) were upregulated in ischemic cortex, beginning at 6 h, peaking at 48 h and declining at 72 h after reperfusion. With a novel immunohistochemical technique, Srivastava et al. [55] examined the temporal and spatial distribution of Nrf2 in rat brain following ischemia-reperfusion injury in a detailed way. They used the nuclear/cytoplasmic Nrf2 ratio to indicate the level of Nrf2 activation of Nrf2 pathway, and demonstrated that nuclear to cytoplasmic Nrf2 ratios in stroke-affected regions were increased after 24 h and then declined after 72 h reperfusion. Moreover, Nrf2 expression was significantly higher in the penumbra than the infarct core. Oligemia is another model used to study the stroke penumbra, in which lipid peroxidation induces an increased oxidative stress and an augmented ▪OH production during the reperfusion phase. Liverman et al. [56] adopted this model in mice by lowering the mean arterial pressure to 30–40 mmHg, and reported a significant elevation of Nrf2 level in neurons in the Purkinje cells of the cerebellar cortex and pyramidal neurons of the cingulate cortex.
In short, piles of evidence verify the indispensable role of Nrf2 pathway as a neuronal endogenous protective mechanism through the induction of its downstream antioxidants, including the direct (small molecular antioxidants) and indirect (predominant phase 2 enzyme) ones. These antioxidants then provide a cytoprotective role against ischemia-induced neuronal death.
2.2 Nrf2 Activations in Nonneuronal Cells: The Contribution of Cell–Cell Interaction
As indicated previously, the neuronal death in ischemic stroke is a not a separate procedure; instead, it is caused by additional dysfunction of other types of cells within and beyond the NVU due to complicated cell–cell interactions. Diverse types of cells in the brain work as a unit to maintain the central nervous system (CNS) homeostasis. This can partially explain the reason why neuroprotection trials have mostly failed, as any disruption within the complicated system might lead to NVU dysfunction after brain ischemia [57]. In this regard, therapeutic strategies should not only aim to neurons, but also to salvage other cells and hence restore function of the NVU [26, 29, 57].
Dual-direction responses have been reported between neurons and endothelial cells in the brain. During brain development, microvessel and neuron are already arranged to grow together along the extracellular matrix paths [58–60]. In adult brain, the regulation of regional cerebral blood flow depends on the activity of neurons [61]. Zonta et al. reported that, in a rat hyperemia model, neuronal afferent stimulation mediates the dilation of cerebral arterioles, which is dependent on the glutamate-mediated [Ca2+]i oscillations in astrocytes [25, 62]. On the other hand, some novel discoveries focusing on microvessel structure, endothelial cells, and astrocyte endothelial adhesion indicated an opposite regulatory direct, from microvessel to the neurons they supply [63, 64]. An interesting research by Mabuchi et al. showed that an ordered and sequential microvessel–neuron relationship existed in contralateral basal ganglia; in ischemic area, neurons more distant from their nearest microvessel are more sensitive to ischemia, indicating that neuronal survival is dependent on the microvascular function [65]. Besides, microglias and macrophages play specific roles in the ischemic brain, functioning as a “double-edged sword” by either cleaning up or inducing local inflammation [66, 67]. In the following context, we will discuss the contribution of non-neuronal Nrf2 pathways to neuronal survival.
2.2.1 Nrf2 Pathway in Astrocyte
Comparing to neurons, astrocytes produce large amount of antioxidants [45, 68], and several lines of evidence have demonstrated that astrocytic Nrf2 contributes to neuronal survival following brain ischemia. Kraft and colleagues reported that the activation of astrocytic Nrf2 protected neurons from hydrogen peroxide and glutamate in cell cultures [69]. This protection disappeared in cultures from Nrf2 knockout animals; and the protection could be restored by infecting cells with a replication-deficient adenovirus that carried Nrf2, indicating an important role of astrocytic Nrf2 in neuronal protection. Shih et al. also studied the astrocyte–neuron interaction using a co-culture model of neurons, naïve astrocytes, and infected astrocytes (Nrf2 overexpression cells) [70]. They reported that astrocytes have higher basal Nrf2 expression and ARE activity than neurons, with an increased expression of GSH. When stimulated, the co-culture with oxidative glutamate toxicity, they found that Nrf2 activation led to an increase in both media and intracellular GSH in astrocytes; experiments on selective inhibition of glial GSH synthesis indicated that an Nrf2-dependent increase in glial GSH synthesis was both necessary and sufficient for the protection of neurons [70]. It is not fully understood how astrocytic GSH protects neurons [71]. It has been reported that GSH can scavenge free radicals in the extracellular area [72]. In addition, GSH can be hydrolyzed on the external surface of astrocytes and thus provide high yield of cysteine and glycine, which can be taken up by neurons and used for intracellular GSH synthesis [73].
Astrocytic Nrf2 also contributes to neuronal protection in a mitochondria-dependent manner [74, 75]. 3-nitropropionic acid (3NP) and malonate are two mitochondrial complex II inhibitors, and they can kill neurons probably by inducing ROS generation in mitochondria. Calkins et al. reported that Nrf2-deficient astrocytes and mice were more vulnerable to 3NP or malonate; and they also found that astrocytes showed increased ARE-regulated transcription [76]. If Nrf2-overexpressing astrocytes were transplanted into the brain before 3NP or malonate treatments, a dramatic protection was noticed against complex II inhibition [76]. To extend these findings, they developed a line of transgenic mice with astrocyte-specific overexpression of Nrf2, and found the transgenic mice was resistant to malonate insults, which was associated with elevation of Nrf2-driven genes such as NQO1, GCLM, and HO-1. Furthermore, they showed that malonate toxicity could also be reduced by striatal transplantation of neuroprogenitor cells overexpressing Nrf2, which differentiated into astrocytes after grafting [77].
