
Abstract
Homing of circulating cells to pre-metastatic niches of the liver and their subsequent growth to a metastasis requires a complex interaction between tumor cells and various normal cellular and matrix components of the future metastatic site. In the course of homing to endothelial surfaces of hepatic blood vessels and vascular invasion, tumor cells interact with platelets and induce platelet adhesion and aggregation. Tumor cell-induced thrombocyte aggregation facilitates early steps of metastasis through increased tumor cell arrest and formation of tumor cell emboli. Platelets participating in tumor cell aggregates promote cell adhesion and invasion and release growth factors for cancer cells. Following their exit from blood vessels, tumor cells engage in complex interactions with stromal cells. The stromal microenvironment profoundly influences growth and invasion of tumor cells in the metastatic site. Stromal cells interacting with tumor cells include cancer-associated fibroblasts and myofibroblasts, vascular cells, tumor-associated macrophages/TAMs, myeloid suppressor cells, and other cells of local immune responses. Cancer cells in turn constantly modulate the cellular composition of stroma, promote angiogenesis, and are subject to epithelial-mesenchymal transition.
Access provided by CONRICYT-eBooks. Download reference work entry PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Platelet Interactions, Platelet Aggregation, and the Formation of Tumor Metastasis
Introduction
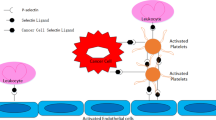
In the course of vascular invasion and the associated endothelial cell damage, cancer cells having contact to streaming blood can induce platelet homing and thrombocyte aggregation. These thrombocyte aggregates, being in contact with the subendothelial space following endothelial cell loss, form a distinct niche for tumor cells that promotes growth and the formation of metastases. Tumor cell-induced platelet aggregation facilitates hematogenous metastatic spread by increasing the arrest of tumor cell clusters and tumor cell emboli in the microcirculation and is also involved in differential homing of platelet-tumor cell aggregates to distinct vascular beds (reviews: Honn et al. 1992; Tsuruo and Fujita 2008; Sharma et al. 2014). Generally, platelets can exert pro-metastatic effects. Platelet-tumor cell interactions are sufficient to prime tumor cells for subsequent metastasis, involving TGF-beta/Smad and NF-kB pathways via platelet-derived TGF-beta (Labelle et al. 2011). Platelets possess a complex granule proteome (Koseoglu and Flaumenhaft 2013) that contains numerous factors that can affect neoplastic cells. Through secretion of granule proteins, platelets modulate growth of tumor cells and the associated angiogenesis (Dovizio et al. 2014; Riedl et al. 2014). Thrombocyte alpha-granules of platelets also contain chemokines that can affect tumor cell behavior (Karshovska et al. 2013). In addition, platelets store laminins 411/421 and 511/521 in non-alpha- or dense-granule compartments and secrete these proteins via microvesicles (Pook et al. 2014).
Thrombocyte-Tumor Cell Interactions
It is known for long that patients with solid cancers can show elevated platelet blood counts in the absence of a paraneoplastic thrombocythemia. This thrombocytosis varies widely in its severity but may be associated with shorter patient survival (review: Buergy et al. 2012) and is associated with a higher metastatic load, e.g., in hepatocellular carcinoma. Elevated circulating thrombocytes enhance cancer cell migration and promote hematogenous metastasis in patients with lung cancer (Li et al. 2014). Neoplastic cells of solid cancers can undergo complex interactions with thrombocytes, whereby these interactions are capable to induce platelet aggregation favoring homing of cancer cells after intravascular spread (see below; Chang et al. 2009) or to affect the biological behavior of neoplastic cells, e.g., promoting their proliferation, cell-matrix adhesion, and/or invasive features. Cancer cells, also those of metastases, can internalize thrombocytes or thrombocyte fragments (Panasci et al. 1980; Bhatia and Dey 2013), a process probably inducing complex signaling pathways mediated by platelet granule factors.
Platelets and Platelet Aggregates as Pacemakers of Metastatic Spread
In the setting of metastatic spread, thrombocytes play a pro-metastatic role involving several steps of the invasion and metastatic cascade (Karpatkin and Pearlstein 1981; Gasic 1984; Mehta 1984; Falanga et al. 2003; Borsig 2008; Erpenbeck and Schön 2010; Jain et al. 2010; Bambace and Holmes 2011; Gay and Felding-Habermann 2011). Interactions between platelets and tumor cells are important for metastatic spread and are mediated by adhesive receptors expressed by both partners of the interaction (Oleksowicz and Dutcher 1995). Thrombocytes promote the adhesion of spreading tumor cells to endothelial cell surfaces (Nierodzik et al. 1995), form aggregates that can store growth factors to be delivered to tumor cells, mediate access to the subendothelial space via collagen-induced viscous metamorphosis, slow down the streaming blood through induction of coagulation, induce a focal angiogenic response, and shield tumor cells from attack of host cells, in particular natural killer cells (Table 1; Yahalom et al. 1985; Eldor et al. 1987; Honn et al. 1992; Tsuruo and Fujita 2008; Gil-Bernabé et al. 2013; Reymond et al. 2013). Promotion of tumor angiogenesis by platelet products is an important mechanism that is not only dependent on VEGF and is the result of a combined action of several molecules that form the angiogenic payload of platelets (Battinelli et al. 2011; Sabrkhany et al. 2011; Radziwon-Balicka et al. 2012; Etulain et al. 2013). Platelet extracts induced growth, migration, and invasion in human hepatocellular carcinoma in vitro (Carr et al. 2014). Direct signaling between thrombocytes and tumor cells also induces epithelial-mesenchymal transition/EMT, an alteration that is critically involved in tumor metastasis (Labelle et al. 2011). Important steps in the process of platelet-induced tumor cell adhesion, homing, and transendothelial migration are mediated by P-selectin. P-selectin plays an important role in liver metastasis even in the absence of natural killer cell function (Coupland et al. 2012).
Tumor Cells Can Promote Platelet Aggregation and Release of Microvesicles and Exosomes
Several tumor types have been shown to produce and secrete the platelet aggregation-inducing factor Aggrus, a protein also termed podoplanin . Aggrus is secreted by various tumors, including CRC (Kato et al. 2003). Aggrus released by tumor cells interacts with a platelet receptor termed CLEC-2, and the ligand-receptor binding induced thrombocyte aggregation (Takagi et al. 2013). Aggrus-induced aggregate formation plays an important role for the changes in blood rheology required for tumor cell homing in the microvascular bed (Jurasz et al. 2004). Platelet aggregates and thrombocyte-induced local blood coagulation cause slowdown of streaming blood in microvessels, enabling tumor cells to engage with endothelia and adhere to the inner vessel wall. In fact, platelet-derived Aggrus/podoplanin was shown to promote metastasis (Kunita et al. 2007; Suzuki-Inoue 2011; Fujita and Takagi 2012; Lowe et al. 2012). One mechanism of metastasis promotion by podoplanin involves an induction of tumor cell migration (Shen et al. 2010; Kunita et al. 2011), whereby serine moieties in the intracellular tail of podoplanin regulate cell motility (Krishnan et al. 2013). In addition, contacts between platelet aggregates induced by tumor cells permit the transfer of granule and nongranule proteins of platelets and of exosomes to adjacent endothelial cells and tumor cells. Platelet-derived microvesicles or microparticles are important participants in intercellular communication and play a role in cancer progression (Varon and Shai 2009; Aatonen et al. 2012). Activated platelets secrete granule proteins and parallel release two types of membrane microvesicles, namely, simple microvesicles by surface shedding and more complex exosomes derived from multivesicular bodies and alpha-granules (Heijnen et al. 1999). Platelet-derived microvesicles (microparticles) possess membrane domains that contain adhesion molecules and diverse receptors and contain signal substances that can stimulate angiogenesis (Varon and Shai 2009) and tissue regeneration (Varon et al. 2012).
Platelet Mimicry of Cancer Cells
Certain cancer cell types acquire a geno-phenotype the closely resembles that of thrombocytes/platelets. Such tumor cells express megakaryocyte genes, including adhesion receptors alpha IIb beta 3, thrombin receptor and PECAM/Cd31, and/or platelet-type 12-LOX. These acquired expression patterns enable the cancer cells to activate the coagulation cascade. It is currently considered that these platelet-like features of cancer cells affect their capability to spread and metastasize (Timar et al. 2005).
The Metastatic Stromal and Vascular Environment of the Liver
Introduction
The liver is known to provide a tissual microenvironment that favors the establishment of metastases of various malignancies, but specifically carcinomas. Part of the tumor cells transported into the liver via the vascular system resists antitumor defense mechanisms and can adhere as viable cells to endothelium of blood vessels, chiefly endothelia of hepatic sinusoids. Here, they start to respond to growth factors produced and secreted by neighboring hepatocytes, perisinusoidal cells, and vascular cells. In the first step, this results in avascular micrometastases in periportal areas of liver lobules. The next step is characterized by stroma formation, a prerequisite for angiogenesis, because newly formed blood vessels depend on being embedded within a mesenchymal matrix with its specific extracellular matrix (ECM) proteins. Fibroblasts from portal tracts become cancer-associated fibroblasts (CAFs) as an important component of stroma, and stromal myofibroblasts are recruited from activated hepatic stellate cells and portal tracts. Together, these cells generate a “private” tumor microenvironment that has a pro-metastatic effect (Vidal-Vanaclocha 2008, 2011).
The Pro-metastatic Effect of Stromal Interactions
The stromal microenvironment profoundly influences many steps of cancer progression, including the ability of cancer cells to metastasize (the ‘stromal factor’; Wernicke et al. 2011). Within stroma, influences of this distinct and complex microenvironment are mediated by bidirectional interactions between epithelial tumor cells and the neighboring stromal cells, these interactions referring to adhesion, migration, proteolysis, angiogenesis, homing strategies, epithelial-mesenchymal transition, immune escape mechanisms, and survival (reviews: Bogenrieder and Herlyn 2003; Orimo and Weinberg 2006).
