
Abstract
This chapter summarizes concepts regarding the effects of hypertension on the cerebral circulation. In relation to end-organ effects, the brain is one of the organs most significantly affected by chronic elevations in arterial pressure. Hypertension is a leading risk factor for cerebrovascular disease, stroke, and cognitive decline. Endothelial cells are a target of hypertension. These cells are normally key determinants of vascular tone, while also protecting against thrombosis and abnormal vascular growth. The impact of acute and chronic hypertension is outlined with emphasis on changes in the biology of the signaling molecule nitric oxide and the role that oxidative stress plays in these changes. The importance of NADPH oxidase and other mechanisms that promote oxidative stress are presented. As part of the discussion, there is an overview regarding vascular alterations in various genetic and pharmacological models of hypertension. Effects of the renin-angiotensin system and angiotensin-II are highlighted. Endothelial cells are also the site of the blood–brain barrier (BBB). The molecular organization of the BBB is summarized along with key changes that occur in response to acute and chronic hypertension. Lastly, the clinical impact of these changes is discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Endothelial function
- Blood–brain barrier
- Cerebral blood flow
- Oxidative stress
- EDCF
- Acute hypertension
- Chronic hypertension
1 Introduction
Hypertension is a common disease that impacts the entire vasculature. It is estimated that approximately 30 % of the adult population (over 77 million people) in the United States are hypertensive and this number is expected to rise significantly over the coming years [1, 2]. Based on current guidelines, a person is diagnosed as hypertensive if he or she has: (1) a systolic blood pressure greater than 140 mmHg and/or diastolic blood pressure greater than 90 mmHg, (2) are currently being treated for hypertension, (3) or have been told by at least two physicians or other healthcare professionals that they have hypertension [1, 2]. The most recent Global Burden of Disease study reported that hypertension is currently the leading risk factor for mortality and disability adjusted life years worldwide [3]. The economic cost of hypertension (both direct and indirect) in the United States was estimated to be $51 billion in 2009 and this is expected to increase to approximately $343 billion by 2030 [1]. Such data highlight the individual and economic toll hypertension has worldwide and the need for a greater understanding of the disease. Because hypertension is controlled suboptimally in many patients, there is also the continued need for improved therapeutic approaches to limit its frequency as well as protecting against end-organ damage.
In relation to end-organ effects of hypertension, the brain is among the most significantly affected organs. Hypertension has profound effects on the cerebral circulation and the brain and is a major risk factor for stroke and dementia, including vascular cognitive impairment and Alzheimer’s disease [4–6]. Within the cerebral circulation, hypertension produces diverse effects including oxidative stress, low-grade inflammation, cerebrovascular remodeling, increased vascular permeability, and impairment of key mechanisms that regulate local cerebral perfusion (e.g., endothelium-dependent vasodilation and neurovascular coupling) [7–10]. In this chapter, we review concepts related to these topics with an emphasis on effects of hypertension on cerebrovascular endothelial function and blood–brain barrier (BBB) integrity and regulation.
2 Endothelial Function
Endothelium plays a critical role in the biology and regulation of blood vessels. These roles include modulating the function of other cells within the vessel wall as well as non-vascular cells (e.g., neurons and astrocytes). Endothelial cells regulate recruitment of leukocytes to tissues by expression of adhesion molecules and cytokine release, and therefore play an important role in inflammation [11]. Polymorphisms in the cytokines IL-6 and IL-10 are associated with endothelial dysfunction and peripheral arterial disease [12]. The endothelial cell is a major cellular site in relation to regulation of coagulation, such that endothelial dysfunction promotes thrombosis [13], thus contributing to ischemic events in brain. Endothelial cells are also a key determinant of vascular permeability and diffusion of substances out of the vascular compartment into the tissues. Tight junction proteins between endothelial cells are a key component of the BBB [14]. Thus, the endothelium is a complex organ with multiple functions that are essential to maintain cardiovascular homeostasis.
One of the most studied roles of endothelial cells involves the regulation of vascular tone via their influence on vascular muscle. Studying vasodilation in response to mechanical (flow-induced) or pharmacological (e.g., acetylcholine) stimuli are very common experimental approaches used to gain insight into endothelial cell function in both experimental models and people [15–17]. In addition to providing insight into regulation of vascular tone, endothelial dysfunction is predictive of clinical events including stroke [11, 18].
Within the cerebral circulation, endothelium-induced vasodilation is mediated largely by the actions of the endothelium-derived relaxing factor (EDRF) nitric oxide (NO) (Fig. 8.1) [17]. In the 1980s a series of studies identified NO as a key substance released by endothelium that then elicits vasodilation. Subsequently, endothelial- and NO-mediated vasodilation has been widely studied in health and disease in many vascular beds. In addition to NO, endothelial cells release other vasoactive molecules that also regulate the tone of underlying vascular muscle. These molecules include vasoconstrictor substances such as endothelin-1 [19] and endothelium-derived contracting factor(s) (EDCF) [20, 21]. Other vasodilator molecules including endothelium-derived hyperpolarizing factors (EDHF) have also been studied [11, 22]. The molecular identity of EDHF is controversial, in part because there appears to be a family of factors that fulfill this role physiologically, including potassium ion and hydrogen peroxide [11, 22, 23]. In addition, in some portions of the vasculature, endothelium-dependent hyperpolarization may be mediated by the spread of hyperpolarization from endothelial cells to vascular muscle, and not the extracellular release of a specific molecule (Fig. 8.1). In summary, endothelium plays a critical role in regulating vasomotor tone by production and release of multiple vasoactive molecules and/or other mechanisms.

Schematic illustration of selected endothelium-dependent effects that are present under normal conditions. Endothelial NO synthase (eNOS) produces NO under basal conditions, and in response to a variety of receptor-mediated agonists or increased shear stress. eNOS-derived NO produces relaxation of vascular muscle by activating soluble guanylate cyclase (sGC) causing production of cGMP from GTP. In addition to effects on vascular tone, NO inhibits thrombosis via inhibitory effects on platelets and other mechanisms. NO also inhibits vascular inflammation via effects on nuclear factor κB, endothelial adhesion molecules, etc. Cyclooxygenase (COX) produces PGH2 which can then be converted into prostacyclin (PGI2) by prostacyclin synthase or thromboxane A2 by thromboxane synthase. PGI2 also inhibits aggregation of platelets thus suppressing thrombosis. COX also produces superoxide (O2 −) during the metabolism of arachidonic acid. Superoxide is converted into H2O2 by superoxide dismutase (SOD). H2O2 can function as an endothelium-derived relaxing factor (EDHF) producing relaxation of underlying vascular muscle via effects on K+ channels. Some studies suggest that K+ may also function as an EDHF. Lastly, hyperpolarization that is initiated within endothelial cells (endothelium-dependent hyperpolarization, EDH) may spread to vascular muscle via myoendothelial gap junctions without the release of an EDHF. ADMA asymmetric dimethylarginine, BH 4 tetrahydrobiopterin, Cav-1 caveolin-1
3 NO Biology
It is well established that endothelium-derived vasoactive factors are crucial regulators of cerebral vascular tone [17]. Once released from the endothelium, these factors cross the subendothelial space and ultimately cause vasodilation or vasoconstriction. Since its discovery as an EDRF in the aorta, NO has been extensively studied and it has become apparent that NO is central to the regulation of vascular tone in many vascular beds including large and small vessels in both animal models and people.
NO is generated by NO synthase (NOS) enzymes. Three isoforms of NOS have been identified, namely inducible NOS (iNOS or NOS1), neuronal NOS (nNOS or NOS2), and endothelial NOS (eNOS or NOS3). iNOS is an inducible, Ca2+-independent enzyme which generates large amounts of NO upon activation [24, 25]. In contrast, eNOS and nNOS generate relatively smaller amounts of NO and their activity is Ca2+-dependent [24, 25]. Both eNOS and nNOS are constitutively expressed in blood vessels in endothelium and perivascular neurons (respectively), whereas iNOS is not expressed under normal conditions in vascular cells. However, expression of iNOS may be induced in the vessel wall in response to a variety of inflammatory stimuli and in disease [26–28]. All NOS isoforms catalyze the reaction between l-arginine and molecular oxygen via a two-step process involving the generation of the unstable intermediate N-hydroxy-l-arginine before ultimately producing NO and l-citrulline [29, 30]. This process involves the presence of nicotinamide adenine dinucleotide phosphate (NADPH), tetrahydrobiopterin (BH4), flavin adenine dinucleotide (FAD), and flavin mononucleotide. Once generated by NOS, NO can rapidly diffuse to other cells where its main target is the cytosolic enzyme, soluble guanylate cyclase (sGC). Upon activation, sGC catalyzes the conversion of guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP), which serves as an important second messenger, mediating most of the vascular effects of NO. cGMP has multiple downstream targets including cGMP-dependent protein kinases (cGKs), phosphodiesterases, and cGMP-modulated cation channels [31, 32]. Activation of cGKs and cGMP-modulated cation channels can lead to a decrease in intracellular Ca2+ and thus cause vasodilation.
