
Abstract
This chapter describes charge generation and recombination dynamics in polymer solar cells. Photovoltaic conversion mechanism in polymer solar cells ranges from femtoseconds to microseconds on a temporal scale. Therefore, time-resolved optoelectronic measurements are powerful tools for studying elementary processes in the photovoltaic conversion mechanism. First, the charge generation dynamics studied by transient absorption spectroscopy is described in order to discuss how polymer crystallinity and energy offsets at the interface have an impact on the charge dissociation in polymer/fullerene blends. In addition, the charge generation dynamics in ternary blends is also described. Next, the bimolecular recombination of free charge carriers studied by transient absorption spectroscopy and transient photovoltage and photocurrent measurements is discussed. Finally, challenging issues to be solved in polymer solar cells are discussed for further improvements in photovoltaic performance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


6.1 Introduction
Polymer solar cells have attracted increasing interest as a next-generation solar cell because they have excellent advantages of colorful, flexible, lightweight, and especially high-throughput and low-cost production by printing techniques. Thus, they have been intensively studied over the world for a long time. As a result, the power conversion efficiency (PCE) has been steadily improved year by year in the last two decades. In 1995, the PCE was less than 1% [1, 2]. Currently, the PCE is more than 17%, which was reported for tandem solar cells in 2018 [3] and is more than 18%, which was reported for single-junction cells in 2020 [4]. This efficiency is even higher than that of amorphous silicon solar cells.
These improvements are partly due to remarkable developments of various photovoltaic materials such as soluble fullerene derivatives like a [6,6]-phenyl-C61-butyric acid methyl ester (PCBM) [5], crystalline conjugated polymers like a regioregular poly(3-hexylthiphene) (RR-P3HT) [6], low-bandgap conjugated polymers like a poly[2,6-(4,4-bis-(2-ethylhexyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (PCPDTBT) [7], and non-fullerene acceptors like a 6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydro-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene (ITIC) [8]. In other words, such diversity of organic materials is one of the great advantages over inorganic solar cells. For fullerene-based polymer solar cells, as described in Sect. 6.4, charge dissociation is likely to be more efficient in polymer solar cells based on highly crystalline conjugated polymers.
In addition, new approaches recently proposed have effectively boosted the PCE of polymer solar cells furthermore. Ternary blend solar cells are promising approaches to expand the light-harvesting wavelength range from visible to near-IR region. For example, we have demonstrated that the short-circuit current density (JSC) can be effectively enhanced by addition of near-IR dye molecules into RR-P3HT/PCBM binary blend solar cells [9, 10]. The key to success of this approach is location of dye molecules in ternary blend films: Dye molecules should be located at a RR-P3HT/PCBM interface in blend films so that they can contribute to photocurrent generation effectively [11, 12]. Interestingly, most dye molecules are spontaneously located at the RR-P3HT/PCBM interface in ternary blend films, which is revealed by transient absorption spectroscopy [13], as described in Sect. 6.4.4.
In order to improve photovoltaic performance rationally, it is of particular importance to gain in-depth understanding of photovoltaic conversion mechanism in polymer solar cells. Thus, we need to directly observe dynamics of transient species such as exciton and charge carriers generated in polymer solar cells. In polymer solar cells, as described in Sect. 6.2, the exciton and charge carrier dynamics ranges from femtoseconds up to microseconds on a temporal scale [14,15,16]. Thus, time-resolved measurement techniques are essential for understanding photovoltaic conversion mechanism. In particular, transient absorption spectroscopy is one of the most powerful methods for studying the exciton and charge carrier dynamics directly. This chapter describes recent progress in understanding of photovoltaic conversion mechanism in polymer solar cells studied by time-resolved optoelectronic measurements.
6.2 Photovoltaic Conversion in Polymer Solar Cells
This section describes elementary processes of photovoltaic conversion in polymer solar cells. Figure 6.1 shows a schematic illustration of the photovoltaic conversion process in bilayered polymer solar cells based on hole-transporting (donor) and electron-transporting (acceptor) materials. Under the solar illumination, a photon is absorbed by either donor or acceptor material. The photon absorption efficiency (ηA) is defined as the ratio of the number of absorbed photons to the number of incident photons at a wavelength. Here, it is considered that a photon is absorbed by the donor material. Roughly speaking, the photon absorption excites an electron in the highest occupied molecular orbital (HOMO) of the donor material into the lowest unoccupied molecular orbital (LUMO), resulting in a hole in the HOMO and an electron in the LUMO. This electron–hole pair is tightly bound because of large Coulomb interaction in organic materials. This bound electron–hole pair is called a singlet exciton. This state is also called singlet excited state, which consists of several different electron configurations more strictly. In polymer solar cells, excitons cannot dissociate into free charge carriers at room temperature because the exciton binding energy is much larger than thermal energy kBT. This is partly because relative permittivity εr is as small as 3–4 in most organic materials, and hence a critical distance rC at which the thermal energy of a charge carrier is equal to the Coulomb attractive potential energy e2/4πεrε0rC would be as long as 14–19 nm at room temperature. Here, kB is the Boltzmann constant, T is the absolute temperature, e is the elementary charge, and ε0 is the vacuum permittivity. In contrast, excitons easily dissociate into free carriers in inorganic solar cells like silicon even at room temperature. This is partly due to large εr of inorganic semiconductors. For example, εr of crystalline silicon is as large as 11.9 and hence rC would be as short as 5 nm at room temperature. This is the most critical difference in photovoltaic conversion mechanism between organic and inorganic solar cells. As such the exciton cannot dissociate into free carriers in polymer solar cells but can migrate randomly in polymer domains. Thus, some excitons can arrive at a donor/acceptor interface and the others cannot before deactivating to the ground state radiatively or non-radiatively. The exciton diffusion efficiency (ηED) is defined as the ratio of the number of excitons arriving at the interface to the number of excitons generated. In most conjugated polymers, the exciton diffusion length is typically as short as ~10 nm. Thus, ηED is sensitive to phase-separated structures in blend films: It would be small in largely phase-separated blend structures and large in finely mixed blend structures. For excitons arriving at the interface, an electron in the LUMO of the donor can transfer to a more stable LUMO of the neighboring acceptor because of the energy offset ΔELL between the two LUMO levels as shown in Fig. 6.2a. Upon photoexcitation of the acceptor instead of the donor, the same is equally true of hole transfer from the HOMO of the acceptor to that of the neighboring donor because of the energy offset ΔEHH between the two HOMO levels as shown in Fig. 6.2b. It has been believed that these energy offsets should be large enough to break the Coulomb binding energy of electron–hole pairs in excitons. As a result, the charge transfer (CT) state is formed at the donor/acceptor interface. The charge transfer efficiency (ηCT) is defined as the ratio of the number of CT states formed at the interface to the number of excitons arriving at the interface. Some CT states recombine to the ground state radiatively or non-radiatively. This monomolecular recombination is called geminate recombination. The other CT states dissociate into free charge carriers. The charge dissociation efficiency (ηCD) is defined as the ratio of the number of CT states dissociating to free charge carriers to the number of CT states generated. After the charge dissociation, hole carriers can migrate in donor domains and electron carriers can migrate in acceptor domains. During the charge migration, some electron carriers encounter hole carriers at the donor/acceptor interface and recombine to the ground state radiatively or non-radiatively. This bimolecular recombination is called non-geminate recombination. Finally, electron and hole carriers are collected to cathode and anode electrodes, respectively. The charge collection efficiency (ηCC) is defined as the ratio of the number of charge carriers collected to the number of charge carriers generated. In summary, photocurrent generation is a final product of a series of elementary processes, which include (1) photon absorption to generate exciton, (2) exciton diffusion into a donor/acceptor interface, (3) charge transfer at the interface, (4) charge dissociation into free charge carriers, and (5) charge collection into each electrode. The charge dissociation is in competition with (4)’ geminate (monomolecular) recombination to the ground state. The charge collection is in competition with (5)’ non-geminate (bimolecular) recombination to the ground state, as shown in Fig. 6.1. Thus, the external quantum efficiency (EQE) of polymer solar cells is equal to the product of the efficiency of each elementary process: EQE = ηA × ηED × ηCT × ηCD × ηCC. The internal quantum efficiency (IQE) is given by IQE = ηED × ηCT × ηCD × ηCC.

