
Abstract
The hepatobiliary tract can develop tumor-like lesions or pseudotumors that consist of hematopoietic tissue or macrophage accumulations. Extramedullary hematopoiesis can occur in the liver and may result in macroscopically visible nodular lesions (nodular extramedullary hematopoiesis). Macroscopically, the lesions may have several cm in diameter and usually show a red-brown or tan color. One variant is characterized by the production of an abundant fibrous stroma (sclerosing extramedullary hematopoietic tumor). The liver can be the site of various eosinophilic pseudotumors and eosinophilic necrosis. Macrophage-rich lesions of the liver that can result in tumor-like nodular lesions comprise lipogranulomas, xanthomatous lesions, rheumatoid nodules, malakoplakia, and accumulations of Gaucher cells in Gaucher’s disease.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
- Gauche Disease
- Pyoderma Gangrenosum
- Hypereosinophilic Syndrome
- Rheumatoid Nodule
- Extramedullary Hematopoiesis
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Hematopoietomas and other Nodular Manifestations of Extramedullary Hematopoiesis in the Liver
Introduction
Extramedullary hematopoiesis (EMH) refers to the production of blood cells outside the bone marrow and is often a compensatory response to cope with marrow infiltration, marrow depletion, or marrow hyperactivity seen in certain chronic anemias (review: O’Malley 2007). Moreover, EMH is a typical feature of several myeloproliferative disorders. Mostly, EMH is a diffuse or “microfocal” process. However, there are situations where EMH presents in the form of gross nodular lesions or tumorous masses. Tumor-like extramedullary hematopoiesis (nodular EMH) was originally described in 1945 (Ask-Upmark 1945) and occurs at several sites, including the spleen, the kidney (Moskovitz et al. 1991; Tamiolakis et al. 2003), the intrathoracic compartment (Malamos et al. 1962), the adrenal gland (Ask-Upmark 1945), and the liver. Based on the cases reported so far, an intrathoracic paravertebral location seems to prevail (Verani et al. 1980). Nodular EMH has been observed in several hematologic and bone marrow disorders, including sickle cell disease (Verani et al. 1980; Lemos et al. 1997), congenital dyserythropoietic anemia (Heimpel et al. 2009), thalassemia major (Papavisiliou and Sfikakis 1964; Kumar et al. 1995), beta-thalassemia intermedia (Wong et al. 1999), beta-thalassemia trait (Ma and Au 1999), hemoglobin E-thalassemia disease (Da Costa et al. 1974), myelofibrosis (Navarro et al. 2000) as a manifestation of thymoma (Hervé et al. 2001), and medullary tuberculosis (Ben Rejeb et al. 1992).
Nodular Extramedullary Hematopoiesis of the Liver
Since the liver is a site for early hematopoiesis, it is not surprising that it is a relatively frequent site of extramedullary hematopoiesis. In pediatric patients, EMH is considered normal up to about 5 weeks of age. Nodular extramedullary hematopoiesis of the liver (NEMH-L) occurs in the form of solitary or multiple lesions of highly variable size, involving one or both liver lobes (Kopecky et al. 1986; Wiener et al. 1987; Abbitt and Teates 1989; Dewar et al. 1990; Bradley and Metreweli 1990; Warshauer and Schiebler 1991; Kumar et al. 1995; Tamm et al. 1995; Lemos et al. 1997; Aytac et al. 1999; Wong et al. 1999; Kwak and Lee 2000; Navarro et al. 2000; Hervé et al. 2001; Gil-Fernandez et al. 2001; Parwani and Ali 2003; Tamiolakis et al. 2004; Gupta et al. 2004; Jelali et al. 2006; Lee et al. 2008). Among eight cases with sufficient data compiled from the literature, five were multiple and three solitary lesions (Wong et al. 1999). NEMH-L can present as a solitary space-occupying lesion mimicking a primary liver neoplasm, mainly HCC, metastatic disease, FNH, or an atypical hemangioma (Wiener et al. 1987; Bradley and Metreweli 1990; Gil-Fernandez et al. 2001; Tamiolakis et al. 2004), thus promoting diagnostic fine needle aspiration (Dardi et al. 1990; Raab et al. 1993; Lemos et al. 1997). Metastatic disease may in particular be suspected in case NEMH-L produces several lesions with nodular hepatomegaly (Hervé et al. 2001). Sizes of the nodules have been reported to range from tiny lesions mimicking microabscesses (Kopecky et al. 1986) to 1.5 cm (Hervé et al. 2001), 3 cm (Wong et al. 1999), 7 cm (Parwani and Ali 2003), and 8 cm (Ma and Au 1999). Massive solitary NEMH-L has been observed in thalassemia (Dewar et al. 1990) and in myelofibrosis (Navarro et al. 2000). The masses may grow rather rapidly; particularly post-splenectomy; in one observation, the first hepatic manifestation was a 3 cm lesion, growing to 6 cm within 3 months (Wong et al. 1999). On the other hand, even very large lesions have been reported to remain unchanged for 4 years (Ma and Au 1999).
At ultrasound examination, the lesions are usually hypoechoic (Bradley and Metreweli 1990; Dardi et al. 1990; Dewar et al. 1990; Aytac et al. 1999; review: Wong et al. 1999). CT shows fairly well-defined, homogeneously low attenuated masses. On contrast-enhanced portal venous phase CT, most of the lesions show intense enhancement (Wong et al. 1999; Kwak and Lee 2000; Lee et al. 2008). The appearance of the lesions on MR consists of slightly increased signal on T2-weighted images with heterogeneous enhancement of some of the lesions during bolus infusion of gadolinium, while the T1-weighted images post-gadolinium showed no delayed enhancement (Warshauer and Schiebler 1991; Jelali et al. 2006). The imaging features for Doppler US (Aytac et al. 1999) and for Technetium-99 m red blood cell SPECT (Tamm et al. 1995) have been reported. When using the latter method, NEMH-L may mimic hepatic hemangioma (Tamm et al. 1995). Radiologically, NEMH-L can present as a fatty lesions, thus causing differential diagnostic difficulties (Gupta et al. 2004).
Macroscopically, NEMH are generally described as nodular and sometimes bulky, rather well-defined lesions, that are of medium consistency and show a cut surface that varies from red-brown or tan (in case of high vascularity and/or iron deposition) to grayish-white. Histologically, NEMH-L is composed of normal-looking hematopoietic tissue consisting of all three lineages (Figs. 1, 2, and 3). Few small cells with a lymphoid morphology may be present. It is not yet known whether such cells may, in addition to a lymphocyte population, represent hematopoietic stem cells. Similar to bone marrow, the hemopoietic tissue contains scattered macrophages with or without stainable iron. In addition, cells with a spindle-shaped morphology are detectable in small numbers (an analogue to marrow stromal cells?). In larger lesions, extracellular matrix may be increased, sometimes with focal fibrosis or sclerosis. In fact, NEMH-L can produce stellate scars visible in MRI and CT (Wong et al. 1999). The adjacent liver substance is compressed in large lesions, and perifocal hepatocytes may show iron overload (Ma and Au 1999). The morphologic differential diagnosis of NEMH-L chiefly includes hepatic myelolipoma and malignant tumors of the hemopoietic lineage.

Hematopoietoma of the liver, a mass-forming variant of extramedullary hematopoiesis. There is focal hemosiderosis (hematoxylin and eosin stain)

Hematopoietoma of the liver. This lesion is associated with formation of adipose tissue (hematoxylin and eosin stain)

Extramedullary hematopoiesis of the liver with production of megakaryocytes (CD61 immunostain)
Sclerosing Extramedullary Hematopoietic Tumor of the Liver
Sclerosing extramedullary hematopoietic tumor (SEMHT) is a rare lesion that typically arises in patients with chronic myeloproliferative disorders (Remstein et al. 2000; Yang et al. 2002; Sukov et al. 2009). Multiple intra-abdominal nodules of SEMHT can develop after splenectomy in patients with chronic myeloproliferative syndrome (Gualco et al. 2010). Macroscopically, these lesions are soft to firm, gray-white to yellow-tan nodular masses. Histology reveals extramedullary hepatopoiesis with atypical megakaryocytes, granulocytic precursors, and erythroid precursors set in a background of dense collagenous sclerosis. SEMHT has been observed in the ligamentum teres of the liver, in one patient in association with chronic idiopathic myelofibrosis (Kwon et al. 2004).
Benign Extramedullary Hematopoietic Proliferations of the Biliary Tract
In very rare instances, benign extramedullary hematopoiesis develops in and around the walls of large bile ducts. These foci of hematopoiesis occur in myeloid neoplasms destroying the bone marrow and in certain other situations of marked secondary extramedullary hematopoiesis. In massive forms, the intrahepatic biliary tract may be encased by the hematopoietic tissue, causing hepatic failure (La Fianza et al. 2010).
Hepatic Myeloid Hyperplasia in Caffey’s Disease
Infantile cortical hyperostosis (ICH; Caffey’s disease; OMIM 114000) was described in 1945 (Caffey and Silverman 1945). The disorder is characterized by cortical thickening of affected bones (mainly diaphysis of long bones) and inflammatory swelling of the contiguous soft tissues (reviews: Glorieux 2005; Kamoun-Goldrat and le Merrer 2008). The acute manifestations are usually inflammatory in nature, with fever and hot swelling of involved bones, clinically suggesting osteomyelitis. ICH usually manifests in young infants (the classical infantile form), but is sometimes also seen in neonates and in fetuses as early as 20 weeks of gestation (Herman 1996; Schweiger et al. 2003). A distinct variant is early-onset prenatal ICH (Lecolier et al. 1992). The classical form reveals autosomal-dominant inheritance (Fried et al. 1981), whereas autosomal recessive inheritance has been suggested for the early-onset prenatal variant. The disorder is assigned to chromosome 17q21-31-q22. Patients with classical ICH have been shown to have a 3040C>>>T mutation in the COL 1A1 gene (Gensure et al. 2005; Cho et al. 2008; Kamoun-Goldrat et al. 2008). Early-onset prenatal ICH, the form which shows liver alterations, is divided into two forms, a severe form with onset before 35 weeks of gestation, associated with polyhydramnios, lung disease, hydrops fetalis, anasarca, hepatomegaly, prematurity, and high lethality, and a mild form, with onset after 35 weeks of gestation and lack of complications (Schweiger et al. 2003). In a detailed autopsy analysis of a patient with early-onset prenatal ICH, hepatomegaly was found to be caused by marked myeloid hyperplasia. Myeloid hyperplasia was most prominent in the tissue surrounding the portal tracts, and myeloid precursors were located within hepatic sinusoids. In contrast, an increase in erythropoiesis was not noted. This extramedullary myeloid hyperplasia was suggested to be caused by the generalized skeletal inflammatory process characterized by ICH, with microabscess formation and widespread granulation tissue at the interface between bone and periosteum (Wright et al. 2005).
Focal Eosinophilic Infiltration of the Liver
Introduction
Hypereosinophilia is associated with several conditions, comprising allergic reactions, parasite infestations, connective tissue diseases, and neoplastic processes, but idiopathic forms are also recognized. Idiopathic hypereosinophilic syndrome (HES) is a heterogeneous group of disorders that has been defined as peripheral eosinophilia >1500/mm3 for at least 6 months and evidence of end-organ involvement in the absence of an identifiable cause (Leiferman and Gleich 2004). A subset of patients with a myeloproliferative variant of hypereosinophilic syndrome (MHES) is characterized by elevated serum tryptase levels, increased atypical mast cells in the bone marrow, tissue fibrosis, and the presence of the fusion tyrosine kinase, Fip1-like 1 (FIP1L1) – PDGFRalpha (generated by a cryptic interstitial deletion on chromosome 4q12; del(4)(q12q112)), a therapeutic target of imatinib mesylate (Griffin et al. 2003; Klion et al. 2003; Cools et al. 2003; Pardanani et al. 2003; Klion et al. 2004a). This novel fusion kinase is detectable in chronic eosinophilic leukemia (CEL), MHES, and systemic mast cell disease (Coutre and Gotlib 2004; Cools et al. 2004; Gotlib et al. 2004). Recently, familial eosinophilia (FE; OMIM 131400) has been characterized, a disorder that can also cause end-organ damage (Klion et al. 2004b).
Liver Alterations in Hypereosinophilic Syndromes
Hypereosinophilic syndrome may cause eosinophil-related tissue alterations in various organs, including the gastrointestinal tract (eosinophilic gastrointestinal disorders, EGID; Rothenberg 2004) and the liver (Table 1).
In a study on 13 patients, mild to moderate hepatomegaly was found in all cases, and seven patients exhibited multiple round or oval hypoechoic or variably echogenic lesions measuring 1–2 cm with poorly defined margins in both lobes of the liver, but four patients showed lesions of up to 4 cm diameter. The number of lesions and the extent of diffuse lesions seemed to be proportional to the degree of eosinophilia (Nam et al. 1999). The lesions may mimic multiple liver metastases or hepatocellular carcinoma (Won et al. 1999; Jang et al. 2002; Kwak et al. 2004). CT demonstrates well-defined, homogeneous, or heterogeneous low attenuation with a straight margin limited to a hepatic lobe, segments, or subsegments, in the latter situation with multiple, ovoid, or wedge-shaped lesions (Lim et al. 2000; Yoo et al. 2003).