Many studies have shown that activation of eNOS and/or the sGC–cGMP pathway leads to cerebral vasodilation [33–35]. This signaling cascade is active under basal conditions, thus influencing resting vascular tone. For example, inhibitors of NOS cause constriction of cerebral arteries, pial arterioles, and parenchymal arterioles resulting in increases in vascular tone and decreases in cerebral blood flow in diverse experimental models and in humans [36–42]. Such findings, along with other lines of evidence indicate that constitutive activity of NOS and levels of NO in the cerebral vasculature are sufficient to influence vascular tone and thus cerebral blood flow under normal conditions.
Endothelium-derived NO mediates responses of cerebral blood vessels to neurotransmitters including acetylcholine and substance P, metabolic factors like insulin and adiponectin, and therapeutic agents such as HMG-CoA reductase inhibitors (statins) [11, 42, 43]. For example, dilation of small cerebral arterioles to acetylcholine is mediated by activation of type-5 muscarinic receptors and eNOS [35, 44]. In addition to these effects, endothelial cells play a critical role in neurovascular coupling [45]. Recent work has shown that selective inhibition of endothelial signaling prevents retrograde (upstream) dilation of cerebral arterioles on the brain surface in response to activation of the somatosensory cortex [45]. In regions such as the basal forebrain, neurovascular coupling is mediated by neuronally released acetylcholine acting on endothelium with subsequent activation of eNOS (see [11]). These mechanisms of blood flow regulation are important for multiple reasons including the fact the basal forebrain is a key region in relation to the development of dementia and Alzheimer’s disease, processes that are accelerated in the presence of hypertension. Other lines of evidence also support a role for eNOS in neurovascular coupling [46].
In addition to its effects on cerebrovascular tone, NO has a significant influence on the structure of cerebral arteries and arterioles [47]. In mice for example, genetic deletion of eNOS or chronic treatment with an inhibitor of NOS produces hypertrophy of cerebral arterioles and rarefaction of collateral arterioles [47, 48]. Lastly, NO inhibits the aggregation of platelets and adhesion of leukocytes to the vessel wall as well as suppressing the metabolism of amyloid precursor protein, a key molecule in the pathogenesis of Alzheimer’s disease [11, 49–51]. Thus, it is clear that NO is central to the regulation of cerebrovascular homeostasis in both large and small vessels.
4 Free Radical Biology, Oxidative Stress, and Endothelial Dysfunction
Reactive oxygen species (ROS) are primarily formed as by-products of cellular metabolism or by specialized oxidases. Common ROS in cells and tissues include superoxide, hydrogen peroxide, and hydroxyl radical. ROS typically oxidize or reduce other molecules and thereby can modify the activity and structure of proteins, lipids, and small molecules including NO [52–55]. Oxidized proteins and lipids may also serve as second messengers in signaling cascades [52].
There are many potential sources of ROS within the cell including the mitochondrial respiration chain [56], xanthine oxidase [57], cyclooxygenases [58, 59], and importantly the family of NADPH oxidases [9, 60, 61]. NADPH oxidases are probably the most studied family of oxidases in the vasculature, and are associated with the modulation of multiple processes including inflammatory responses, cell growth and proliferation, and cellular metabolism [55]. Furthermore, the NADPH oxidase family of enzymes is currently the only known enzymes whose sole purpose is to generate ROS [60]. The other enzymes listed above generate ROS as a by-product of normal metabolism or during a dysfunctional state [62]. Therefore, NADPH oxidases are prime candidates for generating the excessive levels of ROS that has been described during disease, including hypertension. In summary, ROS derived from oxidases modulate multiple cellular processes and exert important effects on vascular homeostasis.
Cells have developed sophisticated systems to regulate levels of ROS both intracellularly and extracellularly, as well as in specific subcellular compartments. Superoxide is readily metabolized by superoxide dismutase (SOD) to hydrogen peroxide. Three isoforms of SOD exist: cytoplasmic (CuZnSOD, SOD1), mitochondrial (MnSOD, SOD2), and extracellular (EcSOD, SOD3) [63, 64]. Hydrogen peroxide is also metabolized by specific enzymatic systems that include catalase and several isoforms of glutathione peroxidase. Enzymatic antioxidant systems are complex, and cooperate with nonenzymatic antioxidants such as vitamin C and vitamin E [65].
Cellular metabolism is adapted to the redox state in cells. Redox state is determined by the balance of oxidative and reductive reactions, and the activity of antioxidant systems described above [66]. This redox state may vary according to the function and biological state of cells and tissues [67]. When the balance of oxidants and antioxidants is lost, the redox state of cells change, and oxidative stress ensues.
Oxidative stress affects vascular physiology and contributes to vascular pathophysiology. NO signaling and metabolism is extremely sensitive to oxidative stress. Oxidation of BH4 by free radicals can impair the function of NOS resulting in the generation of superoxide rather than NO (termed NOS uncoupling) [11, 13]. Although uncoupled eNOS is thought to be a source of superoxide and oxidative stress in some portions of the circulation [53, 68], the overall importance of this mechanism in the cerebral circulation is still unclear. Importantly, superoxide readily reacts with NO to form the reactive nitrogen species (RNS) peroxynitrite [69]. Peroxynitrite reacts with aromatic amino acids in proteins to form nitrotyrosine adducts and that can then modify protein function [54, 69, 70]. Thus, an oxidative environment may change the character of NO metabolism, decreasing NO bioavailability in blood vessels, and promoting further oxidative damage by NOS uncoupling and/or increased formation of RNS. Although the result of these reactions for the vessel can be quite diverse, one of the most studied consequences is impairment of endothelium-dependent vasodilation [11, 69].
5 Hypertension and Cerebrovascular Function
Hypertension affects cerebrovascular structure and function through a number of mechanisms. For example, impairment of endothelium-dependent vasodilator responses are described in many models of hypertension including angiotensin-II (Ang-II)-dependent models, the spontaneously hypertensive rat (SHR), and the stroke-prone SHR (SHRSP) (Table 8.1). Although perhaps the best described mechanism for endothelial dysfunction involves loss of NO bioavailability, endothelial dysfunction extends to other mechanisms as well including NO-independent pathways and generation of EDCFs.
Loss of NO bioavailability most often results from an imbalance between ROS generation and ROS degradation or metabolism (oxidative stress). As the reaction between NO and superoxide is one of the fastest known in biology [71], increased superoxide will reduce NO levels if superoxide is present in close proximity to the NO molecule. Numerous studies have demonstrated that NO-dependent vasodilation (NO-dependent signaling) is impaired during hypertension and this change is thought to be primarily due to scavenging of NO by superoxide resulting in an acute reduction in NO bioavailability.
5.1 Ang-II-Dependent Models of Hypertension
Endothelium-dependent vasodilation is reduced in models of Ang II-dependent hypertension. Most of this work has been done using mice in which hypertension is induced by chronic systemic administration of Ang-II—typically for a few weeks. Although effects on NO-dependent responses are often described in these models, impaired vasodilation can also occur via other endothelium-dependent mechanisms. Reduced NO bioavailability is closely associated with elevated ROS levels. Numerous studies have reported that Ang-II increases superoxide generation in the cerebral vasculature [9, 72–75] and as described above, is likely to scavenge NO, thus reducing its bioavailability.
It has been shown that endothelium-dependent vasodilation is impaired in isolated cerebral arteries following either acute or chronic Ang-II administration as well as in mice with lifelong (genetic) elevations in Ang-II [9, 76–80]. Endogenous NO-dependent vasodilation, as measured by responses to endothelium-dependent vasodilators such as acetylcholine and bradykinin, are significantly impaired in vessels from hypertensive animals compared with controls. Importantly, vasodilation to exogenously administered NO is similar between normotensive and hypertensive animals indicating that vascular muscle can respond normally. An oxidative stress-dependent mechanism accounts for reduced endothelium-dependent vasodilation as treatment with scavengers of superoxide typically restores endothelial function largely to normal [9, 76, 79].
There has been much interest in identifying the underlying cause or source of oxidative stress during hypertension. As discussed above, NADPH oxidases have been postulated to be a major source of elevated ROS levels during disease. Several studies have examined the importance of Nox2 in relation to acute and chronic effects of Ang-II on cerebrovascular function (Fig. 8.2) [8, 9, 69, 72, 73]. For example, a recent study used mice genetically deficient in either Nox1 or Nox2 NADPH oxidase to determine the source of augmented ROS during chronic Ang-II infusion. Following treatment with Ang-II, Nox2-deficient mice were protected against endothelial dysfunction as measured by dilation of basilar arteries to acetylcholine [78]. Interestingly, a role for Nox1 NADPH oxidase was also observed as dilation to acetylcholine was improved compared with arteries from wild-type mice; however, the improvement was less than what was observed in Nox2-deficient mice [78]. Thus, these findings would indicate that the impaired endothelial function in Ang-II infusion models of hypertension is dependent on oxidative stress with contributions by multiple Nox isoforms although Nox2-containing NADPH oxidase appears to be of greatest importance. In relation to signaling pathways, other evidence indicates that activation of signal transducer and activator of transcription 3 (STAT3) plays an important role in these processes. Small molecule inhibitors of STAT3 activation protect against Ang-II-induced oxidative stress and cerebrovascular endothelial dysfunction [81].

