
Abstract
The understanding of the mechanisms involved in pituitary ontogenesis, and leading to combined pituitary hormone deficiency, has made huge progress over the last 20 years. Since the launching of GENHYPOPIT, our large international cohort aimed at identifying etiologies of combined pituitary hormone deficiency (CPHD), several new transcription factors and mechanisms have been identified. Unfortunately, the rate of etiological identification remains low: up to now, it makes sense to consider that a patient has only a 10 % chance of having an explanation for his/her CPHD. Pituitary development is indeed a very complex phenomenon, requiring the temporospatial coordination of expression and inhibition of specific and nonspecific transcription factors and pathways. To try to improve our knowledge, a “gold standard” murine model has been chosen: this model, frequently used to extrapolate on potential roles of transcription factors, does not, however, perfectly follow the development of the pituitary in humans. For instance, inactivation of a transcription factor in mice can be lethal and partial inactivation (heterozygous state) asymptomatic, whereas humans with a heterozygous defective anomaly can have a very severe phenotype. This non-exhaustive chapter will thus focus on the main phenotypic traits induced by some anomalies of pituitary transcription factors in humans and also emphasize the similarities and differences between the murine model and humans. We will not detail the etiologies of isolated pituitary deficiency (somatotroph, thyrotroph, corticotroph, or gonadotroph). There are two main ways to describe the factors involved in pituitary development: by their types or by the phenotype (including, or not, extrapituitary anomalies) they induce.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pituitary Deficiency
- Kallmann Syndrome
- Isolate Growth Hormone Deficiency
- Combine Pituitary Hormone Deficiency
- Pituitary Development
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
The Murine Pituitary Development : A Useful Tool to Decode Human Pituitary Development
Human pituitary development is assumed to follow more or less closely the murine pituitary development, and this is why the murine model currently represents the most appropriate model to determine the major temporospatial interactions between signaling pathways and transcription factors leading to a mature endocrine organ [1, 2]. Pituitary development in humans is imperfectly known, and all the steps described in the following lines are based on our knowledge of murine pituitary development.
Anterior and posterior pituitary lobes have two different embryonic origins: the anterior lobe is derived from oral ectoderm, whereas the posterior lobe is derived from neurectoderm. Even if close connections exist between both structures, we will only focus on the development of the anterior lobe and the mature pituitary. No study to date on human pituitary deficiency has identified strong connections and phenotypic associations that include anterior pituitary deficiencies and congenital diabetes insipidus (except for the only reported aryl hydrocarbon receptor nuclear translocator (ARNT2) mutation, as described later).
Briefly, anterior pituitary ontogenesis begins early during brain neurogenesis, around embryonic day (e) 7.5 in the mouse, corresponding to the first visualization of the pituitary placode [3]. At e9, the placode forms the rudimentary Rathke’s pouch, under the control of signaling molecules issued from the infundibulum (bone morphogenetic protein 4 (Bmp4) and fibroblast growth factor 8 (Fgf8) ). Definitive Rathke’s pouch is observed at e11.5 [4]. Progenitors around the lumen move progressively to the developing pituitary and differentiate under the control of several factors including SRY-box (Sox)2, Sox9, and Isl Lim homeobox (Isl)-1, among others; the majority of these have not been identified as causative factors for pituitary deficiencies, suggesting that they are either crucial (and would lead to early death if abnormal) or that other pathways can be used if they are abnormal [5–7]. This first step leading to terminal differentiation of the pituitary is possible due to a tightly controlled temporospatial gradient of morphogenic factors from different origins, the diencephalon (Bmp4, Fgf8, 10 and 18, Wnt5a), the ectoderm (Isl1, Bmp2, sonic hedgehog (Shh), Wnt 4), the ventral mesoderm (chordin, Bmp2) [8], or the pituitary cells (Table 12.1).
At e11.5, α-subunit is expressed in the rostral tip [9], followed by adrenocorticotropin (ACTH) (e12.5), thyrotropin (TSH)β (e14.5), proopiomelanocortin (Pomc) (e14.5, intermediate lobe), growth hormone (GH) and prolactin (Prl) (e15.5) [10], luteinizing hormone (Lh)β (e16.5), and finally follicle-stimulating hormone (Fsh)β (e17.5). Precise mechanisms leading to this differentiation and the formation of pituitary cell networks remain incompletely understood. Pituitary specific or nonspecific transcription factors are involved in a timely manner during these steps of differentiation, early acting such as LIM homeobox (Lhx)3, Lhx4, paired-like homeodomain transcription factor (Pitx)2, Hesx1 (also known as Rpx), or ARNT2 [11] or late-acting such as prophet of Pit-1 (Prop1) and Pou1f1 (Pit-1). Early acting transcription factors are also involved in the development of other organs (e.g., the eye, inner ear), and their defects lead to extrapituitary anomalies, whereas alterations of late-acting transcription factors usually lead to a pure pituitary phenotype. A summarized scheme of the timing of expression of the transcription factors known to be involved in CPHD is given in Fig. 12.1.

Simplified scheme representing the main transcription factor expression during pituitary development. Note that early transcription factor dysfunction is associated with pituitary and extrapituitary anomalies, whereas late transcription factor (PIT-1, PROP1) dysfunction is associated with pure pituitary phenotype
Early Acting Transcription Factors: The Pituitary Phenotype Is Usually Not Alone
Anomalies of these transcription factors are characterized by a wide range of phenotypes, usually including anterior pituitary hormone deficiencies, extrapituitary abnormalities, and malformations such as pituitary stalk interruption syndrome (PSIS) or midline defects. These complex phenotypes are due to the non-pituitary-specific expression of these transcription factors, which are also involved in the development of the forebrain and related midline structures such as the hypothalamus. To make the description easier, we focused on the phenotypic traits that should guide the clinician to certain transcription factors.
Etiological Possibilities in Patients Carrying Pituitary Deficiency and Midline Anomalies: HESX1, GLI2, FGF8 and FGFR1, PROK2, and PROKR2
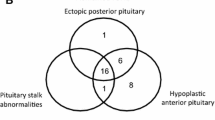
What do we call midline anomalies? It is a large group of diseases from pituitary stalk interruption syndrome to septo-optic dysplasia (SOD) and holoprosencephaly. Pituitary stalk interruption syndrome is defined on brain MRI by the association of an absent or thin pituitary stalk, pituitary hypoplasia, and/or ectopic posterior pituitary [12]. As only 30 % of patients with PSIS have a history of traumatic event, it is likely that a high number of cases are actually due to genetic anomalies. Septo-optic dysplasia is defined by at least two of the following criteria: septum or corpus callosum agenesis, optic nerve hypoplasia, and pituitary deficiencies [13]. Holoprosencephaly is a complex brain malformation, affecting both the brain and face (cyclopia, median or bilateral labial and/or palatal cleft, hypotelorism or a single median incisor in milder cases) due to an abnormal division of the prosencephalon between days 18 and 28. Intellectual disability is frequently associated. Recent studies emphasize the continuum between these different genetic causes leading to phenotypes of variable severity depending on the degree of abnormal development of the anterior brain [14–16]. This likely explains why, for any given pathway or transcription factor, the phenotype can be highly variable from mild to extremely severe. This group mainly includes anomalies of the paired transcription factor HESX1, and few novelties less well known such as GLI2, or pathways previously thought to be only involved in isolated hypogonadotropic hypogonadism. We will see, however, in the next paragraph that other transcription factors, more likely involved in eye development, can also lead to midline anomalies, which makes this classification difficult to perform.
