
Abstract
Purpose of Review
The goal of the review is to summarize the current knowledge on the process of chondrocyte-to-osteoblast transdifferentiation during endochondral bone formation and its potential implications in fracture healing and disease.
Recent Findings
Lineage tracing experiments confirmed the transdifferentiation of chondrocytes into osteoblasts. More recent studies lead to the discovery of molecules involved in this process, as well as to the hypothesis that these cells may re-enter a stem cell-like phase prior to their osteoblastic differentiation.
Summary
This review recapitulates the current knowledge regarding chondrocyte transdifferentiating into osteoblasts, the developmental and postnatal events where transdifferentiation appears to be relevant, and the molecules implicated in this process.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chondrocytes build up the cartilaginous parts of the skeleton. The embryonic skeleton is to a great extent cartilaginous and needs to be remodeled into bone in a process referred to as endochondral bone formation. Based on fate, two types of hyaline cartilage can be distinguished: temporary and permanent cartilage. The articular cartilage in the joints is considered permanent cartilage, as it normally in a healthy joint does not ossify. In contrast, temporary cartilage is gradually replaced by bone, a process that ends in humans at puberty and is referred to as endochondral bone formation. During the process of endochondral ossification, chondrocytes eventually go through hypertrophy. This differentiation step is accompanied by an exit from cell cycle and 10–15-fold volume increase [1,2,3]. In addition, hypertrophic chondrocytes produce a unique extracellular matrix consisting mostly of type 10 collagen fibers [4, 5]. As the hypertrophic chondrocytes mature, their matrix mineralizes. The matrix originally mineralizes in a circumferential pattern as long as the hypertrophic chondrocytes are not organized within the growth plate. Upon the occurrence of the latter, the mineralization pattern changes and only the intercolumnar matrix of the longitudinal septa is mineralized. For the remodeling of the cartilage template into bone, hypertrophic chondrocytes have to be removed. Yet, the final fate of hypertrophic chondrocytes, whether they undergo programmed cell death or survive and differentiate into osteoblasts, has long been under debate. Only recently, lineage-tracing data provided in vivo evidence that chondrocytes derived from the hypertrophic zone continue to live and differentiate into osteoblasts and osteocytes. This process is referred to as chondrocyte-to-osteoblast transdifferentiation, whereby probably mature hypertrophic chondrocytes differentiate into osteoblasts. Transdifferentiation has been proposed to occur in two ways: either directly without an intermediate step of pluripotency or progenitor-like stage in the secondary ossification center (SOC) or indirectly involving intermediate steps of dedifferentiation and re-differentiation [6]. This review will summarize the current knowledge on the process of chondrocyte-derived osteoblast differentiation, the molecules implicated in this process, and the developmental and postnatal events where transdifferentiation may be relevant.
Endochondral Bone Formation in Long Bones
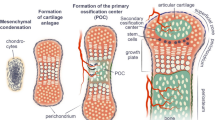
The majority of the skeletal elements are generated during embryonic development by the process of endochondral bone formation. This involves the formation of a cartilage template, which is then remodeled into bone during the late stages of embryonic development and juvenile life. The cellular components of the skeletal elements are chondrocytes producing the hyaline cartilage matrix rich in type 2 collagen and osteoblasts producing as a bone matrix osteoid, which subsequently becomes mineralized. Both cell types are of mesenchymal origin and share a common precursor the osteochondro-progenitor. In endochondral bone formation, chondrocytes differentiate within mesenchymal cellular condensations molding the cartilaginous template of the future skeletal element. These templates then expand by appositional and interstitial growth involving the recruitment of precursors from the surrounding mesenchymal perichondrial cells or the division of chondrocytes within the template, respectively. Once the template reaches a critical size, chondrocytes in the middle begin to differentiate into prehypertrophic chondrocytes, which produce the secreted factor Indian hedgehog (IHH). Amongst others, IHH induces mesenchymal precursor cells in the perichondrium to differentiate into osteoblast precursors [7, 8]. Prehypertrophic chondrocytes then mature further into hypertrophic chondrocytes. The latter produce amongst others vascular endothelial growth factor (VEGF) to attract blood vessels to the central region of the avascular cartilage template [9]. The invasion of blood vessels is necessary for the formation of the bone marrow cavity/primary ossification center (POC). Along with the formation of the bone marrow cavity, the cartilage-to-bone transition zone, also known as the chondro-osseous border, forms. Prior to bone marrow cavity formation, the centrally located hypertrophic chondrocytes produce a mineralized extracellular matrix. During bone marrow cavity formation, hypertrophic chondrocytes in the mineralized zone are removed, while their mineralized matrix stays behind and serves as the primary scaffold for osteoblasts to lay down bone matrix and to build the primary spongiosa. The textbook view has long been that osteoblasts originate from the perichondrium/periosteum; from there, they migrate as precursors together with the blood vessels into the forming bone marrow cavity [10]. The perichondrial/periosteal located precursors are still not fully committed to the osteoblast lineage and differentiate into chondrocytes if certain factors, such as OSTERIX or β-catenin are missing [11,12,13,14]. In addition to the primary ossification center, a SOC forms in the epiphyses of long bones. Dependent on the species this occurs either peri- (e.g., human) or postnatally (e.g., mouse). In all species, cartilage canals are formed prior to SOC formation, with substantial differences regarding the time intervals between the two events [15]. Cartilage canals start off as invaginations from the perichondrium with macrophages at their apical tips resorbing the cartilage matrix. The canals bear vessels and mesenchymal cells. They finally reach the concentric zone of hypertrophic chondrocytes within the epiphysis, which produces VEGF. In mice, several small ossification nuclei appear in the center of the epiphysis and rapidly coalesce into a large SOC [16].