During hypertension and other risk factors for vascular disease, many changes occur within endothelial cells that result in increased vascular tone as well as a prothrombotic environment and low-grade inflammation. There are multiple sources of superoxide (O2 −) in endothelium and vascular muscle (not shown). A major source of O2 − in vascular cells during hypertension is NADPH oxidase. When activated by angiotensin-II (Ang II) acting on AT1 receptors (or other stimuli), O2 − can interact with NO or be converted into other reactive oxygen species including hydroxyl radical (HO.) which is highly reactive and induces mutations in nucleic acids, lipid peroxidation, protein damage, etc. The interaction between O2 − and NO not only reduces NO bioavailability and NO signaling (less activation of sGC), but also results in the formation of peroxynitrite (ONOO−). ONOO− has direct effects of its own but can also reduce formation of NO from eNOS, via effects on tetrahydrobiopterin (BH4). During vascular disease, production of thromboxane A2 and endothelin-1 (ET-1) are increased, contributing to increased vascular tone and platelet aggregation. ECE endothelin-converting enzyme
In addition to studies of isolated cerebral arteries, numerous studies have investigated the effect of hypertension on smaller cerebral arterioles or on changes in local cerebral blood flow in vivo [8–10, 69, 73, 80, 82, 83]. In general, findings obtained using these approaches are similar to those observed in larger cerebral arteries—both acute and chronic Ang-II-dependent hypertension [76] resulted in impairment of endothelial- and NO-dependent vasodilator responses in hypertensive animals compared with controls. Vasodilation to exogenous NO donors and other agonists that act directly on vascular muscle are typically comparable between normotensive and hypertensive animals. In these studies of the microcirculation, oxidative stress was again identified as the key mechanism underlying impaired vasodilator responses as ROS scavengers typically restored endothelium-dependent regulation of vascular tone largely to normal [9, 73, 84]. Importantly, studies using pharmacological inhibitors and genetically modified mice identified Nox2 NADPH oxidase as the source of superoxide [9, 73]. Interestingly, when hypertension was induced by acute administration of the α1-adrenoceptor agonist phenylephrine, endothelium-dependent responses were not impaired [10] suggesting that Ang-II-induced dysfunction is dependent on direct effects of Ang-II and not elevations in blood pressure per se. Consistent with this concept, chronic infusion of a non-pressor dose of Ang-II, or local treatment with Ang-II, also produced endothelial dysfunction in cerebral blood vessels [10, 76]. In relation to this latter point, it has been reported that a dose of Ang-II that elevates blood pressure more gradually (i.e., over a 7-day period vs. ~2–3 days for higher doses of Ang-II) also has profound effects on cerebrovascular function. Like the use of higher doses, this slow-pressor dose of Ang-II promotes oxidative stress and endothelial dysfunction (based on measurements of local cerebral blood flow) before the onset of hypertension [82]. This study also further highlights that many of the effects of hypertension on the cerebral circulation are due to direct effects of Ang-II on cerebrovascular function.
Transgenic mice that genetically express human renin and human angiotensinogen (R+A+ mice) represent a model of truly chronic (lifelong) Ang-II-dependent hypertension. Consistent with data obtained where mice were infused with Ang-II for up to a few weeks, marked superoxide-mediated endothelial dysfunction is present in the basilar artery of these mice [79]. In relation to effects of hypertension on vascular structure, both inward microvascular remodeling and vascular hypertrophy occur in small cerebral arterioles in R+A+ mice [85]. Both pressor and non-pressor doses of Ang-II increased superoxide and produced inward remodeling of cerebral arterioles in wild-type mice, effects that were absent in mice lacking the Nox2 component of NADPH oxidase [75]. These studies provide direct evidence that NADPH oxidase was a key source of superoxide that is required for Ang-II-induced inward remodeling to occur in cerebral arterioles.
In summary, findings to date indicate that in Ang-II-dependent models of hypertension, there is increased oxidative stress within the cerebral vasculature. Elevated superoxide levels overwhelm antioxidant defense mechanisms resulting in impairment of NO-dependent vasodilation, a major signaling pathway for endothelium-dependent effects on vascular muscle and other cell types. Furthermore, Ang-II and superoxide contribute to alterations in cerebrovascular structure, which can further compromise the function of the cerebrovasculature. These effects are likely to have important consequences for regulation of cerebral blood flow.
5.2 Deoxycorticosterone (DOCA)/Salt-Dependent Hypertension
Chronic administration of DOCA with high salt in the drinking water is a model characterized by neurohumoral activation and volume expansion. The DOCA/salt model exhibits suppression of the peripheral renin-angiotensin system but activation of the central renin-angiotensin system [86–88]. Increased activity of the sympathetic nervous system, secretion of vasopressin, and local expression of endothelin-1 are also thought to be key features in the model [86, 87]. Although this model of hypertension has been used widely in studies of the peripheral circulation [89–92], there are relatively few reports of its use to study endothelial function in the cerebral circulation. In one study, responses of the basilar artery to acetylcholine were unaffected after DOCA/salt treatment [93]. In contrast, we found that mild DOCA/salt-induced hypertension profoundly impairs cerebral microvascular endothelial function in mice [94]. Initial work to define mechanisms underlying these changes identified activation of AT1 receptors and Rho kinase as a key contributor to DOCA/salt-induced microvascular endothelial dysfunction in brain. These findings are interesting in relation to recent evidence that activity of Rho kinase is positively associated with cardiovascular events including stroke [95].
5.3 SHR/SHRSP Models of Genetic Hypertension
Endothelial function has been studied on many occasions in the cerebral circulation of SHR and SHRSP. To our knowledge, the first examination of effects of chronic hypertension on endothelial function in cerebral arterioles was performed by Mayhan et al. using SHRSP [96]. Since that initial effort, numerous reports have described impaired endothelium-dependent vasodilation in the cerebral vasculature from SHR/SHRSP compared with vessels from normotensive WKY rats [96–102]. In contrast to effects on endothelium, vascular responses to endothelium-independent agonists are typically normal in these models [97, 99, 100, 102–104]. Studies in SHRSP also indicated that impaired endothelial function in SHRSP extends beyond stimuli that are dependent on NO-signaling but also involved agents that acted on endothelium through receptor-independent mechanisms as well as agonists that act via endothelium but through non-NO-dependent pathways [99, 101, 102]. As with the Ang-II-dependent model of hypertension, the findings from SHR/SHRSP suggest that a major form of vascular dysfunction seen during chronic hypertension occurs at the level of the endothelium, while vascular muscle appears to be able to respond normally. Importantly, genetic links between impaired endothelial function and stroke have been established [18].
A number of studies have attempted to define the mechanisms that contribute to endothelial dysfunction in SHR/SHRSP with different mechanisms mediating dysfunction in different segments of the cerebral circulation (Fig. 8.2). For example, endothelial dysfunction in small cerebral arterioles can be reversed by treatment with a COX inhibitor [100]. Furthermore, inhibition of prostaglandin H2/thromboxane A2 receptors improved endothelium-dependent vasodilation [98]. In contrast, treatment of basilar arteries with a COX inhibitor does not improve endothelium-dependent vasodilation [96, 104].
Similar to what is observed in Ang-II-dependent models of hypertension, increased levels of superoxide are present in cerebral arteries from SHR and SHRSP [105]. Although there is some evidence to the contrary [106], functional studies suggest that oxidative stress contributes to endothelial dysfunction in these models. For example, an inhibitor of 20-hydroxy-5,8,11,14-eicosatetraenoic acid (20-HETE) reduced levels of superoxide and restored endothelial function to normal in middle cerebral arteries from SHR [105]. This suppression of oxidative stress may arise from inhibition of an interaction between 20-HETE and NADPH oxidase, although details for such a mechanism are limited at present. There is also evidence that the SHRSP model of hypertension is dependent on endogenously generated Ang-II. Both acute and long-term treatment of SHRSP with an ACE inhibitor restored endothelium-dependent vasodilation of cerebral arterioles, effects that are independent of changes in arterial pressure [101].
Overall, these studies highlight some of the major and complex effects of hypertension on the cerebral circulation. Different signaling mechanisms appear to play a more prominent role in causing endothelial dysfunction at different points along the cerebrovascular tree.