HESX1
Hesx1 is a paired homeodomain transcription factor that has been well characterized over the last 15 years. It is a major actor in pituitary development as its expression and then inhibition are crucial at given time points to allow the formation of a mature Rathke’s pouch. The expression profile of Hesx1 perfectly illustrates the complexity of pituitary development. For instance, decreased expression of Hesx1 at e13 in mice is necessary for Prop1 and secondarily Pou1f1 expression, two late transcription factors necessary for proper differentiation of GH-, TSH-, and Prl-secreting cells [17–20]. Appropriate expression of other early acting transcription factors such as Lhx1, Lhx3, or Six3 (some being involved in human disease) is also necessary for early proper Hesx1 expression [21]: the lack of Hesx1 in mice (homozygous inactivation Hesx1 −/−) indeed leads to a very severe phenotype with corpus callosum aplasia and ectopic posterior pituitary. In humans, HESX1 mutations can lead to a wide range of phenotypes: 16 HESX1 mutations have been reported [17, 22–30], the homozygous anomalies (40 % cases) usually leading to a more severe phenotype [31]. GH deficiency is constant; other pituitary deficiencies are reported in 50 % cases. Optic nerve anomalies are the other major phenotypic sign, observed in 30 % cases. One should not consider, however, that SOD is always due to HESX1 mutations, as only 1 % cases have actually been linked to this genotype [31–34]. Brain MRI usually reveals pituitary hypoplasia (80 % cases) and midline anomalies such as ectopic or non-visible posterior pituitary in 50–60 % cases and corpus callosum agenesis or hypoplasia in 25 % cases.
Sonic Hedgehog and GLI2
Sonic hedgehog (SHH) signaling pathway is involved in the early steps of pituitary development: SHH mutations have been reported in patients with severe forms of isolated holoprosencephaly [35]. SHH targets, GLI transcription factors, have also been involved in CPHD: GLI2 heterozygous mutations have been reported in patients with holoprosencephaly or with pituitary hormone deficits and less severe midline craniofacial anomalies and pituitary hypoplasia, corpus callosum agenesis, or ectopic posterior pituitary on brain MRI; some individuals also have polydactyly.
Pathways Known to Be Involved in Hypogonadotropic Hypogonadism
FGF8 and FGFR1
FGFR1 and FGF8 heterozygous mutation s were first reported in 10 % of Kallmann syndrome and 7 % of normosmic hypogonadism [36]. Pituitary MRI showed normal or hypoplastic anterior pituitary and inconstant ectopic posterior pituitary. Penetrance was incomplete [37, 38]. However, the expression of Fgf8 and Fgfr1 in the ventral diencephalon is necessary for proper Rathke’s pouch formation, temporospatial pattern of pituitary cell lineages, and the development of extrapituitary structures [39]. This explains why other anomalies were then reported such as ear hypoplasia, dental agenesis, cleft palate, and distal limb malformations. Finally, FGFR1 and FGF8 mutations have also been reported in patients with SOD, with about 4 % prevalence [14].
PROK2 and PROKR2
Prokineticin pathway is known to be involved in portal angiogenesis and neuronal development and migration [40]: this suggested its potential involvement in pituitary stalk development. PROK2 and PROKR2 mutations have recently been reported in a cohort of patients with pituitary deficiencies, anterior pituitary hypoplasia or aplasia, and PSIS [15]. Mutations in these genes were also reported thereafter in patients with SOD, and inconstant additional brain abnormalities, such as cerebellar hypoplasia, Dandy-Walker cyst, or focal abnormality of mesial frontal cortex [16].
Etiological Possibilities in Patients Carrying Pituitary Deficiency and Eye Anomalies: OTX2, SOX2, PITX2, ARNT2
Whereas OTX2 mutations seem to play an important role in CPHD, the other factors reported here have been recently described or do not seem to be involved in a large number of patients. This explains why they are usually not screened in patients, except in case of a specific phenotypic sign associated to CPHD.
OTX2
Otx2 is a paired homeodomain transcription factor involved in the early steps of brain development. In mice, Otx2 is expressed from e10.5 to e14.5 in the ventral diencephalon, where it likely interacts with Hesx1, and from e10.5 to e12.5 in Rathke’s pouch. Otx2 is also involved in gonadotropin-releasing hormone (GnRH) neuronal development [41]. In mice, homozygous inactivation of Otx2 (Otx2 −/−) leads to a severe brain phenotype; heterozygous inactivation leads to a wide range of phenotype, with eye anomalies, inconstant holoprosencephaly, and usually pituitary hypoplasia. This phenotype is close to the one observed in humans: 25 heterozygous de novo OTX2 mutations have been reported, including nine in patients with congenital hypopituitarism; the remaining 16 mutations were reported in patients with ophthalmic diseases and no mention of pituitary deficiency. Individuals can either present with isolated GH deficiency or panhypopituitarism and inconstant hypoplastic pituitary, ectopic posterior pituitary, and Chiari syndrome. There is no genotype/phenotype correlation [42–46].
SOX2
Sox2 is an “HMG DNA-binding domain” (similar to SRY gene) transcription factor. At e9.5, Sox2 expression is observed in the brain, the neural tube, the oral endoderm, the sensorial placodes, and the branchial arcs. At e11.5, Sox2 is expressed in Rathke’s pouch and the future hypothalamus. Sox2 is then expressed in the periluminal proliferative zone where it could be involved in the maintenance and function of pituitary progenitors [47]. At adult age, Sox2 is expressed in the periventricular zone of the lateral ventricles and in the dentate gyrus, but its precise role (promoting the differentiation of stem cells in injured pituitary?) is unknown. Homozygous inactivation is lethal in mice; heterozygous inactivation leads to increased perinatal death, epilepsy, and almost complete panhypopituitarism (corticotroph axis is usually functional); in contrast, eye anomalies are inconstant. The phenotype is different in humans: heterozygous de novo SOX2 mutations have been observed in six patients with hypogonadotropic hypogonadism, bilateral microphthalmia, corpus callosum hypoplasia, and inconstant intellectual disability. Pituitary phenotypes included inconstant GH, TSH, or ACTH deficiencies, and pituitary hypoplasia in 80 % cases. Surprisingly, corpus callosum anomaly has been reported in one case [47].