What are the pathways leading to stromal formation in metastases? Early in the outgrowth phase of murine colorectal carcinoma micrometastases, hepatic Kupffer cells and fibroblasts are recruited and start to invade the metastatic nodules. These transitional metastases are connected by protrusions of fibroblast-rich tissues co-localized with collagen-rich matrix and CD31-positive cells. The latter step is characterized by the generation of a pro-angiogenic niche and the switch of a transitional metastasis to an established, vascularized metastasis (Higashi et al. 2002). Stromal components, including fibroblasts, myofibroblasts, endothelial and other vascular cells, mesenchymal stem cells, granulocytes, and cells of the immune system, interact with carcinoma cells to promote growth, invasion, and metastasis (reviews: Li et al. 2007; Aharinejad et al. 2009; Finger and Giaccia 2010; Pietras and Ostman 2010; Udagawa and Wood 2010). Stroma-derived growth factors and cytokines produced by stromal-resident lymphoid cells and macrophages activate autocrine and paracrine oncogenic signaling pathways, which in turn promote proliferation or spread of epithelial cancer cells. Stromal cell can produce matrix metalloproteinases (MMPs) which are known to operate within the invasion cascade. However, whereas MMP-9 is upregulated in primary CRCs, it is expressed mainly by macrophages in the invasion zone of hepatic metastases and there generally less expressed than in primary tumors (Illemann et al. 2006). Both mesenchymal cells and macrophages are active in the procoagulative environment of tumors, coagulation factors such as fibrinogen and thrombin affecting tumor cell growth and differentiation. Components of stromal cells may also promote the locomotor and migratory activity of carcinoma cells. The intermediate filament protein, vimentin, a constituent of mesenchymal cells, is an AKT1 target mediating motility and invasion (Zhu et al. 2011a). Factors regulating epithelial-mesenchymal transition (EMT) affect the metastatic process. The developmental transcription factor, Six1, induces EMT in dependence of TGF-beta signaling. Six1 is a critical mediator of the switch in TGF-beta signaling from tumor-suppressive to tumor-promotional activity, the TGF-beta type I receptor being the target of Six1 and a critical effector of Six1-induced TGF-beta signaling and EMT (Micalizzi et al. 2010).
Cancer-Associated Fibroblasts
Cancer-associated fibroblasts (CAFs) seem to regulate many aspects of tumorigenesis and cancer spread (reviews: Orimo and Weinberg 2006; Cirri and Chiarugi 2012). Stromal fibroblasts in CRC metastases originate from resident fibroblasts and create an inflammatory microenvironment characterized by TNF-alpha-mediated upregulation of IL-8 via nuclear factor kappaB (Mueller et al. 2007). Colorectal cancer-associated fibroblasts represent a genetically distinct population of stromal cells (Mrazek et al. 2014). Activated CAFs residing in tumor stroma are a source of growth factors for carcinoma cells, factors promoting angiogenesis and lymphangiogenesis, cytokines mediating tumor immunity, and factors involved in epithelial-mesenchymal transition (Räsänen and Vaheri 2010). Primary CAFs from colorectal liver metastases express several inflammatory, tumor-enhancing factors, including IL6 and monocyte-chemoattractant protein (MCP)-1. Both factors are induced by TNF-alpha, which also upregulates ICAM-1. A similar reaction pattern was found for liver-resident, non-tumor-associated fibroblasts (Mueller et al. 2010), which may therefore constitute a cell type playing a role in the generation of a pro-metastatic stroma. The stroma of carcinomas exerts an influence on angiogenesis and antiangiogenesis and by this affects the development of metastases. The growth of metastases is dependent on the growth of blood vessels into the tumor mass. The extracellular matrix of the stroma and its specific proteins, including basement membrane laminin, fibronectin, and tenascin, is a source of both pro-angiogenic factor activation and endogenous angiogenesis inhibitors, such as endostatin (review: Nyberg et al. 2008). Multicellular spheroids of murine colorectal cancer cells mimic micrometastases and their growth pattern activates the pro-angiogenic phenotype of the carcinoma cells (Valcarcel et al. 2008). Antiangiogenic effects and hypoxia-dependent signaling pathways are counteracted by semaphorin 3A produced by cancer cells in metastases (Maione et al. 2012).
Lymphangiogenesis in Hepatic Metastasis: Does It Play a Role?
For the metastatic spread of cancer cells, generation of a lymph vessel network (lymphangiogenesis) plays a crucial role. A dynamic lymph vessel system has an active role in metastatic dissemination, and metastasizing tumor cells are capable to synthesize and secrete factors that promote lymphangiogenesis. In a mouse model of breast cancer, tumor cells produced the homeodomain-containing transcription factor SIX1, which induces lymphangiogenesis and metastasis via upregulation of VEGF-C (Wang et al. 2012a).
Interactions of Metastatic Carcinoma Cells with Hepatic Stellate Cells (HSC)
As stroma plays a significant role in homing and growth of carcinoma metastasis, cells potentially producing stroma in the liver are of specific interest. Apart from fibroblasts and myofibroblasts in portal tracts and vessel walls, the only cell type of the liver capable of producing extracellular matrix is the hepatic stellate cell (HSC) . We have shown that HSCs can interact and form tight adhesions with hepatocytes during the early regeneration phase of the normal rat liver (Mabuchi et al. 2004a, b), suggesting that HSCs can form a epithelial-mesenchymal interactome in the liver. By the use of a nude mouse model, it was demonstrated that intrasplenically injected colon carcinoma cells migrated into the space of Disse and underwent proliferation, in close association with hepatocytes and HSCs. At 14 days, HSCs were accumulated around the emerging small tumor mass and showed transformation into alpha-SMA-positive myofibroblasts. The growing colon carcinoma cells produced PDGF-AB, enhancing proliferation and migration of HSCs, while HSCs produced PDGF-AB, HGF, and TGF-beta and could augment proliferation of carcinoma cells, suggesting complex bidirectional interactions between metastasizing carcinoma cells and activated HSCs (Shimizu et al. 2000). This interaction may result in an “amplification loop” to further metastatic growth in the liver (review: Kang et al. 2011). The transition from HSCs to myofibroblasts in pro-metastatic hepatic stroma is regulated by the fibrillar collagen receptor discoidin domain receptor 2 (DDR2). Downregulation of DDR2 promotes myofibroblast transition and predisposes hepatic tissue to colorectal carcinoma metastasis in mice (Badiola et al. 2011).
Stroma-Associated Inflammatory and Immunologic Effector Cells Control Metastasis Formation: TANs, TAMs, Myeloid-Derived Suppressor Cells (MDSCs), Myeloid Angiogenic Cells (MACs), and Lymphocytes
The stroma of many tumors contains leukocytic infiltrates/inflammatory cells which are instrumental in immune reactions that either inhibit tumors or produce factors that stimulate growth and metastasis. An inflammatory microenvironment induced by myeloid cells is key alteration for promoting tumor invasion and metastatic spread (Grugan et al. 2012; Capece et al. 2013; Smith and Kang 2013; Keskinov and Shurin 2014). A specific role is attributed to tumor-associated macrophages (TAMs) , tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells/MDSCs. The presence of TAMs and TANs has been linked to poor clinical outcomes in solid tumors, as these cells can switch from a tumor-suppressing to a tumor-promoting phenotype (review: Alphonso and Alahari 2009). TANs can play a role in tumor cell detachment (tumor cell individualization as a prerequisite for motility and migration), have a pro-angiogenic activity, and secrete MMP-9 playing a role in tumor invasion (Benson et al. 2012).
TAMs form a major component of tumor stroma, consist of several functionally different subsets, and originate from distinct circulating monocytes that home to the tumor microenvironment (Movahedi et al. 2010). But accumulation of TAMs in stroma is also accomplished by in situ proliferations of TAMs in tumors (Tymoszuk et al. 2014). Local accumulation of monocytes differentiating into TAMs is promoted by the TGF-beta superfamily member, Nodal, which is an embryonal morphogen that is upregulated in numerous tumors (Wang et al. 2014a). The interaction of stromal cells with TMAs results in differential macrophage programming in the tumor microenvironment (Ruffell et al. 2012). TAMs are major players in the tumor microenvironment and exert a strong influence on progression and metastatic spread of cancer (Hao et al. 2012; Galdiero et al. 2013). In the course of tumor progression, TAMs and their hematogenous precursors, the monocytes, are actively recruited into tumor tissue, and particularly its stroma. In their interaction with tumor and stromal cells, TAMs can undergo several functional changes. Whereas classical M1 macrophages are engaged in inflammatory responses, antitumor immunity, and antigen clearance, alternative M2 macrophages are active in anti-inflammatory reactions, wound healing, stroma formation, and tumor progression (reviews: Chanmee et al. 2014; Van Overmeire et al. 2014). TAMs most closely resemble polarized class two macrophages/M2 and are cells that markedly modulate the composition and function of tumor microenvironments (Mantovani et al. 2002). TAMs have upregulated expression of CD206 and CD163 markers of alternative activation but do not have increased expression of classically activated macrophage markers, CCR2 and CCR5. Monocytes as precursors of M2 TAMs are recruited to metastatic sites by distinct homing mechanisms. A major ligand facilitating monocyte accumulation is P-selectin glycoprotein ligand-1/PSGL-1 (Hoos et al. 2014).
The interaction of TAMs with tumor cells is mediated by adhesion molecules and glycosaminoglycans. Hyaluronan synthase HAS2 stimulates the interaction between cancer cells and TAMs and promotes tumor progression and metastasis (Okuda et al. 2012). The interactome of TAMs, lymphoid cells (mainly T cells), and myeloid-derived suppressor cells is active in pro-angiogenic reactions and the preparation of homing vascular surface facilitating tumor cell and platelet adherence and cancer cell transmigration (reviews: Labelle and Hynes 2012; Smith and Kang 2013).
M2 TAMs exhibit a distinct functional profile characterized by the (stimulated) production of a wide array of factors. Activated TAMs can produce a host of cytokines that can modulate tumor cell behavior and affect stromal cell production function. TAMs strongly express interleukin receptor-associated kinase (IRAK) -M, a serine/threonine kinase that is a potent negative regulator of Toll-like receptor signaling (Kobayashi et al. 2002). In tumors, IRAK-M is induced by TGF-beta1 from stromal cells and regulates tumor growth (Standiford et al. 2011). TGF-beta1 plays a central role in myeloid cell regulation (Pang et al. 2013). TAMS can secrete matrix metalloproteinases (MMPs), including MMP-9, whereby differentiation-related expression of MMP depends on an interaction of the ECM component, laminin.-5, with monocytes (Kamoshida et al. 2014). TAMs produce oncostatin M and VEGF (Benson et al. 2012). Tumor-infiltrating monocytes/macrophages produce and secrete the innate immune response mediator, human beta-defensin-3, a factor that inhibits cancer cell migration through downregulation of metastasis-associated 1 family, member 2 (MTA2 9 expression (Uraki et al. 2014)). TAMs are involved in the progression of HCC, as M2 type macrophages can modulate the composition of HCC stroma and modify immunosuppressive functions of immune effector cells (Shirabe et al. 2012). In HCCs, TAMs can promote a stem cell-like property of cancer cells through TGF-beta1-induced epithelial-mesenchymal transition (Fan et al. 2014), a mechanism that favors a metastatic phenotype. TAMs can also induce the expression of the macrophage receptor, CD163, in malignant cells, thus resulting in some sort of “induced receptor sistership”. CD163 expression in a subpopulation of cancer cells may constitute a phenotypic shift associated with epithelial-mesenchymal transition, increased invasion, and a metastatic phenotype (Maniecki et al. 2012). Part of TMAs are involved in the promotion of angiogenesis (Owen and Mohamadzadeh 2013). These specialized cells are termed myeloid angiogenic cells (MACs). MACs are in part Tie-2 expressing monocytes and significantly contribute to angiogenesis and vascular repair through paracrine mechanisms as they lack the capacity to differentiate into endothelial cells (Chambers et al. 2013). Experimental selective ablation of TAMs with MAC features suppresses angiogenesis and metastatic tumor spread (Lin et al. 2013). MAC-induced angiogenesis depends on the expression of the myeloid cell receptor LRP1/CD91 that regulates monocyte recruitment and VEGF availability in tumors (Staudt et al. 2013).