6 COX-Dependent Mechanisms Contribute to Vascular Dysfunction
COX is an important enzyme within the cerebral vasculature and has been linked with cerebrovascular dysfunction in several experimental models (see below). Although recent attention has focused on NADPH oxidase, COX is also an important source of ROS in the vasculature (Fig. 8.1) [58, 59] and there is evidence that COX-dependent ROS production can directly and indirectly (via ROS-dependent scavenging of NO) alter vascular tone. As discussed above, some evidence suggests that endothelial dysfunction in cerebral arterioles of SHRSP is COX-dependent (Fig. 8.2).
COX1-derived prostaglandin E2 and EP1 receptors are required for impaired endothelium-dependent vasodilation during acute hypertension induced by Ang-II infusion [8]. While it was shown that COX1 did not directly generate ROS in response to Ang-II, this COX1 signaling pathway was required for NADPH oxidase-dependent ROS production and subsequent vascular dysfunction during acute hypertension [8]. It will be of interest to determine if similar mechanisms are present during chronic hypertension.
7 Endothelium-Derived Contracting Factors
In addition to impaired endothelium-dependent vasodilator responses, hypertension also alters vascular function via the generation of EDCFs (Fig. 8.2). EDCFs may include COX-derived products, endothelin-1, and others [11]. Relatively few studies have investigated the contribution and identity of EDCFs during hypertension in the cerebral circulation. Evidence from noncerebral arteries suggests that a COX-derived product functions as an EDCF [107, 108] and studies of cerebral blood vessels agree with this concept. For example, responses of cerebral arterioles to ADP, which is an endothelium-dependent vasodilator that signals mainly through NO, are impaired in SHRSP [100]. Mechanistically, evidence suggests that this dysfunction is dependent on a COX product (Fig. 8.2) as treatment with a COX inhibitor or a thromboxane A2 receptor antagonist [98] restored endothelial microvascular responses essentially to normal [100]. Interestingly, while a key functional role is suggested for these pathways in the microcirculation, a similar EDCF does not play such a role in larger cerebral arteries [96].
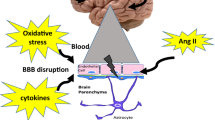
8 Blood–Brain Barrier
The movement of molecules or cells out of the intravascular compartment and into the cerebrospinal fluid and/or brain parenchyma is normally highly regulated. Adjacent cerebral endothelial cells are tightly linked, forming a structural and functional barrier to the diffusion of most molecules, particularly those with poor lipid solubility [109]. This organization of endothelial cells in brain is known as the BBB (Fig. 8.3) [109–112].

Schematic illustration of adjacent endothelial cells that constitute the primary site of the blood–brain barrier. The physical, intercellular barrier between endothelial cells comprises the tight junctions (made up of occludins, claudins, and junctional adhesion molecule) at the apical surface and adherens junctions at the basolateral surface. These intercellular adhesion molecules are connected to the actin cytoskeleton by zonula occludens. Specialized ATP-dependent transporters facilitate the movement of molecules from the circulation into the parenchyma. Cerebral endothelial cells also contain ATP-dependent drug transporters and drug metabolizing enzymes that prevent entry of some drugs into the parenchyma. The maintenance of the BBB requires relatively large amounts of ATP and cerebral endothelial cells contain a relatively high density of mitochondria, presumably to meet this energy demand. Molecules can move across the endothelium by the activity of ATP-dependent transporters. However, some drugs can be exported out of the cells by ATP-dependent efflux pumps, or be metabolized into inactive compounds by drug metabolizing enzymes. ATP adenosine triphosphate, D drug, JAM junctional adhesion molecule, M molecule, ZO zona occludens
The main determinants of BBB permeability include: (1) the close connections between adjacent endothelial cells (mediated by tight junction proteins), (2) limited active transport and diffusion of molecules across endothelial cells, and (3) high expression of drug efflux pumps and metabolizing enzymes [113]. Although the presence of a BBB derives largely from the properties of cerebral endothelial cells, other cell types, including astrocytes and pericytes, play an important role in regulating BBB integrity by modulating endothelial function and expression of BBB components [114–117]. In the following sections we will review selected aspects of our understanding of the structure, molecular organization, and regulation of the BBB. We will then summarize concepts related to effects of acute and chronic hypertension on BBB integrity.
9 The Concept and Molecular Makeup of the BBB
Based on studies utilizing injections of dyes in experimental animals, Erlich in 1885 revealed for the first time that some blood-borne molecules cannot simply diffuse into the brain [118]. Years later, Max Lewandosky postulated that cerebral blood vessels had unique properties that allow them to block the diffusion of molecules—a function he defined as “bluthirnschranke” (blood–brain barrier in German) [109, 113, 118]. With the development of electron microscopy, Reese and Karnovsky demonstrated the fundamental role of endothelial cells in a functional BBB [113, 118, 119] showing that transport of proteins across the endothelial layer was limited by tight junctions between endothelial cells. Many subsequent studies have built upon these seminal findings, increasing our understanding of the molecular makeup of the BBB and its function in health and disease [118].
Endothelial cells of the BBB have properties that distinguish them from vascular endothelium in most other organs as well as from endothelial cells in regions of the brain where the BBB is not present (e.g., circumventricular organs). These differences can be divided into several major categories—structural, molecular, biochemical, and metabolic. Brain endothelial cells lack fenestrations and are tightly connected by interacting proteins that anchor adjacent endothelial cells while also facilitating communication between endothelial cells. These proteins are localized in structures known as tight junctions (in the apical border) and adherent junctions (near the basal border) (Fig. 8.3). Tight junctions closely connect one endothelial cell with adjacent endothelial cells. As a result, there are no structural gaps between endothelial cells in most areas of the brain.
Several families of proteins make up the structural components of tight junctions. Occludin, claudins 5, 3, and 12, and junctional adhesion molecules (JAM) 1 and 4 appear to be particularly enriched in brain endothelial cells [111]. These are integral membrane proteins that bind other tight junction proteins in the extracellular space while also interacting closely with cytoskeletal proteins. The interactions of tight junction and cytoskeletal proteins are facilitated by members of the zonula occludens (ZO) family. ZO-1 and ZO-2 proteins also facilitate regulation of endothelial and tight junction function by acting as scaffolds for signaling molecules at the cell membrane [111, 113]. Modulation of expression and the functional state (e.g., phosphorylation) of occludins, claudins, JAM, and ZO proteins is pivotal for the maintenance of BBB integrity and permeability [120]. Alterations in expression or function can lead to changes in BBB permeability and have been described in various experimental models of neurological disease, stroke, and other forms of brain injury [111, 112, 120, 121].
Endothelial cells in the brain are very metabolically active. Cerebral endothelium has a high rate of pinocytosis and expresses a series of ATP-dependent transporters (in the luminal and basolateral surfaces) that control the active transport of molecules across the cells [109, 113]. Cerebral endothelium also expresses a high level of drug efflux pumps and drug metabolizing enzymes that consume ATP and actively metabolize and transport compounds from the endothelial cell back to the blood stream [109, 113]. Brain endothelial cells characteristically contain a relatively high number of mitochondria, which presumably supports the increased energy expenditure associated with the increased expression of ATP-dependent transporters, drug efflux pumps, and metabolizing enzymes.
Although specific features can change segmentally along the vasculature, the BBB is present in vessels on the brain surface (both arterioles and venules) as well as in parenchymal vessels including capillaries [110, 122]. Capillaries may have the ‘tightest’ BBB, but the permeability of pial vessels in brain is still orders of magnitude less than similar size vessels in the peripheral circulation [122]. Other segmental differences exist in relation to the BBB. For example, the site where many interactions with circulating immune cells occur, sometimes referred to as the immunological BBB, is primarily at post-capillary venules [123], a segment of the microvasculature that can be prominently affected by hypertension (see below).
Disruption of the integrity of the BBB is a common feature of cerebrovascular and neurological disease as well as with aging [111]. Mechanisms responsible for these changes can be diverse and may include interacting oxidant- and immune-related pathways, alterations in cell surface charge, both transcellular and paracellular pathways, phosphorylation of tight junction proteins, and effects on the actin-myosin cytoskeleton [112, 124]. For example, cellular injury may lead to the increased production of matrix metalloproteinases (MMPs) and VEGF, which then degrades the integrity of tight junctions. Expression and activity of several MMPs are elevated in cerebrovascular disease including models of hypertension [121, 125–131]. For decades it has been known that hypertension induces BBB disruption [132], however the mechanisms that link hypertension with BBB disruption are still poorly understood.
