
Abstract
Senescence is an irreversible mitotic arrest of the cell that can result from replicative aging or stressors. It can be beneficial by conferring resistance to apoptosis, or detrimental by inducing pro-inflammatory signaling in the microenvironment. Senescent cells have been observed in both aged and diseased tissue, including the brain. The aging brain undergoes changes such as cortical atrophy and increases in inflammatory and oxidative factors, with decreases in synaptic plasticity and mitochondrial function. Significant neuronal loss is observed and thought to drive the atrophy in the corresponding areas of the brain in neurodegenerative diseases (ND). Despite being terminally differentiated, a senescence-like phenotype is observed in neurons upon stress in vitro and also in neurocognitive disorders like HIV-associated dementia and Alzheimer’s disease in vivo. Aging is also associated with lower regenerative capacity of neural stem and progenitor cells (NSPC). In vivo, their neurogenerative capacity is modulated by a variety of external factors, including growth factors, diet, and inflammation. NSPC have been observed to undergo stress-induced senescence in vitro. Deregulation of other CNS cell types, including oligodendrocytes and microglia occur in aging and ND. Microglia, which are not post-mitotic, senesce in culture in response to replicative or inflammatory stress. Astrocytes, which make up half of all cells in the CNS, maintain and protect neurons. In response to insult or injury however, astrocytes undergo phenotypic changes collectively termed reactive astrogliosis. This response can be both detrimental and beneficial to the neurons, and its downregulation improves disease parameters in a mouse model of AD. We have observed astrocyte senescence in vitro in response to replicative and oxidative stress and Aβ peptides, along with accumulation of senescent astrocytes in aged and AD brain. Given that astrocytes perform a myriad of complex functions in the CNS in order to maintain homeostasis, the loss of astrocyte function or the gain of neuroinflammatory function as a result of senescence could have profound implications for aging brain and neurodegenerative disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cellular senescence
- Astrocyte senescence
- Brain aging
- Brain inflammation
- Neurocognitive disease
- Alzheimer’s disease
1 Introduction
Although the phenomenon of cellular senescence in vitro was originally observed more than 50 years ago (Hayflick and Moorhead 1961; Hayflick 1965), much of our understanding of the underlying mechanisms and physiologic relevance of cellular senescence to aging and disease has only been elucidated within the last decade. Normal human cells undergo a finite number of cell divisions in vitro before their growth is irreversibly arrested. This finite replicative lifespan, known as replicative senescence, was thought to reflect aging at the level of the individual cell (Hayflick 1965). Replicative senescence occurs as a result of telomere attrition from progressive rounds of DNA replication (Stewart et al. 2003); however, the senescence arrest can also be induced by a variety of stressors including oncogene activation, oxidative stress and proteasome inhibition, and is termed stress-induced senescence (Torres et al. 2006; Serrano et al. 1997; Chen and Ames 1994). Overall, the phenotypes elicited by replicative senescence and stress-induced senescence are collectively known as cellular senescence.
In addition to irreversible cell cycle arrest, the senescence phenotype is characterized by several biomarkers that serve to identify senescent cells. Senescent cells are metabolically active, resistant to apoptosis (Wang 1995), and undergo widespread changes in gene expression (Shelton et al. 1999). Alterations in gene expression are thought to increase the secretion of pro-inflammatory mediators and proteases that act on the microenvironment and potentially contribute to age-related declines in organ function (Campisi et al. 2011); this process has been termed the senescence-associated secretory phenotype (SASP) (Coppe et al. 2008). Perhaps one of the most relevant reports indicating the physiologic relevance of cellular senescence to aging is that the selective clearance of cells expressing the senescence biomarker p16INK4a in a mouse model of premature aging delayed and alleviated certain age-related pathologies in several organ systems (Baker et al. 2011). These findings implicate senescent cells in age-related tissue dysfunction and suggest that interventions to prevent the initiation of the senescence phenotype or to target the removal of senescent cells would be both relevant and beneficial.
Replicative senescence of normal human cells in vitro was considered a counterpart to aging in vivo; however, a landmark study by Cristofalo (Cristofalo et al. 1998) failed to establish a correlation between donor age and the proliferative potential of fibroblasts in culture. This finding seemingly challenged the prevailing concept at the time that all cells in an organism undergo senescence. However, the results did not preclude the possibility that there is an accumulation of a subpopulation of senescent cells in aged individuals. In fact, there is ample evidence that cells displaying biomarkers of senescence accumulate in tissues of aged animals and humans, suggesting that at least in vivo, cells undergo senescence prior to their telomeres becoming critically short (Dimri et al. 1995; Jeyapalan and Sedivy 2008). Cells displaying biomarkers of senescence have been identified in tissue biopsies of aged animals and humans (Kreiling et al. 2011; Jeyapalan et al. 2006; Ressler et al. 2006). In addition, cells demonstrating biomarkers of senescence are localized to sites of common aging-related degenerative pathologies including atherosclerotic plaques (Vasile et al. 2001), osteoarthritic joints (Price et al. 2002), hypertensive kidney (Westhoff et al. 2008), pulmonary arteries from patients with chronic obstructive pulmonary disease (COPD) (Noureddine et al. 2011), and are found in cultured fibroblasts isolated from chronic non-healing venous ulcers (Stanley and Osler 2001), and from the lungs of emphysema patients (Müller et al. 2006). These studies associate the appearance of senescent cells with aging and age-related degenerative pathology, but additional work is required to test the more difficult question of causality. Interestingly, much less is known about the role of senescent cells in the brain. In this chapter we will review aging-related changes in the brain, including changes in physiology and the role of senescence-like phenotypes in CNS cell types and their potential impact on brain associated pathology.
2 The Aging Brain
Like many other organs throughout the body, the brain is susceptible to aging-related changes and functional decline. Often these aging-related alterations vary greatly among individuals or are specific to certain brain regions. Here, tissue-wide general features of aging brain and neurodegenerative disease are discussed, while cell-type specific changes will be described in more detail in a subsequent section.
At the most basic structural level, the normal aging brain is associated with cortical atrophy characterized by thinning of the gyri, widening of the sulci, and an expansion of the ventricles that contribute to an overall decrease in brain weight (Magnotta et al. 1999; Apostolova et al. 2012). Age-related structural changes can be evaluated in the brains of living subjects through the use of magnetic resonance imaging (MRI) based neuroimaging techniques (Walhovd et al. 2011). In longitudinal MRI studies, neuroanatomical volume loss follows a unique trajectory in individuals diagnosed with mild cognitive impairment (MCI) compared with normal aging (Driscoll et al. 2009). Brain regions that classically exhibit pathologic features of Alzheimer’s disease (AD) early in the course of disease, such as the hippocampus and temporal gray matter, show accelerated volume loss in individuals with MCI, suggesting that volume loss may be a surrogate biomarker for disease progression (Driscoll et al. 2009).
Aging is the greatest risk factor for cognitive decline; however, not all aspects of cognitive function decline during normal aging (Yankner et al. 2008). Certain aspects of cognitive function such as processing speed and formation of new memories are compromised with normal aging, whereas other aspects including long-term memory and verbal knowledge remain stable (Salthouse 2010). An age-related decrease in functional connectivity between different brain regions is thought to contribute to a decline in cognitive functions related to the hippocampus and pre-frontal cortex (Samson and Barnes 2013).
Several studies have examined the association between systemic inflammatory factors and cognitive performance during aging (Wright et al. 2006; Weaver et al. 2002). Evidence of systemic low-grade inflammation and endothelial dysfunction was associated with impaired attention and processing speed in older adults (Heringa et al. 2014). In addition, higher baseline levels of inflammatory mediators C-reactive protein (CRP) and IL-6 were associated with an increased risk of developing dementia (Yaffe et al. 2003; Dziedzic 2006). Compared with similar-aged controls, patients with AD demonstrated significantly higher levels of plasma inflammatory cytokines including IL-1β and TNF-α (Zuliani et al. 2007). In addition to systemic evidence of inflammation, higher levels of inflammatory mediators are also present in the cerebrospinal fluid (CSF) of individuals with MCI (Galimberti et al. 2006). The CSF inflammatory profile of individuals who are in the pre-clinical stage of AD is similar to that of patients with the prototypical neuroinflammatory disease multiple sclerosis, suggesting that neuroinflammation is an early event in AD pathogenesis (Monson et al. 2014). Studies of post-mortem tissues from AD patients have also overwhelmingly demonstrated evidence of neuroinflammation (Akiyama et al. 2000). Glial cells, including microglia and astrocytes, are likely sources of inflammatory mediators within the CNS during aging and neurodegeneration (Salminen et al. 2011; Jensen et al. 2013). Persistent neuroinflammation by sustained overexpression of IL-1β also exacerbates features of AD pathology in the triple transgenic mouse model of AD (3x-Tg-AD) (Ghosh et al. 2013).