Elementary processes of photovoltaic conversion in polymer solar cells with a bilayered device structure based on donor (light yellow part) and acceptor (light blue part) materials. (1) exciton generation upon photon absorption by the donor material, (2) exciton diffusion into a donor/acceptor interface, (3) charge transfer from the exciton arriving at the interface, (4) charge dissociation into free charge carriers, (4)’ geminate (monomolecular) recombination of an electron–hole pair at the charge transfer state, (5) charge collection of free charge carriers to each electrode, and (5)’ non-geminate (bimolecular) recombination of free charge carriers

Energy diagram of the HOMO and LUMO levels of donor (light yellow) and acceptor (light blue) materials at the heterojunction: a Upon photoexcitation of the donor material, an electron in the HOMO of the donor is excited to the LUMO level, and then transfers to more stable LUMO of the acceptor because of the LUMO energy offset ΔELL. b Upon photoexcitation of the acceptor material, an electron in the HOMO of the acceptor is excited to the LUMO level, and then the remaining hole in the HOMO transfers to more stable HOMO of the donor because of the HOMO energy offset ΔEHH
These elementary processes in photovoltaic conversion range over a wide temporal scale as described below. The photon absorption is an electronic transition and hence occurs on a time scale of femtoseconds (~10−15 s). The exciton diffusion typically occurs on a timescale of picoseconds to sub-nanoseconds (10−12–10−10 s) though it depends on phase-separated structures. The charge transfer has been reported to be promptly completed in the order of tens of femtoseconds (~10−14 s). The geminate recombination typically ranges from picoseconds to nanoseconds (10−12–10−9 s). The charge collection time typically ranges from sub-microseconds to microseconds (10−7–10−6 s), though it depends on the charge mobility, the thickness of the active layer, and the electric field applied to the active layer. In other words, the elementary processes in photovoltaic conversion range from femtoseconds to microseconds (over nine orders of magnitude on a temporal scale). This chapter focuses on these rapid photovoltaic conversion events studied by time-resolved optoelectronic measurements such as transient absorption spectroscopy and discusses the recent findings obtained from the kinetics analyses.
6.3 Optoelectronic Measurements
6.3.1 Transient Absorption Spectroscopy
Transient absorption spectroscopy is one of the most powerful tools for studying the dynamics of short-lived transient species such as excitons and charge carriers. Historically, Norrish and Porter developed the flash photolysis technique around 1950 before the invention of lasers [17, 18]. Their pioneering work enabled us to directly observe such short-lived transient products and therefore opened a new research field. As a result, they were awarded the Nobel Prize in Chemistry 1967 with Eigen for their studies of extremely fast chemical reactions, effected by disturbing the equilibrium by means of very short pulses of energy [19, 20]. Owing to the emergence of short-pulsed lasers, this technique has been further improved in temporal resolution. Currently, we can directly observe ultrafast phenomena with a resolution of femtoseconds. In 1999, Zeweil was awarded the Nobel Prize in Chemistry for his studies of the transition states of chemical reactions using femtosecond spectroscopy [21]. These two typical measurement methods are described below.
For ultrafast measurements up to tens of nanoseconds, pump and probe techniques are typically employed. In this technique, ultrashort laser pulses are split into two pulsed light sources: one is employed as a pump light source for exciting the sample and the other is employed as a probe light source for measuring transmittance of the sample. Figure 6.3 shows a pump and probe femtosecond transient absorption measurement system we employed. As shown in the figure, the probe light passes through the additional path length in the optical delay line and hence arrives at the sample following the pump light. The delay time can be tuned by controlling the optical delay length. For example, when the optical decay length is set at 30 cm, the probe light arrives at the sample 1 ns after the pump light excites the sample. In this case, we can measure the change in optical density ΔOD of the sample 1 ns after the laser excitation. By changing the optical delay length systematically, we can trace the time evolution of transient absorption spectra.

©[2016] IEEE, Reprinted, with permission, from [16]
Block diagram of a pump and probe femtosecond transient absorption measurement systems. This system consists of an ultrashort pulsed laser (regenerative amplified Ti:sapphire laser), wavelength converters, and a pump and probe spectrometer: SHG second harmonic generator, OPA optical parametric amplifier, C chopper, ODL optical delay line, WLG white light generator, S sample, and D detector. As a detector, a CMOS linear sensor is employed for the visible wavelength range and an InGaAs linear diode array sensor is employed for near-IR wavelength range.
For measurements after nanoseconds, laser photolysis techniques are widely employed. In this technique, a short-pulsed laser is employed as a pump light source for exciting the sample and a stable white light such as tungsten or xenon lamp is employed as a probe light source for measuring transmittance of the sample. Figure 6.4 shows the highly sensitive microsecond transient absorption measurement system we employed. At such a later time stage, most transient species have already decayed. A highly sensitive transient absorption measurement system is required to detect small signals. As shown in the figure, a tungsten lamp with a stabilized power source is employed as a probe light to reduce fluctuation of the probe light intensity. To reduce scattering light, stray light, and emission from the sample, two monochromators and appropriate optical filters are placed before and after the sample in the probe light line. The probe light passing through the sample is detected with a Si or InGaAs p-i-n photodiode depending on the measuring wavelength. The signal from the photodiode is pre-amplified, sent to electronic band-pass filters to improve the signal-to-noise ratio, and amplified again with the main amplification system. The amplified signal is collected with a digital oscilloscope, which is synchronized with a trigger signal of the laser pulse from another photodiode. By using this system, the detectable ΔOD is as small as 10−5–10−6 depending on the measuring time domain and the accumulation times.

©[2016] IEEE, Reprinted, with permission, from [16]
Block diagram of a highly sensitive microsecond transient absorption measurement system. This system consists of a pump light source (nanosecond dye laser pumped by N2 laser), a probe light source (power-stabilized tungsten lamp), and a detection system: MC monochromator, S sample, PD p-i-n photodiode to detect a part of the pump laser light as a trigger signal, and PC computer. As a detector for the probe light, a Si p-i-n photodiode is employed for the visible wavelength range and an InGaAs p-i-n photodiode is employed for near-IR wavelength range.
6.3.2 Transient Photovoltage and Photocurrent Measurements
Transient photovoltage (TPV) and transient photocurrent (TPC) measurements have been widely employed to study charge carriers dynamics and charge carrier density in solar cells under operation conditions. For polymer solar cells, Durrant and his co-workers were the first to apply TPV and TPC techniques in order to discuss charge carriers dynamics in polymer solar cells as reported previously [22]. Details have been described in their papers [23, 24]. Here, it will be briefly explained how charge carrier lifetime and density are evaluated by TPV and TPC measurements.
In TPV measurements, polymer solar cells are operated at the open-circuit under white light (simulated solar) illumination and then are excited by a small perturbation pulsed laser light. As a result, photovoltage is slightly increased from the open-circuit voltage (VOC) because of the slight increase in the charge density Δn due to minority excess charges generated by the small perturbation excitation. The minority charges recombine with the majority charges steadily generated by the white light illumination. In other words, the decay of the minority charges follows pseudo-first-order kinetics and hence is given by an exponential function. Thus, the lifetime of the minority charges τΔn is evaluated as a time constant of the exponential decay. Similarly, the lifetimes τΔn for different VOCs can be obtained by changing illumination intensities. The following relationship between τΔn and VOC is given by
where τΔn and ν are obtained from an intersection and a slope in Logarithmic plots of τΔn against VOC, respectively.
In TPC measurements, polymer solar cells are operated at the short-circuit under white light (simulated solar) illumination and then are excited by the same small perturbation pulsed laser light as TPV. Under the short-circuit condition, transient photocurrent decay is ascribed to charge collection to electrodes. As described previously, the total charge carrier density n can be evaluated from TPC analyses [23, 24]. The following relationship between n and VOC is given by
where n0 and m are obtained from an intersection and a slope in Logarithmic plots of n against VOC, respectively. Finally, the lifetime of the total charge carriers τn is given by τn = (1 + λ)τΔn = δτΔn where λ = m/ν and δ = 1 + λ. Here, δ is the empirical reaction order that describes how the recombination rate scales with all charge carrier density in a device including both free and trapped charges [25]. By using Eqs. (6.1) and (6.2), τn can be expressed as a function of the carrier density n.
6.4 Charge Generation Dynamics
This section describes the charge generation dynamics in various polymer/PCBM blend films studied by transient absorption spectroscopy [26,27,28,29,30,31,32,33,34,35,36,37,38]. Figure 6.5 shows the photovoltaic materials studied in this chapter. As shown in the figure, conjugated polymers have similar structures but different crystallinity. For example, RRa-P3HT exhibits the same backbone as RR-P3HT but exhibits totally different crystallinity from RR-P3HT: RRa-P3HT is a typical amorphous conjugated polymer but RR-P3HT is a typical crystalline conjugated polymer. PCPDTBT and PSBTBT have almost the same backbone except for one atom: carbon for PCPDTBT and sulfur for PSBTBT. PSBTBT exhibits higher crystallinity than PCPDTBT. PNTz4T and PNOz4T also have almost the same backbone except for one atom: sulfur for PNTz4T and oxygen for PNOz4T. PNOz4T exhibits higher crystallinity than PNTz4T although both polymers are highly crystalline polymers. Acceptor materials are fixed to fullerene derivatives of PCBM or PC71BM, which have almost the same LUMO level of –3.7 eV. Note that there are wide variations in ΔELL for these polymer/fullerene blends. It will be discussed later how polymer crystallinity impacts on the charge generation efficiency in Sect. 6.6.