9.1 Acute Hypertension
Episodes of acute hypertension can occur under both physiological and pathophysiological conditions. For example, large increases in systemic arterial pressure occur during isometric exercise and sexual activity [133, 134], however there is little indication that these activities normally injure the BBB. In contrast, large increases in arterial pressure that occur in some disease states have a major impact on cerebrovascular function and permeability of the BBB. There are many causes of acute hypertension in pathophysiology including traumatic brain injury, seizures, and severe preeclampsia and eclampsia [135]. Preexisting chronic hypertension is also a risk factor for acute hypertension [135]. BBB changes have been described in all these latter conditions. Hypertensive encephalopathy is a condition characterized by acute and severe hypertension, headache, seizures, and other neurological symptoms including cerebral edema [136]. In a model of hypertensive encephalopathy, inhibition of the delta isoform of protein kinase C prevented hypertension-induced disruption of tight junctions and increased permeability of the BBB [137]. In another study, treatment with TEMPOL (a scavenger of superoxide anion) protected against disruption of the BBB and brain edema in a model of acute hypertension, suggesting a critical role for this key oxygen-derived free radical in the process [138]. Expression of tight junction proteins (e.g., claudin 5) are reduced in response to acute hypertension [139].
Loss of BBB integrity during large acute increases in arterial pressure exhibits regional and segmental differences in the cerebral circulation [138, 140–143]. This heterogeneity likely results from a variety of factors including differences in vascular structure, intrinsic functional properties of blood vessels (autoregulatory capacity), as well as perivascular innervation by sympathetic and sensory neurons. As a result, there are regional differences in local hemodynamics and increases in microvascular pressure which are the main determinants of changes in BBB permeability under these conditions [138, 140–143]. One of the best examples of this concept is the finding that small pial venules are the major site of disruption of the BBB following acute hypertension [142, 143]. Differences in venous microvascular pressure can account for regional (and model-specific) differences that are seen in disruption of the BBB during acute hypertension [138, 140–143].
9.2 Chronic Hypertension and BBB Integrity
Chronic hypertension is associated with BBB disruption in several animal models. In the laboratory, chronic hypertension is often studied using genetic models of hypertension (e.g., SHR/SHRSP) or by inducing hypertension pharmacologically (e.g., chronic infusion of Ang-II or other pressor agents). To study the effects of chronic hypertension on BBB permeability, tracer molecules that do not normally cross the BBB are typically utilized. Intravenous administration substances such as Evans blue, radiolabeled albumin, or fluorescent molecules are common approaches along with quantification of the movement of tracers into brain as an index of changes in BBB permeability.
BBB integrity is compromised in genetic models of hypertension. For example, cerebral arteries of SHRSP and SHR exhibit increased permeability to horseradish peroxidase compared to normotensive controls. In these models, arteries in the hypothalamus appear to be particularly sensitive to the effects of hypertension [144]. Increased permeability of the BBB has been seen in selected brain regions involved in autonomic control in SHR and in renal hypertensive rats [145]. These differences were prevented by an inhibitor of AT1-receptors and interestingly, appeared to be due to direct effects of Ang-II, and not a secondary change caused by increased arterial pressure [145]. In SHRSP on high salt, reductions in cerebral blood flow, decreases in expression of occludin, increased BBB permeability and white matter lesions all occur [146]. These changes are prevented by renal denervation. Although the mechanisms that account for this protective effects are not clear, it may involve reductions in oxidative stress [146]. In brain, small vessel disease accounts for up to 30 % of strokes and is a leading cause of age-related and hypertension-related cognitive decline and its resulting disability [147]. Early endothelial dysfunction, including loss of BBB integrity, is now thought to be key event in the pathogenesis of small vessel disease [147, 148]. Hypertension is a major risk factor for small vessel disease.
The BBB is also disrupted in nongenetic models of hypertension. For example, pharmacologically induced hypertension produces BBB disruption in mice. Angiotensin-II-induced hypertension increases Evans blue extravasation in mouse brain [149, 150]. BBB disruption in mice treated with Ang-II is associated with increased leukocyte recruitment to the vessel wall [149, 150]. Similar to what was described above, Ang-II appears to affect the BBB by its local actions on vascular AT1 receptors. BBB integrity is preserved in AT1-receptor-deficient mice treated with angiotensin-II, but not in mice in whom AT1 receptors are absent in hematopoietic progenitor cells [150]. In another study, increases in BBB permeability in response to chronic treatment with Ang-II in regions like white matter and the hippocampus are augmented with age and are associated with reductions in tight junction protein expression [151]. Similar to changes that have been described in small vessel disease, there is evidence for structural abnormalities and edema formation in white matter in a mouse model of DOCA/salt-induced hypertension [152].
Hypertension-induced impairment of BBB integrity may increase the risk for stroke. Chronically hypertensive mice that received injections of Ang-II to induce acute hypertension exhibited spontaneous intracerebral hemorrhage [131]. Increased MMP expression and oxidative stress, and decreased expression of antioxidant enzymes were found in brain in this model and may have contributed to the intracerebral hemorrhages [131]. Whether disruption of the BBB preceded intracerebral hemorrhages in this model is not clear at this time.
9.3 Clinical Relevance of BBB Disruption
Because the BBB plays an important role in controlling the exposure of the brain to blood-borne molecules, and because the integrity of the BBB requires the participation of several cellular components, it is not surprising that the BBB integrity is compromised in neurological disease. Studies in humans suggest the integrity of the BBB is compromised in the presence of brain trauma, epilepsy, and neurodegenerative diseases including Alzheimer’s disease, multiple sclerosis, Parkinson’s disease, and amyotrophic lateral sclerosis (reviewed by Daneman [111]). Importantly, vascular risk factors, including hypertension, may contribute to the pathogenesis of some of these diseases. Thus, hypertension, directly or indirectly, may be a contributor to multiple forms of neurological disease at least in part because of its effects on BBB structure and function.
Studies of BBB permeability in humans have been more limited, because of the lack of methods to study the BBB in patients. However, some techniques for the visualization of BBB permeability in humans have been developed recently [153–156]. These techniques generally involve the administration of a molecule that can be visualized by conventional imaging techniques and normally will not move across the BBB. Several studies have demonstrated alteration of the BBB permeability induced by hypertension in patients. In elderly patients without clinical history of cerebrovascular disease, and who were evaluated by perfusion-CT, BBB permeability was increased in basal ganglia, especially in patients with hypertension [156]. As noted above, hypertension is a key risk factor for small vessel disease, and hypertension may play a role in the BBB disruption observed in patients with small vessel disease or white matter lesions in the brain [157]. In contrast, another long-term study of BBB permeability in basal ganglia of patients with cerebral small vessel disease did not show a significant effect of hypertension [155]. It should be noted that BBB studies in humans are generally limited to populations of patients with comorbid conditions, and the methods that have been developed may not be sensitive enough to detect BBB alterations induced in earlier stages of hypertensive-induced cerebrovascular disease.
References
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–245.
Roger VL, Go AS, Lloyd-Jones DM, Adams RJ, Berry JD, Brown TM, et al. Heart disease and stroke statistics—2011 update. Circulation. 2011;123:e18–209.
Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2224–60.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Collaboration PS. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–13.
O’Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, et al. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE Study): a case-control study. Lancet. 2010;376:112–23.
Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia. Stroke. 2011;42:2672–713.
Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62:810–7.
Capone C, Faraco G, Anrather J, Zhou P, Iadecola C. Cyclooxygenase 1-derived prostaglandin E2 and EP1 receptors are required for the cerebrovascular dysfunction induced by angiotensin II. Hypertension. 2010;55:911–7.
Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Angiotensin II attenuates endothelium-dependent responses in the cerebral microcirculation through Nox-2-derived radicals. Arterioscler Thromb Vasc Biol. 2006;26:826–32.
Kazama K, Wang G, Frys K, Anrather J, Iadecola C. Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. Am J Physiol Heart Circ Physiol. 2003;285:H1890–9.
Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol. 2011;300:H1566–82.
Stoica AL, Stoica E, Constantinescu I, Uscatescu V, Ginghina C. Interleukin-6 and interleukin-10 gene polymorphism, endothelial dysfunction, and postoperative prognosis in patients with peripheral arterial disease. J Vasc Surg. 2010;52:103–9.
Lentz SR. Mechanisms of homocysteine-induced atherothrombosis. J Thromb Haemost. 2005;3:1646–54.
Neuwelt EA. Mechanisms of disease: the blood-brain barrier. Neurosurgery. 2004;54:131–40.
Dharmashankar K, Welsh A, Wang J, Kizhakekuttu TJ, Ying R, Gutterman DD, et al. Nitric oxide synthase-dependent vasodilation of human subcutaneous arterioles correlates with noninvasive measurements of endothelial function. Am J Hypertens. 2012;25:528–34.
Ganz P, Vita JA. Testing endothelial vasomotor function: nitric oxide, a multipotent molecule. Circulation. 2003;108:2049–53.
Faraci FM, Heistad DD. Regulation of the cerebral circulation: role of endothelium and potassium channels. Physiol Rev. 1998;78:53–97.
