
Abstract
The cell fate of hypertrophic growth plate chondrocytes at the chondro-osseous junction has been a subject of discussion for several decades: On the one hand, there is ample evidence for programmed cell death by apoptosis or other mechanisms in the lower hypertrophic zone; on the other hand, several studies have indicated that some hypertrophic chondrocytes may not be “terminally differentiated” but are able to further differentiate into osteoblasts. Recent lineage tracing studies from four laboratories using genetic markers have now unequivocally demonstrated that a progeny of growth plate chondrocytes is able to give rise to osteoblasts which contribute substantially to trabecular, endosteal, and cortical bone formation during fetal and postnatal long bone development. This shows that not all “terminally” differentiated cells at the chondro-osseous junction are eliminated by programmed cell death but have the option to survive and enter the osteogenic lineage. The ability of chondrocytes to transdifferentiate into bone cells was also confirmed in bone injury healing experiments which demonstrated that the majority of newly formed osteoblasts were derived from chondrocytes of the fracture callus. The molecular mechanism of chondrocyte reprogramming into the osteogenic lineage, however, remains to be elucidated. A detailed analysis of the lower hypertrophic growth plate zone by confocal microscopy revealed small, 4–6 μm reporter gene-positive cells, i.e., hypertrophic chondrocyte-derived cells, which were mitotically active and differentiated to osteoblasts in vitro. They seem to be derived from hypertrophic chondrocytes by condensation, possibly through autophagy, and may represent intermediate stemlike cells that have the option to enter the osteogenic or other bone marrow cell lineages.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
5.1 Introduction
The skeletal body plans of all vertebrates are laid out as cartilaginous models, but only in few species such as sharks or rays the skeleton remains cartilaginous throughout lifetime. In most vertebrates, the cartilage models of long bones, ribs, and vertebrae are subsequently replaced by bone during fetal and postnatal development, while only specialized cartilaginous tissues such as articular cartilage and nasal or tracheal cartilage persist in the adult (Hall 1987, 2005). Cartilage replacement by bone occurs in a dynamic and highly complex process called endochondral ossification, which is one of the most intriguing puzzles in vertebrate development with important implications on our understanding of bone fracture healing, skeletal dysplasias, and degenerative joint diseases (for reviews, see Poole (1991), Mackie et al. (2008), and Ballock and O’Keefe (2003)).
The cartilaginous elements of the vertebrate appendicular skeleton develop by condensation of limb mesenchyme. In the limb bud, hyaline chondrocytes in the center of the blastema proliferate and undergo a series of distinct differentiation steps to proliferating, prehypertrophic, and hypertrophic chondrocytes. The zone of mature hypertrophic cartilage in the diaphysis of long bones is then invaded by vascular sprouts from the perichondrium, resorbed by osteoclasts, and filled with bone marrow and trabecular bone, forming the primary ossification center (POC). A sheath of cortical bone develops around the diaphysis by endesmal ossification, while trabecular bone is formed in the POC by osteoprogenitor cells which invade with endothelial cells from the perichondrium or – as we will see below – are derived from hypertrophic chondrocytes. Both perichondrium-derived and chondrocyte-derived osteoprogenitor cells deposit osteoid on the surface of remaining cartilage spicules which after mineralization provide the bone trabeculae of the primary spongiosa (Olsen et al. 2000; Karsenty et al. 2009). At both the distal and proximal sides of the primary ossification center (POC), growth plates develop in which the process of chondrocyte maturation and replacement by bone proceeds toward both epiphyses (for reviews, see Ballock and O’Keefe (2003), Mackie et al. (2008), Provot and Schipani (2005), Poole (1991), Amizuka et al. (2012), and Tsang et al. (2014)).
Pace and extent of skeletal growth are tightly controlled by multiple, coordinated mechanisms regulating proliferation, differentiation, and maturation of chondrocytes and osteoblasts. The role and the cell fate of hypertrophic chondrocytes at the cartilage–bone marrow interface, also called chondro-osseous junction, in endochondral ossification are complex and controversially discussed. According to common view, late hypertrophic chondrocytes are eliminated by programmed cell death during the cartilage resorption process (for reviews, see Gibson (1998) and Shapiro et al. (2005)). On the other hand, numerous morphological and experimental studies going back almost a century have indicated that hypertrophic chondrocytes may be able to convert into osteoblast-like, bone-forming cells in vitro and in vivo (Yang et al. 2014a, b; Park et al. 2015; Zhou et al. 2014). The relevance of these studies has, however, not been widely accepted. Recently, however, four independent lineage tracing studies in transgenic mice have provided conclusive evidence by using genetic recombination in chondrocytes that hypertrophic growth plate chondrocytes can survive and give rise to a progeny of osteoblasts which contribute substantially to trabecular, endosteal, and cortical bone formation. Furthermore, in two of these studies, it was shown that also in regenerating bone tissue after experimental bone fracture callus formation, the majority of osteoblasts are derived from chondrocytes (Zhou et al. 2014; Yang et al. 2014b). These findings challenge current textbook views on the origin of trabecular, endosteal, and cortical osteoblasts constituting endochondral bone and on the cell fate of hypertrophic growth plate chondrocytes. The new data indicates that hypertrophic chondrocytes have the option to undergo programmed cell death or to survive by reentering the cell cycle and undergoing reprogramming into osteoprogenitor cells and other bone marrow cells.
In this chapter, the authors will present new concepts of the cell fate of hypertrophic growth plate chondrocytes in light of the new genetic lineage tracing studies and discuss the relevance of the results with respect to our understanding of the origin of osteoblasts not only in development but also in bone repair, chondrodysplasias, and degenerative joint diseases. Overall, previous and recent findings indicate that programmed cell death and reprogramming of hypertrophic chondrocytes to osteoblasts are not mutually exclusive cell fates, but two options which are both realized at the chondro-osseous junction, although the mechanism regulating entry into these different pathways is still open. At this point, the authors would like to refer the detailed descriptions of the developing growth plate and the regulation of differentiation and maturation of chondrocytes in Chaps. 4, 8, and 10 in this volume.
5.2 Chondrocyte and Osteoblast Differentiation in the Growth Plate
5.2.1 Chondrocyte Differentiation
In the growth plate, the chondrocytes in the resting, proliferating, prehypertrophic, hypertrophic, and late (“terminal”) hypertrophic zone have each specific roles in endochondral ossification: In the “resting” zone – or better called reserve zone – chondrocytes proliferate and expand to shape the epiphysis which is later also ossified by the secondary ossification center. In the proliferating zone, chondrocytes divide most rapidly and align into vertical columns of flattened, lens-shaped cells which finally differentiate into prehypertrophic and hypertrophic chondrocytes. Proliferating and hypertrophic chondrocytes are primarily responsible for the longitudinal growth of long bones, as cells increase not only in number but also in their volume up to 20-fold, and deposit substantial amounts of extracellular cartilage matrix (for review, see Ballock and O’Keefe (2003), Provot and Schipani (2005), Mackie et al. (2008), and Chap. 4). Maturation into hypertrophic chondrocytes is also characterized by the development of a granular cell surface and protrusion of numerous microvilli which release matrix vesicles that serve as nuclei of cartilage mineralization (Anderson 1985; Wuthier 1988).
Maturation of proliferating to hypertrophic chondrocytes is associated with fundamental matrix remodeling (summarized in Ortega et al. (2004)): The hyaline cartilage matrix which is composed predominantly of aggrecan and collagens II, VI, IX, and XI, deposited by resting and proliferating chondrocytes, is substituted by a calcifying matrix which contains type X collagen, a network forming collagen synthesized only by hypertrophic chondrocytes (Schmid and Conrad 1982; Gibson et al. 1984), as well as alkaline phosphatase (AP), osteopontin, bone sialoprotein (BSP), osteocalcin, and proteoglycans (Ballock and O'Keefe 2003; Cancedda et al. 1995; Mackie et al. 2008). In their late stage of differentiation, hypertrophic chondrocytes secrete MMP13 (collagenase 3), a matrix metalloproteinase which breaks up the matrix of calcified cartilage in preparation for the resorption by osteoclasts (Stickens et al. 2004; Inada et al. 2004; Johansson et al. 1997), and the vascular endothelial growth factor (VEGF) A, which induces capillary invasion and bone marrow formation (Gerber et al. 1999; Zelzer et al. 2004). The remarkable similarity of the gene expression pattern of hypertrophic chondrocytes to that of osteoblasts which includes AP, osteopontin, bone sialoprotein, and osteocalcin (Gerstenfeld and Shapiro 1996) strongly supports the concept of a continuous chondrocyte-to-osteoblast lineage.
Growth and differentiation of chondrocytes in the growth plate are regulated in a complex, synergistic manner by growth factors, hormones, developmental growth factors, and their receptors (for reviews, see, Goldring et al. (2006), Cancedda et al. (2000), Lefebvre and Smits (2005), Kronenberg (2003), and Nishimura et al. (2012)). Most important for growth and proliferation are insulin-like growth factors (IGFs) and thyroxin, growth factors of the fibroblast growth factor (FGF), and bone morphogenetic proteins (BMPs). The critical step of chondrocyte maturation into hypertrophic chondrocytes is also under the control of the regulatory cycle of parathyroid hormone-related protein (PTHrP) and Indian hedgehog (IHH) (see Chap. 8 and reviews by Lanske and Kronenberg (1998), Kronenberg (2006), and Vortkamp (2000)), while various WNT factors regulate chondrogenic and osteogenic differentiation in the growth plate through canonical and noncanonical pathways (see Chap. 10 and reviews by Day and Yang (2008) and Hartmann (2007)).