Pathologically, hypereosinophilia causes several groups of hepatobiliary lesions. In cases of low to moderate eosinophilia, an infiltrate dominated by eosinophils is chiefly identifiable in portal tracts, but the number of these cells is also increased within sinusoids and around the central veins (Fig. 4; Lim et al. 2000). The liver involvement in marked peripheral eosinophilia can present as cholestatic liver disease (Valente et al. 1997). In case of direct hepatic involvement by parasites (fasciola, clonorchis, and other liver flukes; visceral larva migrans, e.g., Toxocara or Ascaris suum; Capillaria; echinococcosis), the dense eosinophilic infiltrate is found in close association with the parasite, sometimes to a degree to form a focal eosinophilic infiltration, an eosinophilic abscess, or an eosinophilic pseudotumor (Pulpeiro et al. 1991; Han et al. 1993, 1996, 1999; Bhatia and Sarin 1994; Murrell et al. 1997; Kim et al. 1999, 2002; Hayashi et al. 1999; Andresen et al. 2000; Azuma et al. 2002; Sakakibara et al. 2002). These lesions are further discussed in another chapter.

Focal eosinophilia of the liver (hematoxylin and eosin stain)
The hypereosinophilic lesions may contain accumulations of Charcot-Leyden crystals (CLC; Fig. 5). These crystals accumulate in any situation where numerous eosinophils are present in tissues, including eosinophilic abscess (Fig. 6). The crystal constituents show indirect lysophospholipase activity (galectin-10, CLC protein; chromosome 19; Mastrianni et al. 1992; Zhou et al. 1992; Leonidas et al. 1995). The galectins form a family of proteins playing a role in myeloid differentiation (Dyer et al. 1997; Abedin et al. 2003) and include, apart from the CLC galectin-10, the placental protein 13 (galectin-13) that also has lysophospholipase activity. The CLC protein, galectin-10, is not a lysophospholipase as such, as has been sometimes reported, but a protein that binds a lysophospholipase inhibitor in a distinct structural fashion (Ackerman et al. 2002). CLC are not only formed by eosinophils but also by basophil granulocytes (Ackerman et al. 1982).

Charcot-Leyden crystalloids in focal eosinophilia of the liver (hematoxylin and eosin stain)

Eosinophilic abscess of the liver. The abscess mainly consists of eosinophil granulocytes, with cell decay and nuclear debris in the center (hematoxylin and eosin stain)
Focal Eosinophilic Necrosis of the Liver (FEN)
A second and distinct situation occurs when eosinophil infiltration of the liver is associated with tissue necrosis, a condition termed focal of eosinophil-related necrosis, eosinophilic hepatic necrosis (Figs. 7, 8, and 9; Ung et al. 2000; Hur et al. 2005; Laghi et al. 2010), focal eosinophilic necrosis (FEN; Jang et al. 2002), or focal eosinophilic liver disease (FELD; Kim et al. 2010). FEN is a descriptive term based on pathologic features and has not yet been clearly defined (Jang et al. 2002). In principle, FEN is a distinct type of parenchymal necrosis (mostly periportal), associated with eosinophilic infiltration. The main difference to focal eosinophilic infiltration and eosinophilic abscess is the presence of necrosis, thought to be caused by cytotoxic proteins released from eosinophils, but necrosis also occurs in abscesses and the separation of the entities is therefore somewhat arbitrary. FEN seems to almost always be accompanied by peripheral eosinophilia, and it occurs in several conditions causing eosinophilia, including lymphoma, leukemia, and carcinomas (Lee et al. 1999, 2011); Jang et al. 2002; Yu et al. 2005; Choi et al. 2009, 2010. In these instances, eosinophil-recruiting facts such as tumor-associated eosinophil-chemotactic factor are thought to be involved (Wasserman et al. 1974).

Focal eosinophilic necrosis of the liver. Collapsed areas of necrosis with eosinophilia and nuclear debris are important features (hematoxylin and eosin stain)

Focal eosinophilic necrosis of the liver. The necrotic aspect of the lesion is more prominent in this case (hematoxylin and eosin stain)

Focal eosinophilic necrosis of the liver. The area of eosinophilic necrosis is demarcated by activated, epithelioid macrophages. Note few Charcot-Leyden crystalloids within the necrosis (hematoxylin and eosin stain)
Pathologically, FEN is characterized by focal coagulative necrosis of hepatic parenchyma associated with an infiltration of eosinophils. The necrosis is, due to high protein content, markedly eosinophilic, but this oxyphilic staining features may also be caused by the high content of the necrosis of eosinophil granules and debris. In the adjacent tissue and particularly in portal tracts, an eosinophilic infiltration is noted.
Xanthomatous Lesions of the Liver
Introduction
Xanthomas are solitary or multiple lesions characterized by grossly yellowish to frankly yellow plaques (Greek: xanthos, yellow) histologically consisting of an accumulation of lipid-rich macrophages/histiocytes. Xanthomas chiefly occur in several types of hyperlipidemia/hyperlipoproteinemia and mainly involve the skin and the soft tissues, but may also involve deep-seated tissues. In periocular areas, the term xanthelasma is employed (Greek: elasma, plaque; yellow plaque). Xanthomas were first described by Rayer under the term “plaques jaunatres des paupières” (what exactly means, “yellowish plaques,” i.e., xanthelasmas; Rayer 1835). In 1851, the same type of lesions was described under the term “vitiligoidea,” with a “plana” and a “tuberosa” variant (Addison and Gull 1851). The term, xanthelasma, was coined in 1863 by Wilson, while the tumor-like appearance of some of the yellow nodules led to the name xanthoma by Smith in 1869.
Liver Xanthomas
Xanthomas and xanthogranulomas of the liver are rare lesions, but may produce multiple nodular changes suggesting manifestations of a neoplastic process (Figs. 10, 11, and 12; Tanino and Ohta 1981). Probably the first description of hepatic xanthoma is the report by Chvostek in 1911, who also used the term xanthoma of the liver (Chvostek 1911). Before, other terms had been used, such as “xanthomatous tubercles” (Hardaway 1884). A similar case has been described in 1938, based on a partial autopsy (performed by Ludwig Aschoff) of a patient with liver cirrhosis and partially eruptive skin xanthomas (Thannhauser and Magendantz 1938). In part of the cases, the hepatic xanthomatous reaction was suggested to be caused by hyperlipidemia. For example, multiple liver xanthomas have been found in multiple myeloma associated with hyperlipidemia (Yim et al. 1995). In proliferative plasmacytic disorders, xanthomas are known to occur in other locations, and lipid deposits may, apart from hyperlipidemia, be caused by paraprotein Ig-mediated complexing of lipoproteins (Moschella 1970; Roberts-Thomson et al. 1975; Taylor et al. 1978). A xanthomatous mass at the liver hilum has been described in a 9-year-old boy with generalized xanthomatosis and xanthoma tuberosum (Weidman and Freeman 1924). Xanthomas may also involve the liver capsule (Weidman and Boston 1937). In hyperlipoproteinemia associated with extensive xanthoma tuberosum, hepatocytes in addition to macrophages may take part in lipid storage, sometimes resulting in parenchymal changes reminiscent of adrenocortical tissue (Weidman and Stokes 1937). Xanthomatous lesions of the liver parenchyma itself also occur in obstructive cholestasis; the lesions are characterized by the accumulation of foamy macrophages chiefly in the periportal zone (“xanthomatosis of the liver,” Thannhauser and Magendantz 1938; Desmet 1995; Woollons and Darley 1998). Multiple xanthomatous foci have been reported to occur in the liver in the systemic form of juvenile xanthogranuloma (Chantranuwat 2004), a disorder further discussed in the chapter of Langerhans cell histiocytosis.

Tumor-like hepatic xanthoma (hematoxylin and eosin stain)

Xanthoma of the liver. Numerous lipid-rich foamy macrophages occupy the tissue, associated with focal lymphocytic infiltration (hematoxylin and eosin stain)

Hepatic xanthoma. In this lesion, numerous cholesterol clefts are noted (hematoxylin and eosin stain)
Xanthomatous Cholangitis (Xanthomatous Cholangiopathy)
An interesting group of lesions has been termed xanthomatous cholangitis , now rarely found in the current literature. The lesion spectrum has been summarized under the term “xanthomatosis of the bile ducts” (Thannhauser and Magendantz 1938). Early reports go back to the end of the nineteenth century (Moxon 1873) and the beginning of the twentieth century (Futcher 1905). We propose the term xanthomatous cholangiopathy, to denote these lesions, as this alteration is not primarily an inflammatory disorder, but rather a lesion characterized by the focal accumulation of lipid-laden macrophages within the inner parts of medium-sized and large bile ducts, albeit usually with some lymphocytic infiltration. This lesion has mainly been seen in patients with biliary cirrhosis or diabetes mellitus and usually eruptive, sometimes papulopustular, skin xanthomas. At gross examination, the lesions are spheroid to oblong, elevated, rather sharply delineated spots or patches, sometimes showing a dimple in the center (molluscum-like lesions). In some patients, the involved bile ducts were dilated, had thickened walls, and were lined by bright yellow granular incrustive material (Weidman and Boston 1937). In an another patient, the walls of the bile ducts were of a deep yellow color, and there were minute yellow flecks in the mucosa of the bile ducts (Futcher 1905). Histology is characterized by a patchy accumulation of foam cells involving the mucosa and, in larger lesions, inner parts of the bile duct muscularis. The lesions may be accompanied by impressive hyperplasia of the peribiliary glands (Weidman and Boston 1937) and by scarring causing stenosis or even obstruction (Weidman and Boston 1937; Thannhauser and Magendantz 1938). The pathology has already been described in early reports. It is difficult to judge of what these patients had suffered, but reading the original works suggests that most of them may have had primary or secondary biliary cirrhosis, or diabetes mellitus. In one patient with xanthomata diabeticorum, the postmortem examination showed dilated bile ducts with white to yellowish, opaque, xanthelasma-looking patches on the mucosal surface of the ducts (Moxon 1873). In another patient, the xanthomatous lesion was located to the bifurcation of the hepatic duct (Thannhauser and Magendantz 1938). The striking resemblance of bile duct xanthomas to the respective skin lesions, or to atheromas, has been acknowledged in an early report (“Patches precisely like those in the eyelids and hands were found on the surface of the spleen and in the mucous membrane of the dilated hepatic ducts.” “The patches in the ducts looked just like atheroma in the artery with which condition, indeed, they correspond histologically”; Pye Smith 1873). The xanthomatous patches may cause stenosis or occlusion of bile ducts involved (Futcher 1905; Weidman and Stokes 1937).
Hepatic Xanthomatous Reactions in Xanthomatous Cholecystitis
In severe xanthogranulomatous cholecystitis, the massive macrophage reaction with production of foamy cells can extend into the gallbladder bed and from there into the liver substance of the right lobe, mimicking cancer (Pinocy et al. 2003; Yang et al. 2007). Xanthogranulomatous cholecystitis, treated in more detail in a separate chapter, was first described in 1970 under the term fibroxanthogranulomatous cholecystitis (Christensen and Ishak 1970), while the term xanthogranulomatous cholecystitis was coined in 1976 (McCoy et al. 1976). Alternative terms include biliary granulomatous cholecystitis, ceroid-like histiocytic granuloma, and ceroid granuloma. The change is found in 1.2 % of cholecystectomy specimens. It is not yet clear whether the incidence of carcinoma is increased in the presence of this distinct type of inflammation (Benbow 1990; Franco et al. 1990; Reed et al. 1994; Parra et al. 2000). Histologically, three different types of xanthogranulomatous cholecystitis are distinguished, i.e., multinodular, focal, and diffuse, the latter being the least common (Franco et al. 1990). Apart from an extension of the xanthomatous (or better pseudo-xanthomatous) reaction into the liver, this type of cholecystitis has been reported to be associated with liver abscess (Eriguchi et al. 2002).
Xanthomatous Inflammatory Pseudotumors
A further group of masses located to the liver and sometimes containing large numbers of lipid-laden, foamy macrophages are the several variants of inflammatory pseudotumor, discussed in more detail in a separate chapter. The accumulation of fatty macrophages can result in xanthomatous features; therefore, these lesions have also been termed xanthogranuloma of the liver. In some situations, the masses are large and present as lobulated, yellowish tumors measuring up to 7 cm in diameter, with septate fibrotic bands, or are manifest as multiple yellow nodules of few cm size, with or without abscesses in the vicinity (Noi et al. 1994; Tai et al. 1998; Yoon et al. 1999; Yoshida et al. 2003). Histology is characterized by a complex mixture of foamy macrophages, plasma cells, lymphocytes, and granulation tissue. A high lipid content of pseudotumors are in part due to accompanying purulent changes with massive decay of lipid-rich granulocytes, such as pyogenic cholangitis (Yoon et al. 1999).
Hepatic Lipogranulomatosis (Farber’s Disease)
Disseminated lipogranulomatosis Farber or Farber disease is a rare inherited autosomal recessive disorder of lipid metabolism in the childhood population due to a genetically determined defect in ceramide degradation (acid ceramidase deficiency), with ceramide accumulation in the lung, liver, bowel, skeletal muscle, cartilage, and bone. Ceramidase splits ceramide into free fatty acids and sphingosine. The storage cells show lysosomal inclusions containing lamellar and curvilinear membrane profiles, zebra structures, and banana bodies (Farber bodies). This storage process is followed by a sometimes massive lipogranulomatous reaction associated with the presence of macrophages, lymphocytes, and fibroblasts. Liver involvement is clinically manifest as hepatomegaly and histologically in the form of dense focal infiltrations of ceramide-containing storage cells (histiocytes/macrophages). Lipid-containing foam cells can form hepatic granuloma-like lesions (Koga et al. 1992). The multiple, up to pinhead-sized lesions in the liver, may already be evident in the newborn period (Schäfer et al. 1996). Type 4 disease (see below) is particularly prone to develop marked lipogranulomatous changes in the liver, sometimes forming tumor-like masses. Liver involvement in type 4 Farber’s disease can cause neonatal liver failure (Willis et al. 2008). Lipid-laden foam cells in Farber’s disease may develop in suture foreign body granulomas of the liver (Koga et al. 1992).