Astrocytic Nrf2 pathway was also reported to play a positive role in brain protection following ischemic preconditioning [78]. Bell et al. reported that Nrf2 could be activated by mild oxidative stress in both rodent and human astrocytes, and that transient ischemic conditions in vitro and in vivo cause an increase in the expression of Nrf2 target genes, especially the GSH system [78]. Astrocytic Nrf2 also contributes to chemical preconditioning induced by resveratrol [79]. Nrf2 pathway was found activated in astrocytes by resveratrol, and loss of Nrf2 reduced resveratrol -mediated neuroprotection in mice. After resveratrol treatment, both wild-type and Nrf2−/− cortical mitochondria produced ROS, and Nrf2−/− cells showed decreased mitochondrial antioxidant expression and failed to elevate cellular antioxidants after preconditioning. These data suggest that astrocytic Nrf2 pathway is critical in preconditioning-induced neuroprotection [79].
It has also been reported that astrocytes can protect endothelial cells in a GSH-dependent manner. Schroeter et al. reported that astrocytic cultures showed higher antioxidative activity than endothelial cultures, indicated by increased levels of SOD, catalase, and glutathione peroxidase [80]. When cultured these types of cells together, they found an increased antioxidative capacity in endothelial cells. In another report, Laird et al. reported that hemin could induce apoptosis in mouse and human endothelial cells; and that astrocyte-conditioned media could rescue the apoptotic endothelia death, in which GSH played a key role as the protection was aborted by prior treatment of astrocyte with DL-buthionine (S, R)-sulfoximine, a GSH-depleting agent [81].
2.2.2 Nrf2 Pathway in Endothelial Cell
In term of structure, endothelial cells are the center of NVU, as they play important roles in nourishing surrounding cells and keeping the integrity of BBB and NVU. Under pathological conditions, however, endothelial cells may generate ROS that may induce neuronal cell death [82]. When their ROS reacts with nitric oxide (NO), peroxynitrite, a much more powerful oxidant than ROS, will form [83, 84]. In addition to damaged surrounding structures, those ROS and RNS will cause the disruption of BBB integrity following ischemia, in the forms of increased passive diffusion or massive cellular infiltration [85, 86].
As a way of self-defense, endothelial cells equip themselves with Nrf2 system, and several studies suggest that Nrf2 and its target enzymes protect endothelial cells from oxidative insults and sustain BBB integrity following brain ischemia. Alfieri et al. [87] pretreated rats with sulforaphane and then subject them to MCAO followed by reperfusion. They found sulforaphane significantly increased HO-1 expression in brain microvessel and that BBB disruption was attenuated after stroke. Bénardais et al. detected the protective effects of dimethylfumarate (DMF), an Nrf2 activator, on BBB integrity and found that both DMF and its primary metabolite monomethylfumarate (MMF) activated Nrf2 pathway and upregulated NQO1 in brain endothelial cells [88]. Moreover, DMF can partially attenuate TNF-α-induced downregulation of tight junction (TJ) protein [88]. DMF may also protect endothelia cells by stabilizing the BBB via preventing TJ disruption and suppressing the activity of matrix metallopeptidase (MMP) [89]. A similar protective effect was reported by Wu et al. [90] with an oral administration of procyanidin B2, a dietary Nrf2 inducer, 3 h after MCAO. They found that procyanidin B2 significantly reduced the infarct volume, brain edema, and neurological deficits; moreover, they also noticed increased levels of TJ proteins in the brain microvessel and sustained BBB integrity indicated by Evans blue leakage. Nrf2 activation may underlie the observed protections as these protections are associated with increased expression of HO-1, NQO1, and GSTα [90].
In addition to ischemic stroke, BBB protection by Nrf2 pathway is also reported in other CNS disorders, such as subarachnoid hemorrhage (SAH) and traumatic injuries to brain and spinal cord.
SAH is another form of stroke with noticeable vasospasm and subsequent ischemia for several days, and BBB disruption is common after SAH that can result in further brain injury. It has been reported that Nrf2-ARE pathway was activated after SAH, and administration of Nrf2 inducers, such as sulforaphane [91] or t-BHQ [92, 93] can significantly attenuate BBB disruption induced by SAH. In addition to these Nrf2 inducers, some other neuroprotective agents can ameliorate BBB disruption through activating Nrf2 and upregulating HO-1, NAD(P)H, NQO-1; such agents include astaxanthin [94], propofol [95], melatonin [96], and recombinant human erythropoietin [97]. On the other hand, Nrf2 knockout exacerbated brain injury after SAH with increased brain edema, BBB deficits, and decreased GSH [98].
In traumatic brain injury, sulforaphane was reported to preserve BBB function in association with reduced loss of TJ protein and endothelial markers [99]. This protection disappeared in Nrf2 knockout mice or when the animals were pretreated with decoy oligonucleotides containing the binding site of Nrf2 [99]. Similar phenomenon was reported in animals subjected to spinal cord injury. For instance, t-BHQ could alleviate spinal cord edema and suppress the expression of tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1β) [100]. Nrf2−/− mice showed more severe edema after spinal cord injury, which is associated with increased levels of MMP-9 and TNF-α probably caused by an increase in BBB disruption [101]. These studies suggest that Nrf2 pathway contributes to endothelial protection in broad-spectrum conditions.
2.2.3 Nrf2 Pathway in Microglia/Macrophage
For a long time, the brain has been regarded as an “immune privileged” organ for its lack of classical immune response [102, 103]. However, it has become increasingly clear that the brain is actually an immune organ, especially with the discovery of lymphatic vascular system in the brain [104–106], suggesting that complicated interactions may exist between CNS and the immune system.
