
Abstract
Gastrointestinal stromal tumor (GIST) is the most commonly diagnosed mesenchymal tumor in the gastrointestinal tract. This tumor type is driven by gain-of-function mutations in receptor tyrosine kinases (such as KIT, PDGFRA, and BRAF) or loss-of-function mutations in succinate dehydrogenase complex subunit genes (SDHx). Molecular studies on GIST have improved our understanding of the biology of the disease and have led to the use of targeted therapy approach, such as imatinib for KIT/PDGFRA-mutated GIST. Recently, microRNAs have emerged as important regulators of KIT expression, cancer cell behavior, and imatinib response in GIST. This chapter aims to provide an overview on current understanding of the biological roles of microRNAs in GIST and possible implications in prognosis and therapeutic response.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
Gastrointestinal stromal tumors (GISTs) comprise one-fifth of soft tissue sarcomas, making them the most common sarcoma of the gastrointestinal tract [1]. The annual incidence of GIST is between 11 and 19.5 per million [2–5], and it has a prevalence of about 130 cases per million population [2–4]. For many years, GISTs were considered as smooth muscle sarcomas based on their morphology, and had been misdiagnosed as leiomyomas, leiomyosarcomas, or leiomyoblastomas. The prognosis of advanced GIST was very poor due to resistance to conventional chemotherapy and radiotherapy prior to the discovery of targeted therapies [6].
In the late 1990s, two groundbreaking discoveries had revolutionized the approach to diagnosis and treatment of GIST: (1) majority of GISTs (>95 %) were found immunohistochemically positive for the tyrosine kinase receptor KIT (also known as CD117) [7], and (2) KIT gene mutations were identified in 70–80 % of GISTs [8]. To date, KIT immunostaining and mutation screening are used as key diagnostic markers in clinical practice for GISTs, and mutant KIT is a clinically important therapeutic target in GISTs. The evolution of understanding the biology of GIST transformed it from a challenging chemotherapy-resistant disease to a model for molecular targeted therapy.
Although the initial events in GIST development are well characterized, the prognosis is clearly influenced by other genetic or epigenetic events that are still poorly understood. Aberrant microRNA expression is common in a wide range of human cancers. Accumulated evidence has shown that microRNAs are associated with clinical and pathological features in GIST, suggesting their important roles in GIST development.
This chapter gives a brief background on clinical features and biology of GIST, and provides an overview of the current knowledge on involvement of microRNAs in GIST tumorigenesis and therapeutic response.
Gastrointestinal Stromal Tumor
GISTs are thought to originate from the interstitial cells of Cajal (ICC) or their stem-like precursors [7, 8]. ICC function as pacemaker in the gastrointestinal tract that controls peristaltic contractions [7]. GISTs can be found anywhere along the gastrointestinal tract, but predominantly occur in the stomach (50–60 %) and the small intestine (30–35 %), less frequently in the colon/rectum (5 %) and esophagus (<1 %) [9]. These tumors can arise at any age, with a median age of diagnosis at 63 years [1, 9]. The tumor size varies between 2 and 30 cm at the time of diagnosis [10].
Oncogenic Mutations
The main initial event in GIST tumorigenesis is gain-of-function mutations in KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) or PDGFRA (platelet-derived growth factor-α) genes. These genes are located on the long arm of chromosome 4 (4q12), and encode transmembrane proteins that belong to the type III tyrosine kinase receptor family.
Under normal physiological conditions, activation of KIT and PDGFRA receptors is controlled by spatial and temporal expression of their respective ligands, SCF and PDGF. Binding of these ligands to the receptors results in homodimerization, transphosphorylation of the tyrosine residues, and kinase activation that initiates signal transduction cascades promoting cell proliferation, growth, and survival [11–13]. About 75 % of GISTs harbor KIT mutations [14], whereas 10 % of GISTs harbor PDGFRA mutations [15, 16]. These mutations disrupt the autoregulatory mechanisms and cause ligand-independent constitutive activation of the encoded tyrosine kinase receptors [17], which results in aberrant cell growth and tumor formation [18]. Activation of KIT or PDGFRA stimulates several downstream signaling pathways such as mitogen-activated protein kinase (MAPK), phosphatidylinositol-3-kinase (PI3K)/AKT/mTOR and signal transducer, and activator of transcription 3 (STAT3) [19–21].