PITX2
PITX2 is not the perfect example of a transcription factor to think about in patients with CPHD. Despite its obvious roles in pituitary development, only three patients have been reported as having GH deficiency and pituitary hypoplasia [48–50]. As shown in mice, it is probably because of compensatory mechanisms, at least in the pituitary, likely due to a close transcription factor, Pitx1. Pitx2 is a paired homeodomain transcription factor expressed in Rathke’s pouch at e10.5 [51, 52] and pituitary anterior and intermediate lobes at e12.5. At adult age, Pitx2 is expressed in thyrotrophs and gonadotrophs [53]. Pitx2 expression is ubiquitous, as it has also been observed in the adult brain, eye, kidney, lungs, testis, and tongue [51, 54]. In humans, PITX2 mutations have been reported in patients with Axenfeld-Rieger syndrome , which is characterized by anomalies in the ocular anterior compartment and systemic malformations (craniofacial dysmorphy, dental, and umbilical anomalies) [55, 56]. PITX2 mutations should thus be screened in patients with this phenotype, keeping in mind that some pituitary deficiencies might be associated. It does not make sense to routinely screen for PITX2 mutations in patients with CPHD.
ARNT2
A recent report described a large consanguineous family with eye abnormalities, congenital hypopituitarism, diabetes insipidus, and renal and central nervous system (CNS) anomalies, related to a defect in the helix-loop-helix transcription factor ARNT2 . ARNT2 is known to be involved in the development of the hypothalamus, other CNS structures, the kidneys, and the eyes. All patients presented with a thin pituitary stalk, hypoplastic anterior pituitary, ectopic or nonvisualized posterior pituitary, hypoplastic frontal and temporal lobes, thin corpus callosum, and delay in brain myelination [11]. Precise roles of ARNT2 during pituitary and extrapituitary structure development are, however, imperfectly determined, and the search for other mutations in patients with CPHD has been negative to date.
Etiological Possibilities in Patients Carrying Pituitary Deficiency and Neurogenesis Anomalies: The LIM Domain Transcription Factors
LHX4 and LHX3 are two close transcription factors belonging to a large family of transcription factors known to be involved in the development of several structures. Several mutations of LHX4 and LHX3 have been reported for the last 10 years in patients with CPHD. In contrast, up to now, no mutation has been identified in patients with a pituitary phenotype in the other LIM domain transcription factors.
LHX4
Lhx4 is involved in the early steps of pituitary ontogenesis. In mice, Lhx4 expression has been reported in Rathke’s pouch at e9.5 and in the anterior part of the pituitary at e12.5. A low expression is still observed at adult age [57, 58]. The phenotype of homozygous inactivation of Lhx4 in mice is lethal due to respiratory distress, whereas heterozygous inactivation is not symptomatic. The main difference with humans is actually the transmission mode of inheritance, as all human LHX4 mutations are in a heterozygous state: 11 sporadic or familial LHX4 mutations have been reported in 17 patients [59], with a wide intra- and interfamilial phenotypic variability in terms of pituitary phenotype (ranging from isolated GH deficiency to complete panhypopituitarism) [60, 61] and brain MRI (pituitary hypoplasia, inconstant ectopic posterior pituitary and sellar hypoplasia, corpus callosum hypoplasia, or Chiari syndrome). Of note, one patient carrying a 1q25 microdeletion (including LHX4 deletion) also presented with a cardiac defect (but it was likely multifactorial).
LHX3
Lhx3 is the perfect example of how extrapolating a human phenotype from a mouse phenotype is complex: while homozygous Lhx3 inactivation in mice is lethal, heterozygous inactivation does not lead to any particular phenotype. In contrast, in humans, all described LHX3 mutations were homozygous, and even if the phenotype was complex, it was never lethal. This discrepancy might be explained by the different weight of compensatory mechanisms performed by Lhx4 in both species, but this remains highly hypothetical [57]. The role of Lhx3 during pituitary development is crucial, as it is necessary for proper expression of several other transcription factors or receptors such as Hesx1 [62], forkhead box (fox)l2, Notch2, splicing factor (SF) 1, T-box (tbx)19 (involved in corticotroph differentiation), GnRH receptor and FSHβ [63–65], and Pou1f1 [66]. In addition to its role during pituitary development, Lhx3 is involved in the development of extrapituitary structures, such as medullar motoneurons [67, 68] (which likely explains neck rotation anomalies in humans with LHX3 mutations) and inner ear [69, 70] (which explains hearing trouble in humans with LHX3 mutations). In humans, 12 homozygous LHX3 mutations have been reported [71–77]. Pituitary phenotype usually includes GH, TSH, and LH/FSH deficiencies, while ACTH deficiency is inconstant (roughly half of the cases). On MRI, pituitary aplasia or hypoplasia is observed in 60 % cases, whereas hyperplasia is observed in 30 % cases. The mechanisms for hyperplasia are unknown but may be close to the ones reported for PROP1 mutations (detailed later in the text). As previously mentioned, extrapituitary phenotype can include abnormal head and neck rotation (70 % cases), vertebral abnormalities (50 % cases), and mild to severe hearing deficits (50 % cases).
Late-Acting Transcription Factors: The Pituitary Phenotype Is Alone
If we only focus on transcription factors with anomalies reported in CPHD, then the list is short: PROP1 and POU1F1 are the only major actors known to be involved in pure pituitary phenotype. It does not mean that the final differentiation of thyrotrophs, for instance, or their function does not require other transcription factors such as GATA2 or maybe ISL1; it only means that no mutation of these genes has been reported so far in humans. Patients with PROP1 or POU1F1 mutations thus present anterior pituitary hormone deficiencies (progressive or not), normal hypothalamo-pituitary morphology at MRI (regardless of the size of the pituitary gland), and no extrapituitary malformations. In such a context, PROP1 mutations remain the most frequently reported genetic defect.
PROP1
Prop1 is a pituitary-specific paired domain transcription factor. In mice, its expression is observed from e10 to e15.5, with a peak around e12 [78]. Prop1 is necessary for proper Pou1f1 expression, leading to somato-lactotroph and thyrotroph cell differentiation [55, 79]. In mice, the phenotype is close to the one reported in humans, except for the lack of ACTH deficiency. The reason why humans might have corticotroph deficiency (seen in about 50 % of cases) remains a mystery, and the large period of appearance (from young age to 40 years old) is another intriguing fact. In humans, at least 25 PROP1 mutations have been reported [80–101]. Homozygous or compound heterozygous PROP1 mutations, transmitted in an autosomal recessive manner, currently represent the most frequently identified etiologies of CPHD [1, 102, 103]. Pituitary phenotype includes GH, TSH, LH/FSH, ACTH, and PRL deficiencies, diagnosed from childhood to adulthood [104]. Pituitary MRI can show transient pituitary hyperplasia and normal or hypoplastic pituitary; pituitary hyperplasia sometimes precedes spontaneous hypoplasia [82, 105–109]. A hypothesis that may account for this phenomenon is that pituitary progenitors might not differentiate in the absence of Prop1, thus accumulating in the intermediate lobe causing hyperplasia, with apoptosis then resulting in final hypoplasia [110].