Microglias are CNS residential immune cells which share a lot of features with monocytes and microphages and provide innate immune responses. They need to consume energy for a broad range of activities, and are thus highly sensitive to energy deficits and local changes in blood perfusion [67]. It is not surprising that microglias are among the first cells to respond to CNS injuries. For example, they are mobilized within 1 h after ischemic stroke, start to proliferate within 72 h, and continue to accumulate for >1 month. The function of microglias in stroke is recognized as a “double-edged sword” [66, 104]; they release cytotoxic cytokines that can kill the other cells including neurons at acute stage after ischemia, but they clear cellular debris via phagocytosis and autophagy and restrict the inflammation at late stage. Therefore, it may be critical to fine-tune the function of microglias and macrophages in order to limit brain damage and enhance restoration functionally and partially structurally following stroke. We will briefly discuss the perspectives of activating microglial Nrf2 pathway in the modulation of inflammatory response, autophagy, and phagocytosis after ischemic stroke.
2.2.3.1 Nrf2 Suppresses Microglial Inflammatory Response
It is widely accepted that inflammatory processes play a pivotal role in neuronal death, especially the delayed one that may last for days and even weeks after ischemic stroke [107]. Microglias and macrophages mediate inflammatory response by releasing pro-inflammatory cytokines such as IL-1β and TNF-α and by releasing other factors such as cyclooxygenase-2 (COX-2), NO and MMPs [107, 108].
Foresti et al. screened 56 small molecules that are potent Nrf2 activators, including t-BHQ, and carnosol, and showed that activation of Nrf2 could significantly inhibit LPS-induced inflammation in mice microglia-like cell line BV2 cells, which could be abrogated by transfection of Nrf2 shRNA and HO-1 shRNA [109]. Dilshara and coworkers reported that α-viniferin could reduce LPS-induced NO and COX-2 production in BV-2 cells, which could also abrogated by Nrf2-siRNA transfection [110]. In addition, several other Nrf2 activators, including adenosine [111], β-lapachone [112], KCHO-1 [113] and tissue inhibitor of metalloproteinase-2 [114], could also reduce inflammatory responses in association with Nrf2 activation and HO-1 upregulation. It is not clear how Nrf2 activation can inhibit inflammatory response. While Foresti [109] and Dilshara’s findings [110] may suggest a direct link between Nrf2 pathway and the inhibition of inflammation, findings of other studies may need to be further investigated.
2.2.3.2 Nrf2 Pathway in Microglial Autophagy
Autophagy is a lysosome-mediated degradation process for non-essential or damaged cellular constituents, which involves more than 30 autophagy-related proteins (ATGs) and 50 lysosomal hydrolases [115]. Physiologically, autophagy also contributes to preserve homeostasis through the removal of unwanted or damaged mitochondria as well as other organelles. Three classes of autophagy have been described based on how protein substrates for degradation reach the lumen of lysosome [115]. (i) Macroautophagy, which is generally referred to as autophagy, is mediated by the formation of double-membrane vesicles known as autophagosomes. When autophagosomes mature, they fuse with and deliver their contents for degradation. (ii) Microautophagy is a process in which lysosomes directly engulf the cytoplasmic material to degrade. (iii) Chaperone-mediated autophagy is more complex and specific, which involves the recognition by the heat shock cognate protein 70 (HSC70)-containing complex. In brief, the target protein must contain the recognition site for HSC70 complex, which will allow it to bind to this chaperone, leading to the formation of the substrate/chaperone complex. The complex will then move to the lysosomal membrane and enter the cell, whereby get degraded by the lysosome. Among the three classes mentioned above, macroautophagy is the most common one and will be our focus in this section.
The overall mechanism of autophagic regulation is almost clear now. Generation of the autophagosomal structure requires the beclin-1-class III PI3K complex and the generation and insertion of light chain 3 (LC3, also known as Atg8)-II complex into the autophagosomal membrane. The Atg genes control autophagosome formation through Atg12-Atg5 and LC3-II complex [116]. Atg12 is conjugated to Agt5 in a ubiquitin-like reaction, which requires Atg7 and Atg10 as enzymes of the reaction. The Atg12-Atg5 conjugate then interacts with Atg16, and together form a large complex with beclin-1-class III PI3K. The generation of LC3-II complex requires Atg3, Agt4, and Atg7 as enzymes of a ubiquitin-like reaction. Sequestosome 1 (SQSTM1, also known as p62), an “adaptor” molecule in selective autophagy, helps the attachment of LC3-II complex to the autophagosome membrane.
There are at least two mechanisms via which Nrf2 pathway regulates autophagy. First, the Atg proteins mentioned above are sensitive to redox signaling, thus redox status regulates autophagy [117]. It is not surprised that Nrf2 can regulate autophagy through regulating the redox status. In addition, Nrf2 has also been reported to upregulate the expression of Sestrin 2 which acts to scavenge ROS and promote autophagy, through enhancement of Sestrin 2 promoter activity [118]. The other mechanisms involve the interaction of Nrf2 with p62. As a matter of fact, positive feedback loop exists between p62 and Nrf2 pathway. It is reported that Nrf2 pathway directly enhances macrophage autophagy through promoting p62 expression [119, 120], because p62 is a target gene of Nrf2 [121, 122]. Nrf2 is also necessary in p62 aggregation. Using mouse RAW264.7 macrophages, Fujita et al. reported that treatment with LPS or E. coli could induce LC3-II and p62 expression, as well as the formation of selective autophagy of aggresome-like induced structures; and this effect is aborted in Nrf2-deficient microphages, indicating the necessity of Nrf2 in p62 aggregation and p62-mediated autophagy [122]. Similar phenomenon is also shown in liver cells [123] and in the septic lung [124]. In this positive feedback pattern, p62 also contributes to Nrf2 activation. There is a Keap1-interacting region (KIR) in p62, and various stresses can induce the phosphorylation of the serine residue in the KIR, which markedly increases its binding affinity to Keap1. Therefore, phosphorylated p62 can completely abrogate the interaction between Nrf2 and Keap1, thus activating Nrf2 [125]. In addition, Keap1 in the complex with phosphorylated p62 is degraded by selective autophagy [121, 126], contributing to the continuous activation of Nrf2 pathway.
