
Abstract
The term cellular senescence was introduced more than five decades ago to describe the state of growth arrest observed in aging cells. Since this initial discovery, the phenotypes associated with cellular senescence have expanded beyond growth arrest to include alterations in cellular metabolism, secreted cytokines, epigenetic regulation and protein expression. Recently, senescence has been shown to play an important role in vivo not only in relation to aging, but also during embryonic development. Thus, cellular senescence serves different purposes and comprises a wide range of distinct phenotypes across multiple cell types. Whether all cell types, including post-mitotic neurons, are capable of entering into a senescent state remains unclear. In this review we examine recent data that suggest that cellular senescence plays a role in brain aging and, notably, may not be limited to glia but also neurons. We suggest that there is a high level of similarity between some of the pathological changes that occur in the brain in Alzheimer’s and Parkinson’s diseases and those phenotypes observed in cellular senescence, leading us to propose that neurons and glia can exhibit hallmarks of senescence previously documented in peripheral tissues.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Senescence, or “to grow old” in Latin, can be observed both systemically and on the level of individual cells. Overall, it can be viewed as a state that is associated with aging, exhibiting a decline in normal function and increased vulnerability to stressors. The concept of cellular senescence (CS) was first introduced more than five decades ago (Hayflick and Moorhead 1961) based on the finding that cells in culture could only undergo a limited number of divisions (the Hayflick limit). It is generally believed to be an alternate cell fate in the absence of apoptosis (programmed cell death) (Bree et al. 2002). Recently, however, it has become clear that senescence is not solely restricted to the loss of replicative ability, but in fact involves changes in cellular metabolism, epigenetic regulation and gene expression. The prototypical molecular changes that occur during senescence, which include altered morphology, expression of pro-inflammatory cytokines, growth factors and proteases, have collectively been termed the senescence-associated secretory phenotype (SASP) by the Campisi Lab (Coppe et al. 2010a). At present, these phenotypic changes along with increased expression of the cell cycle regulating protein p16(INK4a) and β-galactosidase (β-gal) activity are the predominate markers used to identify senescence cells (Carnero 2013; Salama et al. 2014).
The relationship between CS and organismal aging has only just begun to be explored. Markers of senescence have been found to increase progressively with age in most organisms, including mouse and human tissues (see (van Deursen 2014) for a review). However, correlation does not necessarily indicate causality. A recent study examined this question directly utilizing transgenic mice in which senescent cells (defined as those expressing p16(INK4a)) undergo apoptosis (Baker et al. 2011). Crossing these mice with a progeroid mouse model (BubR1H/H) reduced age-related phenotypes including sarcopenia, cataracts and loss of adipose tissue (Baker et al. 2011). Also SASP has been suggested to contribute to several age-related diseases including obesity, diabetes, cancer and cardiovascular dysfunction (See (Tchkonia et al. 2013) for a review). These experiments suggest that CS plays a role in age-related conditions in multiple tissues.
The idea that cellular senescence is only an aging-related phenomenon was recently called into question by the discovery of developmental senescence. This research has demonstrated that during embryonic development cells enter a senescent state, as evidenced by β-gal activity, and exhibit SASP (Munoz-Espin et al. 2013; Storer et al. 2013). This distinctly non-aging and non-insult induced occurrence of SASP suggests that SASP and senescence cannot be viewed merely as proliferation arrest and a “side effect” of aging, but is in itself a selective and purposeful mechanism, i.e. a means of clearing unnecessary cells and modulating the tissue microenvironment.
The observation that senescence is not restricted to aging but occurs during normal development and across multiple tissue types raises many questions. Is aging-associated senescence simply a developmental process gone awry? Do all cells senesce through the same mechanisms and subsequently exhibit a similar senescent phenotype? Growth arrest is traditionally viewed as one of the major hallmarks of senescence. How does senescence in this regard apply to post-mitotic cell populations such as fully differentiated neurons, osteocytes, skeletal and cardiac muscle cells? Are there, and if so to what extent, shared phenotypic traits between aging dividing cells (traditionally described as “senescent cells”) and aging post-mitotic cells (traditionally not believed to undergo senescence)?
As interesting studies are emerging on the role of senescence in different age-related pathological conditions, it seems that a particularly understudied field is that of senescence in age-related diseases of the central nervous system (CNS). In addition to age-related cognitive decline, age is the primary risk factor for many neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD) and frontotemporal dementia (FTD). These diseases are all characterized by dysfunction and death of neurons and glial reactions that create an inflammatory milieu in the affected brain regions. In light of the many recent findings on metabolic and epigenetic alterations in relation to senescence, we will explore different phenotypes and hallmarks of CS, and examine whether similar hallmarks are seen in neurons and glia associated with age-related neurodegenerative disease as well as normal aging.
Hallmarks of cellular senescence
Pathways to cellular senescence
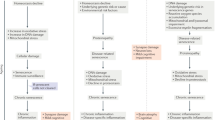
The inducers of a senescent phenotype are typically categorized into three groups (Fig. 1): replicative senescence (RS), stress-induced premature senescence (SIPS) and oncogene-induced senescence (OIS) (Kuilman et al. 2010). In brief, RS—the original idea of senescence proposed by Hayflick and Moorhead (Hayflick and Moorhead 1961)—canonically occurs in response to telomere shortening. This triggers the DNA damage response (DDR) causing activation of the Ataxia-Telangiectasia Mutated (ATM) and Ataxia Telangiectasia and Rad3-related (ATR) pathways that induce cell cycle arrest by activating the cell cycle-regulating protein kinases CDK2 and CDK1, respectively (Di Micco et al. 2008).

Pathways to cellular senescence. This figure summarizes the different stressors (top layer) and molecular mechanisms (top middle layer) involved in mediating each type of senescence (bottom middle layer) collectively termed “cellular senescence” (bottom layer). RS replicative senescence, SIPS stress induced premature senescence, OIS oncogene-induced senescence. Yellow color indicates a type of stressor that is relevant to neurons. Green color indicates a type of stressor only relevant to other cell types. (Color figure online)
In contrast, SIPS occurs independently of telomere length—hence the name premature—and its induction is seen in response to long-term exposure to sub-cytotoxic doses of a number of stressors including oxidative stress (Chen et al. 1995), UVB radiation (Medrano et al. 1995), artificial and inadequate culture conditions ((Ramirez et al. 2001; Sherr and DePinho 2000) for a review; also see Fig. 1). OIS can be considered a special type of SIPS, in which senescence arises due to activation of oncogenes such as K-ras, B-raf, PTEN and NF1 (Larsson 2011). Common to all types of senescence is the up-regulation of one or several of the tumor suppressor genes including p19ARF, p53, p21, p16(INK4a) and the mitogen-activated protein kinase p38MAPK. In particular, p53 and the p16(INK4a)-Rb signaling pathways play a central role in the achievement of growth arrest regardless of inducer, whereas p19ARF is strongly associated with OIS (Larsson 2011; van Deursen 2014). OIS is commonly viewed as a feedback mechanism to suppress oncogenesis, although oncogenic mutations in some cases appear to co-opt a senescence-associated inflammatory response (SIR), a component of SASP to further enhance tumor progression (Pribluda et al. 2013).
Gene expression and epigenetic mechanisms
Altered morphology is one of the major hallmarks of CS across disparate cell types, with cells becoming enlarged and demonstrating an increased cytoplasm to nucleus ratio (Hayflick and Moorhead 1961; Majore et al. 2009). mRNA and miRNA expression profiles of different human tissues during aging reveal the existence of a common pattern of defined transcriptional and epigenetic changes as well as tissue-specific age-related alterations (ElSharawy et al. 2012; Li et al. 2009; Maes et al. 2009; Ren et al. 2012; Santarosa et al. 2009; Serna et al. 2012).