5.2.2 Osteoblast Differentiation
Concomitant with the maturation of chondrocytes in the mid diaphysis into hypertrophic cells, osteoprogenitor cells in the perichondrium surrounding the diaphysis differentiate into osteoblasts which produce the calcified cortical bone sheath consisting of type I collagen as major organic constituent, as well as osteocalcin, bone sialoprotein, osteopontin, matrix GLA protein, alkaline phosphatase, proteoglycans, and other bone constituents (for reviews, see Olsen et al. (2000), Karsenty et al. (2009), and Reddi (1981)). Differentiation of osteoprogenitor cells to osteoblasts is strongly enhanced by factors of the TGFß/BMP family, in particular by BMP-3, BMP-7, and TGFß1, and by vitamin D (1,25(OH)2D3), PTH/PTHrP, glucocorticoids, prostaglandin E2, IGF-I, and IGF-II (Aubin et al. 1995; Karsenty et al. 2009; Lian et al. 2006; Komori 2006). Osteoblasts are characterized by an epithelioid cell shape and cell polarity, directing secretion of bone matrix components to the basal surface facing the bone matrix, and by the development of long cell protrusions (canaliculi) into the bone matrix which are essential for intercellular communication, mechanotransduction, and exchange of metabolites. When epithelioid osteoblasts are embedded into bone matrix, they differentiate into postmitotic osteocytes which terminate bone matrix production, acquire stellate cell shape, and develop numerous canaliculi (Komori 2013). During longitudinal bone growth in the fetus, further osteoprogenitor cells are self-renewing in Ranvier’s groove (Langenskiold 1998) and migrate along the periosteum to produce further cortical bone; some migrate into the bone marrow with onset of capillary invasion though the cortical bone sheath along with endothelial cells (Colnot et al. 2004; Maes et al. 2010). In the primary spongiosa, they differentiate into osteoblasts on the surface of remaining, unresorbed calcified cartilage spicules and form bone trabeculae by deposition of osteoid, the organic part of the bone matrix, which consists of type I collagen and other bone matrix proteins. Recent evidence strongly indicates that further osteoblasts in the primary spongiosa are generated by transdifferentiation of hypertrophic chondrocytes (see below).
5.3 Sox9, Runx2, and Osterix as Key Transcription Factors Regulating Chondrogenic and Osteogenic Differentiation
A continued chondrocyte-to-osteoblast lineage during endochondral ossification becomes plausible when considering the common origin of limb chondrocytes and osteoblasts from Sox9 expressing chondro-osteoprogenitor cells in the limb mesenchyme (Akiyama et al. 2005). Continued Sox9 expression promotes chondrogenic differentiation and maintains the chondrogenic phenotype, whereas RUNX2 and osterix (Osx) drive the progenitor cells into the osteogenic lineage (for reviews, see de Crombrugghe et al. (2001), Lefebvre and Smits (2005), Tsang et al. (2014), Karsenty et al. (2009), Komori (2010), and Lefebvre et al. (2001)). Spatial and temporal aspects of chondrogenic versus osteogenic differentiation in skeletal development are primarily controlled by Wnt/ß-catenin signals (Hartmann 2007; Day and Yang 2008) (see also Chap. 10).
SOX9 is a transcription factor of the SRY family which is essential for sex determination and development of cartilage and neural tissues (Lefebvre and de Crombrugghe 1998; Bi et al. 1999; Tsang et al. 2014). The existence of Sox9-positive chondro-osteoprogenitor cells in the undifferentiated limb mesenchyme was demonstrated by inactivation of Sox9 before mesenchymal condensation under the control of the prx promoter, which abolished not only cartilage formation but also endochondral bone formation (Akiyama et al. 2002). Sox9 controls all steps of chondrocyte differentiation until chondrocytes reach hypertrophy (Leung et al. 2011; Lefebvre and de Crombrugghe 1998). Together with the related Sox5 and Sox6 transcription factors, SOX9 activates the expression of cartilage matrix genes Col2a1, Col9a1, Col11, Agc1 (the gene of aggrecan) and others (Lefebvre et al. 1997; Lefebvre and Smits 2005; Ng et al. 1997). In the growing limb, Sox9 is expressed in resting, proliferating, and early hypertrophic cartilage, but interestingly, it is absent from late hypertrophic chondrocytes (Zhao et al. 1997; Hattori et al. 2010). The complete loss of SOX9 from the late hypertrophic zone is noticeable in light of the enhanced expression of osteogenic genes in this zone, including Runx2, osterix (Osx), osteopontin (Spp1), and bone sialoprotein (BSP), and compatible with the concept of a conversion of hypertrophic chondrocytes into osteoblasts.
RUNX2 (cbfa1, Osf2) is a transcription factor of the runt family which is essential for osteogenic differentiation and bone development (Ducy et al. 1999; Karsenty et al. 1999; Komori 2008). It activates the expression of osteoblast-typical genes such as osteocalcin (Bglap), type I collagen (col1a1 and col1a2), Bsp, Spp1, and Runx2 itself by binding to OSE2 consensus sequences in their promoters (Komori 2010; Ducy et al. 1996, 1997). Inactivation of Runx2 gene allows the development of the cartilaginous anlagen of the skeleton but blocks completely both endesmal and endochondral ossification (Ducy et al. 1997; Komori et al. 1997; Otto et al. 1997). RUNX2 also activates expression of Osx (SP7), a zinc finger transcription factor which also acts as master gene of osteogenic differentiation (Nakashima et al. 2002).
Interestingly, although both Runx2 and Osx are primarily essential for osteogenic differentiation, they are also involved in the regulation of chondrocyte maturation and endochondral ossification. Both Runx2 and Osx are expressed in prehypertrophic chondrocytes (Takeda et al. 2001; Nakashima et al. 2002; Hattori et al. 2010), and Runx2 promotes expression of type X collagen and maturation to hypertrophic chondrocytes (Kim et al. 1999; Wang et al. 2004; Stricker et al. 2002). As a consequence, Runx2-deficient mice lack hypertrophic chondrocytes in growth plates (Komori et al. 1997). Furthermore, Runx2 induces the expression of Vegfa chondrocytes of late hypertrophic cartilage, which is required for capillary invasion into hypertrophic cartilage (Carlevaro et al. 2000; Gerber et al. 1999; Zelzer et al. 2001), and Mmp13 (Hess et al. 2001; Jimenez et al. 1999; Porte et al. 1999; Inada et al. 2004), which loosens up the matrix of hypertrophic cartilage in order to allow invasion of bone marrow sprouts (Inada et al. 2004; Johansson et al. 1997). Thus, while early stages of chondrocyte differentiation are strictly controlled by Sox9, with onset of Runx2 and Osx expression in prehypertrophic chondrocytes, the cell fate of growth plate chondrocytes seems to be successively directed into an osteogenic lineage as heralded by cellular hypertrophy, matrix calcification, and onset of osteogenic gene expression.
5.4 The Cell Fate of Hypertrophic Chondrocytes
5.4.1 Programmed Cell Death
Morphological studies dating back more than a century ago have reported on alterations in hypertrophic growth plate chondrocytes of different species which pointed to cellular degeneration and death (Brachet 1893; Fell 1825; Ham 1952). Also in more recent ultrastructural studies, degenerative changes in growth plate chondrocytes were interpreted as signs of programmed cell death reminiscent of apoptosis (Farnum and Wilsman 1989a; Gibson et al. 1995; Gibson 1998; Bronckers et al. 1996; Aizawa et al. 1997; Zenmyo et al. 1996; Roach et al. 2004; Roach 1997). Apoptosis, the most common form of programmed cell death, is an evolutionary conserved process that serves to eliminate unwanted or superfluous cells during normal tissue development and regeneration of organs and tissues in almost all species (for reviews, see Jacobson et al. (1997) and Fuchs and Steller (2011)). The term apoptosis was coined first by Kerr et al. (1972) in an ultrastructural study where they defined a series of distinct morphological changes in cells dying under physiological conditions. In contrast to cell necrosis in which cells swell and become ruptured in response to stress or tissue injury, during apoptosis, cells retain membrane integrity but shrink by genetically controlled intracellular degradation of organelles, proteins, and DNA, leaving condensed nuclei. The dead cells or cell fragments are rapidly phagocytosed and therefore are visible mostly inside macrophages or other phagocytic cells. Apoptosis is primarily an intrinsic, cell-autonomous process but can be also induced by extrinsic signals, e.g., by phosphate (Mansfield et al. 2001) and NO (Teixeira et al. 2001).
Extensive studies on the cellular and molecular changes in nematodes, flies, and vertebrae led to the discovery of reliable molecular tools suitable to characterize stages of apoptotic cell death, in addition to ultrastructural criteria. Intracellular degradation involves a cascade of caspases, a group of proteases detected originally in C. elegans (Ellis and Horvitz 1986). Today, the immunohistological identification of caspase 3 and of cleaved caspase 7 is commonly used to detect apoptotic cells in tissues, besides the detection of DNA fragmentation using the DNA nick end labeling method (TUNEL method) (Gavrieli et al. 1992; Hatori et al. 1995).
Reports on programmed cell death of hypertrophic chondrocytes were partially divergent, with respect to type, stage, and extent of cell death (for reviews, see Gibson (1998), Shapiro et al. (2005), and Tsang et al. (2014)). For example, Gibson et al. (1995) demonstrated by light microscopy condensation of chondrocytes in resorbing chick sterna at the interface with the invading bone marrow, and electron microscopy demonstrated a range of morphological alterations including retraction from the pericellular matrix, cytoplasmic and nuclear condensation, and vesiculation, suggestive of sequential steps of apoptosis. Similarly, Farnum and Wilsman (1987, 1989b) described condensed hypertrophic chondrocytes in the lower lacunae adjacent to the cartilage–bone marrow interface in Yucatan swine and suggested that these cells might be dying by apoptosis. They reported that only about 12 % of the lowest hypertrophic chondrocytes revealed condensed morphology, whereas in the chick sterna, more than 50 % of hypertrophic chondrocytes at the interface with invading bone marrow showed signs of apoptosis owing to the more intensive vascular invasion of chick cartilage (Gibson et al. 1995).
Evidence for apoptotic death of hypertrophic chondrocytes was also supported by the detection of DNA breaks using the TUNEL method (Hatori et al. 1995; Aizawa et al. 1997; Ohyama et al. 1997; Gibson 1998), but again with diverging results. For example, Hatori et al. (1995) reported that only 9 % of chondrocytes isolated from the hypertrophic region chick growth plate at the vascular interface showed fragmented DNA, whereas other studies suggested that DNA fragmentation occurred earlier in differentiation and was much more extensive (Aizawa et al. 1997), with up to 44 % TUNEL-positive cells in the hypertrophic and 15 % in the proliferative zone (Ohyama et al. 1997).
5.4.2 Regulation and Induction of Apoptosis
Several in vitro studies indicate that programmed cell death of hypertrophic chondrocytes in the growth plate is activated by microenvironmental factors (Shapiro et al. 2005). For example, Mansfield et al. (2001, 2003) have shown that inorganic phosphate (Pi) and Ca2+ which are released during resorption of hydroxyapatite of the calcified cartilage during endochondral ossification are able to induce apoptotic cell death of chick chondrocytes in vitro. Texeira et al. (2001) provided evidence that this process is mediated by nitrogen oxide (NO) since Pi induced a threefold increase in NO concentration, which can kill chondrocytes in vitro; it may activate cell death by causing loss of thiol in the growth plate (Teixeira et al. 2003) and loss of mitochondrial membrane potential (Teixeira et al. 2001).