So far, there is little information about the role of Nrf2 in regulating microglial autophagy in stroke, although two reports showed that hypoxia promote both Nrf2 activation and microglial autophagy [127, 128], without knowing the potential link between these two events. Nevertheless, given that phagocytosing microglia produce large amount of ROS, and that ROS is a strong inducer of Nrf2 activation and autophagy, it is reasonable to speculate that the activation of Nrf2 in microglia may promote its autophagic capacity. It is worth to note that autophagy may also be a “double-edged sword,” with side effects of further induction of inflammation and delayed cell death, etc. The degree and time of autophagy would determine the overall prognosis [129]. In this regard, further research could be helpful and focus on how Nrf2 regulates microglial autophagy after stroke, and how to precisely control the degree and time of Nrf2 activation to achieve protective effects and avoid detrimental ones.
2.2.3.3 Nrf2 Pathway Promotes Microglial Phagocytosis
Phagocytosis is a Greek-derived term. Literally, it means the cellular processes of eating, which includes the recognition, engulfment, and degradation of large (>0.5 μm) particulated organisms or structures [130]. Using a co-culture system and live imaging technique, it has been determined that microglia can eliminate an apoptotic cell in 25–95 min in vivo [131]. In vitro under physiological conditions, the microglial clearance time of apoptotic cells has been estimated from 70–90 min to 1–2 h [132, 133]. Besides clearance of apoptotic cells and debris, which is critical for the CNS homeostasis [133], phagocytosis has other functions [134], which includes: (1) antigen presentation, (2) activation of respiratory burst, which can be triggered by hypoxia/reoxygenation, inducing ROS generation that contributes to killing engulfed microorganisms and degradation of other cargo, and (3) modulation of inflammatory responses.
In stroke, phagocytosis by microglias and macrophages plays a critical role in the clearance of cellular bodies and debris, thus limiting ischemic injury [135, 136] and promoting tissue repair [137]. As demonstrated by Lalancette-Hébert et al., selective ablation of proliferating microglias/macrophages 72 h after MCAO led to a 2.7-fold increase in the number of apoptotic cells, predominantly neurons, and exacerbated ischemic brain injury [136]. In another research carried out by Faustino et al., they reported that depletion of microglia significantly enhanced focal inflammatory responses and ROS expression [135].
Few researches focus on the role of Nrf2 pathway in regulating microglial phagocytosis in the acute or sub-acute phase post the onset of stroke, though Nrf2 pathway has been shown to promote the phagocytosis of macrophages and microglia in several other circumstances. For example, curcumin could enhance the phagocytosis of parasitic protozoans by microphage, playing an antimalarial role; and this process was mediated by the activation of Nrf2 signal pathway but not PPAR-γ [138]. Sulforaphane and benzyl isothiocyanate, two Nrf2 activators, could increase the uptake of 2-μm diameter polystyrene beads by RAW 264.7, a line of murine macrophage-like cells, and this effect was aborted in peritoneal macrophages from Nrf2−/− mice [139]. In the lung, alveolar macrophages demonstrated a decreased ability to recognize and phagocytose bacteria in chronic obstructive pulmonary disease; sulforaphane treatment could restore bacteria recognition and phagocytosis of alveolar macrophages, which was only observed in wild-type mice but not in Nrf2-deficient mice [140]. Gene expression and promoter analysis revealed that Nrf2 increased the phagocytic ability of macrophages by direct transcriptional upregulation of the scavenger receptor MARCO (macrophage receptor with collagenous structure) [140]. Within the CNS, microglial phagocytosis is also under the regulation of Nrf2 pathway. Using an in vitro model of amyloid β-induced toxicity in microglia, Li et al. reported that milk fat globule-EGF factor 8 (MFG-E8) accelerated microglial phagocytosis, associated with enhanced Nrf2 and HO-1 expression [141]. In murine intracranial hemorrhage model, activating Nrf2 pathway with sulforaphane could induce the increased expression of CD36, a phagocytosis-mediating scavenger receptor, and increased hematoma clearance, which is abrogated in Nrf2−/− mice [142]. Adopted primary microglia and red blood cells in a phagocytic study, Zhao and colleges also found that activating Nrf2 pathway could upregulate CD36 expression and enhance red blood cell phagocytosis [142]. It is worth noting that the harmful condition of self-producing ROS and pro-inflammatory mediators makes microglias hard to survive; Nrf2 pathway might enhance the tolerance and survival of microglia under this detrimental milieu. As mentioned above, phagocytosis of microglia could regulate inflammation through releasing anti-inflammatory cytokines, one of which is transforming growth factor (TGF)-β, and reduced production of pro-inflammatory cytokines such as TNF-α [143]. It has been reported that TGF-β is critical for survival of phagocytizing microglia through autocrine suppression of TNF-α and ROS [144], and thus enhance microglial phagocytosis. Therefore, enhancing microglial Nrf2 may promote its phagocytosis through downregulating ROS and prolong the lifetime of phagocytizing microglia, expediting debris cleanup in ischemic brain.
Like autophagy being a “double-edged sword” in microglia/macrophage, phagocytosis in microglia/macrophage could also be recognized as such a “double-edged sword.” Under physiological and regulated conditions, the phagocytosis helps to scavenge the apoptotic and necrotic cells, to clear the debris and thus restrict the inflammation; under pathophysiological circumstances, however, dysregulated microglial phagocytosis may contribute to excess neuronal death after acute or chronic ischemia, leading to delayed neuronal loss, brain atrophy even vascular dementia. In a MCAO research carried out by Neher et al. [145], for example, genetic deficiency of phagocytic protein MFG-E8 or Mer receptor tyrosine kinase (MerTK) could completely prevent long-term functional deficits of motor neurons, and the phagocytic deficiency strongly reduced brain atrophy as a result of inhibiting phagocytosis of neurons. The mechanism might be associated with suppressed phagocytosis of neurons, as neurons reversibly exposed the “eat-me” signal phosphatidylserine (PS) to microglias or macrophages after stroke [145].