In addition to neuroinflammation, oxidative stress is another prominent feature of the aging brain and plays an important role in the pathogenesis of several neurodegenerative diseases. Although low levels of reactive oxygen species (ROS) have physiologic signaling function in the CNS, an imbalance between their generation and detoxification leads to oxidative damage (Wang and Michaelis 2010). Evidence of oxidative damage to macromolecules including protein, nucleic acid, and lipids in the CNS occurs during normal aging and is an early feature of Alzheimer’s disease (Bradley-Whitman et al. 2014; Bradley et al. 2010; Nunomura et al. 2001). The CNS in general and neurons in particular are thought to be vulnerable to oxidative stress because of the high metabolic rate and the propensity for mitochondrial dysfunction (Chen and Zhong 2014). The sources of oxidative stress and the mechanisms surrounding neuronal death and dysfunction during aging and neurodegeneration are active areas of investigation; however, dysfunctional mitochondria in neurons are likely key mediators (Mattson et al. 2008). The blood-brain barrier (BBB) which sits at the interface between the CNS and the periphery is another likely target of inflammatory and oxidative insult (Enciu et al. 2013). Disruption of this barrier is one potential mechanism by which oxidative stress and inflammation contribute to neurodegenerative disease. Changes in BBB permeability positively correlated with disease progression in patients with the Parkinson-like neurodegenerative disorder multiple system atrophy (MSA) (Lee et al. 2013).
Despite the evidence for the role of oxidative stress in the pathogenesis of aging related neurodegenerative disease, the clinical use of antioxidants as therapeutics has been inconclusive in large randomized clinical trials in humans (Kamat et al. 2008). Trials of vitamin E (alpha-tocopherol) failed to improve cognitive function in individuals with MCI, whereas in moderate to severe AD, long-term alpha-tocopherol treatment slowed disease progression (Evans et al. 2014; Sano et al. 1997). In a randomized, placebo-controlled clinical trial by Galasko et al. (2012) in subjects with mild to moderate AD, short-term combination treatment with antioxidants alpha-tocopherol, vitamin C, and α-lipoic acid had no impact on CSF biomarkers of AD pathology; however, they did observe a decrease in the level of F-2 isoprostane, which is a CSF biomarker of oxidative injury in vivo (Paolo et al. 2004). In addition, the effect of alpha-tocopherol alone or in combination with memantine, which is an FDA-approved drug for the treatment of AD, was examined in a recent trial in subjects with mild to moderate AD (Dysken et al. 2014). Subjects receiving alpha-tocopherol alone had slower rates of functional decline compared with placebo or alpha-tocopherol plus memantine (Dysken et al. 2014). There is also evidence that some ROS scavengers may be effective in mitigating cognitive decline in mice if treatment is started early enough. Chronic treatment with a superoxide dismutase/catalase mimetic (EUK-207) decreased oxidative damage to macromolecules and improved cognitive function in aged mice (Clausen et al. 2010), whereas EUK-207 treatment decreased oxidative damage, alleviated features of AD neuropathology, and improved cognitive function in 3x-Tg-AD mice (Clausen et al. 2012).
At the transcriptome level, the aging brain is associated with broad changes in gene expression. Analysis of the gene expression profile of aged mouse (30 months) neocortex revealed increased mRNA levels of genes involved in the inflammatory response and oxidative stress, while genes involved in protein-turnover and trophic support were decreased (Lee et al. 2000). A similar profile is noted in human frontal cortex; beginning in middle age there is a decrease in the expression of genes involved in synaptic plasticity, vesicular transport and mitochondrial function, while the expression of genes involved in the stress response and inflammation was increased (Lu et al. 2004). Interestingly, many of these age down-regulated genes demonstrated oxidative damage to their promoters (Lu et al. 2004). Furthermore, changes in the brain transcriptome are thought to precede the onset of neuropathology in AD (Bossers et al. 2010).
3 Senescence-Related Changes in CNS Cell Types
3.1 Neurons
With very few notable exceptions including the dentate gyrus (DG) and the subventricular zone (SVZ), new neurons are not generated throughout life in the adult brain; therefore the neurons generated early in development must survive and remain functional for the lifetime of the organism (Yankner et al. 2008).
In most brain areas, normal aging is not associated with profound neuronal loss (Rapp and Gallagher 1996; Gazzaley et al. 1997; West et al. 1994); however, the aging brain is characterized by subtle morphological and functional alterations in neurons that contribute to cognitive decline (Burke and Barnes 2006) and may be brain region-specific (Morrison and Baxter 2012). Aged rats with spatial learning deficits related to hippocampal function exhibited a decline in levels of synaptophysin, which is a marker of pre-synaptic vesicles (Smith et al. 2000). Compared with similarly aged cognitively normal controls, subjects with Alzheimer’s disease and to a lesser extent subjects with mild cognitive impairment (MCI) exhibited synaptic loss in the hippocampus suggesting that synaptic loss in this region is associated with cognitive ability (Scheff et al. 2006). In addition to changes in synapses, there is also a dramatic reduction in the number of thin dendritic spines, which are important for neuronal plasticity and learning (Kasai et al. 2003), on pyramidal neurons of the aged non-human primate brain (Dumitriu et al. 2010).
In contrast to normal aging, neurodegenerative disease is associated with profound neuronal loss in specific brain regions. Selective populations of neurons are vulnerable to degeneration in different disorders including dopaminergic neurons in the substantia nigra pars compacta in Parkinson’s disease (PD), medium spiny neurons in the striatum in Huntington’s disease (HD), motor neurons in amyotrophic lateral sclerosis (ALS), and hippocampal and frontal lobe pyramidal neurons in AD (Mattson and Magnus 2006). One of the greatest risk factors for the development of sporadic forms of any of these disorders is aging. General aging-associated changes are qualitatively similar, but often more severe in regions undergoing degeneration (Mattson and Magnus 2006).
Recent evidence suggests that although mature post-mitotic neurons fail to undergo replicative aging, these cells are subject to a stress-induced senescence-like phenotype. Purkinje, hippocampal, and cortical neurons from aged mice (32 months) in situ exhibit features of a p21-dependent senescence-like phenotype (Jurk et al. 2012). This phenotype is characterized by increases in SA β-gal activity, p38 MAPK activation, IL-6 production, lipid peroxidation and DNA damage, and could be mitigated by caloric restriction or exacerbated by telomere dysfunction in the aged mice (Jurk et al. 2012). Interestingly, p21 overexpression protects primary cultured neurons from apoptosis in response to treatment with the neurotoxin ethylcholine aziridinium (AF64A) (Harms et al. 2007).
A senescence-like phenotype is also evident in the neurons of aging rat (24 months) in the CA3 region of the hippocampus or upon prolonged culturing of primary hippocampal neurons by staining for SA β-gal activity, a senescence biomarker that differentiates between senescent and terminally-differentiated cells (Geng et al. 2010; Dimri et al. 1995). Finally, human and rodent neural cell lines undergo ROS-dependent senescence in response to treatment with the environmental toxin TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin) (Wan et al. 2014). A likely trigger of stress-induced senescence in neurons is DNA damage. DNA damage in the form of double strand breaks in neurons is a consequence of normal brain activity, but is increased in aged brain and can be exacerbated by the presence of amyloid-β (Suberbielle et al. 2013; Bhaskar and Rao 1994).
Even though neurons have undergone a permanent exit from the cell cycle, the cyclin-dependent kinase inhibitor p16INK4a is not normally detectable in adult brain (Robertson and Jones 1999), while p21 is not typically expressed in mature neurons (Pechnick et al. 2008). However, immunohistochemical analyses of post-mortem frontal cortex from subjects with HIV-associated dementia (HAD) revealed that p21 is increased in neurons and subcortical glia compared uninfected controls (Jayadev et al. 2007), whereas p16INK4a immunoreactivity was detectable in neurons containing neurofibrillary tangles in AD hippocampus and temporal cortex (McShea et al. 1997; Arendt et al. 1998).
4 Neural Stem/Progenitor Cells
In the mammalian adult brain, two major zones of neurogenesis persist: the subgranular zone of the dentate gyrus (DG) of the hippocampus and the subventricular zone (SVZ) of the lateral ventricles (Alvarez-Buylla and Lim 2004). These regions harbor neural stem and progenitor cells (NSPC). NSPC are capable of self-renewal and differentiation into the major CNS types including astrocytes, oligodendrocytes, and neurons. The first evidence for new neurogenesis in adult human brain came from a landmark study by Eriksson et al. (1998) in post-mortem tissues from cancer patients, who were administered bromodeoxyuridine (BrdU) for diagnostic purposes while alive. They showed that new neurons arise from dividing progenitor cells in the DG. Aging is associated with a major decline in the proliferation and differentiation of NSPC. The steepest decline in neurogenesis occurs from young to middle age and is thought to underlie an aging-related decline in cognition (Hamilton et al. 2013).