Important factors controlling survival and differentiation of growth plate chondrocytes are the hypoxia-induced factors HIF1α and HIF2α (Schipani et al. 2001). HIF factors are essential for chondrocyte survival in the growth plate by activating energy generation through anaerobic glycolysis. They are enhanced under the hypoxic conditions in the center of the hypertrophic zone, but an increase under the oxygen levels at the cartilage erosion zone due to capillary invasion may render hypertrophic chondrocytes to become sensitive to apoptogens (reviewed in Shapiro et al. (2005) and Toledo-Pereyra et al. (2004)).
5.4.3 Paralysis and Chondroptosis, Dark and Light Cell Death
In most tissues, apoptotic cells are normally phagocytosed by macrophages or other adjacent cells, whereas growth plate chondrocytes (with the exception of terminally differentiated chondrocytes in opened lacunae and the chondro-osseous junction) are surrounded by extracellular cartilage matrix and cannot be phagocytosed. Owing to the distinct lack of typical apoptotic morphological changes in the terminal hypertrophic chondrocytes, some authors concluded that chondrocyte death in the growth plate does not follow the classical mechanism of apoptosis (Farnum and Wilsman 1989a; Roach and Clarke 2000; Carames et al. 2010). Instead, the presence of autophagic vacuoles and the expression of autophagy-regulating genes by growth plate chondrocytes suggest that these cells undergo processes more similar to autophagy than apoptosis (Roach and Clarke 1999, 2000; Shapiro et al. 2005; Staines et al. 2013). Roach et al. (1995, 1997) defined special forms of chondrocyte cell death such as paralysis (Roach and Clarke 1999) or chondroptosis (Roach et al. 2004). In an electron microscopic study using an improved fixation technique with ruthenium hexamine trichloride (RHT; Hunziker et al. 1983), Roach and Clarke (2000) distinguished between two forms of cell death in the chick growth plate, one leading to “dark” condensed chondrocytes containing a convoluted nucleus with patchy chromatin condensations and dark cytoplasm with excessive amounts of endoplasmic reticulum. Other cells defined as “light” hypertrophic chondrocytes showed inconsistent chromatin condensation and varying nuclear appearance from pale to uniformly condensed, with progressively disintegrating cytoplasm and organelles, while cell membranes and nuclear membranes were largely preserved. These findings were confirmed in a study on fetal horse growth plates by Ahmed et al. (2007) who distinguished “dark” cell death, marked by a condensed nucleus with progressively convoluted nuclear membranes and cytoplasm fragmented into membrane-bound apoptotic bodies, whereas in “light” cell death, the nucleus was condensed into irregular patches with a convoluted nuclear membrane.
In conclusion, all experimental evidence available supports the notion that hypertrophic chondrocytes undergo some form of programmed cell death at the cartilage erosion front which may be different from the classical apoptotic cell death. But the studies show diverging results with respect to the rate of cell death, morphology, and extent of cellular changes in the growth plate; some of the observed morphological alterations may be indicative of autophagy which may lead to cell death, but in other cases to cell shrinkage, to cell survival, and to reprogramming into a progeny of osteogenic or other stemlike cells with future functions in the primary ossification center.
5.4.4 Evidence for Transdifferentiation of Chondrocytes to Osteoblasts
5.4.4.1 Hypertrophic Chondrocytes Proliferate and Are Metabolically Active
There is ample evidence that hypertrophic chondrocytes have the ability to proliferate, although slowly, and are metabolically active, expressing distinct sets of genes including Col10a1, Opn, ALP, PTHrP, IHH, Osx, Runx2 HIF1a, FGFR3, LOX4, and others in the upper hypertrophic zone and Mmp13, Vegfa, Rankl, BMP6, and Col10a1 in the lower hypertrophic zone (see Chap. 8). Also ultrastructural studies are indicative of a high metabolic activity of chondrocytes even in the lower hypertrophic zone (Holtrop 1972; Hunziker et al. 1984; Farnum and Wilsman 1993). The proliferation rate, however, declines with increasing hypertrophy, and the general view is that the late hypertrophic chondrocytes located in the lower growth plate adjacent to the cartilage erosion zone have left the cell cycle (Farnum and Wilsman 1993). They were therefore defined as “terminally” differentiated, although there is experimental evidence that some of these cells are still mitotic. For example, Crelin and Koch (1967) showed that hypertrophic chondrocytes in the mouse pubic bone cartilage incorporated 3H-thymidine. Evidence that hypertrophic chondrocytes may resume proliferation was also obtained by BrdU labeling of hypertrophic chondrocytes in the chick growth plate (Galotto et al. 1994).
5.4.4.2 Previous Evidence for Chondrocyte-to-Osteoblast Transformation
In their thymidine labeling experiment, Crelin and Koch (1967) observed that not only chondrocytes but also chondroclasts, osteoblasts, and osteocytes had incorporated 3H-thymidine and thus concluded that hypertrophic chondrocytes of the calcified cartilage may transform into chondroclasts and bone-forming cells. A number of further cell and organ culture experiments have supported the concept of a continuous chondrocyte-to-osteoblast transition in the growth plate, indicating that hypertrophic chondrocytes are not terminally differentiated (Fell 1825; Holtrop 1972; Bentley and Greer 1970; Kahn and Simmons 1977). For example, Thesingh et al. (1991) described organ cultures of fetal mouse bones that were stripped of periosteum and observed that the chondrocytes differentiated to hypertrophic cells and further converted into osteoblasts when the bones were cocultured with brain tissue. The potency of hypertrophic chondrocytes to convert into bone-forming cells was also demonstrated in chondrocyte cell cultures: Human chondrocytes changed their gene expression pattern from collagens II and X to collagen I within 4 weeks in the presence of ascorbate (Kirsch et al. 1992). Similarly, chondrocytes isolated from chick cartilage differentiated to hypertrophic cells in suspension culture in the presence of ascorbate and retinoic acid and changed rapidly after transfer to anchorage-dependent culture conditions into calcifying, osteoblast-like cells expressing collagen I and ovotransferrin, a peculiarity of chick chondrocytes (Gentili et al. 1993; Descalzi-Cancedda et al. 1992).
Strong morphological evidence for direct transdifferentiation of hypertrophic chondrocytes to osteoblasts was presented by Roach et al. (Roach 1992; Roach et al. 1995; Roach and Erenpreisa 1996) who demonstrated the appearance of osteoblast-like cells in intact lacunae of hypertrophic cartilage of 14-day chick femora several days after setting a cut through the hypertrophic zone. The cells within the lacunae stained positive for alkaline phosphatase, osteonectin, osteopontin, and type I collagen, and many hypertrophic chondrocytes were still mitotic as shown by tritiated thymidine incorporation. No osteogenic markers were observed in hypertrophic lacunae of uncut femurs; therefore, the authors suggested that the transdifferentiation to osteoblast-like cells was initiated by disruptions of the cell–cell associations (Roach and Erenpreisa 1996). Interestingly, the change from chondrogenic to osteogenic commitment was associated with an asymmetric cell division with diverging fates of the two daughter cells, with one daughter cell remaining viable and the other one dying (Roach et al. 1995).
Despite these and other experimental studies on the cell fate of hypertrophic chondrocytes, the notion of a continuous chondrocyte-to-osteoblast lineage was never fully accepted, either because of unsettled questions concerning the exclusion of contaminating osteoprogenitor or stem cells in the cell or organ culture systems or because of experimentally induced nonphysiological modulations of the microenvironment of hypertrophic chondrocytes. Furthermore, many of the relevant studies were performed with chick chondrocytes or chick organs which show a higher plasticity of the phenotype and different responses to growth factors and hormones. Therefore, the question remained open whether similar transitions of the chondrogenic to the osteogenic phenotype also occur on the mammalian growth plate in situ.
5.4.4.3 Lineage Tracing Experiments
A definite proof of a chondrocyte-to-osteoblast continuum during endochondral ossification requires labeling of chondrocytes with cell-specific and stable cellular markers which are transmitted to all progeny of the cell. Previous attempts to label the progeny of hypertrophic chondrocytes with cherry, the gene for a red fluorescent protein, expressed under the Col10a1 promoter were, however, not entirely conclusive due to the limited half-life of the cherry protein (Maye et al. 2011). Another study using inducible and stable Col2a1Cre-induced reporter gene expression detected a chondrocyte-derived progeny associated with bone trabeculae in the spongiosa underneath the chondro-osseous junction, but these cells did not express bone-specific markers (Hilton et al. 2007). The use of Col2a1Cre-induced recombination is also hampered by the fact that Col2a1 is expressed very early in chondro-osteoprogenitor cells and in the perichondrium and thus will also label cells which follow a separate osteogenic pathway along the perichondrium (Maes et al. 2010; Ono et al. 2014).
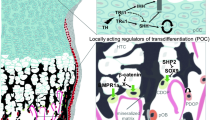
Final affirmation of the concept of chondrocyte-to-osteoblast transdifferentiation was achieved independently in four laboratories in genetic lineage tracing studies based on Cre-induced reporter gene activation. To follow the cell fate of growth plate chondrocytes, hypertrophic chondrocytes were specifically labeled with tamoxifen-inducible or non-inducible Cre recombinases inserted into the Col10a1 gene (Yang et al. 2014a, b) or driven by a BAC Col10 transgene (Golovchenko et al. 2013; Zhou et al. 2014; Park et al. 2015). In addition, the chondrocyte lineage was followed using tamoxifen-inducible Agc1-CreERT2 mouse (Zhou et al. 2014) in which Cre was inserted in the 3′UTR of the aggrecan gene Agc1 (Henry et al. 2009). Col10Cre- or Agc1Cre-induced genetic recombination was made visible by mating with reporter mice bearing LacZ, YFP, or tomato genes under the ROSA26 promoter (Soriano 1999). Using these tools, L. Yang et al. (2014b), G. Yang et al. (2014a), Zhou et al. (2014), and Park et al. (2015) detected reporter gene expression in transgenic mice in hypertrophic chondrocytes but also in trabecular osteoblasts of the primary spongiosa, in the endosteum, and in cortical bone (Figs. 5.1, 5.2, and 5.3). Importantly, no LacZ+ or YFP+ cells were found in calvaria, nor in the perichondrium/periosteum which is the other source for osteoprogenitor cells invading the spongiosa (Colnot et al. 2004; Maes et al. 2010).