Chemical structures of photovoltaic materials studied in this chapter: a RRa-P3HT, b RR-P3HT, c PCPDTBT (X = C), PSBTBT (X = Si), d NP-7, e PNTz4T (X = S), PNOz4T (X = O), f PCBM, g PC71BM, and h SiPc6
6.4.1 Amorphous Polymer Solar Cells
Figure 6.6 shows transient absorption spectra of RRa-P3HT pristine and RRa-P3HT/PCBM blend films excited at 400 nm [27]. For the RRa-P3HT pristine film, a large absorption band was observed at around 1000 nm immediately after the laser excitation. This band is ascribed to polymer singlet excitons, which decays with a time constant of 270 ps under a low excitation intensity, as reported previously [26]. The negative absorption signal is observed at around 500 nm, which is consistent with RRa-P3HT absorption in the ground state and hence is ascribed to ground-state photobleaching (GSB) signals. For the RRa-P3HT/PCBM blend films, on the other hand, the singlet exciton band is reduced to almost half of that observed for the pristine film even at 0 ps. Instead, new absorption bands are observed at around 800 and 1600 nm, which are ascribed to polymer polarons. In other words, polymer polarons are promptly generated even at 0 ps in the blend films. Subsequently, the broadband of singlet excitons completely disappears at 0.2 ps. Instead, a small and sharp absorption band is clearly observed at around 1050 nm, which is ascribed to PCBM radical anion. As described in [27], the singlet exciton signals decay with a time constant of ~0.2 ps while the polaron signals increase with a time constant of ~0.2 ps. This agreement suggests that polymer polarons are rapidly generated from singlet excitons with a time constant of ~0.2 ps, which are more than two orders of magnitude faster than the lifetime of singlet excitons. In other words, polymer polarons are assumed to be generated from singlet excitons with a 100% efficiency from a kinetic point of view. Such a highly efficient polaron generation is consistent with almost 100% quenching efficiency of photoluminescence of RRa-P3HT/PCBM blend films. This is probably because PCBM molecules are likely to be distributed relatively homogeneously in an amorphous polymer matrix like RRa-P3HT. In the case of 50 wt% PCBM, PCBM would be located at intervals of a few nanometers and therefore can quench efficiently singlet excitons before the exciton diffusion. This would be true of other amorphous polymer blends. Indeed, such a prompt polaron generation in a few picoseconds is observed for other amorphous polymer/PCBM blend films like PCPDTBT/PCBM [29] and N-P7/PCBM blends [30].

Adapted with the permission from [27]. Copyright 2010 American Chemical Society
Transient absorption spectra of RRa-P3HT pristine (broken line) and RRa-P3HT/PCBM blend (solid lines) films measured at 0 (black), 0.2 (red), 1, (blue), 100 (light green), and 3000 (orange) ps after the laser excitation. The excitation wavelength was 400 nm (~30 μJ cm−2). The ΔOD was corrected for variations in the absorption at the excitation wavelength.
As shown in Fig. 6.7, the PCBM anion band decays exponentially with a time constant of 813 ps (~70%) and a constant fraction (~30%). On the other hand, the GSB signals recover with the same time constant. This agreement suggests that PCBM anions recombine with polymer polarons to the ground state. In addition, this rise and decay time constant is independent of the excitation intensities ranging from 6 to 120 μJ cm−2, suggesting that the recombination is geminate recombination of CT pairs formed at the RRa-P3HT/PCBM interface. Thus, 70% of polymer polarons and PCBM anions form interfacial CT states and then geminately recombine to the ground state, and 30% of them dissociate into free charge carriers. In other words, the charge dissociation efficiency ηCD is as low as ~30% in RRa-P3HT/PCBM blend films. Similarly, the geminate recombination is one of the major losses in PCPDTBT/PCBM and NP-7/PCBM blend films. As a result, the charge dissociation efficiency is 0.5 and 0.7 for PCPDTBT/PCBM blends without and with additive [29], respectively, and 0.65 for NP-7/PCBM blends [30]. In summary, the exciton diffusion efficiency ηED is almost 100% but the charge dissociation efficiency ηCD is limited by the geminate recombination in amorphous polymer solar cells as will be discussed in Sect. 6.6.

Reprinted with the permission from [27]. Copyright 2010 American Chemical Society
Transient absorption decays of RRa-P3HT/PCBM blend films excited at 400 nm with a fluence of ~12 μJ cm−2, which were measured at a 1030 nm and b 480 nm. The decays were fitted with an exponential function and a constant: ΔOD(t) = AD exp(−t/τD) + B. The broken lines represent the best-fitting curves.
6.4.2 Crystalline Polymer Solar Cells
Figure 6.8 shows the transient absorption spectra of RR-P3HT pristine and RR-P3HT/PCBM blend films excited at 400 or 600 nm [27]. For the RR-P3HT pristine film, a large absorption band was observed at around 1200 nm immediately after the laser excitation. This band is ascribed to polymer singlet excitons, which decay with a time constant of 330 ps under a low excitation intensity, as reported previously [26]. The negative signals at around 500 nm are ascribed to the GSB. For the RR-P3HT/PCBM blend films, as shown in the figure, the singlet exciton band is reduced to ~30–60% of that observed for the pristine film at 0 ps depending on the weight fraction of PCBM: It is more quenched with increasing PCBM fraction. Instead, new absorption bands are observed over 700 and 1100 nm, which are ascribed to polymer polarons. In other words, polymer polarons are promptly generated even at 0 ps in the blend films. This is similar to that observed for RRa-P3HT/PCBM blends. Thus, it would be ascribed to the prompt charge generation in amorphous P3HT domains mixed with PCBM. Subsequently, the broadband of singlet excitons decays and completely disappears at 100 ps. As described in [27], the singlet exciton signals decay with different time constants, which shorten with increasing PCBM fraction and extend after thermal annealing at 140 °C for 30 min. In other words, the decay time is dependent upon P3HT domain size in the blend films. Indeed, the decay time of singlet excitons is dependent upon polymer crystallinity: It was reported to be as short as 0.8 ps for less crystalline PSBTBT and PCBM and as long as 100 ps for highly crystalline PNOz4T and PC71BM blend films. As such, it is ascribed to the delayed charge generation following the exciton diffusion in large crystalline domains. In summary, there are two pathways for the charge generation in crystalline polymer solar cells: one is prompt charge generation at amorphous polymer domains mixed with PCBM or at an interface of crystalline polymer domains and PCBM where no exciton diffusion is needed to generate charge carriers and the other is delayed charge generation following exciton diffusion in large crystalline polymer domains.

Reprinted with the permission from [27]. Copyright 2010 American Chemical Society
Transient absorption spectra of RR-P3HT pristine (broken line in the panel a) and RR-P3HT/PCBM blend (solid lines) films measured at 0 (black), 1 (red), 10, (blue), 100 (light green), and 3000 (orange) ps after the laser excitation. The excitation wavelengths were a–d 400 nm (~15 μJ cm−2) and e 600 nm (~10 μJ cm−2). The PCBM concentrations were a 5, b 20, and c–e 50 wt%. The spectra in the panels d and e were measured after thermal annealing at 140 °C for 30 min. The ΔOD was corrected for variations in the absorption at the excitation wavelength.
At a later time stage after several tens of picoseconds, as shown in Fig. 6.8, singlet excitons disappear and instead polymer polarons are observed from 700 to 1100 nm. As shown in Fig. 6.9, the decay dynamics of these bands is dependent upon the excitation intensity. For the transient absorption signals at 700 and 1000 nm, the absorption decays slower with decreasing excitation intensities, suggesting bimolecular recombination. Interestingly, no decay is observed under lower excitation intensities, suggesting that no geminate recombination is involved: The dissociation efficiency ηCD is almost 100%. For the transient absorption signals at 850 nm, on the other hand, the absorption decays slower with decreasing excitation intensities and finally decays with the same time constant under lower excitation intensities, suggesting geminate recombination. This is similar to the decay observed for RRa-P3HT/PCBM blend films. Therefore, as described in [27], the transient absorption signals are ascribed to delocalized polarons in crystalline P3HT domains for 700 nm, localized polarons loosely bound to PCBM at the interface in disordered amorphous P3HT domains for 850 nm, and localized polarons in disordered P3HT domains for 1000 nm.