Gene expression profiles suggest that senescence is a tightly regulated process, with consistent alterations in heterochromatin in CS across tissues. Senescence-associated heterochromatic foci (SAHF) are dense puncta of DNA observed in vitro following replicative and stress-induced senescence. They are associated with increased methylation of Lys9 on histone H3 (Bannister et al. 2001). Although SAHF are a frequently observed feature of CS and often used as a marker of CS, a recent report found dissociation between SAHF occurrence and other CS-phenotypes in multiple primary cells and cell lines (Kosar et al. 2011). Despite this report, it is still unclear how the presence or absence of SAHF and the underlying alterations in methylation modulate features of the global transcriptional profile of CS including growth arrest (reviewed by Adams (Adams 2007; Zhang et al. 2007)) and SASP. Further research is necessary to discern if global transcriptional phenotypes of CS occur regardless of alterations in heterochromatin or if these phenotypes can be dissociated between different inducers of CS and/or different cell types.
Alterations in H2K4me3 and H3K27me3 methylation are one mechanism through which CS-related changes in the transcriptome can occur (Shah et al. 2013). Altered miRNA expression and disruption of miRNA biogenesis through deletion of the enzyme Dicer, (expression of which declines with age,) can also shift cells into a senescent state (Mori et al. 2012). miRNAs such as miR-29 have been discovered to be master regulators of CS (Hu et al. 2014) and it is possible that individual CS phenotypes such as SASP may also be regulated by specific miRNAs. In C. elegans and Drosophila perturbed miRNA expression can alter lifespan, modulate lipofuscin accumulation in tissues, accelerate a transcriptional profile associated with aging and reduce age-related neurodegeneration (Boehm and Slack 2005; Liu et al. 2012). Mounting evidence suggests that miRNAs also play an important role in mammalian CS, aging and neurodegenerative diseases (Abe and Bonini 2013). However, a single miRNA regulating these processes similarly to those in C. elegans and Drosophila has not yet been discovered.
SASP is a collective term for the altered expression of secreted cytokines observed in CS. SASP is implicated in inflammation, tissue growth and remodeling. Prominent components of SASP include the cytokines IL-6 and IL-8, MMP-1, MMP-3, fibronectin and laminin B (see (Coppe et al. 2010a, b) for a review). The general increase in inflammation associated with aging has also been attributed to SASP, although further experiments are necessary to provide conclusive evidence of this relationship. The emerging evidence suggests that there are specific gene expression and epigenetic mechanisms by which CS has a major effect on the cell microenvironment and, through SASP, CS may mediate organism-wide phenotypes such as systemic inflammation. Network analysis of age related genes revealed multiple connections to CS and systemic inflammation, further supporting the role of CS in organismal aging (Tacutu et al. 2010, 2011).
Senescence-associated metabolic alterations
The disruption of energy metabolism and degradation of macromolecules (proteins and lipids) is frequently observed in senescent cells. Although senescent cells are metabolically active and synthesize macromolecules (Blagosklonny 2011; Blagosklonny and Hall 2009) there are abnormalities in organelle quality and structure. In particular, the mitochondria of senescent cells are abnormally elongated, likely due to increased expression of mitochondrial fusion proteins such as Fis1, DRP1 and OPA1 (Lee et al. 2007; Mai et al. 2010; Yoon et al. 2006). This altered mitochondrial morphology has been proposed to confer protection against oxidative stress (Mai et al. 2010), but it can also result in proton leakage across the mitochondrial inner membrane leading to enhanced compensatory electron transport (Lee et al. 2007). Increased activity of the mitochondrial pyruvate dehydrogenase, which mediates conversion of pyruvate into acetyl-CoA, has been observed in OIS constituting another potential mechanism for the higher reactive oxygen species (ROS) levels and AMP/ATP ratio observed in CS (Zwerschke et al. 2003). The onset of CS can be delayed in low-glucose culture conditions, potentially by reducing glucose-mediated protein and DNA damage, although it is not clear whether glycolysis is also altered in senescent cells (Zwerschke et al. 2003). Overall, the reduced energy level and structural as well as functional abnormalities of mitochondria may lead to enhanced generation of DNA damage while inhibiting energy-intensive repair.
The evidence for compromised cellular repair in CS remains controversial. Although DNA damage is an established inducer of CS, the occurrence of impaired DNA repair pathways in CS have not been well studied. Accumulation of unrepairable double strand DNA breaks were reported in senescent cells while their ability to repair radiation-induced damage was unimpaired (Sedelnikova et al. 2004). Thus, CS may be exhibiting a different type of DNA damage rather than impaired DNA repair.
Interestingly, the classical CS marker β-galactosidase (lacZ) staining (at pH 6) likely reflects altered lysosomal mass—the lysosome being the main cell organelle responsible for degradation of damaged macromolecules (Lee et al. 2006). The lipofuscin and α-fucosidase accumulation observed during CS furthermore suggests an increase in lysosomal biomass (Hohn et al. 2012; Singh and Piekorz 2013). However, it is still controversial if the increase in lysosomal mass indicates enhanced degradation activity or results from impaired lysosomal function. Similarly, reports exist on both enhanced and inhibited autophagy in CS (Grune et al. 2005; Young et al. 2009). Ceramide levels have been shown to increase during CS (Venable et al. 1995), which is interesting since an increase in ceramide concentration can induce a pro-apoptotic autophagy response. Taken together, there appears to be a relationship between autophagy and CS which is understandable in light of the relationship between autophagy and apoptosis.
Senescence in the CNS
As described above, hallmarks of CS encompass more than just irreversible growth arrest (Fig. 2). However, given the typical association of growth arrest with CS, it is interesting to examine whether moderately proliferative cells like astrocytes and oligodendrocyte precursors or even post-mitotic cells like neurons are also subject to CS. Direct experimental evidence for neural cell senescence has been lacking until recently, as some phenotypical markers of senescence have been shown in neurons and astrocytes (Table 1).

Evidence for senescence in CNS cell populations. Both aging neurons (left) and glial cells (right) show phenotypic traits characteristic of cellular senescence (middle)
A senescence-like phenotype has been observed in Purkinje and cortical neurons in response to DNA damage. The mechanism of induction was similar to classical CS and involved a DDR response, and p21 and p38MAPK activation. Other senescence-associated phenotypes were also observed, including elevated β-gal staining and a SASP-like secretion of pro-inflammatory cytokines (Jurk et al. 2012). A recent study found that cultured cerebellar granule neurons (CGNs) exhibit decreased base excision repair and non-homologous end joining repair over a 5-week time course in culture. This reduction in DDR was correlated to an observed increase in beta-galactosidase activity and intracellular calcium level (Bhanu et al. 2010). These findings suggest that the DNA damage that accumulates in aging neurons leads to senescence, although this study did not demonstrate a causal relationship between DDR impairment and neuronal senescence. In addition to DDR-induced senescence, aging neurons also exhibit CS phenotypes after exposure to oxidative and metabolic stress (Jurk et al. 2012). Increased beta-galactosidase activity was observed in hippocampal neurons upon prolonged culture of 20–30 days (Dong et al. 2011) and also in the hippocampus of aging rats (Geng et al. 2010). Dong et al. also report on mitochondrial dysfunction in neurons that result in elevated ROS levels (Dong et al. 2011). While these observations suggest mitochondrial impairment plays a role in neuronal senescence, this evidence is correlative and there is an inherent selection bias as typically many neurons die in prolonged culture conditions.