In addition to TAN and TAM, myeloid-derived suppressor cells (MDSCs ) play a significant role in the function of tumor microenvironment, tumor progression, and metastasis (reviews: Schmid and Varner 2012; Brandau et al. 2013; Diaz-Montero et al. 2014). MDSCs are bone marrow-derived myeloid cells that are recruited to tumors, where they are transformed to potent immunosuppressive cells, which, however, also have functions other than blunting immune reactions (reviews: Mantovani et al. 2009; Dumitru et al. 2012; Schmid et al. 2012; Diaz-Montero et al. 2014). Apart from their role in mediating immunosuppression via strong inhibition of antitumor immune reactions produced by T cells, MDSCs strongly stimulate tumorigenesis, tumor growth, and metastasis (Umansky and Sevko 2013), probably by modulating the cellular composition of stroma in pro-metastatic niches. MDSCs enhance the stemness of cancer cells by inducing micron-101 and suppressing the corepressor CtBP2(C-terminal binding protein-2, a protein that directly targets stem cell core genes resulting in cancer cell stemness and an invasive and metastatic phenotype (Cui et al. 2013)). The function of MDSCs is regulated by microRNA-155. Downregulation of the miR enhances the recruitment and function of MDSCs in the tumor microenvironment (Wang et al. 2014d).
Interactions of Carcinoma Cells with Hepatic Blood Vessel Cells and Vascularization of Metastases
Successful growth of metastases requires the generation of a stroma harboring blood vessels produced in the setting of tumor-induced angiogenesis. The mode of neovascularization of metastases originating from various tumors varies itself considerably, suggesting an “individualized” mode of angiogenesis. A corrosion case study of 22 livers with multiple metastases from different primary neoplasms showed the development of an individual pattern of vascularization in all metastases. The majority of metastase displayed a distinct blood supply by branches of the hepatic artery and portal vein branches, whereby all metastases of the same liver showed an identical vascularization pattern (Strohmeyer et al. 1986). Intra- and peritumoral connections between vessels fed by the arteries and portal vein branches are mediated by sinusoidal connections (Nikfarjam et al. 2003). Also a Microfil injection study in human autopsies with liver metastases showed both an arterial and portal mode of vascularization, although the hepatic artery mode was more important (Lin et al. 1984). Injection studies of human liver metastases exhibited a highly abnormal mode of vascularization, never imitating the normal angioarchitecture of the liver (Strohmeyer et al. 1987). Gelatine-injected specimens of human hepatic metastases also revealed that vascularized micrometastases in the vicinity of macrometastases are more common than anticipated from routine preparations (Haugeberg et al. 1988). A gamma camera imaging study of colorectal hepatic metastases revealed that more than twice as much of test substrate was delivered per volume of tumor relative to liver by the hepatic artery as by the portal vein (Ridge et al. 1987). Portal vein blood played a minor role in a rat model of liver metastases (Archer and Gray 1989). In a murine model, blood feeding metastases was distributed from arteries to capillaries of the metastasis center, from where it flowed to a superficial venous network and then further to hepatic veins (Voboril 2005).
In a murine hepatic metastasis model , sinusoidal endothelial cells are activated by interaction with colon carcinoma cells. These activated endothelial cells display an increased expression of mannose receptor (ManR) and ManR-mediated endocytosis. This effect depends on a two-step mechanism: (1) release of COX-2-dependent IL-1 stimulating factors by LFA1-expressing C26 cells in response to ICAM1, which is expressed by activated sinusoidal endothelial cells and (2) widespread upregulation of ManR in endothelia through tumor cell-induced IL-1. ICAM-1-induced tumor COX-2 decreases antitumor activity during hepatic metastasis through Il-1-induced ManR in endothelial cells, ManR therefore constituting a common mediator for the pro-metastatic effect of Il-1, COX-2, and ICAM-1 (Arteta et al. 2010).
Features of Hepatic Parenchyma That Can Promote Metastatic Growth
Ischemia/reperfusion injury to liver parenchyma can accelerate the outgrowth of preestablished colorectal micrometastases (van der Bilt et al. 2005). With increasing ischemia times, tissue necrosis, and ischemia/reperfusion injury-accelerated tumor growth in mice increased, and in this murine model, outgrowth of micrometastases was further increased by increasing the age of the mice and steatosis of the livers (van der Bilt et al. 2008).
Actions of miRNAs in the Tumor Stroma
In breast cancer metastases, it has been found that miRNA-31 inhibits metastatic growth through the pleiotropic suppression of pro-metastatic target genes that include integrin alpha(5), radixin, and RhoA (Valastyan et al. 2010). Heparanase , a potent protumorigenic and pro-metastatic enzyme which is produced by stromal cells, is targeted and suppressed by miRNA-1258 (Zhang et al. 2011). Epigenetic alterations of miRNA deregulation are common in various tumors. Enhancer of zeste homolog 2 (histone H3 lysine 27 trimethylating enzyme), frequently upregulated in malignancies, epigenetically silences multiple tumor suppressor miRNAs to promote liver cancer metastasis (Au et al. 2012).
Pathogenic Features of Liver Metastases: Epithelial-Mesenchymal Transition (EMT)
Introduction
Epithelial-mesenchymal transition (EMT) is a major process involved in the construction of tissues in early stages of ontogenesis. There, EMT is characterized by the internalization of epithelial cells to give rise to mesodermal/mesenchymal tissues. To arrive at this result, epithelial cells must undergo dramatic changes, including the disconnection of junctions, undergoing polarization and shape change to become migrating cells, and altering cell surface adhesion molecule complements (the “adhesome”). Specifically, distinct expression patterns of integrins are involved in EMT, described as an integrin adhesome (Zaidel-Bar et al. 2007; Zaidel-Bar and Geiger 2010; Winograd-Katz et al. 2014). Other adhesion platforms involved in EMT generation comprise the cadherin adhesome (Zaidel-Bar 2013) and dystroglycan adhesome (Bello et al. 2014). EMT in cancer is characterized by dysregulation of several molecules that comprise, apart from adhesion molecules, intermediate filament proteins, calcium-binding proteins, and transcription factors. Tumor cells in EMT show loss of E-cadherin expression, upregulation of vimentin, induction of alpha-SMA, and expression of Snail1 and Snail2/Slug (Maeng et al. 2014). Typically, cells engaged in EMT fail to express E-cadherin, an important adhesin. The dissolution of the E-cadherin-mediated adherens junction is an important early step in EMT in epithelial malignancies. Downregulation of E-cadherin is accomplished by the action of several transcription factors forming a hierarchical system. Initial transcriptional inducers of EMT are Snail1 and Snail2/Slug, which lead to the activation of ZEB family members, TCF3, TCF4, Twist, Goosecoid, FOXC1, FOXC2, H-Ras, CD44, claudin-1, TGF-beta1, and TNF-alpha. Upregulation of Snail2/Slug and FOXC2 by either Snail1 or Twist does not depend in TGF-beta1 signaling (Taube et al. 2010; Hugo et al. 2011; Qin et al. 2012; Xu et al. 2012; Suh et al. 2013; Caja and Vannucci 2014; Liu et al. 2014b; Okabe et al. 2014). Slug (or now Snail2) is a transcriptional repressor of E-cadherin and is a downstream target of SPARC/osteonectin (Fenouille et al. 2012). Twist1, a basic helix-loop-helix protein transcription factor involved in EMT both in ontogenesis and cancer, is regulated by phosphoregulation (Firulli and Conway 2008) and stabilized by autophagy deficiency via binding of SQSTM1 to Twist1 in order to prevent autophagosomal Twist1 degradation (Qiang and He 2014). A negative regulator in EMT-related TGF-beta signaling is Smurf2, a protein that interacts with the HCV viral protease NS3-4A (Verga-Gérard et al. 2013). The factors activated by the master EMT inducers Snail1 and Slug have themselves distinct targets that mediate their effects in the EMT molecular pathway. For example, ZEB1 has the collagen receptor tyrosine kinase, discoidin domain receptor 1/DDR1 as its transcriptional target, whereby ZEP1 downregulates DDR1 and contributes to an invasive cancer cell phenotype (Koh et al. 2014). Twist1, having a central role as an effector for EMT induction, can also induce endothelial transdifferentiation of cancer cells through a Jagged1-Krüppel-like factor/KLF-4 signaling axis (Chen et al. 2014a). A further transcription factor involved in EMT and metastasis I a cytotoxic T-lymphocyte agonist epitope of brachyury. EMT is also regulated by the Wnt/beta-catenin signaling cascade, which is activated in the setting of EMT by stromal cell-derived factor 1/SDF-1 and its receptor, CXCR4 (Hu et al. 2014). EMT induced by TNF-alpha requires stabilization of Snail1 mediated by Akt/GSK-3beta signaling, GSK-3beta being a Wnt signaling component (Wang et al. 2013a). EMT can be induced by extravasated platelet aggregation, one critical molecular being CD42b coexpressed with Snail1 (Miyashita et al. 2014).
In addition to factors promoting EMT, there are pathways counteracting EMT generation, and it may be anticipated that failure of such inhibitory pathways in tumors might be involved in metastasis. Protocadherin 9, frequently lost in HCC, inhibits EMT and cell migration through activation of the Wnt signaling component, GSK-3beta (Zhu et al. 2014). EMT can be repressed expression of SMAR1 which represses Snail2/Slug transcription and inhibits E-cadherin degradation in cancer cells (Adhikary et al. 2014). SMAR1 is a nuclear protein involved in chromatin homeostasis that binds to nuclear matrix attachment regions/MARs, regions crucial for proper periodic chromatin arrangement.
Epithelial-Mesenchymal Transition as a Metastasis-Promoting Alteration in Cancer
EMT plays a central role in the generation of a proinvasive tumor microenvironment and metastatic spread of cancer (reviews: Chaffer and Weinberg 2011; Jing et al. 2011; Meng and Wu 2012; Jung et al. 2015). EMT provides cancer cells with migratory, invasive, stroma interactive, and stem cell properties that enable them to disseminate and settle at remote sites. Advanced cancer stage and metastatic spread in HCC are associated with a typical EMT-related cellular alteration, i.e., downregulation of E-cadherin (Zhai et al. 2014). Abnormal expression of EMT-related proteins in HCC, i.e., loss of E-cadherin and overexpression of vimentin and S100 proteins, is correlated with an aggressive metastatic phenotype of this tumor (Zhai et al. 2014). EMT in HCC is related to hepatitis virus infection, viral proteins promoting EMT pathways by various mechanisms (Wang et al. 2014d). For example, HCV-induced expression of osteopontin, a pro-metastatic phosphoprotein overexpressed in HCC, is associated with EMT via the activation of the GSK-3beta signaling pathway (Iqbal et al. 2013).