Reprinted with the permission from [27]. Copyright 2010 American Chemical Society
Transient absorption signals measured at a 700, b 850, and c 1000 nm of RR-P3HT/PCBM (50 wt%) blend films excited at 400 nm with a fluence of 6, 12, 24, 48, 72, and 120 μJ cm−2 from bottom to top in each panel.
Figure 6.10 shows the time evolution of the GSB measured at 400 and 610 nm. As mentioned above, transient species observed are not singlet excitons but polymer polarons at this later time stage. Thus, these negative signals are also ascribed to the GSB due to polymer polarons. In the steady-state absorption, RR-P3HT crystalline films exhibit a broadband due to amorphous phase at shorter wavelengths and vibronic bands due to the crystalline phase at around 600 nm. Thus, the GSB bands at 400 and 610 nm are ascribed to polymer polarons in amorphous and crystalline domains, respectively. As shown in Fig. 6.10, the GSB band at 400 nm recovers with a time constant of 250 ps while the GSB band at 610 nm is negatively increased with the same time constant (250 ps). This agreement suggests hole transfer from amorphous to more stable crystalline domains. This would improve the charge dissociation efficiency in amorphous domains. If no hole transfer is involved, polymer polarons generated in amorphous domains would suffer from the geminate recombination as is observed for RRa-P3HT/PCBM blend films. This hole transfer is probably due to cascade energy structures at the P3HT/PCBM interface, which consists of three phases: P3HT crystalline domains, P3HT amorphous domains mixed with PCBM, and PCBM aggregated domains. A similar hole transfer has been reported for crystalline polymer/PCBM blends with three-phase structures [39]. In other words, cascade energy structures at the interface would be beneficial for the efficient charge dissociation in crystalline polymer solar cells like RR-P3HT/PCBM. The charge dissociation efficiency in other crystalline polymer solar cells will be discussed in Sect. 6.6.

Adapted with the permission from [27]. Copyright 2010 American Chemical Society
Transient absorption signals of RR-P3HT/PCBM (50 wt%) blend films excited at 400 nm (~12 μJ cm−2), measured at a 480 and b 610 nm, which were fitted by ΔOD(t) = AR[1 − exp(−t/τR)] + B and ΔOD(t) = AD exp(−t/τD) + B, respectively. The broken lines represent the best-fitting curves
6.4.3 Polymer Solar Cells with Small Photon Energy Loss
Figure 6.11 shows spectroscopic data of PNTz4T and PNOz4T pristine and PNTz4T/PC71BM and PNOz4T/PC71BM blend films [36]. As shown in Fig. 6.11a and d, the photoluminescence is significantly quenched to a few % for PNTz4T/PC71BM but modestly quenched to ~35% for PNOz4T/PC71BM blend films. These findings suggest that the exciton diffusion efficiency ηED is almost 100% for PNTz4T/PC71BM but as low as ~65% for PNOz4T/PC71BM blend films. As shown in Fig. 6.11 b and e, transient absorption spectra of PNTz4T/PC71BM and PNOz4T/PC71BM blend films exhibit slightly complicated spectral changes with time but can be resolved to two components of polymer singlet excitons and polarons as described in Ref. 36. Figure 6.11c and f show the time evolution of polymer singlet excitons, polarons, and the total of them. As shown in the figure, polymer polarons are generated at 0 ps more efficiently in PNTz4T/PC71BM than in PNOz4T/PC71BM blend films, suggesting that PNTz4T/PC71BM blends have a larger fraction of mixed regions and or small domains. Subsequently, polymer singlet excitons decay and polymer polarons rise with the same time constant: 5.5 ps for PNTz4T/PC71BM and 100 ps for PNOz4T/PC71BM blend films. These time constants are consistent with the photoluminescence quenching efficiency mentioned above because the singlet exciton lifetime is as long as a few hundreds of picoseconds. For PNTz4T/PC71BM, the polaron signals decay after 100 ps, which is ascribed to the geminate recombination because the decay is independent of the excitation intensities. For PNOz4T/PC71BM, on the other hand, the polaron signals do not decay at all on this time scale, suggesting efficient charge dissociation. In summary, polymer polarons are generated from singlet excitons with >95% but ~30% of them recombine geminately in PNTz4T/PC71BM blend films. In other words, the exciton diffusion efficiency ηED is as high as >95% but the charge dissociation efficient ηCD is limited to ~70% in PNTz4T/PC71BM blend films. On the other hand, ~35% of singlet excitons radiatively deactivate to the ground state and hence only ~65% of them are converted into polymer polarons with a dissociation efficiency of ~100% in PNOz4T/PC71BM blend films. In other words, the exciton diffusion efficiency ηED is as low as ~65% but the charge dissociation efficient ηCD is as high as ~100%. The difference in ηED is due to the difference in crystalline domain size, which is revealed by TEM and AFM images of the blend films. As described in [36], the LUMO–LUMO energy offset is as large as 0.31 eV for PNTz4T/PC71BM and as small as 0.12 eV for PNOz4T/PC71BM, which are estimated by CV measurements. The small energy offset in PNOz4T/PC71BM is consistent with an effective bandgap energy evaluated from temperature dependence of VOC and electroluminescence being similar to the photoluminescence of PNOz4T pristine films. These results show that the charge dissociation efficiency ηCD is as high as ~100% even for such a small energy offset of ~0.1 eV. As a result, photon energy loss, which is defined by the difference between the optical bandgap Eg and the open-circuit voltage energy eVOC, is as small as 0.56 eV for PNOz4T/PC71BM solar cells. As described in Sect. 6.2, it has been believed that the energy offset should be large enough to break the Coulomb binding energy of electron–hole pairs in excitons, which has been considered to be more than 0.3 eV for efficient devices [40], although there was a study reporting that the minimum driving force is as small as 0.1 eV for the charge transfer from singlet excitons to the CT state [41]. In other words, our finding clearly contradicts the conventional photovoltaic conversion mechanism. After this study, similar small photon energy losses have been reported for many systems [42]. Thus, further studies are needed for revising the conventional photovoltaic conversion mechanism.

Reprinted by permission from Macmillan Publishers Ltd [36]. Copyright 2015
Spectroscopic data of PNTz4T and PNOz4T pristine and PNTz4T/PC71BM and PNOz4T/PC71BM blend films. Photoluminescence spectra of a PNTz4T and d PNOz4T pristine films (red dots) and a PNTz4T/PC71BM and d PNOz4T/PC71BM blend films (black dots). Transient absorption spectra of b PNTz4T and e) PNOz4T pristine films at 0 ps (black dots) and b) PNTz4T/PC71BM and e PNOz4T/PC71BM blend films measured at 0 (blue), 1 (light blue), 10 (green), 100 (light brown), 1000 (orange), and 3000 ps (red dots). Time evolution of singlet excitons (red circles), polarons (blue squares), and the total of singlet excitons and polarons (green triangles) in c PNTz4T/PC71BM and f PNOz4T/PC71BM blend films.
6.4.4 Ternary Blend Polymer Solar Cells
Figure 6.12 shows transient absorption spectra of RR-P3HT/PCBM binary and RR-P3HT/PCBM/SiPc6 ternary blend films upon the laser excitation of the P3HT absorption at 400 nm. As described before, the large absorption band at around 1250 nm is ascribed to polymer singlet excitons. The negative signals at around 500 nm are ascribed to the GSB of P3HT. A broad depression is observed from 650 to 850 nm, which is ascribed to stimulated emission from P3HT. At 100 ps, broad absorption bands are observed from 650 to 1000 nm, which is ascribed to polymer polarons. For the RR-P3HT/PCBM binary blends, polymer singlet excitons decay with a time constant of 25 ps (85%) and 330 ps (15%). Polymer polarons are generated with the same time constant of 25 ps. Thus, this is ascribed to the charge generation following exciton diffusion. The longer and minor time constant is ascribed to the lifetime of polymer singlet excitons. For the RR-P3HT/PCBM/SiPc6 ternary blends, on the other hand, polymer singlet excitons decay with a time constant of 4 ps (50%) and the same time constants observed for the binary blends. Polymer polarons are generated with a time constant of 6 ps. This disagreement in the two time constants suggests that polymer polarons are not generated directly from polymer singlet excitons. There should be an intermediate state. As shown in Fig. 6.12c, a small but sharp depression is observed at around 680 nm only for the RR-P3HT/PCBM/SiPc6 ternary blends. This depression band is ascribed to the GSB of SiPc6 dye molecules because the wavelength is the same as that of the dye absorption. As described in [13], the depression fraction increases with a time constant of 4 ps, which is in good agreement with the lifetime of polymer singlet excitons observed for the ternary blends. Note that SiPc6 has negligible absorption at 400 nm and hence cannot be generated directly by the laser excitation at 400 nm. Therefore, the rapid decay of polymer singlet excitons (4 ps) is ascribed to an efficient energy transfer from polymer singlet excitons to SiPc6 dye molecules. This is consistent with an effective spectral overlap between P3HT emission and SiPc6 absorption bands. In summary, polymer singlet excitons migrate to a donor/acceptor interface with a time constant of 25 ps followed by charge generation in RR-P3HT/PCBM binary blend films upon the photoexcitation of the P3HT absorption band at 400 nm. On the other hand, polymer singlet excitons are efficiently transferred to SiPc6 dye molecules by long-range energy transfer with a time constant of 4 ps, and then polymer polarons are generated from the dye singlet excitons with a time constant of 6 ps, which includes the energy transfer time.