A bone marrow cavity forms in the POC and the SOC. Yet, the process of bone marrow cavity formation is still a kind of a mystery. One prerequisite for it to occur is that hypertrophic chondrocytes need to be removed/disappear. With the re-discovery of programmed cell death in the 1990s, it was generally assumed that chondrocyte removal involves apoptosis [17, 18]. Other studies suggest that besides classical apoptosis other mechanisms of physiological cell death must be at play, as apoptotic chondrocytes are only sparsely detected in vivo [19,20,21,22]. However, the idea that the hypertrophic chondrocytes eventually all undergo cell death has been opposed by careful histological analyses revealing that some of the hypertrophic chondrocytes survive after blood vessels penetrated their lacunae [23,24,25]. In line with this, numerous in vitro observations ranging from histology over explant/cell cultures suggested that hypertrophic chondrocytes have the potential to differentiate into osteoblasts (reviewed in [26,27,28]). In addition, an alternative model, the concept of the “borderline chondrocyte,” has been proposed, whereby the early hypertrophic chondrocytes located in the mid-diaphysis directly bordering the perichondrial osteogenic cells are being exposed to a special microenvironment that fosters their differentiation into osteoblasts, while hypertrophic chondrocytes located further away would undergo apoptosis [29, 30]. Together, this led to the controversial discussion about the final fate of chondrocytes; do they die or have a second career as osteoblasts?
The “Second Career” of Hypertrophic Chondrocytes
A chondrogenic origin of osteoblast has previously been observed in various culture and graft experiments [29, 31,32,33]. Molecularly, conditional inactivation of the Sox9 gene in chondrocytes resulted in the observation of an aberrant differentiation of osteoblasts potentially originating from chondrocytes, which appear to skip full hypertrophic differentiation, suggesting that Sox9 is a negative regulator of the chondrocyte-to-osteoblast differentiation process [34]. Yet, as no lineage-tracer was used in that study, the chondrogenic origin of the osteoblastic cells observed was based on the transient detection of overlapping expressions of Col2a1, a marker for proliferating hyaline chondrocytes, with osteoblastic markers. More recently, cell-lineage tracing experiments by different labs provided further evidence that chondrocytes, in particular, chondrogenic cells originating from the Col10a1-expressing hypertrophic zone of the growth plate, differentiate into osteoblasts [35, 36••, 37••, 38••]. Chondrocyte-derived osteoblasts are found in the trabeculae of the POC and at the endosteal layer. Like perichondrial-derived osteoblasts, they differentiate further into osteocytes. However, they are never found in the periosteal layer [35, 36••, 38••]. Interestingly, periosteal bone formation resembles, to a certain extent, intramembranous ossification, and in the latter, chondrocyte-derived bone cells have not been observed in either of the lineage studies [37••]. Yet, according to all lineage-tracing studies, not all of the chondrocyte-derived cells observed in the bone marrow cavity differentiate into osteoblasts. Only 70–80% of all hypertrophic chondrocyte-derived cells co-expressed bone-markers such as Col1a1 or OSTERIX [35, 39••]. Some of the chondrocyte-derived cells may contribute to other lineages as well, such as the adipogenic and pericyte lineages as suggested by Yang and colleagues [36••].
According to the lineage studies, the amount of chondrocyte-derived osteoblast precursors (CDOPs) is highest at early developmental stages reaching approximately 40% of the total OSTERIX-positive precursors at embryonic day 16.5 and decreases as development progresses reaching a steady state of about 20% from postnatal day 1 (P1) through P7 [38••]. Given that the lifespan of a murine osteoblast is approximately 12 days, the postnatal rate of newly derived CDOPs is estimated to be around 10% [38••, 40]. In a previous study, the fluorescent dye mCherry, a monomeric form of the tetrameric fluorophore DSRed, was expressed directly under the control of the Col10a1 regulatory elements [41]. Here, the fluorescent reporter was detected in hypertrophic chondrocytes and in cells inside the trabeculae. Yet, based on co-localization studies using a second transgenic osteoblast-specific line, the authors came to the conclusion that the trabecular mCherry signal is due to the persistence of hypertrophic chondrocytes within the trabeculae and not caused by transdifferentiation into osteoblasts [41]. Contrary to the lineage-tracing studies mentioned above, in which the progeny of Col10a1 expressing cells is labeled using either a constitutive or tamoxifen-inducible form of Cre-recombinase under the control of the Col10a1 regulatory elements, in the study by Maye and colleagues, primarily the cells that actively express Col10a1 are labeled and only if the label is stable enough, a signal can be detected in the progeny as well [35, 36••, 37••, 41].
In the long bones, chondrocyte-to-osteoblast transdifferentiation also occurs postnatally in the epiphysis during SOC formation [42••]. Aghajanian and colleagues report that osteoblast differentiation in the SOC proceeds vascular invasion, which occurs around postnatal day (P) 9 [42••]. Similarly, mesenchymal stem cells arrived in the SOC only later together with the invading blood vessels. Given the relatively fast temporal occurrence of osteoblastic differentiation, Agahajanian and colleagues hypothesize that the hypertrophic chondrocytes, in this context, should rather be considered as preosteoblasts. Yet alternatively, these hypertrophic SOC cells may have some kind of stem cell character as they express high levels of the BMP antagonist, Gremlin 1, which was recently proposed to mark skeletal stem cells in the bone marrow [43, 44]. In conclusion, Aghajanian and colleagues proposed based on their work that epiphyseal chondrocytes are the major source of SOC-osteoblasts and raise the question to what extent mesenchymal stem cells contribute to the osteoblast population [42••]. At least at P15, we found that about 41% percent of the OSX-positive cells were negative for the Col10a1-Cre lineage marker, YFP, and hence should be of mesenchymal stem cell origin, while on average 59% percent of the OSX-positive cells co-expressed YFP (Wolff and Hartmann, unpublished observation). As such, the percentage of CDOPs in the SOC at P15 corresponds to the percentage of CDOPs in the POC at E16.5. As such, our unpublished data points towards a potentially higher contribution of chondrocyte-derived osteoblasts in the SOC. Nevertheless, based on our unpublished data, there still is a considerable contribution from a non-chondrogenic preosteoblast population in the SOC.