Farber’s lipogranulomatosis (Farber disease; ceramidase deficiency; OMIM 228000) is a rare genetic, autosomal recessive lysosomal storage disease caused by acid ceramidase (E.C. 3.5.1.23) deficiency, which leads to ceramide accumulation in several organs (Koch et al. 1996; review: Ehlert et al. 2007). Farber’s disease presents with six phenotypes. Type 1 is the classic variant with early subcutaneous nodules and joint involvement, and often progressive neurologic involvement and lung disease. Type 2 and 3 patients show only slight alterations of the central nervous system, but severe manifestations in the soft tissues. Type 4 patients show severe neurologic deterioration and marked hepatosplenomegaly already in the neonatal period. Type 5 is characterized by progressive CNS dysfunction, beginning already at 1–2 ½ years of life. Finally, type 6 is a combination of Farber’s disease and Sandhoff’s disease. Ceramide is an important cellular lipid involved in signal transduction, the synthesis of complex sphingolipids, and in apoptosis pathways (el Bawab et al. 2002; Pettus et al. 2002). The Farber phenotype has been divided into seven different subgroups that differ in the age of onset, severity of symptoms and signs, and tissue involvement of ceramide storage. The acid ceramidase gene has 14 exons and 13 introns, and mutations so far described in humans have recently been reviewed (Zhang et al. 2000; Bar et al. 2001; Muramatsu et al. 2002). In the mouse, insertional mutagenesis of the acid ceramidase gene cause early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes (Li et al. 2002).
Differential Diagnosis
Hepatic xanthomas have to be distinguished from situations where visible depositions of cholesterol-rich lipids involve liver cell systems in a diffuse distribution pattern (e.g., “hepatic cholesterolosis” in hepatic cholesterol storage disease; a minor variant of Wolman’s disease; Verola et al. 1983). This distinct type of lipid accumulation in several hepatic cell systems has also been observed in the respective animal model, the Wolman’s disease rat or Yoshida rat (Kuriwaki and Yoshida 1999).
Lipogranulomas, Lipogranulomatosis, and Mineral Oil Granulomas of the Liver
Hepatic Lipogranuloma
Hepatic lipogranulomas in a wider sense of the term are focal accumulations of macrophages that have pinocytosed neutral fat and other lipids. Lipogranulomas are well known to occur in adipose tissue following adipocyte damage with release of neutral fat from injured or dead cells, but they also occur in fatty livers subsequent to steatotic hepatocyte injury and death. In case of numerous such lesions, the term lipogranulomatosis is sometimes used. Clusters of hepatic lipogranulomas can be identified in vivo by means of modern imaging techniques and may present as small tumor-like lesions.
The morphologic definition of lipogranulomas led to the inclusion, apart from those caused by neutral fat, also of reactions to exogenous substances inducing a similar or the same histology, e.g., mineral oil. Lipogranulomas sensu stricto represent a macrophage- and lymphocyte-driven reaction to triglycerides (neutral fat) (Iversen et al. 1970; Christoffersen et al. 1971). They mainly occur in the setting of hepatic steatosis of any cause (Christoffersen et al. 1971; Andersen et al. 1984; Ferrell et al. 1995). In a study of 61 morbidly obese patients, patients showing lipogranulomas in liver biopsies were significantly more obese than patients without this change (Andersen et al. 1984). In a study comparing patients with nonalcoholic steatohepatitis (NASH) and alcoholic steatohepatitis (ASH), lipogranulomas were more remarkable in the ASH group (Morita et al. 2005). The etiology of hepatic lipogranulomas depends, however, markedly on epidemiological factors. Lipogranulomas are also a features of some types of chronic HCV infection. Among 376 sequential, archival liver biopsy specimens examined in New York, 58 (15.4 %) had hepatic lipogranulomas, including 46 patients with hepatitis C and 14 patients with fatty liver disease. Hepatic lipogranulomas were more often seen in patients with hepatitis C (21 %) and fatty liver disease (18 %). Therefore, hepatitis C, which can be associated with hepatic steatosis, is an additional important cause of lipogranulomas (Zhu et al. 2010).
The prevalence of hepatic lipogranulomas is only partially known, and the data are markedly related to the sampling used (biopsies vs. autopsies) and to the histologic methods employed (paraffin sections vs. lipid staining). Part of the investigations include lesions that may have been caused by exogenous oily substances, such as mineral oil, hence not representing true lipogranulomas. In liver biopsies, lipogranulomas have an estimated frequency of 2.4–5 % (Delladetsima et al. 1987; Scheuer and Lefkowitch 2006). Portal tract lipogranulomas were recognized in 2.4 % of biopsies and were consistently associated with hepatic steatosis (Delladetsima et al. 1987). In an autopsy study (465 autopsies), they were found in 48 %, which is a very high figure that may markedly depend on the autopsy material and the criteria used (Wanless and Geddie 1985). In this study, hepatic lipogranulomas were more common in portal tracts than adjacent to the terminal hepatic venules and more frequent in older subjects (especially men). These two findings suggest that, in addition to neutral fat, mineral oil exposure (see below) may have played a significant role in the etiology of the lipogranulomas found in this investigation (what also the authors took into consideration).
Pathology
In case of larger lipogranulomas, the yellowish cut surface of steatotic livers may show discrete spots of a darker yellow, usually of the size of 1–2 mm. Numerous lipogranulomas forming dense clusters appear as yellow granular collections of coalescing lesions. As in other organs and tissues, lipogranulomas of the liver are histologically defined as roughly spheroid accumulations of activated macrophages and lymphocytes. In early lesions, the macrophages (Kupffer cells) surround extracellular lipid droplets (vacuoles in paraffin sections). The macrophages may show discrete epithelioid changes in some cases. The periphery of lipogranulomas mostly exhibits only few lymphocytes. Older lesions reveal less vacuoles, but the macrophages have pinocytosed the lipid and have become lipophages (foamy cells). Some lipogranulomas may contain multinucleated giant foam cells of the Touton type (Touton giant cells; see below), although Touton cells are more frequently encountered in xanthomatous lesions described in the previous chapter. Hepatic lipogranulomas are usually microscopic, but they may grow to macroscopically recognizable size and, in case they form conglomerate lesions, may be seen radiologically. Rarely, lipogranulomas result in a generalized process in the liver, progressively replacing the liver parenchyma and portal tracts.
Where are lipogranulomas typically located within the liver? Many authors noted that hepatic lipogranulomas are usually located in portal tracts (Stryker 1941; Goldberg and Saphir 1960; Boitnott and Margolis 1970; Nochomovitz et al. 1975; Blewitt et al. 1977), which may more often represent mineral oil granulomas, while other authors found most lipogranulomas in the pericentral lobule zone 3 (Christoffersen et al. 1971; Klatskin 1977; Dincsoy et al. 1982). True neutral fat lipogranulomas will probably develop at sites where most steatotic hepatocytes are damaged or are decaying. Ultrastructurally, fat droplets in severely fatty livers are encircled by lymphoid cells and histiocytoid cells. There was evidence that fat droplets protruded through the cell membranes of hepatocytes, followed by settling of macrophages/histiocytes and lymphocytes around such fat protrusions. Remnants of liver cells may be seen between the fat droplets and the macrophages (Petersen and Christoffersen 1979). Lipogranulomas consist of lipid-storing macrophages that are CD68-immunoreactive. It is not yet known whether cells of the hepatic macrophage system, i.e., Kupffer cells, are recruited to form granulomas, or whether blood-borne monocytes attracted to lipid accumulations are involved. However, prominently enlarged and aggregated Kupffer cells were observed in steatohepatitis, with a predominantly perivenular distribution, suggesting a potential direct Kupffer cell role in hepatic lipid processing (Lefkowitch et al. 2002).
Hepatic Phlebocentric Lipogranulomatosis
This is a very rare disorder of unknown cause that has first been described based on autopsy findings in two patients with no history of liver disease (Keen et al. 1985). The livers showed congestive alterations caused by a process mimicking veno-occlusive disease. In both patients, central veins were occluded by an inflammatory and fibrosing process, with a striking accumulation of lipid-containing macrophages showing large fat vacuoles (Oil Red O-positive and osmiophilic). Giant cells were also noted. In one of the patients, the process had extended into the sublobular venous radicles, where intimal proliferation and sclerosis were present. The etiology of these lesions remained unknown, but an environmental insult such as a drug or toxin were suspected.
Other Types of Granulomas Containing Neutral Fat
An increased prevalence of hepatic lipogranulomas (56 %) was observed in patients with rheumatoid arthritis who had been routinely biopsied to evaluate side effects of methotrexate therapy. The lipogranulomas contained a dark pigment shown, in few cases, to represent gold-containing particles (chrysiasis). It was, therefore, assumed that lipogranulomas were caused by the frequent administration of gold in an oily vehicle (Landas et al. 1992).
Mineral Oil Granulomas
Lipogranulomas in the liver can be induced by exogenous lipoid-like substances or substances histologically mimicking lipid accumulation, such as mineral oil (Stryker 1941; Boitnott and Margolis 1970; Nochomovitz et al. 1975; Dincsoy et al. 1982; Fleming et al. 1998) or paraffin oil (Trivalle et al. 1991). This issue has already been discussed briefly above. Mineral oil has been used or is still used for the treatment of constipation and enters the food chain owing to its use as a lubricant for machines, e.g., in bakeries.
What Are Touton Giant Cells?
Touton giant cells are large and multinucleated cells resembling foreign body-type giant cells, but usually with a ringlike arrangement of the nuclei and a central vacuolated area containing numerous lipid droplets. Although not investigated so far, it is assumed that these giant cells, similar to other multinuclear giant cells, develop via fusion of macrophages mediated by several cytokines/lymphokines (McNally and Anderson 1995; Anderson 2000). The eponymic designation goes back to Karl Touton (1858–1934), a German dermatologist. Student of Ernst Ziegler, Moriz Kaposi, Albert Neisser, and Isidor Neumann, and after studies in Augsburg and Breslaw, he settled in Wiesbaden as a dermatologist and built up an international reputation (Herxheimer 1935; Gougerot 1935; Gans 1964; Holubar 1990). In this function, he published several articles on phytodermatosis and described his “xanthelasmic giant cell” (Touton 1885; Aterman et al. 1988). Touton wrote his survey on xanthomas when he was only 25 years of age. Touton described the characteristic features of multinucleated giant cells occurring in xanthomatous lesions, but the presence of such cells in lesions, then called “endothelioma adiposo,” had been noted earlier already by an Italian author, who noted large cells showing numerous nuclei “da impartire loro la forma de celluli giganti” (“rendering them the feature of giant cells”; De Vincentiis 1883; reviewed in Aterman et al. 1988). Apart from Touton’s activities as a physician and professor of medicine, he was an outstanding botanist specialized mainly in the composite genus, Hieracium. He published numerous botanical works and left a Hieracium herbarium consisting of about 20,000 specimens, now kept in the Botanical Museum Berlin-Dahlem, Germany (Vogt 1998). Among this genus of hawkweed, one species has his name (Hieracium toutoni).
Necrobiotic Granulomas of the Liver (Rheumatoid Nodules, Rheumatoid Nodulosis, Rheumatismus Nodosus Hepatis)
Introduction
Patients with seropositive rheumatoid arthritis develop rheumatoid nodules (RN; Hart 1994). RN have already been recognized as a soft tissue manifestation of rheumatoid arthritis in the nineteenth century (Hilliers in 1868 and Jacoud in 1874), but the first detailed description is by Meynet (1875). The pathology of rheumatism was studied in detail by the pathologists Aschoff, Klinge, and Gräff (review: Keitel 2008). The term, rheumatismus nodosus, had been coined by Rehn. Classic RN occur in approximately 20–25 % of patients with seropositive rheumatoid arthritis and are the most common extra-articular manifestation of this disease (Kaye et al. 1984; Ziff 1990). A much greater incidence (75 %) is observed in patients with rheumatoid arthritis-associated Felty syndrome. The HLA-DR4 haplotype is predictive of the risk of subcutaneous RN in rheumatoid arthritis. Homozygosity for HLA-DRB1 *0401 makes rheumatoid arthritis patients susceptible to major organ involvement. These nodules may evolve very rapidly, within few hours to days, and grow up to few cm in diameter, may form conglomerate lesions, and are known to also develop in visceral organs and tissues. The nodules can undergo accelerated growth (subsequent to therapy, e.g., with methotrexate), but can also regress spontaneously (McCarty 1991; Garcia-Patos 2007; Highton et al. 2007).
Rheumatoid Nodules of the Hepatobiliary Tract (“Rheumatismus Nodosus Hepatis”)
RNs (nodulosis) in the liver are very rare (Fig. 13). An autopsy report referring to a patient with chronic rheumatoid polyarthritis documented, apart from widespread amyloidosis, numerous grayish-white nodules with a peripheral zone scattered throughout the liver, their maximum diameter being 5 mm. Histologic analysis of these nodules disclosed a central necrosis surrounded by histiocytes/macrophages in a palisade arrangement, and peripheral fibrosis associated with chronic inflammation. This morphology closely resembled that of typical rheumatoid nodules in observed in the subcutis (Smits and Kooijman 1986). RN rarely also develop in the mesentery and may mimic an intra-abdominal malignancy (Thinda and Tomlinson 2009).

Rheumatoid nodule of the liver. A focus of fibrinoid necrosis (center) is surrounded by a palisading granuloma (hematoxylin and eosin stain)
What Are Rheumatoid Nodules?