SIRT1, a deacetylase associated with longevity and metabolic regulation, has been shown to attenuate CS in several cell types including neurons. Aging neurons, particularly those that participate in wakefulness activity (hypothalamic orexinergic neurons, locus ceruleus neurons, and mesopontine cholinergic and dopaminergic neurons) have been found to have accumulation of lipofuscin, another commonly used hallmark of CS and indicator of senescence-associated metabolic dysfunction. These aged neurons presented with morphological alteration including reduced neurotransmitter synthesis and dendritic complexity (Panossian et al. 2011). In two neuron-like cell lines (P12 and SH-SY5Y) exposure to the neurotoxin TCDD (2, 3, 7, 8-tetrachlorodibenzo-P-dioxin), which is known to cause mitochondrial dysfunction and accumulation of ROS, also induced a senescence-like phenotype including β-gal staining, increased expression of p16 and p21, reduced p-Rb expression and γ-H2AX foci. TCDD-induced neuronal senescence was dependent on oxidative stress and the senescent phenotype was attenuated by the ROS scavenger N-acetylcysteine (NAC) (Wan et al. 2014). In addition to stress-induced senescence, there is also evidence for CS in neuroblastoma cells as a result of altered gene expression. Knockdown of MECP2, a chromatin-modifying protein that mediates gene silencing increases β-gal staining in a neuroblastoma cell line (Squillaro et al. 2012). In neurons, MECP2 mediates the expression of immediate early gene expression, a set of genes important for synaptic plasticity and memory formation (Deng et al. 2014). This suggests that an increase in β-gal staining could be the result of individual gene expression or a response to an aberrant transcriptional network. While these data raise the possibility of a direct connection between MECP2 status, synaptic plasticity and CS, the experiments were performed in neuroblastoma cells, which are proliferative and thus fundamentally different from neurons.
While the concept of neuronal senescence is new, it is well established that neuronal function declines with advancing age. Disruption of normal calcium homeostasis, with increased intracellular resting levels of Ca2+ and an impaired ability to remove excess Ca2+ in response to glutamate stimulation is one extensively studied hallmark of aged neurons (Raza et al. 2007; Verkhratsky et al. 1994). The impaired calcium homeostasis may be the result of an age-related reduction in glutathione, a major antioxidant protein that was also found to contribute to the oxidative stress-dependent CS (Belrose et al. 2012). Impaired neuronal calcium homeostasis, neurotransmitter release and cognitive impairment is also likely exacerbated by glial dysfunction, as these support cells of the brain play a central role in maintaining homeostasis crucial for neuronal function including regulation of metabolites, neurotransmitter uptake and synaptic pruning.
Glial cells include astrocytes, oligodendrocytes and microglia. Microglia are macrophage-like myeloid cells, which serve as the primary innate immune cells of the CNS. As opposed to astrocytes and oligodendrocytes microglia are of non-neural tube origin (Ginhoux et al. 2010). Aging microglia cells show altered morphology distinct from the reactive microglia (Conde and Streit 2006b). This dystrophic microglia morphology is observed with an increasing frequency in older individuals and in relation to several neuropathologic conditions supporting the notion that dystrophy is age-related and followed by altered functionality of the cells (Conde and Streit 2006a; Streit 2006; Streit et al. 2004). Flanary et al. reported that microglia grown in- vitro were subject to replicative senescence as assessed by telomere shortening and decreased proliferation (Flanary and Streit 2004). Additional telomere and telomerase analysis studies in vivo support this hypothesis (Flanary et al. 2007; Miller et al. 2007; Miller and Streit 2007).
Astrocytes and oligodendrocytes originate from CNS stem cells and have an important role in regulating and supporting neuronal function (Rowitch and Kriegstein 2010). Neural stem cells (NSCs) themselves where shown to undergo senescence in response to extensive proliferation and stressors in vitro (Ferron et al. 2004). Interestingly, amyloid β-peptide (Aβ) which accumulates in the brain in AD and can be neurotoxic induced NSC senescence possibly through oxidative stress and/or the formylpeptide receptor 2 (FPR2) (He et al. 2013). In vivo NSC express several markers of CS with age including telomere shortening, up-regulation of cell cycle genes and ROS accumulation with age (Bose et al. 2010; Ferron et al. 2009).
Differentiated NSC to astrocytes and oligodendrocytes can also undergo CS. Differences exist between these two populations of glial cells with respect to replicative senescence. While astrocytes have been reported to senesce as a result of protracted proliferation (Bitto et al. 2010), oligodendrocyte precursor cells (OPCs) were observed to be resistant to RS in vitro (Tang et al. 2001). On the other hand, in OPCs both stress and serum-starvation were shown to induce CS mediated by the esophageal cancer-related gene 4 (Ecrg4). In addition, recombinant Ecrg4 was sufficient to induce CS in vitro, and the level of Ecrg4 was reported to increase with age in mice (Kujuro et al. 2010).
More studies have been performed on senescence in relation to astrocytes and suggest that astrocytes are subject to stress-induced CS in vitro, with clear similarities to the phenotype observed in senescent fibroblasts: SAHF, p53, p21, p16, SA β-gal activity and secretion of pro-inflammatory cytokines similar to SASP (Bhat et al. 2012; Bitto et al. 2010; Evans et al. 2003) (Fig. 2). Due to the prominent role of astrocytes in CNS homeostasis, CS of astrocytes will likely have implications for impaired maintenance of the BBB, regulation of CNS vasculature, neurotransmitter uptake and many other critical CNS functions that are known to decline with age (Sofroniew and Vinters 2010). Taken together, it appears that both neurons and glia are subject to stress-induced CS as a function of either accumulated DNA damage or oxidative stress, both of which increase during CNS aging and in neurodegenerative disease.
The concept of neuronal CS in response to accumulating stress is emerging (Fig. 1). Moreover, it seems that several phenotypes of aging neuronal cells are similar to those observed in proliferative cells (Fig. 2). It has also been suggested that neuronal senescence could result from stress due to proteopathies such as aggregates of Aβ or misfolded proteins such as α-synuclein (Golde and Miller 2009). Hence, further research is needed to clarify the matter and determine whether neuronal and glial senescence could be a result of distinct molecular mechanisms and if neuronal CS presents with a unique phenotype that may contribute to age-related decline and neurodegeneration.
Do hallmarks of cellular senescence occur in neurodegenerative disease?
Senescent cells are characterized by morphological abnormalities, altered gene and protein expression, changes in global methylation patterns and SAHF (Carnero 2013; Salama et al. 2014; van Deursen 2014), but do similar abnormalities occur in neurodegenerative disease? Evidence for telomere shortening, an established marker of cellular senescence, in the CNS of aged individuals and AD and PD patients is inconclusive (Eitan et al. 2014), which indicates that RS is unlikely to play an important role in the etiology of these diseases.
Interestingly, similar to senescent cells, increased expression of p16 and p21 were observed in the CNS during aging and in neurons of AD patient tissue (Luth et al. 2000; McShea et al. 1997) (Table 1). However, other cell cycle proteins like cyclin B, cyclin D and PCNA that are not necessarily related to CS are also elevated in neurons of patients with mild cognitive impairment and AD (Yang et al. 2003). It is therefore unknown if these alteration lead to CS or apoptosis and, in that regard, it is interesting that Aβ disrupts signaling by PAK3, a kinase downstream of p21 that mediates neuronal apoptosis and DNA synthesis (McPhie et al. 2003). Inhibition of PAK3 signaling results in disrupted dendritic morphology both in primary hippocampal neurons and in vivo in APPswe mice (Zhao et al. 2006). Curiously, in contrast to senescent fibroblasts neurons in AD may exhibit elevated levels of p16 and p21, and an increase in DNA synthesis. This has contributed to the idea that post-mitotic neurons aberrantly enter the cell cycle due to DNA damage, which results in an endpoint of programmed cell death (Kruman 2004; Kruman et al. 2004). Reduced DNA synthesis in senescent fibroblasts can be restored by blocking both p53 (Gire and Wynford-Thomas 1998) and p21 (Ma et al. 1999). It is possible that this divergence in DNA synthesis phenotype in AD neurons expressing classical senescence markers could be due to the unique post-mitotic environment of neurons, but more thorough in vitro and in vivo work will be required to answer this question.