PIT-1/POU1F 1
Pit-1 was the first pituitary-specific transcription factor identified in Snell mice and then in humans (POU1F1, human ortholog of Pit-1) [111]. Pou1f1 expression is first observed at e13.5 during pituitary development. Pou1f1 is necessary for thyrotroph, somatotroph, and lactotroph differentiation and remains expressed in these cell lineages at adult age. In humans, POU1F1 mutations can be transmitted as an autosomal recessive or dominant trait. Complete TSH and GH deficiencies are usually observed during childhood, whereas gonadotroph and corticotroph axes remain functional. Brain MRI can be normal or show pituitary hypoplasia.
Conclusions and Perspectives
The identification of almost all genes identified to date in CPHD was based on the murine model. Even if it is clear that having a close animal model is crucial, the discrepancy between mice and humans might explain why only 10 % of the etiologies of CPHD have been identified today. This is an issue, as identifying the etiologies of congenital hypopituitarism is of major importance to better diagnose and treat the patients, in particular in the differential diagnosis of a pituitary mass on MRI, or to identify the patients at risk of developing delayed corticotroph deficiency and as a prenatal diagnosis to decrease the risk of early death (undiagnosed corticotroph deficiency, for instance).
Another possibility to explain this poor rate of identification is the limits in the detection techniques that we have: classical Sanger sequencing has, for instance, inherent limits with the impossibility to identify large deletions or insertions or intronic alterations leading to splicing anomalies. The development of new techniques in the recent years should dramatically improve the rate of identification of etiologies of congenital hypopituitarism: array comparative genomic hybridization (aCGH) has been created for identifying segmental genomic copy number variations (gain or loss) such as structural rearrangements (deletions, duplications, insertions, translocations) or complex chromosomal aneuploidies; it can be designed in a whole genome approach, where the array targets are equally spaced with coverage of 100 to 1000 kb. Another promising approach is whole-exome sequencing, which is based on the assumption that 85 % of mutations are located in coding regions of the genome. This technique should be of great interest in highly penetrant Mendelian diseases. However, reporting new variants in a single patient does not mean pathogenicity and requires confirmation by a similar finding in other persons presenting with similar phenotypes. Moreover, confirmatory steps by bioinformatics analysis after a usually large dataset of results can be highly challenging.
To summarize, in one sentence, huge progress has been made over the last 20 years, but we are only at the beginning of the path. Thinking differently might likely help explaining the majority of yet unknown causes of CPHD.
References
Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev. 2009;30(7):790–829. doi:10.1210/er.2009-0008.
Castinetti F, Reynaud R, Saveanu A, Quentien MH, Albarel F, Barlier A, Enjalbert A, Brue T. Clinical and genetic aspects of combined pituitary hormone deficiencies. Ann Endocrinol. 2008;69(1):7–17. doi:10.1016/j.ando.2008.01.001.
Schlosser G. Induction and specification of cranial placodes. Dev Biol. 2006;294(2):303–51. doi:10.1016/j.ydbio.2006.03.009.
Rizzoti K, Lovell-Badge R. Early development of the pituitary gland: induction and shaping of Rathke's pouch. Rev Endocr Metab Disord. 2005;6(3):161–72. doi:10.1007/s11154-005-3047-7.
Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci U S A. 2008;105(8):2907–12. doi:10.1073/pnas.0707886105.
Gleiberman AS, Michurina T, Encinas JM, Roig JL, Krasnov P, Balordi F, Fishell G, Rosenfeld MG, Enikolopov G. Genetic approaches identify adult pituitary stem cells. Proc Natl Acad Sci U S A. 2008;105(17):6332–7. doi:10.1073/pnas.0801644105.
Garcia-Lavandeira M, Quereda V, Flores I, Saez C, Diaz-Rodriguez E, Japon MA, Ryan AK, Blasco MA, Dieguez C, Malumbres M, Alvarez CV. A GRFa2/Prop1/stem (GPS) cell niche in the pituitary. PLoS One. 2009;4(3), e4815. doi:10.1371/journal.pone.0004815.
Gleiberman AS, Fedtsova NG, Rosenfeld MG. Tissue interactions in the induction of anterior pituitary: role of the ventral diencephalon, mesenchyme, and notochord. Dev Biol. 1999;213(2):340–53.
Japon MA, Rubinstein M, Low MJ. In situ hybridization analysis of anterior pituitary hormone gene expression during fetal mouse development. J Histochem Cytochem. 1994;42(8):1117–25.
Lamolet B, Pulichino AM, Lamonerie T, Gauthier Y, Brue T, Enjalbert A, Drouin J. A pituitary cell-restricted T box factor, Tpit, activates POMC transcription in cooperation with Pitx homeoproteins. Cell. 2001;104(6):849–59.
Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, Andoniadou CL, Banyan A, Alsawaid A, Alrifai MT, Alahmesh MA, Balwi M, Mousavy-Gharavy SN, Lukovic B, Burke D, McCabe MJ, Kasia T, Kleta R, Stupka E, Beales PL, Thompson DA, Chong WK, Alkuraya FS, Martinez-Barbera JP, Sowden JC, Dattani MT. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain. 2013;136(Pt 10):3096–105. doi:10.1093/brain/awt218.
Di Iorgi N, Allegri AE, Napoli F, Bertelli E, Olivieri I, Rossi A, Maghnie M. The use of neuroimaging for assessing disorders of pituitary development. Clin Endocrinol. 2012;76(2):161–76. doi:10.1111/j.1365-2265.2011.04238.x.
Kelberman D, Dattani MT. Genetics of septo-optic dysplasia. Pituitary. 2007;10(4):393–407. doi:10.1007/s11102-007-0055-5.
Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, Sykiotis GP, Gregory LC, Diaczok D, Tziaferi V, Elting MW, Padidela R, Plummer L, Martin C, Feng B, Zhang C, Zhou QY, Chen H, Mohammadi M, Quinton R, Sidis Y, Radovick S, Dattani MT, Pitteloud N. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012;97(4):E694–9. doi:10.1210/jc.2011-2938.
Reynaud R, Jayakody SA, Monnier C, Saveanu A, Bouligand J, Guedj AM, Simonin G, Lecomte P, Barlier A, Rondard P, Martinez-Barbera JP, Guiochon-Mantel A, Brue T. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab. 2012;97(6):E1068–73. doi:10.1210/jc.2011-3056.
McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O, Rondard P, Masumoto KH, Nagano M, Shigeyoshi Y, Pfeifer M, Hulse T, Buchanan CR, Pitteloud N, Martinez-Barbera JP, Dattani MT. Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab. 2013;98(3):E547–57. doi:10.1210/jc.2012-3067.
Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19(2):125–33. doi:10.1038/477.
Martinez-Barbera JP, Rodriguez TA, Beddington RS. The homeobox gene Hesx1 is required in the anterior neural ectoderm for normal forebrain formation. Dev Biol. 2000;223(2):422–30. doi:10.1006/dbio.2000.9757.
