
Abstract
Adipose tissue in human can be divided into two main categories, being white adipose tissue and brown adipose tissue. White adipose tissue (WAT) is located in the subcutis (subcutaneous) and in intra-abdominal locations in association with the viscera (visceral). WAT functions as a storage facility, sequestering energy in the form of triglyceride, which is found in the cytoplasmic, unilocular lipid droplet within mature adipocytes. Accumulation or mobilisation of these fat stores occurs in the face of varying energy requirements.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Adipose Tissue: Definition
Adipose tissue in human can be divided into two main categories, being white adipose tissue and brown adipose tissue. White adipose tissue (WAT) is located in the subcutis (subcutaneous) and in intra-abdominal locations in association with the viscera (visceral). WAT functions as a storage facility, sequestering energy in the form of triglyceride, which is found in the cytoplasmic, unilocular lipid droplet within mature adipocytes. Accumulation or mobilisation of these fat stores occurs in the face of varying energy requirements.
Brown adipose tissue (BAT) is responsible for ‘non-shivering’ thermogenesis, which is the production of heat from glucose and fat. In human infants, the interscapular, neck, axillae, mediastinal, para-aortic and perirenal regions are depots for BAT [1]. In adult humans, brown adipocytes are found in the cervical-supraclavicular region in the ventral neck and may also extend inferiorly along the thoracic and abdominal paraspinal region [2]. Sympathetic nerve stimulation of these adipocytes results in uncoupled oxidative phosphorylation within their abundant mitochondria to generate heat. In contrast to WAT, the triglyceride in these cells is stored in numerous small cytoplasmic droplets, enabling rapid mobilisation of fuel for heat production [3].
2 White Adipose Tissue
Adipose tissue has functions more complex and far reaching than its role in fat storage. In addition to mature adipocytes, WAT contains stromal vascular cells. This stromal component consists of adipocyte precursors (preadipocytes), fibroblasts, endothelial cells and immune cells, including macrophages and lymphocytes. Both the adipocyte and stromal fractions of WAT have secretory functions.
Healthy adipose tissue, when unchallenged by excess nutrition and a positive energy balance, contains macrophages which are of M2 morphology, or ‘alternately activated’. These secrete interleukin-10 (IL-10), which is an insulin-sensitising cytokine.
The difference between subcutaneous and visceral deposits may extend beyond anatomical location. It is the visceral adiposity which correlates more strongly with obesity-associated disease [4]. Reduction of subcutaneous fat mass via liposuction does not improve the metabolic profile of patients; however, bariatric surgery that includes visceral fat reduction does produce improvements in glucose metabolism [4].
Thus, in addition to storage, the secretory function of WAT enables it to have significant impact on metabolism, circadian body clocks, energy homeostasis and inflammation. As the fat mass expands, the inflammatory consequences become significant.
The observation of the inflammatory changes that occur in the presence of excessive fat stores has helped define adipose tissue as a key regulator of immunity and inflammation and an endocrine organ in its own right.
3 Adipose Tissue: Chronic Inflammation
Adipose tissue is the connection between metabolism, energy regulation and inflammation. The visceral compartment of white adipose tissue is believed to be most related to alterations of metabolic health. Visceral adipose tissue contains significant numbers of resident leucocytes, including T and B lymphocytes, regulatory T cells (Tregs), natural killer T cells, eosinophils, mast cells and macrophages. Whether it be during times of starvation and illness, when mobilisation of fat stores is required to meet increased intrinsic energy needs or when chronic overfeeding resulting excessive fat storage, signalling between the resident leucocytes and adipocytes is a vital component of the metabolic and inflammatory processes which occur within adipose tissue [5].
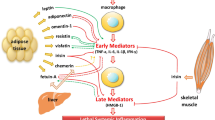
Expansion of adipose tissue in the obese state results in WAT inflammation. This inflammation that develops in the growing fat stores is likely to be multifactorial in aetiology. Contributing factors are postulated to be lipotoxicity, endoplasmic reticulum stress, local tissue hypoxia and Toll-like receptor activation [5]. The ‘stressed’ and now insulin-resistant adipocytes secrete proinflammatory mediators such as TNF-α, monocyte chemoattractant protein (MCP) and fatty acids. These proinflammatory substances result in the activation of resident leucocytes, increased local inflammation and an increased macrophage population with an influx of ‘classically’ activated or ‘M1’ macrophages into adipose tissue and transformation of resident M2 macrophages to the M1 phenotype. M1 macrophages produce proinflammatory cytokines TNF-α and IL-6. This then feeds back upon local adipocytes and a cycle of adipose tissue dysfunction and inflammation is established (see Fig. 14.1).