Another feature of both age-related neurological disease and senescence is the expression of p38MAPK and chronic inflammatory signaling. Expression of the p38MAPK in fibroblasts was sufficient to induce SASP by up-regulating NF-κB at the transcriptional level (Freund et al. 2011). This induction of SASP may be independent of DDR and other senescence-inducing mechanisms because inhibition of p38MAPK is sufficient to mitigate the response (Freund et al. 2011). Activity of p38MAPK is increased in neurons of AD patients with neurofibrillary tangles and precedes Aβ plaques in mouse models of AD (Pei et al. 2001; Savage et al. 2002; Sun et al. 2003). Soluble APPα has also been found to activate microglia in a p38-dependent manner (Bodles and Barger 2005) and histopathological studies have suggested a role for dystrophic microglia in tau pathology (Streit et al. 2009), further supporting the notion of senescent cells being implicated in AD pathogenesis. Interestingly, p38MAPK hyper-phosphorylation or its inhibition blocks Aβ-induced inhibition of LTP (Li et al. 2003; Wang et al. 2004). It is important to note, however, that p38MAPK also plays a central role in immune activation (Cuenda and Rousseau 2007) that is unrelated to CS and it is therefore difficult to conclude how much of its observed activity in the CNS is due to CS.
Cytokines such as IL-6 that are characteristic of SASP are found elevated in AD and PD patient tissue and CSF (Bauer et al. 1991b; Blum-Degen et al. 1995; Huell et al. 1995; Wood et al. 1993). Similarly, elevation of IL-6 in the CNS is also observed in normal aging and as a response to chronic psychological stress, as seen for example in AD caregivers (Kiecolt-Glaser et al. 2003). Interestingly, expression of IL-6 in transgenic mice is sufficient to induce neurodegeneration (Campbell et al. 1993). At present it is not clear if the cytokines are produced due to CS or other aspects of disease etiology.
Interestingly, elevated levels of transforming growth factor β (TGFβ) mRNA have been observed in AD patient brain tissue (Luterman et al. 2000). TGFβ released by astrocytes increases neuronal expression of the complement protein c1q (Bialas and Stevens 2013), which can mediate synapse elimination (Stevens et al. 2007) and is known to increase with age in the CNS (Stephan et al. 2013). Hence, AD-related TGFβ signaling could be a central mechanism mediating synapse loss. TGFβ signaling has also been shown to induce senescence in vitro (Acosta et al. 2013; Cipriano et al. 2011; Senturk et al. 2010). Furthermore, transgenic mice with elevated astrocyte TGFβ signaling show accelerated Aβ plaque deposition (Mattson et al. 1997; Wyss-Coray et al. 1997), while in an AD mouse model in which TGFβ signaling was blocked, there was a reduction in Aβ pathology (Town et al. 2008). Taken together, these data suggest a direct role for TGFβ and other SASP-related cytokines in both synaptic loss and neurodegeneration.
In AD patient tissue compared to age-matched control subjects a global decrease in euchromatin was observed (Crapper et al. 1979), and another study employed electron microscopy to provide evidence of structural alterations in the nuclear envelope (Metuzals et al. 1988). A more recent study found that tau induces aberrant gene expression through ROS-mediated chromatin relaxation in both human AD patient tissue and in tau transgenic Drosophila and mice (Frost et al. 2014). Methylation patterns in AD patient brain tissue have been examined (see (Mattson 2003) for a review) but have focused mostly on methylation status of specific pathology-related genes rather than global methylation patterns. One study found a global decrease in methylation in entorhinal cortex tissue from AD patients, and a decrease in the methylation maintenance factors MBD2 and DNMT1 (Mastroeni et al. 2010). How this global decrease in methylation relates to the recent observation of senescence-induced methylation “mesas” (H3K4me3 and H3K27me3, e.g. methylation enriched regions) and “canyons” (H3K27me3, e.g. methylation-depleted regions) is unclear, and more rigorous global methylation studies need to be performed in tissue from relevant brain regions of patients with AD or other neurodegenerative disorders. It will also be important to establish changes that occur in brain cells during normal aging.
Lysosomal dysfunction in senescent cells results in two of the most utilized markers of senescence, namely, the accumulation of senescence-associated beta-galactosidase (SA β-gal) and lipofuscin (Carnero 2013). As noted above, increased β-gal activity was observed in rat hippocampus during aging (Geng et al. 2010). Interestingly, most of the 40 different lysosomal storage diseases frequently result in neurodegeneration (see (Settembre et al. 2008) for a review). Moreover, accumulation of autophagosomes has been observed in several neurodegenerative diseases including AD, PD, Huntington’s disease and amyotrophic lateral sclerosis (ALS) (Nixon 2013; Wong and Cuervo 2010). In AD the accumulation of autophagosomes appears to result from lysosomal dysfunction and rather than an increase in autophagy initiation (Lee et al. 2010). Ceramide has also been shown to increase in senescent cells as well as aging brain and brains of AD and ALS patients (Cutler and Mattson 2001; Cutler et al. 2002; Haughey et al. 2010). More work is needed to elucidate the connection between ceramide, autophagy, apoptosis and CS in brain cells.
Similar to the alterations observed in senescence, cellular energy metabolism is perturbed in AD and PD which manifests as altered mitochondrial function and increased ROS production ((Demetrius and Driver 2013) see (Mattson et al. 1999) for a review). Evidence from in vitro cybrid experiments in which native mitochondria are replaced with AD and PD patient mitochondria demonstrated enlarged mitochondria, altered calcium homeostasis and reduced mitochondrial membrane potential as measured by JC-1 when compared to cells populated with mitochondria from age-matched controls (Sheehan et al. 1997a, b; Trimmer et al. 2000). A more recent study examining levels of mitochondrial fission and fusion proteins found a reduction of mitochondrial fission proteins (Drp1, Opa1, Mfn1, Mfn2) and an increase in the fusion protein Fis1 (Wang et al. 2009). In PD diminished mitochondrial function and increased ROS clearly plays a role in pathology as familial PD mutations such as DJ-1, PINK1 and Parkin1 directly impact mitochondrial health and function, and environmentally-induced PD is typically due to toxins that act on mitochondria such as MPTP (see (Scarffe et al. 2014) for a review). It is possible that the altered mitochondrial function and morphology found in AD tissue and models could be a consequence of Aβ proteopathy (Mattson and Goodman 1995; Wang et al. 2009) or mutations in genes such as presenilin-1 (AD, (Keller et al. 1998)) or DJ-1 (PD, (Wang et al. 2012)) rather than a consequence of neuronal or glial senescence. While the etiology still needs further investigation, it is clear that brain cells in both AD and PD share similar mitochondrial phenotypes to peripheral senescent cells including altered mitochondrial morphology and enhanced ROS production (Fig. 3).

Evidence for senescence in neurodegenerative disease. Several hallmarks of cellular senescence are found to be increased in normal aging as well as in neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases
Conclusions
Hitherto, limited evidence exists on whether post-mitotic neurons can enter into a senescent state. The potential role of neuronal and glial senescence in neurodegenerative disease is similarly underexplored. However, evidence is beginning to emerge. In both AD and PD there is evidence for several CS hallmarks including: aberrant expression of cell cycle proteins (Luth et al. 2000), nuclear abnormalities (Metuzals et al. 1988), lysosomal and autophagic dysfunction (Nixon 2013), impaired mitochondrial function that leads to enhanced ROS generation (Mattson et al. 1999), and production of pro-inflammatory cytokines such as IL-6 (Wood et al. 1993). While it is still possible that this collection of senescent-like phenotypes found in CNS cell populations of aging, AD and PD brains could all be due to unique aspects of disease etiology, it is suggestive that CS does indeed take place in the CNS during normal aging as well as in age-related disease. Currently, it is not clear whether the observed senescent-like phenotypes occur in NSCs, glial cells, neurons, or all of these cell types. It seems likely that CS in the CNS is predominantly of the SISP type as even glia cells are not highly proliferative compared to peripheral cells that undergo RS. The potential stress origin of CNS CS may also explain the increase in hallmarks observed during neurodegenerative disease. The recent discovery, that senescence is not solely an age-related process but in addition a developmental mechanism for clearance of unnecessary cells and recruiting an immune response re-frames the process of CS (Munoz-Espin et al. 2013; Storer et al. 2013). In this regard it may be viewed as a signal for recruiting blood-borne immune cells ((Schwartz and Shechter 2010) for a review), which can exert either beneficial or devastating effects during neurodegenerative diseases. In addition, CNS senescence could be a downstream feature activated as a result of proteopathy-inducing genetic factors or environmental insults that occur with aging and not necessarily a causal force in neurodegeneration. Finally, the possibility that CS in brain aging and neurodegenerative disorders represents an adaptive response should be investigated.