One potential reason for the decline in neurogenesis with aging is the senescence of NSPC. The reduced regenerative capacity and frequency of neural stem cells in the SVZ correlates with increased p16INK4a expression in aged mice (Molofsky et al. 2006). Cultured NSPC undergo senescence in response to diverse stimuli. These include DNA-damage-induced senescence upon treatment with hydroxyurea (HU) (Dong et al. 2014) or ionizing radiation (Schneider et al. 2013), ROS-induced senescence in response to treatment with Aβ1–42 oligomers (He et al. 2013), and oncogene-induced senescence with BRAF V600E in a model of pilocytic astrocytoma (Raabe et al. 2011). In order to maintain stem cell function, a proper balance of factors that promote stem cell self-renewal yet contribute to neoplasia relative to factors that reduce stem cell regenerative potential, decreasing the propensity for cancer but contributing to aging, is required (Molofsky et al. 2006).
NSPC self-renewal and differentiation are regulated by external cues from the specialized microenvironment or “niche” in which they reside (Alvarez-Buylla and Lim 2004). In contrast to the cell-intrinsic impact of stem cell senescence, age-related changes in the stem cell microenvironment and organismal systemic milieu also impair stem cell function and neurogenesis (Villeda et al. 2011). These non-cell autonomous challenges to the neurogenic niche can be classified into two main categories: the loss of neurogenesis-promoting cues or gain of neurogenesis-inhibitory signals (Hamilton et al. 2013). For example, brain-derived neurotrophic factor (BDNF) is an important neurogenesis-promoting factor in the adult brain (Lee et al. 2002). Physical exercise, an intervention that improves overall health and increases hippocampal neurogenesis in aged mice also increases the level of BDNF, presumably contributing to the enhanced neurogenesis (van Praag et al. 2005). Like BDNF, insulin-like growth factor-I (IGF-I) is another growth factor that mediates the effect of exercise on neurogenesis in the adult brain (Cotman et al. 2007; Ding et al. 2006).
IGF-I increases the proliferation and differentiation of NSPC (Anderson et al. 2002), and circulating levels of IGF-I are increased in response to exercise (Trejo et al. 2001), but are decreased during aging (Gong et al. 2014). In contrast, a high-fat diet decreases hippocampal neurogenesis through an increase in lipid peroxidation and a decrease in BDNF (Park et al. 2010). Similarly, chronic inflammation inhibits neurogenesis in the adult brain although the precise mechanism is not yet clear (Ekdahl et al. 2003).
5 Oligodendrocytes
The major function of oligodendrocytes is the production of myelin, which ensheathes axons and is a major component of white matter in the vertebrate central nervous system (CNS). The myelin sheath facilitates conduction of signals along axons and provides metabolic support for axons in form of lactate (Fünfschilling et al. 2012). The process of myelination occurs in distinct regions of the human brain throughout life and reaches a pinnacle at middle-age. Demyelination occurs when axons lose their myelin sheath as a result of injury to the oligodendrocyte and results in a functional deficit (Franklin and Ffrench-Constant 2008). An age-related loss in the amount of myelin, but not the protein composition of myelin was observed in post-mortem analyses of human frontal white matter and corpus callosum (Berlet and Volk 1980). Increased degeneration of white matter has also been observed histologically in the frontal lobe of AD and dementia with Lewy bodies (DLB) compared with similar aged-controls (Ihara et al. 2010).
Mature oligodendrocytes are generated from the proliferation and differentiation of oligodendrocyte precursor cells (OP) in the early post-natal period and throughout adulthood (Kang et al. 2010; Young et al. 2013). While OP retain the ability to proliferate indefinitely and fail to undergo replicative senescence in culture (Tang et al. 2001), mature oligodendrocytes are post-mitotic.
Oligodendrocytes demonstrate morphological changes in aged brain which include the swelling of cell processes, and the presence of inclusions in the cell body and cell processes (Peters 2002), while a reduction in oligodendrocyte nuclear diameter is a feature of AD and DLB (Gagyi et al. 2012). These morphological alterations correlate with a decline in myelination rate by oligodendrocytes in aged brain and neurodegenerative disease. Neuroimaging studies with magnetic resonance imaging (MRI) permit an in vivo assessment of white matter and confirm the loss of white matter volume and structural integrity accompanying aging and neurodegenerative disease (Bartzokis et al. 2004). In neuroimaging studies of healthy subjects, individuals carrying the ApoE4+ risk allele for the development of AD showed increased evidence of myelin breakdown (Bartzokis et al. 2006). In addition, myelination abnormalities are evident prior to the appearance of amyloid and tau pathology in the brains of a triple-transgenic AD mouse model (3x-Tg-AD) (Desai et al. 2009). In contrast, white matter integrity is preserved in the offspring of nonagenarians compared with age-matched controls, suggesting that familial longevity has a role in maintaining white matter health (Altmann-Schneider et al. 2013).
Like many repair processes during aging, remyelination efficiency following injury declines (Shields et al. 1999). The decline in remyelination in chronic multiple sclerosis lesions and following toxin-induced demyelination in rats was attributed to impaired recruitment and differentiation of OP (Sim et al. 2002; Kuhlmann et al. 2008). The aging-associated defect in remyelination can be rescued by over-expression of the anti-aging protein Klotho (Chen et al. 2013) or exposure to a more youthful systemic milieu via heterochronic parabioisis (Ruckh et al. 2012).
6 Microglia
In contrast to other major cell types in the CNS with neuroectodermal origins, microglia are derived from an erythromyeloid progenitor of mesodermal origin and migrate into the CNS very early in development (Alliot et al. 1999). Local proliferation of CNS resident parenchymal microglia is the sole source of microglia in adult CNS (Ajami et al. 2007). As the resident immune cells of the brain, microglial cells actively survey the microenvironment with their processes, and perform housekeeping functions through the phagocytosis of debris, clearance of apoptotic cells, and maintenance of synapses (Nimmerjahn et al. 2005; Wake et al. 2009). In response to CNS injury or to damage signaling with ATP, microglia migrate to the site of damage and proliferate. Although data from numerous in vitro studies support a neurotoxic role for microglia (Boje and Arora 1992; Burguillos et al. 2011) and microglia are found at the sites of infectious and sterile inflammatory CNS lesions, the role of microglia as mediators of neurotoxicity has been recently re-evaluated (Biber et al. 2014). The microglial response to injury, termed “activation”, was once thought to be primarily detrimental and a major cause of neurodegeneration; however, recent findings have challenged this concept and highlighted beneficial roles of microglia (Biber et al. 2014).
Compared with young microglia, aged microglia function very differently under normal resting conditions and in response to injury. For example, microglia isolated from aged brains and cultured ex vivo constitutively secrete excess cytokines including IL-6 and TNF-α and resist stimulation with LPS (Njie et al. 2012). Aged microglia demonstrate a blunted surveillance program and a delayed, but sustained response to injury in situ in aged mice (Damani et al. 2011; Wynne et al. 2010). The combination of constitutive inflammation and deregulated function in aged microglia has profound implications for both aged brain and neurodegenerative disease. Interestingly, transcriptome profiling studies of microglia isolated from healthy aged mice demonstrate increased expression of genes involved in neuroprotection relative to young adult mice (Hickman et al. 2013).
In human tissues, microglia exhibit “dystrophic” morphological changes in both aged brain and in association with tau pathology in AD brain (Streit et al. 2004, 2009). These dystrophic changes are characterized by a fragmented cytoplasm, gnarled cell processes, and the absence of features of microglial activation (Streit et al. 2004). The altered morphology seen in microglial cells with aging and neurodegenerative disease is thought to occur as these cells become senescent.
Microglial cells initiate a senescence program in response to replicative stress following overstimulation with mitogen (Flanary and Streit 2004) or repeated inflammatory activation with lipopolysaccharide (LPS) in culture (Yu et al. 2012). Reduced telomere length and telomerase activity were also observed in microglia purified from aged rat cortex, while telomere length was reduced in microglia isolated from post-mortem tissue from AD brain tissue (Flanary et al. 2007). In contrast, in response to acute nerve injury, microglia increase telomerase activity by upregulating the expression of telomerase protein up to 7 days post-injury (Flanary and Streit 2005). These findings suggest that the degree and timing of insult as well as the microglial proliferative response affect the ability of these cells to maintain telomere length.
7 Astrocytes
During mid- to late embryonic development, astrocytes are generated from NSC in the ventricular zone (VZ) (Namihira and Nakashima 2013) via a differentiation process that involves epigenetic de-repression of astrocyte-related genes (Namihira et al. 2004; Takizawa et al. 2001) and activation of several signaling pathways including JAK/STAT-3 (Bonni et al. 1997). In contrast, the majority of astrocytes in the postnatal CNS arise from the local symmetric division of differentiated, cortical astrocytes rather than from the differentiation and migration of progenitors (Ge et al. 2012).