EMT promoting invasion and metastatic spread in cancers, including liver cancer, is induced by aberrant expression of various pro-EMT factors. Metastasis in HCC is strongly promoted by overexpression of the pro-EMT transcription factor, Snail2/Slug (Sun et al. 2014). TGF-beta1 plays a central role in EMT induction and metastasis in HCC (Reichl et al. 2012). TGF-beta1, which induces EMT in HCC, is induced in these tumors by tumor-associated macrophages/TAMS which also promote cancer stem cell-like features in HCC cells (Fan et al. 2014). Tumor-derived secretory clusterin, a protein involved in the regulation of TGF-beta1-Smad3 signaling, facilitates HCC metastasis via induction of EMT (Wang et al. 2012b). Another member of the TGF-beta family of proteins, bone morphogenetic protein-9/BMP-9, also induces EMT in HCC (Li et al. 2013). Upregulation of hepatocyte growth factor/HGF promotes carcinogenesis and EMT in HCC through Akt and COX-2 pathways, whereby HGF causes loss of cell surface E-cadherin (Ogunwobi and Liu 2011). Expression of the Hedgehog signaling effector, Gli1, in HCC promotes EMT, invasion, and metastasis (Chen et al. 2014b). ZEB1, a second rank effector of EMT-inducing factors, reduces E-cadherin expression in HCC cells and increased expression of N-cadherin and vimentin, promoting EMT and metastasis in HCC (Zhou et al. 2012). Forkhead box Q1/FOXQ1, a master regulator of tumor metastasis, promotes HCC metastasis by transactivating the EMT inducer ZEB2 (Xia et al. 2014). Loss of expression of functional p53 in HCC is correlated with EMT via regulation of the beta-catenin signaling pathway (Wang et al. 2013b). Downregulation of lipocalin-22 by TGF-beta1 is associated with Tist1 expression and EMT in HCC (Wang et al. 2013b). Stress-activated protein kinase (SAPK)-interacting protein1 or SIN1 which is a key regulator of Akt is overexpressed in HCC and promotes metastasis via induction of EMT (Xu et al. 2013). Activated Wnt/beta-catenin signaling enhances hypoxia-induced EMT in HCC via crosstalk with hypoxia inducing factor-1alpha signaling (Zhang et al. 2013b). Hypoxia and hypoxia-inducible factor-1alpha as such induce EMT in HCCs through activation of Snail1 (Zhang et al. 2013c). EMT-related upregulation of hypoxia-inducible factor-1alpha induces the transcription factor PROX1, resulting in HCC metastasis (Liu et al. 2013). Sirtuin 1/SIRT1, a protein implicated I telomere maintenance and growth in HCC, can mediate EMT in these neoplasms by GSK-3beta/beta-catenin signaling (Chen et al. 2013). Metastasis of HCC induced by EMT is promoted by elevated expression of myeloid differentiation factor 88/MyD88, a protein interacting with p85, a regulatory subunit of phosphoinositide 3-kinase/PI3-K, causing Akt activation, subsequent GSK-3beta phosphorylation, and stabilization of the pro-EMT factor Snail1 (Jia et al. 2014). HCC expresses mortalin, a member of the HSP70 heat shock protein family involved in stress response regulation. Overexpression of mortalin is associated with EMT, angiogenesis metastasis in HCC (Yi et al. 2008; Chen et al. 2014c). In HCC, EMT is induced metadherin, a protein known to increase metastasis in these tumors (Zhu et al. 2011b). In various cancer types, including HCC, EMT as a mechanism involved in metastasis is modulated through the expression of members of a calcium-binding family, the S100 proteins. S100 proteins are critically involved in tumor cell-stromal cell interactions, promotion of EMT, and the generation of a proinflammatory tumor microenvironment promoting invasion and spread (Lukanidin and Sleeman 2012). Induction of EMT by S100A4 is regulated by the Sonic Hedgehog-Gli1 signaling pathway, whereby the Hedgehog effector Gli1 modulates S100A4 expression via cis-acting elements (Xu et al. 2014). S100A4 induces migration and invasion in HCC cells through the induction of an NF-kappaB-dependent MMP-9 signal (Zhang et al. 2013d). In HCC, abnormal expression of EMT-related S100A4 is correlated with an aggressive tumor biology (Zhai et al. 2014). The metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to have protumorigenic features (Bettum et al. 2014). In addition to S100A4, a second S100 protein can induce EMT, i.e., S100A2, acting through functional interaction with Smad3 which is enhanced in the presence of high calcium and TGF-beta activity (Naz et al. 2014). In HCC, ectopic expression of metastasin, a further calcium-binding protein, induces typical EMT with decreased E-cadherin and upregulation of vimentin via upregulation of SNAI1 (Zheng et al. 2013). Several EMT-associated proteins are regulated by BTB/POZ domain-containing protein 7/BTBD7 in HCC (Tao et al. 2013). In cancers, EMT is connected with the induction of an inflammatory microenvironment that plays a role both in favoring invasion and spread and tumor control by immune mechanisms. TGF-beta1 contributes to an inflammatory niche through promotion of immune cell homing and switching the phenotypes of tumor-infiltrating immune cells (Fuxe and Karlsson 2012).
Regulation of Epithelial-Mesenchymal Transition
EMT as a motor of the metastatic pathway is regulated by several mechanisms that control expression of pro- and anti-EMT factors. Numerous factors are regulated by epigenetic promoter methylation, with a complex EMT-related or even EMT-specific DNA methylome. EMT factors are controlled by micro-RNAs. MiR-29c mediates EMT in human CRC metastases via modulation of beta-catenin signaling (Zhang et al. 2014a). MiR-424-5p can reverse EMT in HCC by targeting the beta-catenin inhibitor, ICAT (Zhang et al. 2014b). MiR-122 triggers EMT and suppresses HCC cell motility, migration, and invasion via targeting RhoA (Wang et al. 2014b). Downregulation of microRNA-30a in HCC facilitates migration and invasion through promotion of EMT (Liu et al. 2014a). This miR can, however, also inhibit EMT via targeting the pro-EMT factor Snail1/Snai1, an effect circumvented by its downregulation in cancers (Kumarswamy et al. 2012). MicroRNA-26b inhibits EMT by targeting USP9X, a protein which in turn affects EMT through Smad4 and the TGF-beta signaling pathway (Shen et al. 2014). MicroRNA-148a suppresses EMT and metastasis of HCC by targeting Snail signaling (Zhang et al. 2014c). MiR-491 is involved in the regulation of HCC metastasis by blocking EMT via increase of surface E-cadherin expression (Zhou et al. 2013). Overexpression of microRNA-106b promotes cell migration and metastasis in HCC via promoting EMT via overexpression of the RhoGTPases, RhoA, and RhoC (Yau et al. 2013). Micro-RNA-490-3p modulates HCC cell growth and EMT by targeting endoplasmic reticulum-Golgi intermediate compartment protein 3/ERGIC3 (Zhang et al. 2013). The invasive and metastatic behavior of HCC is suppressed by microRNA-612 which inhibits EMT (Tao et al. 2013). Metastasis regulated by EMT is also subject to modulation by long noncoding RNAs that affect transcriptional and posttranscriptional steps in synthesis of critical factors (Serviss et al. 2014). A long noncoding RNA activated by TGF-beta promotes the invasion-metastatic cascade in HCC (Yuan et al. 2014).
The Concept of the Pre-metastatic Niche
Introduction
The establishment of a microenvironment that favors homing and growth of cancer cells is a critical condition for the generation of metastasis. The biogenesis of metastases requires a distinct tumor cell-host organ interface in early phases of the metastatic process (Gassmann and Haier 2008). The cellular basis for this pathway of metastasis has been reviewed (Oppenheimer 2006) and has resulted in the concept of the pre-metastatic niche (pre-MN). A pre-MN is defined as a distinct variant of microenvironment that is induced in target organs of metastasis and facilitates homing, adhesion, growth, invasion, and survival of metastatic tumor cells (Kaplan et al. 2006; Perelmuter and Manskikh 2012; Zoccoli et al. 2012). In this sense, the pre-MN is a structure that precedes, and eventually anticipates, the generation of a metastasis, a novel concept for the understanding of a metastasis cascade. Also for the liver, which is a major metastasis-susceptible organ, a pre-or pro-metastatic microenvironment has been discussed (review: Vidal-Vanaclocha 2008).
The Cellular Preparation of the Pre-metastatic Niche
It was demonstrated that bone marrow-derived cells/BMDC home to the pre-MN via the action of adhesion molecule and chemokine signaling, and there establish clusters that precede the arrival of even single metastasizing cancer cells. Interaction of these BMDC with local cells promotes the upregulation of several factors that will be important for cancer cell growth, including signaling chemokines, stromal differentiation factors, and angiogenic factors (review: Kaplan et al. 2006). Bone marrow-derived myeloid progenitor cells can induce mesenchymal-epithelial transition/MET in remote tissues (Gao et al. 2012b).
Cancer Progenitor/Stem Cells in a Pre-metastatic Niche
Cancer stem cells are thought to possess distinct tissue and cell selection capabilities allowing them to home to a pre-MN (Malanchi et al. 2011). In their interaction with microenvironmental structures, cancer stem cells are not a fixed set of cells but rather a flexible population of cells that can differentiate to typical tumor cells and revert to a stemlike state. There is strong evidence that this switching is regulated by interactions with mesenchymal cells of the niche (Fessler et al. 2013), these cells themselves being programmed to later become true stromal cells, including cancer-associated fibroblasts and myofibroblasts. Within a pre-MN, cancer stem cells dwell in some sort of a “CSC niche” which controls their self-renewal, differentiation, and departure for other niches (Borovski et al. 2011).
Cross-Talk Between Tumor Cells and Elements of the Pre-metastatic Niche
In addition to several classes of stem cells that can prepare the pre-MN, differentiated tumor cells themselves interact in a complex manner with elements of the pre-MN. The interaction with cells of the pre-MN can induce genetic and epigenetic alterations in the homing tumor cells, changes that can themselves promote a metastatic phenotype of the cancer cells (Carlini et al. 2011). The offspring of tumor cells that have undergone such pre-MN-induced changes may be primed to transfer these acquired changes to other potential metastatic sites, eventually to induce novel pre-MNs. On the other hand, tumor cells that have settled in the niche can prime the further development of cells active in the niche (Deng et al. 2012) and induce the evolution of the pre-MN to a complete and active, vascularized stroma (the concept of induced stromal progression).
Programming of the Pre-metastatic Niche by Exosomes
The mechanisms as to how homing cells induce resident cells to become a pre-metastatic microenvironment and then a tumor stroma and an angiogenic field are still not well known. It is assumed that direct contact of homing bone marrow-derived cells and cancer stem cells and differentiated tumor cells with tissue can elicit a differentiation switch of mesenchymal and vascular cells in situ. On the other hand, exosomes delivered by tumor cells and harboring genetic information or signaling molecules can drive pre-MN formation (Alderton 2012). Exosomes contain membrane anchors that allow them to attach to target cells in a niche, to deliver their information cargo (Pant et al. 2012). Exosomes released from melanoma cells and transported to sentinel lymph nodes prepare these nodes for metastasis through remodeling of the lymphonodular microenvironment (Hood et al. 2011). Exosomes released from malignant cells, e.g., melanoma cells, can also educate noncancer progenitor cells toward a pre-metastatic phenotype through the action of the receptor tyrosine kinase Met (Peinado et al. 2012). For this pathway to work, certain conditions must be present in resident cells to interact with exosomes, such as expression of the CD44v6 isoform of CD44, this factor assembling a soluble matrix for exosomes to allow cell embedding and growth (Jung et al. 2009).