Rheumatoid nodules (RN, tumefactive necrobiotic granulomas; nodulosis) are histologically characterized by a central, so-called fibrinoid, necrosis showing a typical, “geographic” shape, a demarcation zone consisting of epithelioid macrophages (sometimes arranged in a typical palisading, radial, or corona-like pattern), and a peripheral rim of fibrosclerotic tissue (Ziff 1990). The histopathologic progression of RN occurs in three stages: an acute inflammatory stage, a granulomatous stage, and finally a necrotic stage. In the acute inflammatory stage, a nonspecific granulation tissue develops as a reparation response. The second or granulomatous stage is the stage of massive macrophage reaction with palisading. Fibrinoid necrosis develops in the preexisting collagenous tissue, but also involves the macrophages of the granulomas. The most prominent cells of the rheumatoid nodule belong to the monocyte/macrophage lineage. These cells migrate from the vascular periphery of the nodule toward the central palisade layer (Palmer et al. 1987a). The migrating cells undergo differentiation to epithelioid macrophages (so-called maturation) and release cytokines and the MRP-8/MRP-14 heterodimer, labeling them as newly arrived activated macrophages (Palmer et al. 1987b; De Rycke et al. 2005). The immigrating macrophages and their epithelioid offspring accumulate in the palisade interspersed with myofibroblasts and fibroblasts. In addition to monocytes /macrophages, lymphocytes are present in and around the RN. Both CD4 and CD8 subtypes are detectable in the nodules, and these cells tend to accumulate around blood vessels and in the area immediately outside the palisade (Highton et al. 2007). Lymphoid cells in this tissue space are attracted by chemokines produced by the nodules, including CCL21, CXCL12, and CXCL13. The peri-palisade compartment also contains dendritic cells (Highton et al. 2000). In contrast to T cells, B lymphocyte density around RN is more variable, and B cells in RN are mostly sparse and sometimes almost undetectable, but B cell signaling and the formation of germinal centers has been reported. The markedly eosinophilic necrosis was initially interpreted to contain large amounts of fibrin (Fahr 1918), finally resulting in the term fibrinoid necrosis to denote the fibrin-like staining of the central necrosis. The name “fibrinoid” goes back 1880, when Neumann described a new picrocarmine staining to demonstrate the features of “fibrinoid degeneration” (Neumann 1880; reviewed in Bajema and Bruijn 2000). A change from “degeneration” to “necrosis” appeared in 1932 (Klinger 1932), and still later, several morphologic variants of fibrinoid necrosis were distinguished (Zeek 1952). In regard to the composition of fibrinoid necrosis, it was first assumed that precipitation of fibrin and/or other coagulation factors, necrosis of collagen, and coagulation of the intercellular ground substance, or a combination of these, were involved (Altshuler and Angevine 1949). In fact, fibrin is one component that is detectable in this type of lesions (Gitlin et al. 1957). But later it was found that the eosinophilic material of fibrinoid at least in part consists of fibronectin isoforms indicating that the lesion is actually part of a wound-healing process (Bajema et al. 1998). RN are also termed tumefactive necrobiotic granulomas, or nodulosis (Riedlinger et al. 2005), and rheumatoid nodulosis employed for variants of rheumatoid arthritis with numerous soft tissue nodules and an unusual, rather benign course (Ginsberg et al. 1975; Wisnieski and Askari 1981; Couret et al. 1988; Goni et al. 1992).
Malakoplakia of the Hepatobiliary Tract
Introduction
Malakoplakia (Greek: soft plaque; frequently used term: malacoplakia, von Hansemann’s disease) is an unusual inflammatory process that was first described in the early 1900s (Michaelis and Gutmann 1902), although the discovery of the lesion as such goes back to Michaelis and Gutmann’s senior colleague, von Hansemann (reviews: Damjanov and Moriber Katz 1981; Stanton and Maxted 1981; Dasgupta et al. 1999). Malakoplakia is mostly known from the urinary tract, but it may also involve extra-urinary sites (Yousef and Naghibi 2007), including the appendix, colon, spleen, skin, frontal and ethmoidal sinuses, tongue, lungs, lymph nodes, and brain.
The eponymic designations go back to Carl Gutmann (born 1872), a German physician, and Leonor Michaelis (1875–1947). Leonor Michaelis, an assistant of Paul Ehrlich, is particularly known for his role in the formulation of the Michaelis-Menten law, equation, and constant in enzymology (1913). Maud Leonora Menten (1879–1960), one of the first Canadian women to become a doctor, came to Berlin in 1912 to work together with Michaelis on invertase enzyme activity, and together they developed their now famous equation. Michaelis also discovered Janus green as a supravital stain for mitochondria. Together with Sam Granick, he furthermore characterized ferritin and apoferritin. However, the first human case of malakoplakia was in fact seen by von Hansemann in 1901, who had read about a similar disease described by Olt in a veterinary magazine in 1900. von Hansemann provided his assistant, Dr. Gutmann, with the details of his case, as Dr. Michaelis had agreed to investigate the disease in a collaborative approach. von Hansemann himself published this index case 1 year after Michaelis and Gutmann (Von Hansemann 1903; review: Dasgupta et al. 1999). Malakoplakia may, hence, be called von Hansemann’s disease.
Malakoplakia of the Hepatobiliary Tract
Malakoplakia of the liver is a rare condition (Moldavskii and Rustamov 1984; De Saint-Maur and Gallot 1990; Robertson et al. 1991; Boucher et al. 1994; Chen et al. 1994; Hartman et al. 2002; Botros et al. 2014). Hepatic malakoplakia may be detected as an incidental alteration at autopsy (Botros et al. 2014). Reported cases of liver malakoplakia were associated with immunosuppression and liver abscess (Robertson et al. 1991), perforated colonic diverticulum (Boucher et al. 1994), small bowel ileus in conjunction with Klebsiella sepsis (Hartman et al. 2002), hepatic invasion by renal parenchymal malakoplakia (Chen et al. 1994), hepatic multichamber pseudocyst in miliary tuberculosis (Moldavskii and Rustamov 1984), infected polycystosis (De Saint-Maur and Gallot 1990), and systemic lupus erythematodes (Botros et al. 2014). It is seen that in part of cases, bacterial infection seems to have been involved. Malakoplakia also occurs in the bile duct system and in the gallbladder (Sahel et al. 1976; Charpentier et al. 1983; Hide et al. 2001; Regragui et al. 2003; Agnarsdottir et al. 2004; Di Tommaso et al. 2005). Interestingly, renal and intestinal malakoplakia has been repeatedly observed in patients after liver transplantation (Rull et al. 1995; Kamishima et al. 2000; Weinrach et al. 2004), but this association is likely to be based on immunosuppression, because a similar association has been seen in renal graft recipients (Ourahma et al. 1996; Berney et al. 1999).
Lesions of malakoplakia are variably sized yellow to brownish plaques or nodules that are usually soft (malakos, soft) and friable, sometimes with central liquefaction and frequently with a peripheral rim of inflammatory hyperemia. Malakoplakia in the liver appears to follow a sequence of changes also seen in other locations of this disorder. There are three phases in the histopathologic evolution, originally defined for urinary tract malakoplakia (Smith 1965). In the early phase, plasma cells and the distinct macrophages (the so-called von Hansemann cells) prevail, and classic Michaelis-Gutmann bodies are not usually seen. In the second or classic (granulomatous) phase, large macrophages with Michaelis-Gutmann bodies, lymphocytes, and occasional giant cells are found. The third phase is the “healing” phase, which consists of fibroblasts and collagen deposits around macrophages, along with rather few and sometimes extracellular Michaelis-Gutmann bodies. In the liver, the fully developed lesion shows a rather circumscribed accumulation of histiocytes/macrophages with an admixture of plasma cells and lymphocytes. Already in the H&E preparation, targetoid cytoplasmic inclusions are seen in the macrophages, which represent the Michaelis-Gutmann bodies which have distinct histochemical and ultrastructural features (McClure et al. 1981; Robertson et al. 1991). In a liver biopsy specimen, portal and periportal regions showed scattered small, round to oval targetoid structures consistent with Michaelis-Gutmann bodies that stained with PAS (with and without diastase treatment), von Kossa calcium stain, and colloidal iron stain. CD 68 immunohistochemistry identified the bodies to be situated within macrophages (Hartman et al. 2002). In late stage, fibrosed lesions of malakoplakia and Michaelis-Gutmann bodies tend to accumulate in the extracellular space, both in hepatic malakoplakia (Robertson et al. 1991) and in extrahepatic malakoplakia (Esparza et al. 1981). In malakoplakia of the gallbladder wall, yellowish plaques or nodules are noted, with a histology as outlined above, whereby the targetoid Michaelis-Gutmann bodies may be crowed just beneath the mucosal epithelium (Di Tommaso et al. 2005).
Etiology and Pathogenesis
Malakoplakia is usually associated with bacterial or fungal infections, in particular infections with Klebsiella and Escherichia coli, suggesting a pathogenetic relationship between bacterial antigen and a functional disorder of the macrophage system. Apart from Klebsiella and Escherichia coli, germs found in conjuncture with malakoplakia include mycobacterium tuberculosis, Rhodococcus equi (mainly pleuropulmonary lesions in AIDS patients), Shigella boydii, and paracoccidioidomycosis (Yuoh et al. 1996; Rocha et al. 1997; Raguin et al. 1999; Altwegg and Hinrickson 1999; Guerrero et al. 1999; Caterino-de-Araujo et al. 2000). The reason why macrophages react in a particular way in infected patients developing malakoplakia is not known, but functional and structural monocyte abnormalities are suspected to play a role (van Crevel et al. 1998). The morphogenesis of Michaelis-Gutmann bodies has been studied (Ho 1979; Yuoh et al. 1996). Immature or early Michaelis-Gutmann bodies consist of a circular, electron-dense core containing bacteria and parts thereof, whereas mature Michaelis-Gutmann bodies have a concentric, trilaminated structure with a central mineralized core and usually no remnants of bacterial cell walls. These findings suggested that the bodies are formed around undigested or incompletely digested bacteria as a seemingly alternative pathway for bacterial destruction (Yuoh et al. 1996).
Hepatic Gaucher Cell Pseudotumors (“Gaucheroma”)
Introduction
Gaucher disease is autosomal recessive glucocerebrosidosis and is the most common lysosomal storage disease (reviews: Rosenbloom and Weinreb 2013). It is caused by deficiency of the lysosomal enzyme, beta-glucocerebrosidase, leading to the accumulation of the substrate, glucocerebroside, in the monocyte/macrophage system of many tissues and organs. Glucocerebrosidase normally splits the glycolipid, glucocerebroside, into ceramide and glucose (Chen and Wang 2008). Based on the age of manifestation and clinical presentation, three types of Gaucher disease are recognized, all localized to the same chromosomal region and involving the same gene. Type 1 (the non-neuropathic type; OMIM 230800) is the most frequent form of the disease. Type 2 (acute infantile neuropathic Gaucher disease; OMIM 230900) typically begins within 6 months of birth and has a rapidly progressive course with unfavorable outcome. Type 3 (chronic neuropathic Gaucher disease; OMIM 231000) begins at any time in childhood or even adulthood. It has a milder phenotype than type 2 and shows a slow progression. Type 3 predominates in the northeastern part of Sweden, Norrbotten (Norrbottnian type; Erikson 1986). Around 300 unique mutations have been reported in the glucocerebrosidase gene (GBA), with a distribution that spans the entire gene.
More than 200 of these mutations are missense mutations, while nonsense mutations and splice junction mutations each account for less than 20 mutation types. Thirty-six small insertions or deletions that lead to either frameshifts or in-frame alterations have been found. Recombination events with a highly homologous pseudogene downstream of the GBA locus have been identified, resulting from gene conversion, fusion, or duplication (Hruska et al. 2008). Mutant glucocerebrosidase variants present variable degrees of endoplasmic reticulum (ER) retention and undergo ER-associated degradation (ERAD) in the proteasome (Bendikov-Bar and Horowitz 2012). The degree of ERAD of the mutant enzyme correlates with and is one of the factors that determine Gaucher disease severity (Ron et al. 2010). Gaucher cells typically accumulate in the bone marrow, causing severe bone complications (Poll et al. 2010).
Liver Manifestations of Gaucher’s Disease
Among 500 patients with Gaucher disease, 7.8 % had sonographic evidence of hepatic disease. Hepatomegaly is a well-known feature and is sometimes marked with rounded or blunt liver margins (Gruber 1930). The color of the involved liver is variable, in children sometimes pink yellow in case of marked. In long-standing cases, the liver is grayish yellow, tan, or, rarely, chocolate brown.
Gaucher Cell Infiltration
Pick noted a characteristic whitish network in the parenchyma resulting in a geographic lesion pattern. Focal accumulations of Gaucher cells can produce nodules of variable diameter. These lesions are usually hyperechoic and slowly evolving (Hadas-Halpem et al. 2010).
Accumulation of cerebroside-laden macrophages (the Gaucher cells) in hepatic sinusoids and portal tracts are a typical feature of this storage disorder. In the course of the disease, chiefly in patients without adequate therapy, inflammation, septal fibrosis, and cholestatic liver cirrhosis may develop (Bothe et al. 2013; Debnath et al. 2013). Under enzyme replacement therapy, the number of Gaucher cells is markedly decreased (Hulkova et al. 2009). Large accumulations of Gaucher cells with nodules formation may develop central necrosis (Gruber 1930).
Patients with Gaucher disease type 1 show a frequent iron overload of both hepatocytes and Kupffer cells (Bohte et al. 2013). This alteration is associated with hyperferritinemia (with a mean 3.7-elevation of serum ferritin), and prior splenectomy is associated with most severe hyperferritinemia. This iron overload is not related to HFE mutations (Stein et al. 2010).