Adapted with the permission from [13]. Copyright 2010 American Chemical Society
Transient absorption spectra of a RR-P3HT/PCBM and b RR-P3HT/PCBM/SiPc6 blend films measured at 0 (black), 1 (red), 10, (blue), and 100 (light green) ps after the laser excitation. The excitation wavelength and intensity were 400 nm and 6 μJ cm−2, respectively. c Transient absorption spectra of RR-P3HT/PCBM (black line) and RR-P3HT/PCBM/SiPc6 (red line) blend films measured at 10 ps
Figure 6.13 shows transient absorption spectra of RR-P3HT/SiPc6 binary, PCBM/SiPc6 binary, and RR-P3HT/PCBM/SiPc6 ternary blend films upon the laser excitation of the SiPc6 absorption at 680 nm. The dye concentration was fixed to 3.4 wt%. In other words, SiPc6 molecules are isolated in RR-P3HT/SiPc6 or PCBM/SiPc6 binary blend films. As shown in Fig. 6.13a, a flat absorption band is observed first, which is ascribed to SiPc6 singlet excitons because of the selective excitation at 680 nm. Subsequently, a broad absorption and a small but sharp absorption bands are observed at around 850–1050 nm and at 940 nm, respectively. As described in [13], the broad and sharp absorption bands are ascribed to polymer polarons and dye anions, respectively. These two bands increase with a time constant of 2 ps and then decay with a time constant of 2 ns. In summary, polymer polarons are generated with a time constant of 2 ps in RR-P3HT/SiPc6 binary blends. As shown in Fig. 6.13b, a large absorption and a small and broad absorption are observed at 855 and 1030 nm, respectively. As described in [13], the large and small absorption bands are ascribed to SiPc6 anion and PCBM anion, respectively. These two bands are observed even at 0 ps and then decay with time constants of 35 ps (40%) and 2 ns (60%). In summary, polymer polarons are promptly generated within a laser pulse width of 100 fs in PCBM/SiPc6 binary blends. In other words, charge generation dynamics is dependent on the location of dye molecules in blend films.

Adapted with the permission from [13]. Copyright 2010 American Chemical Society
Transient absorption spectra of a RR-P3HT/SiPc6, b PCBM/SiPc6 binary, and c RR-P3HT/PCBM/SiPc6 ternary blend films measured at 0 (black), 1 (red), 10, (blue), and 200 (light green) ps after the laser excitation. The excitation wavelength and intensity were 680 nm and 7 μJ cm−2, respectively.
For RR-P3HT/PCBM/SiPc6 ternary blend films excited at the dye absorption, as shown in Fig. 6.13c, a flat absorption band due to SiPc6 singlet excitons is observed first immediately after the laser excitation, and then a broad absorption band with a small and sharp absorption peak, which is due to P3HT polarons and SiPc anions, respectively, increase with a time constant of 2 ps. This charge generation dynamics is the same as that observed for RR-P3HT/SiPc6 binary blends, suggesting that SiPc6 dye molecules are basically located in P3HT domains. If SiPc6 dye molecules were isolated in P3HT domains as is the case for RR-P3HT/SiPc6 binary blends, polymer polarons and SiPc6 anions recombine geminately with a time constant of 2 ns. However, this is not the case. As shown in Fig. 6.13c, the absorption at 940 nm does not decay at all. Instead, the absorption peak is shifted from 940 to 1000 nm with time. The absorption at 1000 nm is ascribed to P3HT polarons and PCBM anions observed for RR-P3HT/PCBM blends as described in Sect. 6.4.2. In other words, this peak shift is indicative of charge shift (electron transfer) from SiPc6 anions to PCBM.
The time evolution of these transient species can be obtained from the spectral simulation. As shown in Fig. 6.14, SiPc6 anions rapidly decay with a time constant of 50 ps, which is much faster than the geminate recombination of polymer polarons and dye anions observed for RR-P3HT/SiPc6 blend films. Instead, PCBM anions increase with the same time constant, suggesting a charge shift from SiPc6 to PCBM. This finding suggests that SiPc6 dye molecules contact with PCBM as well as P3HT in RR-P3HT/PCBM/SiPc6 ternary blend films. In other words, SiPc6 dye molecules are located at the P3HT/PCBM interface in the ternary blend films. As described in [13], only a small part of polymer polarons decay during the charge shift, suggesting that a majority of dye molecules are located at the interface and a small minor part of them are isolated in P3HT domains. Interestingly, no decay of polymer polarons is observed at all during the charge shift for RR-P3HT/PCBM/SiPc6 ternary blend films fabricated by solvent annealing. In this case, almost all the dye molecules are spontaneously located at the P3HT/PCBM interface in the ternary blend films. As discussed in [11], this is partly because dye molecules are expelled from polymer crystalline or PCBM aggregated domains to disordered mixed domains, and partly because dye molecules have an intermediate surface energy between RR-P3HT and PCBM. In other words, dye locations can be controlled by careful design of dye molecules as demonstrated in [43].

Adapted with the permission from [13]. Copyright 2010 American Chemical Society
The fraction of SiPc6 anions (red circles), PCBM anions (blue circles), and P3HT polarons (open circles) in RR-P3HT/PCBM/SiPc6 ternary blend films after the laser excitation of SiPc6 at 680 nm with a fluence of 7 μJ cm−2.
6.5 Charge Recombination Dynamics
This section describes the bimolecular recombination dynamics of dissociated charge carriers on a timescale of microseconds in polymer/fullerene blend films. Here, our attention is focused on bimolecular recombination dynamics in RR-P3HT/PCBM blend films. Figure 6.15 shows photovoltaic materials studied in this chapter [38, 44,45,46]. It will be discussed later how bimolecular recombination is reduced in these polymer/fullerene blend films in Sect. 6.6.

Chemical structures of photovoltaic materials studied in this chapter: a RR-P3HT, b DT-PDPP2T-TT, c PSBTBT, d PNTz4T (X = Y = H), PNTz4TF2 (X = H, Y = F), PNTz4TF4 (X = Y = F), e PCBM, and f PC71BM
6.5.1 Bimolecular Recombination Dynamics
Figure 6.16 shows the transient absorption spectra of RR-P3HT/PCBM blend films excited at 400 nm measured on a time scale of microseconds [44]. On this time stage, singlet excitons completely disappear but triplet excitons may be observed. However, this is not the case because these bands are not quenched at all under an oxygen atmosphere. As shown in the figure, two large absorption bands are observed at around 700 and 1000 nm. The absorption band at 700 nm is larger than that at 1000 nm at an early time stage of <10 μs but smaller than that at 1000 nm at a later time stage of >20 μs. In other words, the absorption band at 700 nm decays faster than that at 1000 nm. This finding suggests that there are at least two different polymer polarons in the RR-P3HT/PCBM blend film. As described in Sect. 6.4.2, the band at 700 nm is ascribed to delocalized polarons in crystalline domains and the band at 1000 nm is ascribed to localized polarons in disordered domains.

Adapted with the permission from [44]. Copyright 2010 American Chemical Society
Transient absorption spectra of RR-P3HT/PCBM blend films excited at 400 nm with a fluence of 30 μJ cm−2. The spectra were measured at 0.5 (black), 1 (red), 2 (blue), 5, (light blue), 10 (light green), 20 (green), and 100 μs (brown).
As shown in Fig. 6.17, these two bands decay faster at an early time stage under higher excitation intensities, suggesting bimolecular recombination. The decay dynamics can be well-fitted with an empirical power-law equation

Reprinted with the permission from [44]. Copyright 2010 American Chemical Society
Charge density decays of RR-P3HT/PCBM blend films excited at 400 nm with a fluence of 0.8 (blue lines), 1.8 (red lines), and 4.7 μJ cm−2 (black lines). The spectra were measured at a 700 nm and b 1000 nm. The broken lines represent fitting curves with an empirical equation: n(t) = n(0)/(1 + at)α.
where n(t) is the charge carrier density at time t, n(0) is the initial carrier density at t = 0, and a and α are kinetic parameters. This power-law decay is characteristic of trap-limited charge recombination in a medium with an energetic disorder [47, 48]. For diffusion-limited bimolecular recombination, the rate equation is given by
By substituting Eq. (6.4) into Eq. (6.5), the time-dependent bimolecular recombination rate γ(t) is obtained as
By substituting Eq. (6.4) into Eq. (6.6) again, the carrier density-dependent bimolecular recombination rate γ(n) is obtained as
From the fitting curves in Fig. 6.17, the exponent α is evaluated to be unity for the delocalized polaron observed at 700 nm and ~0.5 for the localized polaron observed at 1000 nm. As shown in Fig. 6.18, γ(t) and γ(n) are constant for the delocalized polaron. This finding suggests that the decay kinetics is ascribed to bimolecular recombination of free carriers. In other words, delocalized polarons can diffuse freely as free charge carriers. On the other hand, γ(t) and γ(n) obey a power-law with a slope of ~0.5 and ~1, respectively, for the localized polaron. This finding suggests that the decay kinetics is ascribed to trap-limited bimolecular recombination. In other words, localized polarons are trapped in disordered domains with an energetic disorder. At an early time stage, an effective trap depth is shallower because most trap sites are filled with more charge carriers, and hence, the recombination rate is faster. At a later time stage, an effective trap depth is deeper because trap sites are partly filled with less charge carriers, and hence, the recombination rate is slower. These assignments are consistent with the temperature dependence of the decay kinetics as described in [44]. The activation energy for delocalized polarons is as small as ~0.078 eV, which is independent of the carrier density. On the other hand, the activation energy for localized polarons decreases exponentially from 0.178 to 0.097 eV with increasing carrier density.