Chondrocyte-to-osteoblast transdifferentiation is also observed in other skeletal elements such as the mandibular condyle [28, 45••]. The mandibular condylar cartilage is considered “secondary” cartilage and differs in its developmental origin from that of primary cartilage as found, for example, in the limbs [46]. Structurally and histologically, the process of bone formation in secondary cartilage is reminiscent to that in epiphyseal cartilage [46]. The percentage of chondrocyte-derived bone cells in the mandibular condyle is relatively high reaching 80% in the immediately subchondral (superior) zone. Yet, similar to the situation in the long bones, not all of these chondrocyte-derived bone cells correspond to osteoblasts, as only half of them co-express type I collagen. A more detailed examination using earlier osteoblastic markers such as RUNX2 and OSTERIX may allow for an even more accurate estimation of the percentage of chondrocyte-derived osteoblasts in the mandibular condyle.
A unique form of chondrocyte-to-osteoblast transformation has been observed postnatally in the calvarial cartilage anlagen of MT1-MMP knockout animals. Here, MT1-MMP deficiency results in the persistence of distinct, devitalized, ghost cartilages. Most of the chondrocytes retained therein undergo delayed apoptosis, yet, some chondrocytes escape this fate. These chondrocytes initiate cell division and switch to an osteogenic program expressing type I collagen and osteocalcin [47]. Interestingly, distinct from the POC, SOC, and mandibular condyle, these chondrocytes never expressed type X collagen, the marker for hypertrophic chondrocytes [47].
At last, chondrocyte-to-osteoblast transformation also occurs during fracture repair as reported by several groups [28, 35, 37••, 48,49,50,51, 52••].
Mechanisms Underlying Chondrocyte-to-Osteoblast Transdifferentiation
The molecular mechanisms underlying the process of chondrocyte-to-osteoblast differentiation are not well understood yet. As mentioned already, hypertrophic chondrocytes increase their volume by about 10-fold. In contrast, osteoblast precursors are much smaller in size. Hence, either some cells expressing the hypertrophic marker Col10a1 within the hypertrophic zone remain small in size or the hypertrophic cells would need to shrink again during the process of transdifferentiation into osteoblasts. As for the latter, one potential mechanism could be asymmetric cell division. This has been reported previously to occur at the cartilage to bone marrow edge in cultured cut femoral pieces from the chicken leg [53, 54]. Results from the in vitro experiments in chicken further suggest that within the long bone growth plate, only hypertrophic chondrocytes are capable to transdifferentiate [55]. One early event during transdifferentiation appears to be cell division. Hypertrophic chondrocytes are normally characterized as cells that no longer undergo mitosis, yet, in culture, some cells, located directly at the chondro-osseous border, have been observed to re-enter the cell cycle and divide [55,56,57]. Interestingly, the electron microscopic study by Yoshioka and Yagi demonstrated in the rat mandibular condylar cartilage that hypertrophic chondrocytes at the chondro-osseous border have been released from their lacunae into the area of the primary spongiosa [58]. BrdU-labeling of newborn mouse pups 12 h prior to delivery revealed that within the growth plate Col10a1-derived YFP+ cells located directly at the chondro-osseous border are mitotically active [38••]. Furthermore, the Col10a1-derived YFP+ cells co-stained in culture with the stem cell marker SCA1 and this cell population expressed additional stem cell markers, such as CD34, c-Myc, and Sox2 [38••]. The latter suggests that one potential mechanism for transdifferentiation may be that CDOPs revert to a more stem cell-like state prior to differentiating into osteoblasts. Similarly, chondrocytes contributing to bone formation during fracture healing re-enter the cell cycle and react positively for the pluripotency markers OCT4A, NANOG, and SOX2 [49, 52, 59]. Hu and colleagues demonstrated a functional role for Sox2 in fracture repair. In Sox2-deficient animals, 14 days after fracture, the callus is smaller and a compositional shift towards a lower percentage of bone and a higher percentage of cartilage occurs [52••]. Regarding the functional analysis of the Sox2-deficient animals, it would have been interesting to see additional time points during fracture healing being examined. Hu and colleagues suggested further that the vasculature plays an important role in triggering transdifferentiation [52••]. Yet, another study in rabbit provided evidence that the vasculature is not required for chondrocyte-to-osteoblast transdifferentiation or may even negatively influence this process under physiological conditions [60•]. Here, vascular invasion of the mineralized hypertrophic chondrocytes was constrained by the insertion of a membranous permeable filter into the hypertrophic zone. Chondrocytes above the membrane survived, re-entered the cell cycle, produced an osteoblast-like extracellular matrix and eventually stopped to transcribe the Col10a1 gene, yet they maintain a chondrocyte-like appearance [60•]. Nevertheless, under this condition, molecules produced by the vascular endothelial cells can still permeate the membrane and thus may still be involved in the transdifferentiation of chondrocytes into osteoblast precursors. As shown recently, there are different types of blood vessels in bone. The H-type vessels and the L-type vessels in the embryo that are both Endomucinhigh and PCAM/CD31high have been shown to support osteoblastogenesis producing pro-osteogenic factors such as PDGFα/β, TGFβ1/3, and FGF1 [61, 62]. Hence, it would be interesting to see whether the insertion of an impermeable membrane would still allow transdifferentiation to occur. This kind of experiment has been performed in the 1950s by Harris and Dayle, who inserted a Teflon membrane into the growing rabbit growth plate [63]. Yet, no detailed phenotypic or histological analysis of this experiment was published. Only one data point was reported in the review by Salter and Harris showing that the chondrocytes above the membrane continued to grow and that the region above the membrane underwent endochondral ossification 3 weeks after the manipulation [63]. Yet, it is not known whether the hypertrophic chondrocytes above the membrane differentiated “abnormally” or if they transdifferentiated under these conditions or whether blood vessels invaded the new region undergoing endochondral ossification.