The consequences of an expanding fat mass. As the result of a prolonged positive energy balance, there is an expansion of fat mass, with both adipocyte hypertrophy and hyperplasia. It is the increase in cell volume seen in adipocyte hypertrophy that is most strongly associated with adipocyte dysfunction. This hypertrophy places adipocytes under ‘stress’ and subsequent activation of signalling pathways, which results in the transformation and accumulation of inflammatory cells in adipose tissue and the subsequent release of proinflammatory cytokines and adipokines with local insulin resistance. Development of the ‘metabolic syndrome’ – a systemic manifestation of adipose tissue dysfunction – ensues, characterised by obesity, insulin resistance, dyslipidaemia and hypertension
As these hypertrophic, dysfunctional adipocytes produce a local milieu of insulin resistance, impaired lipid handling and inflammation, the consequence becomes systemic metabolic dysregulation, manifest as systemic insulin resistance, type 2 diabetes, dyslipidaemia and cardiovascular disease. Secondary organ damage occurs in the liver, pancreas, skeletal muscle and brain [4]. A ‘spillover’ effect likely contributes to a low-grade systemic inflammatory state, with elevated circulating inflammatory markers in the obesity [6].
In addition to resident leucocyte cytokine adipocyte production and secretion of proinflammatory cytokines (e.g. TNF-α, IL-6, MCP-1), adipocytes secrete a multitude of bioactive molecules, known as ‘adipokines’. Over 600 have been described. Many adipokines have net proinflammatory effects by promoting and/or maintaining inflammation. These include leptin, resistin, retinol-binding protein 4 (RBP4), lipocalin 2, angiopoietin-like protein 2 (ANGPTL2), visfatin, CC-chemokine ligand 2 (CCL2) and CXC-chemokine ligand 5 (CXCL5) [7]. A smaller family of ‘anti-inflammatory’ adipokines includes adiponectin and secreted frizzled-related protein 5 (sFRP5) [7].
The role of adipokines in the chronic inflammation of obesity and its associated metabolic perturbations have led to increased interest and awareness of adipokines as potential signalling molecules in the acute inflammatory response.
4 Adipose Tissue in Critical Illness
Sepsis induces a state of tissue catabolism, during which fat is the preferred source of energy. Experimental endotoxaemia in humans is associated with increased lipolysis within WAT [8].
It has long been recognised that lipolysis liberates free fatty acids (FFAs) to act as a circulating energy source, but it is now clear that FFAs have direct signalling effects via a range of receptors in multiple tissues. Furthermore, WAT releases a multitude of adipokines that, during catabolism, directly influence multiple organ systems.
The metabolic syndrome and critical illness share several key physiological perturbations, being insulin resistance, dysregulation of the hypothalamic-pituitary and gonadal axes and inflammation as evidenced by increased circulating proinflammatory cytokines [9]. How this includes adipokine dysregulation is the subject of ongoing research.
There is a huge amount of literature reporting on the multitude of adipokines and acute inflammation. Adipokines described here are those whose potential role in sepsis is most clearly defined at this point.
5 The Adipokines
5.1 Leptin
Leptin is a peptide hormone synthesized and secreted mostly by adipocytes of WAT. Circulating leptin is positively associated with body fat mass and can be considered a reflection of adipose tissue mass.
Encoded in humans by the obese (ob) gene on chromosome 7, leptin is an adipokine belonging to the type I cytokine family, which includes growth hormone and prolactin [10].
Low leptin levels signal starvation. Leptin acts via central and peripheral mechanisms to regulate food intake, appetite, glucose metabolism and energy expenditure. It acts upon the leptin receptor within the hypothalamus to regulate appetite and body weight. During fasting, insulin and then leptin levels decline, which stimulate appetite and feeding. In the fed state, insulin and leptin levels increase, promoting decreased appetite and food intake and increased energy expenditure [10].
In addition to the central nervous system, leptin receptors are widely expressed in peripheral locations, including the pancreas, liver, adipose tissue and various immune cell types (neutrophils, monocytes, macrophages, lymphocytes, mast cells, dendritic cells and NK cells) [11]. Leptin levels act as a signal to the brain regarding the status of available energy for various biological pathways, including those of the reproductive and immune systems.
Alterations in circulating leptin levels contribute to the immune dysfunction that is a feature of both obesity and malnutrition. Interest in a role for leptin in inflammation was in part due to the observation that it may interact with CRP [12].
Leptin acts to promote, maintain and regulate the immune response. Leptin regulates both innate and adaptive immunities. It induces cytokine secretion by inflammatory cells, stimulates macrophage activation and phagocytosis, acts as a chemoattractant and inhibits immune cell apoptosis. Its anti-apoptotic actions on T lymphocytes maintain the thymic parenchyma and it regulates T-cell function [13].
Leptin induces the expression of acute phase proteins such as lipocalin-2, tissue plasminogen activator (tPA) and fibrinogen β [14].
Acute inflammatory states are associated with elevated leptin levels. Leptin stimulates the release of proinflammatory cytokines such as TNF-α and IL-6, which in turn stimulate the release of leptin from adipocytes, thus establishing a proinflammatory cycle.
Animal studies suggest that leptin controls body temperature and duration and extent of the immune response in sepsis [15]. Interestingly, it may be that the elevated levels of leptin seen in the class 1 obese state (BMI 30–34.9) improve the cellular immune response and contribute to improved outcomes [15]. Exogenous leptin treatment has been reported to attenuate the development of acute lung injury in mice [16].
How this translates into the clinical setting of human sepsis remains unclear, so too, is the usefulness of serum leptin concentration as a marker of inflammation or disease severity in sepsis.