Thomas PQ, Johnson BV, Rathjen J, Rathjen PD. Sequence, genomic organization, and expression of the novel homeobox gene Hesx1. J Biol Chem. 1995;270(8):3869–75.
Webb GC, Thomas PQ, Ford JH, Rathjen PD. Hesx1, a homeobox gene expressed by murine embryonic stem cells, maps to mouse chromosome 14, bands A3-B. Genomics. 1993;18(2):464–6. doi:10.1006/geno.1993.1505.
Gaston-Massuet C, Andoniadou CL, Signore M, Sajedi E, Bird S, Turner JM, Martinez-Barbera JP. Genetic interaction between the homeobox transcription factors HESX1 and SIX3 is required for normal pituitary development. Dev Biol. 2008;324(2):322–33. doi:10.1016/j.ydbio.2008.08.008.
Corneli G, Vivenza D, Prodam F, Di Dio G, Vottero A, Rapa A, Bellone S, Bernasconi S, Bona G. Heterozygous mutation of HESX1 causing hypopituitarism and multiple anatomical malformations without features of septo-optic dysplasia. J Endocrinol Invest. 2008;31(8):689–93.
Sobrier ML, Maghnie M, Vie-Luton MP, Secco A, di Iorgi N, Lorini R, Amselem S. Novel HESX1 mutations associated with a life-threatening neonatal phenotype, pituitary aplasia, but normally located posterior pituitary and no optic nerve abnormalities. J Clin Endocrinol Metab. 2006;91(11):4528–36. doi:10.1210/jc.2006-0426.
Sobrier ML, Netchine I, Heinrichs C, Thibaud N, Vie-Luton MP, Van Vliet G, Amselem S. Alu-element insertion in the homeodomain of HESX1 and aplasia of the anterior pituitary. Hum Mutat. 2005;25(5):503. doi:10.1002/humu.9332.
Tajima T, Hattorri T, Nakajima T, Okuhara K, Sato K, Abe S, Nakae J, Fujieda K. Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and optic nerve hypoplasia and combined pituitary hormone deficiency in a Japanese patient. J Clin Endocrinol Metab. 2003;88(1):45–50. doi:10.1210/jc.2002-020818.
Cohen RN, Cohen LE, Botero D, Yu C, Sagar A, Jurkiewicz M, Radovick S. Enhanced repression by HESX1 as a cause of hypopituitarism and septooptic dysplasia. J Clin Endocrinol Metab. 2003;88(10):4832–9. doi:10.1210/jc.2002-021868.
Carvalho LR, Woods KS, Mendonca BB, Marcal N, Zamparini AL, Stifani S, Brickman JM, Arnhold IJ, Dattani MT. A homozygous mutation in HESX1 is associated with evolving hypopituitarism due to impaired repressor-corepressor interaction. J Clin Invest. 2003;112(8):1192–201. doi:10.1172/JCI18589.
Mitchell LA, Thomas PQ, Zacharin MR, Scheffer IE. Ectopic posterior pituitary lobe and periventricular heterotopia: cerebral malformations with the same underlying mechanism? Am J Neuroradiol. 2002;23(9):1475–81.
Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F, Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10(1):39–45.
Brickman JM, Clements M, Tyrell R, McNay D, Woods K, Warner J, Stewart A, Beddington RS, Dattani M. Molecular effects of novel mutations in Hesx1/HESX1 associated with human pituitary disorders. Development. 2001;128(24):5189–99.
Kelberman D, Dattani MT. Septo-optic dysplasia – novel insights into the aetiology. Horm Res. 2008;69(5):257–65. doi:10.1159/000114856.
McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou A, Keller E, Keller A, Haufs N, Krude H, Shalet SM, Dattani MT. HESX1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol Metab. 2007;92(2):691–7. doi:10.1210/jc.2006-1609.
Reynaud R, Gueydan M, Saveanu A, Vallette-Kasic S, Enjalbert A, Brue T, Barlier A. Genetic screening of combined pituitary hormone deficiency: experience in 195 patients. J Clin Endocrinol Metab. 2006;91(9):3329–36. doi:10.1210/jc.2005-2173.
Webb EA, Dattani MT. Septo-optic dysplasia. Eur J Hum Genet. 2009;18(4):393–7. doi:10.1038/ejhg.2009.125.
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;2:8. doi:10.1186/1750-1172-2-8.
Raivio T, Sidis Y, Plummer L, Chen H, Ma J, Mukherjee A, Jacobson-Dickman E, Quinton R, Van Vliet G, Lavoie H, Hughes VA, Dwyer A, Hayes FJ, Xu S, Sparks S, Kaiser UB, Mohammadi M, Pitteloud N. Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2009;94(11):4380–90. doi:10.1210/jc.2009-0179.
Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Soussi-Yanicostas N, Coimbra RS, Delmaghani S, Compain-Nouaille S, Baverel F, Pecheux C, Le Tessier D, Cruaud C, Delpech M, Speleman F, Vermeulen S, Amalfitano A, Bachelot Y, Bouchard P, Cabrol S, Carel JC, Delemarre-van de Waal H, Goulet-Salmon B, Kottler ML, Richard O, Sanchez-Franco F, Saura R, Young J, Petit C, Hardelin JP. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33(4):463–5. doi:10.1038/ng1122.
Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118(8):2822–31. doi:10.1172/JCI34538.
Treier M, Gleiberman AS, O’Connell SM, Szeto DP, McMahon JA, McMahon AP, Rosenfeld MG. Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev. 1998;12(11):1691–704.
Martin C, Balasubramanian R, Dwyer AA, Au MG, Sidis Y, Kaiser UB, Seminara SB, Pitteloud N, Zhou QY, Crowley Jr WF. The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev. 2011;32(2):225–46. doi:10.1210/er.2010-0007.
Larder R, Mellon PL. Otx2 induction of the gonadotropin-releasing hormone promoter is modulated by direct interactions with Grg co-repressors. J Biol Chem. 2009;284(25):16966–78. doi:10.1074/jbc.M109.002485.
Diaczok D, Romero C, Zunich J, Marshall I, Radovick S. A novel dominant negative mutation of OTX2 associated with combined pituitary hormone deficiency. J Clin Endocrinol Metab. 2008;93(11):4351–9. doi:10.1210/jc.2008-1189.
Dateki S, Fukami M, Sato N, Muroya K, Adachi M, Ogata T. OTX2 mutation in a patient with anophthalmia, short stature, and partial growth hormone deficiency: functional studies using the IRBP, HESX1, and POU1F1 promoters. J Clin Endocrinol Metab. 2008;93(10):3697–702. doi:10.1210/jc.2008-0720.
Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K, Fujieda K. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocrinol Metab. 2009;94(1):314–9. doi:10.1210/jc.2008-1219.