Taken together, several lines of evidence suggest that activation of Nrf2 pathway may contribute to the inhibition of inflammatory responses after stroke. Considering that cell death, either in the form of apoptosis or necrosis, is an inevitable event in ischemic stroke, enhancing phagocytic capacity of microglia and macrophage will expedite the clean-up process and enhance tissue repair.
3 Conclusion
In summary, Nrf2 is sequestered under physiological conditions and degraded through ubiquitination. Ischemic stroke induces severe oxidative stress that activate Nrf2 pathway by disassociating Nrf2 from Keap1. After nuclear translocation, Nrf2 induces the expression of antioxidants, mainly phase 2 enzymes, which then protect neurons against oxidative injury. It is worth noting that Nrf2 may protect not only through cellular self-defense, but also through cell–cell interaction. Therefore, Nrf2 pathway could be a promising therapeutic target to treat ischemic stroke.
References
Motohashi H, O’Connor T, Katsuoka F, et al. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294(1–2):1–12.
Jain AK, Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem. 2007;282(22):16502–10.
Itoh K, Chiba T, Takahashi S, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–22.
Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol. 2011;85(4):241–72.
Motohashi H, Yamamoto M. Nrf2–Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10(11):549–57.
Yamamoto T, Suzuki T, Kobayashi A, et al. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol. 2008;28(8):2758–70. Pubmed Central PMCID: 2293100.
McMahon M, Thomas N, Itoh K, et al. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem. 2006;281(34):24756–68.
Jain AK, Bloom DA, Jaiswal AK. Nuclear import and export signals in control of Nrf2. J Biol Chem. 2005;280(32):29158–68.
Li W, Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog. 2009;48(2):91–104. Pubmed Central PMCID: 2631094.
Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277(45):42769–74. PubMed PMID: WOS:000179081200046. English.
Jain AK, Jaiswal AK. Phosphorylation of tyrosine 568 controls nuclear export of Nrf2. J Biol Chem. 2006;281(17):12132–42.
Rada P, Rojo AI, Chowdhry S, et al. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol. 2011;31(6):1121–33. Pubmed Central PMCID: 3067901.
Itoh K, Igarashi K, Hayashi N, et al. Cloning and characterization of a novel erythroid cell-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol Cell Biol. 1995;15(8):4184–93. Pubmed Central PMCID: 230657.
Rushmore TH, Morton MR, Pickett CB. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J Biol Chem. 1991;266(18):11632–9.
Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu Rev Pharmacol Toxicol. 2003;43:233–60.
Rada P, Rojo AI, Evrard-Todeschi N, et al. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol. 2012;32(17):3486–99. Pubmed Central PMCID: 3422007.
Rojo AI, Medina-Campos ON, Rada P, et al. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: role of glycogen synthase kinase-3. Free Radic Biol Med. 2012;52(2):473–87.
Lee OH, Jain AK, Papusha V, et al. An auto-regulatory loop between stress sensors INrf2 and Nrf2 controls their cellular abundance. J Biol Chem. 2007;282(50):36412–20.
Niture SK, Jaiswal AK. Prothymosin-alpha mediates nuclear import of the INrf2/Cul3 Rbx1 complex to degrade nuclear Nrf2. J Biol Chem. 2009;284(20):13856–68. Pubmed Central PMCID: 2679486.
Holtzclaw WD, Dinkova-Kostova AT, Talalay P. Protection against electrophile and oxidative stress by induction of phase 2 genes: the quest for the elusive sensor that responds to inducers. Adv Enzyme Regul. 2004;44(1):335–67.
Zhang M, An C, Gao Y, et al. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog Neurobiol. 2013;100:30–47.
Zhang Y, Talalay P, Cho C-G, et al. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A. 1992;89(6):2399–403.
Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25(44):10321–35.
Kraft AD, Lee JM, Johnson DA, et al. Neuronal sensitivity to kainic acid is dependent on the Nrf2-mediated actions of the antioxidant response element. J Neurochem. 2006;98(6):1852–65.
del Zoppo GJ. The neurovascular unit in the setting of stroke. J Intern Med. 2010;267(2):156–71. Pubmed Central PMCID: Pmc3001328, Epub 2010/02/24. eng.
Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4(5):399–414.
Zhang JH, Badaut J, Tang J, et al. The vascular neural network—a new paradigm in stroke pathophysiology. Nat Rev Neurol. 2012;8(12):711–6. Pubmed Central PMCID: Pmc3595043, Epub 2012/10/17. eng.
Silva-Islas C, Colín-González AL, Maldonado PD, et al. Nrf2 activation, an innovative therapeutic alternative in cerebral ischemia. INTECH Open Access; 2012.
Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–98.
Terasaki Y, Liu Y, Hayakawa K, et al. Mechanisms of neurovascular dysfunction in acute ischemic brain. Curr Med Chem. 2014;21(18):2035.
Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21(1):2–14.
Danton GH, Dietrich WD. Inflammatory mechanisms after ischemia and stroke. J Neuropathol Exp Neurol. 2003;62(2):127–36.
Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med. 2005;39(4):429–43.
Crack PJ, Taylor JM. Reactive oxygen species and the modulation of stroke. Free Radic Biol Med. 2005;38(11):1433–44.
Gouriou Y, Demaurex N, Bijlenga P, et al. Mitochondrial calcium handling during ischemia-induced cell death in neurons. Biochimie. 2011;93(12):2060–7.
Zhang Y, Wang H, Li J, et al. Peroxynitrite-induced neuronal apoptosis is mediated by intracellular zinc release and 12-lipoxygenase activation. J Neurosci. 2004;24(47):10616–27.
Moro MA, Almeida A, Bolaños JP, et al. Mitochondrial respiratory chain and free radical generation in stroke. Free Radic Biol Med. 2005;39(10):1291–304.