Gaucher Cell Pseudotumors (So-Called Gaucheromas)
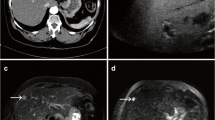
Gaucher cell pseudotumor is defined as a mass-like accumulation of Gaucher cells (cerebroside-containing macrophages). This lesion occurs in the liver, where pseudotumors of this type within or adjacent to parenchymal tissue are a common finding (Poll et al. 2000). Sometimes, gaucheromas grow to important size. In a 23-year-old male patient with Gaucher disease presented with hepatomegaly, an ultrasound of the abdomen showed a hyperechoic mass of up to 9 cm in diameter. Liver MRI demonstrated a well-delineated mass, which appeared hyperintense in comparison to adjacent liver substance on T2-weighted images. A central hypointense scar was detected, suggestive of focal nodular hyperplasia. Biopsy of the lesion revealed accumulation of typical Gaucher cells (Poll and vom Dahl 2009).
Differential Diagnosis
Storing macrophages resembling Gaucher cells can occur in the bone marrow and probably also in visceral organs of some patients with hematologic malignancy or anemia. These cells are termed pseudo-Gaucher cells. The cells have also been found in large numbers in the liver following chemotherapy and bone marrow transplantation (Stenzel and Weeks 2013). In case of large Gaucher cell nodules, hepatocellular carcinoma (HCC) should be excluded in this situation, because HCC without preexisting cirrhosis is increased in Gaucher disease (Breiden-Langen et al. 1991; Xu et al. 2005; De Fost et al. 2006). Cholangiocarcinoma has also been reported (Hulkova et al. 2009).
Neutrophilic Dermatoses/Systemic Neutrophilic Diseases: Manifestations in the Liver
Introduction
Neutrophilic dermatoses (systemic neutrophilic diseases) constitute a group of related and in part hereditary disorders which comprise pyoderma gangrenosum, Sweet’s syndrome, erythema elevatum diutinum, subcorneal pustular dermatosis, acute generalized exanthematous pustulosis (AGEP), pustular vasculitis, and CANDLE syndrome. There are phenotypical overlaps between part of these diseases (review: Cohen 2009). The most important members of this group of disorders are pyoderma gangrenosum and Sweet’s syndrome, both characterized by dense dermal inflammatory infiltrates composed of neutrophils, usually with little evidence of a primary vasculitis (review: Lear et al. 1997). Part of these disorders are associated with visceral aseptic abscesses, including the liver. Such abscesses are deep, sterile, round lesions consisting of neutrophils that do not respond to antibiotics but improve rapidly with corticosteroids.
Pyoderma Gangrenosum
Pyoderma gangrenosum (PG) is a neutrophilic dermatosis of unknown etiology (review: Wollina and Haroske 2011). In its classical clinical presentation, the main feature of PG are pustules or plaques that rapidly progress painful skin ulcers with a necrotic center and red to violaceous undermined margins. PG is typically associated with systemic inflammatory disease, but particularly with inflammatory bowel disease and arthritis, including juvenile rheumatoid arthritis. It has been reported that PG develops in 30–60 % of patients with inflammatory bowel disease (Powell et al. 1996). PG can be associated with visceral manifestations, including cavitating nodular pulmonary inflammation (the lung being the most frequent internal organ site of PG), aseptic splenic abscess, pancreatitis, and focal liver inflammation.
Involvement of the Liver and the Biliary Tract
In PG, focal and sometimes multiple liver lesions representing aseptic abscesses can develop (Urano et al. 1995; Ferrazzi et al. 1996; Hastier et al. 1996; Rodot et al. 1996; Vadillo et al. 1999; Delbrel et al. 2001; Matsumura et al. 2011). At imaging the nodular liver lesions show up as cystic, well-circumscribed foci with cystic change (Vadillo et al. 1999). Aseptic abscesses of the liver in PG are often associated with analogous lesions in the spleen (hepatosplenic abscesses; Vadillo et al. 1999). Fine needle aspiration of the liver lesions yielded purulent, neutrophil-rich fluid, Gram and Ziehl-Neelsen stains being negative (Vadillo et al. 1999).
PG is sometimes associated with other hepatic disorders, including chronic active hepatitis (Byrne et al. 1976; Marshall et al. 1978; Burns and Sarkany 1979; Sampson et al. 1982; Banerjee 1990) and chronic persistent hepatitis (Green et al. 1985).
References
Abbitt PL, Teates CD (1989) The sonographic appearance of extramedullary hematopoiesis in the liver. J Clin Ultrasound 17:280–282
Abedin et al. 2003. http://www.ncbi.nlm.nih.gov/pubmed/12714580
Ackerman et al. 1982. http://www.ncbi.nlm.nih.gov/pubmed/7042896
Ackerman et al. 2002. http://www.ncbi.nlm.nih.gov/pubmed/11834744
Addison T, Gull W (1851) On a certain affection of the skin, Vitiligoidea – A. plana, B. tuberosa, with remarks. Guys Hosp Rev 7:265
Agnarsdottir M, Willen R, El Hag IA (2004) Three cases of malacoplakia of the gallbladder. Ups J Med Sci 109:255–259
Altshuler CH, Angevine DM (1949) Histochemical studies on the pathogenesis of fibrinoid. Am J Pathol 25:1061–1077
Altwegg M, Hinrickson HP (1999) Shigella boydii as cause of malakoplakia in a human immunodeficiency virus-infected patient. Clin Infect Dis 29:1602
Andersen T, Christoffersen P, Gluud C (1984) The liver in consecutive patients with morbid obesity: a clinical, morphological, and biochemical study. Int J Obes 8:107–115
Anderson JM (2000) Multinucleated giant cells. Curr Opin Hematol 7:40–47
Andresen B, Blum J, von Weymarn A, Burge M, Steinbrich W, Duewell S (2000) Hepatic fascioliasis: report of two cases. Eur Radiol 10:1713–1715
Ask-Upmark E (1945) Tumours simulating intrathoracic heterotopia of bone marrow. Acta Radiol 26:425–440
Aterman K, Remmele W, Smith M (1988) Karl Touton and his “xanthelamatic giant cell”. A selective review of multinucleated giant cells. Am J Dermatopathol 10:257–269
Aytac S, Fitoz S, Akyar S, Atasoy C, Erekul S (1999) Focal intrahepatic extramedullary hematopoiesis: color Doppler US and CT findings. Abdom Imaging 24:366–368
Azuma et al. 2002. http://www.ncbi.nlm.nih.gov/pubmed/12041704
Bajema IM, Bruijn JA (2000) What stuff is this! A historical perspective on fibrinoid necrosis. J Pathol 191:235–238
Bajema IM, Hagen EC, de Heer E, Bruijn JA (1998) The PAF-Halmi staining distinguishes two types of glomerular fibrinoid necrosis in systemic vasculitis. J Am Soc Nephrol 9:532A
Banerjee 1990. http://www.ncbi.nlm.nih.gov/pubmed/2317437
Bar J, Linke T, Ferlinz K, Neumann U, Schuchman EH, Sandhoff K (2001) Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum Mutat 17:199–209
Ben Rejeb A, Haouala H, Ben Hammadi F, Ben Othman M, Khelil A, Gueddiche M, Ben Cheikh M, Rokbani L (1992) Pseudotumor extramedullary hematopoiesis. Report of 3 cases and review of literature (in French). Ann Pathol 12:183–187
Benbow EW (1990) Xanthogranulomatous cholecystitis. Br J Surg 77:255–256
Bendikov-Bar I, Horowitz M (2012) Gaucher disease paradigm: from ERAD to comorbidity. Hum Mutat 33:1398–1407
Berney T, Chautems R, Ciccarelli O, Latinne D, Pirson Y, Squifflet JP (1999) Malakoplakia of the caecum in a kidney-transplant recipient: presentation as acute tumoral perforation and fatal outcome. Transpl Int 12:293–296
Bhatia and Sarin 1994. http://www.ncbi.nlm.nih.gov/pubmed/8147371
Blewitt RW, Bradbury K, Greenall MJ et al (1977) Hepatic damage associated with mineral oil deposits. Gut 18:476–479
Bohte et al. 2013. http://www.ncbi.nlm.nih.gov/pubmed/23554863
Burns and Sarkany 1979. http://www.ncbi.nlm.nih.gov/pubmed/535175
Byrne et al. 1976. http://www.ncbi.nlm.nih.gov/pubmed/999311
Boitnott JK, Margolis S (1970) Saturated hydrocarbons in human tissues: III. Oil droplets in the liver and spleen. Johns Hopkins Med J 127:65–78
Bothe AE, van Dussen L, Akkerman EM, Nederveen AJ, Sinkus R, Jansen PL, Stoker J et al (2013) Liver fibrosis in type I Gaucher disease: magnetic resonance imaging, transient elastography and parameters of iron storage. PLoS One 8:e57507
Botros N, Yan SR, Wanless IR (2014) Malakoplakia of liver: report of two cases. Pathol Res Pract 210:459–462
Boucher LD, Aoki M, Lee EY, Cibull ML (1994) Malakoplakia of liver associated with a perforated colonic diverticulum. A case report and review of the literature. J Clin Gastroenterol 19:318–320
Bradley MJ, Metreweli C (1990) Sonography of extramedullary hematopoiesis of the liver. AJR Am J Roentgenol 154:900–901
Breiden-Langen et al. 1991. http://www.ncbi.nlm.nih.gov/pubmed/16511436
Caffey J, Silverman WA (1945) Infantile cortical hyperostosis: preliminary report on a new syndrome. AJR Am J Roentgenol 54:1–16
Caterino-de-Araujo A, de los Santos-Fortuna E, Zandona-Meleiro MC, Calore EE, Perez Calore NM (2000) Detection of the 20-kDA virulence-associated antigen of Rhodococcus equi in malakoplakia-like lesion in pleural tissue obtained from an AIDS patient. Pathol Res Pract 196:321–327
Chantranuwat C (2004) Systemic form of juvenile xanthogranuloma: report of a case with liver and bone marrow involvement. Pediatr Dev Pathol 7:646–648
Charpentier P, Prade M, Bognel C, Gadenne C, Duvillard P (1983) Malacoplakia of the gallbladder. Hum Pathol 14:827–828
Chen M, Wang J (2008) Gaucher disease: review of the literature. Arch Pathol Lab Med 132:851–853
Chen CS, Lai MK, Hsueh S, Hwang TL, Chuang CK (1994) Renal malacoplakia with secondary hepato-duodenal involvement. J Urol 151:982–985
Cho TJ, Moon HJ, Cho DY, Park MS, Lee DY, Yoo WJ, Chung CY, Choi IH (2008) The c.3040C > T mutation in COL1A1 is recurrent in Korean patients with infantile cortical hyperostosis (Caffey disease). J Hum Genet 53:947–949
Cho ES, Yu JS, Kim MJ, Kim JH, Chung JJ, Kim KW (2010) Focal eosinophilic necrosis on superparamagnetic iron oxide-enhanced MRI. AJR Am J Roentgenol 194:1296–1302
Choi JI, Lee JM, Kim SH, Lee JY, Lee MW, Han CK, Choi BI (2009) Differentiating focal eosinophilic necrosis of the liver from hepatic metastases using unenhanced and portal venous phase computed tomographic imagings: results of univariate and multivariate statistical analyses. J Comput Assist Tomogr 33:705–709
Christensen AH, Ishak KG (1970) Benign tumors and pseudotumors of the gallbladder. Report of 180 cases. Arch Pathol 90:423–432
Christoffersen P, Braendstrup O, Juhl E, Poulsen H (1971) Lipogranulomas in human liver biopsies with fatty change. A morphological, biochemical and clinical investigation. Acta Pathol Microbiol Scand A 79:150–158
Chvostek F (1911) Xanthelasma und Ikterus. Ztschr f Klein Med 73:476
Cohen PR (2009) Neutrophilic dermatoses: a review of current treatment options. Am J Clin Dermatol 10:301–312
Cools J, DeAngelo DJ, Gotlib J, Stover EH, Legare RD, Cortes J, Kutok J, Clark J, Galinsky I, Griffin JD, Cross NC et al (2003) A tyrosine kinase created by fusion of the PDGFRA and FIP1L1 genes as a therapeutic target of imatinib in idiopathic hypereosinophilic syndrome. N Engl J Med 348:1201–1214
Cools J, Stover EH, Wlodarska I, Marynen P, Gilliland DG (2004) The FIP1L1-PDGFRalpha kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia. Curr Opin Hematol 11:51–57
Couret M, Combe B, Chuong VT, Leroux JL, Blotman F, Sany J (1988) Rheumatoid nodulosis: report of two cases and discussion of diagnostic criteria. J Rheumatol 15:1427–1430
Coutre S, Gotlib J (2004) Targeted treatment of hypereosinophilic syndromes and chronic eosinophilic leukemias with imatinib mesylate. Semin Cancer Biol 14:23–31
Da Costa JL, Loh YS, Hanam E (1974) Extramedullary hemopoiesis with multiple tumor-simulating mediastinal masses in hemoglobin E-thalassemia disease. Chest 65:210–212