Over the past years, a few factors have been identified that are involved in chondrocyte-to-osteoblast transdifferentiation: one of them being β-catenin, a cytoplasmic molecule that functions on the one hand as a transcriptional co-factor in the canonical Wnt/β-catenin pathway and on the other hand it is an integral component of the cadherin cell adhesion complex linking cadherins to the cytoskeleton [64]. Chondrocyte-to-osteoblast transdifferentiation in the long bones is almost completely blocked in mice lacking β-catenin activity in hypertrophic chondrocytes [39••]. The observation that a few cells still transdifferentiate may be due to the fact that a conditional system was used; hence, in some cells, Cre-mediated recombination may not have occurred. The results from gain-of-function experiments, in which β-catenin is stabilized in a subset of hypertrophic chondrocytes, suggest that β-catenin drives transdifferentiation in a non-cell-autonomous fashion as the transdifferentiated OSX+;YFP+ cells in the bone marrow near the chondro-osseous front did not contain high levels of β-catenin protein [39••]. This factor X, which is controlled by β-catenin and which promotes chondrocyte-to-osteoblast differentiation, remains to be uncovered (Fig. 1). Similar observations regarding the role of β-catenin in transdifferentiation were also reported in the mandibular condyle cartilage [65•]. In periosteal osteoblast precursor cells, β-catenin activity is required to repress the pro-chondrogenic transcription factor Sox9 [13]. Yet, this appears not to be the mechanism by which β-catenin in hypertrophic chondrocytes influences chondrocyte-to-osteoblast transdifferentiation, as Sox9 levels or persistence were not altered in hypertrophic chondrocytes lacking β-catenin [39••]. Another molecule implicated in transdifferentiation is the tyrosine-protein phosphatase non-receptor type 11 (PTPN11) protein also known as SH2 domain-containing protein tyrosine phosphatase 2 (SHP2). This cytoplasmic phosphatase is ubiquitously expressed. Conditional mutants lacking SHP2 activity specifically in Col10a1-Cre expressing cells are osteopenic and display a reduction of transdifferentiating chondrocytes by about 1.3-fold accompanied by the reduction of osteogenic markers [66••]. Mechanistic studies revealed that SOX9 was more abundant, while β-catenin abundance was reduced in the hypertrophic zone of conditional Shp2 mutants (Fig. 1) [66••]. Furthermore, Wang and colleagues showed genetically that removal of one Sox9 allele restored the expression of osteogenic markers and the β-catenin levels [66••]. It has been shown previously in chondrocytes that an increase in SOX9 protein promotes phosphorylation of β-catenin in the nucleus and its subsequent degradation [67]. Whether the reverse is also true is still under debate [67, 68]. Currently, it is unclear if or to what extent the downregulation of β-catenin contributes to the reduced chondrocyte-to-osteoblast transdifferentiation in the conditional Shp2 mutants [66••]. In a follow-up study, the Yang group showed that SHP2 negatively regulated the phosphorylation and sumoylation of SOX9 affecting its stability and transcriptional activity [69].

Sites of chondrocyte-to-osteoblast transdifferentiation during endochondral ossification in the mouse long bone and signaling pathways and molecules implicated in the regulation of this process at the POC and SOC. For further details, see text. CDOP chondrocyte-derived osteoblast precursor, cOB chondrocyte-derived osteoblast, HTC hypertrophic chondrocytes, pCC proliferating chondrocytes, PDOP perichondrium-derived osteoblast precursor, pOB perichondrium-derived osteoblast
Another factor implicated in the transition from chondrocytes to osteoblasts is BMPR1a, a receptor for bone morphogenetic proteins [70•]. Postnatal inactivation of BMPR1a using the inducible Acan-Cre line results in the nearly complete absence of hypertrophic chondrocytes in the growth plate, absence of osteoblasts and osteocytes in the primary spongiosa, and absence of the SOC. Yet, immature chondrocyte-derived osteoblasts were still present in the SOC [70•]. The authors propose a biphasic model of a continuous skeletal program, with chondrogenesis being phase one and osteogenesis phase two which strictly depends on BMPR1a in Aggrecan-positive cells [70•].
