
Abstract
In this chapter we review the basic principles of photocurrent generation in bulk heterojunction organic solar cells, discuss the loss channels limiting their efficiency, and present case studies of several polymer–fullerene blends. Using steady-state and transient, optical, and electrooptical techniques, we create a precise picture of the fundamental processes that ultimately govern solar cell efficiency.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
1 Photophysics of an Organic Solar Cell
Organic photovoltaic (OPV) devices have recently exceeded power conversion efficiencies (PCEs) of 10 % in single-junction cells [1] and 13.2 % in a multijunction device using small molecules [2]. The tenfold efficiency increase has been achieved in less than a decade by fine-tuning material processing and bandgap engineering. To further improve photovoltaic performance and to move forward to commercial applications, we are now urged to critically assess and revise the current understanding of photophysical processes occurring in OPV devices, in particular those that limit efficiency. For instance, the mechanism of charge carrier generation via dissociation of photogenerated excitons is still debated: In the past it was often assumed that bound charge-transfer (CT) states are formed at the donor–acceptor (D–A) interfaces and dissociate into free-charge carriers in a field-dependent separation process. Recent studies, however, indicate that several systems exhibit a field-independent charge generation with an internal quantum efficiency close to unity. A multitude of explanations for the efficient charge separation has been proposed, such as hot CT-state dissociation [3, 4], delocalization of charges during the ultrafast CT process [5], high initial values of charge mobilities and the presence of energy cascades [6], an interplay of entropic and enthalpic contributions [7], and long-range electrostatic forces at intermixed D–A interfaces [8, 9].
The controversial interpretation of experimental results hints at an approaching paradigm shift. New models that try to account for coherent CT, heterogeneity of blends, and nonequilibrium processes urgently require further development and validation. Once established, they will help us to understand the interplay among the chemical composition, bulk and interfacial morphologies, energetic landscapes for excitons, as well as CT and charge-separated states. This understanding is the key for the successful design of new materials.
The purpose of this review is threefold: first, to summarize the current understanding of processes that take place during photocurrent generation in an organic solar cell; second, to link them to the morphology and chemical composition for several polymer–fullerene blends; and finally, to generalize the results to other bulk heterojunction solar cells.
1.1 Charge Carrier Photogeneration
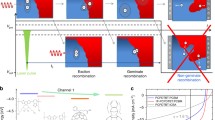
We start by listing the steps leading to photocurrent generation. After photon absorption in the donor/acceptor materials and consequent exciton generation, the exciton diffuses toward the D–A interface, where an intermediate CT state is formed after an ultrafast CT reaction. The CT state then splits into free charges, which drift-diffuse in the bulk materials and are eventually extracted at the electrodes. These steps are depicted in Fig. 1. Each step is accompanied by loss processes, which are also shown in Fig. 1. The different loss channels include the following:

Photophysical processes in organic solar cells leading to photocurrent generation (green arrows) and photocurrent loss (red arrows): (1) exciton generation by photon absorption in the donor and/or acceptor material; (2) exciton diffusion-limited and ultrafast charge transfer (CT) at the interface, potentially creating an intermediate CT state at the interface; (3) separation into free-charge carriers, followed by charge transport through the bulk materials; (4) charge carrier extraction at the electrodes; (5) exciton recombination; (6) geminate recombination of CT states; and (7) nongeminate recombination of free charges
-
1.
Incomplete photon absorption in the photoactive layer, mainly for three reasons:
-
a.
The optical gap of organic materials is typically larger than 1.4 eV; hence, photons with lower energies are not absorbed by the photoactive layer. This issue can be addressed in part by using low-bandgap materials with an absorption in the near-infrared (NIR) region. Such materials, however, often exhibit lower quantum efficiencies [10, 11].
-
b.
The photoactive layer is rather thin (100 nm) and thus captures only a fraction of the above-bandgap photons because of its finite optical density. To enhance absorption, the thickness of the photoactive layer can be increased. This, however, often leads to a lower fill factor and to a reduction in the PCE, mainly as a result of the low charge carrier mobility of organic materials [12].
-
c.
The fullerene derivatives, frequently used as acceptors, contribute only little to the total absorbance of the photoactive layer, as their absorption cross sections are small in the visible spectrum. Moreover, low-bandgap polymers often require an excess of the fullerene component for optimum device performance, which further dilutes the absorbance of the photoactive layer. This issue can be partially circumvented by using (much more costly) [6,6]-phenyl C71-butyric acid methyl ester (PC70BM), which has a stronger absorption in the visible spectrum than [6,6]-phenyl-C61-butyric acid methyl ester (PC60BM). Another alternative is to use non-fullerene acceptors. Here, efficiencies approaching those obtained with fullerene acceptors were achieved only recently [13].
-
a.
-
2.
The exciton diffusion length in organic materials is limited to about 10 nm [14]. This is a consequence of the short exciton lifetime, which is typically in the range of several hundred picoseconds, and the incoherent exciton motion process, which is best described by the hopping of the exciton between chromophores with different energy values. Hence, exciton harvesting is most efficient if the exciton has to diffuse only a short distance before encountering the interface, which requires a nanoscale phase separation in bulk heterojunction solar cells. The intrinsically short exciton diffusion length also limits the performance of bilayer solar cells. Here, the organic materials with an enhanced exciton diffusion length could help to reduce the recombination of free charges compared with bulk heterojunction devices [15] since the bilayer device has a much smaller area of D–A interfaces.
-
3.
Interfacial CT will not necessarily yield free charges but may lead to the formation of bound electron–hole pairs, in which the two oppositely charged particles have not entirely escaped their mutual Coulomb attraction. Such pairs are often called geminate pairs (GPs), where “geminate” reflects the fact that the electron and the hole are the product of the same primary photoexcitation. As charges are further separated from each other across the heterojunction, the binding energy of the GP reduces.
-
4.
The mechanism of charge photogeneration is still debated. Either it is directly by excitons in the neat materials in process (3), or it is via the GP dissociation. An important question is whether “hot charge transfer states” dominate the charge photogeneration or thermally relaxed GPs. Also, tightly bound CT states may represent an ultimate loss channel because of geminate recombination, which is still discussed controversially.
-
5.
Charge carrier recombination during the transport to the electrodes is often the main loss mechanism for state-of-the-art solar cells [15, 16]. In disordered organic semiconductor thin films, charge transport occurs via hopping of charge carriers between transport sites within the density of states [17]. In a device it is often described by drift-diffusion models using the classical semiconductor band theory as a simplified starting point. The work function difference of the device electrodes creates an internal (built-in) electric field in the organic layer, which causes the charge carriers to drift to their respective electrodes. However, charge carriers may get trapped either in tail states of the density of states or in deeper-lying trap states. Trapping leads to both slower charge transport and trap-assisted recombination equivalent to Shockley–Read–Hall recombination in inorganic semiconductors. Furthermore, free-charge carriers can encounter each other again at the D–A interface and undergo nongeminate recombination during the transport to the electrodes if not extracted prior to their encounter and recombination. Therefore, the competition between recombination and extraction rates needs to be tuned in favor of the latter. Recently, this competition was analyzed by the groups of Koster, Würfel, and Neher [18, 19]. It was shown that nongeminate recombination affects the device efficiency even for reasonably high mobilities of 10−3 cm2/Vs and that electron and hole mobilities in excess of 10−2 cm2/Vs are needed when aiming at an efficient extraction of charges out of thick active layers.
-
6.