Appearance of non-chondrocytic reporter + cells in the primary spongiosa of tamoxifen-treated Agc1-CreERT2 embryos that are derived from mature chondrocytes. (a) The LacZ-stained femur section of E16.5 Agc1-CreERT2;ROSA26R embryo treated with tamoxifen at E11.5. The black arrows indicate the non-chondrocytic LacZ+ cells in the primary spongiosa (ps). Black brackets hypertrophic zone. No LacZ+ cells were detected within the perichondrium (red arrows) and periosteum (red arrowheads). (b) Higher magnification of black square in (a). (From Zhou et al. (2014), with kind permission by PLOS genetics). (c) Similarly, in BACol10Cre;RosaLACZ transgenic mice in which the ROSA29 locus is activated under the Col10a promoter, LacZ-positive cells appear in the primary ossification center (here: ribs of a P5 mouse). The LacZ + cells have typical shape and location of osteoblasts adherent to bone trabeculae (From Park et al. (2015), with kind permission from The Company of Biologists)

Dual origin of trabecular osteoblasts. BACCol10Cre-induced recombination of the ROSA 26YFP locus in hypertrophic chondrocytes activates YFP expression in hypertrophic chondrocytes (hc in a) and a progeny of hc in the primary spongiosa and endosteum (E). Immunofluorescence double staining of a E18.5 tibia of a BACCol10Cre;R26RYFP mouse for YFP (a) and osterix (b) shows that about 30 % of the Osx + osteogenic cells are also positive for YFP (yellow cells in c), i.e., derived from hypertrophic chondrocytes. (c) The rest of the Osx+ cells in the spongiosa is derived from perichondrium (red cells, periosteal osteoblasts po). Note the complete absence of YFP+ cells from periosteum/perichondrium (P). White arrow chondro-osseous junction (Modified from Park et al. (2015))

Upper panel (a–c) Lineage tracing of hypertrophic chondrocytes using Col10a1-Cre;Osx flox/+ mice (Zhou et al. 2014). Double immunofluorescence (DIF) experiments revealed that the EGFP+ (Osx−/+) cells (yellow arrows) in the primary spongiosa and endosteum are mature chondrocyte-derived osteoblasts. DIF experiment with anti-Ocn and anti-GFP using femur frozen sections of 1-month-old Col10a1-Cre;Osx flox/+ mice. Green arrows EGFP + Ocn- cells, red arrows Ocn + EGFP- cells. Lower panel (a–c) Lineage tracing using Agc1-CreERT2;2.3Col1-GFP;ROSA-tdTomato triple transgenic mice (Zhou et al. 2014). Cellular co-localization of the chondrocyte-derived tomato marker and the osteoblast-specific 2.3Col1-GFP marker shows collagen 1-expressing cells (yellow arrows) in the spongiosa of P1 femora that are derived from chondrocytes. Tamoxifen treatment was done at E14.5. Green cells are Tomato− EGFP+ cells, yellow arrows Tomato+EGFP+ cells. Only EGFP+ cells but no Tomato + cells were present in the perichondrium (between white arrows) and periosteum (between white arrowheads) (From Zhou et al. (2014), with kind permission from PLOS Genetics) white brackets growth plate (gp)
These studies unanimously provided conclusive evidence that growth plate chondrocytes give rise to a progeny of osteoblasts which contribute substantially to endochondral bone formation. This conclusion relies in part on the premise that the Col10Cre activity used in the various transgenic mouse lines is restricted to hypertrophic chondrocytes. Lack of Cre expression in calvarial bone or any other tissue outside hypertrophic cartilage was demonstrated in all Col10Cre knock-in mice (Yang et al. 2014a, b) or BACCol10Cre transgenic mice by in situ hybridization (Zhou et al. 2014; Park et al. 2015; Golovchenko et al. 2013).
Comprehensive histological investigations of Col10cre- and Agc1Cre-induced reporter gene expression during skeletal development demonstrated specific expression of LacZ, YFP, or Tomato reporter genes coinciding with Cre expression in the zone of hypertrophic cartilage (Yang et al. 2014a, b; Park et al. 2015; Zhou et al. 2014) of long bones prior to capillary invasion and bone marrow formation, which begins at about E14–E15 in the mouse tibia anlage. With formation of the primary ossification center, strong expression of reporter genes appeared in the spongiosa in cells that were associated with bone trabeculae or lining the endosteal surface (Yang et al. 2014a, b; Zhou et al. 2014; Park et al. 2015). Many of the reporter gene-positive cells showed epithelioid, osteoblast-like morphology (Fig. 5.1) and co-expressed osteogenic markers including osterix and/or collagen I and osteocalcin, as shown by immunofluorescence double or triple staining, by immunohistochemistry or combinations with in situ hybridization (Fig. 5.2). Increasing numbers of reporter gene-positive cells, i.e., hypertrophic chondrocytes (HC)-derived osteogenic cells, were observed in the primary and secondary ossification centers of all long bones, ribs, and vertebrae (Park et al. 2015) in fetal, postnatal, and even adult bones (Yang et al. 2014a, b), again confirming the notion that hypertrophic chondrocytes give rise to a non-chondrocytic, osteogenic progeny.
In light of remaining uncertainties with respect to antibody specificity, further efforts were made to ensure the osteogenic phenotype of HC-derived cells in the spongiosa. For example, Zhou et al. (2014) identified osteogenic cells in the spongiosa unequivocally by expressing EGFP+ under the Col10Cre-recombined Osx promoter (Agc1-CreERT2 Osx +/EGFP) (Fig. 5.3). An elegant and specific way to mark Col1a1-expressing cells in an antibody-independent way was the insertion of the 2.3-Col1;GFP transgene which is expressed specifically in osteoblasts but not in fibroblasts (Kalajzic et al. 2002) into Col10Cre;tomato or Agc1Cre;tomato mice (Zhou et al. 2014) (Fig. 5.3).
The osteogenic character of YFP+, i.e., HC-derived cells in the spongiosa of P5 BACCol10Cre;ROSAYFP bones, was further confirmed by qRT-PCR analysis of FACS-sorted YFP+ trabecular osteoblasts which revealed expression of Col1a1, bsp (bone sialoprotein), and Runx2 (Park et al. 2015). Similarly, cultured endosteal cells isolated from 3-week-old BACCOL10Cre; ROSAYFP bones and sorted for YFP fluorescence expressed Osx, Runx2, osteocalcin, and Col1a1 as shown by RT-PCR (Park et al. 2015).
5.4.4.4 Tam-Inducible Cre Expression
Despite the results of in situ hybridization analysis showing the absence of Cre expression in cells other than hypertrophic chondrocytes or in calvarial bone (Yang et al. 2014a, b; Zhou et al. 2014; Park et al. 2015), questions remained whether minute levels of Col10Cre expression below the detection limit of in situ hybridization can be excluded in osteogenic cells of the primary spongiosa. Further strong evidence for the specificity of Col10Cre activity was, however, provided by L. Yang et al. (2014b) and Zhou et al. (2014) who confirmed the chondrocyte-to-osteoblast continuation in the growth plate by generating tamoxifen-inducible Col10CreERt (Yang et al. 2014b) or Agc1CreERT2 (Zhou et al. 2014) reporter lines. CreERt and CreERT2 are Cre recombinases bearing mutated estrogen receptor binding domains (ERT) which do not respond to endogenous estradiols but are activated through nuclear localization by the estrogen analog 4-hydroxy-tamoxifen (Feil et al. 2014). The advantage of this system is that it allows time-controlled pulses of cre activation by administering tamoxifen (TAM) to the pregnant mothers at distinct stages of development. If the pulse is started in a defined window prior to onset of bone marrow formation, it will mark only hypertrophic chondrocytes, and the time course of reporter gene expression in hypertrophic chondrocytes and their progeny including osteogenic cells can be tracked by analyzing the offspring at various days after TAM injection. For example, when pregnant Col10CreErt;LacZ+ mothers were injected with tamoxifen at stage E 13.5, after a time lag of 2 days, many LacZ+ chondrocytes were detected in the hypertrophic zone of the humerus and some in the tibia at E15.5, whereas at E16.5, fewer LacZ+ cells were seen in the hypertrophic zone, but more in the primary ossification center (Yang et al. 2014b). This excluded unspecific Col10Cre-induced recombination of the ROSA26 locus in osteoblasts, because in that case, one would expect LacZ+ cells appearing in the primary spongiosa simultaneously with hypertrophic chondrocytes.
Col2a1Cre-driven reporter gene expression is activated already in early chondro-osteoprogenitor cells in the cartilage blastema as well as in the perichondrium and may therefore also mark the perichondrium-derived osteogenic lineage. In contrast, Agc1 (aggrecan) is expressed in resting chondrocytes but not in chondro-osteoprogenitor cells nor in the perichondrium (Henry et al. 2009) and is therefore a suitable marker to follow the cell fate of mature (resting and proliferating) chondrocytes. Agc1;CreERT2 activity was detected in limb chondrocytes already at embryonic stage E12.5 (Zhou et al. 2014). The activation window in Agc1CreERT2; ROSALacZ mice was 24–48 h after TAM injection. When TAM was injected at E11.5, LacZ+ chondrocytes were already detected at E13.5 (Zhou et al. 2014), and at E15.5, with onset of ossification in the primary spongiosa, cre-inducing activity of tamoxifen was no longer retained in the tissue, but at E16.5, LacZ+ cells were seen in the primary spongiosa. The presence of osteogenic cells derived from mature chondrocytes was also confirmed by analyzing Agc1-CreERT2;Osx +/EGFP mice which revealed chondrocyte-derived, osterix-positive (Osx + EGFP+) cells in the spongiosa at E15.5 2 days after tamoxifen treatment (Zhou et al. 2014).
5.4.4.5 Quantitative Contribution of Chondrocyte-Derived Osteoblasts to Endochondral Bone
These findings challenged not only the common view that late hypertrophic chondrocytes are eliminated at the chondro-osseous junction of the growth plate but also the concept that all trabecular and endosteal osteoblasts in the primary and secondary ossification center originate from osteoprogenitor cells invading from the periosteum along with bone marrow capillaries (Colnot et al. 2004; Maes et al. 2010; Ono et al. 2014). An important issue was therefore the question to what extent chondrocyte-derived osteoblasts contributed to endochondral bone formation in relation to periosteum-derived osteoblasts. Depending on the type of genetic lineage tracing, the markers to define osteoblasts (osteoprogenitors, osteoblasts, or mature osteocytes), and the stage and types of bones investigated, the estimated contributions of chondrocyte-derived osteoblasts to total endochondral bone cells varied between 8.5 and 30 % in the trabecular and endosteal zone of the spongiosa at embryonic stage E18.5 or newborns and 40–60 % in postnatal cortical bone (Yang et al. 2014a, b; Zhou et al. 2014; Park et al. 2015).