In the SOC, thyroid hormone (TH) has been implicated in SOC formation and subsequently in the regulation of chondrocyte-to-osteoblast transdifferentiation [42••, 71]. Pharmaceutical inhibition of triiodothyronine (T3) and thyroxine (T4) production interferes with SOC formation and transdifferentiation at P14 [42••]. Similarly, SOC formation was not apparent in P7-P10 hypothyroid Tshrhyt/hyt mice. Yet, in these mice, chondrocyte maturation was also abnormal and no vascular invasion occurred complicating the interpretation. Agajanian and colleagues proposed based on previous knowledge and in vitro experiments a model whereby TH through TRβ1, expressed in prehypertrophic chondrocytes, regulates the Ihh and Shh expression in a positive and negative manner, respectively (Fig. 1). TRα1-dependent signaling, on the other hand, promotes the expression of Shh [72]. In line with previous reports that IHH positively regulates chondrocyte hypertrophy independent of parathyroid hormone and that forced SHH expression in chondrocytes maintains them in an immature state, Agajanian and colleagues proposed a mechanistic model for TH activity in SOC formation (Fig. 1) [42••, 73, 74]. Here, TH through IHH promotes chondrocyte maturation, opposed by SHH activity maintaining their immature state. Yet, the situation may be more complicated, as ablation of Ihh also affects chondrocyte proliferation. In the embryo, germline as well as conditional chondrocyte-specific ablation of Ihh initially reduces chondrocyte proliferation and delays chondrocyte maturation [7, 75]. Similarly, knockdown of Ihh in cultured chondrocytes affects their proliferation [76]. Postnatal conditional deletion of Ihh in Col2a1-expressing cells also results in a decreased chondrocyte proliferation associated with accelerated chondrocyte maturation and consequently a premature closure of the growth plate. In addition, abnormalities regarding SOC formation are observed in these mice, with an abnormal or premature blood vessel invasion into the epiphysis of P7 specimens and subsequent loss of the SOC due to the premature growth plate closure [77].
Conclusions and Outlook
The concept that mature chondrocytes can take on a new fate and differentiate into osteoblasts contributing to trabecular and endosteal bone formation is now accepted in the field. This process has also been referred to as the chondro-osseous cellular continuum [70•, 78]. Regarding SOC formation, the Mohan group even proposed to rename hypertrophic chondrocytes as preosteoblasts [42••]. Currently, it is not known whether the osteoblasts from the different sources are absolutely identical. There are already some hints in the literature that there may be differences between them [79, 80]. Furthermore, although recently, some molecules have been identified that are involved in this process, we are far from understanding how this exactly occurs at the mechanistic and cellular level. The process of chondrocyte-to-osteoblast transformation may be primarily relevant during embryonic and early postnatal endochondral ossification since it does not involve extensive cell migration, as it is the case for perichondrium-derived osteoblasts. This is reflected by the observations that the ratio of chondrocyte- to perichondrium-derived osteoblasts declines during the later stages of embryonic endochondral bone formation. This process is also relevant in postnatal events, such as in fracture repair, a process recapitulating to a certain extent embryonic endochondral bone formation. Whether the process of chondrocyte-to-osteoblast transdifferentiation also plays a role in pathologic events such as osteoarthritis or hereditary exostosis remains to be seen in the future.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Farnum CE, Lee R, O’Hara K, Urban JP. Volume increase in growth plate chondrocytes during hypertrophy: the contribution of organic osmolytes. Bone. 2002;30(4):574–81.
Miller JP, Yeh N, Vidal A, Koff A. Interweaving the cell cycle machinery with cell differentiation. Cell Cycle. 2007;6(23):2932–8. https://doi.org/10.4161/cc.6.23.5042.
Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW, Tabin CJ. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 2013;495(7441):375–8. https://doi.org/10.1038/nature11940.
Schmid TM, Conrad HE. A unique low molecular weight collagen secreted by cultured chick embryo chondrocytes. J Biol Chem. 1982;257(20):12444–50.
Grant WT, Sussman MD, Balian G. A disulfide-bonded short chain collagen synthesized by degenerative and calcifying zones of bovine growth plate cartilage. J Biol Chem. 1985;260(6):3798–803.
Aghajanian P, Mohan S. The art of building bone: emerging role of chondrocyte-to-osteoblast transdifferentiation in endochondral ossification. Bone Res. 2018;6:19. https://doi.org/10.1038/s41413-018-0021-z.
St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13(16):2072–86.
Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–6. https://doi.org/10.1038/nature01657.
Gerber HP, Vu TH, Ryan AM, Kowalski J, Werb Z, Ferrara N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat Med. 1999;5(6):623–8. https://doi.org/10.1038/9467.
Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19(2):329–44. https://doi.org/10.1016/j.devcel.2010.07.010.
Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29.
Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5):739–50. https://doi.org/10.1016/j.devcel.2005.03.016.
Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38. https://doi.org/10.1016/j.devcel.2005.02.013.
Rodda SJ, McMahon AP. Distinct roles for hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. https://doi.org/10.1242/dev.02480.
Blumer MJ, Longato S, Fritsch H. Structure, formation and role of cartilage canals in the developing bone. Ann Anat. 2008;190(4):305–15. https://doi.org/10.1016/j.aanat.2008.02.004.
Blumer MJ, Longato S, Schwarzer C, Fritsch H. Bone development in the femoral epiphysis of mice: the role of cartilage canals and the fate of resting chondrocytes. Dev Dyn. 2007;236(8):2077–88. https://doi.org/10.1002/dvdy.21228.
Hung RW, Chow AW. Apoptosis: molecular mechanisms, regulation and role in pathogenesis. Can J Infect Dis. 1997;8(2):103–9.
Gibson G. Active role of chondrocyte apoptosis in endochondral ossification. Microsc Res Tech. 1998;43(2):191–204. https://doi.org/10.1002/(SICI)1097-0029(19981015)43:2<191::AID-JEMT10>3.0.CO;2-T.
Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7(3):253–66. https://doi.org/10.1002/tera.1420070306.
Roach HI, Clarke NM. Physiological cell death of chondrocytes in vivo is not confined to apoptosis. New observations on the mammalian growth plate. J Bone Joint Surg Br. 2000;82(4):601–13.
Roach HI, Aigner T, Kouri JB. Chondroptosis: a variant of apoptotic cell death in chondrocytes? Apoptosis. 2004;9(3):265–77.