Astrocytes are the most abundant population of cells within the human brain, accounting for about half of the total CNS cell number (Azevedo et al. 2009). The population of astrocytes within the human brain is more structurally complex and diverse compared with astrocytes from other mammalian species (Oberheim et al. 2006). This unique structural complexity of astrocytes is thought to underlie cognitive abilities that are uniquely human (Oberheim et al. 2009).
Astrocytes perform a diverse array of complex functions within the CNS to maintain homeostasis: regulation of ions, water, and neurotransmitter homeostasis (Simard and Nedergaard 2004; Schousboe et al. 2013), maintenance of the blood-brain barrier (BBB) (Abbott et al. 2006), contribution to CNS metabolism and participation in synaptic transmission as part of the tripartite synapse (Perea et al. 2009; Sofroniew and Vinters 2010). In addition to the pleiotropic array of functions they perform in normal brain, astrocytes acquire additional functions and release mediators that are known to result in neuronal toxicity (Bi et al. 2013).
Astrocytes undergo a spectrum of molecular and functional changes in order to respond to all forms of CNS insult and injury, which is collectively referred to as reactive astrogliosis (Sofroniew 2009). Recent studies have underscored the heterogeneity of reactive astrogliosis in response to different forms of insult (Zamanian et al. 2012) or distance from the site of injury (Wanner et al. 2013); however, typical features of this phenotype include increased expression of the intermediate filament proteins GFAP and vimentin, and variable cell hypertrophy in mild to moderate stages, while astrocyte proliferation, glial scar formation, and tissue reorganization are indicative of later stages (Sofroniew and Vinters 2010). Often this spectrum of changes is reversible upon removal of the insult, while glial scar formation is a terminal endpoint (Sofroniew 2009). While some forms of reactive astrogliosis are detrimental in some contexts, this response can also be beneficial (Hamby and Sofroniew 2010). For example, glial scar formation inhibits axon regeneration following injury (Iseki et al. 2012); however, the glial scar also surrounds and insulates damaged tissue to promote neuroprotection and repair (Bush et al. 1999). Several pathways including STAT3 (Herrmann et al. 2008), NF-κB (Brambilla et al. 2005), and p38MAPK (Roy Choudhury et al. 2014) have been implicated in reactive astrogliosis. The outcome of inhibition of these signalling pathways in astrocytes is often context dependent.
The interplay between changes that occur with reactive astrogliosis and changes that occur with astrocytes during aging is not entirely clear. Often, an increase in GFAP expression is used as a marker for evidence of reactive astrogliosis in tissues (Sofroniew and Vinters 2010). Astrocytes demonstrating evidence of this reactive phenotype can be found with increasing frequency at sites of virtually every CNS pathologic lesion regardless of etiology (Sofroniew 2009). Although several studies have demonstrated an increase in GFAP expression in aged brain especially in the hippocampus, there is also support for a decrease in GFAP expression during aging (Rodríguez et al. 2014). In contrast to increases in GFAP and hypertrophy, astroglial atrophy is an alternate pathologic feature of AD brain that has been observed in an AD mouse model (Olabarria et al. 2010; Kulijewicz-Nawrot et al. 2012; Yeh et al. 2011).
In contrast to neurons which are post-mitotic, glial cells are capable of undergoing cell division in vitro and in rodent brain tissues (Pontén and Macintyre 1968; Smart and Leblond 1961). Recent studies have also confirmed the proliferative potential of mature astrocytes in adult human brain tissue (Colodner et al. 2005). Remarkably, prolonged culture of glial cells results in a decline in their proliferative capacity (Blomquist et al. 1980), which in normal human astrocytes could not be rescued by expression of hTERT (Evans et al. 2003). This form of in vitro aging by prolonged culture of astrocytes results in a decline in their functional properties including a loss of neuroprotective capacity (Pertusa et al. 2007), impaired glutamate uptake in response to oxidative stress (Gottfried et al. 2002), and impaired synaptic transmission in neurons in a co-culture system (Kawano et al. 2012). Astrocytes isolated from adult rodent cortex are also capable of undergoing replication in culture and are subject to dysfunction from oxidative stress (Souza et al. 2013).
Recently there has been a paradigm shift toward understanding the integral role of astrocyte function and dysfunction in the initiation and progression of neurodegenerative disease and cognitive decline with aging (Verkhratsky et al. 2012). The presence of astrocytes enhances tau phosphorylation and accelerates Aβ induced neurotoxicity in primary neuronal cultures (Garwood et al. 2011). This effect was mediated by secreted factors from astrocytes and could be abrogated upon treatment of the astrocytes with the anti-inflammatory agent minocycline (Garwood et al. 2011). In addition, selective targeting of calcineurin/NFAT (nuclear factor of activated T-cells) signalling in astrocytes improved disease parameters in vivo as evidenced by reduced astrocyte activation, lower Aβ levels, and improved cognitive function in a mouse model of AD (APP/PS1) (Furman et al. 2012). Aging-related changes in secreted factors from astrocytes impair NSPC proliferation in the neurogenic niche leading to a decline in neurogenesis with aging (Miranda et al. 2012).
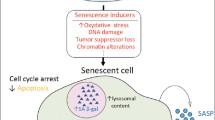
We have demonstrated that in response to oxidative stress and exhaustive replication, human astrocytes activate a senescence program accompanied by the expression of p16, p21, p53, 53BP1; G1 cell cycle arrest; a reduction in telomere length; and increased co-localization of the histone chaperone HIRA and the promyelocytic leukemia PML proteins, a requirement for the formation of senescence-associated heterochromatin foci (Bitto et al. 2010). The importance of senescent astrocytes in age-related dementia has been the subject of recent discussion (Salminen et al. 2011), but to date, there is little evidence to suggest that senescent astrocytes accumulate in the brain. By examining brain tissue we have observed an increase in the number of astrocytes expressing the senescence marker p16 in aged brains, and in AD patient brains (Bhat et al. 2012). Furthermore, since Aβ peptides induce mitochondrial dysfunction, oxidative stress, and alterations in the metabolic phenotype of astrocytes (Abramov et al. 2004; Rhein et al. 2009; Allaman et al. 2010) we examined whether Aβ peptides initiate the senescence response in these cells. In vitro, we found that exposure of astrocytes to Aβ1–42 triggers senescence and that senescent astrocytes produce high quantities of interleukin-6 (IL-6), which seems to be regulated by p38MAPK. IL-6 is a cytokine known to be increased in the CNS of AD patients (Glass et al. 2010). Based on this evidence, we have proposed that accumulation of senescent astrocytes may be one age-related risk factor for sporadic AD.
8 Discussion
Despite the realization that senescent cells play a causal role in many aging-related phenotypes (Baker et al. 2011), senescence has been largely understudied in the brain both in vitro and in tissues (Yeoman et al. 2012). A major challenge for the study of senescence in CNS-derived cell types in the adult has been the availability of tissue and the lack of a source for adult human CNS-derived cells including astrocytes, while studies of cellular senescence and its physiologic relevance to aging and disease in the periphery have been facilitated by tissue biopsies from living human donors. Therefore, the majority of tissues obtained for the study of CNS pathologic processes rely on tissues obtained post-mortem. In addition, a major conceptual challenge in the study of CNS senescence is the relationship between terminal differentiation and cellular senescence, which had been considered mutually exclusive but appear to have a much more complex relationship (Campisi and d’Adda di Fagagna 2007). Astrocytes have the potential to impact CNS function at multiple levels and senescence in this cell population has implications well beyond the simple loss of parenchyma. A model of the impact of astrocyte senescence is presented in Fig. 14.1.

Model. Astrocytes interact with a variety of cell types in the CNS and perform critical functions in order to maintain homeostasis. In response to classic stressors and stressors relevant to the aging brain, human astrocytes exhibit prototypical biomarkers of the senescent phenotype in vitro. Astrocyte senescence is also accompanied by profound changes in the transcriptome including the loss of brain-expressed transcripts, which suggests the loss of differentiated function in senescent astrocytes. The loss of differentiated function in senescent astrocytes and/or the gain of neuroinflammatory function as a result of the SASP have profound implications for the brain tissue microenvironment and the potential to impact virtually every CNS cell type (e.g. neurons, oligodendrocytes, microglia, brain microvascular endothelial cells, and neuronal stem and precursor cells (NSPC)) and every facet of normal CNS function
Our findings demonstrate that human astrocytes undergo cellular senescence in response to a variety of stressors including replicative stress, oxidative stress, and small oligomers of amyloid- beta (Aβ1–42) in vitro. Consistent with our studies in human astrocytes, others have characterized the senescent phenotype in response to treatment with Aβ in vitro in endothelial cells (Donnini et al. 2010) as well as neuronal stem/precursor cells (NSPC) from model organisms (He et al. 2013). Further support for Aβ-induced senescence comes from studies performed in human retinal pigment epithelium (RPE), in which treatment with a low concentration of oligomeric Aβ (0.3 μM) causes RPE senescence and increased secretion of IL-8 and matrix metalloproteinase-9 (MMP-9) (Cao et al. 2013).