Hallenbeck JM, Dutka AJ, Tanishima T, et al. Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke. 1986;17(2):246–53.
Clark R, Lee E, Fish C, et al. Development of tissue damage, inflammation and resolution following stroke: an immunohistochemical and quantitative planimetric study. Brain Res Bull. 1993;31(5):565–72.
Campanella M, Sciorati C, Tarozzo G, et al. Flow cytometric analysis of inflammatory cells in ischemic rat brain. Stroke. 2002;33(2):586–92.
Shichita T, Ago T, Kamouchi M, et al. Novel therapeutic strategies targeting innate immune responses and early inflammation after stroke. J Neurochem. 2012;123(s2):29–38.
Heiss W-D. Stroke—acute interventions. Berlin: Springer; 2002.
Hou ST, MacManus JP. Molecular mechanisms of cerebral ischemia-induced neuronal death. Int Rev Cytol. 2002;221:93–148.
Dinkova-Kostova A, Cheah J, Samouilov A, et al. Phenolic Michael reaction acceptors: combined direct and indirect antioxidant defenses against electrophiles and oxidants. Med Chem. 2007;3(3):261–8.
Lindenau J, Noack H, Possel H, et al. Cellular distribution of superoxide dismutases in the rat CNS. Glia. 2000;29(1):25–34.
Dinkova‐Kostova AT, Talalay P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol Nutr Food Res. 2008;52(S1):S128–38.
Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116.
Kobayashi A, Kang M-I, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–9.
Peters O, Back T, Lindauer U, et al. Increased formation of reactive oxygen species after permanent and reversible middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1998;18(2):196–205.
Christensen T, Bruhn T, Balchen T, et al. Evidence for formation of hydroxyl radicals during reperfusion after global cerebral ischaemia in rats using salicylate trapping and microdialysis. Neurobiol Dis. 1994;1(3):131–8.
Liu S, Liu M, Peterson S, et al. Hydroxyl radical formation is greater in striatal core than in penumbra in a rat model of ischemic stroke. J Neurosci Res. 2003;71(6):882–8.
Tanaka N, Ikeda Y, Ohta Y, et al. Expression of Keap1–Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011;1370:246–53.
Dang J, Brandenburg L-O, Rosen C, et al. Nrf2 expression by neurons, astroglia, and microglia in the cerebral cortical penumbra of ischemic rats. J Mol Neurosci. 2012;46(3):578–84.
Li M, Zhang X, Cui L, et al. The neuroprotection of oxymatrine in cerebral ischemia/reperfusion is related to nuclear factor erythroid 2-related factor 2 (nrf2)-mediated antioxidant response: role of nrf2 and hemeoxygenase-1 expression. Biol Pharm Bull. 2011;34(5):595–601.
Srivastava S, Alfieri A, Siow R, et al. Temporal and spatial distribution of Nrf2 in rat brain following stroke: quantification of nuclear to cytoplasmic Nrf2 content using a novel immunohistochemical technique. J Physiol. 2013;591(14):3525–38.
Liverman CS, Cui L, Yong C, et al. Response of the brain to oligemia: gene expression, c-Fos, and Nrf2 localization. Mol Brain Res. 2004;126(1):57–66.
Gladstone DJ, Black SE, Hakim AM. Toward wisdom from failure lessons from neuroprotective stroke trials and new therapeutic directions. Stroke. 2002;33(8):2123–36.
Herken R, Götz W, Thies M. Appearance of laminin, heparan sulphate proteoglycan and collagen type IV during initial stages of vascularisation of the neuroepithelium of the mouse embryo. J Anat. 1990;169:189.
Engvall E, Davis GE, Dickerson K, et al. Mapping of domains in human laminin using monoclonal antibodies: localization of the neurite-promoting site. J Cell Biol. 1986;103(6):2457–65.
Grant D, Kleinman H. Regulation of capillary formation by laminin and other components of the extracellular matrix. Regulation of angiogenesis. Berlin: Springer; 1997. p. 317–33.
Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5(5):347–60.
Zonta M, Angulo MC, Gobbo S, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6(1):43–50.
del Zoppo GJ, Mabuchi T. Cerebral microvessel responses to focal ischemia. J Cereb Blood Flow Metab. 2003;23(8):879–94.
del Zoppo GJ, Milner R. Integrin–matrix interactions in the cerebral microvasculature. Arterioscler Thromb Vasc Biol. 2006;26(9):1966–75.
Mabuchi T, Lucero J, Feng A, et al. Focal cerebral ischemia preferentially affects neurons distant from their neighboring microvessels. J Cereb Blood Flow Metab. 2005;25(2):257–66.
Hu X, Leak RK, Shi Y, et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. 2015;11(1):56–64.
Fumagalli S, Perego C, Pischiutta F, et al. The ischemic environment drives microglia and macrophage function. Front Neurol. 2015;6:81.
Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. Eur J Biochem. 2000;267(16):4912–6.
Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24(5):1101–12.
Shih AY, Johnson DA, Wong G, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23(8):3394–406.
Chen Y, Vartiainen NE, Ying W, et al. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem. 2001;77(6):1601–10.
Halliwell B. Role of free radicals in the neurodegenerative diseases. Drugs Aging. 2001;18(9):685–716.
Vargas MR, Johnson JA. The Nrf2–ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17.
Bambrick L, Kristian T, Fiskum G. Astrocyte mitochondrial mechanisms of ischemic brain injury and neuroprotection. Neurochem Res. 2004;29(3):601–8.
Swanson RA, Ying W, Kauppinen TM. Astrocyte influences on ischemic neuronal death. Curr Mol Med. 2004;4(2):193–205.
Calkins MJ, Jakel RJ, Johnson DA, et al. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005;102(1):244–9.
Calkins MJ, Vargas MR, Johnson DA, et al. Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol Sci. 2010;115(2):557–68.
Bell KF, Fowler JH, Al-Mubarak B, et al. Activation of Nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxid Med Cell Longev. 2011;2011:689524.