Finally, at the electrodes the charge carriers are extracted from the photoactive layer. Extraction of the wrong carrier type (which is equivalent to recombination [20]) can be countered by adding hole- or electron-blocking layers. Note that energy barriers at the metal–organic interface (or materials of low conductivity for the majority carrier type) can hinder charge extraction, leading to charge accumulation and to so-called S-shaped current–voltage characteristics [21, 22].
The aforementioned loss channels reduce the photocurrent of the solar cell, the fill factor of the device (at least if charge carriers are involved), and the open-circuit voltage, V OC. In fact, the open-circuit voltage of an organic solar cell is typically much lower than the energy of the absorbed photons. Several loss mechanisms contribute to the V OC loss: 1. Absorption of above-bandgap photons creates losses because of the fast internal conversion of the exciton and the dissipation of the excess energy into heat. 2. Charge transfer at the interface adds a loss because of the energy-level offset between donor and acceptor. 3. The nongeminate recombination of charges [23, 24] determines the charge density and with that the Fermi-level splitting under open-circuit conditions. Together with the broadening of the density of states, this recombination results in a V OC loss of at least 0.5 V [8].
The low open-circuit voltage is a significant drawback of organic solar cells; improving V OC would definitely boost the commercialization of organic solar cells [25].
1.2 Charge-Transfer States and Geminate Charge Recombination
In this section we discuss in more detail the role of CT states. At the D–A interface, only a fraction of electrons and holes escape their Coulomb attraction and split into free charges. Electron–hole pairs that have not managed to fully separate will remain bound and form interfacial GPs. These electron–hole pairs might either reside directly at the interface, forming CT states [26], or be at a larger distance with negligible wave function overlap. In the latter case, terms such as “bound” and “loosely bound polaron pairs,” or “dark states,” are often used [27, 28]. Interfacial electron–hole pairs that have relaxed into CT states can undergo geminate recombination to the ground state. Geminate recombination of CT states has been experimentally observed by time-resolved photoluminescence (PL) spectroscopy and exhibits characteristic PL at an energy corresponding roughly to the energy gap between the ionization potential of the donor and the electron affinity of the acceptor, both in a solid state. The lifetime of the emission has been determined to be on the order of several hundred picoseconds up to several nanoseconds depending on the investigated material system [29]. Another way to excite CT states is by driving the solar cell as a light-emitting diode [29]. In this case, the radiative recombination of CT states leads to a low-energy emission, with a significant redshift of the emission peak with regard to the emission from the pure donor and acceptor components. Interestingly, charge-transfer photoluminescence (CT-PL) and electroluminescence (CT-EL) spectra do not fully overlap, with the EL peaking at a lower energy. It has therefore been proposed that the PL is from CT states on D–A pairs within mixed domains (molecular heterojunctions) while CT-EL is caused by the recombination of CT states formed at the interface between donor and acceptor domains (domain heterojunction) [30]. Due to a detailed balance, free-charge generation upon illumination, which is the inverse of the EL process, must involve the same CT states that are formed in EL [24]. Indeed, it has been demonstrated that quenching of the CT-state PL by an electric field requires fields that are substantially larger than those usually present in operating photovoltaic devices [30, 31], which questions whether the CT states leading to PL are precursors to free charges. The upper limit of the binding energy of the CT state in poly[2-methoxy-5-(3′,7′-dimethyloctyloxy)-1,4-phenylenevinylene]:1-(3-methoxycarbonyl)propyl-1-phenyl[6,6]C61 (MDMO-PPV:PCBM) has been experimentally determined to be on the order of 200 meV [32], which corresponds to several times k B T at room temperature and thus makes thermally assisted splitting unlikely. Therefore, the interpretation of CT-PL data with regard to the efficiency of free-charge generation should be considered with care. On the other hand, Vandewal and coworkers [33] were able to demonstrate efficient free-charge generation even when exciting D–A blends at the energy maximum of CT-EL, suggesting a thermally relaxed interfacial CT state as the precursor to free charges.
Further evidence of geminate recombination has been obtained using transient absorption (TA) spectroscopy (TAS) [34, 35]. In TA pump-probe experiments, geminate recombination can be identified as a pump-intensity–independent signal decay of both the ground-state bleach and charge-induced absorption that can be fitted by a single exponential with inverse rate constants on the order of picoseconds to nanoseconds [36]. The intensity-independent character of the recombination process is a consequence of the localization of the CT states at the interface since they are much less mobile than free charges. Thus, interactions between CT states or between CT states and other excited species occur only at very high excitation densities. The quasi single-exponential recombination dynamics of CT states, often observed in TA experiments, is, however, not straightforward to interpret. In principle, the disordered nature of bulk heterojunction D–A blends should cause a distribution of electron–hole pair distances across the interface, in turn causing a distribution of CT-state lifetimes, resulting in a stretched exponential rather than a single exponential decay. However, one may argue that the spectroscopically observed CT states are relaxed and similar in nature, resulting in a narrow distribution of lifetimes, which can be approximated by a single exponential.
1.3 Free Charges and Nongeminate Recombination
Electron–hole pairs, which escaped their mutual Coulomb attraction, form charge-separated states. Charge-separated states contribute to the device's photocurrent if their spatially separated carriers are extracted at the electrodes. During charge transport to the electrodes, however, free electrons and holes can encounter and recombine. Since the recombining charge carriers originate from different photoexcitations, this mechanism is termed nongeminate recombination. The rate of nongeminate recombination, R, depends on both the carrier concentration n and the effective lifetime τ(n). Usually, the higher the charge carrier concentration is, the faster the recombination, \( R\sim n/\tau (n) \), will be. For most state-of-the-art organic solar cells, the nongeminate recombination of already separated electrons and holes is the dominant loss mechanism.
The simplest description of charge carrier recombination is Langevin’s model. In this model, the Coulomb attraction between electrons and holes results in a recombination rate of \( R=e/\epsilon {\epsilon}_0\left({\mu}_e+{\mu}_h\right)np \) [37]. Here e is the elementary charge, ϵϵ 0 is the material dielectric constant, and n/p and μ e/h are the electron and hole densities and mobilities, respectively. If the electron and hole concentrations under illumination are similar (for instance, in systems without doping), then \( n\approx p \), the recombination process is of the second order, and the carrier concentration decay is inversely proportional to the time, \( n\sim {t}^{-1} \).
In polymer–fullerene systems, deviations from Langevin’s recombination have been observed. For instance, Pivrikas et al. [38] noted a reduction in the recombination rate by orders of magnitude for poly(3-hexylthiophene-2,5-diyl): 1-(3-methoxycarbonyl)propyl-1-phenyl[6,6]C61 (P3HT:PCBM). This reduction is often taken into account by multiplying Langevin’s recombination rate by a prefactor varying within the range from 10−3 to 10−1, depending on the material system, and is termed a reduced Langevin recombination. For P3HT:PCBM the reported values are of the order of 0.02 or less, whereas almost no reduction is measured in MDMO-PPV:PCBM [39]. Reduced recombination can be explained by the redissociation of bound pairs [40], the presence of the charge carrier concentration gradients [41], and spatial localization of recombination zones to D–A interfaces [42, 43].
Apart from the recombination rate reduction, time-dependent experiments, for example, transient absorption, demonstrated that the decay of the carrier concentration can be different from the \( {t}^{-1} \) behavior predicted by Langevin’s model [44], leading to a power law decay rate, \( R\sim {n}^{\lambda +1} \). It was found that the exponent λ increases with lower temperatures [45, 46], indicating a relationship to thermal activation. Inspired by transient absorption experiments on pristine polymers [47] and using random walk modeling [48], the power law decay has been related to the trapping of charge carriers in the tail states of the density of states [17]. This effect can be accounted for by assuming the carrier concentration-dependent mobility, \( R\sim {\mu}_{\mathrm{eff}}(n){n}^2 \). Other effects influencing the recombination exponent include the injection of majority carriers by electrodes with small injection barriers [49] and doping [50].