In summary, CS is characterized by more than just cell cycle arrest serving as a tumor suppressor mechanism. Emerging evidence indicates that CS take place in the aging brain, and probably even in post-mitotic neurons. Current evidence indicates that at least several of the neuronal senescent phenotypes are similar to those observed in proliferative cells, while others, like calcium homeostasis may be neuron-specific. The contribution of CS to brain aging and age-related neurodegenerative disease is still not clear. Further investigation of senescence markers in the aging brain and in tissue samples from patients suffering from neurodegenerative diseases can elucidate the type of CS cells of the CNS undergo and the underlying mechanisms, and may help assess the contribution of senescence to disease etiology.
Future directions
Our review of the literature has revealed that only indirect evidence exists to suggest the possibility of CS in post-mitotic cells such as neurons. We feel that experiments addressing four critical questions will shed light on the contribution of CS to CNS aging and neurodegenerative disease:
Does inhibiting cellular senescence in the brain in either neuronal or glial populations attenuate age-related cognitive decline and progression of neurodegeneration?
Conversely, does artificially inducing senescence in either neuronal or glial populations produce enhanced cognitive decline and accelerated neurodegeneration?
Are the common phenotypes observed in both classical CS and neurodegenerative disease representative of a common mechanism of senescence or simply indicators of cellular stress and dysfunction?
What is the occurrence of neuronal senescence in vivo under normal cognitive aging and in neurological disease?
Recent developments have now enabled researchers to answer these critical questions. Progress in single cell analysis techniques now enables researchers to examine neuronal populations expressing SAβ-gal using techniques like single-cell PCR to determine similarities between neuronal senescence and senescence in mitotic populations. A more thorough understanding of the mechanisms involved in CNS senescence will be critical to understanding the contribution of CS to age-related neurodegenerative disease. Such work will contribute to understanding of age-related cognitive decline and could help in the development of novel therapeutic interventions in age-related neurodegenerative disease.
Abbreviations
- Aβ:
-
αmyloid β-peptide
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- ATM:
-
Ataxia telangiectasia mutated
- ATR:
-
Ataxia telangiectasia and Rad3-related
- β-gal:
-
Β-galactosidase
- CGNs:
-
Cerebellar granule neurons
- CNS:
-
Central nervous system
- CS:
-
Cellular senescence
- CSF:
-
Cerebrospinal fluid
- DDR:
-
DNA damage response
- Ecrg4:
-
Esophageal cancer-related gene 4
- FTD:
-
Frontotemporal dementia
- HD:
-
Huntington’s disease
- LTP:
-
Long-term potentiation
- MiRNA:
-
Microrna
- NSCs:
-
Neural stem cells
- OIS:
-
Oncogene-induced senescence
- OPCs:
-
Oligodendrocyte precursor cells
- PD:
-
Parkinson’s disease
- ROS:
-
Reactive oxygen species
- RS:
-
Replicative senescence
- SA β-gal:
-
Senescence-associated β-gal
- SAHF:
-
Senescence-associated heterochromatic foci
- SASP:
-
Senescence-associated secretory phenotype
- SIPS:
-
Stress induced replicative senescence
- SIR:
-
Senescence-associated inflammatory response
- TGFβ:
-
Transforming growth factor β
References
Abe M, Bonini NM (2013) MicroRNAs and neurodegeneration: role and impact. Trends Cell Biol 23:30–36
Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M et al (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol 15:978–990
Adams PD (2007) Remodeling chromatin for senescence. Aging cell 6:425–427
Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC (1996) Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA 93:13742–13747
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479:232–236
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120–124
Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Yolk B, Berger M (1991a) Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett 285:111–114
Bauer J, Strauss S, Volk B, Berger M (1991b) IL-6-mediated events in Alzheimer’s disease pathology. Immunol Today 12:422
Belrose JC, Xie YF, Gierszewski LJ, MacDonald JF, Jackson MF (2012) Loss of glutathione homeostasis associated with neuronal senescence facilitates TRPM2 channel activation in cultured hippocampal pyramidal neurons. Mol brain 5:11
Berciano MT, Andres MA, Calle E, Lafarga M (1995) Age-induced hypertrophy of astrocytes in rat supraoptic nucleus: a cytological, morphometric, and immunocytochemical study. Anat Record 243:129–144
Bhanu MU, Mandraju RK, Bhaskar C, Kondapi AK (2010) Cultured cerebellar granule neurons as an in vitro aging model: topoisomerase IIbeta as an additional biomarker in DNA repair and aging. Toxicol Vitro 24:1935–1945
Bhat R, Crowe EP, Bitto A, Moh M, Katsetos CD, Garcia FU, Johnson FB, Trojanowski JQ, Sell C, Torres C (2012) Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 7:e45069
Bialas AR, Stevens B (2013) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16:1773–1782
Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S, Torres C (2010) Stress-induced senescence in human and rodent astrocytes. Exp Cell Res 316:2961–2968
Blagosklonny MV (2011) Cell cycle arrest is not senescence. Aging 3:94–101
Blagosklonny MV, Hall MN (2009) Growth and aging: a common molecular mechanism. Aging 1:357–362
Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P (1995) Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett 202:17–20
Bodles AM, Barger SW (2005) Secreted beta-amyloid precursor protein activates microglia via JNK and p38-MAPK. Neurobiol Aging 26:9–16
Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE (1998) Extension of life-span by introduction of telomerase into normal human cells. Science 279:349–352
Boehm M, Slack F (2005) A developmental timing microRNA and its target regulate life span in C. elegans. Science 310:1954–1957
Bonifati V (2014) Genetics of Parkinson’s disease–state of the art, 2013. Parkinsonism Relat Disord 20(Suppl 1):S23–S28
Bose R, Moors M, Tofighi R, Cascante A, Hermanson O, Ceccatelli S (2010) Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis 1:e92
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211
Bree RT, Stenson-Cox C, Grealy M, Byrnes L, Gorman AM, Samali A (2002) Cellular longevity: role of apoptosis and replicative senescence. Biogerontology 3:195–206
Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L (1993) Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci USA 90:10061–10065
Carnero A (2013) Markers of cellular senescence. Methods Mol Biol 965:63–81
Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T et al (2012) Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 47:203–214
Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN (1995) Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci USA 92:4337–4341
Cherif H, Tarry JL, Ozanne SE, Hales CN (2003) Ageing and telomeres: a study into organ- and gender-specific telomere shortening. Nucleic Acids Res 31:1576–1583
Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–398
Cipriano R, Kan CE, Graham J, Danielpour D, Stampfer M, Jackson MW (2011) TGF-beta signaling engages an ATM-CHK2-p53-independent RAS-induced senescence and prevents malignant transformation in human mammary epithelial cells. Proc Natl Acad Sci USA 108:8668–8673
Conde JR, Streit WJ (2006a) Effect of aging on the microglial response to peripheral nerve injury. Neurobiol Aging 27:1451–1461
Conde JR, Streit WJ (2006b) Microglia in the aging brain. J Neuropathol Exp Neurol 65:199–203
Coppe JP, Desprez PY, Krtolica A, Campisi J (2010a) The senescence-associated secretory phenotype: the dark side of tumor suppression. Ann Rev Pathol 5:99–118
Coppe JP, Patil CK, Rodier F, Krtolica A, Beausejour CM, Parrinello S, Hodgson JG, Chin K, Desprez PY, Campisi J (2010b) A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 5:e9188