Volpe M, Iaccarino G, Vecchione C, Rizzoni D, Russo R, Rubattu S, et al. Association and cosegregation of stroke with impaired endothelium-dependent vasorelaxation in stroke prone, spontaneously hypertensive rats. J Clin Invest. 1996;98:256–61.
Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–5.
Barton M. The discovery of endothelium-dependent contraction: the legacy of Paul M. Vanhoutte. Pharmacol Res. 2011;63:455–62.
Vanhoutte PM. Inhibition by acetylcholine of adrenergic neurotransmission in vascular smooth muscle. Circ Res. 1974;34:317–26.
Garland CJ, Hiley CR, Dora KA. EDHF: spreading the influence of the endothelium. Br J Pharmacol. 2011;164:839–52.
Takaki A, Morikawa K, Murayama Y, Yamagishi H, Hosoya M, Ohashi J, et al. Roles of endothelial oxidases in endothelium-derived hyperpolarizing factor responses in mice. J Cardiovasc Pharmacol. 2008;52:510–7.
Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: Structure, function and inhibition. Biochem J. 2001;357:593–615.
Michel T, Feron O. Nitric oxide synthases: which, where, how, and why? J Clin Invest. 1997;100:2146–52.
Perrotta I, Brunelli E, Sciangula A, Conforti F, Perrotta E, Tripepi S, et al. iNOS induction and PARP-1 activation in human atherosclerotic lesions: an immunohistochemical and ultrastructural approach. Cardiovasc Pathol. 2010;20:195–203.
Daneshtalab N, Smeda JS. Alterations in the modulation of cerebrovascular tone and blood flow by nitric oxide synthases in SHRsp with stroke. Cardiovasc Res. 2010;86:160–8.
Panayiotou CM, Baliga R, Stidwill R, Taylor V, Singer M, Hobbs AJ. Resistance to endotoxic shock in mice lacking natriuretic peptide receptor-A. Br J Pharmacol. 2010;160:2045–54.
Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–42.
Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res. 1999;43:521–31.
Feil R, Lohmann SM, de Jonge H, Walter U, Hofmann F. Cyclic GMP-dependent protein kinases and the cardiovascular system: insights from genetically modified mice. Circ Res. 2003;93:907–16.
Conti M, Beavo J. Biochemistry and physiology of cyclic nucleotide phosphodiesterases: essential components in cyclic nucleotide signaling. Annu Rev Biochem. 2007;76:481–511.
Didion SP, Heistad DD, Faraci FM. Mechanisms that produce nitric oxide-mediated relaxation of cerebral arteries during atherosclerosis. Stroke. 2001;32:761–6.
Bai N, Moien-Afshari F, Washio H, Min A, Laher I. Pharmacology of the mouse-isolated cerebral artery. Vasc Pharmacol. 2004;41:97–106.
Sobey CG, Faraci FM. Effects of a novel inhibitor of guanylyl cyclase on dilator responses of mouse cerebral arterioles. Stroke. 1997;28:837–43.
Dietrich HH, Kimura M, Dacey Jr RG. N omega-nitro-L-arginine constricts cerebral arterioles without increasing intracellular calcium levels. Am J Physiol Heart Circ Physiol. 1994;266:H1681–6.
Cipolla MJ, Bullinger LV. Reactivity of brain parenchymal arterioles after ischemia and reperfusion. Microcirculation. 2008;15:495–501.
Yamashiro K, Milsom AB, Duchene J, Panayiotou C, Urabe T, Hattori N, et al. Alterations in nitric oxide and endothelin-1 bioactivity underlie cerebrovascular dysfunction in ApoE-deficient mice. J Cereb Blood Flow Metab. 2010;30:1494–503.
Faraci FM. Role of nitric oxide in regulation of basilar artery tone in vivo. Am J Physiol Heart Circ Physiol. 1990;259:H1216–21.
Iadecola C, Li J, Ebner TJ, Xu X. Nitric oxide contributes to functional hyperemia in cerebellar cortex. Am J Physiol. 1995;268:R1153–62.
McPherson RW, Kirsch JR, Ghaly RF, Traystman RJ. Effect of nitric oxide synthase inhibition on the cerebral vascular response to hypercapnia in primates. Stroke. 1995;26:682–7.
Faraci FM, Sobey CG. Role of potassium channels in regulation of cerebral vascular tone. J Cereb Blood Flow Metab. 1998;18:1047–63.
Osuka K, Watanabe Y, Yasuda M, Takayasu M. Adiponectin activates endothelial nitric oxide synthase through AMPK signaling after subarachnoid hemorrhage. Neurosci Lett. 2012;514:2–5.
Yamada M, Lamping KG, Duttaroy A, Zhang W, Cui Y, Bymaster FP, et al. Cholinergic dilation of cerebral blood vessels is abolished in M5 muscarinic acetylcholine receptor knockout mice. Proc Natl Acad Sci U S A. 2001;98:14096–101.
Chen BR, Kozberg MG, Bouchard MB, Shaik MA, Hillman EM. A critical role for the vascular endothelium in functional neurovascular coupling in the brain. J Am Heart Assoc. 2014;3:s.
Stobart JLL, Lu L, Anderson HDI, Mori H, Anderson CM. Astrocyte-induced cortical vasodilation is mediated by D-serine and endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2013;110:3149–54.
Baumbach GL, Sigmund CD, Faraci FM. Structure of cerebral arterioles in mice deficient in expression of the gene for endothelial nitric oxide synthase. Circ Res. 2004;95:822–9.
Dai X, Faber JE. Endothelial nitric oxide synthase deficiency causes collateral vessel rarefaction and impairs activation of a cell cycle gene network during arteriogenesis. Circ Res. 2010;106:1870–81.
Bermejo E, Saenz DA, Alberto F, Rosenstein RE, Bari SE, Lazzari MA. Effect of nitroxyl on human platelets function. Thromb Haemost. 2005;94:578–84.
Mondoro TH, Ryan BB, Hrinczenko BW, Schechter AN, Vostal JG, Alayash AI. Biological action of nitric oxide donor compounds on platelets from patients with sickle cell disease. Br J Haematol. 2001;112:1048–54.
Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35:888–94.
Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95.
Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–37.
Chen AF, Chen D-D, Daiber A, Faraci FM, Li H, Rembold CM, et al. Free radical biology of the cardiovascular system. Clin Sci. 2012;123:73–91.
Touyz RM, Briones AM. Reactive oxygen species and vascular biology: implications in human hypertension. Hypertens Res. 2011;34:5–14.
Narayanan D, Xi Q, Pfeffer LM, Jaggar JH. Mitochondria control functional CaV1.2 expression in smooth muscle cells of cerebral arteries. Circ Res. 2010;107:631–41.
Kinugawa S, Huang H, Wang Z, Kaminski PM, Wolin MS, Hintze TH. A defect of neuronal nitric oxide synthase increases xanthine oxidase-derived superoxide anion and attenuates the control of myocardial oxygen consumption by nitric oxide derived from endothelial nitric oxide synthase. Circ Res. 2005;96:355–62.
Niwa K, Haensel C, Ross ME, Iadecola C. Cyclooxygenase-1 participates in selected vasodilator responses of the cerebral circulation. Circ Res. 2001;88:600–8.
Didion S, Hathaway C, Faraci F. Superoxide levels and function of cerebral blood vessels after inhibition of CuZn-SOD. Am J Physiol Heart Circ Physiol. 2001;281:H1697–703.
Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–71.
Miller AA, De Silva TM, Judkins CP, Diep H, Drummond GR, Sobey CG. Augmented superoxide production by Nox2-containing NADPH oxidase causes cerebral artery dysfunction during hypercholesterolemia. Stroke. 2010;41:784–9.
Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol Ther. 2008;120:254–91.
Heistad DD, Wakisaka Y, Miller J, Chu Y, Pena-Silva R. Novel aspects of oxidative stress in cardiovascular diseases. Circ J. 2009;73:201–7.
Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vac Biol. 2004;24:1367–73.
Du J, Cullen JJ, Buettner GR. Ascorbic acid: chemistry, biology and the treatment of cancer. Biochim Biophys Acta. 2012;1826:443–57.
Buettner GR, Wagner BA, Rodgers VG. Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell Biochem Biophys. 2013;67:477–83.
Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–212.
Santhanam AVR, d’Uscio LV, Smith LA, Katusic ZS. Uncoupling of eNOS causes superoxide anion production and impairs NO signaling in the cerebral microvessels of hph-1 mice. J Neurochem. 2012;122:1211–8.
Girouard H, Park L, Anrather J, Zhou P, Iadecola C. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol. 2007;27:303–9.
Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4:161–77.
Thomson L, Trujillo M, Telleri R, Radi R. Kinetics of cytochrome C2+ oxidation by peroxynitrite: implications for superoxide measurements in nitric oxide-producing biological systems. Arch Biochem Biophys. 1995;319:491–7.