A quantitative analysis of the ratio of chondrocyte-derived osteogenic cells (defined by nuclear staining for osterix) to total osteogenic cells in the primary spongiosa of E16.5 to P7 BACCol10cre;ROSAYFP + tibiae indicated that chondrocyte-derived (YFP+) Osx+ cells contributed 30 % to all Osx+ cells in the spongiosa at E18.5, leveling off at 18 % in the postnatal stages (Park et al. 2015). Similarly, L. Yang et al. (2014b) reported that 16 % of all Col1-positive cells in the primary ossification center of E18.5 tibiae of BACCol10CreErt;ROSALacZ + embryos were positive for LacZ. This corresponded to the 30 % ratio reported above (Park et al. 2015), considering that in Col10Cre knock-in mice, only 50 % of the hypertrophic chondrocytes were LacZ+ after TAM-induced reporter gene activation (Yang et al. 2014b), whereas 100 % of the hypertrophic chondrocytes in growth plates in BACCol10Cre;ROSALAcZ bone were LacZ positive (Gebhard et al. 2008; Park et al. 2015). Divergent results were published on postnatal stages: G. Yang et al. (2014a) reported that about 30 % of all Col1-positive cells in metaphyses of 20-day-old Col10Cre;ROSALacZ tibiae were LacZ+, as compared to 21 % in the trabecular zone. Zhou et al. (2014), however, reported much higher rates with 60 % chondrocyte-derived Ocn+ Col1a1+ osteoblasts in the trabecular zone, 68 % in the endosteal zone of 3- to 4-week-old Col10a1Cre;Osxflox/EGFP or Col10a1-cre;2.3Col1-GFP; ROSA Tomato tibiae, and 69 % in the cortical zone. Similarly, a rate of 40–50 % LacZ-positive, matrix-embedded mature osteocytes expressed the LacZ marker in cortical bone of 4 week of Col10Cre;ROSA LacZ mice (Park et al. 2015). These studies also provided strong evidence for a dual origin of cortical bone in fetal and postnatal stages forming by appositional growth, with chondrocyte-derived osteoblasts accumulating matrix from the endosteal side and periosteum-derived osteoblasts providing the outer part of the cortical bone (Fig. 5.4).

(a) Cell fate analysis of hypertrophic chondrocytes reveals transdifferentiation to osteoblasts: Lineage tracing of hypertrophic chondrocytes with Col10Cre-activated reporter genes (here: YFP) revealed that in the fetal spongiosa, a substantial fraction of trabecular osteoblasts is derived from hypertrophic chondrocytes (green cells), whereas the majority of osteoblasts are derived from perichondrial osteoprogenitor cells (red cells) invading the spongiosa through capillaries (Modified after Park et al. (2015)). (b) Dual pathways but common origin of trabecular osteoblasts: Chondrogenic and osteogenic lineages start from a common Sox9+ osteo-chondroprogenitor cell in early limb development. In the growth plate, chondrocytes mature in several distinct differentiation steps to hypertrophic cells which either undergo programmed cell death (apoptosis or chondroptosis) or shrink to small osteoprogenitor cells (CDOP) which may be reprogrammed into the osteogenic lineage and other bone marrow cells
5.5 Transdifferentiation During Fracture Callus Formation and Bone Repair
Bone fracture healing involves in most cases a cartilaginous callus intermediate that is replaced by bone in a process analogous to endochondral ossification. To elucidate whether chondrocytes of the fracture callus also give rise to bone cells during the repair process, L. Yang et al. (2014b) grafted pieces of hypertrophic cartilage isolated from Col10Cre;RLacZ pups into bone injury sites generated by drilling holes in the tibia of 3-month-old adult mice. Within 8 days post operation, LacZ-positive cartilage in the graft decreased and was replaced by bone containing LacZ+ osteoblast- and osteocyte-like cells.
In another study, healing of semi-stabilized tibia fractures was investigated in 2- to 3-month-old Agc1-CreERT2;ROSA-tdTomato mice (Zhou et al. 2014). Seven days post surgery, Agc1- mice were treated with tamoxifen, and at day 9, when chondrocyte differentiation occurred in the repair callus, Tomato+ fluorescence was completely matching the areas of Safranin-O-stained chondrocytes in the callus, suggesting that the Tomato+ cells in the cartilage callus were in fact chondrocytes. On day 14, the repair callus was partially ossified, showing a mixture of bone and cartilage, and almost all cells in the repair callus, both in the cartilage and in the bone regions, were positive for Tomato, suggesting the presence of non-chondrocytic Tomato+ cells in the repair callus. In the repair callus of a post-surgery day 14 Agc1-CreERT2;2.3Col1-GFP;ROSA-tdTomato mouse treated with tamoxifen at post-surgery day 6, many of the Tomato+ cells were also positive for GFP (Tomato+ GFP+), implying that the mature chondrocytes present in the repair callus have the ability to become Col1a1-expressing bone-forming osteoblasts. At day 29 post surgery, the number of Tomato+ GFP+ cells was substantially increased, and ossification was almost complete (Zhou et al. 2014). Thus, both studies provide convincing evidence that chondrocytes in the repair callus are a source of osteoblasts involved in bone fracture healing.
5.6 Mechanism of Chondrocyte-to-Osteoblast Conversion: Transdifferentiation or Redifferentiation and Reprogramming?
Despite the overwhelming evidence for a continuation of chondrocyte differentiation into osteogenic cells, many questions remain open concerning the mechanism and regulation of conversion of hypertrophic chondrocytes into osteoblasts. Do hypertrophic chondrocytes give rise to osteoblasts or osteoprogenitor cells by direct transdifferentiation, or does this process involve redifferentiation of hypertrophic chondrocytes to stemlike cells and reprogramming into osteoblasts? What is the sequence of gene expression changes during transdifferentiation? Are the osteogenic cells generated by asymmetric division of hypertrophic chondrocytes as proposed by Roach et al. (1995), with one daughter cell undergoing cell death and the other entering an osteogenic transdifferentiation program? Is the process of transdifferentiation a cell-autonomous process or regulated by the microenvironment of the hypertrophic chondrocytes in the calcifying lacunae adjacent to invading bone marrow capillaries? Which extracellular factors are involved in this case, and which factors are critical for the decision of transdifferentiation versus programmed cell death? How can small osteoprogenitor cells emerge from the substantially larger hypertrophic chondrocytes with diameters of 15–20 μm, do they shrink by autophagy or by other mechanisms?
5.6.1 Identification of Chondrocyte-Derived Osteoprogenitor Cells in the Growth Plate
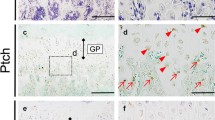
In attempts to identify the origin of the Col10Cre-induced YFP-labeled osteoprogenitor cells at the chondro-osseous junction, growth plates isolated by microdissection of P5-P7 Col10CreYFP + tibiae, femora, and humeri were analyzed by confocal microscopy (Park et al. 2015). Detailed analysis of the chondro-osseous junction at serial levels along the longitudinal axis (z-axis) revealed the presence of small chondrocyte-derived (YFP+) Col1+ cells and YFP+Osx+ cells of 4–6 μm diameters in the lowest zone of hypertrophic chondrocytes adjacent to the cartilage–spongiosa interface. Importantly, these cells which were named chondrocyte-derived osteoprogenitor (CDOP) were mitotically active, as shown by BrdU injection into pregnant Col10Cre;YFP + mice 1 day before delivery (Park et al. 2015). Size and position of these cells resembled the “condensed hypertrophic chondrocytes” described ultrastructurally by Farnum and Wilsman (1987, 1989b) in the lowest row of closed lacunae of hypertrophic growth plate cartilage. These cells have a condensed nucleus and extensive cytoplasmic vacuolization and fill only a small part of the space of its lacuna. Initial attachments to the pericellular and territorial matrices eventually disappear, except at the last transverse septum (Farnum and Wilsman 1987, 1989b). Whether the CDOP cells identified by Park et al. (2015) actually represent these condensed cells, however, remains to be confirmed.
To confirm the ability of CDOP cells to differentiate into osteoblasts, CDOP cells were isolated from P5-P7 Col10CreYFP + growth plates by sequential enzymatic digestion of isolated growth plates with trypsin and collagenase, and three fractions were obtained which were cultured on fibronectin-coated culture dishes. After 7–12 days in monolayer culture, immunofluorescence analysis of fraction 2 cells with diameters of 4–5 μm revealed numerous small YFP+ cells staining for the stem cell marker Sca1. When cells were expanded for 10 days and YFP+ and YFP− cells were separated by FACS, almost all YFP+ cells were positive for Osx. RT-PCR analysis of RNA isolated from sorted YFP+ fraction 2 cells revealed expression of both stem cell-typical genes such as CD34, sca1, sox2, and c-myc and genes of the osteogenic lineage including col1a1, osx, and Runx2. After additional 14 days in culture, mRNA levels of Col1a1, Ocn, and Runx2 in YFP+CDOP cells were comparable to those of differentiating MC3T3 osteoblasts, confirming the ability of CDOP cells to differentiate into osteoblasts (Park et al. 2015). Altogether, these findings confirm that the population of small, 4–6 μm cells in fraction 2 isolated from growth plates contained chondrocyte-derived cells with osteoprogenitor character. They represent most likely the CDOP cells observed by confocal microscopy in the chondro-osseous junction of the growth plate.