To conclude this section, we emphasize that fast charge carrier extraction from the photoactive layer and the suppression of nongeminate recombination are prerequisite in order to obtain a high performance from photovoltaic devices. In a working solar cell, the nongeminate recombination of spatially-separated charge carriers always competes with charge carrier extraction during the drift–diffusion of charges toward the electrodes [51]. If charge generation is efficient but charge extraction is slower than nongeminate recombination, the buildup of high charge carrier concentrations can lead to the device having a reduced fill factor and lower PCE [18].
2 Case Studies
In what follows, we present a detailed photophysical characterization of four polymer blends, the chemical structures of which are shown in Fig. 2.

Structures of polymers discussed in this chapter: poly(2,5-bis(3-hexadecylthiophen-2-yl)thieno[3,2-b] thiophene (PBTTT); poly((4,8-bis((2-ethylhexyl)oxy)benzo (1,2-b:4,5-b’)dithiophene-2,6-diyl) (3-fluoro-2-((2-ethylhexyl)carbonyl) thieno(3,4-b)thiophenediyl)) (PTB7); poly[(4,8-bis-(2-ethylhexyloxy)-benzo(1,2-b:4,5-b’)dithiophene)-2,6-diyl-alt-(4-(2-ethylhexanoyl)-thieno[3,4-b]thiophene)-2-6-diyl)] (PBDTTT-C); poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b’]dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] (C-PCPDTBT), and poly[2,6-(4,4-bis-(2-ethylhexyl)dithieno[3,2-b:2’,3’-d]silole)-alt-4,7-(2,1,3-benzothiadiazole)] (Si-PCPDTBT). Blends were made using either [6,6]-phenyl-C61-butyric acid methyl ester (PC60BM) or [6,6]-phenyl-C71-butyric acid methyl ester (PC70BM)
2.1 Effect of Solvent Additives on Charge Carrier Recombination in PTB7:PCBM
Blends of the donor polymer thieno[3,4-b]thiophene-alt-benzodithiophene (PTB7) and the fullerene acceptor PC70BM are among the most efficient OPV materials, with a reported record PCE of 9.2 % [52]. The photovoltaic performance of these blends improves dramatically upon adding 1,8-diiodooctane (DIO) to the solution. DIO has a strong impact on the active layer morphology [53–56]: Pristine blends comprise large (100-nm) clusters consisting almost entirely of fullerene embedded in mixed polymer-rich domains. DIO suppresses the formation of large fullerene clusters without affecting the overall crystallinity or composition of domains. As a consequence, the short-circuit current and the fill factor of the photovoltaic cell increase [52] because of the more efficient generation of free charges or/and reduced nongeminate recombination.
To disentangle these two contributions, the effect of DIO on the generation and recombination of charges was investigated by a combination of transient photovoltage, charge extraction, and time-delayed collection field (TDCF) experiments [15]. Two sets of PTB7:PC70BM 1:1.5 blend solar cells were compared: one prepared from pure chlorobenzene (CB) solution and another from CB with a small amount of DIO as co-solvent. The current–voltage characteristics for both sets are shown in Fig. 3. As expected, the corresponding PCE increases from 3.6 to 7.0 % when DIO is added.

Measured and reconstructed current–voltage response of PTB7:PC70BM 1:1.5 solar cells processed with (a) and without (b) 1,8-diiodooctane (DIO) as cosolvent for different illumination intensities at 300 K. The solar cell with DIO (a) is only limited by nongeminate recombination, which is responsible for the entire shape of the current–voltage characteristics. The device without DIO (b) is additionally limited by field-dependent photogeneration, yielding the generation current j gen as determined from time-delayed collection field measurements. [Adapted from Adv. Funct. Mater. 24, 1306 (2014), reprinted by permission of John Wiley & Sons, Inc.]
Pre-bias-dependent TDCF measurements [57, 58] were performed to investigate the field dependence of the photogeneration yield, that is, the efficiency of the CT-state dissociation. Devices processed without DIO show a pronounced field dependence in conjunction with a lower carrier yield, whereas devices prepared with DIO exhibit a higher carrier yield in line with a weak field dependence. It appears that the well-balanced presence of mixed domains and neat material domains in the blend prepared with DIO assists the charge photogeneration by promoting a field-independent CT separation. This observation is also in agreement with theoretical calculations predicting that well-ordered domains of the donor and acceptor phases with intermixed interfaces are required in order to split CT states [9].
In contrast, the nongeminate recombination in both sets of devices (see Fig. 4) shows comparable charge carrier lifetimes at low charge carrier densities, resulting in a recombination order of about 3.5 at room temperature. If the density of tail state distribution were exponential, this would correspond to a characteristic energy of the tail states of about 50 meV. For carrier concentrations generated at 1 sun illumination, however, only the blend with DIO shows a recombination order of 2, that means a second-order recombination mechanism.

Effective lifetime in dependence of charge carrier concentration of PTB7:PC70BM 1:1.5 solar cells processed with and without 1,8-diiodooctane (DIO). Both devices show trap-limited losses with a recombination order of about 3.5 at low carrier concentrations. Around one sun illumination, only the device with DIO shows second-order recombination, as described in the text. [Adapted from Adv. Funct. Mater. 24, 1306 (2014), reprinted by permission of John Wiley & Sons, Inc.]
The relative contributions of geminate and nongeminate recombinations are shown in Fig. 5, in the reconstructed current–voltage characteristics. The device prepared with DIO is limited only by nongeminate recombination, while the device prepared without DIO also exhibits a field-dependent photogeneration as described above. We propose that the discrepancy between the measured and reconstructed curves in the device prepared without DIO is caused by the trapping of electrons in isolated fullerene clusters from which they cannot escape but from which they can still contribute to recombination. This scenario is consistent with the lower electron mobility reported for devices prepared without DIO [59].

J–V characteristics at different light intensities relative to one sun for the device prepared with (a) and without (b) 1,8-diiodooctane. Symbols are the experimental data and solid lines are drift-diffusion simulations based on experimentally determined parameters. [Reprinted with permission from Phys. Chem. C 119, 8310 (2015). Copyright American Chemical Society]
To address this point in more detail, we combined TDCF with bias-assisted charge extraction (BACE) [60], space charge limited current measurements, and numerical device simulations [61]. In the BACE technique, charges are extracted at a reverse bias, thus reducing the amount of charge being lost by nongeminate recombination or trapping.
It turned out that the nongeminate recombination coefficient of the pristine blend, γ = 4 × 10−17 m3/s, is larger than in the DIO-processed blend, \( {\gamma}_{\mathrm{DIO}} = 1.5\kern0.75em \times \kern0.75em {10}^{-17}\kern0.5em {\mathrm{m}}^3/\mathrm{s} \). This can be attributed to the presence of large intermixed regions in the blend prepared without the additive. Interestingly, the electron mobility is an order of magnitude lower in the pristine blend, which seems to be counterintuitive to the fact that this blend comprises large fullerene domains, with diameters comparable to the active layer thickness. However, a recent study by Hedley et al. [62] suggested that these domains consist of smaller fullerene spheres, of the order of 20–60 nm. While the electron mobility within these spheres may be quite high, the macroscopic transport across the entire fullerene domain can be limited by transitions between small spheres, explaining the low mobilities and charge trapping.