De Silva TM, Broughton BRS, Drummond GR, Sobey CG, Miller AA. Gender influences cerebral vascular responses to angiotensin II through Nox2-derived reactive oxygen species. Stroke. 2009;40:1091–7.
Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA, et al. Angiotensin II impairs neurovascular coupling in neocortex through NADPH oxidase-derived radicals. Circ Res. 2004;95:1019–26.
Miller A, Drummond G, Schmidt H, Sobey C. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–62.
Chan SL, Baumbach GL. Deficiency of Nox2 prevents angiotensin II-induced inward remodeling in cerebral arterioles. Front Physiol. 2013;4:133.
Chrissobolis S, Faraci FM. Sex differences in protection against angiotensin II-induced endothelial dysfunction by manganese superoxide dismutase in the cerebral circulation. Hypertension. 2010;55:905–10.
Gerzanich V, Ivanova S, Zhou H, Simard JM. Mislocalization of eNOS and upregulation of cerebral vascular Ca2+ channel activity in angiotensin-hypertension. Hypertension. 2003;41:1124–30.
Chrissobolis S, Banfi B, Sobey CG, Faraci FM. Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol. 2012;113:184–91.
Faraci FM, Lamping KG, Modrick ML, Ryan MJ, Sigmund CD, Didion SP. Cerebral vascular effects of angiotensin II: new insights from genetic models. J Cereb Blood Flow Metab. 2006;26:449–55.
Capone C, Faraco G, Peterson JR, Coleman C, Anrather J, Milner TA, et al. Central cardiovascular circuits contribute to the neurovascular dysfunction in angiotensin II hypertension. J Neurosci. 2012;32:4878–86.
Johnson AW, Kinzenbaw DA, Modrick ML, Faraci FM. Small-molecule inhibitors of signal transducer and activator of transcription 3 protect against angiotensin II–induced vascular dysfunction and hypertension. Hypertension. 2013;61:437–42.
Capone C, Faraco G, Park L, Cao X, Davisson RL, Iadecola C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol Heart Circ Physiol. 2011;300:H397–407.
Girouard H, Lessard A, Capone C, Milner TA, Iadecola C. The neurovascular dysfunction induced by angiotensin II in the mouse neocortex is sexually dimorphic. Am J Physiol Heart Circ Physiol. 2008;294:H156–63.
Kitayama J, Yi C, Faraci FM, Heistad DD. Modulation of dilator responses of cerebral arterioles by extracellular superoxide dismutase. Stroke. 2006;37:2802–6.
Baumbach GL, Sigmund CD, Faraci FM. Cerebral arteriolar structure in mice overexpressing human renin and angiotensinogen. Hypertension. 2003;41:50–5.
Yemane H, Busauskas M, Burris SK, Knuepfer MM. Neurohumoral mechanisms in deoxycorticosterone acetate (DOCA)-salt hypertension in rats. Exp Physiol. 2010;95:51–5.
Schenk J, McNeill JH. The pathogenesis of DOCA-salt hypertension. J Pharmacol Toxicol Meth. 1992;27:161–70.
Grobe JL, Grobe CL, Beltz TG, Westphal SG, Morgan DA, Xu D, et al. The brain renin-angiotensin system controls divergent efferent mechanisms to regulate fluid and energy balance. Cell Metab. 2010;12:431–42.
Toque HA, Nunes KP, Rojas M, Bhatta A, Yao L, Xu Z, et al. Arginase 1 mediates increased blood pressure and contributes to vascular endothelial dysfunction in deoxycorticosterone acetate-salt hypertension. Front Immunol. 2013;4:219.
Somers MJ, Mavromatis K, Galis ZS, Harrison DG. Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation. 2000;101:1722–8.
Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ, et al. Endothelin-1 increases vascular superoxide via endothelin-NADPH oxidase pathway in low-renin hypertension. Circulation. 2003;107:1053–8.
Viel EC, Benkirane K, Javeshghani D, Touyz RM, Schiffrin EL. Xanthine oxidase and mitochondria contribute to vascular superoxide anion generation in DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2008;295:H281–8.
Soltis EE, Bohr DF. Cerebral vascular responsiveness in deoxycorticosterone acetate-salt hypertensive rats. Am J Physiol. 1987;252:H198–203.
De Silva TM, Lynch CM, Grobe JL, Faraci FM. Activation of the central renin angiotensin system (RAS) causes selective cerebrovascular dysfunction (Abstract). FASEB J. 2015;29:646.4.
Kajikawa M, Noma K, Maruhashi T, Mikami S, Iwamoto Y, Iwamoto A, et al. Rho-associated kinase activity is a predictor of cardiovascular outcomes. Hypertension. 2014;63:856–64.
Mayhan WG. Impairment of endothelium-dependent dilatation of basilar artery during chronic hypertension. Am J Physiol. 1990;259:H1455–62.
Kitazono T, Heistad DD, Faraci FM. Enhanced responses of the basilar artery to activation of endothelin-B receptors in stroke-prone spontaneously hypertensive rats. Hypertension. 1995;25:490–4.
Mayhan WG. Role of prostaglandin H2-thromboxane A2 in responses of cerebral arterioles during chronic hypertension. Am J Physiol. 1992;262:H539–43.
Mayhan WG, Faraci FM, Heistad DD. Impairment of endothelium-dependent responses of cerebral arterioles in chronic hypertension. Am J Physiol. 1987;253:H1435–40.
Mayhan WG, Faraci FM, Heistad DD. Responses of cerebral arterioles to adenosine 5′-diphosphate, serotonin, and the thromboxane analogue U-46619 during chronic hypertension. Hypertension. 1988;12:556–61.
Yang ST, Faraci FM, Heistad DD. Effects of cilazapril on cerebral vasodilatation in hypertensive rats. Hypertension. 1993;22:150–5.
Yang ST, Mayhan WG, Faraci FM, Heistad DD. Endothelium-dependent responses of cerebral blood vessels during chronic hypertension. Hypertension. 1991;17:612–8.
Riedel MW, Anneser F, Haberl RL. Different mechanisms of l-arginine induced dilation of brain arterioles in normotensive and hypertensive rats. Brain Res. 1995;671:21–6.
Kitazono T, Faraci FM, Heistad DD. L-Arginine restores dilator responses of the basilar artery to acetylcholine during chronic hypertension. Hypertension. 1996;27:893–6.
Toth P, Csiszar A, Sosnowska D, Tucsek Z, Cseplo P, Springo Z, et al. Treatment with the cytochrome P450 ω-hydroxylase inhibitor HET0016 attenuates cerebrovascular inflammation, oxidative stress and improves vasomotor function in spontaneously hypertensive rats. Br J Pharmacol. 2013;168:1878–88.
Pires PW, Girgla SS, McClain JL, Kaminski NE, van Rooijen N, Dorrance AM. Improvement in middle cerebral artery structure and endothelial function in stroke-prone spontaneously hypertensive rats after macrophage depletion. Microcirculation. 2013;20:650–1.
Ge T, Hughes H, Junquero DC, Wu KK, Vanhoutte PM, Boulanger CM. Endothelium-dependent contractions are associated with both augmented expression of prostaglandin H synthase-1 and hypersensitivity to prostaglandin H2 in the SHR aorta. Circ Res. 1995;76:1003–10.
Virdis A, Ghiadoni L, Taddei S. Human endothelial dysfunction: EDCFs. Pflugers Arch. 2010;459:1015–23.
Saunders NR, Habgood MD, Dziegielewska KM. Barrier mechanisms in the brain, I. Adult brain. Clin Exp Pharmacol Physiol. 1999;26:11–9.
Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11.
Daneman R. The blood-brain barrier in health and disease. Ann Neurol. 2012;72:648–72.
Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011;37:3–23.
Weidenfeller C, Svendsen CN, Shusta EV. The blood-brain barrier. In: Aird WC, editor. Endothelial biomedicine. Cambridge: Cambridge University Press; 2007. p. 1124–39.
Lippmann ES, Weidenfeller C, Svendsen CN, Shusta EV. Blood-brain barrier modeling with co-cultured neural progenitor cell-derived astrocytes and neurons. J Neurochem. 2011;119:507–20.
Zehendner CM, Luhmann HJ, Kuhlmann CR. Studying the neurovascular unit: an improved blood-brain barrier model. J Cereb Blood Flow Metab. 2009;29:1879–84.
Shayan G, Choi YS, Shusta EV, Shuler ML, Lee KH. Murine in vitro model of the blood-brain barrier for evaluating drug transport. Eur J Pharm Sci. 2011;42:148–55.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–6.
Ribatti D, Nico B, Crivellato E, Artico M. Development of the blood-brain barrier: a historical point of view. Anatomical Record Part B New Anatomist. 2006;289:3–8.
Reese TS, Karnovsky MJ. Fine structural localization of a blood-brain barrier to exogenous peroxidase. J Cell Biol. 1967;34:207–17.