Dateki S, Kosaka K, Hasegawa K, Tanaka H, Azuma N, Yokoya S, Muroya K, Adachi M, Tajima T, Motomura K, Kinoshita E, Moriuchi H, Sato N, Fukami M, Ogata T. Heterozygous orthodenticle homeobox 2 mutations are associated with variable pituitary phenotype. J Clin Endocrinol Metab. 2009;95(2):756–64.
Henderson RH, Williamson KA, Kennedy JS, Webster AR, Holder GE, Robson AG, FitzPatrick DR, van Heyningen V, Moore AT. A rare de novo nonsense mutation in OTX2 causes early onset retinal dystrophy and pituitary dysfunction. Mol Vis. 2009;15:2442–7.
Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JM, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson IC, Dattani MT. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116(9):2442–55. doi:10.1172/JCI28658.
Sadeghi-Nejad A, Senior B. A familial syndrome of isolated “aplasia” of the anterior pituitary. Diagnostic studies and treatment in the neonatal period. J Pediatr. 1974;84(1):79–84.
Feingold M, Shiere F, Fogels HR, Donaldson D. Rieger’s syndrome. Pediatrics. 1969;44(4):564–9.
Mammi I, De Giorgio P, Clementi M, Tenconi R. Cardiovascular anomaly in Rieger syndrome: heterogeneity or contiguity? Acta Ophthalmol Scand. 1998;76(4):509–12.
Hjalt TA, Semina EV, Amendt BA, Murray JC. The Pitx2 protein in mouse development. Dev Dyn. 2000;218(1):195–200.
Lin CR, Kioussi C, O'Connell S, Briata P, Szeto D, Liu F, Izpisua-Belmonte JC, Rosenfeld MG. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401(6750):279–82. doi:10.1038/45803.
Charles MA, Suh H, Hjalt TA, Drouin J, Camper SA, Gage PJ. PITX genes are required for cell survival and Lhx3 activation. Mol Endocrinol. 2005;19(7):1893–903. doi:10.1210/me.2005-0052.
Gage PJ, Camper SA. Pituitary homeobox 2, a novel member of the bicoid-related family of homeobox genes, is a potential regulator of anterior structure formation. Hum Mol Genet. 1997;6(3):457–64.
Tumer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009;17(12):1527–39. doi:10.1038/ejhg.2009.93.
Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14(4):392–9. doi:10.1038/ng1296-392.
Raetzman LT, Ward R, Camper SA. Lhx4 and Prop1 are required for cell survival and expansion of the pituitary primordia. Development. 2002;129(18):4229–39.
Sheng HZ, Moriyama K, Yamashita T, Li H, Potter SS, Mahon KA, Westphal H. Multistep control of pituitary organogenesis. Science. 1997;278(5344):1809–12.
Tajima T, Ishizu K, Nakamura A. Molecular and clinical findings in patients with LHX4 and OTX2 mutations. Clin Pediatr Endocrinol. 2013;22(2):15–23. doi:10.1292/cpe.22.15.
Castinetti F, Saveanu A, Reynaud R, Quentien MH, Buffin A, Brauner R, Kaffel N, Albarel F, Guedj AM, El Kholy M, Amin M, Enjalbert A, Barlier A, Brue T. A novel dysfunctional LHX4 mutation with high phenotypical variability in patients with hypopituitarism. J Clin Endocrinol Metab. 2008;93(7):2790–9. doi:10.1210/jc.2007-2389.
Pfaeffle RW, Hunter CS, Savage JJ, Duran-Prado M, Mullen RD, Neeb ZP, Eiholzer U, Hesse V, Haddad NG, Stobbe HM, Blum WF, Weigel JF, Rhodes SJ. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab. 2008;93(3):1062–71. doi:10.1210/jc.2007-1525.
Chou SJ, Hermesz E, Hatta T, Feltner D, El-Hodiri HM, Jamrich M, Mahon K. Conserved regulatory elements establish the dynamic expression of Rpx/HesxI in early vertebrate development. Dev Biol. 2006;292(2):533–45. doi:10.1016/j.ydbio.2005.12.053.
McGillivray SM, Bailey JS, Ramezani R, Kirkwood BJ, Mellon PL. Mouse GnRH receptor gene expression is mediated by the LHX3 homeodomain protein. Endocrinology. 2005;146(5):2180–5. doi:10.1210/en.2004-1566.
Granger A, Bleux C, Kottler ML, Rhodes SJ, Counis R, Laverriere JN. The LIM-homeodomain proteins Isl-1 and Lhx3 act with steroidogenic factor 1 to enhance gonadotrope-specific activity of the gonadotropin-releasing hormone receptor gene promoter. Mol Endocrinol. 2006;20(9):2093–108. doi:10.1210/me.2005-0184.
West BE, Parker GE, Savage JJ, Kiratipranon P, Toomey KS, Beach LR, Colvin SC, Sloop KW, Rhodes SJ. Regulation of the follicle-stimulating hormone beta gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology. 2004;145(11):4866–79. doi:10.1210/en.2004-0598.
Girardin SE, Benjannet S, Barale JC, Chretien M, Seidah NG. The LIM homeobox protein mLIM3/Lhx3 induces expression of the prolactin gene by a Pit-1/GHF-1-independent pathway in corticotroph AtT20 cells. FEBS Lett. 1998;431(3):333–8.
Thaler JP, Lee SK, Jurata LW, Gill GN, Pfaff SL. LIM factor Lhx3 contributes to the specification of motor neuron and interneuron identity through cell-type-specific protein-protein interactions. Cell. 2002;110(2):237–49.
Sharma K, Sheng HZ, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff SL. LIM homeodomain factors Lhx3 and Lhx4 assign subtype identities for motor neurons. Cell. 1998;95(6):817–28.
Huang M, Sage C, Li H, Xiang M, Heller S, Chen ZY. Diverse expression patterns of LIM-homeodomain transcription factors (LIM-HDs) in mammalian inner ear development. Dev Dyn. 2008;237(11):3305–12. doi:10.1002/dvdy.21735.
Hume CR, Bratt DL, Oesterle EC. Expression of LHX3 and SOX2 during mouse inner ear development. Gene Expr Patterns. 2007;7(7):798–807. doi:10.1016/j.modgep.2007.05.002.
Pfaeffle RW, Savage JJ, Hunter CS, Palme C, Ahlmann M, Kumar P, Bellone J, Schoenau E, Korsch E, Bramswig JH, Stobbe HM, Blum WF, Rhodes SJ. Four novel mutations of the LHX3 gene cause combined pituitary hormone deficiencies with or without limited neck rotation. J Clin Endocrinol Metab. 2007;92(5):1909–19. doi:10.1210/jc.2006-2177.
Savage JJ, Hunter CS, Clark-Sturm SL, Jacob TM, Pfaeffle RW, Rhodes SJ. Mutations in the LHX3 gene cause dysregulation of pituitary and neural target genes that reflect patient phenotypes. Gene. 2007;400(1–2):44–51. doi:10.1016/j.gene.2007.05.017.