Given the fact that the use of the additive affects all relevant parameters (mobilities, the nongeminate recombination coefficient, the field dependence on photogeneration), drift-diffusion simulations of the J–V characteristics were performed to identify the main reason for the large difference in performance. The simulation results are compared to the experimental J–V curves in Fig. 5. The detailed analysis of these simulations revealed that the low performance of the devices prepared without DIO cannot be explained solely on the basis of a field-dependent generation and a higher coefficient for nongeminate recombination. Instead, the effect of the additive can only be quantified when the reduced electron mobility in the device processed without DIO is taken into account, which causes inefficient electron extraction and a poor fill factor (see Fig. 5b). This result emphasizes the importance of efficient charge extraction for reaching high currents and fill factors.
In fact, a similar situation is encountered in as-prepared P3HT:PCBM blends. These devices suffer from poor performance, mainly because of a low fill factor. The efficiency can be largely improved by thermal annealing or solvent treatment. Our recent experimental and simulation work revealed that annealing affects all relevant parameters, but it is mainly the low mobility of holes in the as-prepared blend that causes its poor performance [63].
Bartesaghi et al. [18] recently showed that the fill factor of organic bulk heterojunction devices can be related to a dimensionless quantity, θ, which is proportional to the total loss current caused by nongeminate recombination divided by the extraction current at short-circuit conditions:
where θ itself is a function of the nongeminate recombination coefficient γ, the generation rate G, the active layer thickness d, the mobilities of electrons and holes, μ e and μ h, and the built-in bias V int. We find that \( \theta =0.16 \) for the pristine PTB7:PCBM blend and \( {\theta}_{\mathrm{DIO}}=0.018 \) for the device processed with DIO. An order-of-magnitude reduction upon the addition of DIO is in agreement with the observed increase in fill factor from 49.7 to 71.5 % and is the combined effect of a reduced nongeminate recombination coefficient and a higher electron mobility.
2.2 Photophysics of C- and Si-PCPDTBT Blends
Poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b:3,4-b′] dithiophene)-alt-4,7-(2,1,3-benzothiadiazole)] (C-PCPDTBT) and poly[2,6-(4,4-bis(2-ethylhexyl)dithieno[3,2-b:2,3-d]silole)-alt-4,7-(2,1,3 benzothiadiazole)] (Si-PCPDTBT or PSBTBT) are D–A low-bandgap polymers that have demonstrated photovoltaic efficiencies exceeding 5 %. Peet et al. [64] showed that the efficiency of the carbon-bridged polymer C-PCPDTBT blended with PC70BM increases drastically when a small amount of an alkanedithiole such as 1,8-octanedithiol (ODT) is added to the solution prior to spin coating. It was proposed that ODT selectively dissolves fullerenes and promotes the aggregation of polymer chains into more ordered supramolecular structures prior to complete drying of the photoactive layer [65]. Substitution of the bridging carbon by silicon increased the interchain order of Si-PCPDTBT. As a result, Si-PCPDTBT/PC70BM solar cells could reach conversion efficiencies over 5 % without the use of any additives.
The performance differences caused by different film preparation conditions and the substitution of the carbon by silicon atoms have been addressed by several research groups. For instance, Durrant and coworkers [66] used TAS to determine the amplitude of the charge-induced absorption signal and thereby the free-charge carrier yield in the absence and presence of an electric field. They concluded that charge generation in both C- and Si-PCBTBT with PCBM is field independent at room temperature. Furthermore, based on the observation of a lower charge carrier yield in TAS experiments, the authors assigned the poorer performance of the C-PCPDTBT:PCBM blend processed without ODT to a lower efficiency for photogeneration, caused by geminate recombination of interfacial CT states in highly intermixed blends. The polaron dynamics at longer timescales was explained by the nongeminate recombination of free carriers competing with charge extraction. Interestingly, the polaron dynamics did not show any appreciable dependence on bias even at longer timescales, where the carrier dynamics is mostly determined by the competition between recombination and extraction. As some of these data have been recorded for a very high excitation density of 85 μJ/cm2, screening of the external electric field by space charge effects likely occurred. In fact, in a later study Neher’s group [67, 68] showed that in C-PCPDTBT:PCBM and Si-PCBTBT:PCBM charge generation is field dependent and the magnitude of the field dependence decreases in the order C-PCPDTBT:PCBM (without additive) > C-PCPDTBT:PCBM (with additive) > Si-PCBTBT:PCBM, explaining the pronounced differences in fill factor observed for these systems. The field dependence on generation was shown to scale inversely with the polymer domain size, highlighting the need for sufficiently large and preferably pure domains for efficient free-charge generation [69, 70].
Blom and coworkers [71] also studied the effect of solvent additives on the charge carrier yield in C-PCPDTBT:PCBM photovoltaic devices. They succeeded to model the entire device’s J–V curves using Braun–Onsager’s model, including a field-assisted separation of rather long-lived CT states. They proposed that adding ODT reduces the rate of geminate recombination of the CT state from 1.7 ⋅ 107 s−1 to 3 ⋅ 106 s−1. However, the rate constants are very sensitive to the underlying model and thus may lag behind physical interpretation. In fact, a recent paper by Jarzab et al. [72] indicates that the lifetime of photogenerated CT states in C-PCPDTBT:PCBM is only about 500 ps as measured by time-resolved fluorescence spectroscopy.
The early time charge-generation processes in C-PCPDTBT:PCBM have also been studied by Heeger et al. [73] using ultrafast pump-probe spectroscopy. Their results indicate ultrafast exciton dissociation and charge generation from CT states on a timescale of several picoseconds. Rao et al. and Chow et al. [74, 75] recently presented ns-μs TA studies on C-PCPDTBT:PCBM blends and demonstrated that the polymer’s triplet state is populated by the nongeminate recombination of free charges created by exciton dissociation after photoexcitation. Interestingly, the blend processed with a solvent additive showed more triplet excitons, supporting the assignment of triplet formation to a nongeminate recombination mechanism; in other words, the more free charges that were created, the higher was the yield of triplets. In fact, for optimized blends an almost quantitative recombination of charges into the triplet state was observed under pulsed laser excitation as typically used in TA experiments [75].
In our own studies on C-PCPDTBT:PCBM blends processed with and without solvent additives, we confirmed that processing with solvent additives leads to enhanced demixing of the blend’s components [76]. Whereas in the blend processed without additive the CT upon excitation of the polymer was exclusively ultrafast, the blend processed with an additive showed ultrafast charge separation as well as a diffusion-limited component extending to several picoseconds. Interestingly, a diffusion-limited CT component becomes visible in the blend processed without DIO when exciting the fullerene acceptor, and it is even more pronounced in the blend processed with an additive, indicating enhanced demixing. Additionally, processing with an additive was found to increase the yield of free charges by reducing the geminate recombination of interfacial CT states. As mentioned above, parallel work by the Cambridge group demonstrated that both blends create triplet states upon the nongeminate recombination of free charges, with the extent to which triplets were created depending on the microstructure of the blend controlled by the processing conditions. In our own work we compared the creation of triplets in C/Si-PCPDTBT:PCBM blends by a combination of TAS and multivariate curve resolution alternating least-squares (MCR-ALS) data analysis [77]. Typical ps-ns NIR TA data of a Si-PCPDTBT:PCBM blend along with the component-associated spectra and dynamics of singlet excitons, charge carriers, and triplet states (obtained by MCR-ALS) are shown in Fig. 6. We have also looked into the triplet-state formation in C-PCPDTBT:PCBM blends processed with and without adding DIO. The triplet yield increases upon processing the C-PCPDTBT:PCBM with cosolvents because of increased free-charge generation, in line with the results reported by the Cambridge group. However, triplet generation was found to be reduced in Si-PCBTBT:PCBM blends despite a further increased yield of free charges compared to C-PCPDTBT:PCBM (see Fig. 7). The reason for the decreased triplet formation is not yet fully understood; it could be related to the blend’s microstructure and interfacial morphology as well as the slightly different triplet energy levels of both material systems. Further experiments on related systems are ongoing to elucidate the interplay among energetics, morphology, and triplet-state formation.