The timing of the appearance of senescent cells relative to the development of dysfunction and disease needs to be more thoroughly investigated especially in the context of the aging human CNS. Increased inflammation, oxidative damage, and transcriptional changes precede the onset of CNS dysfunction in AD (Monson et al. 2014; Bradley et al. 2010; Bradley-Whitman et al. 2014; Bossers et al. 2010); however, whether cellular senescence is a cause or a consequence of these insults remains to be determined. Overall, this suggests that in addition to being mediators and drivers of CNS dysfunction and disease during aging, senescent astrocytes could accumulate and persist in very old individuals with AD. By comparing brain regions that are selectively vulnerable to dysfunction and/or degeneration with areas that are relatively spared within the same subject, we can begin to establish whether CNS cellular senescence is a general feature of aged brain or localized to specific to sites of pathology. These studies could be expanded to tissues from other neurodegenerative disorders that increase with age including Parkinson’s disease, ALS, and fronto-temporal lobar degeneration (FTLD) and HIV-associated neurocognitive disorders (HAND).
In addition to astrocytes, the possibility exists that other CNS cell types including brain microvascular endothelial cells, microglia, NSPC, and possibly neurons elicit certain aspects of the senescent phenotype (see model) (Streit et al. 2009; Dong et al. 2014; Molofsky et al. 2006; Jurk et al. 2012). While it was once thought that cellular senescence and terminal differentiation were mutually exclusive (Campisi and d’Adda di Fagagna 2007), recent evidence for a senescence-like phenotype in post-mitotic cells including megakaryocytes (Besancenot et al. 2010), and neurons (Jurk et al. 2012) has challenged this viewpoint. In response to damage, the direction of cell fate toward senescence and away from apoptosis may be a better strategy for potentially irreplaceable cells in order to maintain tissue function, although the senescent phenotype is associated with dysfunction (Naylor et al. 2013). Senescence could also be viewed as compensatory mechanism to avoid cell-cycle reentry-mediated cell death in neurons (Herrup and Yang 2007).
References
Abbott N, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7:41–53
Abramov AY, Canevari L, Duchen MR (2004) Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575
Ajami B, Bennett J, Krieger C, Tetzlaff W, Rossi F (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole G, Cooper N, Eikelenboom P, Emmerling M, Fiebich B, Finch C, Frautschy S, Griffin W, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie I, Mcgeer P, O’Banion M, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, van Muiswinkel F, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421
Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ (2010) Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338
Alliot F, Godin I, Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117:145–152
Altmann-Schneider I, de Craen A, Veer I, van den Berg-Huysmans A, Slagboom P, Westendorp R, van Buchem M, van der Grond J, Leiden Longevity Study G (2013) Preserved white matter integrity is a marker of familial longevity. Ann Neurol 74:883–892
Alvarez-Buylla A, Lim D (2004) For the long run: maintaining germinal niches in the adult brain. Neuron 41:683–686
Anderson M, Aberg M, Nilsson M, Eriksson P (2002) Insulin-like growth factor-I and neurogenesis in the adult mammalian brain. Brain Res Dev Brain Res 134:115–122
Apostolova L, Green A, Babakchanian S, Hwang K, Chou Y-Y, Toga A, Thompson P (2012) Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment (MCI), and Alzheimer disease. Alzheimer Dis Assoc Disord 26:17–27
Arendt T, Holzer M, Gärtner U (1998) Neuronal expression of cycline dependent kinase inhibitors of the INK4 family in Alzheimer’s disease. J Neural Transm (Vienna) 105:949–960
Azevedo F, Carvalho L, Grinberg L, Farfel J, Ferretti R, Leite R, Jacob Filho W, Lent R, Herculano-Houzel S (2009) Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513:532–541
Baker D, Wijshake T, Tchkonia T, Lebrasseur N, Childs B, van de Sluis B, Kirkland J, van Deursen J (2011) Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature 479(7372):232–236
Bartzokis G, Lu P, Mintz J (2004) Quantifying age-related myelin breakdown with MRI: novel therapeutic targets for preventing cognitive decline and Alzheimer’s disease. J Alzheimers Dis 6:9
Bartzokis G, Lu P, Geschwind D, Edwards N, Mintz J, Cummings J (2006) Apolipoprotein E genotype and age-related myelin breakdown in healthy individuals: implications for cognitive decline and dementia. Arch Gen Psychiatry 63:63–72
Berlet H, Volk B (1980) Studies of human myelin proteins during old age. Mech Ageing Dev 14:211–222
Besancenot R, Chaligne R, Tonetti C, Pasquier F, Marty C, Lecluse Y, Vainchenker W, Constantinescu SN, Giraudier S (2010) A senescence-like cell-cycle arrest occurs during megakaryocytic maturation: implications for physiological and pathological megakaryocytic proliferation. PLoS Biol 8(9). pii: e1000476. doi: 10.1371/journal.pbio.1000476. PMID:20838657
Bhaskar M, Rao K (1994) Altered conformation and increased strand breaks in neuronal and astroglial DNA of aging rat brain. Biochem Mol Biol Int 33:377–384
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C (2012) Astrocyte senescence as a component of Alzheimer’s disease. PLoS One 7:e45069
Bi F, Huang C, Tong J, Qiu G, Huang B, Wu Q, Li F, Xu Z, Bowser R, Xia X-G, Zhou H (2013) Reactive astrocytes secrete lcn2 to promote neuron death. Proc Natl Acad Sci U S A 110:4069–4074
Biber K, Owens T, Boddeke E (2014) What is microglia neurotoxicity (Not)? Glia 62(6):841–854
Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S, Torres C (2010) Stress-induced senescence in human and rodent astrocytes. Exp Cell Res 316:2961–2968
Blomquist E, Westermark B, Pontén J (1980) Ageing of human glial cells in culture: increase in the fraction of non-dividers as demonstrated by a minicloning technique. Mech Ageing Dev 12:173–182
Boje K, Arora P (1992) Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res 587:250–256
Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank D, Rozovsky I, Stahl N, Yancopoulos G, Greenberg M (1997) Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science 278:477–483
Bossers K, Wirz K, Meerhoff G, Essing A, van Dongen J, Houba P, Kruse C, Verhaagen J, Swaab D (2010) Concerted changes in transcripts in the prefrontal cortex precede neuropathology in Alzheimer’s disease. Brain 133:3699–3723
Bradley M, Markesbery W, Lovell M (2010) Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med 48:1570–1576
Bradley-Whitman M, Timmons M, Beckett T, Murphy M, Lynn B, Lovell M (2014) Nucleic acid oxidation: an early feature of Alzheimer’s disease. J Neurochem 128:294–304
Brambilla R, Bracchi-Ricard V, Hu W-H, Frydel B, Bramwell A, Karmally S, Green E, Bethea J (2005) Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 202:145–156
Burguillos M, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero J, Joseph B (2011) Caspase signalling controls microglia activation and neurotoxicity. Nature 472:319–324
Burke S, Barnes C (2006) Neural plasticity in the ageing brain. Nat Rev Neurosci 7:30–40
Bush T, Puvanachandra N, Horner C, Polito A, Ostenfeld T, Svendsen C, Mucke L, Johnson M, Sofroniew M (1999) Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron 23:297–308
Campisi J, D’Adda Di Fagagna F (2007) Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 8:729–740
Campisi J, Andersen JK, Kapahi P, Melov S (2011) Cellular senescence: a link between cancer and age-related degenerative disease? Semin Cancer Biol 21:354–359
Cao L, Wang H, Wang F, Xu D, Liu F, Liu C (2013) Aβ-induced senescent retinal pigment epithelial cells create a proinflammatory microenvironment in AMD. Invest Ophthalmol Vis Sci 54:3738–3750
Chen Q, Ames BN (1994) Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc Natl Acad Sci U S A 91:4130–4134
Chen Z, Zhong C (2014) Oxidative stress in Alzheimer’s disease. Neurosci Bull 30(2):271–281
Chen C-D, Sloane J, Li H, Aytan N, Giannaris E, Zeldich E, Hinman J, Dedeoglu A, Rosene D, Bansal R, Luebke J, Kuro-O M, Abraham C (2013) The antiaging protein Klotho enhances oligodendrocyte maturation and myelination of the CNS. J Neurosci 33:1927–1939