Crapper DR, Quittkat S, de Boni U (1979) Altered chromatin conformation in Alzheimer’s disease. Brain 102:483–495
Cuenda A, Rousseau S (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773:1358–1375
Cutler RG, Mattson MP (2001) Sphingomyelin and ceramide as regulators of development and lifespan. Mech Ageing Dev 122:895–908
Cutler RG, Pedersen WA, Camandola S, Rothstein JD, Mattson MP (2002) Evidence that accumulation of ceramides and cholesterol esters mediates oxidative stress-induced death of motor neurons in amyotrophic lateral sclerosis. Annals Neurol 52:448–457
De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM (2013) Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 12:247–256
Demetrius LA, Driver J (2013) Alzheimer’s as a metabolic disease. Biogerontology 14:641–649
Deng JV, Wan Y, Wang X, Cohen S, Wetsel WC, Greenberg ME, Kenny PJ, Calakos N, West AE (2014) MeCP2 phosphorylation limits psychostimulant-induced behavioral and neuronal plasticity. J Neurosci 34:4519–4527
Di Micco R, Cicalese A, Fumagalli M, Dobreva M, Verrecchia A, Pelicci PG, di Fagagna F (2008) DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle 7:3601–3606
Dimri GP, Hara E, Campisi J (1994) Regulation of two E2F-related genes in presenescent and senescent human fibroblasts. J Biol Chem 269:16180–16186
Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O et al (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92:9363–9367
Dong W, Cheng S, Huang F, Fan W, Chen Y, Shi H, He H (2011) Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med Sci Monit 17:91–96
Eitan E, Hutchison ER, Mattson MP (2014) Telomere shortening in neurological disorders: an abundance of unanswered questions. Trends Neurosci 37:256–263
ElSharawy A, Keller A, Flachsbart F, Wendschlag A, Jacobs G, Kefer N, Brefort T, Leidinger P, Backes C, Meese E et al (2012) Genome-wide miRNA signatures of human longevity. Aging Cell 11:607–616
Evans RJ, Wyllie FS, Wynford-Thomas D, Kipling D, Jones CJ (2003) A P53-dependent, telomere-independent proliferative life span barrier in human astrocytes consistent with the molecular genetics of glioma development. Cancer Res 63:4854–4861
Ferron S, Mira H, Franco S, Cano-Jaimez M, Bellmunt E, Ramirez C, Farinas I, Blasco MA (2004) Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 131:4059–4070
Ferron SR, Marques-Torrejon MA, Mira H, Flores I, Taylor K, Blasco MA, Farinas I (2009) Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. J Neurosci 29:14394–14407
Flanary BE, Streit WJ (2004) Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 45:75–88
Flanary BE, Sammons NW, Nguyen C, Walker D, Streit WJ (2007) Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 10:61–74
Freund A, Patil CK, Campisi J (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30:1536–1548
Freund A, Laberge RM, Demaria M, Campisi J (2012) Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 23:2066–2075
Frost B, Hemberg M, Lewis J, Feany MB (2014) Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci 17:357–366
Geng YQ, Guan JT, Xu XH, Fu YC (2010) Senescence-associated beta-galactosidase activity expression in aging hippocampal neurons. Biochem Biophys Res Commun 396:866–869
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Gire V, Wynford-Thomas D (1998) Reinitiation of DNA synthesis and cell division in senescent human fibroblasts by microinjection of anti-p53 antibodies. Mol Cell Biol 18:1611–1621
Golde TE, Miller VM (2009) Proteinopathy-induced neuronal senescence: a hypothesis for brain failure in Alzheimer’s and other neurodegenerative diseases. Alzheimers Res Ther 1:5
Grune T, Merker K, Jung T, Sitte N, Davies KJ (2005) Protein oxidation and degradation during postmitotic senescence. Free Radic Biol Med 39:1208–1215
Harley CB, Futcher AB, Greider CW (1990) Telomeres shorten during ageing of human fibroblasts. Nature 345:458–460
Haughey NJ, Bandaru VV, Bae M, Mattson MP (2010) Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim Biophys Acta 1801:878–886
Hayflick L, Moorhead PS (1961) The serial cultivation of human diploid cell strains. Exp Cell Res 25:585–621
He N, Jin WL, Lok KH, Wang Y, Yin M, Wang ZJ (2013) Amyloid-beta(1-42) oligomer accelerates senescence in adult hippocampal neural stem/progenitor cells via formylpeptide receptor 2. Cell Death Dis 4:e924
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M et al (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21:3017–3023
Hohn A, Sittig A, Jung T, Grimm S, Grune T (2012) Lipofuscin is formed independently of macroautophagy and lysosomal activity in stress-induced prematurely senescent human fibroblasts. Free Radic Biol Med 53:1760–1769
Hu Z, Klein JD, Mitch WE, Zhang L, Martinez I, Wang XH (2014) MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging 6:160–175
Huell M, Strauss S, Volk B, Berger M, Bauer J (1995) Interleukin-6 is present in early stages of plaque formation and is restricted to the brains of Alzheimer’s disease patients. Acta Neuropathol 89:544–551
Iwasa H, Han J, Ishikawa F (2003) Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes cells 8:131–144
Jordan-Sciutto KL, Dorsey R, Chalovich EM, Hammond RR, Achim CL (2003) Expression patterns of retinoblastoma protein in Parkinson disease. J Neuropathol Exp Neurol 62:68–74
Jurk D, Wang C, Miwa S, Maddick M, Korolchuk V, Tsolou A, Gonos ES, Thrasivoulou C, Saffrey MJ, Cameron K et al (2012) Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11:996–1004
Kang HT, Lee KB, Kim SY, Choi HR, Park SC (2011) Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS ONE 6:e23367
Keller JN, Guo Q, Holtsberg FW, Bruce-Keller AJ, Mattson MP (1998) Increased sensitivity to mitochondrial toxin-induced apoptosis in neural cells expressing mutant presenilin-1 is linked to perturbed calcium homeostasis and enhanced oxyradical production. J Neurosci 18:4439–4450
Kiecolt-Glaser JK, Preacher KJ, MacCallum RC, Atkinson C, Malarkey WB, Glaser R (2003) Chronic stress and age-related increases in the proinflammatory cytokine IL-6. Proc Natl Acad Sci USA 100:9090–9095
Kim HC, Bing G, Jhoo WK, Kim WK, Shin EJ, Park ES, Choi YS, Lee DW, Shin CY, Ryu JR et al (2002) Oxidative damage causes formation of lipofuscin-like substances in the hippocampus of the senescence-accelerated mouse after kainate treatment. Behav Brain Res 131:211–220
Kosar M, Bartkova J, Hubackova S, Hodny Z, Lukas J, Bartek J (2011) Senescence-associated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16(ink4a). Cell Cycle 10:457–468
Kruman II (2004) Why do neurons enter the cell cycle? Cell Cycle 3:769–773
Kruman II, Wersto RP, Cardozo-Pelaez F, Smilenov L, Chan SL, Chrest FJ, Emokpae R Jr, Gorospe M, Mattson MP (2004) Cell cycle activation linked to neuronal cell death initiated by DNA damage. Neuron 41:549–561
Kuilman T, Michaloglou C, Mooi WJ, Peeper DS (2010) The essence of senescence. Genes Dev 24:2463–2479
Kujuro Y, Suzuki N, Kondo T (2010) Esophageal cancer-related gene 4 is a secreted inducer of cell senescence expressed by aged CNS precursor cells. Proc Natl Acad Sci USA 107:8259–8264
Kulju KS, Lehman JM (1995) Increased p53 protein associated with aging in human diploid fibroblasts. Exp Cell Res 217:336–345
Larsson LG (2011) Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin Cancer Biol 21:367–376
Ledda M, Barni L, Altieri L, Pannese E (2000) Decrease in the nucleo-cytoplasmic volume ratio of rabbit spinal ganglion neurons with age. Neurosci Lett 286:171–174
Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES (2006) Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5:187–195
Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, Youle RJ, Cho H (2007) Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem 282:22977–22983
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G et al (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141:1146–1158
Lesuisse C, Martin LJ (2002) Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J Neurobiol 51:9–23
Li Y, Liu L, Barger SW, Griffin WS (2003) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23:1605–1611