Contour plot of ps-ns near-infrared transient absorption data of a Si-PCPDTBT:PCBM blend, component spectra (right panel), and component dynamics (bottom panel) obtained by multivariate curve resolution analysis. [Reproduced from Energy Environ. Sci. 8, 1511 (2015) with permission from the Royal Society of Chemistry]

Singlet-, charge carrier, and triplet-state dynamics in Si-PCPDTBT:PCBM (upper panel) and optimized C-PCPDTBT:PCBM blends (lower panel) obtained by multivariate curve resolution analysis of ps-ns near-infrared transient absorption data. Note that under similar excitation conditions charge carrier recombination (dashed lines) is slower and fewer triplet states (solid lines) are created in the Si-PCPDTBT:PCBM system compared to PCPDTBT:PCBM. [Reproduced from Energy Environ. Sci. 8, 1511 (2015) with permission from the Royal Society of Chemistry]
2.3 Effect of Morphology on Charge Generation in PBTTT:PCBM
The conjugated polymer poly(2,5-bis(3-hexadecylthiophen-2-yl)thieno[3,2-b] thiophene (PBTTT-C16) is a versatile model system for OPVs, as it allows a systematic variation of the blend morphology when the type and fraction of the fullerene acceptor are changed. Depending on the size of the fullerene, either the acceptor molecules can intercalate between the polymer side chains, leading to an intimate mixing of donor and acceptor on a local scale, or they are too large to intercalate, so that separate material domains are formed. These properties explain why this material system is interesting for studying the relationship between morphology and the fundamental processes in organic bulk heterojunction solar cells. The intercalation of fullerene molecules between polymer side chains and the formation of a bimolecular crystal (cocrystal) were previously investigated on PBTTT:fullerene blends by McGehee’s group [78, 79]. PBTTT polymer chain spacing increases when blended with PC60BM relative to the neat polymer, indicating the formation of a closely intermixed cocrystal in which fullerene molecules intercalate polymer side chains. For excess concentrations of fullerene molecules beyond a 1:1 ratio, pure fullerene phases are formed in addition to the intercalated/cocrystal phase. In contrast, bis-PC60BM–based blends do not exhibit increased chain spacing, as the fullerene molecules bearing two side chains cannot intercalate for steric reasons. We recently confirmed these results using photothermal deflection spectroscopy [80]: Here a clear sub-bandgap absorption below 1.65 eV is an exclusive feature of the polymer–fullerene blend—that is, it is not seen in either of the neat materials—and corresponds to the CT-state absorption, which is created by the close interaction between the donor and acceptor molecules. In comparison, the CT-state absorption of PBTTT:bis-PCBM is reduced by at least one order of magnitude compared to PBTTT:PCBM. This confirms the reduced interaction of the donor and acceptor molecules at the interface because of the lack of intercalation in the bis-PCBM–based blend.
TDCF measurements were performed on these model systems at very low laser fluence to exclusively study the competition between free-charge generation and geminate recombination while avoiding any unwanted carrier losses from nongeminate recombination [80]. Between open-circuit conditions and −4V bias photogenerated charges are extracted, where their field dependence is a measure of the competition between field-dependent charge generation and its counterprocess, namely, geminate recombination. For 1:0.7 PBTTT:PC60BM, the field dependence changes by a factor of 10, indicating that the separation yield at low internal voltages is very weak and cannot compete with geminate recombination. For the fully intercalated 1:1 blend, the photogeneration yield still changes by a factor of 3. In contrast, the 1:4 blend shows a comparably weak field dependence by a factor of about 1.3. Only in the latter case (namely, the intercalated blend with excess fullerene molecules that create a separate pure phase) is a reasonable photovoltaic performance achieved. Hence, for an effective dissociation of bound charge carrier pairs in an intercalated system, connected to a high photogeneration yield and weak field dependence, an excess fullerene phase is necessary to ensure charge generation. This allows the spatial separation of the GP, which is directly related to delocalization [81, 82].
Concerning the already-mentioned debate on the role of hot CT complexes on the photogeneration yield, we compared spectrally resolved quasi–steady-state photocurrent spectroscopy to pulsed ns-laser excitation by a TDCF technique on PBTTT:PC60BM blends. In the time-resolved experiment, two different photon energies were used, leading to excitation of singlet excitons in the donor and acceptor at 2.34 eV as well as direct subgap excitation at 1.17 eV, which is exclusively absorbed by the CT complexes [80]. With both experimental techniques we found that the influence of the excitation energy (above- or subgap) on the overall field dependence for photogeneration is very weak and cannot be traced to an effect of hot CT complexes.
Another aspect related to morphology becomes important. Comparing the best of the above intercalated systems, the 1:4 PBTTT:PC60BM blend to a nonintercalated 1:1 bis-PCBM blend, we found that the overall amount of photogeneration is more favorable in the former, whereas the latter has a more efficient extraction of photogenerated charge carriers [80]. Between short- and open-circuit conditions, the normalized photogenerated charge of PBTTT:bis-PCBM exceeds PBTTT:PCBM, resulting in an overall field dependence of about 15 % for the bis-PCBM-based solar cell. We expect that the lower extraction yield in the PBTTT:PC60BM device is caused by the extraction of electrons from the intercalated phase into the pure fullerene phase. This extra step is not required in the bis-PCBM–based blend, where the bound charge carriers, which can only be separated at the D–A interface, can more easily delocalize into the respective nonintercalated material phases. Laquai’s group [83] recently performed ps-μs TA experiments on PBTTT:PC60BM blends using a blending ratio of 1:1, corresponding to the cocrystal, and a 1:4 ratio as optimized for device performance. The TA experiments confirmed substantial fast sub-ns geminate recombination in the 1:1 blend, in line with the low photocurrent observed in devices (Fig. 8). Furthermore, geminate recombination was found to be substantially reduced in 1:4 blends, in line with higher photocurrents. These experiments complement the TDCF experiments presented by Zusan et al. [85] and support their findings regarding the importance of extended fullerene-rich regions for efficient charge carrier separation.

(a) Transient absorption spectra at 1 ps and 1 ns after photoexcitation of a PBTTT:PCBM blend with a 1:1 (upper panel) and 1:4 (lower panel) blending ratio and (b) corresponding kinetics of the ground-state bleach for a 1:1 (upper panel) and 1:4 (lower panel) blending ratio. Note the fast and intensity-independent signal decay in the 1:1 blend indicating geminate recombination of charge-transfer states. [From Macromol. Rapid Commun. 36, 1054 (2015), reprinted by permission of John Wiley & Sons, Inc.]