Damjanov I, Moriber Katz S (1981) Malakoplakia. Pathol Annu 16:103–126
Dardi LE, Marzano M, Froula E (1990) Fine needle aspiration cytologic diagnosis of focal intrahepatic extramedullary hematopoiesis. Acta Cytol 34:567–569
Dasgupta P, Womack C, Turner AG, Blackford HN (1999) Malacoplakia: von Hansemann’s disease. BJU Int 84:464–469
De Fost M, vom Dahl S, Weverling GJ, Brill N, Brett S, Häussinger D et al (2006) Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol Dis 36:53–58
De Rycke L, Baeten D, Foell D et al (2005) Differential expression and response to anti-TNF alpha treatment by infiltrating versus resident tissue macrophage subsets in autoimmune arthritis. J Pathol 206:17–27
De Saint-Maur P, Gallot D (1990) Hepatic malacoplakia as a complication of polycystosis (in French). Ann Pathol 10:50–51
De Vincentiis C (1883) Endothelioma adiposo. Riv Clin 35:481–501
Debnath CR, Debnath MR, Nabi N, Khan NA, Chakraborty S (2013) A case of Gaucher’s disease progressing to liver cirrhosis. Mymensingh Med J 22:394–396
Delbrel X, Germain P, Fach J, Etienne G, Beylot C, Parrens M, De Mascarel A, Le Bras M et al (2001) Cutaneous pyoderma gangrenosum with hepatosplenic localization and monoclonal gammopathy. A case report (in French). Ann Med Interne (Paris) 152:65–67
Delladetsima JK, Horn T, Poulsen H (1987) Portal tract lipogranulomas in liver biopsies. Liver 7:9–17
Desmet VJ (1995) Histopathology of cholestasis. Verh Dtsch Ges Pathol 79:233–240
Dewar G, Leung NWY, Ng HK, Bradley M, Li AKC (1990) Massive, solitary, intrahepatic, extramedullary hematopoietic tumor in thalassemia. Surgery 107:704–707
Di Tommaso L, Arizzi C, Roncalli M (2005) Malacoplakia of the gallbladder. Histopathology 46:474–475
Dincsoy HP, Weesner RE, MacGee J (1982) Lipogranulomas in non-fatty human livers. A mineral oil induced environmental disease. Am J Clin Pathol 78:35–41
Dyer et al. 1997. http://www.ncbi.nlm.nih.gov/pubmed/9119387
Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J (2007) Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol 5:15
el Bawab S, Mao C, Obeid LM, Hannun YA (2002) Ceramidases in the regulation of ceramide levels and function. Subcell Biochem 36:187–205
Eriguchi N, Aoyagi S, Horiuchi H, Tamae T, Uchida S, Hiraki M, Nishimura K, Kawabata M, Hamada S (2002) Xanthogranulomatous cholecystitis with a liver abscess and metastatic endophthalmitis: report of a case. Surg Today 32:285–288
Erikson A (1986) Gaucher disease – Norrbottnian type (III). Neuropaediatric and neurobiological aspects of clinical patterns and treatment. Acta Paediatr Scand Suppl 326:1–42
Esparza AR, Mckay DB, Cronan JJ et al (1981) Renal parenchymal malakoplakia. Histologic spectrum and its relationship to megalocytic interstitial nephritis and xanthogranulomatous pyelonephritis. Am J Surg Pathol 13:225–236
Fahr Th (1918) Zur Frage des Rheumatismus nodosus. Zbl Path 29:625
Ferrazzi V, Rivière S, Sany J (1996) Association of pyoderma gangrenosum with hepato-pancreatic involvement in rheumatoid polyarthritis (in French). Rev Med Interne 17:266–267
Ferrell LD, Lee R, Brixko C, Bass NM, Lake JR, Roberts JP, Ascher N, Rabkin J (1995) Hepatic granulomas following liver transplantation. Clinicopathologic features in 42 patients. Transplantation 60:926–933
Fleming KA, Zimmerman H, Shubnik P (1998) Granulomas in the livers of humans and Fischer rats associated with the ingestion of mineral hydrocarbons: a comparison. Regul Toxicol Pharmacol 27:75–81
Franco V, Aragona F, Genova G, Florena AM, Stella M, Campesi G (1990) Xanthogranulomatous cholecystitis. Histopathological study and classification. Pathol Res Pract 186:383–390
Fried K, Manor A, Pajewski M, Starinsky R, Vure E (1981) Autosomal dominant inheritance with incomplete penetrance in Caffey disease (infantile cortical hyperostosis). Clin Genet 19:271–274
Futcher TB (1905) Xanthelasma and chronic jaundice. Am J Med Sci 130:939–951
Gans O (1964) Zur Geschichte der Vereinigung südwestdeutscher Dermatologen. Hautarzt 15:623–625
Garcia-Patos V (2007) Rheumatoid nodule. Semin Cutan Med Surg 26:100–107
Gensure RC, Mäkitie O, Barcklay C, Chan C, DePalma SR, Bastepe M, Abuzahra H et al (2005) A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest 115:1250–1257
Gil-Fernandez JJ, Martinez-Chamorro C, Tomas JF (2001) The irreplaceable image: a giant hepatic mass of myeloid metaplasia in a patient without myelofibrosis. Haematologica 86:445
Ginsberg MH, Genant HK, Yu TF, McCarty DJ (1975) Rheumatoid nodulosis: an unusual variant of rheumatoid disease. Arthritis Rheum 18:49–58
Gitlin D, Craig JM, Janeway CA (1957) Studies on the nature of fibrinoid in the collagen diseases. Am J Pathol 33:55–77
Glorieux FH (2005) Caffey disease: an unlikely collagenopathy. J Clin Invest 115:1142–1144
Goldberg GM, Saphir O (1960) Lipidosis of the liver portal spaces: I. A study of its relationship to the lymphatics of the liver. Arch Pathol Lab Med 69:586–593
Goni MA, Scheines EJ, Paira SO, Barcelo HA, Maldonado Cocco JA (1992) Rheumatoid nodulosis: a puzzling variant of rheumatoid arthritis. Clin Rheumatol 11:396–401
Gotlib J, Cools J, Malone JM, Schrier SL, Gilliland DG, Coutre SE (2004) The FIP1L1-PDGFR(alpha) fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: implications for diagnosis, classification, and managment. Blood 104:2879–2891
Gougerot M (1935) Karl Touton. Bull Soc Fr Dermatol Syphiligr 42:413–415
Green et al. 1985. http://www.ncbi.nlm.nih.gov/pubmed/4067029
Griffin JH, Leung J, Bruner RJ, Caligiuri MA, Briesewitz R (2003) Discovery of a fusion kinase in EOL-1 cells and idiopathic hypereosinophilic syndrome. Proc Natl Acad Sci U S A 100:7830–7835
Gruber GB (1930) 6. Die Leber bei Erkrankungen des blut-und lymphbildenden Gewebs-Apparates. In: Henke F, Lubarsch O (eds) Handbuch der speziellen pathologischen Anatomie und Histologie, Vol. 5, Verdauungsdrüsen, Erster Teil, Leber. Julius Springer, Berlin, pp 669–672
Gualco G, Ojopi EB, Chioato L, Cordeiro DL, Negretti F, Bacchi CE (2010) Postsplenectomy sclerosing extramedullary hematopoietic tumor with unexpected good clinical evolution: morphologic, immunohistochemical, and molecular analysis of one case and review of the literature. Appl Immunohistochem Mol Morphol 18:291–295
Guerrero MF, Ramos JM, Renedo G, Gadea I, Alix A (1999) Pulmonary malacoplakia associated with Rhodococcus equi infection in patients with AIDS: case report and review. Clin Infect Dis 28:1334–1336
Gupta P, Naran A, Auh YH, Chung JS (2004) Focal intrahepatic extramedullary hematopoiesis presenting as fatty lesions. AJR Am J Roentgenol 182:1031–1032
Hadas-Halpem I, Deeb M, Abrahamov A, Zimran A, Elstein D (2010) Gaucher disease: spectrum of sonographic findings in the liver. J Ultrasound Med 29:727–733
Han JK, Choi BI, Cho JM, Chung KB, Han MC, Kim CW (1993) Radiological findings of human fascioliasis. Abdom Imaging 18:261–264
Han JK, Han D, Choi BI, Han MC (1996) MR findings in human fascioliasis. Trop Med Int Health 1:367–372
Han JK, Jang HJ, Choi BI, Kim SH, Kim TK, Won HJ, Kim YI, Cho SY (1999) Experimental hepatobiliary fascioliasis in rabbits: a radiology-pathology correlation. Invest Radiol 34:99–108
Hardaway W (1884) Xanthoma multiplex. J Cutan Vener Dis 354:20
Hart FD (1994) Rheumatoid nodules. Ann Rheum Dis 53:83
Hartman G, Yong SL, Chejfec G (2002) Malakoplakia of liver diagnosed by a needle core biopsy: a case report and review of the literature. Arch Pathol Lab Med 126:372–374
Hastier P, Caroli-Bosc FX, Bartel HR, Harris AG, Maes B, Arpurt JP, Taillan B, Bodock I et al (1996) Pyoderma gangrenosum with hepatopancreatic manifestations in a patient with rheumatoid arthritis. Dig Dis Sci 41:594–597
Hayashi et al. 1999. http://www.ncbi.nlm.nih.gov/pubmed/10475929
Heimpel H, Dührsen U, Hofbauer P, Rigamonti-Wermlinger V, Kreuser ED, Schwarz K et al (2009) Bulky extramedullary hematopoiesis is not a rare complication of congenital dyserythropoietic anemia. Ann Hematol 88:937–941
Herman TE (1996) Antenatal-onset cortical hyperostosis and nonimmune hydrops. J Perinatol 16:137–139
Hervé S, Savoye G, Savoye-Collet C, Behbahani A, Auliac JB, Bota S, François A, Lerebours E (2001) Intrahepatic extramedullary hematopoiesis as a manifestation of a malignant thymoma: an unusual cause of nodular hepatomegaly (in French). Gastroenterol Clin Biol 25:711–713
Herxheimer K (1935) Karl Touton. Dermatol Zschr 70:247–248
Hide G, Desai S, Bloxham CA (2001) Malakoplakia of the gallbladder: imaging and histological features. Clin Radiol 56:326–328
Highton J, Kean A, hessian P, Wilsher M (2000) Cells expressing dendritic cell markers are present in the rheumatic nodule. J Rheumatol 27:339–346
Highton J, Hessian PA, Stamp L (2007) The rheumatoid nodule: peripheral or central to rheumatoid arthritis? Rheumatology (Oxford) 46:1385–1387
Ho KJ (1979) Michaelis-Gutmann bodies. Arch Pathol Lab Med 103:488–489
Holubar K (1990) Looking back…Paul Langerhans, Theodor Langhans, and Carl Touton – contemporary histopathologists. Am J Dermatopathol 12:534–535
Hruska KS, LaMarca ME, Scott CR, Sidransky E (2008) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29:567–583
Hulkova H, Ledvinova J, Poupetova H, Kohout A, Malinova V, Elleder M (2009) Autopsy case of Gaucher disease type I in a patient on enzyme replacement therapy. Comments on the dynamics of persistent storage process. J Inherit Metab Dis 32:551–559
Hur J, Park MS, Yu JS, Lim JS, Hong SW, Kim KW (2005) Focal eosinophilic necrosis versus metastasis in the liver: the usefulness of two-phase dynamic CT. AJR Am J Roentgenol 184:1085–1090
Iversen K, Christoffersen P, Poulsen H (1970) Epithelioid cell granulomas in liver biopsies. Scand J Gastroenterol Suppl 7:61–67
Jang HJ, Lee WJ, Lee SJ, Kim SH, Lim HK, Lim JH (2002) Focal eosinophilic necrosis of the liver in patients with underlying gastric or colorectal cancer: CT differentiation from metastasis. Korean J Radiol 3:240–244
Jelali MA, Luciani A, Kobeiter H, Zafrani S, Anglade MC, Zegai B et al (2006) MRI features of intrahepatic extramedullary haematopoiesis in sickle cell anaemia. Cancer Imaging 6:182–185
Kamishima T, ito K, Awaya H, Mitchell DG (2000) MR imaging of bilateral renal malakoplakia after liver transplantation. AJR Am J Roentgenol 175:919–920
Kamoun-Goldrat A, le Merrer M (2008) Infantile cortical hyperostosis (Caffey disease). A review. J Oral Maxillofac Surg 66:2145–2150
Kamoun-Golsdrat A, Martinovic J, Saada J, Sonigo-Cohen P, Razavi F, Munnich A, Le Mercier M (2008) Prenatal cortical hyperostosis with COL 1A1 gene mutation. Am J Med Genet A 146A:1820–1824
Kaye BR, Kaye RL, Bobrove A (1984) Rheumatoid nodules. Review of the spectrum of associated conditions and proposal of a new classification, with a report of four seronegative cases. Am J Med 76:279–292
Keen ME, Engstrand DA, Hafez GR (1985) Hepatic lipogranulomatosis simulating veno-occlusive disease of the liver. Arch Pathol Lab Med 109:70–72
Keitel W (2008) The pathologists: Aschoff, Klinge and Gräff (in German). Z Rheumatol 67:165–171
Kim KA, Lim HK, Kim SH, Lee WJ, Lim JH (1999) Necrotic granuloma of the liver by human fascioliasis: imaging findings. Abdom Imaging 24:462–464
Kim et al. 2002. http://www.ncbi.nlm.nih.gov/pubmed/12191679
Kim WH, Kim SH, Kim YH, Youn SH, Kang WJ, Kim MA, Han JK, Choi BI (2010) Fluorine-18-FDG PET findings of focal eosinophilic liver disease: correlation with CT and/or MRI, laboratory, and pathologic findings. Abdom Imaging 35:437–446
Klatskin G (1977) Hepatic granulomata: problems in interpretation. Mt Sinai J Med 44:798–812
Klinger H (1932) Grenzformen der Periarteriitis nodosa. Frankf Z Pathol 42:455–480