Clausen A, Doctrow S, Baudry M (2010) Prevention of cognitive deficits and brain oxidative stress with superoxide dismutase/catalase mimetics in aged mice. Neurobiol Aging 31:425–433
Clausen A, Xu X, Bi X, Baudry M (2012) Effects of the superoxide dismutase/catalase mimetic EUK-207 in a mouse model of Alzheimer’s disease: protection against and interruption of progression of amyloid and tau pathology and cognitive decline. J Alzheimers Dis 30:183–208
Colodner KJ, Montana RA, Anthony DC, Folkerth RD, de Girolami U, Feany MB (2005) Proliferative potential of human astrocytes. J Neuropathol Exp Neurol 64:163–169
Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6:2853–2868
Cotman C, Berchtold N, Christie L-A (2007) Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci 30:464–472
Cristofalo V, Allen R, Pignolo R, Martin B, Beck J (1998) Relationship between donor age and the replicative lifespan of human cells in culture: a reevaluation. Proc Natl Acad Sci U S A 95:10614–10619
Damani M, Zhao L, Fontainhas A, Amaral J, Fariss R, Wong W (2011) Age-related alterations in the dynamic behavior of microglia. Aging Cell 10:263–276
Desai M, Sudol K, Janelsins M, Mastrangelo M, Frazer M, Bowers W (2009) Triple-transgenic Alzheimer’s disease mice exhibit region-specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 57:54–65
Dimri G, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano E, Linskens M, Rubelj I, Pereira-Smith O (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92:9363–9367
Ding Q, Vaynman S, Akhavan M, Ying Z, Gomez-Pinilla F (2006) Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 140:823–833
Dong CM, Wang sXL, Wang GM, Zhang WJ, Zhu L, Gao S, Yang DJ, Qin Y, Liang QJ, Chen YL, Deng HT, Ning K, Liang AB, Gao ZL, Xu J (2014) A stress-induced cellular aging model with postnatal neural stem cells. Cell Death Dis 5:e1116. doi: 10.1038/cddis.2014.82
Donnini S, Solito R, Cetti E, Corti F, Giachetti A, Carra S, Beltrame M, Cotelli F, Ziche M (2010) Abeta peptides accelerate the senescence of endothelial cells in vitro and in vivo, impairing angiogenesis. FASEB J 24:2385–2395
Driscoll I, Davatzikos C, An Y, Wu X, Shen D, Kraut M, Resnick S (2009) Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology 72:1906–1913
Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen W, Lou W, Rapp P, Morrison J (2010) Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci 30:7507–7515
Dysken M, Sano M, Asthana S, Vertrees J, Pallaki M, Llorente M, Love S, Schellenberg G, McCarten J, Malphurs J, Prieto S, Chen P, Loreck D, Trapp G, Bakshi R, Mintzer J, Heidebrink J, Vidal-Cardona A, Arroyo L, Cruz A, Zachariah S, Kowall N, Chopra M, Craft S, Thielke S, Turvey C, Woodman C, Monnell K, Gordon K, Tomaska J, Segal Y, Peduzzi P, Guarino P (2014) Effect of vitamin E and memantine on functional decline in Alzheimer disease: the TEAM-AD VA cooperative randomized trial. JAMA 311:33–44
Dziedzic T (2006) Systemic inflammatory markers and risk of dementia. Am J Alzheimers Dis Other Demen 21:258–262
Ekdahl C, Claasen J-H, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100:13632–13637
Enciu A-M, Gherghiceanu M, Popescu B (2013) Triggers and effectors of oxidative stress at blood-brain barrier level: relevance for brain ageing and neurodegeneration. Oxid Med Cell Longev 2013:297512
Eriksson P, Perfilieva E, Björk-Eriksson T, Alborn A, Nordborg C, Peterson D, Gage F (1998) Neurogenesis in the adult human hippocampus. Nat Med 4:1313–1317
Evans R, Wyllie F, Wynford-Thomas D, Kipling D, Jones C (2003) A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res 63:4854–4861
Evans D, Morris M, Rajan K (2014) Vitamin E, memantine, and Alzheimer disease. JAMA 311:29–30
Flanary B, Streit W (2004) Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 45:75–88
Flanary B, Streit W (2005) Effects of axotomy on telomere length, telomerase activity, and protein in activated microglia. J Neurosci Res 82:160–171
Flanary B, Sammons N, Nguyen C, Walker D, Streit W (2007) Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 10:61–74
Franklin R, Ffrench-Constant C (2008) Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci 9:839–855
Fünfschilling U, Supplie L, Mahad D, Boretius S, Saab A, Edgar J, Brinkmann B, Kassmann C, Tzvetanova I, Möbius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda M, Moraes C, Frahm J, Goebbels S, Nave K-A (2012) Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 485:517–521
Furman J, Sama D, Gant J, Beckett T, Murphy M, Bachstetter A, van Eldik L, Norris C (2012) Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci 32:16129–16140
Gagyi E, Kormos B, Castellanos K, Valyi-Nagy K, Korneff D, Lopresti P, Woltjer R, Valyi-Nagy T (2012) Decreased oligodendrocyte nuclear diameter in Alzheimer’s disease and Lewy body dementia. Brain Pathol 22:803–810
Galasko D, Peskind E, Clark C, Quinn J, Ringman J, Jicha G, Cotman C, Cottrell B, Montine T, Thomas R, Aisen P, Alzheimer’s Disease Cooperative, S (2012) Antioxidants for Alzheimer disease: a randomized clinical trial with cerebrospinal fluid biomarker measures. Arch Neurol 69:836–841
Galimberti D, Schoonenboom N, Scheltens P, Fenoglio C, Bouwman F, Venturelli E, Guidi I, Blankenstein M, Bresolin N, Scarpini E (2006) Intrathecal chemokine synthesis in mild cognitive impairment and Alzheimer disease. Arch Neurol 63:538–543
Garwood C, Pooler A, Atherton J, Hanger D (2011) Noble W (2011) Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture. Cell Death Dis 2:e167. doi:10.1038/cddis.2011.50.PMID:21633390
Gazzaley A, Thakker M, Hof P, Morrison J (1997) Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neurobiol Aging 18:549–553
Ge W-P, Miyawaki A, Gage F, Jan Y, Jan L (2012) Local generation of glia is a major astrocyte source in postnatal cortex. Nature 484:376–380
Geng Y-Q, Guan J-T, Xu X-H, Fu Y-C (2010) Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem Biophys Res Commun 396:866–869
Ghosh S, Wu M, Shaftel S, Kyrkanides S, Laferla F, Olschowka J, O’Banion M (2013) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci 33:5053–5064
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934
Gong Z, Kennedy O, Sun H, Wu Y, Williams G, Klein L, Cardoso L, Matheny R, Hubbard G, Ikeno Y, Farrar R, Schaffler M, Adamo M, Muzumdar R, Yakar S (2014) Reductions in serum IGF-1 during aging impair health span. Aging Cell 69(3):408–418
Gottfried C, Tramontina F, Gonçalves D, Gonçalves C, Moriguchi E, Dias R, Wofchuk S, Souza D (2002) Glutamate uptake in cultured astrocytes depends on age: a study about the effect of guanosine and the sensitivity to oxidative stress induced by H(2)O(2). Mech Ageing Dev 123:1333–1340
Hamby ME, Sofroniew MV (2010) Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 7:494–506
Hamilton L, Joppé S, Cochard LM, Fernandes K (2013) Aging and neurogenesis in the adult forebrain: what we have learned and where we should go from here. Eur J Neurosci 37:1978–1986
Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hübner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, Von Harsdorf R, Endres M, Hörtnagl H (2007) Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci 27:4562–4571
Hayflick L (1965) The limited in vitro lifetime of human diploid strains. Exp Cell Res 37:614–636
Hayflick L, Moorhead P (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621
He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ (2013) Amyloid-β(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis 4:e924. doi: 10.1038/cddis.2013.437. PMID: 24263098
Heringa S, van den Berg E, Reijmer Y, Nijpels G, Stehouwer C, Schalkwijk C, Teerlink T, Scheffer P, van den Hurk K, Kappelle L, Dekker J, Biessels G (2014) Markers of low-grade inflammation and endothelial dysfunction are related to reduced information processing speed and executive functioning in an older population – the Hoorn study. Psychoneuroendocrinology 40:108–118
Herrmann J, Imura T, Song B, Qi J, Ao Y, Nguyen T, Korsak R, Takeda K, Akira S, Sofroniew M (2008) STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci 28:7231–7243
Herrup K, Yang Y (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat Rev Neurosci 8:368–378
Hickman S, Kingery N, Ohsumi T, Borowsky M, Wang L-C, Means T, El Khoury J (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16:1896–1905
Ihara M, Polvikoski T, Hall R, Slade J, Perry R, Oakley A, Englund E, O’Brien J, Ince P, Kalaria R (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 119:579–589