Adapted with the permission from ref. [44]. Copyright 2010 American Chemical Society
From Eq. (6.7), the carrier lifetime τn is given by
By comparing Eq. (6.8) with Eq. (6.3) for TPV/TPC measurements, the following relationship is obtained.
Thus, the empirical equation Eq. (6.4) can be expressed by using kinetic parameters for TPV/TPC measurements.
Durrant and his co-workers demonstrated that the decay dynamics observed by transient absorption spectroscopy is consistent with that observed by TPV/TPC measurements [23]. At a charge density of ~1017 cm−3, which corresponds to 1 sun illumination condition, both charge carriers exhibit a similar bimolecular recombination rate of ~10−12 cm3 s−1 at an early time stage of <1 μs as shown in Fig. 6.18. This is because localized polarons also contribute to charge transport owing to trap filling. More interestingly, the recombination rate is two orders of magnitude smaller than the Langevin recombination rate (γL ≈ 10−10 cm3 s−1). Here, γL is estimated by γL = eμh/εrε0 with a slower charge mobility [49] of μh ≈ 10−4 cm2 V−1 s−1 [50]. Such a slow recombination rate would be the key for highly efficient charge collection in RR-P3HT/PCBM solar cells as described in the next section.
6.5.2 Charge Carrier Lifetime
For highly efficient charge collection, the charge carrier lifetime should be longer than the charge collection time. The charge carrier lifetime can be estimated by transient absorption spectroscopy as mentioned above. The carrier density has been reported to be ~1017 cm−3 in RR-P3HT/PCBM blend films under 1 sun illumination. Thus, the carrier lifetime τn is estimated to be τn = 14 μs from Eq. (6.8) with kinetic parameters a and α, which are obtained by fitting analysis for the decay curve in Fig. 6.17. On the other hand, the charge collection time τCC is roughly estimated by Eq. (6.12)
where L is the thickness of the active layer, μ is the charge mobility, and V is the applied voltage, which is typically assumed to be VOC at the short circuit. For RR-P3HT/PCBM blend films, τCC is estimated to be τCC = 0.8 μs for a thickness of 100 nm. In other words, the charge carrier lifetime τn is two orders of magnitude longer than the charge collection time τCC. This is consistent with highly efficient charge collection in RR-P3HT/PCBM blends. This would be true for RR-P3HT/PCBM solar cells under device operation conditions. We note that the charge carrier dynamics observed by transient absorption with pulsed laser excitations might be different from the charge carrier dynamics under steady-state solar illumination. TPV/TPC measurements are a useful method for estimating charge carrier lifetime under device operation conditions.
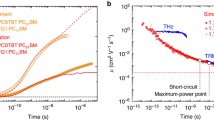
Figure 6.19 shows TPV and TPC decays of RR-P3HT/PCBM solar cells under 1 sun illumination [45]. Under steady-state illumination, the photovoltage is constant at VOC. Upon a small perturbation laser pulse excitation, minority excess charges are generated, resulting in a small increase in the photovoltage ΔV0 at a decay time of t = 0. As shown in Fig. 6.19a, TPV signals decay exponentially because the decay of the minority excess charges follows pseudo-first-order kinetics as mentioned in Sect. 6.3. From the slope in the figure, the lifetime of minority excess charges τΔn can be estimated to be τΔn = 4.8 μs. With increasing VOC, τΔn decreases exponentially as shown in Eq. (6.1). From the slope in Logarithmic plots of τΔn against VOC, ν is estimated to be ν = 1.5. As shown in Fig. 6.19b, TPC signals decay with a time constant of 0.5 μs, which is almost one order of magnitude faster than TPV signals due to bimolecular recombination in the blend film. The decay time constant is rather consistent with the charge collection time τCC = 0.8 μs. Thus, the rapid TPC decay is ascribed to the charge collection to the electrode because of the shirt-circuit condition. Consequently, the amount of the minority excess charges Δq can be evaluated from the integral of the TPC decay signals over time. As described in [23, 24], the differential capacitance dC is defined as follows:

Adapted with the permission from ref. [45]. Copyright 2016 The Society of Photopolymer Science and Technology
a TPV and b TPC decays of RR-P3HT/PCBM solar cells under 1 sun illumination.
where ΔV0(VOC) is the initial increment in the TPV signals observed under the same condition as the TPV measurement. Consequently, the charge carrier density n is given by Eq. (6.14)
where A and L are the area and the thickness of the active layer, respectively. With increasing VOC, n increases exponentially as shown in Eq. (6.2). Under 1 sun condition, n and m are evaluated to be n = 5.6 × 1016 cm−3 and m = 3.6, respectively. Thus, λ is estimated to be λ = m/ν = 2.4. Finally, the charge carrier lifetime τn is estimated to be τn = (1 + λ)τΔn = 16 μs, which is in good agreement with τn = 14 μs estimated from the transient absorption spectroscopy described above. On the other hand, bimolecular recombination rate γ is estimated to be γ = 1/τnn = 1.1 × 1012 cm3 s−1, which is also consistent with that evaluated from the transient absorption spectroscopy. Thus, the reduction factor ζ is estimated to be ζ = 8.7 × 10−3 for RR-P3HT/PCBM solar cells. This good agreement indicates that charge carriers generated by pulsed laser light are in thermal equilibrium in RR-P3HT/PCBM blends on a timescale of microseconds.
6.6 Challenging Issues and Concluding Remarks
This section describes challenging issues to be solved for further improvements in photovoltaic performance of polymer solar cells. As described in Sect. 6.4, polymer morphology has impact on photovoltaic conversion efficiencies. Table 6.1. summarizes ηED and ηCD in polymer/fullerene blends with different polymer crystallinities. As shown in the table, ηED is almost unity for amorphous polymer/fullerene blends. This is probably because fullerene small molecules are likely to be distributed relatively homogeneously in amorphous matrices. For crystalline polymers, ηED decreases with increasing polymer crystallinity. This is because a part of excitons cannot arrive at a donor/acceptor interface because of large crystalline domains. On the other hand, ηCD increases with increasing polymer crystallinity. One possible explanation is that Coulomb binding energy would be reduced for polymer polarons delocalized in crystalline domains. Assuming an effective dielectric constant of 3.5, the Coulomb binding energy is as large as 0.41 eV for an electron–hole pair at a separation distance of 1 nm, but decreases to 0.14 eV at a separation distance of 3 nm. This is still much larger than the thermal energy at room temperature (kBT = 26 meV). Durrant and his co-workers have suggested that the effective Coulomb binding energy would be comparable to the entropy term by considering the number of states for the charge separation [56, 57]. Gregg has demonstrated that such an entropy effect would be larger for three-dimensional aggregates such as fullerene molecules [58]. As shown in Fig. 6.20, the entropy term –TΔS would be dominant at longer separation distances. As a result, the Gibbs free energy ΔG exhibits a maximum at a separation distance of around a few nanometers. In other words, electron–hole pairs can dissociate into free charge carriers at the threshold distance. Deibel and his co-workers have demonstrated by kinetic Monte Carlo simulation that polaron pairs can dissociate efficiently by considering delocalized charge carriers along conjugated polymer chain segments [59]. On the other hand, it has been suggested that fullerene aggregates also have impact on efficient charge dissociation [60, 61]. As such, charge delocalization in polymer crystalline domains or fullerene aggregates would be one of the key factors for highly efficient charge dissociation in polymer solar cells. As described in Sect. 6.4.3, recent studies have shown that singlet excitons can efficiently dissociate into free charge carriers even for an energy offset as small as <0.1 eV. We should understand why such efficient charge dissociation is possible for small energy offset and also the minimum energy offset for efficient charge generation in polymer solar cells.