Programming the Pre-metastatic Niche by Soluble Factors from Progenitor Cells and Tumor Cells
Apart from direct interaction with future stromal cells and release of exosomes, tumor cells can also secrete various factors that “prepare” the future niche, and in particular its endothelial cells, for cancer cell homing. Such secreted factors include vascular endothelial growth factors, TGF-beta and TNF-alpha. These molecules can elicit the release of secondary signal substances from the niche, e.g., the chemokines S100A8 and S100A9 (Rafii and Lyden 2006). The latter two chemokines are also produced by macrophages, including those located in pre-MNs (Maru 2007). Proteins of the S100A family, including S100A4, are metastasis-associated proteins (Hemandas et al. 2006). Myeloid progenitor cells from the bone marrow can secrete the proteoglycan, which is capable to regulate remote microenvironments to become of pre-MN (Gao et al. 2012a). Carcinoma cells secreting vascular endothelial growth factor C into lymph, draining the cancer, can induce characteristic changes in locoregional lymph nodes, including an increase in lymphatic vessels and the induction of high endothelial venules, before the arrival of tumor cells (Chung et al. 2012).
Counteracting the Function of the Pre-metastatic Niche
There are several mechanisms that may abolish the generation and functions of pre-MN. Mesenchymal cells and endothelial cells in certain tissues and organs may be refractory to pre-MN-inducing cells and factors, being one explanation why certain tissues are only seldom involved by metastases, e.g., skeletal muscle. Tumor cells having settled and grown in the area of the pre-MN can later destroy this microenvironment (Bidard et al. 2008), rendering it non-functional. A third mechanism depends on the formation of tumor-entrained neutrophils (TENs), cells that circulate in the blood as a response to malignancies but are absent in healthy subjects. TENs inhibit metastatic seeding by blunting the function of pre-MNs via secretion of hydrogen peroxide (Granot et al. 2011).
References
Aatonen M, Grönholm M, Siljander PR (2012) Platelet-derived microvesicles: multitalented participants in intercellular communication. Semin Thromb Hemost 38:102–113
Adhikary A, Chakraborty S, Mazumdar M, Ghosh S, Mukherjee S, Manna A, Mohanty S et al (2014) Inhibition of epithelial to mesenchymal transition by E-cadherin up-regulation via repression of slug transcription and inhibition of E-cadherin degradation: dual role of SMAR1 in breast cancer cells. J Biol Chem 289:25431–25444
Aharinejad S, Sioud M, Lucas T, Abraham D (2009) Targeting stromal-cancer cell interactions with siRNAs. Methods Mol Biol 487:243–266
Alderton GK (2012) Metastasis. Exosomes drive premetastatic niche formation. Nat Rev Cancer 12:447
Alphonso A, Alahari SK (2009) Stromal cells and integrins: conforming to the needs of the tumor microenvironment. Neoplasia 11:1264–1271
Archer SG, Gray BN (1989) Vascularization of small liver metastases. Br J Surg 76:545–548
Arteta B, Lasuen N, Lopategi A, Sveinbjörnsson B, Smedsrod B, Vidal-Vanaclocha F (2010) Colon carcinoma cell interaction with liver sinusoidal endothelium inhibits organ-specific antitumor immunity through interleukin-1-induced mannose receptor in mice. Hepatology 51:2172–2182
Au SL, Wong CC, Lee JM, Fan DN, Tsang FH, Ng IO, Wong CM (2012) Enhancer of zeste homolog 2 epigenetically silences multiple tumor suppressor microRNAs to promote liver cancer metastasis. Hepatology 56(2):622–631
Badiola I, Olaso E, Crende O, Friedman SL, Vidal-Vanaclocha F (2011) Discoidin domain receptor 2 deficiency predisposes hepatic tissue to colon carcinoma metastasis. Gut 26:971–978
Bambace NM, Holmes CE (2011) The platelet contribution to cancer progression. J Thromb Haemost 9:237–249
Battinelli EM, Markens BA, Italiano JE (2011) Release of angiogenesis regulatory proteins from platelet alpha granules: modulation of physiologic and pathologic angiogenesis. Blood 118:1359–1369
Bello V, Moreau N, Sirour C, Hidalgo M, Buisson N, Darribère T (2014) The dystroglycan: nestled in an adhesome during embryonic development. Dev Biol. doi:10.1016/j.ydbio.2014.07.006
Benson DD, Meng X, Fullerton DA, Moore EE, Lee JH, Ao L, Silliman CC, Barnett CC (2012) Activation state of stromal inflammatory cells in murine metastatic pancreatic adenocarcinoma. Am J Physiol Regul Integr Comp Physiol 302:R1067–R1075
Bettum IJ, Vasiliauskaine K, Nygaard V, Clancy T, Pettersen SJ, Tenstad E, Maelandsmo GM et al (2014) Metastasis-associated protein S100A4 induces a network of inflammatory cytokines that activate stromal cells to acquire pro-tumorigenic properties. Cancer Lett 344:28–39
Bhatia A, Dey P (2013) Platelet internalization within the cancer cells: what is the significance? Diagn Cytopathol 41:659–660
Bidard FC, Pierga JY, Vincent-Salomon A, Poupon MF (2008) A “class action” against the microenvironment: do cancer cells cooperate in metastasis? Cancer Metastasis Rev 27:5–10
Bogenrieder T, Herlyn M (2003) Axis of evil: molecular mechanisms of cancer metastasis. Oncogene 22:6524–6536
Borovski T, De Sousa E, Melo F, Vermeulen L, Medema JP (2011) Cancer stem cell niche: the place to be. Cancer Res 71:634–639
Borsig L (2008) The role of platelet activation in tumor metastasis. Expert Rev Anticancer Ther 8:1247–1255
Brandau S, Moses K, Lang S (2013) The kinship of neutrophils and granulocytic myeloid-derived suppressor cells in cancer: cousins, siblings or twins? Semin Cancer Biol 23:171–182
Buergy D, Wenz F, Groden C, Brockmann MA (2012) Tumor-platelet interaction in solid tumors. Int J Cancer 130:2747–2760
Caja F, Vannucci L (2014) TGFβ: a player on multiple fronts in the tumor microenvironment. J Immunotoxicol 20:1–8
Capece D, Fischietti M, Verzella D, Gaggiano A, Cicciarelli G, Tessitore A, Zazzeroni F et al (2013) The inflammatory microenvironment in hepatocellular carcinoma: a pivotal role for tumor-associated macrophages. Biomed Res Int 2013:187204
Carlini MJ, De Lorenzo MS, Puricelli L (2011) Cross-talk between tumor cells and the microenvironment at the metastatic niche. Curr Pharm Biotechnol 12:1900–1908
Carr BI, Cavallini A, D Allessandro R, Refolo MG, Lippolis C, Mazzocca A, Messa C (2014) Platelet extracts induced growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer 14:43
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331:1559–1564
Chambers SE, O’Neill CL, O’Doherty TM, Medina RJ, Stitt AW (2013) The role of immune-related myeloid cells in angiogenesis. Immunobiology 218:1370–1375
Chang MC, Chan CP, Ho YS, Lee JJ, Lin PS, Lin BR, Huang YL, Hahn LJ, Yeh HW et al (2009) Signaling pathways for induction of platelet aggregation by SAS tongue cancer cells – a mechanism of hematogenous metastasis. J Oral Pathol Med 38:434–440
Chanmee T, Ontong P, Konno K, Itano N (2014) Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) 6:1670–1690
Chen J, Chan AW, To KF, Chen W, Zhang Z, Ren J, Song C, Cheung YS, Lai PB et al (2013) SIRT2 overexpression in hepatocellular carcinoma mediates epithelial to mesenchymal transition by protein kinase B/glycogen synthase kinase-3β/β-catenin signaling. Hepatology 57:2287–2298
Chen HF, Huang CH, Liu CJ, Hung JJ, Hsu CC, Teng SC, Wu KJ (2014a) Twist1 induces endothelial differentiation of tumour cells through the Jagged1-KLF4 axis. Nat Commun 5:4697
Chen JS, Li HS, Huang JQ, Zhang LJ, Chen XL, Wang Q, Lei J, Feng JT, Liu Q et al (2014b) Down-regulation of Gli-1 inhibits hepatocellular carcinoma cell migration and invasion. Mol Cell Biochem 393:283–291
Chen J, Liu WB, Jia WD, Xu GL, Ma JL, Huang M, Deng YR, Li JS (2014c) Overexpression of Mortalin in hepatocellular carcinoma and its relationship with angiogenesis and epithelial to mesenchymal transition. Int J Oncol 44:247–255
Chung MK, Do IG, Jung E, Son YI, Jeong HS, Baek CH (2012) Lymphatic vessels and high endothelial venules are increased in the sentinel lymph nodes of patients with oral squamous cell carcinoma before the arrival of tumor cells. Ann Surg Oncol 19:1595–1601
Cirri P, Chiarugi P (2012) Cancer-associated fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer Metastasis Rev 31:195–208
Coupland LA, Chong BH, Parish CR (2012) Platelets and P-selectin control tumor cell metastasis in an organ-specific manner and independently of NK cells. Cancer Res 72:4662–4671
Cui TX, Kryczek I, Zhao L, Zhao E, Kuick R, Roh MH, Vatan L, Szeliga W, Mao Y et al (2013) Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 39:611–621
Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M et al (2012) S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell 21:642–654
Diaz-Montero CM, Finke J, Montero AJ (2014) Myeloid-derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol 41:174–184
Dovizio M, Alberti S, Guillem-Llobat P, Patrignani P (2014) Role of platelets in inflammation and cancer: novel therapeutic strategies. Basic Clin Pharmacol Toxicol 114:118–127
Dumitru CA, Moses K, Trellakis S, Lang S, Brabdau S (2012) Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother 61:1155–1167
Eldor A, Bar-Ner M, Yahalom J, Fuks Z, Vlodavsky I (1987) Role of heparanase in platelet and tumor cell interactions with the subendothelial extracellular matrix. Semin Thromb Hemost 13:475–488
Erpenbeck L, Schön MP (2010) Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 115:3427–3436
Etulain J, Fondevila C, Negrotto S, Schattner M (2013) Platelet-mediated angiogenesis is independent of VEGF and fully inhibited by aspirin. Br J Pharmacol 170:255–265
Falanga A, Marchetti M, Vignoli A, Balducci D (2003) Clotting mechanisms and cancer: implications in thrombus formation and tumor progression. Clin Adv Hematol Oncol 1:673–678
Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, Li R, Zhao QD, Yang Y, Lu ZH et al (2014) Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett 352:160–168