Klion AD, Noel P, Akin C, Law MA, Gilliland DG, Cools J, Metcalfe DD, Nutman TB (2003) Elevated serum tryptase levels identify a subset of patients with a myeloproliferative variant of idiopathic hypereosinophilic syndrome associated with tissue fibrosis, poor prognosis, and imatinib responsiveness. Blood 101:4660–4666
Klion AD, Robyn J, Akin C, Noel P, Brown M, Law M, Metcalfe DD, Dunbar C, Nutman TB (2004a) Molecular remission and reversal of myelofibrosis in response to imatinib mesylate treatment in patients with the myeloproliferative variant of hypereosinophilic syndrome. Blood 103:473–478
Klion AD, Law MA, Riemenschneider W, McMaster ML, Brown MR, Horne M, Karp B, Robinson M, Sachdev V et al (2004b) Familial eosinophilia: a benign disorder? Blood 103:4050–4055
Koch J, Gartner S, Li CM, Quintern LE, Bernardo K, Levran O, Schnabel D, Desnick RJ, Schuchman EH, Sandhoff K (1996) Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. J Biol Chem 271:33110–33115
Koga M, Ishihara T, Uchino F, Fujiwaki T (1992) An autopsy case of Farber’s lipogranulomatosis in a Japanese boy with gastrointestinal involvement. Acta Pathol Jpn 42:42–48
Kopecky KK, Moriarty AT, Antony AC, Baker MK (1986) Extramedullary hematopoiesis in acute lymphocytic leukemia masquerading as hepatic, renal, and splenic microabscesses. AJR Am J Roentgenol 147:846–847
Kumar A, Aggarwal S, de Tilly LN (1995) Case of the season: thalassemia major with extramedullary hematopoiesis in the liver. Semin Roentgenol 2:99–101
Kuriwaki K, Yoshida H (1999) Morphological characteristics of lipid accumulation in liver-constituting cells of acid lipase deficiency rats (Wolman’s disease model rats). Pathol Int 49:291–297
Kwak HS, Lee JM (2000) CT findings of extramedullary hematopoiesis in the thorax, liver and kidneys, in a patient with idiopathic myelofibrosis. J Korean Med Sci 15:460–462
Kwak HS, Han YM, Lee JM (2004) Focal eosinophilic infiltration of the liver mimicking hepatocellular carcinoma; case reports. Clin Imaging 28:36–39
Kwon Y, Yu E, Huh J, Lee SK, Ro JY (2004) Sclerosing extramedullary hematopoietic tumor involving the lesser omentum and ligamentum teres in liver explant. Ann Diagn Pathol 8:227–232
La Fianza A, van der Byl G, Maccabelli G, Torretta L, Calliada F (2010) CT and MR findings in extramedullary haematopoiesis with biliary system encasement: a case report. J Radiol Case Rep 4:1–8
Laghi F, Catalano O, Maresca M, Sandomenico F, Siani A (2010) Indeterminate, subcentimetric focal liver lesions in cancer patients: additional role of contrast-enhanced ultrasound. Ultraschall Med 31:283–288
Landas SK, Mitros FA, Furst DE, LaBrecque DR (1992) Lipogranulomas and gold in the liver in rheumatoid arthritis. Am J Surg Pathol 16:171–174
Lear JT, Atherton MT, Byrne JP (1997) Neutrophilic dermatosis: pyoderma gangrenosum and Sweet’s syndrome. Postgrad Med J 73:65–68
Lecolier B, Bercau G, Gonzalez M, Afriat R, Rambaud D, Mulliez N, de Kermadec S (1992) Radiographic, haematological, and biochemical findings in a fetus with Caffey disease. Prenat Diagn 12:637–641
Lee WJ, Lim HK, Lim JH, Kim SH, Choi SH, Lee SJ (1999) Foci of eosinophil-related necrosis in the liver: imaging findings and correlation with eosinophilia. AJR Am J Roentgenol 172:1255–1261
Lee IJ, Kim SH, Kim DS, Lee JM, Han JK, Choi BI (2008) Intrahepatic extramedullary hematopoiesis mimicking a hypervascular hepatic neoplasm on dynamic – and SPIO-enhanced MRI. Korean J Radiol 9(Suppl):S34–S38
Lee MH, Kim SH, Kim H, Lee MW, Lee WJ (2011) Differentiating focal eosinophilic infiltration from metastasis in the liver with gadoxetic acid-enhanced magnetic resonance imaging. Korean J Radiol 12:439–449
Lefkowitch JH, Haythe JH, Regent N (2002) Kupffer cell aggregation and perivenular distribution in steatohepatitis. Mod Pathol 15:699–704
Leiferman KM, Gleich GJ (2004) Hypereosinophilic syndrome: case presentation and update. J Allergy Clin Immunol 113:50–58
Lemos LB, Baliga M, Benghuzzi HA, Cason Z (1997) Nodular hematopoiesis of the liver diagnosed by fine-needle aspiration cytology. Diagn Cytopathol 16:51–54
Leonidas DD, Elbert BL, Zhou Z, Leffler H, Ackerman SJ, Acharya KR (1995) Crystal structure of human Charcot-Leyden crystal protein, an eosinophil lysophospholipase, identifies it as a new member of the carbohydrate-binding family of galectins. Structure 3:1379–1393
Li CM, Park JH, Simonaro CM, He X, Gordon RE, Friedman AH, Ehleiter D, Paris F, Manova K, Hepbildikler S et al (2002) Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics 79:218–224
Lim JH, Lee WJ, Lee DH, Nam KJ (2000) Hypereosinophilic syndrome: CT findings in patients with hepatic lobar or segmental involvement. Korean J Radiol 1:98–103
Ma SK, Au WY (1999) Images in haematology. Hepatic haemopoiesis in beta-thalassemia trait. Br J Haematol 107:1
Malamos B, Papavisiliou C, Avramidis A (1962) Tumor-simulating intrathoracic extramedullary hemopoiesis. Acta Radiol 57:227–231
Marshall et al. 1978. http://www.ncbi.nlm.nih.gov/pubmed/285688
Mastrianni DM, Eddy RL, Rosenberg HF, Corrette SE, Shows TB, Tenen DG, Ackerman SJ (1992) Localization of the human eosinophil Charcot-Leyden crystal protein (lysophospholipase) gene (CLC) to chromosome 19 and the human ribonuclease 2 (eosinophil-derived neurotoxin) and ribonuclease 3 (eosinophil cationic protein) genes (RNS2 and RNS3) to chromosome 1. Genomics 13:240–242
Matsumura Y, Nishiwaki F, Morita N, Kore-Eda S, Miyachi Y (2011) Pyoderma gangrenosum in a patient with myelodysplastic syndrome followed by possible extracutaneous manifestations in the gallbladder, liver, bone and lung. J Dermatol 38:1102–1105
McCarty DJ (1991) Complete reversal of rheumatoid nodulosis. J Rheumatol 18:736–737
McClure J, Cameron CHS, Garrett R (1981) The ultrastructural features of malakoplakia. J Pathol 134:13–25
McCoy JJ, Vila R, Petrossian G, McCall RA, Reddy KS (1976) Xanthogranulomatous cholecystitis. Report of two cases. J S C Med Assoc 72:78–79
McNally AK, Anderson JM (1995) Interleukin-4 induces foreign body giant cells from human monocytes/macrophages. Differential lymphokine regulation of macrophage fusion leads to morphological variants of multinucleated giant cells. Am J Pathol 147:1487–1499
Meynet C (1875) Rhumatisme articulaire subaigu avec productions de tumeures multiples dans les tissus fibreux périarticulaires et sur le périoste d’un grand nombre d’os. Lyon Med 20:495
Michaelis L, Gutmann C (1902) Ueber Einschlüsse in Blasentumoren. Z Klin Med 47:208–215
Moldavskii MI, Rustamov IR (1984) Multichamber pseudocyts of the liver with malacoplakia (in Russian). Klin Khir 9:64–65
Morita Y, Ueno T, Sasaki N, Kuhara K, Yoshioka S, Tateishi Y, Nagata E, Kage M, Sata M (2005) Comparison of liver histology between patients with non-alcoholic steatohepatitis and patients with alcoholic steatohepatitis in Japan. Alcohol Clin Exp Res 29(12 Suppl):277S–281S
Moschella SL (1970) Xanthomatosis, associated with multiple myeloma. Arch Dermatol 102:121–123
Moskovitz B, Malberger E, Brenner B, Gaitini D, Vardi Y, Bolkier M (1991) Renal extramedullary hematopoiesis simulating hypernephroma. Eur Urol 19:343–345
Moxon W (1873) Simple stricture of hepatic duct, causing chronic jaundice and xanthelasma. Trans Path Soc Lond 129:129–134
Muramatsu T, Sakai N, Yanagihara I, Yamada M, Nishigaki T, Kokubu C, Tsukamoto H, Ito M, Inui K (2002) Mutation analysis of the acid ceramidase gene in Japanese patients with Farber disease. J Inherit Metab Dis 25:585–592
Murrell et al. 1997. http://www.ncbi.nlm.nih.gov/pubmed/9105307
Nam KJ, Jung WJ, Choi JC, Koo BS, Park BH, Lee KN, Han SY, Shin WW, Han SS (1999) Hepatic involvement in hypereosinophilia: sonographic findings. J Ultrasound Med 18:475–479
Navarro M, Crespo C, Perez L, Martinez C, Galant J, Gonzalez I (2000) Massive intrahepatic extramedullary hematopoiesis in myelofibrosis. Abdom Imaging 25:184–186
Neumann E (1880) Die Picrocarminfärbung und ihre Anwendung auf die Entzündungslehre. Arch Mikrosk Anat 18:130–150
Nochomovitz LE, Uys CJ, Epstein S (1975) Massive deposition of mineral oil after prolonged ingestion. S Afr Med J 49:2187–2190
Noi I, Loberant N, Cohen I (1994) Inflammatory pseudotumor of the liver. Clin Imaging 18:283–285
O’Malley DP (2007) Benign extramedullary myeloid proliferations. Mod Pathol 20:405–415
Ourahma S, Barrou B, Bitker MO, Sylla C, Mouquet C, Chatelain C (1996) Rectal malakoplakia in a renal transplant recipient. Transplant Proc 28:2824
Palmer DG, Hogg N, Highton J, Hessian PA, Denholm I (1987a) Macrophage migration and maturation within rheumatoid nodules. Arthritis Rheum 30:729–736
Palmer DG, Hogg N, Allen CA, Highton J, Hessian PA (1987b) A mononuclear phagocyte subset associated with cell necrosis in rheumatoid nodules: identification with monoclonal antibody 5.5. Clin Immunol Immunopathol 45:17–28
Papavisiliou C, Sfikakis P (1964) Tumour-simulating intrathoracic marrow heterotopia in thalassaemia major. Thorax 19:121–124
Pardanani A, Ketterling RP, Brockman SR, Flynn HC, Paternoster SF, Shearer BM et al (2003) CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood 102:3093–3096
Parra JA, Acinas O, Bueno J, Guezmes A, Fernandez MA, Farinas MC (2000) Xanthogranulomatous cholecystitis: clinical, sonographic, and CT findings in 26 patients. AJR Am J Roentgenol 174:979–983
Parwani AV, Ali SZ (2003) Pathologic quiz case. A 52-year-old woman with a liver mass. Arch Pathol Lab Med 127:631–632
Petersen P, Christoffersen P (1979) Ultrastructure of lipogranulomas in human fatty liver. Acta Pathol Microbiol Scand A 87:45–49
Pettus BJ, Chalfant CE, Hannun YA (2002) Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta 1585:114–125
Pinocy J, Lange A, Konig C, Kaiserling E, Becker HD, Krober SM (2003) Xanthogranulomatous cholecystitis resembling carcinoma with extensive tumorous infiltration of the liver and colon. Langenbecks Arch Surg 388:48–51
Poll LW, vom Dahl S (2009) Image of the month. Hepatic gaucheroma mimicking focal nodular hyperplasia. Hepatology 50:985–986
Poll LW, Koch JA, vom Dahl S, Loxtermann E, Sarbia M, Niederau C et al (2000) Extraosseous manifestation of Gaucher’s disease type I: MR and histological appearance. Eur Radiol 10:1660–1663
Poll LW, Willers R, Häussinger D, Mödder U, vom Dahl S (2010) MRI bone marrow findings in 63 patients with type I Gaucher disease. Rofo 182:979–985
Powell FC, Su WP, Pemy HO (1996) Pyoderma gangrenosum: classification and management. J Am Acad Dermatol 34:395–409
Pulpeiro JR, Armesto V, Varela J, Corredoira J (1991) Fascioliasis: findings in 15 patients. Br J Radiol 64:798–801
Pye Smith PH (1873) Xanthelasma (Vitiligoidea plana) of skin, peritoneum and mucous membrane, associated with jaundice: autopsy. Trans Path Soc Lond 24:250–253
Raab SS, Silverman JF, McLeod DL, Geisinger KR (1993) Fine-needle aspiration cytology of extramedullary hematopoiesis (myeloid metaplasia). Diagn Cytopathol 9:522–526
Raguin G, nemeth J, Wassef M et al (1999) Identification of Shigella boydii in colonic malakoplakia by universal bacterial 16S ribosomal DNA-based amplification in a human immunodeficiency virus-infected patient. Clin Infect Dis 28:142–143
Rayer PFO (1835) Traité théorique et pratique des maladies de la peau. Baillière, Paris
Reed A, Ryan C, Schwartz SI (1994) Xanthogranulomatous cholecystitis. J Am Coll Surg 179:249–252
Regragui A, Amrani M, Gamra L, Alaoui Belabbas M (2003) Malacoplakia of the biliary tract and duodenum. Case report (in French). Gastroenterol Clin Biol 27:655–656