Alterations in circulating leptin concentrations in critical illness have not been conclusively defined. Increased leptin levels have been reported in sepsis and may correlate with proinflammatory markers and illness severity and be useful in distinguishing between systemic inflammatory response syndrome (SIRS) and sepsis [17, 18]. Serum leptin levels have been reported to be as effective as a diagnostic marker for sepsis as traditional biomarkers such as CRP, procalcitonin and body temperature [17]. However, not all studies of septic patients have shown significant alterations in serum leptin concentrations [19].
5.2 Visfatin/NAMPT/PBEF
Visfatin (the term used in this review) also known as nicotinamide phosphoribosyltransferase (NAMPT) or pre-B-cell colony-enhancing factor (PBEF) is a protein ubiquitously expressed in almost all human tissues. It is, however, highly expressed by adipocytes of visceral WAT.
The major function of intracellular NAMPT is as a biosynthetic enzyme in the pathway responsible for the generation of nicotinamide adenine dinucleotide (NAD). NAMPT generates nicotinamide mononucleotide (NMN) from nicotinamide. NMN is then converted to NAD [20].
NAD is a critical coenzyme which is utilised in cellular redox reactions and as an enzyme substrate.
By the regulation of the cellular pool of NAD, NAMPT is able to influence NAD-dependent enzymes, many of which are involved in mediation of the inflammatory response, cellular metabolism and circadian rhythms [21].
NAMPT was termed ‘visfatin’ following the observation that it was secreted by visceral fat [22]. Initially believed to be an insulin-mimicking adipokine, further research suggests that visfatin’s role in insulin-signalling pathways is far more complex [20].
It is not yet clear if NAMPT has predominantly proinflammatory or anti-inflammatory actions. The expression of NAMPT in adipocytes is upregulated by exposure to proinflammatory cytokines, and NAMPT exposure stimulates the production of proinflammatory cytokines in adipocytes, suggesting a positive feedback loop. It is secreted from neutrophils and macrophages in response to inflammatory stimuli and may act to prolong neutrophil survival by inhibition of apoptosis [23].
There is much interest in the potential role of PBEF in the development of acute lung injury and ventilator-induced lung injury. Increased PBEF expression in inflammatory lung injury is associated with mechanical stretch, proinflammatory cytokine production and inflammatory cell recruitment in the lung [24]. Its proinflammatory actions are mediated in part by activation of pathways involving Toll-like receptors (TLRs) and NF-κB [25], which are involved in innate immune responses.
Increased levels of PBEF in bronchoalveolar fluid, serum and lung tissues from animal and human, acute lung injury (ALI) models [26] suggest it may be used as a biomarker for acute lung injury. In sepsis, elevated levels of circulating visfatin (PBEF) are associated with scores of illness severity [27, 28] and mortality in ventilated patients [28]. Visfatin levels have been shown to be increased in patients admitted to hospital with community-acquired pneumonia, with levels correlating with mortality and scores of illness severity [29].
5.3 Lipocalin 2
Lipocalin 2 (LPN2, neutrophil gelatinase-associated lipocalin, siderocalin, uterocalin, p25, 24p3) is an adipokine produced and secreted by both adipocytes and macrophages. It is a 198 amino acid, secreted glycoprotein. Originally isolated from neutrophil granules, LPN2 is a member of the lipocalin family of carrier/transporter proteins. These are small, soluble proteins which are characterised by a unique structure known as the ‘lipocalin fold’. This is a cup-shaped fold in the protein to which a ligand binds. Amino acid variations within the fold impart ligand specificity. Lipocalins associate with and act as carriers for various lipophilic or hydrophobic substances such as steroids, bilins and retinoids [30].
Lipocalin 2 is also expressed by a number of sources of adult human tissue including the kidney, liver, trachea, lungs, small intestine, breast and salivary glands [31].
Lipocalin 2 has bacteriostatic functions, mediated through its ability to bind to bacterial siderophores and transport them into mammalian cells. Siderophores are iron-chelating compounds secreted by iron-utilising bacteria into the extracellular environment with the purpose of iron scavenging. Siderophores have a greater affinity for iron than our endogenous chelators (transferrin, ferritin and lactoferrin). Lipocalin 2 is able to bind to siderophores in both their iron-free and iron-laden states and, via the lipocalin 2 receptor, transport the siderophore into mammalian cells. By depriving the bacteria of iron, they are unable to proliferate [31].
In addition to its bacteriostatic functions, LPN2 may act as a chemoattractant for neutrophils, have antioxidant properties and play a key role in the splenic immune response [31].
Increased circulating and urinary LPN2 is a feature of acute and chronic renal impairment.
The precise mechanism underlying this elevation is unknown, but LPN2 is produced along several segments of the nephron in response to stressors such as ischaemia and toxins. Also, as glomerular filtration fails, less LPN2 may be filtered from the blood to the urine. Alternatively, filtered LPN2 may not be reabsorbed by damaged tubules, contributing to elevated urinary levels [31]. Regardless of the mechanism, an elevation in circulating LPN2 is considered a reliable indicator of acute kidney injury and precedes any derangement of plasma creatinine levels [32].