Narayanan SV, Dave KR, Saul I, et al. Resveratrol preconditioning protects against cerebral ischemic injury via nuclear erythroid 2–related factor 2. Stroke. 2015;46(6):1626–32.
Schroeter ML, Mertsch K, Giese H, et al. Astrocytes enhance radical defence in capillary endothelial cells constituting the blood-brain barrier. FEBS Lett. 1999;449(2):241–4.
Laird MD, Ramesh SS, Alleyne CH, et al. Astrocyte-derived glutathione attenuates hemin-induced cytotoxicity in murine cerebral microvessel. FASEB J. 2009;23(1_MeetingAbstracts):614.12.
Lee BJ, Egi Y, van Leyen K, et al. Edaravone, a free radical scavenger, protects components of the neurovascular unit against oxidative stress in vitro. Brain Res. 2010;1307:22–7.
Chrissobolis S, Banfi B, Sobey CG, et al. Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol. 2012;113(2):184–91.
Pacher P, Szabo C. Role of the peroxynitrite-poly (ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173(1):2–13.
Weiss N, Miller F, Cazaubon S, et al. The blood-brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta. 2009;1788(4):842–57.
Posada-Duque RA, Barreto GE, Cardona-Gomez GP. Protection after stroke: cellular effectors of neurovascular unit integrity. Front Cell Neurosci. 2014;8:231.
Alfieri A, Srivastava S, Siow RC, et al. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med. 2013;65:1012–22.
Bénardais K, Pul R, Singh V, et al. Effects of fumaric acid esters on blood–brain barrier tight junction proteins. Neurosci Lett. 2013;555:165–70.
Kunze R, Urrutia A, Hoffmann A, et al. Dimethyl fumarate attenuates cerebral edema formation by protecting the blood–brain barrier integrity. Exp Neurol. 2015;266:99–111.
Wu S, Yue Y, Li J, et al. Procyanidin B2 attenuates neurological deficits and blood–brain barrier disruption in a rat model of cerebral ischemia. Mol Nutr Food Res. 2015;59(10):1930–41.
Chen G, Fang Q, Zhang J, et al. Role of the Nrf2‐ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J Neurosci Res. 2011;89(4):515–23.
Li T, Sun K-J, Wang H-D, et al. Tert-butylhydroquinone ameliorates early brain injury after experimental subarachnoid hemorrhage in mice by enhancing Nrf2-independent autophagy. Neurochem Res. 2015;40(9):1829–38.
Wang Z, Ji C, Wu L, et al. Tert-butylhydroquinone alleviates early brain injury and cognitive dysfunction after experimental subarachnoid hemorrhage: role of Keap1/Nrf2/ARE pathway. PLoS One. 2014;9(5):e97685.
Wu Q, Zhang X-S, Wang H-D, et al. Astaxanthin activates nuclear factor erythroid-related factor 2 and the antioxidant responsive element (Nrf2-ARE) pathway in the brain after subarachnoid hemorrhage in rats and attenuates early brain injury. Mar Drugs. 2014;12(12):6125–41.
Shi S-S, Zhang H-B, Wang C-H, et al. Propofol attenuates early brain injury after subarachnoid hemorrhage in rats. J Mol Neurosci. 2015;7(4):538–45.
Wang Z, Ma C, Meng CJ, et al. Melatonin activates the Nrf2‐ARE pathway when it protects against early brain injury in a subarachnoid hemorrhage model. J Pineal Res. 2012;53(2):129–37.
Zhang J, Zhu Y, Zhou D, et al. Recombinant human erythropoietin (rhEPO) alleviates early brain injury following subarachnoid hemorrhage in rats: possible involvement of Nrf2–ARE pathway. Cytokine. 2010;52(3):252–7.
Li T, Wang H, Ding Y, et al. Genetic elimination of Nrf2 aggravates secondary complications except for vasospasm after experimental subarachnoid hemorrhage in mice. Brain Res. 2014;1558:90–9.
Zhao J, Moore AN, Redell JB, et al. Enhancing expression of Nrf2-driven genes protects the blood–brain barrier after brain injury. J Neurosci. 2007;27(38):10240–8.
Jin W, Ni H, Hou X, et al. Tert-butylhydroquinone protects the spinal cord against inflammatory response produced by spinal cord injury. Ann Clin Lab Sci. 2014;44(2):151–7.
Mao L, Wang H, Qiao L, et al. Disruption of Nrf2 enhances the upregulation of nuclear factor-kappaB activity, tumor necrosis factor-, and matrix metalloproteinase-9 after spinal cord injury in mice. Mediators Inflamm. 2010;2010:238321.
Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci. 2003;358(1438):1669–77.
Bechmann I, Galea I, Perry VH. What is the blood–brain barrier (not)? Trends Immunol. 2007;28(1):5–11.
Molina‐Holgado E, Molina‐Holgado F. Mending the broken brain: neuroimmune interactions in neurogenesis. J Neurochem. 2010;114(5):1277–90.
Aspelund A, Antila S, Proulx ST, et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med. 2015;212(7):991–9. PubMed PMID: WOS:000357117200004. English.
Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41. Pubmed Central PMCID: 4506234.
Stoll G, Jander S, Schroeter M. Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol. 1998;56(2):149–71.
Amantea D, Nappi G, Bernardi G, et al. Post-ischemic brain damage: pathophysiology and role of inflammatory mediators. FEBS J. 2009;276(1):13–26.
Foresti R, Bains SK, Pitchumony TS, et al. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol Res. 2013;76:132–48.
Dilshara MG, Lee K-T, Kim HJ, et al. Anti-inflammatory mechanism of α-viniferin regulates lipopolysaccharide-induced release of proinflammatory mediators in BV2 microglial cells. Cell Immunol. 2014;290(1):21–9.