Remstein ED, Kurtin PJ, Nascimento AG (2000) Sclerosing extramedullary hematopoietic tumor in chronic myeloproliferative disorders. Am J Surg Pathol 24:51–55
Riedlinger WF, Lairmore TC, Balfe DM, Dehner LP (2005) Tumefactive necrobiotic granulomas (nodulosis) of the pancreas in an adult with long-standing rheumatoid arthritis. Int J Surg Pathol 13:207–210
Robertson SJ, Higgins RB, Powell C (1991) Malacoplakia of liver: a case report. Hum Pathol 22:1294–1295
Roberts-Thomson PJ, Venables GS, Onitiri AC, Lewis B (1975) Polymeric IgA myeloma, hyperlipidemia and xanthomatosis: a further case and review. Postgrad Med J 51:44–51
Rocha N, Suguiama EH, Maia D, Costa H, Coelho KI, Franco M (1997) Intestinal malakoplakia associated with paracoccidiodomycosis: a new association. Histopathology 30:79–83
Rodot S, Lacour JP, Van Elslande L, Cognard C, Castanet J, Ortonne JP (1996) Extracutaneius manifestations of neutrophilic dermatosis (in French). Ann Dermatol Venereol 123:129–134
Ron et al. 2010. http://www.ncbi.nlm.nih.gov/pubmed/20643691
Rosenbloom BE, Weinreb NJ (2013) Gaucher disease: a comprehensive review. Crit Rev Oncog 18:163–1975
Rothenberg ME (2004) Eosinophilic gastrointestinal disorders (EGID). J Allergy Clin Immunol 113:11–28
Rull R, Grande L, Garcia-Valdecasas JC, Bombi JA, Alos LL, Fuster J, Lacy AM, Cugat E et al (1995) Malakoplakia in the gastrointestinal tract of a liver transplant recipient. Transplantation 59:1492–1494
Sahel J, Scheiner C, Lebreuil G, Sarles H (1976) Malacoplasia of the cystic duct (in French). Arch Fr Mal App Dig 65:585–590
Sakakibara et al. 2002. http://www.ncbi.nlm.nih.gov/pubmed/12132528
Sampson et al. 1982. http://www.ncbi.nlm.nih.gov/pubmed/7183305
Schäfer A, Harzer K, Kattner E, Schafer HJ, Stoltenburg G, Lietz H (1996) Disseminated lipogranulomatosis (Farber disease) with hydrops fetalis (in German). Pathologe 17:145–149
Scheuer PJ, Lefkowitch JH (2006) Liver biopsy interpretation, 7th edn. Elsevier Sanders, Philadelphia
Schweiger S, Chaoui R, Tennstedt K, Mudlos S, Tinschert S (2003) Antenatal onset of cortical hyperostosis (Caffey disease): case report and review. Am J Med Genet 120A:547–552
Smith B (1965) Malakoplakia of the urinary tract: a study of twenty-four cases. Am J Clin Pathol 43:409–417
Smits JG, Kooijman CD (1986) Rheumatoid nodules in liver. Histopathology 10:1211–1213
Stanton JM, Maxted W (1981) Malacoplakia: a study of the literature and current concepts of pathogenesis, diagnosis and treatment. J Urol 125:139–146
Stein P, Yu H, Jain D, Mistry PK (2010) Hyperferritinemia and iron overload in type 1 Gaucher disease. Am J Hematol 85:472–476
Stenzel P, Weeks DA (2013) Abundant hepatic Gaucher-like cells following chemotherapy and bone marrow transplantation for hematologic malignancy: report of two cases. Int J Surg Pathol 21:89–92
Stryker WA (1941) Absorption of liquid petrolatum (mineral oil) from the intestine: a histologic and chemical study. Arch Pathol Lab Med 29:691–699
Sukov WR, Remstein ED, Nascimento AG, Sethi S, Lewin M (2009) Sclerosing extramedullary hematopoietic tumor: emphasis on diagnosis by renal biopsy. Ann Diagn Pathol 13:127–131
Tai YS, Lin PW, Chen SG, Chang KC (1998) Inflammatory pseudotumor of the liver in a patient with human immunodeficiency virus infection. Hepatogastroenterology 45:1760–1763
Tamiolakis D, Ptassolopoulos P, Simopoulos C, Kotini A, Venizelos J, Alexiadis G, Manavis J, Papadopoulos N (2003) Renal extramedullary hematopoietic tumor diagnosed by fine needle aspiration: a case report. Acta Clin Belg 58:299–301
Tamiolakis D, Venizelos J, Prassopoulos P, Simopoulos S, Bolioti S, Tsiapali M, Papadopoulos N (2004) Intrahepatic extramedullary hematopoietic tumor mimicking metastatic carcinoma from a colonic primary. Onkologie 27:65–67
Tamm EP, Rabushka LS, Fishman EK, Hruban RH, Diehl AM, Klein A (1995) Intrahepatic, extramedullary hematopoiesis mimicking hemangioma on technetium-99m red blood cell SPECT examination. Clin Imaging 19:88–91
Tanino M, Ohta G (1981) An autopsy case of xanthogranuloma in the liver (in Japanese). Nippon Shokakibyo Zasshi 78:1803–1806
Taylor JS, Lewis LA, Battle JD, Butkus A, Robertson AL, Deodhar S, Roenigk HH (1978) Plane xanthoma and multiple myeloma with lipoprotein-paraprotein complexing. Arch Dermatol 114:425–431
Thannhauser SJ, Magendantz H (1938) The different clinical groups of xanthomatous diseases: a clinical physiological study of 22 cases. Ann Intern Med 11:1662–1746
Thinda S, Tomlinson JS (2009) Mesenteric rheumatoid nodules masquerading as an intraabdominal malignancy: a case report and review of the literature. World J Surg Oncol 7:59
Touton K (1885) Ueber das Xanthom, insbesondere dessen Histiologie und Histiogenese. Vierteljahrschr Dermatol Syph 12:3.53
Trivalle C, Profit P, Bonnet B, Manchon ND, Ducastelle T, Hemet J, Bercoff E, Bourreille J (1991) Liver lipogranulomas with long-term fever caused by paraffin oil (in French). Gastroenterol Clin Biol 15:551–553
Ung KA, Remotti H, Olsson R (2000) Eosinophilic hepatic necrosis in hypereosinophilic syndrome. J Clin Gastroenterol 31:323–327
Urano S, Kodama H, Kato K, Nogura K (1995) Pyoderma gangrenosum with systemic involvement. J Dermatol 22:515–519
Vadillo M, Jucgla A, Podzamczer D, Rufi G, Domingo A (1999) Pyoderma gangrenosum with liver, spleen and bone involvement in a patient with chronic myelomonocytic leukaemia. Br J Dermatol 141:541–543
Valente AI, Pinto HC, Ramalho F, Cabrita PF, Catarino C, Serejo F, Baptista A, Saragoca A, Bordalo O, Moura MC (1997) Idiopathic hypereosinophilic syndrome presenting as cholestatic liver disease. Eur J Gastroenterol Hepatol 9:815–817
Van Crevel R, Curfs J, van der Ven AJ, Assmann K, Meis JF, van der Meer JW (1998) Functional and morphological monocyte abnormalities in a patient with malakoplakia. Am J Med 105:74–77
Verani R, Olson J, Moak JL (1980) Intrathoracic extramedullary hematopoiesis: report of a case in a patient with sickle-cell disease-beta-thalassemia. Am J Clin Pathol 73:133–137
Verola O, de Roquancourt A, Chanu B, Rouffy J, Brocheriou C (1983) Hepatic cholesterolosis. Histological, histochemical and ultrastructural study of 2 cases (in French). Sem Hop 59:1753–1759
Vogt R (1998) Das Hieracium-Herbar von Karl Touton. Willdenowia 28:253–261
Von Hansemann D (1903) Ueber Malakoplakie der Harnblase. Virchows Arch Pathol Anat 173:302–308
Wanless IR, Geddie WR (1985) Mineral oil lipogranulomata in liver and spleen. A study of 465 autopsies. Arch Pathol Lab Med 109:283–286
Warshauer DM, Schiebler ML (1991) Intrahepatic extramedullary hematopoiesis: MR, CT and sonographic appearance. J Comput Assist Tomogr 15:683–685
Wasserman SI, Goetzl EJ, Ellman L, Austen KF (1974) Tumor-associated eosinophilotactic factor. N Engl J Med 21:420–424
Weidman FD, Boston LN (1937) Generalized xanthoma tuberosum with xanthomatous changes in fresh scars of an intercurrent zoster. Arch Intern Med 59:793–822
Weidman FD, Freeman W (1924) Xanthoma tuberosum. Two necropsies disclosing lesions of the central nervous system and other tissues. Arch Derm Syphilol 9:149–175
Weidman FD, Stokes J (1937) Extensive xanthoma tuberosum in childhood due to infectious cirrhosis of the liver. Am J Dis Child 53:1230–1244
Weinrach DM, Wang KL, Cisler JJ, Diaz LK (2004) Pathologic quiz case: a 54-year-old liver transplant recipient with diffuse thickening of the sigmoid colon. Malakoplakia of the colon associated with liver transplant. Arch Pathol Lab Med 128:e133–e134
Wiener MD, Halvorsen RA, Vollmer RT, Foster WL, Roberts L (1987) Focal intrahepatic extramedullary hematopoiesis mimicking neoplasm. AJR Am J Roentgenol 149:1171–1172
Willis A, Vanhuse C, Newton KP, Wasserstein M, Morotti RA (2008) Farber’s disease type IV presenting with cholestasis and neonatal liver failure: report of two cases. Pediatr Dev Pathol 11:305–308
Wisnieski JJ, Askari AD (1981) Rheumatoid nodulosis. A relatively benign rheumatoid variant. Arch Intern Med 141:615–619
Wollina U, Haroske G (2011) Pyoderma gangraenosum. Curr Opin Rheumatol 23:50–56
Won JH, Kim BM, Ji H, Chung JJ, Yoo HS, Lee JT, Park YN, Hong SW (1999) Focal eosinophilic infiltration of the liver: a mimic of hepatic metastasis. Abdom Imaging 24:369–372
Wong Y, Chen F, Tai KS, Yip LK, Tsang KW, Chan FL, Ooi GC (1999) Imaging features of focal intrahepatic extramedullary haematopoiesis. Br J Radiol 72:906–910
Woollons A, Darley CR (1998) Xanthoma disseminatum: a case with hepatic involvement, diabetes insipidus and type IIb hyperlipidemia. Clin Exp Dermatol 23:277–280
Wright JR, Van den Hof MC, Macken MB (2005) Prenatal infantile cortical hyperostosis (Caffey’s disease): a ‘hepatic myeloid hyperplasia-pulmonary hypoplasia sequence’ can explain the lethality of early onset cases. Prenat Diagn 25:939–944
Xu R, Mistry P, McKenna G, Emre S, Schiano T, Bu-Ghanim M, Levi G, Fiel MI (2005) Hepatocellular carcinoma in type 1 Gaucher disease: a case report with review of the literature. Semin Liver Dis 25:226–229
Yang X, Bhuiya T, Esposito M (2002) Sclerosing extramedullary hematopoietic tumor. Ann Diagn Pathol 6:183–187
Yang T, Zhang BH, Zhang J, Zhang YJ, Jiang XQ, Wu MC (2007) Surgical treatment of xanthogranulomatous cholecystitis: experience in 33 cases. Hepatobiliary Pancreat Dis Int 6:504–508
Yim H, Ahn HJ, Park C, Cheon JY (1995) Xanthoma of the liver in a patient with multiple myeloma associated with hyperlipidemia. A case report. J Korean Med Sci 10:453–456
Yoo SY, Han JK, Kim YH, Kim TK, Choi BI, Han MC (2003) Focal eosinophilic infiltration in the liver: radiologic findings and clinical course. Abdom Imaging 28:326–332
Yoon KH, Ha HK, Lee JS, Suh JH, Kim MH, Kim PN, Lee MG, Yun KJ, Choi SC, Nah YH, Kim CG, Won JJ, Auh YH (1999) Inflammatory pseudotumor of the liver in patients with recurrent pyogenic cholangitis: CT-histopathologic correlation. Radiology 211:373–379
Yoshida T, Nishimori I, Kumon M, Kohsaki T, Taniuchi K, Ohtsuki Y, Onishi S (2003) Inflammatory pseudotumor of the liver: report of a case diagnosed by needle biopsy. Hepatol Res 27:83–86
Yousef GM, Naghibi B (2007) Malakoplakia outside the urinary tract. Arch Pathol Lab Med 131:297–300
Yu JS, Yoon SW, Park MS, Lee JH, Kim KW (2005) Eosinophilic hepatic necrosis: magnetic resonance imaging and computed tomography comparison. J Comput Assist Tomogr 29:765–771
Yuoh G, Hove MG, Wen J, Haque AK (1996) Pulmonary malakoplakia in acquired immunodeficiency syndrome: an ultrastructural study of morphogenesis of Michaelis-Gutmann bodies. Mod Pathol 9:476–483
Zeek PM (1952) Periarteritis nodosa: a critical review. Am J Clin Pathol 22:777–790
Zhang Z, Mandal AK, Mital A, Popescu N, Zimonjic D, Moser A, Moser H, Mukherjee AB (2000) Human acid ceramidase gene: novel mutations in Farber disease. Mol Genet Metab 70:301–309
Zhou et al. 1992. http://www.ncbi.nlm.nih.gov/pubmed/1464731
Zhu H, Bodenheimer HC, Clain DJ, Min AD, Theise ND (2010) Hepatic lipogranulomas in patients with chronic liver disease: association with hepatitis C and fatty liver disease. World J Gastroenterol 16:5065–5069
Ziff M (1990) The rheumatoid nodule. Arthritis Rheum 33:761–767
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this entry
Cite this entry
Zimmermann, A. (2017). Hepatobiliary Pseudotumors Consisting of Nonneoplastic Hematopoietic Cells and Cells of the Macrophage Lineage. In: Tumors and Tumor-Like Lesions of the Hepatobiliary Tract. Springer, Cham. https://doi.org/10.1007/978-3-319-26956-6_125
Download citation
DOI: https://doi.org/10.1007/978-3-319-26956-6_125
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26954-2
Online ISBN: 978-3-319-26956-6
eBook Packages: MedicineReference Module Medicine