Given the prevalence of acute kidney injury in sepsis, the quantification of LPN2 levels as an indication of impending or developing renal dysfunction may prove to be a useful tool.
There may also be a role for lipocalin 2 as an acute phase protein and biomarker for acute inflammation. However, the wide range of potential tissue sources of LPN2 makes the interpretation of an elevated level in the presence of more than one pathological process challenging.
Mouse studies show that exposure to TNF-α, IL-1β and IL-6 induces LCN2 production in adipocytes, as does prolonged exposure to noradrenaline under fasting conditions [33]. LCN2 attenuates the proinflammatory effects of TNF-α on adipocytes by reducing TNF-α-induced IL-6 and MCP-1 production by adipocytes and is able to reverse the TNF-α-induced suppression of adiponectin and leptin production by adipocytes [34].
Human studies of critical illness show circulating LCN2 levels are significantly elevated during acute peritonitis [35] and severe acute pancreatitis [36].
LCN2 gene expression as measured in whole blood increases in patients with sepsis-related acute respiratory distress syndrome (ARDS) and correlates with LCN2 plasma levels [37]. Sepsis is associated with higher urinary LCN2 levels [38] and strongly correlates with elevated plasma LCN2 levels, independent of renal dysfunction [39]. It may also be a useful biomarker to distinguish between bacterial sepsis and SIRS [39].
An elevated LCN2 level may confer prognostic significance as an independent predictor of mortality and multiorgan dysfunction in severe sepsis [40].
5.4 Adiponectin
Adiponectin, a hormone with structural homology to complement C1q, is secreted almost exclusively from adipocytes. Adiponectin is an insulin-sensitising, proinflammatory and cardioprotective hormone and reduced circulating levels are a marker of metabolic syndrome [41]. Lower levels of adiponectin are observed in obesity and diabetes [42]. The circulating adiponectin fraction comprises several isoforms – high molecular weight (HMW), medium molecular weight (MMW) and low molecular weight (LMW). The HMW fraction is reported to confer the metabolic benefits of adiponectin [43]. The circulating levels of adiponectin vary significantly over a day in both health [44] and in a critical illness [45], and a daily temporal rhythm may exist [46] .
The biological effects of adiponectin are mediated via its association with receptors on a wide variety of target cells.
The two main transmembrane cell receptors for adiponectin are AdipoR1 and AdipoR2. These two receptors are ubiquitous in their distribution. AdipoR1 is most abundantly expressed in the skeletal muscle and AdipoR2 in the liver [47, 48]. However, these receptors are also present in pancreatic beta cells [49], inflammatory cells [50], cardiac tissues [51] and white adipose tissue [52]. Both receptors are expressed widely throughout the central nervous system by neurons within the cortex, hypothalamus, pituitary gland, brainstem and hippocampus [53]. Studies of sections of the human brain show adiponectin localised within the anterior pituitary gland, with AdipoR1 and R2 receptor localisation in the pars distalis. In addition, strong staining for AdipoR1 was identified in neurons of the lateral hypothalamic area and the nucleus basalis of Meynert, which are important central regulators of feeding and energy expenditure [54]. This central expression of adiponectin receptors is of particular interest when considering the role of adiponectin and inflammation.
T-cadherin is also a possible third receptor, or binding protein, for adiponectin. The association between adiponectin and T-cadherin is reported to confer the cardioprotective effects of the hormone [55].
The immune regulatory functions of adiponectin involve multiple pathways [41].
Adiponectin and TNF-α share an opposing physiological relationship, characterised by a cycle of ‘negative feedback’ [41]. There is an inverse relationship between the expression and secretion of adiponectin and proinflammatory TNF-α by adipocytes and stromal macrophages within adipose tissue, with a negative association between circulating adiponectin and TNF-α levels.
Adiponectin has anti-inflammatory actions mediated partly via its interaction with NF-κB-signalling pathways. Adiponectin inhibits the activity of NF-kB. Treatment with adiponectin of lipopolysaccharidase (LPS)-treated adipocytes downregulates the activated NF-kB pathway and reduces IL-6 release [56]. Adiponectin treatment of macrophages was found to decrease LPS-stimulated TNF-a production [57]. However, short-term exposure of macrophages to globular adiponectin activated NF-KB, producing in a rapid increase in TNF-a, which resulted in IL-10 release, with the eventual result being the ‘desensitisation’ of macrophages to LPS exposure [57]. Therefore, the attenuation of the inflammatory response by adiponectin may involve complex interplay between short-term and longer term effects.
Adiponectin has direct actions on inflammatory cells. As mentioned above, adiponectin is able to influence signalling pathways of macrophages, with the net effect being downregulation of their inflammatory response. Adiponectin receptors are present on monocytes, T and B lymphocytes and on NK cells [58] and may effect neutrophil migration [59].
Actions of adiponectin within the central nervous system may contribute to its modulation of inflammatory pathways.