5.6.2 Autophagy in Hypertrophic Chondrocytes: Preparation for Apoptosis or Cell Survival?
A major question arising from these observations relates to the dramatic changes in cell size during the transdifferentiation process: How do the large hypertrophic chondrocytes shrink to osteoprogenitor cells with an approximately 20- to 50-fold lesser volume? In light of increasing evidence for autophagy occurring in growth plate and articular chondrocytes (Bohensky et al. 2009; Srinivas et al. 2009a, b; Carames et al. 2010), it is likely that cell shrinkage occurs by autophagy. Autophagy is primarily considered a cell-protective mechanism that permits cells to survive under stress conditions such as nutrient deficiency or hypoxia (Carames et al. 2010). This includes that “the cell cannibalizes itself to generate energy” (Srinivas et al. 2009b; Yang and Klionsky 2010). By sequestering dysfunctional organelles, proteins, and membranes into double-membrane vesicles called autophagosomes which later fuse with lysosomes, cells generate energy and remain viable and complete their life cycle (Kimura et al. 2007; Srinivas et al. 2009b; Yang and Klionsky 2009). In fact, most YFP-positive hypertrophic chondrocytes in the prehypertrophic and hypertrophic zone of BACCol10CreYFP mice showed a punctate fluorescence after both anti-beclin-1 and anti-LC3B staining (Park et al. 2015), characteristic of autophagic vacuoles (Srinivas et al. 2009b; Carames et al. 2010). Beclin-1 is a protein that participates in the nucleation of autophagic vesicles (Thorburn 2008; Zhang et al. 2013) and LC3B is required for the expansion of autophagosomes (Weidberg et al. 2011).
On the other hand, autophagy is also closely related to apoptosis caused by stress or hypoxia (Wang 2015; Li et al. 2015). Generally, autophagy blocks the induction of apoptosis and inhibits the activation of apoptosis-associated caspases which could reduce cellular injury (Wu et al. 2014). Thus, autophagy is very likely the first step in the condensation process of hypertrophic chondrocytes and either may lead to cell death or permit cell survival and reprogramming to osteogenic cells depending on the microenvironmental conditions of the cells (Carames et al. 2010). Identification of these conditions which may include hypoxic conditions, reactive oxygen species (ROS) (Poillet-Perez et al. 2015), or pH changes appears an important question to be solved.
5.6.3 Programmed Cell Death or Transdifferentiation?
As the current data strongly suggest that hypertrophic chondrocytes have the option to undergo programmed cell death, or to reenter the cell cycle and activate an osteogenic fate (Fig. 5.4), the question arises at which stage of the chondrocyte differentiation program this decision is made and which factors of the microenvironment of the growth plate are involved in the control of this decision. As hypertrophic chondrocytes are arranged in vertical columns, each representing most likely one clone derived from a single prehypertrophic chondrocyte at the top of the column, the decision for cell death versus reprogramming after onset of autophagy may occur at any stage before reaching the lowest lacuna in the growth plate. Using the multicolor Confetti reporter system (Muzumdar et al. 2007) activated by Col2a1Cre ERT-mediated recombination, G. Yang et al. (2014a) observed few chondrocyte columns 5 days after tamoxifen activation, each labeled uniformly with different colors, confirming the clonal character of the chondrocyte columns. Several days later, the colored columns extended into the spongiosa underneath the growth plate, indicating direct conversion of some chondrocytes into osteogenic cells and other bone marrow cells. This powerful lineage tracing system could be extended to investigate whether different types of clonal chondrocyte columns could be distinguished, perhaps on the basis of differentially expressed genes, which would undergo cell death rather than reprogramming into osteogenic cells. The clonal character of hypertrophic chondrocytes within a column and the heterogeneity of the cell fate in individual columns are consistent with the report by Farnum and Wilsman (1987, 1989b) who described the appearance of small “condensed” cells in some, but not all lowest intact lacunae of hypertrophic chondrocytes.
For the decision of cell death by apoptosis versus survival, the expression levels of Bcl2 and BAX have been shown to be instrumental. Bcl-2 and BAX are members of an emerging family (Bcl family) of proteins which are involved in the regulation of programmed cell death (Vaux et al. 1988; Korsmeyer 1992). Bcl-2, an inhibitor of apoptosis, and BAX, an apoptosis inducer, form heterodimers which neutralize the anti-apoptotic effect of Bcl-2, causing accelerated cell death. Within a cell, it is the ratio of Bcl-2 to Bax that determines whether a cell dies or not (Amling et al. 1998; Sedlak et al. 1995; Oshima et al. 2008; Yin et al. 1994).
Very little is known, however, on the heterogeneity of the microenvironment in different zones or different columns of hypertrophic chondrocytes in the growth plate. Gibson et al. (1995) have reported enhanced apoptotic activity in hypertrophic chick sternal chondrocytes located close to invading bone marrow vessels, and also in the mammalian growth plate, the bone marrow invasion front is rather irregular, suggesting that different hypertrophic chondrocytes along the chondro-osseous junction may be exposed to different levels of invading cytokines or growth factors and to different pH or oxygen levels. Elucidation of these parameters and their influence on autophagy, cell death, or cell survival and transdifferentiation will be one of the demanding challenges in the future to clarify this issue.
5.7 Conclusion
The recognition of a continued chondrocyte-to-osteoblast lineage sheds new light on the role of hypertrophic chondrocytes in endochondral ossification. We now understand why such seemingly intricate and complex mechanisms are necessary to regulate chondrocyte differentiation in the growth plate, leading at the end to a hypertrophic chondrocyte which not only produces a specialized calcified cartilage matrix but also induces invasion of vascular sprouts, expresses bone-specific genes, and finally gives rise to a progeny of osteoblasts. Numerous previous studies reporting complex alterations of endochondral bone formation after genetic deletion or overexpression of osteogenic or chondrogenic regulatory genes in chondrocytes may have to be reconsidered in light of the concept of a continued chondrocyte-to-osteoblast lineage, not only in transgenic animal studies but also in human chondrodysplasias.
References
Ahmed YA, Tatarczuch L, Pagel CN, Davies HM, Mirams M, Mackie EJ (2007) Physiological death of hypertrophic chondrocytes. Osteoarthritis Cartilage 15(5):575–586
Aizawa T, Kokubun S, Tanaka Y (1997) Apoptosis and proliferation of growth plate chondrocytes in rabbits. J Bone Joint Surg Br 79(3):483–486
Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B (2002) The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev 16(21):2813–2828
Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, de Crombrugghe B (2005) Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A 102(41):14665–14670
Amizuka N, Hasegawa T, Oda K, Luiz de Freitas PH, Hoshi K, Li M, Ozawa H (2012) Histology of epiphyseal cartilage calcification and endochondral ossification. Front Biosci (Elite Ed) 4:2085–2100
Amling M, Posl M, Hentz MW, Priemel M, Delling G (1998) PTHrP and Bcl-2: essential regulatory molecules in chondrocyte differentiation and chondrogenic tumors [In Process Citation]. Verh Dtsch Ges Pathol 82(160–9):160–169
Anderson HC (1985) Matrix vesicle calcification: a review and uptake. Bone Miner 3:109–130
Aubin JE, Liu F, Malaval L, Gupta AK (1995) Osteoblast and chondroblast differentiation. Bone 17(2 Suppl):77S–83S
Ballock RT, O’Keefe RJ (2003) The biology of the growth plate. J Bone Joint Surg Am 85-A(4):715–726
Bentley G, Greer RB (1970) The fate of chondrocytes in endochondral ossification in the rabbit. J Bone Joint Surg Br 52(3):571–577
Bi W, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B (1999) Sox9 is required for cartilage formation. Nat Genet 22(1):85–89
Bohensky J, Terkhorn SP, Freeman TA, Adams CS, Garcia JA, Shapiro IM, Srinivas V (2009) Regulation of autophagy in human and murine cartilage: hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum 60(5):1406–1415
Brachet A (1893) Etude sur la resorption du cartilage et le developpement des os longs chez les oiseaux. J Anat Physiol 10:391–417
Bronckers AL, Goei W, Luo G, Karsenty G, D’Souza RN, Lyaruu DM, Burger EH (1996) DNA fragmentation during bone formation in neonatal rodents assessed by transferase-mediated end labeling. J Bone Miner Res 11(9):1281–1291. doi:10.1002/jbmr.5650110913
Cancedda R, Castagnola P, Cancedda FD, Dozin B, Quarto R (2000) Developmental control of chondrogenesis and osteogenesis. Int J Dev Biol 44(6 Spec No):707–714
Cancedda R, Descalci-Cancedda F, Castagnola P (1995) Chondrocyte differentiation. Int Rev Cytol 159:265–358
Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M (2010) Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 62(3):791–801
Carlevaro MF, Cermelli S, Cancedda R, Descalzi Cancedda F (2000) Vascular endothelial growth factor (VEGF) in cartilage neovascularization and chondrocyte differentiation: auto-paracrine role during endochondral bone formation. J Cell Sci 113(Pt 1):59–69
Colnot C, Lu C, Hu D, Helms JA (2004) Distinguishing the contributions of the perichondrium, cartilage, and vascular endothelium to skeletal development. Dev Biol 269(1):55–69
Crelin ES, Koch WE (1967) An autoradiographic study of chondrocyte transformation into chondroclasts and osteocytes during bone formation in vitro. Anat Rec 158(4):473–483
Day TF, Yang Y (2008) Wnt and hedgehog signaling pathways in bone development. J Bone Joint Surg Am 90(Suppl 1):19–24
de Crombrugghe B, Lefebvre V, Nakashima K (2001) Regulatory mechanisms in the pathways of cartilage and bone formation. Curr Opin Cell Biol 13(6):721–727
Descalzi-Cancedda F, Gentili C, Manduca P, Cancedda R (1992) Hypertrophic chondrocytes undergo further differentiation in culture. J Cell Biol 117:427–435
Ducy P, Geoffroy V, Karsenty G (1996) Study of osteoblast-specific expression of one mouse osteocalcin gene: characterization of the factor binding to OSE2. Connect Tissue Res 35(1–4):7–14