Iseki K, Hagino S, Nikaido T, Zhang Y, Mori T, Yokoya S, Hozumi Y, Goto K, Wanaka A, Tase C (2012) Gliosis-specific transcription factor OASIS coincides with proteoglycan core protein genes in the glial scar and inhibits neurite outgrowth. Biomed Res 33:345–353
Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison R, Garden G (2007) The glial response to CNS HIV infection includes p53 activation and increased expression of p53 target genes. J Neuroimmune Pharm 2:359–370
Jensen C, Massie A, de Keyser J (2013) Immune players in the CNS: the astrocyte. J Neuroimmune Pharm 8:824–839
Jeyapalan JC, Sedivy JM (2008) Cellular senescence and organismal aging. Mech Ageing Dev 129:467–474
Jeyapalan J, Ferreira M, Sedivy J, Herbig U (2006) Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev 128:36–80
Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos E, Thrasivoulou C, Saffrey M, Cameron K, von Zglinicki T (2012) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11:996–1004
Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN, Hensley K (2008) Antioxidants in central nervous system diseases: preclinical promise and translational challenges. J Alzheimers Dis 15:473–493
Kang S, Fukaya M, Yang J, Rothstein J, Bergles D (2010) NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68:668–681
Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H (2003) Structure-stability-function relationships of dendritic spines. Trends Neurosci 26:360–368
Kawano H, Katsurabayashi S, Kakazu Y, Yamashita Y, Kubo N, Kubo M, Okuda H, Takasaki K, Kubota K, Mishima K, Fujiwara M,Harata N, Iwasaki K (2012) Long-term culture of astrocytes attenuates the readily releasable pool of synaptic vesicles. PLoS One 7(10):e48034. doi:10.1371/journal.pone.0048034. Epub 2012 Oct 26 (Erratum in: PLoS One 7(11). doi:10.1371/annotation/9dd1f25a-55e9-4968-9f70-929d1b8d5064. PMID:23110166)
Kreiling J, Tamamori-Adachi M, Sexton A, Jeyapalan J, Munoz-Najar U, Peterson A, Manivannan J, Rogers E, Pchelintsev N, Adams P, Sedivy J (2011) Age-associated increase in heterochromatic marks in murine and primate tissues. Aging Cell 10:292–304
Kuhlmann T, Miron V, Cui Q, Cuo Q, Wegner C, Antel J, Brück W (2008) Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 131:1749–1758
Kulijewicz-Nawrot M, Verkhratsky A, Chvátal A, Syková E, Rodríguez J (2012) Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J Anat 221:252–262
Lee C, Weindruch R, Prolla T (2000) Gene-expression profile of the ageing brain in mice. Nat Genet 25:294–297
Lee J, Duan W, Mattson M (2002) Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J Neurochem 82:1367–1375
Lee J, Song S, Hong J, Sunwoo M-K, Park H-J, Sohn Y, Lee P (2013) Changes in the blood-brain barrier status closely correlate with the rate of disease progression in patients with multiple system atrophy: a longitudinal study. Parkinsonism Relat Disord 19:450–452
Lu T, Pan Y, Kao S-Y, Li C, Kohane I, Chan J, Yankner B (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429:883–891
Magnotta V, Andreasen N, Schultz S, Harris G, Cizadlo T, Heckel D, Nopoulos P, Flaum M (1999) Quantitative in vivo measurement of gyrification in the human brain: changes associated with aging. Cereb Cortex 9:151–160
Mattson M, Magnus T (2006) Ageing and neuronal vulnerability. Nat Rev Neurosci 7:278–294
Mattson M, Gleichmann M, Cheng A (2008) Mitochondria in neuroplasticity and neurological disorders. Neuron 60:748–766
Mcshea A, Harris P, Webster K, Wahl A, Smith M (1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol 150:1933–1939
Miranda C, Braun L, Jiang Y, Hester M, Zhang L, Riolo M, Wang H, Rao M, Altura R, Kaspar B (2012) Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell 11:542–552
Molofsky A, Slutsky S, Joseph N, He S, Pardal R, Krishnamurthy J, Sharpless N, Morrison S (2006) Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443:448–452
Monson N, Ireland S, Ligocki A, Chen D, Rounds W, Li M, Huebinger R, Munro Cullum C, Greenberg B, Stowe A, Zhang R (2014) Elevated CNS inflammation in patients with preclinical Alzheimer’s disease. J Cereb Blood Flow Metab 34:30–33
Montuschi P, Barnes PJ, Roberts LJ 2nd (2004 Dec) Isoprostanes: markers and mediators of oxidative stress. FASEB J 18(15):1791–800
Morrison J, Baxter M (2012) The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 13:240–250
Müller KC, Welker L, Paasch K, Feindt B, Erpenbeck V, Hohlfeld J, Krug N, Nakashima M, Branscheid D, Magnussen H, Jörres R, Holz O (2006) Lung fibroblasts from patients with emphysema show markers of senescence in vitro. Respir Res 7:32
Namihira M, Nakashima K (2013) Mechanisms of astrocytogenesis in the mammalian brain. Curr Opin Neurobiol 23:921–927
Namihira M, Nakashima K, Taga T (2004) Developmental stage dependent regulation of DNA methylation and chromatin modification in a immature astrocyte specific gene promoter. FEBS Lett 572:184–188
Naylor RM, Baker DJ, van Deursen JM (2013) Senescent cells: a novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther 93:105–116
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Njie E, Boelen E, Stassen F, Steinbusch H, Borchelt D, Streit W (2012) Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging 33:1950–1912
Noureddine H, Gary-Bobo G, Alifano M, Marcos E, Saker M, Vienney N, Amsellem V, Maitre B, Chaouat A, Chouaid C, Dubois-Rande J-L, Damotte D, Adnot S (2011) Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease. Circ Res 109:543–553
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60:759–767
Oberheim N, Wang X, Goldman S, Nedergaard M (2006) Astrocytic complexity distinguishes the human brain. Trends Neurosci 29:547–553
Oberheim N, Takano T, Han X, He W, Lin J, Wang F, Xu Q, Wyatt J, Pilcher W, Ojemann J, Ransom B, Goldman S, Nedergaard M (2009) Uniquely hominid features of adult human astrocytes. J Neurosci 29:3276–3287
Olabarria M, Noristani H, Verkhratsky A, Rodríguez J (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58:831–838
Paolo M, Peter JB, Jackson LR (2004) Isoprostanes: markers and mediators of oxidative stress. FASEB J 18(15):1791–1800
Park H, Park M, Choi J, Park K-Y, Chung H, Lee J (2010) A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett 482:235–239
Pechnick R, Zonis S, Wawrowsky K, Pourmorady J, Chesnokova V (2008) p21Cip1 restricts neuronal proliferation in the subgranular zone of the dentate gyrus of the hippocampus. Proc Natl Acad Sci U S A 105:1358–1363
Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32:421–431
Pertusa M, Garcia-Matas S, Rodriguez-Farre E, Sanfeliu C, Cristofol R (2007) Astrocytes aged in vitro show a decreased neuroprotective capacity. J Neurochem 101:794–805
Peters A (2002) The effects of normal aging on myelin and nerve fibers: a review. J Neurocytol 31:581–593
Pontén J, Macintyre E (1968) Long term culture of normal and neoplastic human glia. Acta Pathol Microbiol Scand 74:465–486
Price J, Waters J, Darrah C, Pennington C, Edwards D, Donell S, Clark I (2002) The role of chondrocyte senescence in osteoarthritis. Aging Cell 1:57–65
Raabe E, Lim K, Kim J, Meeker A, Mao X-G, Nikkhah G, Maciaczyk J, Kahlert U, Jain D, Bar E, Cohen K, Eberhart C (2011) BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res 17:3590–3599
Rapp P, Gallagher M (1996) Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proc Natl Acad Sci U S A 93:9926–9930
Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Dürr P, Wlaschek M (2006) p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 5:379–468
Rhein V, Baysang G, Rao S, Meier F, Bonert A, Muller-Spahn F, Eckert A (2009) Amyloid-beta leads to impaired cellular respiration, energy production and mitochondrial electron chain complex activities in human neuroblastoma cells. Cell Mol Neurobiol 29:1063–1071
Robertson K, Jones P (1999) Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene 18:3810–3820
Rodríguez J, Yeh C-Y, Terzieva S, Olabarria M, Kulijewicz-Nawrot M, Verkhratsky A (2014) Complex and region-specific changes in astroglial markers in the aging brain. Neurobiol Aging 35:15–23
Roy Choudhury G, Ryou M-G, Poteet E, Wen Y, He R, Sun F, Yuan F, Jin K, Yang S-H (2014) Involvement of p38 MAPK in reactive astrogliosis induced by ischemic stroke. Brain Res 1551:45–58
Ruckh J, Zhao J-W, Shadrach J, van Wijngaarden P, Rao T, Wagers A, Franklin R (2012) Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 10:96–103
Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H (2011) Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci 34:3–11
Salthouse T (2010) Selective review of cognitive aging. J Int Neuropsychol Soc 16:754–760
Samson R, Barnes C (2013) Impact of aging brain circuits on cognition. Eur J Neurosci 37:1903–1915
Sano M, Ernesto C, Thomas R, Klauber M, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman C, Pfeiffer E, Schneider L, Thal L (1997) A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. Alzheimers Dis Coop Study N Engl J Med 336:1216–1222
Scheff S, Price D, Schmitt F, Mufson E (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384
Schneider L, Pellegatta S, Favaro R, Pisati F, Roncaglia P, Testa G, Nicolis S, Finocchiaro G, D’Adda Di Fagagna F (2013) DNA damage in mammalian neural stem cells leads to astrocytic differentiation mediated by BMP2 signaling through JAK-STAT. Stem Cell Rep 1:123–138
Schousboe A, Bak L, Waagepetersen H (2013) Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol 4:102
Serrano M, Lin A, Mccurrach M, Beach D, Lowe S (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88:593–602
Shelton D, Chang E, Whittier P, Choi D, Funk W (1999) Microarray analysis of replicative senescence. Curr Biol 9:939–945
Shields S, Gilson J, Blakemore W, Franklin R (1999) Remyelination occurs as extensively but more slowly in old rats compared to young rats following gliotoxin-induced CNS demyelination. Glia 28:77–83
Sim F, Zhao C, Penderis J, Franklin R (2002) The age-related decrease in CNS remyelination efficiency is attributable to an impairment of both oligodendrocyte progenitor recruitment and differentiation. J Neurosci 22:2451–2459
Simard M, Nedergaard M (2004) The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 129:877–896
Smart I, Leblond CP (1961) Evidence for division and transformations of neuroglia cells in the mouse brain, as derived from radioautography after injection of thymidine-H3. J Comp Neurol 116:349–367
Smith T, Adams M, Gallagher M, Morrison J, Rapp P (2000) Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J Neurosci 20:6587–6593
Sofroniew M (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32:638–647
Sofroniew M, Vinters H (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Souza D, Bellaver B, Souza D, Quincozes-Santos A (2013) Characterization of adult rat astrocyte cultures. PLoS One 8:e60282
Stanley A, Osler T (2001) Senescence and the healing rates of venous ulcers. J Vasc Surg 33:1206–1211
Stewart S, Ben-Porath I, Carey V, O’Connor B, Hahn W, Weinberg R (2003) Erosion of the telomeric single-strand overhang at replicative senescence. Nat Genet 33:492–496
Streit W, Sammons N, Kuhns A, Sparks D (2004) Dystrophic microglia in the aging human brain. Glia 45:208–212
Streit W, Braak H, Xue Q-S, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118:475–485
Suberbielle E, Sanchez P, Kravitz A, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer A, Mucke L (2013) Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci 16:613–621
Takizawa T, Nakashima K, Namihira M, Ochiai W, Uemura A, Yanagisawa M, Fujita N, Nakao M, Taga T (2001) DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev Cell 1:749–758
Tang D, Tokumoto Y, Apperly J, Lloyd A, Raff M (2001) Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science 291:868–871
Torres C, Lewis L, Cristofalo VJ (2006) Proteasome inhibitors shorten replicative life span and induce a senescent-like phenotype of human fibroblasts. J Cell Physiol 207:845–853
Trejo J, Carro E, Torres-Aleman I (2001) Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci 21:1628–1634
van Praag H, Shubert T, Zhao C, Gage F (2005) Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci 25:8680–8685
Vasile E, Tomita Y, Brown L, Kocher O, Dvorak H (2001) Differential expression of thymosin beta-10 by early passage and senescent vascular endothelium is modulated by VPF/VEGF: evidence for senescent endothelial cells in vivo at sites of atherosclerosis. FASEB J 15:458–466
Verkhratsky A, Sofroniew MV, Messing A, Delanerolle NC, Rempe D, Rodriguez Arellano JJ, Nedergaard M (2012) Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro 4(3):399–412
Villeda S, Luo J, Mosher K, Zou B, Britschgi M, Bieri G, Stan T, Fainberg N, Ding Z, Eggel A, Lucin K, Czirr E, Park J-S, Couillard-Després S, Aigner L, Li G, Peskind E, Kaye J, Quinn J, Galasko D, Xie X, Rando T, Wyss-Coray T (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477:90–94
Wake H, Moorhouse A, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29:3974–3980
Walhovd K, Westlye L, Amlien I, Espeseth T, Reinvang I, Raz N, Agartz I, Salat D, Greve D, Fischl B, Dale A, Fjell A (2011) Consistent neuroanatomical age-related volume differences across multiple samples. Neurobiol Aging 32:916–932
Wan C, Liu J, Nie X, Zhao J, Zhou S, Duan Z, Tang C, Liang L, Xu G (2014) 2, 3, 7, 8-Tetrachlorodibenzo-P-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLoS One 9(2):e89811. doi: 10.1371/journal.pone.0089811. eCollection 2014. PMID:24587053
Wang E (1995) Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res 55:2284–2292
Wang X, Michaelis E (2010) Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci 2:12
Wanner I, Anderson M, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew M (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33:12870–12886
Weaver JD, Huang MH, Albert M, Harris T, Rowe JW, Seeman TE (2002) Interleukin-6 and risk of cognitive decline: MacArthur studies of successful aging. Neurology 59:371–378
West M, Coleman P, Flood D, Troncoso J (1994) Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344:769–772
Westhoff J, Hilgers K, Steinbach M, Hartner A, Klanke B, Amann K, Melk A (2008) Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 52:123–129
Wright C, Sacco R, Rundek TR, Delman JB, Rabbani LE, Elkind MSV (2006) Interleukin-6 is associated with cognitive function: the northern Manhattan study. J Stroke Cerebrovasc Dis 15:34–38
Wynne A, Henry C, Huang Y, Cleland A, Godbout J (2010) Protracted downregulation of CX3CR1 on microglia of aged mice after lipopolysaccharide challenge. Brain Behav Immun 24:1190–1201
Yaffe K, Lindquist K, Penninx BW, Simonsick EM, Pahor M, Kritchevsky S, Launer L, Kuller L, Rubin S, Harris T (2003) Inflammatory markers and cognition in well-functioning African-American and white elders. Neurology 61:76–80
Yankner B, Lu T, Loerch P (2008) The aging brain. Annu Rev Pathol 3:41–66
Yeh C-Y, Vadhwana B, Verkhratsky A, Rodríguez J (2011) Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3:271–279
Yeoman M, Scutt G, Faragher R (2012) Insights into CNS ageing from animal models of senescence. Nat Rev Neurosci 13:435–445
Young K, Psachoulia K, Tripathi R, Dunn S-J, Cossell L, Attwell D, Tohyama K, Richardson W (2013) Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron 77:873–885
Yu H-M, Zhao Y-M, Luo X-G, Feng Y, Ren Y, Shang H, He Z-Y, Luo X-M, Chen S-D, Wang X-Y (2012) Repeated lipopolysaccharide stimulation induces cellular senescence in BV2 cells. Neuroimmunomodulation 19:131–136
Zamanian J, Xu L, Foo L, Nouri N, Zhou L, Giffard R, Barres B (2012) Genomic analysis of reactive astrogliosis. J Neurosci 32:6391–6410
Zuliani G, Ranzini M, Guerra G, Rossi L, Munari M, Zurlo A, Volpato S, Atti A, Blè A, Fellin R (2007) Plasma cytokines profile in older subjects with late onset Alzheimer’s disease or vascular dementia. J Psychiatr Res 41:686–693
Acknowledgements
This work was supported by grants 1RO1NS078283 (CT), R21AG046943 (CT), the Commonwealth of Pennsylvania Universal Research Enhancement Grant (CT), the Drexel University College of Medicine Research Program Planning Grant (CT); and the Drexel Aging Initiative. Research reported in this publication is also supported by the National Institute on Aging of the National Institutes of Health under Award Number F30AG043307 (EPC). The content of this chapter is solely the responsibility of the authors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Crowe, E.P. et al. (2016). Implications of Cellular Senescence on Aging and Disease in the Brain. In: Rattan, S., Hayflick, L. (eds) Cellular Ageing and Replicative Senescence. Healthy Ageing and Longevity. Springer, Cham. https://doi.org/10.1007/978-3-319-26239-0_14
Download citation
DOI: https://doi.org/10.1007/978-3-319-26239-0_14
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-26237-6
Online ISBN: 978-3-319-26239-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)