The central regulation and response to systemic inflammation occurs via neuronal and humoral pathways. The neural pathway involves communication between visceral sensory afferent fibres of the vagus nerve, the solitary tract nucleus (STN) of the medulla and the hypothalamus. The humoral pathway involves the circumventricular organs (CVOs). The eight circumventricular organs act as communication points between the blood, CSF and brain. These structures lack a blood-brain barrier, rather, demonstrating distinct histological features, with fenestrated capillaries, looser glial cell apposition and larger perivascular spaces [60]. Sensory circumventricular organs – the subfornical organ (SFO), organum vasculosum of the lamina terminalis (OVLT) and the area postrema (AP) – are vital structures in the regulation of metabolic, endocrine and autonomic functions. They have connections with the hypothalamus, autonomic regulatory systems and the dorsal vagal complex. The interaction of circulating inflammatory mediators with Toll-like receptors on macrophage-like cells within the CVOs results in the activation of endothelial cells and microglial cells within the CVOs, which subsequently produce proinflammatory cytokines, including TNF-α and prostaglandin E2 (PGE2). Stress activation of the hypothalamic-pituitary axis (HPA) involves inflammatory mediator activation of CVOs, the rostral ventrolateral medulla (RVLM) and the STN, with subsequent release of corticotropin-releasing hormone (CRH) from paraventricular neurons of the hypothalamus [60].
Adiponectin receptors are expressed in circumventricular organs (CVOs) within the brain. Neurons of both the area postrema (AP) and the subfornical organ (SFO) of rats are responsive to adiponectin [61, 62]. Food deprivation alters the neural response of the SFO to adiponectin [62] and exposure of the AP to adiponectin results in cardiovascular changes [61].
Animal models have shown upregulation of AdipoR2 in the hypothalamus following LPS-induced sepsis [63].
Both AdipoR1 and AdipoR2 are expressed in the cells of the STN of rats and adiponectin affects the electrical activity of neurons within the STN [64]. Introduction of adiponectin into the STN has been observed to reduce systemic blood pressure [64].
The role of adiponectin in the acute inflammatory process of sepsis has not been clearly defined.
The observations that adiponectin antagonised the inflammatory effects of TNF-α negatively correlated with plasma TNF-α levels and showed structural homology to complement factor C1q, leading researchers to further investigate the relationship between adiponectin and lipopolysaccharidase (LPS). Adiponectin was found to directly bind LPS and suppress limulus amoebocyte lysate (LAL) in vitro [65]. In rats with induced polymicrobial sepsis, plasma adiponectin levels have been shown to negatively correlate with plasma endotoxin and TNF-α levels [65].
Studies in human subjects are inconclusive. Lower levels of adiponectin have been found in critically ill patients [66–68]. Additionally, Venkatesh et al. described a strong association between plasma cortisol and adiponectin, an inverse correlation between plasma CRP and adiponectin, as well as a linear response between sickness severity and plasma adiponectin [67]. A study of patients following aneurysmal subarachnoid haemorrhage showed lower levels of adiponectin on days 3 and 7 of their admission when compared to controls. Also, those who developed delayed cerebral ischaemia (DCI) due to vasospasm as evidenced by a clinical deterioration or a new cerebral infarct on imaging had significantly lower adiponectin levels during their admission when compared to those who did not develop DCI [69].
No significant changes in circulating adiponectin levels have been observed in studies of induced endotoxaemia in human subjects [70, 71].
In addition, higher levels of adiponectin in critical illness have been associated with increased risk of mortality [72, 73].
5.5 Resistin
Resistin is a protein first identified in the circulation of mice in 2001 and associated with insulin resistance in this species. Similarly to adiponectin, resistin circulates in a number of higher-order multimers which may confer different levels of biological activity. However, mouse resistin differs from the human form in two very significant ways. Firstly, there is significant disparity between the genomic organisation and composition of the different species. Secondly, the primary site of production in the mouse is the adipocyte. In humans, the circulating resistin fraction is primarily derived from peripheral blood mononuclear cells, macrophages and the bone marrow [74]. Differences in structure and function between the species add additional complexity to the understanding of the biological role of resistin.
In humans, resistin likely targets a wide range of tissues, including human myeloid cells, monocytes, epithelial cells and endothelial cells, and may include hypothalamic regulation of inflammation. Resistin’s mechanism of action involves the modulation of proinflammatory-signalling pathways (including NF-kB and MAPK), which then mediate the expression of proinflammatory genes of IL-6, TNF-alpha and MCP-1.
Elevated levels of resistin have been observed in patients with non-septic critical illness [75], sepsis and septic shock [76], with correlation between resistin levels and proinflammatory cytokines [75, 76]. However, it remains unclear whether there exists a relationship between resistin levels and disease severity and/or prognosis (Table 14.1).
6 Summary
The therapeutic manipulation of circulating biomarkers, including adipokines, for clinical benefit in sepsis remains the ultimate end goal. The functional role and the prognostic significance of adipokine fluctuations in critical illness remain largely undefined. Also, peculiarities and complexities of the molecular structures and circulating forms create additional challenges. For example, there are no current strategies for the therapeutic administration of adiponectin, with its complex structure and very high circulating concentrations making supplementation impractical.