Bhangoo AP, Hunter CS, Savage JJ, Anhalt H, Pavlakis S, Walvoord EC, Ten S, Rhodes SJ. Clinical case seminar: a novel LHX3 mutation presenting as combined pituitary hormonal deficiency. J Clin Endocrinol Metab. 2006;91(3):747–53. doi:10.1210/jc.2005-2360.
Sloop KW, Parker GE, Hanna KR, Wright HA, Rhodes SJ. LHX3 transcription factor mutations associated with combined pituitary hormone deficiency impair the activation of pituitary target genes. Gene. 2001;265(1-2):61–9.
Howard PW, Maurer RA. A point mutation in the LIM domain of Lhx3 reduces activation of the glycoprotein hormone alpha-subunit promoter. J Biol Chem. 2001;276(22):19020–6. doi:10.1074/jbc.M101782200.
Netchine I, Sobrier ML, Krude H, Schnabel D, Maghnie M, Marcos E, Duriez B, Cacheux V, Moers A, Goossens M, Gruters A, Amselem S. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25(2):182–6. doi:10.1038/76041.
Kristrom B, Zdunek AM, Rydh A, Jonsson H, Sehlin P, Escher SA. A novel mutation in the LIM homeobox 3 gene is responsible for combined pituitary hormone deficiency, hearing impairment, and vertebral malformations. J Clin Endocrinol Metab. 2009;94(4):1154–61. doi:10.1210/jc.2008-0325.
Sornson MW, Wu W, Dasen JS, Flynn SE, Norman DJ, O'Connell SM, Gukovsky I, Carriere C, Ryan AK, Miller AP, Zuo L, Gleiberman AS, Andersen B, Beamer WG, Rosenfeld MG. Pituitary lineage determination by the Prophet of Pit-1 homeodomain factor defective in Ames dwarfism. Nature. 1996;384(6607):327–33. doi:10.1038/384327a0.
Ikeshita N, Kawagishi M, Shibahara H, Toda K, Yamashita T, Yamamoto D, Sugiyama Y, Iguchi G, Iida K, Takahashi Y, Kaji H, Chihara K, Okimura Y. Identification and analysis of prophet of Pit-1-binding sites in human Pit-1 gene. Endocrinology. 2008;149(11):5491–9. doi:10.1210/en.2008-0030.
Kelberman D, Turton JP, Woods KS, Mehta A, Al-Khawari M, Greening J, Swift PG, Otonkoski T, Rhodes SJ, Dattani MT. Molecular analysis of novel PROP1 mutations associated with combined pituitary hormone deficiency (CPHD). Clin Endocrinol. 2009;70(1):96–103. doi:10.1111/j.1365-2265.2008.03326.x.
Zimmermann A, Schenk JP, Grigorescu Sido P, Pfaffle R, Lazea C, Zimmermann T, Heinrich U, Weber MM, Bettendorf M. MRI findings and genotype analysis in patients with childhood onset growth hormone deficiency–correlation with severity of hypopituitarism. J Pediatr Endocrinol Metab. 2007;20(5):587–96.
Vieira TC, da Silva MR, Abucham J. The natural history of the R120C PROP1 mutation reveals a wide phenotypic variability in two untreated adult brothers with combined pituitary hormone deficiency. Endocrine. 2006;30(3):365–9.
Nose O, Tatsumi K, Nakano Y, Amino N. Congenital combined pituitary hormone deficiency attributable to a novel PROP1 mutation (467insT). J Pediatr Endocrinol Metab. 2006;19(4):491–8.
Lemos MC, Gomes L, Bastos M, Leite V, Limbert E, Carvalho D, Bacelar C, Monteiro M, Fonseca F, Agapito A, Castro JJ, Regateiro FJ, Carvalheiro M. PROP1 gene analysis in Portuguese patients with combined pituitary hormone deficiency. Clin Endocrinol. 2006;65(4):479–85. doi:10.1111/j.1365-2265.2006.02617.x.
Abrao MG, Leite MV, Carvalho LR, Billerbeck AE, Nishi MY, Barbosa AS, Martin RM, Arnhold IJ, Mendonca BB. Combined pituitary hormone deficiency (CPHD) due to a complete PROP1 deletion. Clin Endocrinol. 2006;65(3):294–300. doi:10.1111/j.1365-2265.2006.02592.x.
Reynaud R, Barlier A, Vallette-Kasic S, Saveanu A, Guillet MP, Simonin G, Enjalbert A, Valensi P, Brue T. An uncommon phenotype with familial central hypogonadism caused by a novel PROP1 gene mutant truncated in the transactivation domain. J Clin Endocrinol Metab. 2005;90(8):4880–7. doi:10.1210/jc.2005-0119.
Lebl J, Vosahlo J, Pfaeffle RW, Stobbe H, Cerna J, Novotna D, Zapletalova J, Kalvachova B, Hana V, Weiss V, Blum WF. Auxological and endocrine phenotype in a population-based cohort of patients with PROP1 gene defects. Eur J Endocrinol. 2005;153(3):389–96. doi:10.1530/eje.1.01989.
Voutetakis A, Maniati-Christidi M, Kanaka-Gantenbein C, Dracopoulou M, Argyropoulou M, Livadas S, Dacou-Voutetakis C, Sertedaki A. Prolonged jaundice and hypothyroidism as the presenting symptoms in a neonate with a novel Prop1 gene mutation (Q83X). Eur J Endocrinol. 2004;150(3):257–64.
Tatsumi KI, Kikuchi K, Tsumura K, Amino N. A novel PROP1 gene mutation (157delA) in Japanese siblings with combined anterior pituitary hormone deficiency. Clin Endocrinol. 2004;61(5):635–40. doi:10.1111/j.1365-2265.2004.02147.x.
Reynaud R, Chadli-Chaieb M, Vallette-Kasic S, Barlier A, Sarles J, Pellegrini-Bouiller I, Enjalbert A, Chaieb L, Brue T. A familial form of congenital hypopituitarism due to a PROP1 mutation in a large kindred: phenotypic and in vitro functional studies. J Clin Endocrinol Metab. 2004;89(11):5779–86. doi:10.1210/jc.2003-032124.
Bottner A, Keller E, Kratzsch J, Stobbe H, Weigel JF, Keller A, Hirsch W, Kiess W, Blum WF, Pfaffle RW. PROP1 mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis. J Clin Endocrinol Metab. 2004;89(10):5256–65. doi:10.1210/jc.2004-0661.
Paracchini R, Giordano M, Corrias A, Mellone S, Matarazzo P, Bellone J, Momigliano-Richiardi P, Bona G. Two new PROP1 gene mutations responsible for compound pituitary hormone deficiency. Clin Genet. 2003;64(2):142–7.
Arroyo A, Pernasetti F, Vasilyev VV, Amato P, Yen SS, Mellon PL. A unique case of combined pituitary hormone deficiency caused by a PROP1 gene mutation (R120C) associated with normal height and absent puberty. Clin Endocrinol. 2002;57(2):283–91.