Ahmed YA, Tatarczuch L, Pagel CN, Davies HM, Mirams M, Mackie EJ. Physiological death of hypertrophic chondrocytes. Osteoarthr. Cartil. 2007;15(5):575–86. https://doi.org/10.1016/j.joca.2006.10.016.
Hunziker EB, Schenk RK. Cartilage ultrastructure after high pressure freezing, freeze substitution, and low temperature embedding. II. Intercellular matrix ultrastructure—preservation of proteoglycans in their native state. J Cell Biol. 1984;98(1):277–82.
Farnum CE, Turgai J, Wilsman NJ. Visualization of living terminal hypertrophic chondrocytes of growth plate cartilage in situ by differential interference contrast microscopy and time-lapse cinematography. J Orthop Res. 1990;8(5):750–63. https://doi.org/10.1002/jor.1100080517.
Takechi M, Itakura C. Ultrastructural and histochemical studies of the epiphyseal plate in normal chicks. Anat Rec. 1995;242(1):29–39. https://doi.org/10.1002/ar.1092420105.
Cancedda R, Descalzi Cancedda F, Castagnola P. Chondrocyte differentiation. Int Rev Cytol. 1995;159:265–358.
Tsang KY, Chan D, Cheah KS. Fate of growth plate hypertrophic chondrocytes: death or lineage extension? Develop Growth Differ. 2015;57(2):179–92. https://doi.org/10.1111/dgd.12203.
Hinton RJ, Jing Y, Jing J, Feng JQ. Roles of chondrocytes in endochondral bone formation and fracture repair. J Dent Res. 2017;96(1):23–30. https://doi.org/10.1177/0022034516668321.
Galotto M, Campanile G, Robino G, Cancedda FD, Bianco P, Cancedda R. Hypertrophic chondrocytes undergo further differentiation to osteoblast-like cells and participate in the initial bone formation in developing chick embryo. J Bone Miner Res. 1994;9(8):1239–49. https://doi.org/10.1002/jbmr.5650090814.
Bianco P, Cancedda FD, Riminucci M, Cancedda R. Bone formation via cartilage models: the “borderline” chondrocyte. Matrix Biol. 1998;17(3):185–92.
Holtrop ME. The potencies of the epiphyseal cartilage in endochondral ossification. Proc K Ned Akad Wet C. 1967;70(1):21–8.
Kahn AJ, Simmons DJ. Chondrocyte-to-osteocyte transformation in grafts of perichondrium-free epiphyseal cartilage. Clin Orthop Relat Res. 1977;129:299–304.
Descalzi Cancedda F, Gentili C, Manduca P, Cancedda R. Hypertrophic chondrocytes undergo further differentiation in culture. J Cell Biol. 1992;117(2):427–35.
Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, et al. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell. 2012;22(3):597–609. https://doi.org/10.1016/j.devcel.2011.12.024.
•• Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. 2014;111(33):12097–102. https://doi.org/10.1073/pnas.1302703111 The use of a tamoxifen-inducible Col10a1-Cre line being exclusively active in hypertrophic chondrocytes enabled the authors to trace the fate of hypertrophic chondrocytes at different stages of development. Furthermore, grafting experiments showed that descendents of hypertrophic chondrocytes contributed to bone repair.
•• Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B, et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014. https://doi.org/10.1038/cr.2014.111 The results from this study using the confetti mouse in combination with an inducible Col2Cre mouse line to trace the fate of chondrocytes allowed to follow the clonal progeny of a chondrocyte showing the presence of chondrocyte-derived clones in the bone marrow cavity.
•• Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice. PLoS genetics. 2014;10(12):e1004820. https://doi.org/10.1371/journal.pgen.1004820 This study showed that chondrocyte-to-osteoblast transdifferentiation occurred not only during normal endochondral bone formation but also in the bone repair callus.
•• Park J, Gebhardt M, Golovchenko S, Branguli F, Hattori T, Hartmann C, et al. Dual pathways to endochondral osteoblasts: a novel chondrocyte-derived osteoprogenitor cell identified in hypertrophic cartilage. Biol Open. 2015;4(5). https://doi.org/10.1242/bio.201411031 The results from this study suggest that transdifferentiating chondrocytes reenter the cell cycle at the chondro-osseous border and are immunopositive for the mesenchymal stem cell marker Sca1.
•• Houben A, Kostanova-Poliakova D, Weissenbock M, Graf J, Teufel S, von der Mark K, et al. Beta-catenin activity in late hypertrophic chondrocytes locally orchestrates osteoblastogenesis and osteoclastogenesis. Development. 2016;143(20):3826–38. https://doi.org/10.1242/dev.137489 The results from this study revealed an essential role for beta-catenin in chondrocyte-to-osteoblast transdifferentiation and suggested that β-catenin must act via a mechanism different from the one described for perichondrial osteoblastogenesis.
Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J Clin Invest. 1998;102(2):274–82. https://doi.org/10.1172/JCI2799.
Maye P, Fu Y, Butler DL, Chokalingam K, Liu Y, Floret J, et al. Generation and characterization of Col10a1-mcherry reporter mice. Genesis. 2011;49(5):410–8. https://doi.org/10.1002/dvg.20733.
•• Aghajanian P, Xing W, Cheng S, Mohan S. Epiphyseal bone formation occurs via thyroid hormone regulation of chondrocyte to osteoblast transdifferentiation. Sci Rep. 2017;7(1):10432. https://doi.org/10.1038/s41598-017-11050-1 The results from this study suggest that in the SOC, thyroid hormone regulates chondrocyte-derived osteoblastogenesis via opposing Ihh- and Shh-mediated signaling pathways.