Leptin is the single adipokine approved for therapeutic administration in a clinical setting. This adipokine is administered for therapeutic purposes in rare patients with leptin deficiency associated with lipodystrophy. In the context of critical illness, the administration of leptin may augment immune cell function and optimise central responses to systemic inflammation and infection. There may be a potential, practical use for leptin in the intensive care setting.
As our understanding of adipokines increases, the modulation of these diverse signalling proteins to improve patient outcomes in critical illness may become possible.
References
Aherne W, Hull D (1966) Brown adipose tissue and heat production in the newborn infant. J Pathol Bacteriol 91(1):223–234
Cypess AM, Lehman S, Williams G et al (2009) Identification and importance of brown adipose tissue in adult humans. N Engl J Med 360(15):1509–1517
Smorlesi A, Frontini A, Giordano A, Cinti S (2012) The adipose organ: white-brown adipocyte plasticity and metabolic inflammation. Obes Rev 13(Suppl 2):83–96
Kloting N, Bluher M (2014) Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord 15(4):277–287
Exley MA, Hand L, O'Shea D, Lynch L (2014) Interplay between the immune system and adipose tissue in obesity. J Endocrinol 223(2):R41–R48
Park HS, Park JY, Yu R (2005) Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF-α and IL-6. Diabetes Res Clin Pract 69(1):29–35
Ouchi N, Parker JL, Lugus JJ, Walsh K (2011) Adipokines in inflammation and metabolic disease. Nat Rev Immunol 11(2):85–97
Wellhoener P, Vietheer A, Sayk F, Schaaf B, Lehnert H, Dodt C (2011) Metabolic alterations in adipose tissue during the early phase of experimental endotoxemia in humans. Horm Metab Res 43(11):754–759
Robinson K, Kruger P, Prins J, Venkatesh B (2011) The metabolic syndrome in critically ill patients. Best Pract Res Clin Endocrinol Metab 25(5):835–845
Otero M, Lago R, Lago F et al (2005) Leptin, from fat to inflammation: old questions and new insights. FEBS Lett 579(2):295–301
Gheorghita V, Barbu AE, Gheorghiu ML, Caruntu FA (2015) Endocrine dysfunction in sepsis: a beneficial or deleterious host response? Germs 5(1):17–25
Chen KK (2006) Induction of leptin resistance through direct interaction of C-reactive protein with leptin. Nat Med 12(4):425–432
Paz-Filho G, Mastronardi C, Franco CB, Wang KB, Wong ML, Licinio J (2012) Leptin: molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq Bras Endocrinol Metabol 56(9):597–607
Hekerman P, Zeidler J, Korfmacher S et al (2007) Leptin induces inflammation-related genes in RINm5F insulinoma cells. BMC Mol Biol 8:41
Siegl D, Annecke T, Johnson BL 3rd et al (2014) Obesity-induced hyperleptinemia improves survival and immune response in a murine model of sepsis. Anesthesiology 121(1):98–114
Landgraf MA, Silva RC, Correa-Costa M et al (2014) Leptin downregulates LPS-induced lung injury: role of corticosterone and insulin. Cell Physiol Biochem 33(3):835–846
Chen M, Wang B, Xu Y et al (2014) Diagnostic value of serum leptin and a promising novel diagnostic model for sepsis. Exp Ther Med 7(4):881–886
Yousef AA, Amr YM, Suliman GA (2010) The diagnostic value of serum leptin monitoring and its correlation with tumor necrosis factor-alpha in critically ill patients: a prospective observational study. Crit Care 14(2):R33
Koch A, Weiskirchen R, Zimmermann HW, Sanson E, Trautwein C, Tacke F. Relevance of serum leptin and leptin-receptor concentrations in critically ill patients. Mediators Inflamm. 2010;2010. doi:10.1155/2010/473540
Garten A, Petzold S, Schuster S, Korner A, Kratzsch J, Kiess W (2011) Nampt and its potential role in inflammation and type 2 diabetes. Handb Exp Pharmacol 203:147–164
Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W (2015) Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol 11(9):535–546
Fukuhara A, Matsuda M, Nishizawa M et al (2005) Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science 307(5708):426–430
Jia SH, Li Y, Parodo J et al (2004) Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J Clin Invest 113(9):1318–1327
Hong SB, Huang Y, Moreno-Vinasco L et al (2008) Essential role of pre-B-cell colony enhancing factor in ventilator-induced lung injury. Am J Respir Crit Care Med 178(6):605–617
Camp SM, Ceco E, Evenoski CL et al (2015) Unique toll-like receptor 4 activation by NAMPT/PBEF induces NFkappaB signaling and inflammatory lung injury. Sci Rep 5:13135
Ye SQ, Simon BA, Maloney JP et al (2005) Pre-B-cell colony-enhancing factor as a potential novel biomarker in acute lung injury. Am J Respir Crit Care Med 171(4):361–370