The Coulomb potential energy ΔH (blue line), the entropy contribution –TΔS (red line), and the Gibbs free energy ΔG (black line) for an electron–hole pair at a separation distance r. The dielectric constant is assumed to be 3.5. The entropy term is calculated for 3D aggregates with a lattice constant of 1 nm
Another challenging issue to be solved is to understand the origin of long-lived charge carriers due to reduced bimolecular recombination observed for several highly efficient polymer solar cells. Table 6.2 summarizes kinetic parameters for bimolecular recombination and device parameters in several polymer/fullerene solar cells. As shown in the table, the reduction factor is seemingly not closely related to FF of the devices. This is partly because FF depends not only on the reduction factor but also other photovoltaic and diode parameters. Among them, the most effectively suppressed reduction factors are found in RR-P3HT/PCBM, DT-PDPP2T-TT/PC71BM, and PNTz4T/PC71BM solar cells. For these solar cells, FF remains high even for thick devices. For the other devices, FF monotonically decreases with increasing thickness. As such, the reduced bimolecular recombination would be one of the key factors for realizing high FF even for thick devices. These polymers are all highly crystalline polymers and hence would be likely to form cascade energy structures at an interface due to three phases: polymer crystalline domains, polymer/fullerene mixed domains, and fullerene aggregated domains. Such cascaded energy structures might suppress bimolecular recombination effectively. Indeed, recent studies have shown that the lifetime of charge carriers is longer in ternary devices with cascade energy structures than in binary devices [12, 64]. We need to clarify further how bimolecular recombination is suppressed in polymer solar cells.
Change history
09 March 2021
In the original version of the book, the following chapters corrections have been incorporated:
The erratum chapters and book have been updated with the changes.
References
Yu, G., Gao, J., Hummelen, J.C., Wudl, F., Heeger, A.J.: Polymer photovoltaic cells: Enhanced efficiencies via a network of internal donor–acceptor heterojunctions. Science 270, 1789–1791 (1995)
Hall, J.J.M., Walsh, C.A., Greenham, N.C., Marseglia, E.A., Friend, R.H., Moratti, S.C., Holmes, A.B.: Efficient photodiodes from interpenetrating polymer networks. Nature 376, 498–500 (1995)
Meng, L., Zhang, Y. Wan, X., Li, C., Zhang, X., Wang, Y., Ke, X., Xiao, Z., Ding, L., Xia, R., Yip, H.L., Cao, Y., Chen, Y.: Organic and solution-processed tandem solar cells with 17.3% efficiency. Science 361, 1094–1098 (2018)
Liu, Q., Jiang, Y., Jin, K., Qin, J., Xu, J., Li, W., Xiong, J., Liu, J., Xiao, Z., Sun, K., Yang, S., Zhang, X., Ding, L.: 18% Efficiency organic solar cells. Bull. Sci. 65, 272–275 (2020)
Shaheen, S.E., Brabec, C.J., Sariciftci, N.S., Padinger, F., Fromherz, T., Hummelen, J.C.: 2.5% Efficient organic plastic solar cells. Appl. Phys. Lett. 78, 841–843 (2001)
Padinger, F., Rittberger, R.S., Sariciftci, N.S.: Effects of postproduction treatment on plastic solar cells. Adv. Funct. Mater. 13, 85–88 (2003)
Peet, J., Kim, J.Y., Coates, N.E., Ma, W.L., Moses, D., Heeger, A.J., Bazan, G.C.: Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat. Mater. 6, 497–500 (2007)
Lin, Y., Wang, J., Zhang, Z.-G., Bai, H., Li, Y., Zhu, D., Zhan, X.: An electron acceptor challenging fullerenes for efficient polymer solar cells. Adv. Mater. 27, 1170–1174 (2015)
Honda, S., Nogami, T., Ohkita, H., Benten, H., Ito, S.: Improvement of the light-harvesting efficiency in polymer/fullerene bulk heterojunction solar cells by interfacial dye modification. ACS Appl. Mater. Interf. 1, 804–810 (2009)
Honda, S., Ohkita, H., Benten, H., Ito, S.: Multi-colored dye sensitization of polymer/fullerene bulk heterojunction solar cells. Chem. Commun. 46, 6596–6598 (2010)
Honda, S., Ohkita, H., Benten, H., Ito, S.: Selective dye loading at the heterojunction in polymer/fullerene solar cells. Adv. Energy Mater. 1, 588–598 (2011)
Xu, H., Ohkita, H., Tamai, Y., Benten, H., Ito, S.: Interface engineering for ternary blend polymer solar cells with a heterostructured near-IR dye. Adv. Mater. 27, 5868–5874 (2015)
Honda, S., Yokoya, S., Ohkita, H., Benten, H., Ito, S.: Light-harvesting mechanism in polymer/fullerene/dye ternary blend studied by transient absorption spectroscopy. J. Phys. Chem. C 115, 11306–11317 (2011)
Ohkita, H., Ito, S.: Transient absorption spectroscopy of polymer-based thin-film solar cells. Polymer 52, 4397–4417 (2011)
Ohkita, H., Ito, S.: Exciton and charge dynamics in polymer solar cells studied by transient absorption spectroscopy. In: Choy, W.C.H. (ed.) Organic Solar Cells, pp. 103–137. Springer-Verlag, London (2013)
Ohkita, H., Tamai, Y., Benten, H., Ito, S.: Transient absorption spectroscopy for polymer solar cells. IEEE J. Sel. Top Quantum Electron. 22, 100–111 (2016)
Norrish, R.G.W., Porter, G.: Chemical reactions produced by very high light intensities. Nature 164, 658 (1949)
Porter, G.: Flash photolysis and spectroscopy. A new method for the study of free radical reactions. Proc. R. Soc. London, Ser. A 200, 284–300 (1950)
Norrish, R.G.W.: Some fast reactions in gases studied by flash photolysis and kinetic spectroscopy. The official website of the Nobel Prize. https://www.nobelprize.org/prizes/chemistry/1967/norrish/lecture/ (1967). Accessed 8 November 2019
Porter, G.: Flash photolysis and some of its applications. The official website of the Nobel Prize. https://www.nobelprize.org/prizes/chemistry/1967/porter/lecture/ (1967). Accessed 8 November 2019
Zewail, A.: Femtochemistry: Atomic-scale dynamics of the chemical bond using ultrafast lasers. The official website of the Nobel Prize. https://www.nobelprize.org/prizes/chemistry/1999/zewail/lecture/ (1999). Accessed 8 November 2019
Shuttle, C.G., O’Regan, B., Ballantyne, A.M., Nelson, J., Bradley, D.D.C., de Mello, J., Durrant, J.R.: Experimental determination of the rate law for charge carrier decay in a polythiophene:fullerene solar cell. Appl. Phys. Lett. 92, 093311 (2008)
Maurano, A., Shuttle, C.G., Hamilton, R., Ballantyne, A.M., Nelson, J., Zhang, W., Heeney, M., Durrant, J.R.: Transient optoelectronics analysis of charge carrier losses in a selenophene/fullerene blend solar cell. J. Phys. Chem. C 115, 5947–5957 (2011)
Credgington, D., Durrant, J.R.: Insights from transient optoelectronic analyses on the open-circuit voltage of organic solar cells. J. Phys. Chem. Lett. 3, 1465–1478 (2012)
Kirchartz, T., Nelson, J.: Meaning of reaction orders in polymer: fullerene solar cells. Phys. Rev. B 86, 165201 (2012)
Guo, J., Ohkita, H., Benten, H., Ito, S.: Near-IR femtosecond transient absorption spectroscopy of ultrafast polarons and triplet formation in polythiophene films with different regioregularities. J. Am. Chem. Soc. 131, 16860–16880 (2009)
Guo, J., Ohkita, H., Benten, H., Ito, S.: Charge generation and recombination dynamics in poly(3-hexylthiophene)/fullerene blend films with different regioregularites and morphologies. J. Am. Chem. Soc. 132, 6154–6164 (2010)
Ohkita, H., Kosaka, J., Guo, J., Benten, H., Ito, S.: Charge generation dynamics in polymer/polymer solar cells studied by transient absorption spectroscopy. J. Photon. Energy 1, 011118 (2011)
Yamamoto, S., Ohkita, H., Benten, H., Ito, S.: Role of interfacial charge transfer state in charge generation and recombination in low-bandgap polymer solar cells. J. Phys. Chem. C 116, 14804–14810 (2012)
Yamamoto, S., Ohkita, H., Benten, H., Ito, S., Yamamoto, S., Kitazawa, D., Tsukamoto, J.: Efficient charge generation and collection in amorphous polymer-based solar cells. J. Phys. Chem. C 117, 10277–10284 (2013)