Fenouille N, Tichet M, Dufies M, Pottier A, Mogha A, Soo JK, Rocchi S, Mallavialle A et al (2012) The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS One 7:e40378
Fessler E, Dijgraaf FE, Melo FD, Medema JP (2013) Cancer stem cell dynamics in tumor progression and metastasis: is the microenvironment to blame? Cancer Lett 341:97–104
Finger EC, Giaccia AJ (2010) Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev 29:285–293
Firulli AB, Conway SJ (2008) Phosphoregulation of Twist1 provides a mechanism of cell fate control. Curr Med Chem 15:2641–2647
Fujita and Takagi (2012) http://www.ncbi.nlm.nih.gov/pubmed/22992842
Fuxe J, Karlsson MC (2012) TGF-β-induced epithelial-mesenchymal transition: a link between cancer and inflammation. Semin Cancer Biol 22:455–461
Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A (2013) Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol 228:1404–1412
Gao D, Vahdat LT, Wong S, Chang JC, Mittal V (2012a) Microenvironment regulation of epithelial-mesenchymal transitions in cancer. Cancer Res 72:4883–4889
Gao D, Joshi N, Choi H, Ryu S, Hahn M, Catena R, Sadik H, Argani P, Wagner P et al (2012b) Myeloid progenitor cells in the premetastatic lung promote metastasis by inducing mesenchymal to epithelial transition. Cancer Res 72:1384–1394
Gasic GJ (1984) Role of plasma, platelets, and endothelial cells in tumor metastasis. Cancer Metastasis Rev 3:99–114
Gassmann P, Haier J (2008) The tumor cell-host organ interface in the early onset of metastatic organ colonisation. Clin Exp Metastasis 25:171–181
Gay LJ, Felding-Habermann B (2011) Contribution of platelets to tumour metastasis. Nat Rev Cancer 11:123–134
Gil-Bernabé AM, Lucotti S, Muschel RJ (2013) Coagulation and metastasis: what does the experimental literature tell us? Br J Haematol 162:433–441
Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R (2011) Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20:300–314
Grugan KD, McCabe FL, Kinder M, Greenplate AR, Harman BC, Ekert JE, van Rooijen N et al (2012) Tumor-associated macrophages promote invasion while retaining Fc-dependent anti-tumor function. J Immunol 189:5457–5466
Hao NB, Lü MH, Fan YH, Cao YL, Zhang ZR, Yang SM (2012) Macrophages in tumor microenvironments and the progression of tumors. Clin Dev Immunol 2012:948098
Haugeberg G, Strohmeyer T, Lierse W, Böcker W (1988) The vascularization of liver metastases. Histological investigation of gelatine-injected liver specimens with special regard to the vascularization of micrometastases. J Cancer Res Clin Oncol 114:415–419
Heijnen et al (1999) http://www.ncbi.nlm.nih.gov/pubmed/10572093
Hemandas AK, Salto-Tellez M, Maricar SH, Leong AF, Leow CK (2006) metastasis-associated protein S100A4 – a potential prognostic marker for colorectal cancer. J Surg Oncol 93:498–503
Higashi N, Ishii H, Fujiwara T, Morimoto-Tomita M, Irimura T (2002) Redistribution of fibroblasts and macrophages as micrometastases develop into established liver metastases. Clin Exp Metastasis 19:631–638
Honn KV, Tang DG, Crissman JD (1992) Platelets and cancer metastasis: a causal relationship? Cancer Metastasis Rev 11:325–351
Hood JL, San RS, Wickline SA (2011) Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res 71:3792–3801
Hoos A, Protsyuk D, Borsig L (2014) Metastatic growth progression caused by PSGL-1-mediated recruitment of monocytes to metastatic sites. Cancer Res 74:695–704
Hu et al (2014) http://www.ncbi.nlm.nih.gov/pubmed/25150783
Hugo HJ, Kokkinos MI, Blick T, Ackland ML, Thompson EW, Newgreen DF (2011) Defining the E-cadherin repressor interactome in epithelial-mesenchymal transition: the PMC42 model as a case study. Cells Tissues Organs 193:23–40
Illemann M, Bird, Majeed A, Sehested M, Laerum OD, Lund LR, Dano K, Nielsen BS (2006) MMP-9 is differentially expressed in primary human colorectal adenocarcinomas and their metastases. Mol Cancer Res 4:293–302
Iqbal J, McRae S, Banaudha K, Mai T, Waris G (2013) Mechanism of hepatitis C virus (HCV)-induced osteopontin and its role in epithelial to mesenchymal transition of hepatocytes. J Biol Chem 288:36994–37009
Jain S, Harris J, Ware J (2010) Platelets: linking hemostasis and cancer. Arterioscler Thromb Vasc Biol 30:2362–2367
Jia RJ, Cao L, Zhang L, Jing W, Chen R, Zhu MH, Guo SW, Wu GB, Fan XY, Wang H et al (2014) Enhanced myeloid differentiation factor 88 promotes tumor metastasis via induction of epithelial-mesenchymal transition in human hepatocellular carcinoma. Cell Death Dis 5:e1103
Jing Y, Han Z, Zhang S, Liu Y, Wei L (2011) Epithelial-mesenchymal transition in tumor microenvironment. Cell Biosci 1:29
Jung T, Castellana D, Klingbeil P, Cuesta Hernandez I, Vitacolonna M, Orlicky DJ et al (2009) CD44v6 dependence of premetastatic niche preparation by exosomes. Neoplasia 11:1093–1105
Jung HY, Fattet L, Yang J (2015) Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res 21:962–968
Jurasz et al (2004) http://www.ncbi.nlm.nih.gov/pubmed/15492016
Kamoshida G, Ogawa T, Oyanagi J, Sato H, Komiya E, Higashi S, Miyazaki K, Tsuji T (2014) Modulation of matrix metalloproteinase-9 secretion from tumor-associated macrophage-like cells by proteolytically processed laminin-322 (laminin-5). Clin Exp Metastasis 31:285–291
Kang N, Gores GJ, Shah VH (2011) Hepatic stellate cells: partners in crime for liver metastases? Hepatology 54:707–713
Kaplan RN, Rafii S, Lyden D (2006) Preparing the “soil”: the premetastatic niche. Cancer Res 66:11089–11093
Karpatkin S, Pearlstein E (1981) Role of platelets in tumor cell metastases. Ann Intern Med 95:636–641
Karshovska E, Weber C, von Hundelshausen P (2013) Platelet chemokines in health and disease. Thromb Haemost 110:894–902
Kato et al (2003) http://www.ncbi.nlm.nih.gov/pubmed/14588983
Keskinov AA, Shurin MR (2014) Myeloid regulatory cells in tumor spreading and metastasis. Immunobiology. doi:10.1016/j.imbio.2014.07.017
Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Medzhitov R, Flavell RA (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110:191–202
Koh M, Woo Y, Valiathan RR, Jung HY, Park SY, Kim YN, Kim HR, Fridman R, Moon A (2014) Discoidin domain receptor 1 is a novel transcriptional target of ZEB1 in breast epithelial cells undergoing H-Ras-induced epithelial to mesenchymal transition. Int J Cancer. doi:10.1002/ijc.29154
Koseoglu S, Flaumenhaft R (2013) Advances in platelet granule biology. Curr Opin Hematol 20:464–471
Krishnan et al (2013) http://www.ncbi.nlm.nih.gov/pubmed/23530051
Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M et al (2012) MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snail1 and is downregulated in non-small cell lung cancer. Int J Cancer 130:2044–2053
Kunita et al (2007) http://www.ncbi.nlm.nih.gov/pubmed/17392172
Kunita et al (2011) http://www.ncbi.nlm.nih.gov/pubmed/21801875
Labelle M, Hynes RO (2012) The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov 2:1091–1099
Labelle M, Begum S, Hynes RO (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20:576–590
Li H, Fan X, Houghton J (2007) Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 101:805–815
Li Q, Gu X, Weng H, Ghafoory S, Liu Y, Feng T, Dzieran J, Li L, Ilkavets I, Kruithof-de Julio M et al (2013) Bone morphogenetic protein-9 induces epithelial to mesenchymal transition in hepatocellular carcinoma cells. Cancer Sci 104:398–408
Li Y, Miso LY, Xiao YL, Cai HR, Zhang DP (2014) Elevated platelets enhance cancer cell migration, promote hematogenous metastasis and associated with a poor prognosis in advanced non-small cell lung cancer cases. Asian Pac J Cancer Prev 15:139–143
Lin G, Hägerstrand I, Lunderquist A (1984) Portal blood supply of liver metastases. AJR Am J Roentgenol 143:53–55
Lin Y, Wei C, Liu Y, Qiu Y, Liu C, Guo F (2013) Selective ablation of tumor-associated macrophages suppresses metastasis and angiogenesis. Cancer Sci 104:1217–1225
Liu Y, Zhang JB, Qin Y, Wang W, Wie L, Teng Y, Guo L, Zhang B, Lin Z, Liu J, Ren ZG et al (2013) PROX1 promotes hepatocellular carcinoma metastasis by way of up-regulating hypoxia-inducible factor 1α expression and protein stability. Hepatology 58:692–705
Liu Z, Tu K, Liu Q (2014a) Effects of microRNA-30a on migration, invasion and prognosis of hepatocellular carcinoma. FEBS Lett 588:3089–3097
Liu L, Dai Y, Chen J, Zeng T, Li Y, Chen L, Zhu YH, Li J, Li Y, Ma S, Xie D, Yuan YF et al (2014b) Maelstrom promotes hepatocellular carcinoma metastases by inducing epithelial-mesenchymal transition by way of Akt/GSK-3β/Snail signaling. Hepatology 59:531–543
Lowe et al (2012) http://www.ncbi.nlm.nih.gov/pubmed/22682130
Lukanidin E, Sleeman JP (2012) Building the niche: the role of the S100 proteins in metastatic growth. Semin Cancer Biol 22:216–225
Mabuchi A, Mullaney I, Sheard PW, Hessian PA, Mallard BL, Tawadrous MN, Zimmermann A et al (2004a) Role of hepatic stellate cell/hepatocyte interaction and activation of hepatic stellate cells in the early phase of liver regeneration in the rat. J Hepatol 40:910–916
Mabuchi A, Mullaney I, Sheard P, Hessian P, Zimmermann A, Wheatley AM (2004b) Role of hepatic stellate cells in the early phase of liver regeneration in rat: formation of tight adhesion to parenchymal cells. Comp Hepatol 3(Suppl 1):S29
Maeng YI, Kim KH, Kim JY, Lee SJ, Sung WJ, Lee CK, Park JB, Park KK (2014) Transcription factors related to epithelial mesenchymal transition in tumor center and margin in invasive lung adenocarcinoma. Int J Clin Exp Pathol 7:4095–4103
Maione F, Capano S, Regano D, Zentilin L, Giacca M, Casanovas O, Bussolino F et al (2012) Semaphorin 3A overcomes cancer hypoxia and metastatic dissemination induced by antiangiogenic treatment in mice. J Clin Invest 122:1832–1848
Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J (2011) Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481:85–89
Maniecki MB, Etzeroth A, Ulhoi BP, Steiniche T, Borre M, Dyrskjot L, Ornoft TF, Moestrup SK et al (2012) Tumor-promoting macrophages induce the expression of the macrophage-specific receptor CD163 in malignant cells. Int J Cancer 131:2320–2331
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Mantovani A, Sica A, Allavena P, Garlanda C, Locati M (2009) Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol 70:325–330
Maru Y (2007) Which came first, tumor cells or macrophages? Cell Adh Migr 1:107–109
Mehta P (1984) Potential role of platelets in the pathogenesis of tumor metastasis. Blood 63:55–63
Meng F, Wu G (2012) The rejuvenated scenario of epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Metast Rev 31:455–467
Micalizzi DS, Wang CA, Farabaugh SM, Schiemann WP, Ford HL (2010) Homeoprotein Six1 increases TGF-beta type I receptor and converts TGF-beta signaling from suppressive to supportive for tumor growth. Cancer Res 70:10371–10380
Miyashita T, Tajima H, Makino I, Nakagawara H, Kitagawa H, Fushida S, Harmon JW et al (2014) Metastasis-promoting role of extravasated platelet activation in tumor. J Surg Res. doi:10.1016/j.jss.2014.07.037
Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, Mack M et al (2010) Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res 70:5728–5739
Mrazek AA, Carmical JR, Wood TG, Hellmich MR, Eltorky M, Bohanon FJ, Chao C (2014) Colorectal cancer-associated fibroblasts are genotypically distinct. Curr Cancer Ther Rev 10:197–218
Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, Walter J, Karnatz N et al (2007) Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol 171:1608–1618
Mueller L, von Seggern L, Schumacher J, Goumas F, Wilms C, Braun F, Broering DC (2010) TNF-alpha similarly induces IL-6 and MCP-1 in fibroblasts from colorectal liver metastases and normal liver fibroblasts. Biochem Biophys Res Commun 397:586–591
Naz S, Bashir M, Ranganathan P, Bodapati P, Santosh V, Kondaiah P (2014) Protumorigenic actions of S100A2 involve regulation of PI3/Akt signaling and functional interaction with Smad3. Carcinogenesis 35:14–23
Nierodzik ML, Klepfish A, Karpatkin S (1995) Role of platelets, thrombin, integrin IIb-IIa, fibronectin and von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thromb Haemost 74:282–290
Nikfarjam N, Muralidharan V, Malcontenti-Wilson C, Christophi C (2003) Scanning electron microscopy study of the blood supply of human colorectal liver metastases. EJSO 29:856–861
Nyberg P, Salo T, Kalluri R (2008) Tumor microenvironment and angiogenesis. Front Biosci 13:6537–6553
Ogunwobi OO, Liu C (2011) Hepatocyte growth factor upregulation promotes carcinogenesis and epithelial-mesenchymal transition in hepatocellular carcinoma via Akt and COX-2 pathways. Clin Exp Metastasis 28:721–731
Okabe H, Ishimoto T, Mima K, Nakagawa S, Hayashi H, Kuroki H, Imai K, Nitta H et al (2014) CD44s signals the acquisition of the mesenchymal phenotype required for anchorage-independent cell survival in hepatocellular carcinoma. Br J Cancer 110:958–966
Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S, Xing F, Liu W, Pandey PR et al (2012) Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res 72:537–547
Oleksowicz L, Dutcher JP (1995) Adhesive receptors expressed by tumor cells and platelets: novel targets for therapeutic anti-metastatic strategies. Med Oncol 12:95–102
Oppenheimer SB (2006) Cellular basis of cancer metastasis: a review of fundamentals and new advances. Acta Histochem 108:327–334
Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5:1597–1601
Owen JL, Mohamadzadeh M (2013) Macrophages and chemokines as mediators of angiogenesis. Front Physiol 4:159
Panasci LC, Comis R, Ginsberg S, Rudolph A, Theodorakis M, Klag M, Fitzpatrick A et al (1980) Pharmacokinetics of vinblastine-loaded platelets utilized in the treatment of platelet-phagocytizing tumors. Cancer Treat Rep 64:1227–1233
Pang Y, Gara SK, Achyut BR, Li Z, Yan HH, Day CP, Weiss JM, Trinchieri G, Morris JC et al (2013) TGF-β signaling in myeloid cells is required for tumor metastasis. Cancer Discov 3:936–951
Pant S, Hilton H, Burczynski ME (2012) The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol 83:1484–1494
Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M et al (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 18:883–891
Perelmuter VM, Manskikh VN (2012) Preniche as missing link of the metastatic niche concept explaining organ-preferential metastasis of malignant tumors and the type of metastatic disease. Biochemistry (Mosc) 77:111–118
Pietras K, Ostman A (2010) Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 316:1324–1331
Pook M, Tamming L, Padarai K, Tiido T, Maimets T, Patarroyo M, Juronen E, Jaks V, Ingerpuu S (2014) Platelets store laminins 411/421 and 511/521 in compartments distinct from α- or dense granules and secrete these proteins via microvesicles. J Thomb Haemost 12:519–527
Qiang L, He YY (2014) Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy 10:1864–1865
Qin Q, Xu Y, He T, Qin C, Xu J (2012) Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res 22:90–106
Radziwon-Balicka A, Moncada de la Rosa C, Jurasz P (2012) Platelet-associated angiogenesis regulating factors: a pharmacological perspective. Can J Physiol Pharmacol 90:679–688
Rafii S, Lyden D (2006) S100 chemokines mediate bookmarking of premetastatic niches. Nat Cell Biol 8:1321–1323
Räsänen K, Vaheri A (2010) Activation of fibroblasts in cancer stroma. Exp Cell Res 316:2713–2722
Reichl P, Haider C, Grubinger M, Mikulits W (2012) TGF-β in epithelial to mesenchymal transition and metastasis of liver carcinoma. Curr Pharm Des 18:4135–4147
Reymond N, d’Agua BB, Ridley AJ (2013) Crossing the endothelial barrier during metastasis. Nat Rev Cancer 13:858–870
Ridge JA, Bading JR, Gelbard AS, Benua RS, Daly JM (1987) Perfusion of colorectal hepatic metastases. Relative distribution of flow form the hepatic artery and portal vein. Cancer 59:1547–1553
Riedl J, Pabinger I, Ay C (2014) Platelets in cancer and thrombosis. Hamostaseologie 34:54–62
Ruffell B, Affara NI, Coussens LM (2012) Differential macrophage programming in the tumor microenvironment. Trends Immunol 33:119–126
Sabrkhany S, Griffioen AW, Oude Egrbrink MG (2011) The role of blood platelets in tumor angiogenesis. Biochim Biophys Acta 1815:189–196
Schmid MC, Varner JA (2012) Myeloid cells in tumor inflammation. Vasc Cell 4:14
Serviss JT, Johnsson P, Grandér D (2014) An emerging role for long non-coding RNAs in cancer metastasis. Front Genet 5:234
Sharma D, Brummel-Ziedins KE, Bouchard BA, Holmes CE (2014) Platelets in tumor progression: a host factor that offers multiple potential targets in the treatment of cancer. J Cell Physiol 229:1005–1015
Shen et al (2010) http://www.ncbi.nlm.nih.gov/pubmed/20123990
Shen G, Lin Y, Yang X, Zhang J, Xu Z, Jia H (2014) MicroRNA-26b inhibits epithelial-mesenchymal transition in hepatocellular carcinoma by targeting USP9X. BMC Cancer 14:393
Shimizu S, Yamada N, Swada T, Ikeda K, Nakatani K, Seki S, Kaneda K, Hirakawa K (2000) Ultrastructure of early phase hepatic metastasis of human colon carcinoma cells with special reference to desmosomal junctions with hepatocytes. Pathol Int 50:953–959
Shirabe K, Mano Y, Muto J, Matono R, Motomura T, Toshima T, Takeishi K, Ichiyama H et al (2012) Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today 42:1–7
Smith HA, Kang Y (2013) The metastasis-promoting roles of tumor-associated immune cells. J Mol Med (Berl) 91:411–429
Standiford TJ, Kuick R, Bhan U, Chen J, Newstead M, Keshamouni VG (2011) TGF-β-induced IRAK-M expression in tumor-associated macrophages regulates lung tumor growth. Oncogene 30:2475–2484
Staudt ND, Jo M, Hu J, Bristow JM, Pizzo DP, Gaultier A, VandenBerg SR, Gonias SL (2013) Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res 73:3902–3912
Strohmeyer T, Haugeberg G, Lierse W (1986) Vascularization of liver metastases: a corrosion cast study (in German). Acta Anat (Basel) 126:172–176
Strohmeyer T, Haugeberg G, Lierse W (1987) Angioarchitecture and blood supply of micro-and macrometastases in human livers. An anatomic-pathological investigation using injection-techniques. J Hepatol 4:181–189
Suh Y, Yoon CH, Kim RK, Lim EJ, Oh YS, Hwang SG, An S, Yoon G, Gye MC et al (2013) Claudin-1 induces epithelial-mesenchymal transition through activation of the c-Abl-ERK signaling pathway in human liver cells. Oncogene 32:4873–4882
Sun Y, Song GD, Sun N, Chen JQ, Yang SS (2014) Slug overexpression induces stemness and promotes hepatocellular carcinoma cell invasion and metastasis. Oncol Lett 7:1936–1940
Suzuki-Inoue (2011) http://www.ncbi.nlm.nih.gov/pubmed/21693546
Takagi et al (2013) http://www.ncbi.nlm.nih.gov/pubmed/23991201
Tao YM, Huang JL, Zeng S, Zhang S, Fan XG, Wang ZM, Yang XH, Yuan XH et al (2013) BTB/POZ domain-containing protein 7: epithelial-mesenchymal transition promoter and prognostic biomarker of hepatocellular carcinoma. Hepatology 57:2326–2337
Taube JH, Herschkowitz JI, Komurov K, Zhou AY, Gupta S, Yang J, Hartwell K, Onder TT et al (2010) Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107:15449–15454
Timar et al (2005) http://www.ncbi.nlm.nih.gov/pubmed/16138000
Tsuruo T, Fujita N (2008) Platelet aggregation in the formation of tumor metastasis. Proc Jpn Acad Ser B Phys Biol Sci 84:189–198
Tymoszuk P, Evens H, Marzola V, Wachowicz K, Wasmer MH, Datta S, Müller-Holzner E et al (2014) In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur J Immunol 44:2247–2262
Udagawa T, Wood M (2010) Tumor-stromal cell interactions and opportunities for therapeutic intervention. Curr Opin Pharmacol 10:369–374
Umansky V, Sevko A (2013) Tumor microenvironment and myeloid-derived suppressor cells. Cancer Microenviron 6:169–177