Lee KA, Gong MN (2011) Pre-B-cell colony-enhancing factor and its clinical correlates with acute lung injury and sepsis. Chest 140(2):382–390
Lee K, Huh JW, Lim CM, Koh Y, Hong SB (2013) Clinical role of serum pre-B cell colony-enhancing factor in ventilated patients with sepsis and acute respiratory distress syndrome. Scand J Infect Dis 45(10):760–765
Hu W, Liu CW, Su J, Lu J, Zhu Y, Liu BW (2013) Elevated plasma visfatin concentrations in patients with community-acquired pneumonia. Peptides 43:8–12
Ferreira AC, Da Mesquita S, Sousa JC et al (2015) From the periphery to the brain: Lipocalin-2, a friend or foe? Prog Neurobiol 131:120–136
Chakraborty S, Kaur S, Guha S, Batra SK (2012) The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta 1826(1):129–169
Martensson J, Bell M, Oldner A, Xu S, Venge P, Martling CR (2010) Neutrophil gelatinase-associated lipocalin in adult septic patients with and without acute kidney injury. Intensive Care Med 36(8):1333–1340
Zhang Y, Foncea R, Deis JA, Guo H, Bernlohr DA, Chen X (2014) Lipocalin 2 expression and secretion is highly regulated by metabolic stress, cytokines, and nutrients in adipocytes. PLoS One 9(5):e96997
Zhang J, Wu Y, Zhang Y, Leroith D, Bernlohr DA, Chen X (2008) The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol Endocrinol 22(6):1416–1426
Axelsson L, Bergenfeldt M, Ohlsson K (1995) Studies of the release and turnover of a human neutrophil lipocalin. Scand J Clin Lab Invest 55(7):577–588
Chakraborty S, Kaur S, Muddana V et al (2010) Elevated serum neutrophil gelatinase-associated lipocalin is an early predictor of severity and outcome in acute pancreatitis. Am J Gastroenterol 105(9):2050–2059
Kangelaris KN, Prakash A, Liu KD et al (2015) Increased expression of neutrophil-related genes in patients with early sepsis-induced ARDS. Am J Physiol Lung Cell Mol Physiol 308(11):L1102–L1113
Bell M, Larsson A, Venge P, Bellomo R, Martensson J (2015) Assessment of cell-cycle arrest biomarkers to predict early and delayed acute kidney injury. Dis Markers 2015:158658
Martensson J, Bell M, Xu S et al (2013) Association of plasma neutrophil gelatinase-associated lipocalin (NGAL) with sepsis and acute kidney dysfunction. Biomarkers Biochem Indicators Expo Response Susceptibility Chem 18(4):349–356
Wang B, Chen G, Zhang J, Xue J, Cao Y, Wu Y (2015) Increased NGAL is associated with mortality and multiple organ dysfunction syndrome in severe sepsis and septic shock. Shock 44:234–238
Robinson K, Prins J, Venkatesh B (2011) Clinical review: adiponectin biology and its role in inflammation and critical illness. Crit Care 15(2):221
Comuzzie AG, Funahashi T, Sonnenberg G et al (2001) The genetic basis of plasma variation in adiponectin, a global endophenotype for obesity and the metabolic syndrome. J Clin Endocrinol Metab 86(9):4321–4325
Seino Y, Hirose H, Saito I, Itoh H (2007) High molecular weight multimer form of adiponectin as a useful marker to evaluate insulin resistance and metabolic syndrome in Japanese men. Metabolism 56(11):1493–1499
Scheer FA, Chan JL, Fargnoli J et al (2010) Day/night variations of high-molecular-weight adiponectin and lipocalin-2 in healthy men studied under fed and fasted conditions. Diabetologia 53(11):2401–2405
Robinson K, Jones M, Ordonez J et al (2013) Random measurements of adiponectin and IL-6 may not be indicative of the 24-h profile in critically ill patients. Clin Endocrinol (Oxf) 79(6):892–898
Gavrila A, Peng C-K, Chan JL, Mietus JE, Goldberger AL, Mantzoros CS (2003) Diurnal and ultradian dynamics of serum adiponectin in healthy men: comparison with leptin, circulating soluble leptin receptor, and cortisol patterns. J Clin Endocrinol Metab 88(6):2838–2843
Yamauchi T, Nio Y, Maki T et al (2007) Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 13(3):332–339
Yamauchi T, Kamon J, Ito Y et al (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423(6941):762–769
Kharroubi I, Rasschaert J, Eizirik DL, Cnop M (2003) Expression of adiponectin receptors in pancreatic beta cells. Biochem Biophys Res Commun 312(4):1118–1122
Chinetti G, Zawadski C, Fruchart JC, Staels B (2004) Expression of adiponectin receptors in human macrophages and regulation by agonists of the nuclear receptors PPARalpha, PPARgamma, and LXR. Biochem Biophys Res Commun 314(1):151–158
Ding G, Qin Q, He N et al (2007) Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor gamma. J Mol Cell Cardiol 43(1):73–84
Nannipieri M, Bonotti A, Anselmino M et al (2007) Pattern of expression of adiponectin receptors in human adipose tissue depots and its relation to the metabolic state. Int J Obes 31(12):1843–1848