Li G, Luna C, Qiu J, Epstein DL, Gonzalez P (2009) Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev 130:731–741
Li W, Prazak L, Chatterjee N, Gruninger S, Krug L, Theodorou D, Dubnau J (2013) Activation of transposable elements during aging and neuronal decline in Drosophila. Nat Neurosci 16:529–531
Lin DT, Wu J, Holstein D, Upadhyay G, Rourk W, Muller E, Lechleiter JD (2007) Ca2 + signaling, mitochondria and sensitivity to oxidative stress in aging astrocytes. Neurobiol Aging 28:99–111
Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR et al (2010) Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci USA 107:14164–14169
Liu N, Landreh M, Cao K, Abe M, Hendriks GJ, Kennerdell JR, Zhu Y, Wang LS, Bonini NM (2012) The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482:519–523
Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429:883–891
Luterman JD, Haroutunian V, Yemul S, Ho L, Purohit D, Aisen PS, Mohs R, Pasinetti GM (2000) Cytokine gene expression as a function of the clinical progression of Alzheimer disease dementia. Arch Neurol 57:1153–1160
Luth HJ, Holzer M, Gertz HJ, Arendt T (2000) Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21ras and p16INK4a. Brain Res 852:45–55
Ma Y, Prigent SA, Born TL, Monell CR, Feramisco JR, Bertolaet BL (1999) Microinjection of anti-p21 antibodies induces senescent Hs68 human fibroblasts to synthesize DNA but not to divide. Cancer Res 59:5341–5348
Maes OC, Sarojini H, Wang E (2009) Stepwise up-regulation of microRNA expression levels from replicating to reversible and irreversible growth arrest states in WI-38 human fibroblasts. J Cell Physiol 221:109–119
Mai S, Klinkenberg M, Auburger G, Bereiter-Hahn J, Jendrach M (2010) Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J Cell Sci 123:917–926
Majore I, Moretti P, Hass R, Kasper C (2009) Identification of subpopulations in mesenchymal stem cell-like cultures from human umbilical cord. Cell Commun Signal 7:6
Martinelli C, Sartori P, Ledda M, Pannese E (2006) A study of mitochondria in spinal ganglion neurons during life: quantitative changes from youth to extremely advanced age. Tissue Cell 38:93–98
Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J (2010) Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging 31:2025–2037
Mattson MP (2003) Methylation and acetylation in nervous system development and neurodegenerative disorders. Ageing Res Rev 2:329–342
Mattson MP, Goodman Y (1995) Different amyloidogenic peptides share a similar mechanism of neurotoxicity involving reactive oxygen species and calcium. Brain Res 676:219–224
Mattson MP, Barger SW, Furukawa K, Bruce AJ, Wyss-Coray T, Mark RJ, Mucke L (1997) Cellular signaling roles of TGF beta, TNF alpha and beta APP in brain injury responses and Alzheimer’s disease. Brain Res Brain Res Rev 23:47–61
Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S (1999) Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer’s and Parkinson’s diseases. Ann NY Acad Sci 893:154–175
McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, Kozlowski MR, Neve KA, Neve RL (2003) DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J Neurosci 23:6914–6927
McShea A, Harris PL, Webster KR, Wahl AF, Smith MA (1997) Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer’s disease. Am J Pathol 150:1933–1939
Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF (1993) Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol 34:609–616
Medrano EE, Im S, Yang F, Abdel-Malek ZA (1995) Ultraviolet B light induces G1 arrest in human melanocytes by prolonged inhibition of retinoblastoma protein phosphorylation associated with long-term expression of the p21Waf-1/SDI-1/Cip-1 protein. Cancer Res 55:4047–4052
Metuzals J, Robitaille Y, Houghton S, Gauthier S, Leblanc R (1988) Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s disease. J Neurocytol 17:827–833
Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL, Barrett JC et al (2008) SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452:492–496
Miller KR, Streit WJ (2007) The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol 3:245–253
Miller BA, Crum JM, Tovar CA, Ferguson AR, Bresnahan JC, Beattie MS (2007) Developmental stage of oligodendrocytes determines their response to activated microglia in vitro. J Neuroinflamm 4:28
Mori MA, Raghavan P, Thomou T, Boucher J, Robida-Stubbs S, Macotela Y, Russell SJ, Kirkland JL, Blackwell TK, Kahn CR (2012) Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab 16:336–347
Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S, Rodriguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155:1104–1118
Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113:703–716
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19:983–997
Noda A, Ning Y, Venable SF, Pereira-Smith OM, Smith JR (1994) Cloning of senescent cell-derived inhibitors of DNA synthesis using an expression screen. Exp Cell Res 211:90–98
Oenzil F, Kishikawa M, Mizuno T, Nakano M (1994) Age-related accumulation of lipofuscin in three different regions of rat brain. Mech Ageing Dev 76:157–163
Panossian L, Fenik P, Zhu Y, Zhan G, McBurney MW, Veasey S (2011) SIRT1 regulation of wakefulness and senescence-like phenotype in wake neurons. J Neurosci 31:4025–4036
Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF (2001) Localization of active forms of C-jun kinase (JNK) and p38 kinase in Alzheimer’s disease brains at different stages of neurofibrillary degeneration. J Alzheimer’s Dis 3:41–48
Peinado MA, Martinez M, Pedrosa JA, Quesada A, Peinado JM (1993) Quantitative morphological changes in neurons and glia in the frontal lobe of the aging rat. Anatom Record 237:104–108
Pertusa M, Garcia-Matas S, Rodriguez-Farre E, Sanfeliu C, Cristofol R (2007) Astrocytes aged in vitro show a decreased neuroprotective capacity. J Neurochem 101:794–805
Peters A, Moss MB, Sethares C (2000) Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J Comp Neurol 419:364–376
Pribluda A, Elyada E, Wiener Z, Hamza H, Goldstein RE, Biton M, Burstain I, Morgenstern Y, Brachya G, Billauer H et al (2013) A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell 24:242–256
Rafols JA, Cheng HW, McNeill TH (1989) Golgi study of the mouse striatum: age-related dendritic changes in different neuronal populations. J Comp Neurol 279:212–227
Ramirez RD, Morales CP, Herbert BS, Rohde JM, Passons C, Shay JW, Wright WE (2001) Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev 15:398–403
Ranganathan S, Scudiere S, Bowser R (2001) Hyperphosphorylation of the retinoblastoma gene product and altered subcellular distribution of E2F-1 during Alzheimer’s disease and amyotrophic lateral sclerosis. J Alzheimer’s Dis 3:377–385
Raza M, Deshpande LS, Blair RE, Carter DS, Sombati S, DeLorenzo RJ (2007) Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci Lett 418:77–81
Ren G, Feng J, Datar I, Yeung AH, Saladi SV, Feng Y, de la Serna I, Yeung KC (2012) A Micro-RNA connection in BRaf(V600E)-mediated premature senescence of human melanocytes. Int J Cell Biol 2012:913242
Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11:973–979
Rodrigues HF, Souza TA, Ghiraldini FG, Mello ML, Moraes AS (2014) Increased age is associated with epigenetic and structural changes in chromatin from neuronal nuclei. J Cell Biochem 115:659–665
Rowitch DH, Kriegstein AR (2010) Developmental genetics of vertebrate glial-cell specification. Nature 468:214–222
Salama R, Sadaie M, Hoare M, Narita M (2014) Cellular senescence and its effector programs. Genes Dev 28:99–114
Santarosa M, Del Col L, Tonin E, Caragnano A, Viel A, Maestro R (2009) Premature senescence is a major response to DNA cross-linking agents in BRCA1-defective cells: implication for tailored treatments of BRCA1 mutation carriers. Mol Cancer Ther 8:844–854
Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW (2002) Activation of c-Jun N-terminal kinase and p38 in an Alzheimer’s disease model is associated with amyloid deposition. J Neurosci 22:3376–3385