2.4 Charge Carrier Photogeneration and Triplet Exciton Formation in PBDTTT-C:PCBM
PBDTTT-C is another example from the family of benzodithiophene-based low-bandgap polymers, to which PTB7 also belongs. Laquai’s group [84] recently reported fs-μs TA experiments on PBDTTT-C:PC60BM blends. Ultrafast CT, following photoexcitation of the polymer, was observed. The assignment of the ps TA spectrum to charges was confirmed by comparing the TA signal to the absorption spectrum of a chemically oxidized polymer film. Interestingly, the NIR photoinduced absorption spectrum showed a pronounced redshift on the ps-ns timescale, indicating the formation of a second component. By MCR-ALS analysis, as outlined above for the PCPDTBT:PC60BM material system, two components were identified that constituted the experimentally measured TA data matrix (see Fig. 9). Here, the second, spectrally redshifted, component was identified as the polymer’s triplet state by comparison of the component spectrum to the triplet absorption obtained independently on a metal-porphyrin–sensitized polymer. Furthermore, MCR analysis showed that triplets were the dominant excited state at 1 ns after photoexcitation and the analysis revealed a strong fluence dependence of the charge carrier decay dynamics and triplet-state formation. This clearly points to a nongeminate recombination of free charges as the origin of triplet formation in this system, which is in line with results obtained on other low-bandgap polymer–fullerene blends [74, 75, 77]. However, on the ns-μs timescale, the MCR analysis showed a regeneration of the charge carrier population. We attributed this to the annihilation of triplet states, which leads to the generation of higher-excited singlet and triplet excitons that now have sufficient energy to undergo charge separation at the polymer–fullerene interface and thereby recreate a small pool of charges. This study demonstrates that triplet-state formation is not exclusive to the aforementioned low-bandgap systems but may be a more general phenomenon in polymer–fullerene blends in which the polymer’s triplet level is sufficiently low to be populated from interfacial triplet CT states that are created during nongeminate charge recombination. In fact, as there may be an equilibrium between free charges and CT states in working devices as previously discussed by Burke et al. [25], any additional channel such as triplet energy transfer from the CT state to the polymer’s triplet state could have an impact on device performance even under solar illumination conditions, where the carrier concentration is lower than under pulsed laser excitation as typically used in TA experiments.

Normalized contour plot of the ps-μs near-infrared transient absorption data obtained on a PBDTTT-C:PCBM blend at an excitation density of 11.5 μJ/cm2. The panel on top shows the component spectra and the panel on the right side the ps-μs component dynamics obtained by multivariate curve resolution analysis of the experimental data. Note the fast and intensity-dependent recombination of charge carriers and the concomitant population of the triplet state on the sub-ns timescale. On the ns-μs timescale, triplet states undergo annihilation and repopulate the pool of charge carriers. [Reprinted with permission from J. Phys. Chem. C 119, 13509 (2015). Copyright American Chemical Society]
In a related investigation, the fraction of the cosolvent DIO in the preparation of PBDTTT-C:PC71BM solar cells was systematically increased. Zusan et al. [85] found that DIO has effects on the blend microstructure which influence charge photogeneration and recombination. The impact of the cosolvent on the current–voltage characteristics, including the contributions of geminate and nongeminate recombination, is shown in Fig. 10. Without the cosolvent, the blend morphology constitutes large PC71BM agglomerates in a PBDTTT-C–rich matrix, in accordance with reports by Collins et al. [86]. The photogeneration was poor because of the accordingly limited interfacial area, also leading to singlet exciton losses within the large fullerene domains. Already small amounts of DIO (0.6 %) led a reduction in the PC71BM domain size and a higher interfacial area between the donor and acceptor. The PCE is approximately doubled from 2.6 to 5.3 % because of the more efficient exciton dissociation; as a result of a strongly enhanced D–A interface, the PCE of the 0.6 % DIO device reaches a moderate level of only 5.3 %. However, the field-dependent photogeneration of the CT complexes and the inefficient collection of free-charge carriers in the too finely intermixed D–A matrix are problematic. Only an increase in the DIO content up to 3 % allows the charge photogeneration and collection to be improved by further decreasing the PC71BM domains and growing pure PBDTTT-C regions. With similar light harvesting, the resulting maximum PCE is 6.9 %. It is limited by a dominant nongeminate recombination and by substantially reduced but still present geminate losses. This study demonstrates that the photovoltaic performance is controlled by a crucial balance between local photogeneration and charge carrier transport due to the blend morphology.

J–V characteristics (left axis) and normalized extracted charge carrier density q tot obtained using time-delayed collection field (right axis) of PBDTTT-C:PC71BM solar cells with varying DIO content. The illumination intensity was 1 sun. The difference between J–V and q tot corresponds to nongeminate recombination losses as indicated by the shaded area using the example of 1 % DIO. [Figure reprinted by permission from Macmillan Publishers Ltd., Sci. Rep. 5, 8286 (2015)]
3 Summary and Discussion
The study presented of four polymer blends supports the accepted viewpoint that the morphology of the blend, which is determined by the (post)processing conditions, is crucial to its photophysical properties. This is consistent with the view that low-energy (thermalized) rather than hot CT states are the main precursors to free charges [33]. As a consequence, the efficiency for free-charge photogeneration is inherently connected to the energy of the relaxed CT state and that of the charge-separated state, with these energies being influenced by the molecular packing and orientation at the D–A heterojunction and within the charge-transporting domains. Given the complexity of the systems, it is also clear that the adequate understanding of photophysics can only be achieved by combining several experimental techniques and theoretical modeling. It is, however, significantly more difficult to provide a clear link between morphology changes and photophysics at an atomic level, which is needed to design new materials. Here we identify several such structure–morphology–property relationships.
First, we shall argue that the role of the additive (or a good solvent) is to provide better local packing (π stacking) of polymer chains as well as to achieve the required sizes of polymer/fullerene domains. This conclusion has been supported by other studies, which also point out that most conjugated polymers are polymorphs [87–90], and additives can help to select the required polymorph. Better local ordering naturally improves charge transport because of reduced paracrystallinity and stronger electronic coupling elements [90–93]. A less trivial observation is that long-range ordering also influences the relative energy-level alignment as well as can help to reduce geminate and nongeminate recombination by contributing to the electrostatic forces acting on CT and charge-separated states at the interface [8, 9]. Detailed analysis of the long-range electrostatic effects teaches us that this comes at a price: While helping to split CT states and reduce nongeminate recombination, electrostatic effects also reduce the open-circuit voltage [9]. Since electrostatic forces, to a large extent, are determined by the quadrupole moment of a polymer repeat unit and its orientation with respect to the interface, one should look for a suitable compromise when tuning the chemistry of the polymer backbone.
Second, a far less trivial observation is that one should still allow for a certain degree of intermixing at the D–A interface. The argument here is that the localized electron and hole at an ideally flat interface are always bound electrostatically, with a binding energy on the order of 0.3 eV. If, however, a donor molecule protrudes into the acceptor phase (or vice versa), its gas-phase ionization potential and electron affinity become “dressed” by the electrostatic fields typical for the acceptor side of the interface. This reduces the binding energy of the CT state and makes it possible to split this state thermally [9].
With these design rules in mind, we can outline the future targets for OPV research. It has already been mentioned that the two main obstacles that prevent OPV commercialization are (1) limited by a thin photoactive layer with low external quantum efficiency and (2) low open-circuit voltages, in spite of large photovoltaic gaps. In both cases the potential strategies can be readily identified: To improve the external quantum efficiency we need materials with higher charge carrier mobilities, which facilitate extraction prior to recombination and thus allow for thicker photoactive layers. Furthermore, acceptors with an absorption stronger than that of fullerenes should be actively investigated.