Thundyil J, Pavlovski D, Sobey CG, Arumugam TV (2012) Adiponectin receptor signalling in the brain. Br J Pharmacol 165(2):313–327
Psilopanagioti A, Papadaki H, Kranioti EF, Alexandrides TK, Varakis JN (2009) Expression of adiponectin and adiponectin receptors in human pituitary gland and brain. Neuroendocrinology 89(1):38–47
Parker-Duffen JL, Walsh K (2014) Cardiometabolic effects of adiponectin. Best Pract Res Clin Endocrinol Metab 28(1):81–91
Lira FS, Rosa JC, Pimentel GD et al (2012) Both adiponectin and interleukin-10 inhibit LPS-induced activation of the NF-kappaB pathway in 3T3-L1 adipocytes. Cytokine 57(1):98–106
Park PH, McMullen MR, Huang H, Thakur V, Nagy LE (2007) Short-term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor-alpha (TNF-alpha) expression via ERK1/2 activation and Egr-1 expression: role of TNF-alpha in adiponectin-stimulated interleukin-10 production. J Biol Chem 282(30):21695–21703
Pang TT, Narendran P (2008) The distribution of adiponectin receptors on human peripheral blood mononuclear cells. Ann N Y Acad Sci 1150:143–145
Lang K, Ratke J (2009) Leptin and Adiponectin: new players in the field of tumor cell and leukocyte migration. Cell Commun Signal 7:27
Siso S, Jeffrey M, Gonzalez L (2010) Sensory circumventricular organs in health and disease. Acta Neuropathol 120(6):689–705
Fry M, Smith PM, Hoyda TD et al (2006) Area postrema neurons are modulated by the adipocyte hormone adiponectin. J Neurosci 26(38):9695–9702
Alim I, Fry WM, Walsh MH, Ferguson AV (2010) Actions of adiponectin on the excitability of subfornical organ neurons are altered by food deprivation. Brain Res 1330:72–82
Iwasa T, Matsuzaki T, Matsui S et al (2014) The effects of LPS-induced endotoxemia on the expression of adiponectin and its receptors in female rats. Endocr J 61(9):891–900
Hoyda TD, Smith PM, Ferguson AV (2009) Adiponectin acts in the nucleus of the solitary tract to decrease blood pressure by modulating the excitability of neuropeptide Y neurons. Brain Res 1256:76–84
Tsuchihashi H, Yamamoto H, Maeda K et al (2006) Circulating concentrations of adiponectin, an endogenous lipopolysaccharide neutralizing protein, decrease in rats with polymicrobial sepsis. J Surg Res 134(2):348–353
Jernas M, Olsson B, Sjoholm K et al (2009) Changes in adipose tissue gene expression and plasma levels of adipokines and acute-phase proteins in patients with critical illness. Metabolism 58(1):102–108
Venkatesh B, Hickman I, Nisbet J, Cohen J, Prins J (2009) Changes in serum adiponectin concentrations in critical illness: a preliminary investigation. Crit Care 13(4):R105
Langouche L, Vander Perre S, Frystyk J, Flyvbjerg A, Hansen TK, Van den Berghe G (2009) Adiponectin, retinol-binding protein 4, and leptin in protracted critical illness of pulmonary origin. Crit Care 13(4):R112
Takeuchi S, Wada K, Otani N, Osada H, Nagatani K, Mori K (2014) Temporal profile of plasma adiponectin level and delayed cerebral ischemia in patients with subarachnoid hemorrhage. J Clin Neurosci 21(6):1007–1010
Keller P, Moller K, Krabbe KS, Pedersen BK (2003) Circulating adiponectin levels during human endotoxaemia. Clin Exp Immunol 134(1):107–110
Anderson PD, Mehta NN, Wolfe ML et al (2007) Innate immunity modulates adipokines in humans. J Clin Endocrinol Metab 92(6):2272–2279
Kaplan JM, Denenberg A, Monaco M, Nowell M, Wong H, Zingarelli B (2010) Changes in peroxisome proliferator-activated receptor-gamma activity in children with septic shock. Intensive Care Med 36(1):123–130
Walkey AJ, Rice TW, Konter J et al (2010) Plasma adiponectin and mortality in critically ill subjects with acute respiratory failure. Crit Care Med 38(12):2329–2334
Codoner-Franch P, Alonso-Iglesias E (2015) Resistin: insulin resistance to malignancy. Clin Chim Acta 438:46–54
Koch A, Gressner OA, Sanson E, Tacke F, Trautwein C (2009) Serum resistin levels in critically ill patients are associated with inflammation, organ dysfunction and metabolism and may predict survival of non-septic patients. Crit Care 13(3):R95
Sunden-Cullberg J, Nystrom T, Lee ML et al (2007) Pronounced elevation of resistin correlates with severity of disease in severe sepsis and septic shock. Crit Care Med 35(6):1536–1542
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Robinson, K., Prins, J., Venkatesh, B. (2016). Adipokines in Critical Illness. In: Preiser, JC. (eds) The Stress Response of Critical Illness: Metabolic and Hormonal Aspects. Springer, Cham. https://doi.org/10.1007/978-3-319-27687-8_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-27687-8_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-27685-4
Online ISBN: 978-3-319-27687-8
eBook Packages: MedicineMedicine (R0)