Shapiro F, Flynn E, Calicchio ML. Molecular differentiation in epiphyseal and physeal cartilage. Prominent role for gremlin in maintaining hypertrophic chondrocytes in epiphyseal cartilage. Biochem Biophys Res Commun. 2009;390(3):570–6. https://doi.org/10.1016/j.bbrc.2009.10.006.
Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, et al. Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell. 2015;160(1–2):269–84. https://doi.org/10.1016/j.cell.2014.11.042.
•• Jing Y, Zhou X, Han X, Jing J, von der Mark K, Wang J, et al. Chondrocytes directly transform into bone cells in mandibular condyle growth. J Dent Res. 2015;94(12):1668–75. https://doi.org/10.1177/0022034515598135 This study shows that chondrocyte-to-osteoblast transdifferentiation also occurs during bone formation in secondary cartilage.
Durkin JF. Secondary cartilage: a misnomer? Am J Orthod. 1972;62(1):15–41.
Holmbeck K, Bianco P, Chrysovergis K, Yamada S, Birkedal-Hansen H. MT1-MMP-dependent, apoptotic remodeling of unmineralized cartilage: a critical process in skeletal growth. J Cell Biol. 2003;163(3):661–71. https://doi.org/10.1083/jcb.200307061.
Scammell BE, Roach HI. A new role for the chondrocyte in fracture repair: endochondral ossification includes direct bone formation by former chondrocytes. J Bone Miner Res. 1996;11(6):737–45. https://doi.org/10.1002/jbmr.5650110604.
Bahney CS, Hu DP, Taylor AJ, Ferro F, Britz HM, Hallgrimsson B, et al. Stem cell-derived endochondral cartilage stimulates bone healing by tissue transformation. J Bone Miner Res Off J Am Soc Bone Miner Res. 2014;29(5):1269–82. https://doi.org/10.1002/jbmr.2148.
Wong SA, Hu D, Miclau T, Bahney C, Marcucio R. Trans differentiation of chondrocytes to osteoblasts during endochondral ossification in the healing mandible. FASEB J. 2016;30. (1) Supplement, Abstract Number:1039.11
Marcucio R, Hu D, Yang F, Bahney C, Miclau T. Transdifferentiation of chondrocytes to osteoblasts during bone fracture healing. FASEB J. 2016;30. (1) Supplement. Abstract Number:223.1
•• Hu DP, Ferro F, Yang F, Taylor AJ, Chang W, Miclau T, et al. Cartilage to bone transformation during fracture healing is coordinated by the invading vasculature and induction of the core pluripotency genes. Development. 2017;144(2):221–34. https://doi.org/10.1242/dev.130807 The results from this study suggest that during fracture repair chondrocytes are induced to dedifferentiate into a pluripotent state prior to differentiating into osteoblasts.
Roach HI, Erenpreisa J, Aigner T. Osteogenic differentiation of hypertrophic chondrocytes involves asymmetric cell divisions and apoptosis. J Cell Biol. 1995;131(2):483–94.
Roach HI, Erenpreisa J. The phenotypic switch from chondrocytes to bone-forming cells involves asymmetric cell division and apoptosis. Connect Tissue Res. 1996;35(1–4):85–91.
Roach HI. Trans-differentiation of hypertrophic chondrocytes into cells capable of producing a mineralized bone matrix. Bone Miner. 1992;19(1):1–20.
Crelin ES, Koch WE. An autoradiographic study of chondrocyte transformation into chondroclasts and osteocytes during bone formation in vitro. Anat Rec. 1967;158(4):473–83. https://doi.org/10.1002/ar.1091580410.
Weiss A, von der Mark K, Silbermann M. Fully differentiated chondrocytes alter their phenotypic expression in vitro and transform into bone cells. In: Hurwitz S, Sela J, editors. Current advances in skeletogenesis 3. Jerusalem: Heiliger Publishing; 1997. p. 40–3.
Yoshioka C, Yagi T. Electron microscopic observations on the fate of hypertrophic chondrocytes in condylar cartilage of rat mandible. J Craniofac Genet Dev Biol. 1988;8(3):253–64.
Bais M, McLean J, Sebastiani P, Young M, Wigner N, Smith T, et al. Transcriptional analysis of fracture healing and the induction of embryonic stem cell-related genes. PLoS One. 2009;4(5):e5393. https://doi.org/10.1371/journal.pone.0005393.
• Enishi T, Yukata K, Takahashi M, Sato R, Sairyo K, Yasui N. Hypertrophic chondrocytes in the rabbit growth plate can proliferate and differentiate into osteogenic cells when capillary invasion is interposed by a membrane filter. PloS one. 2014;9(8):e104638. https://doi.org/10.1371/journal.pone.0104638 The results from this study suggest that chondrocyte-to-osteoblast transdifferentiation can occur in the absence of capillary invasion.
Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. 2014;507(7492):323–8. https://doi.org/10.1038/nature13145.
Langen UH, Pitulescu ME, Kim JM, Enriquez-Gasca R, Sivaraj KK, Kusumbe AP, et al. Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat Cell Biol. 2017;19(3):189–201. https://doi.org/10.1038/ncb3476.
Salter RB, Harris WR. Injuries involving the epiphyseal plate. J Bone Joint Surg Am. 1963;45(3):587–622. https://doi.org/10.2106/00004623-196345030-00019.
Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2(2):a002915. https://doi.org/10.1101/cshperspect.a002915.
• Jing Y, Jing J, Wang K, Chan K, Harris SE, Hinton RJ, et al. Vital roles of beta-catenin in trans-differentiation of chondrocytes to bone cells. Int J Biol Sci. 2018;14(1):1–9. https://doi.org/10.7150/ijbs.23165 This study confirms that β-catenin plays an important role in chondrocyte-to-osteoblast transdifferentiation.