The problem of the low open-circuit voltage is more intricate since arguments that account only for the relative energy-level alignment are insufficient to formulate appropriate design rules. It seems that we need to gain a better control of interfacial ordering (including intermixing) in order to balance the binding energy of CT states and losses in the open-circuit voltage. In addition, we need to look for materials with a small energetic disorder since the significant part of reducing the photovoltaic gap to the open-circuit voltage is caused by pinning the chemical potentials of holes and electrons in (rather broad) densities of states of the donor and the acceptor.
Overall, we now better understand the subtle interplay among molecular architecture, morphology, and electrostatic effects that jointly produces a high-performing solar cell, and we are ready to move forward assisted by the design rules we formulated.
References
Liu Y, Zhao J, Li Z, Mu C, Ma W, Hu H, Jiang K, Lin H, Ade H, Yan H (2014) Nat Commun 5:5293
Bakulin A, Rao A, Pavelyev V, van Loosdrecht P, Pshenichnikov M, Niedzialek D, Cornil J, Beljonne D, Friend RH (2012) Science 335:1340
Grancini G, Maiuri M, Fazzi D, Petrozza A, Egelhaaf H-J, Brida D, Cerullo G, Lanzani G (2013) Nat Mater 12:29
Gagorik AG, Mohin JW, Kowalewski T, Hutchison GR (2015) Adv Funct Mater 25:1996
Burke TM, McGehee MD (2014) Adv Mater 26:1923
Monahan NR, Williams KW, Kumar B, Nuckolls C, Zhu X-Y (2015) Phys Rev Lett 114:247003
Poelking C, Tietze M, Elschner C, Olthof S, Hertel D, Baumeier B, Würthner F, Meerholz K, Leo K, Andrienko D (2015) Nat Mater 14:434
Poelking C, Andrienko D (2015) J Am Chem Soc 137:6320
Scharber MC, Koppe M, Gao J, Cordella F, Loi MA, Denk P, Morana M, Egelhaaf H-J, Forberich K, Dennler G, Gaudiana R, Waller D, Zhu Z, Shi X, Brabec CJ (2010) Adv Mater 22:367
Vandewal K, Ma Z, Bergqvist J, Tang Z, Wang E, Henriksson P, Tvingstedt K, Andersson MR, Zhang F, Inganäs O (2012) Adv Funct Mater 22:3480
Bartelt JA, Lam D, Burke TM, Sweetnam SM, McGehee MD (2015) Adv Energy Mater 5:1500577
Zhao J, Li Y, Lin H, Liu Y, Jiang K, Mu C, Ma T, Lin Lai JY, Hu H, Yu D, Yan H (2015) Energy Environ Sci 8:520
Mikhnenko OV, Azimi H, Scharber M, Morana M, Blom PWM, Loi MA (2012) Energy Environ Sci 5:6960
Foertig A, Kniepert J, Gluecker M, Brenner T, Dyakonov V, Neher D, Deibel C (2014) Adv Funct Mater 24:1306
Kirchartz T, Pieters BE, Kirkpatrick J, Rau U, Nelson J (2011) Phys Rev B 83:115209
Baranovskii SD (2014) Phys Status Solidi 251:487
Bartesaghi D, Pérez IDC, Kniepert J, Roland S, Turbiez M, Neher D, Koster LJA (2015) Nat Commun 6:7083
Würfel U, Neher D, Spies A, Albrecht S (2015) Nat Commun 6:6951
Strobel T, Deibel C, Dyakonov V (2010) Phys Rev Lett 105:266602
Wagenpfahl A, Rauh D, Binder M, Deibel C, Dyakonov V (2010) Phys Rev B 82:115306
Tress W, Leo K, Riede M (2011) Adv Funct Mater 21:2140
Rau U (2007) Phys Rev B 76:085303
Vandewal K, Tvingstedt K, Gadisa A, Inganäs O, Manca JV (2009) Nat Mater 8:904
Burke TM, Sweetnam S, Vandewal K, McGehee MD (2015) Adv Energy Mater 5
Deibel C, Strobel T, Dyakonov V (2010) Adv Mater 22:4097
Shoaee S, Subramaniyan S, Xin H, Keiderling C, Tuladhar PS, Jamieson F, Jenekhe SA, Durrant JR (2013) Adv Funct Mater 23:3286
Dimitrov SD, Durrant JR (2014) Chem Mater 26:616
Scharber MC, Lungenschmied C, Egelhaaf H-J, Matt G, Bednorz M, Fromherz T, Gao J, Jarzab D, Loi MA (2011) Energy Environ Sci 4:5077
Tvingstedt K, Vandewal K, Zhang F, Inganäs O (2010) J Phys Chem C 114:21824
Inal S, Schubert M, Sellinger A, Neher D (2010) J Phys Chem Lett 1:982
Kern J, Schwab S, Deibel C, Dyakonov V (2011) Phys Status Solidi—Rapid Res Lett 5:364
Vandewal K, Albrecht S, Hoke ET, Graham KR, Widmer J, Douglas JD, Schubert M, Mateker WR, Bloking JT, Burkhard GF, Sellinger A, Fréchet JMJ, Amassian A, Riede MK, McGehee MD, Neher D, Salleo A (2014) Nat Mater 13:63
Hodgkiss JM, Campbell AR, Marsh RA, Rao A, Albert-Seifried S, Friend RH (2010) Phys Rev Lett 104:177701
Howard IA, Mauer R, Meister M, Laquai F (2010) J Am Chem Soc 132:14866
Howard IA, Laquai F (2010) Macromol Chem Phys 211:2063
Langevin PMP (1903) Ann Chim Phys 28:433
Pivrikas A, Sariciftci NS, Juška G, Österbacka R (2007) Prog Photovolt Res Appl 15:677
Mingebach M, Walter S, Dyakonov V, Deibel C (2012) Appl Phys Lett 100:193302
Koster LJA, Mihailetchi VD, Ramaker R, Blom PWM (2005) Appl Phys Lett 86:123509
Deibel C, Wagenpfahl A, Dyakonov V (2009) Phys Rev B 80:075203
Koster LJA, Mihailetchi VD, Blom PWM (2006) Appl Phys Lett 88:052104
Heiber MC, Baumbach C, Dyakonov V, Deibel C (2015) Phys Rev Lett 114:136602
Shuttle CG, O’Regan B, Ballantyne AM, Nelson J, Bradley DDC, de Mello J, Durrant JR (2008) Appl Phys Lett 92:093311
Foertig A, Baumann A, Rauh D, Dyakonov V, Deibel C (2009) Appl Phys Lett 95:052104
Gorenflot J, Heiber MC, Baumann A, Lorrmann J, Gunz M, Kämpgen A, Dyakonov V, Deibel C (2014) J Appl Phys 115:144502
Montanari I, Nogueira AF, Nelson J, Durrant JR, Winder C, Loi MA, Sariciftci NS, Brabec C (2002) Appl Phys Lett 81:3001
Nelson J (2003) Phys Rev B 67:155209
Deibel C, Rauh D, Foertig A (2013) Appl Phys Lett 103:043307
Deledalle F, Kirchartz T, Vezie MS, Campoy-Quiles M, Shakya Tuladhar P, Nelson J, Durrant JR (2015) Phys Rev X 5:011032
Baranovski S (2006) Charge transport in disordered solids with applications in electronics, 1 edn. Wiley, 498 p. ISBN: 978-0-470-09504-1
He Z, Zhong C, Su S, Xu M, Wu H, Cao Y (2012) Nat Photonics 6:593