Vallette-Kasic S, Barlier A, Teinturier C, Diaz A, Manavela M, Berthezene F, Bouchard P, Chaussain JL, Brauner R, Pellegrini-Bouiller I, Jaquet P, Enjalbert A, Brue T. PROP1 gene screening in patients with multiple pituitary hormone deficiency reveals two sites of hypermutability and a high incidence of corticotroph deficiency. J Clin Endocrinol Metab. 2001;86(9):4529–35. doi:10.1210/jcem.86.9.7811.
Pernasetti F, Toledo SP, Vasilyev VV, Hayashida CY, Cogan JD, Ferrari C, Lourenco Jr DM, Mellon PL. Impaired adrenocorticotropin-adrenal axis in combined pituitary hormone deficiency caused by a two-base pair deletion (301-302delAG) in the prophet of Pit-1 gene. J Clin Endocrinol Metab. 2000;85(1):390–7. doi:10.1210/jcem.85.1.6324.
Agarwal G, Bhatia V, Cook S, Thomas PQ. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J Clin Endocrinol Metab. 2000;85(12):4556–61. doi:10.1210/jcem.85.12.7013.
Rosenbloom AL, Almonte AS, Brown MR, Fisher DA, Baumbach L, Parks JS. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the PROP1 gene. J Clin Endocrinol Metab. 1999;84(1):50–7. doi:10.1210/jcem.84.1.5366.
Deladoey J, Fluck C, Buyukgebiz A, Kuhlmann BV, Eble A, Hindmarsh PC, Wu W, Mullis PE. “Hot spot” in the PROP1 gene responsible for combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1999;84(5):1645–50. doi:10.1210/jcem.84.5.5681.
Wu W, Cogan JD, Pfaffle RW, Dasen JS, Frisch H, O’Connell SM, Flynn SE, Brown MR, Mullis PE, Parks JS, Phillips 3rd JA, Rosenfeld MG. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18(2):147–9. doi:10.1038/ng0298-147.
Fofanova O, Takamura N, Kinoshita E, Parks JS, Brown MR, Peterkova VA, Evgrafov OV, Goncharov NP, Bulatov AA, Dedov II, Yamashita S. Compound heterozygous deletion of the PROP-1 gene in children with combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1998;83(7):2601–4. doi:10.1210/jcem.83.7.5094.
Cogan JD, Wu W, Phillips 3rd JA, Arnhold IJ, Agapito A, Fofanova OV, Osorio MG, Bircan I, Moreno A, Mendonca BB. The PROP1 2-base pair deletion is a common cause of combined pituitary hormone deficiency. J Clin Endocrinol Metab. 1998;83(9):3346–9. doi:10.1210/jcem.83.9.5142.
Vieira TC, Boldarine VT, Abucham J. Molecular analysis of PROP1, PIT1, HESX1, LHX3, and LHX4 shows high frequency of PROP1 mutations in patients with familial forms of combined pituitary hormone deficiency. Arq Bras Endocrinol Metabol. 2007;51(7):1097–103.
Halasz Z, Toke J, Patocs A, Bertalan R, Tombol Z, Sallai A, Hosszu E, Muzsnai A, Kovacs L, Solyom J, Fekete G, Racz K. High prevalence of PROP1 gene mutations in Hungarian patients with childhood-onset combined anterior pituitary hormone deficiency. Endocrine. 2006;30(3):255–60. doi:10.1007/s12020-006-0002-7.
Fluck C, Deladoey J, Rutishauser K, Eble A, Marti U, Wu W, Mullis PE. Phenotypic variability in familial combined pituitary hormone deficiency caused by a PROP1 gene mutation resulting in the substitution of Arg→Cys at codon 120 (R120C). J Clin Endocrinol Metab. 1998;83(10):3727–34. doi:10.1210/jcem.83.10.5172.
Voutetakis A, Sertedaki A, Livadas S, Xekouki P, Bossis I, Dacou-Voutetakis C, Argyropoulou MI. Pituitary size fluctuation in long-term MR studies of PROP1 deficient patients: a persistent pathophysiological mechanism? J Endocrinol Investig. 2006;29(5):462–6.
Voutetakis A, Argyropoulou M, Sertedaki A, Livadas S, Xekouki P, Maniati-Christidi M, Bossis I, Thalassinos N, Patronas N, Dacou-Voutetakis C. Pituitary magnetic resonance imaging in 15 patients with Prop1 gene mutations: pituitary enlargement may originate from the intermediate lobe. J Clin Endocrinol Metab. 2004;89(5):2200–6. doi:10.1210/jc.2003-031765.
Riepe FG, Partsch CJ, Blankenstein O, Monig H, Pfaffle RW, Sippell WG. Longitudinal imaging reveals pituitary enlargement preceding hypoplasia in two brothers with combined pituitary hormone deficiency attributable to PROP1 mutation. J Clin Endocrinol Metab. 2001;86(9):4353–7. doi:10.1210/jcem.86.9.7828.
Fofanova O, Takamura N, Kinoshita E, Vorontsov A, Vladimirova V, Dedov I, Peterkova V, Yamashita S. MR imaging of the pituitary gland in children and young adults with congenital combined pituitary hormone deficiency associated with PROP1 mutations. AJR Am J Roentgenol. 2000;174(2):555–9. doi:10.2214/ajr.174.2.1740555.
Mendonca BB, Osorio MG, Latronico AC, Estefan V, Lo LS, Arnhold IJ. Longitudinal hormonal and pituitary imaging changes in two females with combined pituitary hormone deficiency due to deletion of A301, G302 in the PROP1 gene. J Clin Endocrinol Metab. 1999;84(3):942–5. doi:10.1210/jcem.84.3.5537.
Himes AD, Raetzman LT. Premature differentiation and aberrant movement of pituitary cells lacking both Hes1 and Prop1. Dev Biol. 2009;325(1):151–61. doi:10.1016/j.ydbio.2008.10.010.
Bodner M, Castrillo JL, Theill LE, Deerinck T, Ellisman M, Karin M. The pituitary-specific transcription factor GHF-1 is a homeobox-containing protein. Cell. 1988;55(3):505–18.
Acknowledgments
This work was supported by grants from the “Agence Nationale de la Recherche” (ANR), # 08-GENOPAT-026 and from the “Association pour le Développement de la Recherche au centre hospitalier de Marseille” (A.DE.RE.M). For inquiries on genetic screening strategies for individual patients, contact defhy@ap-hm.fr and visit the website www.ap-hm.fr/defhy/ for information on withdrawal and shipment guidelines.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Castinetti, F., Brue, T. (2016). Combined Pituitary Hormone Deficiency. In: Cohen, L. (eds) Growth Hormone Deficiency. Springer, Cham. https://doi.org/10.1007/978-3-319-28038-7_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-28038-7_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-28036-3
Online ISBN: 978-3-319-28038-7
eBook Packages: MedicineMedicine (R0)