Ducy P, Starbuck M, Priemel M, Shen J, Pinero G, Geoffroy V, Amling M, Karsenty G (1999) A Cbfa1-dependent genetic pathway controls bone formation beyond embryonic development. Genes Dev 13(8):1025–1036
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G (1997) Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation [see comments]. Cell 89(5):747–754
Ellis HM, Horvitz HR (1986) Genetic control of programmed cell death in the nematode C. elegans. Cell 44(6):817–829
Farnum CE, Wilsman NJ (1987) Morphologic stages of the terminal hypertrophic chondrocyte of growth plate cartilage. Anat Rec 219(3):221–232
Farnum CE, Wilsman NJ (1989a) Cellular turnover at the chondro-osseous junction of growth plate cartilage: analysis by serial sections at the light microscopical level. J Orthop Res 7(5):654–666
Farnum CE, Wilsman NJ (1989b) Condensation of hypertrophic chondrocytes at the chondro-osseous junction of growth plate cartilage in yucatan swine relationship to long-bone growth. Am J Anat 186:346–358
Farnum CE, Wilsman NJ (1993) Determination of proliferative characteristics of growth plate chondrocytes by labeling with bromodeoxyuridine. Calcif Tissue Int 52(2):110–119
Feil S, Krauss J, Thunemann M, Feil R (2014) Genetic inducible fate mapping in adult mice using tamoxifen-dependent Cre recombinases. Methods Mol Biol 1194:113–139. doi:10.1007/978-1-4939-1215-5_6
Fell HB (1825) The histogenesis of cartilage and bone in the long bones of the embryonic fowl. J Morphol 40:417–457
Fuchs Y, Steller H (2011) Programmed cell death in animal development and disease. Cell 147(4):742–758. doi:10.1016/j.cell.2011.10.033
Galotto M, Campanile G, Robino G, Cancedda FD, Bianco P, Cancedda R (1994) Hypertrophic chondrocytes undergo further differentiation to osteoblast-like cells and participate in the initial bone formation in developing chick embryo. J Bone Miner Res 9(8):1239–1249
Gavrieli Y, Sherman Y, Ben-Sasson SA (1992) Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119(3):493–501
Gebhard S, Hattori T, Bauer E, Schlund B, Bosl MR, de Crombrugghe B, von der Mark K (2008) Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biol 27(8):693–699
Gentili C, Bianco P, Neri M, Malpeli M, Campanile G, Castagnola P, Cancedda R, Cancedda FD (1993) Cell proliferation, extracellular matrix mineralization, and ovotransferrin transient expression during in vitro differentiation of chick hypertrophic chondrocytes into osteoblast-like cells. J Cell Biol 122(3):703–712
Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N (1999) VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med 5(6):623–628
Gerstenfeld LC, Shapiro FD (1996) Expression of bone-specific genes by hypertrophic chondrocytes: implication of the complex functions of the hypertrophic chondrocyte during endochondral bone development. J Cell Biochem 62(1):1–9
Gibson G (1998) Active role of chondrocyte apoptosis in endochondral ossification. Microsc Res Tech 43(2):191–204
Gibson GJ, Beaumont BW, Flint MH (1984) Synthesis of a low molecular weight collagen by chondrocytes from the presumptive calcification region of the embryonic chick sterna: the influence of culture with collagen gels. J Cell Biol 84:208–216
Gibson GJ, Kohler WJ, Schaffler MB (1995) Chondrocyte apoptosis in endochondral ossification of chick sterna. Dev Dyn 203(4):468–476
Goldring MB, Tsuchimochi K, Ijiri K (2006) The control of chondrogenesis. J Cell Biochem 97(1):33–44
Golovchenko S, Hattori T, Hartmann C, Gebhardt M, Gebhard S, Hess A, Pausch F, Schlund B, von der Mark K (2013) Deletion of beta catenin in hypertrophic growth plate chondrocytes impairs trabecular bone formation. Bone 55(1):102–112
Hall BK (1987) Earliest evidence of cartilage and bone development in embryonic life. Clin Orthop Rel Res 225:255–272
Hall BK (2005) Bones and cartilage, 1st edn. Elsevier, San Diego
Ham AW (1952) Some histophysiological problems peculiar to calcified tissues. J Bone Joint Surg Am 24a:701–728
Hartmann C (2007) Skeletal development–Wnts are in control. Mol Cells 24(2):177–184
Hatori M, Klatte KJ, Teixeira CC, Shapiro IM (1995) End labeling studies of fragmented DNA in the avian growth plate: evidence of apoptosis in terminally differentiated chondrocytes. J Bone Miner Res 10(12):1960–1968
Hattori T, Muller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bosl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B, von der Mark K (2010) SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification. Development 137(6):901–911
Henry SP, Jang CW, Deng JM, Zhang Z, Behringer RR, de Crombrugghe B (2009) Generation of aggrecan-CreERT2 knockin mice for inducible Cre activity in adult cartilage. Genesis 47(12):805–814. doi:10.1002/dvg.20564
Hess J, Porte D, Munz C, Angel P (2001) AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J Biol Chem 276(23):20029–20038
Hilton MJ, Tu X, Long F (2007) Tamoxifen-inducible gene deletion reveals a distinct cell type associated with trabecular bone, and direct regulation of PTHrP expression and chondrocyte morphology by Ihh in growth region cartilage. Dev Biol 308(1):93–105. doi:10.1016/j.ydbio.2007.05.011
Holtrop ME (1972) The ultrastructure of the epiphyseal plate. II The hypertrophic chondrocyte. Calcif Tissue Res 9(2):140–151
Hunziker EB, Herrmann W, Schenk RK (1983) Ruthenium hexammine trichloride (RHT)-mediated interaction between plasmalemmal components and pericellular matrix proteoglycans is responsible for the preservation of chondrocytic plasma membranes in situ during cartilage fixation. J Histochem Cytochem 31(6):717–727
Hunziker EB, Herrmann W, Schenk RK, Mueller M, Moor H (1984) Cartilage ultrastructure after high pressure freezing, freeze substitution, and low temperature embedding. I Chondrocyte ultrastructure–implications for the theories of mineralization and vascular invasion. J Cell Biol 98(1):267–276
Inada M, Wang Y, Byrne MH, Rahman MU, Miyaura C, Lopez-Otin C, Krane SM (2004) Critical roles for collagenase-3 (Mmp13) in development of growth plate cartilage and in endochondral ossification. Proc Natl Acad Sci U S A 101(49):17192–17197
Jacobson MD, Weil M, Raff MC (1997) Programmed cell death in animal development. Cell 7(88):347–354
Jimenez MJ, Balbin M, Lopez JM, Alvarez J, Komori T, Lopez-Otin C (1999) Collagenase 3 is a target of Cbfa1, a transcription factor of the runt gene family involved in bone formation. Mol Cell Biol 19(6):4431–4442
Johansson N, Saarialho-Kere U, Airola K, Herva R, Nissinen L, Westermarck J, Vuorio E, Heino J, Kahari VM (1997) Collagenase-3 (MMP-13) is expressed by hypertrophic chondrocytes, periosteal cells, and osteoblasts during human fetal bone development. Dev Dyn 208(3):387–397
Kahn AJ, Simmons DJ (1977) Chondrocyte-to-osteocyte transformation in grafts of perichondrium-free epiphyseal cartilage. Clin Orthop Relat Res 129:299–304
Kalajzic Z, Liu P, Kalajzic I, Du Z, Braut A, Mina M, Canalis E, Rowe DW (2002) Directing the expression of a green fluorescent protein transgene in differentiated osteoblasts: comparison between rat type I collagen and rat osteocalcin promoters. Bone 31(6):654–660
Karsenty G, Ducy P, Starbuck M, Priemel M, Shen J, Geoffroy V, Amling M (1999) Cbfa1 as a regulator of osteoblast differentiation and function. Bone 25(1):107–108
Karsenty G, Kronenberg HM, Settembre C (2009) Genetic control of bone formation. Annu Rev Cell Dev Biol 25:629–648. doi:10.1146/annurev.cellbio.042308.113308
Kerr JF, Wyllie AH, Currie AR (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26(4):239–257
Kim IS, Otto F, Zabel B, Mundlos S (1999) Regulation of chondrocyte differentiation by Cbfa1. Mech Dev 80(2):159–170
Kimura S, Noda T, Yoshimori T (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3(5):452–460
Kirsch T, Swoboda B, von der Mark K (1992) Ascorbate-independent differentiation of human chondrocytes in vitro: simultaneous expression of types I and X collagen and matrix mineralization. Differentiation 51:89–100
Komori T (2006) Regulation of osteoblast differentiation by transcription factors. J Cell Biochem 99(5):1233–1239. doi:10.1002/jcb.20958
Komori T (2008) Regulation of bone development and maintenance by Runx2. Front Biosci 13:898–903
Komori T (2010) Regulation of bone development and extracellular matrix protein genes by RUNX2. Cell Tissue Res 339(1):189–195. doi:10.1007/s00441-009-0832-8
Komori T (2013) Functions of the osteocyte network in the regulation of bone mass. Cell Tissue Res 352(2):191–198. doi:10.1007/s00441-012-1546-x
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89(5):755–764
Korsmeyer SJ (1992) Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood 80(4):879–886
Kronenberg HM (2003) Developmental regulation of the growth plate. Nature 423(6937):332–336
Kronenberg HM (2006) PTHrP and skeletal development. Ann N Y Acad Sci 1068:1–13
Langenskiold A (1998) Role of the ossification groove of Ranvier in normal and pathologic bone growth: a review. J Pediatr Orthop 18(2):173–177
Lanske B, Kronenberg HM (1998) Parathyroid hormone-related peptide (PTHrP) and parathyroid hormone (PTH)/PTHrP receptor. Crit Rev Eukaryot Gene Expr 8(3–4):297–320
Lefebvre V, Behringer RR, de Crombrugghe B (2001) L-Sox5, Sox6 and Sox9 control essential steps of the chondrocyte differentiation pathway. Osteoarthritis Cartilage 9(Suppl A):S69–S75
Lefebvre V, de Crombrugghe B (1998) Toward understanding SOX9 function in chondrocyte differentiation. Matrix Biol 16(9):529–540
Lefebvre V, Huang W, Harley VR, Goodfellow PN, de Crombrugghe B (1997) SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol Cell Biol 17(4):2336–2346
Lefebvre V, Smits P (2005) Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today 75(3):200–212
Leung VY, Gao B, Leung KK, Melhado IG, Wynn SL, Au TY, Dung NW, Lau JY, Mak AC, Chan D, Cheah KS (2011) SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet 7(11):e1002356. doi:10.1371/journal.pgen.1002356
Li M, Tan J, Miao Y, Lei P, Zhang Q (2015) The dual role of autophagy under hypoxia-involvement of interaction between autophagy and apoptosis. Apoptosis 20(6):769–777. doi:10.1007/s10495-015-1110-8
Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW (2006) Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord 7(1–2):1–16. doi:10.1007/s11154-006-9001-5
Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M (2008) Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol 40(1):46–62
Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, Carmeliet G, Kronenberg HM (2010) Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell 19(2):329–344
Mansfield K, Pucci B, Adams CS, Shapiro IM (2003) Induction of apoptosis in skeletal tissues: phosphate-mediated chick chondrocyte apoptosis is calcium dependent. Calcif Tissue Int 73(2):161–172. doi:10.1007/s00223-002-1056-z
Mansfield K, Teixeira CC, Adams CS, Shapiro IM (2001) Phosphate ions mediate chondrocyte apoptosis through a plasma membrane transporter mechanism. Bone 28(1):1–8
Maye P, Fu Y, Butler DL, Chokalingam K, Liu Y, Floret J, Stover ML, Wenstrup R, Jiang X, Gooch C, Rowe D (2011) Generation and characterization of Col10a1-mcherry reporter mice. Genesis 49(5):410–418. doi:10.1002/dvg.20733
Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L (2007) A global double-fluorescent Cre reporter mouse. Genesis 45(9):593–605. doi:10.1002/dvg.20335
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B (2002) The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108(1):17–29
Ng LJ, Wheatley S, Muscat GE, Conway-Campbell J, Bowles J, Wright E, Bell DM, Tam PP, Cheah KS, Koopman P (1997) SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol 183(1):108–121
Nishimura R, Hata K, Ono K, Amano K, Takigawa Y, Wakabayashi M, Takashima R, Yoneda T (2012) Regulation of endochondral ossification by transcription factors. Front Biosci 17:2657–2666
Ohyama K, Farquharson C, Whitehead CC, Shapiro IM (1997) Further observations on programmed cell death in the epiphyseal growth plate: comparison of normal and dyschondroplastic epiphyses. J Bone Miner Res 12(10):1647–1656. doi:10.1359/jbmr.1997.12.10.1647
Olsen BR, Reginato AM, Wang W (2000) Bone development. Annu Rev Cell Dev Biol 16:191–220
Ono N, Ono W, Nagasawa T, Kronenberg HM (2014) A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat Cell Biol 16(12):1157–1167
Ortega N, Behonick DJ, Werb Z (2004) Matrix remodeling during endochondral ossification. Trends Cell Biol 14(2):86–93. doi:10.1016/j.tcb.2003.12.003
Oshima Y, Akiyama T, Hikita A, Iwasawa M, Nagase Y, Nakamura M, Wakeyama H, Kawamura N, Ikeda T, Chung UI, Hennighausen L, Kawaguchi H, Nakamura K, Tanaka S (2008) Pivotal role of Bcl-2 family proteins in the regulation of chondrocyte apoptosis. J Biol Chem 283(39):26499–26508. doi:10.1074/jbc.M800933200
Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ (1997) Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89(5):765–771
Park J, Gebhardt M, Golovchenko S, Perez-Branguli F, Hattori T, Hartmann C, Zhou X, deCrombrugghe B, Stock M, Schneider H, von der Mark K (2015) Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open 4(5):608–621. doi:10.1242/bio.201411031
Poillet-Perez L, Despouy G, Delage-Mourroux R, Boyer-Guittaut M (2015) Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol 4:184–192. doi:10.1016/j.redox.2014.12.003
Poole A (1991) The growth plate: cellular physiology, cartilage assembly and mineralization. In: Cartilage: “molecular aspects”. CRC Press, Boca Raton
Porte D, Tuckermann J, Becker M, Baumann B, Teurich S, Higgins T, Owen MJ, Schorpp-Kistner M, Angel P (1999) Both AP-1 and Cbfa1-like factors are required for the induction of interstitial collagenase by parathyroid hormone. Oncogene 18(3):667–678
Provot S, Schipani E (2005) Molecular mechanisms of endochondral bone development. Biochem Biophys Res Commun 328(3):658–665
Reddi AH (1981) Cell biology and biochemistry of endochondral bone development. Collagen Rel Res 1:209–226
Roach HI (1992) Trans-differentiation of hypertrophic chondrocytes into cells capable of producing a mineralized bone matrix. Bone Miner 19(1):1–20
Roach HI (1997) New aspects of endochondral ossification in the chick: chondrocyte apoptosis, bone formation by former chondrocytes, and acid phosphatase activity in the endochondral bone matrix. J Bone Miner Res 12(5):795–805
Roach HI, Aigner T, Kouri JB (2004) Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis 9(3):265–277
Roach HI, Clarke NM (1999) “Cell paralysis” as an intermediate stage in the programmed cell death of epiphyseal chondrocytes during development. J Bone Miner Res 14(8):1367–1378
Roach HI, Clarke NM (2000) Physiological cell death of chondrocytes in vivo is not confined to apoptosis. New observations on the mammalian growth plate. J Bone Joint Surg Br 82(4):601–613
Roach HI, Erenpreisa J (1996) The phenotypic switch from chondrocytes to bone-forming cells involves asymmetric cell division and apoptosis. Connect Tissue Res 35(1–4):85–91
Roach HI, Erenpreisa J, Aigner T (1995) Osteogenic differentiation of hypertrophic chondrocytes involves asymmetric cell divisions and apoptosis. J Cell Biol 131(2):483–494
Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS (2001) Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev 15(21):2865–2876
Schmid TM, Conrad HE (1982) A unique low-molecular weight collagen secreted by cultured chick-embryo chondrocytes. J Biol Chem 257:2444–2450
Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB, Korsmeyer SJ (1995) Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc Natl Acad Sci U S A 92(17):7834–7838
Shapiro IM, Adams CS, Freeman T, Srinivas V (2005) Fate of the hypertrophic chondrocyte: microenvironmental perspectives on apoptosis and survival in the epiphyseal growth plate. Birth Defects Res C Embryo Today 75(4):330–339
Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21(1):70–71
Srinivas V, Bohensky J, Shapiro IM (2009a) Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs 189(1–4):88–92
Srinivas V, Bohensky J, Zahm AM, Shapiro IM (2009b) Autophagy in mineralizing tissues: microenvironmental perspectives. Cell Cycle 8(3):391–393
Staines KA, Pollard AS, McGonnell IM, Farquharson C, Pitsillides AA (2013) Cartilage to bone transitions in health and disease. J Endocrinol 219(1):R1–R12. doi:10.1530/JOE-13-0276
Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z (2004) Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development 131(23):5883–5895
Stricker S, Fundele R, Vortkamp A, Mundlos S (2002) Role of Runx genes in chondrocyte differentiation. Dev Biol 245(1):95–108
Takeda S, Bonnamy JP, Owen MJ, Ducy P, Karsenty G (2001) Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev 15(4):467–481
Teixeira CC, Mansfield K, Hertkorn C, Ischiropoulos H, Shapiro IM (2001) Phosphate-induced chondrocyte apoptosis is linked to nitric oxide generation. Am J Physiol Cell Physiol 281(3):C833–C839
Teixeira CC, Rajpurohit R, Mansfield K, Nemelivsky YV, Shapiro IM (2003) Maturation-dependent thiol loss increases chondrocyte susceptibility to apoptosis. J Bone Miner Res 18(4):662–668. doi:10.1359/jbmr.2003.18.4.662
Thesingh CW, Groot CG, Wassenaar AM (1991) Transdifferentiation of hypertrophic chondrocytes into osteoblasts in murine fetal metatarsal bones, induced by co-cultured cerebrum. Bone Miner 12(1):25–40
Thorburn A (2008) Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 13(1):1–9
Toledo-Pereyra LH, Toledo AH, Walsh J, Lopez-Neblina F (2004) Molecular signaling pathways in ischemia/reperfusion. Exp Clin Transpl Off J Middle East Soc Organ Transpl 2(1):174–177
Tsang KY, Tsang SW, Chan D, Cheah KS (2014) The chondrocytic journey in endochondral bone growth and skeletal dysplasia. Birth Defects Res (Part C) 192:52–73
Vaux DL, Cory S, Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335(6189):440–442. doi:10.1038/335440a0
Vortkamp A (2000) The Indian hedgehog–PTHrP system in bone development. Ernst Schering Res Found Workshop 29:191–209
Wang K (2015) Autophagy and apoptosis in liver injury. Cell Cycle 14(11):1631–1642. doi:10.1080/15384101.2015.1038685
Wang X, Manner PA, Horner A, Shum L, Tuan RS, Nuckolls GH (2004) Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage 12(12):963–973
Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z (2011) LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 20(4):444–454
Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A, Wu Q, Zhang J, Hong Y (2014) Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci 10(9):1072–1083. doi:10.7150/ijbs.9719
Wuthier RF (1988) Mechanism of matrix-vesicle mediated mineralization of cartilage. ISI Atlas Sci Biochem 1:231–241
Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, Teng Y, Yang X (2014a) Osteogenic fate of hypertrophic chondrocytes. Cell Res 2014(22):111
Yang L, Tsang KY, Tang HC, Chan D, Cheah KS (2014b) Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A 111(33):12097–12102
Yang Z, Klionsky DJ (2009) An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol 335:1–32
Yang Z, Klionsky DJ (2010) Eaten alive: a history of macroautophagy. Nat Cell Biol 12(9):814–822
Yin XM, Oltvai ZN, Korsmeyer SJ (1994) BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature 369(6478):321–323. doi:10.1038/369321a0
Zelzer E, Glotzer DJ, Hartmann C, Thomas D, Fukai N, Soker S, Olsen BR (2001) Tissue specific regulation of VEGF expression during bone development requires Cbfa1/Runx2. Mech Dev 106(1–2):97–106
Zelzer E, Mamluk R, Ferrara N, Johnson RS, Schipani E, Olsen BR (2004) VEGFA is necessary for chondrocyte survival during bone development. Development 131(9):2161–2171
Zenmyo M, Komiya S, Kawabata R, Sasaguri Y, Inoue A, Morimatsu M (1996) Morphological and biochemical evidence for apoptosis in the terminal hypertrophic chondrocytes of the growth plate. J Pathol 180(4):430–433
Zhang M, Zhang J, Lu L, Qiu ZY, Zhang X, Yu SB, Wu YP, Wang MQ (2013) Enhancement of chondrocyte autophagy is an early response in the degenerative cartilage of the temporomandibular joint to biomechanical dental stimulation. Apoptosis 18(4):423–434
Zhao Q, Eberspaecher H, Lefebvre V, de Crombrugghe B (1997) Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev Dyn 209(4):377–386
Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B (2014) Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS Genet 10(12):e1004820. doi:10.1371/journal.pgen.1004820
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
von der Mark, K., Zhou, X. (2016). Cell Fate of Growth Plate Chondrocytes in Endochondral Ossification: Cell Death or Reprogramming into Osteogenic Lineage?. In: Grässel, S., Aszódi, A. (eds) Cartilage. Springer, Cham. https://doi.org/10.1007/978-3-319-29568-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-29568-8_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-29566-4
Online ISBN: 978-3-319-29568-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)