Nishitsuji K, Hosono T, Nakamura T, Bu G, Michikawa M. Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J Biol Chem. 2011;286:17536–42.
Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323–8.
Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62.
Nacer A, Movila A, Baer K, Mikolajczak SA, Kappe SH, Frevert U. Neuroimmunological blood brain barrier opening in experimental cerebral malaria. PLoS Pathogens. 2012;8:e1002982.
Knowland D, Arac A, Sekiguchi KJ, Hsu M, Lutz SE, Perrino J, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82:603–17.
Ueno M, Wu B, Nishiyama A, Huang CL, Hosomi N, Kusaka T, et al. The expression of matrix metalloproteinase-13 is increased in vessels with blood-brain barrier impairment in a stroke-prone hypertensive model. Hypertens Res. 2009;32:332–8.
Lakhan SE, Kirchgessner A, Tepper D, Leonard A. Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol. 2013;4:32.
Pons M, Cousins SW, Alcazar O, Striker GE, Marin-Castano ME. Angiotensin II-induced MMP-2 activity and MMP-14 and basigin protein expression are mediated via the angiotensin II receptor type 1-mitogen-activated protein kinase 1 pathway in retinal pigment epithelium: implications for age-related macular degeneration. Am J Pathol. 2011;178:2665–81.
Nakai K, Kawato T, Morita T, Iinuma T, Kamio N, Zhao N, et al. Angiotensin II induces the production of MMP-3 and MMP-13 through the MAPK signaling pathways via the AT1 receptor in osteoblasts. Biochimie. 2013;95:922–33.
Odenbach J, Wang X, Cooper S, Chow FL, Oka T, Lopaschuk G, et al. MMP-2 mediates angiotensin II-induced hypertension under the transcriptional control of MMP-7 and TACE. Hypertension. 2011;57:123–30.
Walter A, Etienne-Selloum N, Sarr M, Kane MO, Beretz A, Schini-Kerth VB. Angiotensin II induces the vascular expression of VEGF and MMP-2 in vivo: preventive effect of red wine polyphenols. J Vasc Res. 2008;45:386–94.
Wakisaka Y, Chu Y, Miller JD, Rosenberg GA. Spontaneous intracerebral hemorrhage during acute and chronic hypertension in mice. J Cereb Blood Flow Metab. 2009;30:56–69.
Johansson BB. Experimental models of altering the blood-brain barrier. Prog Brain Res. 1992;91:171–5.
Reynolds MR, Willie JT, Zipfel GJ, Dacey RG. Sexual intercourse and cerebral aneurysmal rupture: potential mechanisms and precipitants. J Neurosurg. 2011;114:969–77.
MacDougall JD, Tuxen D, Sale DG, Moroz JR, Sutton JR. Arterial blood pressure response to heavy resistance exercise. J Appl Physiol. 1985;58:785–90.
Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet. 2000;356:411–7.
Heistad DD, Faraci FM, Talman WT. Pathogenesis of acute hypertensive encephalopathy. In: Izzo JL, Sica DA, Black HR, editors. Hypertension primer. Philadelphia: Lippincott Williams & Wilkins; 2008. p. 217–9.
Qi X, Inagaki K, Sobel RA, Mochly-Rosen D. Sustained pharmacological inhibition of deltaPKC protects against hypertensive encephalopathy through prevention of blood-brain barrier breakdown in rats. J Clin Invest. 2008;118:173–82.
Zhang XM, Ellis EF. Superoxide dismutase reduces permeability and edema induced by hypertension in rats. Am J Physiol. 1990;259:H497–503.
Mohammadi MT, Dehghani GA. Acute hypertension induces brain injury and blood-brain barrier disruption through reduction of claudins mRNA expression in rat. Pathol Res Pract. 2014;210:985–90. doi:10.1016/j.prp.2014.05.007.
Baumbach GL, Heistad DD. Heterogeneity of brain blood flow and permeability during acute hypertension. Am J Physiol. 1985;249:H629–37.
Mayhan WG, Faraci FM, Heistad DD. Disruption of the blood-brain barrier in cerebrum and brain stem during acute hypertension. Am J Physiol. 1986;251:H1171–5.
Mayhan WG, Faraci FM, Heistad DD. Mechanisms of protection of the blood-brain barrier during acute hypertension in chronically hypertensive rats. Hypertension. 1987;9:III101–5.
Mayhan WG, Heistad DD. Role of veins and cerebral venous pressure in disruption of the blood-brain barrier. Circ Res. 1986;59:216–20.
Ueno M, Sakamoto H, Liao YJ, Onodera M, Huang CL, Miyanaka H, et al. Blood-brain barrier disruption in the hypothalamus of young adult spontaneously hypertensive rats. Histochem Cell Biol. 2004;122:131–7.
Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension. 2014;63:572–9.
Nakagawa T, Hasegawa Y, Uekawa K, Ma M, Katayama T, Sueta D, et al. Renal denervation prevents stroke and brain injury via attenuation of oxidative stress in hypertensive rats. J Am Heart Assoc. 2013;2:e000375.
Joutel A, Faraci FM. Cerebral small vessel disease: insights and opportunities from mouse models of collagen IV-related small vessel disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke. 2014;45:1215–21.
Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 2013;12:483–97.
Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–8.
Vital SA, Terao S, Nagai M, Granger DN. Mechanisms underlying the cerebral microvascular responses to angiotensin II-induced hypertension. Microcirculation. 2010;17:641–9.
Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732–42.
Rosenblum WI, Donnenfeld H, Aleu F. Effects of increased blood pressure on cerebral vessels in mice. Arch Neurol. 1966;14:631–43.
Manfre L, Midiri M, Giuffre G, Mangiameli A, Cardella G, Ponte F, et al. Blood-ocular barrier damage: use of contrast-enhanced MRI. Eur Radiol. 1997;7:110–4.
Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology. 2007;68:1809–14.
Wardlaw JM, Doubal FN, Valdes-Hernandez M, Wang X, Chappell FM, Shuler K, et al. Blood-brain barrier permeability and long-term clinical and imaging outcomes in cerebral small vessel disease. Stroke. 2013;44:525–7.
Dankbaar JW, Hom J, Schneider T, Cheng SC, Lau BC, van der Schaaf I, et al. Age- and anatomy-related values of blood-brain barrier permeability measured by perfusion-CT in non-stroke patients. J Neuroradiol. 2009;36:219–27.
Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H. Blood-brain barrier dysfunction in Binswanger’s disease: an immunohistochemical study. Acta Neuropathol. 1998;95:78–84.
Wei EP, Kontos HA, Christman CW, DeWitt DS, Povlishock JT. Superoxide generation and reversal of acetylcholine-induced cerebral arteriolar dilation after acute hypertension. Circ Res. 1985;57:781–7.
Chan SL, Sweet JG, Cipolla MJ. Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. FASEB J. 2013;27:3917–27.
Sobey CG, Moffatt JD, Cocks TM. Evidence for selective effects of chronic hypertension on cerebral artery vasodilatation to protease-activated receptor-2 activation. Stroke. 1999;30:1933–40.
Capone C, Faraco G, Coleman C, Young CN, Pickel VM, Anrather J, et al. Endothelin 1-dependent neurovascular dysfunction in chronic intermittent hypoxia. Hypertension. 2012;60:106–13.
Chrissobolis S, Drummond GR, Faraci FM, Sobey CG. Chronic aldosterone administration causes Nox2-mediated increases in reactive oxygen species production and endothelial dysfunction in the cerebral circulation. J Hypertens. 2014;32:1815–21.
Moreau P, Takase H, Kung CF, van Rooijen MM, Schaffner T, Luscher TF. Structure and function of the rat basilar artery during chronic nitric oxide synthase inhibition. Stroke. 1995;26:1922–8.
Chan SL, Baumbach GL. Nox2 deficiency prevents hypertension-induced vascular dysfunction and hypertrophy in cerebral arterioles. Int J Hypertens. 2013;2013:793630.
Acknowledgments
Work summarized in this chapter was supported by research grants from the National Institute of Health (HL-38901, NS-24621, HL-62984, and HL-113863), the Department of Veteran’s Affair’s (BX001399), and the Fondation Leducq (Transatlantic Network of Excellence on the Pathogenesis of Cerebral Small Vessel Disease in the Brain). TMD was the recipient of an Overseas Post-doctoral Fellowship from the National Health and Medical Research Council of Australia (1053786). RP received a North Shore University Hospital Chair of Research Award from the Brain Aneurysm Foundation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
De Silva, T.M., Silva, R.A.P., Faraci, F.M. (2016). Endothelium, the Blood–Brain Barrier, and Hypertension. In: Girouard, H. (eds) Hypertension and the Brain as an End-Organ Target. Springer, Cham. https://doi.org/10.1007/978-3-319-25616-0_8
Download citation
DOI: https://doi.org/10.1007/978-3-319-25616-0_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-25614-6
Online ISBN: 978-3-319-25616-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)