Uraki S, Sugimoto K, Shiraki K, Tameda M, Inagaki Y, Ogura S, Kasai C, Nojiri K et al (2014) Human β-defensin-3 inhibits migration of colon cancer cells via downregulation of metastasis-associated 1 family, member 2 expression. Int J Oncol 45:1059–1064
Valastyan S, Chang A, Benaich N, Reinhardt F, Weinberg RA (2010) Concurrent suppression of integrin alpha5, radixin, and RhoA phenocopies the effects of miR-31 on metastasis. Cancer Res 70:5147–5154
Valcarcel M, Arteta B, Jaureguibeitia A, Lopatregi A, Martinez I, Mendoza L, Muruzabal FJ et al (2008) Three-dimensional growth as multicellular spheroid activates the proangiogenic phenotype of colorectal carcinoma cells via LFA-1-dependent VEGF: implications on hepatic micrometastasis. J Transl Med 6:57
van der Bilt JD, Kranenburg O, Nijkamp MW, Smakman N, Veenendaal LM, Te Velde EA et al (2005) Ischemia/reperfusion accelerates the outgrowth of hepatic micrometastases in a highly standardized murine model. Hepatology 42:165–175
van der Bilt JD, Kranenburg O, Borren A, van Hillegersberg R, Borel Rinkes IH (2008) Ageing and hepatic steatosis exacerbate ischemia/reperfusion-accelerated outgrowth of colorectal micrometastases. Ann Surg Oncol 15:1392–1398
Van Overmeire E, Laoui D, Keirsse J, Bonelli S, Lahmar Q, Van Ginderachter JA (2014) STAT of the union: dynamics of distinct tumor-associated macrophage subsets governed by STAT1. Eur J Immunol 44:2238–2242
Varon and Shai (2009) http://www.ncbi.nlm.nih.gov/pubmed/20040277
Varon et al (2012) http://www.ncbi.nlm.nih.gov/pubmed/23026678
Verga-Gérard A, Porcherot M, Meyniel-Schicklin L, André P, Lotteau V, Perrin-Cocon L (2013) Hepatitis C virus/human interactome identifies SMURF2 and the viral protease as critical elements for the control of TGF-β signaling. FASEB J 27:4027–4040
Vidal-Vanaclocha F (2008) The prometastatic microenvironment of the liver. Cancer Microenviron 1:113–129
Vidal-Vanaclocha F (2011) The liver prometastatic reaction of cancer patients: implications for microenvironment-dependent colon cancer gene regulation. Cancer Microenviron 4:163–180
Voboril R (2005) Blood supply of metastatic liver tumors: an experimental study. Int Surg 90:71–77
Wang CA, Jedlicka P, Patrick AN, Micalizzi DS, Lemmer KC, Deitsch E, Casas-Selves M et al (2012a) SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest 122:1895–1906
Wang C, Jiang K, Kang X, Gao D, Sun C, Li Y, Sun L, Zhang S, Liu X, Wu W, Yang P et al (2012b) Tumor-derived secretory clusterin induces epithelial-mesenchymal transition and facilitates hepatocellular carcinoma metastasis. Int J Biochem Cell Biol 44:2308–2320
Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, Zhang G, Bu XZ, Cai SH et al (2013a) Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS 8:e56664
Wang Z, Jiang Y, Guan D, Li J, Yin H, Pan Y, Xie D, Chen Y (2013b) Critical roles of p53 in epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma cells. PLoS One 8, e72846
Wang XF, Wang HS, Zhang F, Guo Q, Wang H, Wang KF, Zhang G, Bu XZ, Cai SH, Du J (2014a) Nodal promotes the generation of M2-like macrophages and downregulates the expression of IL-12. Eur J Immunol 44:173–183
Wang SC, Lin XL, Li J, Zhang TT, Wang HY, Shi JW, Yang S, Zhao WT, Xie RY et al (2014b) MicroRNA-122 triggers mesenchymal-epithelial transition and suppresses hepatocellular carcinoma cell motility and invasion by targeting RhoA. PLoS One 9:e101330
Wang T, Jin Y, Zhao R, Wu Y, Zhang Y, Wu D, Kong D, Jin X, Zhang F (2014c) High load hepatitis B virus replication inhibits hepatocellular carcinoma cell metastasis through regulation of epithelial-mesenchymal transition. Int J Infect Dis 20:37–41
Wang J, Yu F, Jia X, Iwanowycz S, Wang Y, Huang S, Ai W, Fan D (2014d) MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int J Cancer. doi:10.1002/ijc.29151
Wernicke M, Roitman P, Manfre D, Stern R (2011) Breast cancer and the stromal factor. The “prometastatic healing process” hypothesis. Medicina (B Aires) 71:15–21
Winograd-Katz SE, Fässler R, Geiger B, Legate KR (2014) The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol 15:273–288
Xia L, Huang W, Tian D, Zhang L, Qi X, Chen Z, Shang X, Nie Y, Wu K (2014) Forkhead box Q1 promotes hepatocellular carcinoma metastasis by transactivating ZEB2 and VersicanV1 expression. Hepatology 59:958–973
Xu ZY, Ding SM, Zhou L, Xie HY, Chen KJ, Zhang W, Xing CY, Guo HJ, Zheng SS (2012) FOXC1 contributes to microvascular invasion in primary hepatocellular carcinoma via regulating epithelial-mesenchymal transition. Int J Biol Sci 8:1130–1141
Xu J, Li X, Yang H, Chang R, Kong C, Yang L (2013) SIN1 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition. Cancer 119:2247–2257
Xu et al (2014) http://www.ncbi.nlm.nih.gov/pubmed/25072505
Yahalom J, Eldor A, Biran S, Fuks Z, Vlodavsky I (1985) Platelet-tumor cell interaction with the subendothelial extracellular matrix: relationship to cancer metastasis. Radiother Oncol 3:211–225
Yau WL, Lam CS, Ng L, Chow AK, Chan ST, Chan JY, Wo JY, Ng KT, Man K et al (2013) Over-expression of miR-106b promotes cell migration and metastasis in hepatocellular carcinoma by activating epithelial-mesenchymal transition process. PLoS One 8:e57882
Yi X, Luk JM, Lee NP, Peng J, Leng X, Guan XY, Lau GK, Beretta L, Fan ST (2008) Association of mortalin (HSPA9) with liver cancer metastasis and prediction for early tumor recurrence. Mol Cell Proteomics 7:315–325
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT et al (2014) A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25:666–681
Zaidel-Bar R (2013) Cadherin adhesome at a glance. J Cell Sci 126:373–378
Zaidel-Bar R, Geiger B (2010) The switchable integrin adhesome. J Cell Sci 123:1385–1388
Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B (2007) Functional atlas of the integrin adhesome. Nat Cell Biol 9:858–867
Zhai X, Zhu H, Wang W, Zhang S, Zhang Y, Mao G (2014) Abnormal expression of EMT-related proteins, S100A4, vimentin and E-cadherin, is correlated with clinicopathological features and prognosis in HCC. Med Oncol 31:970
Zhang L, Sullivan PS, Goodman JC, Gunaratne PH, Marchetti D (2011) MicroRNA-1258 suppresses breast cancer brain metastasis by targeting heparanase. Cancer Res 71:645–654
Zhang LY, Liu M, Tang H (2013a) miR-490-3p modulates cell growth and epithelial to mesenchymal transition of hepatocellular carcinoma cells by targeting endoplasmic reticulum-Golgi intermediate compartment protein 3 (ERGIC3). J Biol Chem 288:4035–4047
Zhang Q, Bai X, Chen W, Ma T, Hu Q, Liang C, Xie S, Chen C, Hu L, Xu S, Liang T (2013b) Wnt/β-catenin signaling enhances hypoxia-induced epithelial-mesenchymal transition in hepatocellular carcinoma via crosstalk with hif-1α signaling. Carcinogenesis 34:962–973
Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X et al (2013c) Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor-1α in hepatocellular carcinoma. BMC Cancer 13:108
Zhang J, Zhang DL, Jiao XL, Dong Q (2013d) S100A4 regulates migration and invasion in hepatocellular carcinoma HepG2 cells via NF-kB-dependent MMP-9 signal. Eur Rev Med Pharmacol Sci 17:2372–2382
Zhang JX, Mai SJ, Huang XX, Wang FW, Liao YJ, Lin MC, Kung HF, Zeng YX et al (2014a) MiR-29c mediates epithelial-to-mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of β-catenin signaling. Ann Oncol 25:2196–2204
Zhang Y, Li T, Guo P, Kang J, Wie Q, Jia X, Zhao W, Huai W, Qiu Y, Sun L, Han L (2014b) MiR-424-5p reversed epithelial-mesenchymal transition of anchorage-independent HCC cells by directly targeting ICAT and suppressed HCC progression. Sci Rep 4:6248
Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM (2014c) MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene 33:4069–4076
Zheng X, Gai X, Wu Z, Liu Q, Yao Y (2013) Metastasin leads to poor prognosis of hepatocellular carcinoma through partly inducing EMT. Oncol Rep 29:1811–1818
Zhou YM, Cao L, Li B, Zhang RX, Sui CJ, Yin ZF, Yang JM (2012) Clinicopathological significance of ZEB1 protein in patients with hepatocellular carcinoma. Ann Surg Oncol 19:1700–1706
Zhou Y, Li Y, Ye J, Jiang R, Yan H, Yang X, Liu Q, Zhang J (2013) MicroRNA-491 is involved in metastasis of hepatocellular carcinoma by inhibitions of matrix metalloproteinase and epithelial to mesenchymal transition. Liver Int 33:1271–1280
Zhu QS, Rosenblatt K, Huang KL, Lahat G, Brobey R, Bolshakov S, Nguyen T, Ding Z et al (2011a) Vimentin is a novel AKT1 target mediating motility and invasion. Oncogene 30:457–470
Zhu K, Dai Z, Pan Q, Wang Z, Yang GH, Yu L, Ding ZB, Shi GM, Ke AW, Yang XR et al (2011b) Metadherin promotes hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clin Cancer Res 17:7294–7302
Zhu P, Lv J, Yang Z, Guo L, Zhang L, Li M, Han W, Chen X, Zhuang H, Lu F (2014) Protocadherin 9 inhibits epithelial-mesenchymal transition and cell migration through activating GSK-3β in hepatocellular carcinoma. Biochem Biophys Res Commun 452:567–574
Zoccoli A, Iuliani M, Pantano F, Imperatori M, Intagliata S, Vincenzi B, Marchetti P et al (2012) Premetastatic niche: ready for new therapeutic interventions? Expert Opin Ther Targets 16(Suppl 2):S119–S129
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2017 Springer International Publishing Switzerland
About this entry
Cite this entry
Zimmermann, A. (2017). Pathogenic Features of Liver Metastasis: Mechanisms Involving Platelets, Tumor Stroma, Epithelial-Mesenchymal Transition, and the Premetastatic Niche. In: Tumors and Tumor-Like Lesions of the Hepatobiliary Tract. Springer, Cham. https://doi.org/10.1007/978-3-319-26956-6_113
Download citation
DOI: https://doi.org/10.1007/978-3-319-26956-6_113
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26954-2
Online ISBN: 978-3-319-26956-6
eBook Packages: MedicineReference Module Medicine