Min KJ, Kim JH, Jou I, et al. Adenosine induces heme oxygenase-1 expression in microglia through the activation of phosphatidylinositol-3-kinase and nuclear factor E2-related factor 2. Glia. 2008;56(9):1028–37.
Lee EJ, Ko HM, Jeong YH, et al. beta-Lapachone suppresses neuroinflammation by modulating the expression of cytokines and matrix metalloproteinases in activated microglia. J Neuroinflammation. 2015;12:133. Pubmed Central PMCID: Pmc4502557. Epub 2015/07/16. eng.
Lee D-S, Ko W, Yoon C-S, et al. KCHO-1, a novel antineuroinflammatory agent, inhibits lipopolysaccharide-induced neuroinflammatory responses through Nrf2-mediated heme oxygenase-1 expression in mouse BV2 microglia cells. Evid Based Complement Alternat Med. 2014;2014:357154.
Lee E-J, Kim H-S. The anti-inflammatory role of tissue inhibitor of metalloproteinase-2 in lipopolysaccharide-stimulated microglia. J Neuroinflammation. 2014;11:116.
Jisun L, Samantha G, Jianhua Z. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441(2):523–40.
Chen Y, Klionsky DJ. The regulation of autophagy–unanswered questions. J Cell Sci. 2011;124(2):161–70.
Cooper CE, Patel RP, Brookes PS, et al. Nanotransducers in cellular redox signaling: modification of thiols by reactive oxygen and nitrogen species. Trends Biochem Sci. 2002;27(10):489–92.
Bae SH, Sung SH, Oh SY, et al. Sestrins activate Nrf2 by promoting p62-dependent autophagic degradation of Keap1 and prevent oxidative liver damage. Cell Metab. 2013;17(1):73–84.
Vadlamudi RK, Joung I, Strominger JL, et al. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271(34):20235–7.
Bjørkøy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–14.
Jain A, Lamark T, Sjøttem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–91.
Fujita K-I, Maeda D, Xiao Q, et al. Nrf2-mediated induction of p62 controls Toll-like receptor-4–driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A. 2011;108(4):1427–32.
Riley BE, Kaiser SE, Shaler TA, et al. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol. 2010;191(3):537–52.
Chang AL, Ulrich A, Suliman HB, et al. Redox regulation of mitophagy in the lung during murine Staphylococcus aureus sepsis. Free Radic Biol Med. 2015;78:179–89.
Ichimura Y, Waguri S, Sou Y-S, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51(5):618–31.
Taguchi K, Fujikawa N, Komatsu M, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A. 2012;109(34):13561–6.
Yang Z, Zhao T-Z, Zou Y-J, et al. Hypoxia induces autophagic cell death through hypoxia-inducible factor 1α in microglia. PLoS One. 2014;9(5):e96509.
Yang Z, Zhong L, Zhong S, et al. Hypoxia induces microglia autophagy and neural inflammation injury in focal cerebral ischemia model. Exp Mol Pathol. 2015;98(2):219–24.
Chen W, Sun Y, Liu K, et al. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res. 2014;9(12):1210.
Mukherjee S, Ghosh RN, Maxfield FR. Endocytosis. Physiol Rev. 1997;77(3):759–803.
Parnaik R, Raff MC, Scholes J. Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr Biol. 2000;10(14):857–60.
Sierra A, Encinas JM, Deudero JJ, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–95.
Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27(5):244–50.
Sierra A, Abiega O, Shahraz A, et al. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6.
Faustino JV, Wang X, Johnson CE, et al. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J Neurosci. 2011;31(36):12992–3001.
Lalancette-Hébert M, Gowing G, Simard A, et al. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27(10):2596–605.
Manoonkitiwongsa PS, Jackson-Friedman C, McMillan PJ, et al. Angiogenesis after stroke is correlated with increased numbers of macrophages: the clean-up hypothesis. J Cereb Blood Flow Metab. 2001;21(10):1223–31.
Mimche PN, Thompson E, Taramelli D, et al. Curcumin enhances non-opsonic phagocytosis of Plasmodium falciparum through up-regulation of CD36 surface expression on monocytes/macrophages. J Antimicrob Chemother. 2012;67(8):1895–904.
Suganuma H, Fahey JW, Bryan KE, et al. Stimulation of phagocytosis by sulforaphane. Biochem Biophys Res Commun. 2011;405(1):146–51.
Harvey CJ, Thimmulappa RK, Sethi S, et al. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med. 2011;3(78):78ra32.
Li E, Noda M, Doi Y, et al. The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity. J Neuroinflammation. 2012;9(148):2657–66.
Zhao X, Sun G, Ting SM, et al. Cleaning up after ICH: the role of Nrf2 in modulating microglia function and hematoma clearance. J Neurochem. 2015;133(1):144–52.
De Simone R, Ajmone-Cat MA, Tirassa P, et al. Apoptotic PC12 cells exposing phosphatidylserine promote the production of anti-inflammatory and neuroprotective molecules by microglial cells. J Neuropathol Exp Neurol. 2003;62(2):208–16.
Ryu K-Y, Cho G-S, Piao HZ, et al. Role of TGF-β in survival of phagocytizing microglia: autocrine suppression of TNF-α production and oxidative stress. Exp Neurobiol. 2012;21(4):151–7.
Neher JJ, Emmrich JV, Fricker M, et al. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A. 2013;110(43):E4098–107.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Yang, T., Sun, Y., Zhang, F. (2016). The Role of Nonneuronal Nrf2 Pathway in Ischemic Stroke: Damage Control and Potential Tissue Repair. In: Chen, J., Zhang, J., Hu, X. (eds) Non-Neuronal Mechanisms of Brain Damage and Repair After Stroke. Springer Series in Translational Stroke Research. Springer, Cham. https://doi.org/10.1007/978-3-319-32337-4_18
Download citation
DOI: https://doi.org/10.1007/978-3-319-32337-4_18
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-32335-0
Online ISBN: 978-3-319-32337-4
eBook Packages: MedicineMedicine (R0)