•• Wang L, Huang J, Moore DC, Zuo C, Wu Q, Xie L, et al. SHP2 Regulates the osteogenic fate of growth plate hypertrophic chondrocytes. Scientific reports. 2017;7(1):12699. https://doi.org/10.1038/s41598-017-12767-9 Results from this study implemented a role for the phosphatase SHP2 in chondrocyte-to-osteoblast transdifferentiation through inhibition of SOX9 activity.
Topol L, Chen W, Song H, Day TF, Yang Y. Sox9 inhibits Wnt signaling by promoting beta-catenin phosphorylation in the nucleus. J Biol Chem. 2009;284(5):3323–33. https://doi.org/10.1074/jbc.M808048200.
Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18(9):1072–87. https://doi.org/10.1101/gad.1171104.
Zuo C, Wang L, Kamalesh RM, Bowen ME, Moore DC, Dooner MS, et al. SHP2 regulates skeletal cell fate by modifying SOX9 expression and transcriptional activity. Bone Res. 2018;6:12. https://doi.org/10.1038/s41413-018-0013-z.
•• Jing Y, Jing J, Ye L, Liu X, Harris SE, Hinton RJ, et al. Chondrogenesis and osteogenesis are one continuous developmental and lineage defined biological process. Sci Rep. 2017;7(1):10020. https://doi.org/10.1038/s41598-017-10048-z This study shows that BMPR1 signaling plays an important role in chondrocyte-to-osteoblast diffferentiation and suggests that chondrogenesis and osteoblastogenesis are a continuous process.
Xing W, Cheng S, Wergedal J, Mohan S. Epiphyseal chondrocyte secondary ossification centers require thyroid hormone activation of Indian hedgehog and osterix signaling. J Bone Miner Res Off J Am Soc Bone Miner Res. 2014;29(10):2262–75. https://doi.org/10.1002/jbmr.2256.
Kim HY, Mohan S. Role and mechanisms of actions of thyroid hormone on the skeletal development. Bone Res. 2013;1(2):146–61. https://doi.org/10.4248/BR201302004.
Tavella S, Biticchi R, Schito A, Minina E, Di Martino D, Pagano A, et al. Targeted expression of SHH affects chondrocyte differentiation, growth plate organization, and Sox9 expression. J Bone Miner Res Off J Am Soc Bone Miner Res. 2004;19(10):1678–88. https://doi.org/10.1359/JBMR.040706.
Mak KK, Kronenberg HM, Chuang PT, Mackem S, Yang Y. Indian hedgehog signals independently of PTHrP to promote chondrocyte hypertrophy. Development. 2008;135(11):1947–56. https://doi.org/10.1242/dev.018044.
Razzaque MS, Soegiarto DW, Chang D, Long F, Lanske B. Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J Pathol. 2005;207(4):453–61. https://doi.org/10.1002/path.1870.
Deng A, Zhang H, Hu M, Liu S, Gao Q, Wang Y, et al. Knockdown of Indian hedgehog protein induces an inhibition of cell growth and differentiation in osteoblast MC3T3E1 cells. Mol Med Rep. 2017;16(6):7987–92. https://doi.org/10.3892/mmr.2017.7669.
Maeda Y, Nakamura E, Nguyen MT, Suva LJ, Swain FL, Razzaque MS, et al. Indian hedgehog produced by postnatal chondrocytes is essential for maintaining a growth plate and trabecular bone. Proc Natl Acad Sci U S A. 2007;104(15):6382–7. https://doi.org/10.1073/pnas.0608449104.
Javaheri B, Caetano-Silva SP, Kanakis I, Bou-Gharios G, Pitsillides AA. The chondro-osseous continuum: is it possible to unlock the potential assigned within? Front Bioeng Biotechnol. 2018;6:28. https://doi.org/10.3389/fbioe.2018.00028.
Clarkin C, Olsen BR. On bone-forming cells and blood vessels in bone development. Cell Metab. 2010;12(4):314–6. https://doi.org/10.1016/j.cmet.2010.09.009.
Shah M, Gburcik V, Reilly P, Sankey RA, Emery RJ, Clarkin CE, et al. Local origins impart conserved bone type-related differences in human osteoblast behaviour. Eur Cells Mater. 2015;29:155–75 discussion 75-6.
Acknowledgments
The authors acknowledge the contributions of all researchers regarding the fate of hypertrophic chondrocytes in the field of skeletogenesis and apologize for not citing all the original research regarding the previous in vitro observations on transdifferentiation.
Funding
Research by the authors is supported by grants from the Deutsche Forschungsgemeinschaft (HA 4767/2-1, HA 4767/4-2, HA 4767/5-1) and from the Federal Ministry of Education and Science (01EC1408E), as part of the OVERLOAD-Prev-OP consortium. The authors further acknowledge institutional support by a grant from the Interdisciplinary Center of Clinical Research (IZKF, Har2/002/14).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Christine Hartmann reports grants from the German research foundation (DFG) and grants from the German Federal Ministry of Education and Science, during the conduct of the study. Lena Wolff declares no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human subjects performed by the authors. Animal studies performed by the authors regarding chondrocyte-to-osteoblast transdifferentiation were in accordance with local, institutional, and national regulations and licenses (AZ: 84-02.05.2012.261; 84-02.04.2015.128; 84-02.05.50.15.022).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Skeletal Development
Rights and permissions
About this article

Cite this article
Wolff, L.I., Hartmann, C. A Second Career for Chondrocytes—Transformation into Osteoblasts. Curr Osteoporos Rep 17, 129–137 (2019). https://doi.org/10.1007/s11914-019-00511-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11914-019-00511-3