Ide, M., Koizumi, Y., Saeki, A., Izumiya, Y., Ohkita, H., Ito, S., Seki, S.: Near-infrared absorbing thienoisoindigo-based copolymers for organic photovoltaics. J. Phys. Chem. C 117, 26859–26870 (2013)
Ie, Y., Karakawa, M., Jinnai, S., Yoshida, H., Saeki, A., Seki, S., Yamamoto, S., Ohkita, H., Aso, Y.: Electron-donor function of methanofullerenes in donor–acceptor bulk heterojunction systems. Chem. Commun. 50, 4123–4125 (2014)
Yamamoto, S., Yasuda, H., Ohkita, H., Benten, H., Ito, S., Miyanishi, S., Tajima, K., Hashimoto, K.: Charge generation and recombination in fullerene-attached poly(3-hexylthiophene)-based diblock copolymer films. J. Phys. Chem. C 118, 10584–10589 (2014)
Tamai, Y., Tsuda, K., Ohkita, H., Benten, H., Ito, S.: Charge-carrier generation in organic solar cells using crystalline donor polymers. Phys. Chem. Chem. Phys. 16, 20338–20346 (2014)
Mori, D., Benten, H., Ohkita, H., Ito, S.: Morphology-limited free carrier generation in donor/acceptor polymer blend solar cells composed of poly(3-hexylthiophene) and fluorene-based copolymer. Adv. Energy Mater. 5, 1500304 (2015)
Kawashima, K., Tamai, Y., Ohkita, H., Osaka, I., Takimiya, K.: High-efficiency polymer solar cells with small photon energy loss. Nat. Commun. 6, 10085 (2015)
Tamai, Y., Ohkita, H., Namatame, M., Marumoto, K., Shimomura, S., Yamanari, T., Ito, S.: Light-induced degradation mechanism in poly(3-hexylthiophene)/fullerene blend solar cells. Adv. Energy Mater. 6, 1600171 (2016)
Kawashima, K., Fukuhara, T., Suda, Y., Suzuki, Y., Koganezawa, T., Yoshida, H., Ohkita, H., Osaka, I., Takimiya, K.: Implication of fluorine atom on electronic properties, ordering structures, and photovoltaic performance in naphthobisthiadiazole-based semiconducting polymers. J. Am. Chem. Soc. 138, 10265–10275 (2016)
Gallaher, J.K., Prasad, S.K.K., Uddin, M.A., Kim, T., Kim, J.Y., Woo, H.Y., Hodgkiss, J.M.: Spectroscopically tracking charge separation in polymer: fullerene blends with a three-phase morphology. Energy Environ. Sci. 8, 2713–2724 (2015)
Dennler, G., Scharber, M.C., Brabec, C.J.: Polymer-fullerene bulk-heterojunction solar cells. Adv. Mater. 21, 1323–1338 (2009)
Veldman, D., Meskers, S.C.J., Janssen, R.A.: The energy of charge-transfer states in electron donor–acceptor blends: Insight into the energy losses in organic solar cells. Adv. Funct. Mater. 19, 1939–1948 (2009)
Wang, Y., Qian, D., Cui, Y., Zhang, H., Hou, J., Vandewal, K., Kirchartz, T., Gao, F.: Optical gaps of organic solar cells as a reference for comparing voltage losses. Adv. Energy Mater. 8, 1801352 (2018)
Xu, H., Wada, T., Ohkita, H., Benten, H., Ito, S.: Molecular design of near-IR dyes with different surface energy for selective loading to the heterojunction in blend films. Sci. Rep. 5, 9321 (2015)
Guo, J., Ohkita, H., Yokoya, S., Benten, H., Ito, S.: Bimodal polarons and hole transport in poly(3-hexylthiophene):fullerene blend films. J. Am. Chem. Soc. 132, 9631–9637 (2010)
Fukuhara, T., Osaka, M., Tamai, Y., Ohkita, H., Benten, H., Ito, S.: Reduced bimolecular recombination in polymer solar cells. J. Photopolym. Sci. Technol. 29, 575–580 (2016)
Fukuhara, T., Tamai, Y., Osaka, I., Ohkita, H.: Bimolecular recombination and fill factor in crystalline polymer solar cells. Jpn. J. Appl. Phys. 57, 08RE01 (2018)
Nelson, J.: Diffusion-limited recombination in polymer–fullerene blends and its influence on photocurrent collection. Phys. Rev. B 67, 155209 (2003)
Tachiya, M., Seki, K.: Theory of bulk electron–hole recombination in a medium with energetic disorder. Phys. Rev. B 82, 085201 (2010)
Koster, L.J.A., Mihailetchi, V.D., Blom, P.W.M.: Bimolecular recombination in polymer/fullerene bulk heterojunction solar cells. Appl. Phys. Lett. 88, 052104 (2006)
Pivrikas, A., Juška, G., Mozer, A., Scharber, M., Arlauskas, K., Sariciftici, N.S., Stubb, H., Österbacka, R.: Bimolecular recombination coefficient as a sensitive testing parameter for low-mobility solar-cell materials. Phys. Rev. Lett. 94, 176806 (2005)
Chen, D., Nakahara, A., Wei, D., Nordlund, D., Russell, T.P.: P3HT/PCBM bulk heterojunction organic photovoltaics: correlating efficiency and morphology. Nano Lett. 11, 561–567 (2011)
Salleo, A., Kline, R.J., DeLongchamp, D.M., Chabinyc, M.L.: Microstructural characterization and charge transport in thin films of conjugated polymers. Adv. Mater. 22, 3812–3838 (2010)
Motaung, D.E., Malgas, G.F., Arendse, C.J.: Correlation between the morphology and photo-physical properties of P3HT:fullerene blends. J. Mater. Sci. 45, 3276–3283 (2010)
Chen, H.-Y., Hou, J., Hayden, A.E., Yang, H., Houk, K.N., Yang, Y.: Silicon atom substitution enhances interchain packing in a thiophene-based polymer system. Adv. Mater. 22, 371–375 (2010)
Guilbert, A.A., Frost, J.M., Agostinelli, T., Pires, E., Lilliu, S., Macdonald, J.E., Nelson, J.: Influence of bridging atom and side chains on the structure and crystallinity of cyclopentadithiophene-bennzothiadiazole polymers. Chem. Mater. 26, 1226–1233 (2014)
Ohkita, H., Cook, S., Astuti, Y., Duffy, W., Tierney, S., Zhang, W., Heeney, M., McCulloch, I., Nelson, J., Bradley, D.D.C., Durrant, J.R.: Charge carrier formation in polythiophene/fullerene blend films studied by transient absorption spectroscopy. J. Am. Chem. Soc. 130, 3030–3042 (2008)
Clarke, T.M., Durrant, J.R.: Charge photogeneration in organic solar cells. Chem. Rev. 110, 6736–6767 (2010)
Gregg, B.A.: Entropy of charge separation in organic photovoltaic cells: The benefit of higher dimensionality. J. Phys. Chem. Lett. 2, 3013–3015 (2011)
Deibel, C., Strobel, T., Dyakonov, V.: Origin of the efficient polaron-pair dissociation in polymer-fullerene blends. Phys. Rev. Lett. 103, 036402 (2009)
Savoie, B.M., Rao, A., Bakulin, A.A., Gelinas, S., Movaghar, B., Friend, R.H., Marks, T.J., Ratner, M.A.: Unequal partnership: asymmetric roles of polymeric donor and fullerene acceptor in generating free charge. J. Am. Chem. Soc. 136, 2876–2884 (2014)
Gélinas, S., Rao, A., Kumar, A., Smith, S.L., Chin, A.W., Clark, J., van der Poll, T.S., Bazan, G.C., Friend, R.H.: Ultrafast long-range charge separation in organic semiconductor photovoltaic diodes. Science 343, 512–516 (2014)
Li, G., Shrotriya, V., Huang, J., Yao, Y., Moriarty, T., Emery, K., Yang, Y.: High-efficiency solution processable polymer photovoltaic cells by self-organization of polymer blends. Nat. Mater. 4, 864–868 (2005)
Li, W., Hendriks, K.H., Roelofs, W.S.C., Kim, Y., Wienk, M.M., Janssen, R.A.J.: Efficient small bandgap polymer solar cells with high fill factors for 300 nm thick films. Adv. Mater. 25, 3182–3186 (2013)
Izawa, S., Nakano, K., Suzuki, K., Hashimoto, K., Tajima, K.: Dominant effects of first monolayer energetics at donor/acceptor interfaces on organic photovoltaics. Adv. Mater. 27, 3025–3031 (2015)
Acknowledgements
Most of the studies in this chapter were conducted with Professor Emeritus Ito at Kyoto University and Associate Professor Benten at Nara Institute of Science and Technology. They have been done mainly by Dr Guo, Dr Yamamoto, Dr Honda, and Dr Tamai. They were partly supported by the JST PRESTO program (Photoenergy and Conversion Systems and Materials for the Next-Generation Solar Cells) and the JST ALCA program (Solar Cell and Solar Energy Systems: JPMJAL1404).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ohkita, H. (2021). Charge Carrier Dynamics in Polymer Solar Cells. In: Hiramoto, M., Izawa, S. (eds) Organic Solar Cells. Springer, Singapore. https://doi.org/10.1007/978-981-15-9113-6_6
Download citation
DOI: https://doi.org/10.1007/978-981-15-9113-6_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9112-9
Online ISBN: 978-981-15-9113-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)