Liang Y, Xu Z, Xia J, Tsai S-T, Wu Y, Li G, Ray C, Yu L (2010) Adv Mater 22, E135
Chen W, Xu T, He F, Wang W, Wang C, Strzalka J, Liu Y, Wen J, Miller DJ, Chen J, Hong K, Yu L, Darling SB (2011) Nano Lett 11:3707
Yan H, Collins BA, Gann E, Wang C, Ade H, McNeill CR (2012) ACS Nano 6:677
Lou SJ, Szarko JM, Xu T, Yu L, Marks TJ, Chen LX (2011) J Am Chem Soc 133:20661
Mort J, Chen I, Troup A, Morgan M, Knights J, Lujan R (1980) Phys Rev Lett 45:1348
Kniepert J, Schubert M, Blakesley JC, Neher D (2011) J Phys Chem Lett 2:700
Foster S, Deledalle F, Mitani A, Kimura T, Kim K-B, Okachi T, Kirchartz T, Oguma J, Miyake K, Durrant JR, Doi S, Nelson J (2014) Adv Energy Mater 4:1400311
Lange I, Kniepert J, Pingel P, Dumsch I, Allard S, Janietz S, Scherf U, Neher D (2013) J Phys Chem Lett 4:3865
Kniepert J, Lange I, Heidbrink J, Kurpiers J, Brenner TJK, Koster LJA, Neher D (2015) J Phys Chem C 119:8310
Hedley GJ, Ward AJ, Alekseev A, Howells CT, Martins ER, Serrano LA, Cooke G, Ruseckas A, Samuel IDW (2013) Nat Commun 4:2867
Kniepert J, Lange I, van der Kaap NJ, Koster LJA, Neher D (2014) Adv Energy Mater 4:1301401
Peet J, Kim JY, Coates NE, Ma WL, Moses D, Heeger AJ, Bazan GC (2007) Nat Mater 6:497
Peet J, Cho NS, Lee SK, Bazan GC (2008) Macromolecules 41:8655
Jamieson FC, Agostinelli T, Azimi H, Nelson J, Durrant JR (2010) J Phys Chem Lett 1:3306
Albrecht S, Schindler W, Kurpiers J, Kniepert J, Blakesley JC, Dumsch I, Allard S, Fostiropoulos K, Scherf U, Neher D (2012) J Phys Chem Lett 3:640
Albrecht S, Vandewal K, Tumbleston JR, Fischer FSU, Douglas JD, Fréchet JMJ, Ludwigs S, Ade H, Salleo A, Neher D (2014) Adv Mater 26:2533
Albrecht S, Janietz S, Schindler W, Frisch J, Kurpiers J, Kniepert J, Inal S, Pingel P, Fostiropoulos K, Koch N, Neher D (2012) J Am Chem Soc 134:14932
Albrecht S, Tumbleston JR, Janietz S, Dumsch I, Allard S, Scherf U, Ade H, Neher D (2014) J Phys Chem Lett 5:1131
Moet DJD, Lenes M, Morana M, Azimi H, Brabec CJ, Blom PWM (2010) Appl Phys Lett 96:213506
Jarzab D, Cordella F, Gao J, Scharber M, Egelhaaf H-J, Loi MA (2011) Adv Energy Mater 1:604
Hwang I-W, Cho S, Kim JY, Lee K, Coates NE, Moses D, Heeger AJ (2008) J Appl Phys 104:033706
Rao A, Chow PCY, Gélinas S, Schlenker CW, Li C-Z, Yip H-L, Jen AK-Y, Ginger DS, Friend RH (2013) Nature 500:435
Chow PCY, Gélinas S, Rao A, Friend RH (2014) J Am Chem Soc 136:3424
Etzold F, Howard IA, Forler N, Cho DM, Meister M, Mangold H, Shu J, Hansen MR, Müllen K, Laquai F (2012) J Am Chem Soc 134:10569
Etzold F, Howard IA, Forler N, Melnyk A, Andrienko D, Hansen MR, Laquai F (2015) Energy Environ Sci 8:1511
Miller NC, Gysel R, Miller CE, Verploegen E, Beiley Z, Heeney M, McCulloch I, Bao Z, Toney MF, McGehee MD (2011) J Polym Sci Part B Polym Phys 49:499
Miller NC, Cho E, Junk MJN, Gysel R, Risko C, Kim D, Sweetnam S, Miller CE, Richter LJ, Kline RJ, Heeney M, McCulloch I, Amassian A, Acevedo-Feliz D, Knox C, Hansen MR, Dudenko D, Chmelka BF, Toney MF, Brédas J-L, McGehee MD (2012) Adv Mater 24:6071
Zusan A, Vandewal K, Allendorf B, Hansen NH, Pflaum J, Salleo A, Dyakonov V, Deibel C (2014) Adv Energy Mater 4:1400922
Veldman D, Ipek O, Meskers SCJ, Sweelssen J, Koetse MM, Veenstra SC, Kroon JM, van Bavel SS, Loos J, Janssen RAJ (2008) J Am Chem Soc 130:7721
Deibel C, Strobel T, Dyakonov V (2009) Phys Rev Lett 103:036402
Gehrig DW, Howard IA, Sweetnam S, Burke TM, McGehee MD, Laquai F (2015) Macromol Rapid Commun 36:1054
Gehrig DW, Howard IA, Laquai F (2015) J Phys Chem C 119:13509
Zusan A, Gieseking B, Zerson M, Dyakonov V, Magerle R, Deibel C (2015) Sci Rep 5:8286
Collins BA, Li Z, Tumbleston JR, Gann E, McNeill CR, Ade H (2013) Adv Energy Mater 3:65
Fischer FSU, Trefz D, Back J, Kayunkid N, Tornow B, Albrecht S, Yager KG, Singh G, Karim A, Neher D, Brinkmann M, Ludwigs S (2015) Adv Mater 27:1223
Fischer FSU, Kayunkid N, Trefz D, Ludwigs S, Brinkmann M (2015) Macromolecules 48:3974
Scharsich C, Fischer FSU, Wilma K, Hildner R, Ludwigs S, Köhler A (2015) J Polym Sci Part B Polym Phys 53:1416
Poelking C, Daoulas K, Troisi A, Andrienko D (2014) P3HT revisited – from molecular scale to solar cell devices Adv Polym Sci 265:139–180
Poelking C, Andrienko D (2013) Macromolecules 46:8941
Gemünden P, Poelking C, Kremer K, Daoulas K, Andrienko D (2015) Macromol Rapid Commun 36:1047
Andrienko D (2014) Supramolecular materials for opto-electronics. Royal Society of Chemistry, Cambridge
Acknowledgments
This work was partially supported by Deutsche Forschungsgemeinschaft (DFG) under the Priority Program “Elementary Processes of Organic Photovoltaics” (SPP 1355), BMBF grant MESOMERIE (FKZ 13N10723) and MEDOS (FKZ 03EK3503B), and DFG program IRTG 1404. The project has received funding from the NMP-20-2014—“Widening Materials Models” program under grant agreement number 646259 (MOSTOPHOS). F. Laquai thanks the Max Planck Society for funding the Max Planck Research Group.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Laquai, F., Andrienko, D., Deibel, C., Neher, D. (2017). Charge Carrier Generation, Recombination, and Extraction in Polymer–Fullerene Bulk Heterojunction Organic Solar Cells. In: Leo, K. (eds) Elementary Processes in Organic Photovoltaics. Advances in Polymer Science, vol 272. Springer, Cham. https://doi.org/10.1007/978-3-319-28338-8_11
Download citation
DOI: https://doi.org/10.1007/978-3-319-28338-8_11
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28336-4
Online ISBN: 978-3-319-28338-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)