Scarffe LA, Stevens DA, Dawson VL, Dawson TM (2014) Parkin and PINK1: much more than mitophagy. Trends Neurosci 37:315–324
Schwartz M, Shechter R (2010) Systemic inflammatory cells fight off neurodegenerative disease. Nature Rev Neurol 6:405–410
Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC (2004) Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6:168–170
Senturk S, Mumcuoglu M, Gursoy-Yuzugullu O, Cingoz B, Akcali KC, Ozturk M (2010) Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology 52:966–974
Serna E, Gambini J, Borras C, Abdelaziz KM, Belenguer A, Sanchis P, Avellana JA, Rodriguez-Manas L, Vina J (2012) Centenarians, but not octogenarians, up-regulate the expression of microRNAs. Sci Rep 2:961
Settembre C, Fraldi A, Rubinsztein DC, Ballabio A (2008) Lysosomal storage diseases as disorders of autophagy. Autophagy 4:113–114
Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T et al (2013) Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27:1787–1799
Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB (1997a) Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci 17:4612–4622
Sheehan JP, Swerdlow RH, Parker WD, Miller SW, Davis RE, Tuttle JB (1997b) Altered calcium homeostasis in cells transformed by mitochondria from individuals with Parkinson’s disease. J Neurochem 68:1221–1233
Sherr CJ, DePinho RA (2000) Cellular senescence: mitotic clock or culture shock? Cell 102:407–410
Shimi T, Butin-Israeli V, Adam SA, Hamanaka RB, Goldman AE, Lucas CA, Shumaker DK, Kosak ST, Chandel NS, Goldman RD (2011) The role of nuclear lamin B1 in cell proliferation and senescence. Genes Dev 25:2579–2593
Singh M, Piekorz RP (2013) Senescence-associated lysosomal alpha-L-fucosidase (SA-alpha-Fuc): a sensitive and more robust biomarker for cellular senescence beyond SA-beta-Gal. Cell Cycle 12:1996
Sitte N, Merker K, Grune T, von Zglinicki T (2001) Lipofuscin accumulation in proliferating fibroblasts in vitro: an indicator of oxidative stress. Exp Gerontol 36:475–486
Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119:7–35
Squillaro T, Alessio N, Cipollaro M, Melone MA, Hayek G, Renieri A, Giordano A, Galderisi U (2012) Reduced expression of MECP2 affects cell commitment and maintenance in neurons by triggering senescence: new perspective for Rett syndrome. Mol Biol Cell 23:1435–1445
Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH et al (2013) A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33:13460–13474
Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131:1164–1178
Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155:1119–1130
Streit WJ (2006) Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci 29:506–510
Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45:208–212
Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol 118:475–485
Sun A, Liu M, Nguyen XV, Bing G (2003) P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol 183:394–405
Tacutu R, Budovsky A, Fraifeld VE (2010) The NetAge database: a compendium of networks for longevity, age-related diseases and associated processes. Biogerontology 11:513–522
Tacutu R, Budovsky A, Yanai H, Fraifeld VE (2011) Molecular links between cellular senescence, longevity and age-related diseases - a systems biology perspective. Aging 3:1178–1191
Tang DG, Tokumoto YM, Apperly JA, Lloyd AC, Raff MC (2001) Lack of replicative senescence in cultured rat oligodendrocyte precursor cells. Science 291:868–871
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL (2013) Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Investig 123:966–972
Tiribuzi R, Orlacchio A, Crispoltoni L, Maiotti M, Zampolini M, De Angeliz M, Mecocci P, Cecchetti R, Bernardi G, Datti A et al (2011) Lysosomal beta-galactosidase and beta-hexosaminidase activities correlate with clinical stages of dementia associated with Alzheimer’s disease and type 2 diabetes mellitus. J Alzheimer’s Dis 24:785–797
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA (2008) Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med 14:681–687
Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP Jr, Miller SW, Davis RE, Parker WD Jr (2000) Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol 162:37–50
van Deursen JM (2014) The role of senescent cells in ageing. Nature 509:439–446
van Dijk KD, Persichetti E, Chiasserini D, Eusebi P, Beccari T, Calabresi P, Berendse HW, Parnetti L, van de Berg WD (2013) Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov Disord 28:747–754
Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM (1995) Role of ceramide in cellular senescence. J Biol Chem 270:30701–30708
Verkhratsky A, Shmigol A, Kirischuk S, Pronchuk N, Kostyuk P (1994) Age-dependent changes in calcium currents and calcium homeostasis in mammalian neurons. Ann NY Acad Sci 747:365–381
Vogt M, Haggblom C, Yeargin J, Christiansen-Weber T, Haas M (1998) Independent induction of senescence by p16INK4a and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ 9:139–146
Wan C, Liu J, Nie X, Zhao J, Zhou S, Duan Z, Tang C, Liang L, Xu G (2014) 2, 3, 7, 8-Tetrachlorodibenzo-P-dioxin (TCDD) induces premature senescence in human and rodent neuronal cells via ROS-dependent mechanisms. PLoS ONE 9:e89811
Wang E, Gundersen D (1984) Increased organization of cytoskeleton accompanying the aging of human fibroblasts in vitro. Exp Cell Res 154:191–202
Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R (2004) Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci 24:3370–3378
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurochem 29:9090–9103
Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X (2012) Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem 121:830–839
Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13:805–811
Wood JA, Wood PL, Ryan R, Graff-Radford NR, Pilapil C, Robitaille Y, Quirion R (1993) Cytokine indices in Alzheimer’s temporal cortex: no changes in mature IL-1 beta or IL-1RA but increases in the associated acute phase proteins IL-6, alpha 2-macroglobulin and C-reactive protein. Brain Res 629:245–252
Wyss-Coray T, Masliah E, Mallory M, McConlogue L, Johnson-Wood K, Lin C, Mucke L (1997) Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer’s disease. Nature 389:603–606
Yamada T, Sawada R, Tsuchiya T (2008) The effect of sulfated hyaluronan on the morphological transformation and activity of cultured human astrocytes. Biomaterials 29:3503–3513
Yang AJ, Chandswangbhuvana D, Margol L, Glabe CG (1998) Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res 52:691–698
Yang Y, Mufson EJ, Herrup K (2003) Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer’s disease. J Neurochem 23:2557–2563
Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, Malka F, Jou MJ, Martinou JC, Yoon G (2006) Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol 209:468–480
Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM et al (2009) Autophagy mediates the mitotic senescence transition. Genes Dev 23:798–803
Yu WH, Go L, Guinn BA, Fraser PE, Westaway D, McLaurin J (2002) Phenotypic and functional changes in glial cells as a function of age. Neurobiol Aging 23:105–115
Zhang R, Chen W, Adams PD (2007) Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 27:2343–2358
Zhang W, Ji W, Yang J, Yang L, Chen W, Zhuang Z (2008) Comparison of global DNA methylation profiles in replicative versus premature senescence. Life Sci 83:475–480
Zhao L, Ma QL, Calon F, Harris-White ME, Yang F, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE et al (2006) Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nat Neurosci 9:234–242
Zwerschke W, Mazurek S, Stockl P, Hutter E, Eigenbrodt E, Jansen-Durr P (2003) Metabolic analysis of senescent human fibroblasts reveals a role for AMP in cellular senescence. Biochem J 376:403–411
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Tan, F.C.C., Hutchison, E.R., Eitan, E. et al. Are there roles for brain cell senescence in aging and neurodegenerative disorders?. Biogerontology 15, 643–660 (2014). https://doi.org/10.1007/s10522-014-9532-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-014-9532-1