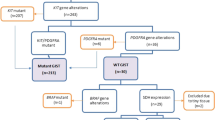
About 10–15 % of GISTs do not harbor KIT or PDGFRA mutations. These tumors display mutations in multiple cancer genes, including succinate dehydrogenase complex subunit genes (SDHA, SDHB, SDHC, and SDHD) (50 %) [22, 23], BRAF V600E substitution (13 %) [24], neurofibromin 1 (NF1) (7 %) [25, 26], and RAS family members [27]. Different signaling pathways in GIST are illustrated in Fig. 4.1.

Signaling pathways in GIST. (a) KIT and PDGFRA signaling pathways. Mutations in KIT or PDGFRA activate MAPK, PI3K/AKT/mTOR, and STAT3 pathways. The overall percentage of specific mutation sites is given in parentheses. (b) Signaling pathways in “wild-type” GISTs. Mutations in NF1, BRAF, or RAS lead to increased MAPK signaling. Mutations in one of the SDH genes (SDHA, SDHB, SDHC, or SDHD) lead to succinate accumulation, which inhibits prolyl hydroxylase-mediated HIF1α degradation and thereby increased HIF1α-mediated transcription of VEGF and IGF. P Phosphate group (Modified from Akcakaya P, thesis for doctoral degree 2015, ISBN 978-91-7549-730-3)
Unlike adult GISTs, pediatric GISTs (1–2 % of all GISTs) are rarely positive for KIT or PDGFRA mutations, despite expressing KIT at similar levels as adult GISTs [28]. Gene expression pattern of these tumors is also different from adult GISTs [29, 30], suggesting alternative mechanisms of KIT activation or distinct pathways in pediatric GISTs.
Chromosomal Changes in GIST
Cytogenetic studies demonstrated that about 65 % of GISTs have either monosomy of chromosome 14 or partial loss of 14q [31–33]. Loss of heterozygosity and comparative genomic hybridization studies identified two hotspot regions (14q11.2 and 14q32), pointing tumor suppressor genes at these loci might be important for GIST development [32, 34]. Several candidate genes are suggested within these regions, such as PARP2, APEX1, and NDRG2 genes at 14q11.2, SIVA [35] and microRNA clusters at 14q32 [36, 37].
Several chromosomal abnormalities have been associated with malignant behavior in GIST. Loss of the long arm of chromosome 22 is observed in approximately 50 % of GISTs and associated with malignancy [31, 33, 38]. Chromosome 9p21 deletion causes inactivation of the tumor suppressor gene CDKN2A and associated with metastatic behavior [39–42]. Gains on chromosomes 8q (including MYC), 3q (including SMARCA3) and 17q are associated with metastasis [32, 43–45].
Treatment of GIST
Surgical resection is the main therapy for localized GIST, with the goal of complete resection and avoidance of tumor rupture [46]. However, surgery is sometimes not applicable for metastatic GISTs or clinically unresectable GISTs. A small molecule tyrosine kinase receptor inhibitor, such as imatinib mesylate, is used for the treatment of advanced GISTs.
Imatinib can selectively block the enzymatic activity of both transmembrane receptor tyrosine kinases KIT and PDGFRA [47, 48]. It competes with ATP for the ATP-binding pocket located in the kinase domain, and blocks the phosphorylation of the tyrosine kinase receptors. Binding of imatinib inhibits the activation of downstream survival pathways such as PI3K-mTOR and MAPK [19], and induces cell apoptosis through BIM [49] and soluble histone H2AX [50]. In addition, imatinib reduces the expression of indoleamine 2,3-dioxygenase (IDO) [51], which is an enzyme that produces immunosuppressive metabolites. Reduction of IDO causes depletion of regulatory T cells and increase of tumor-infiltrating CD8+ T cells. Thus, imatinib stimulates an anticancer immune response by diminishing IDO-mediated immunosuppression.
The majority of GIST patients with advanced disease get a clinical benefit from imatinib treatment. Imatinib achieved disease control in 70–85 % of patients with advanced GIST, median progression-free survival increased from 8–10 months to 20–24 months, and median overall survival increased from 18–20 months to 50 months [52–54]. However, resistance to imatinib is one of the biggest obstacles in current GIST clinical practice.
Approximately 10 % of patients progress within 6 months of initial therapy, which is defined as primary resistance to imatinib [53–56]. Primary resistance shows stronger correlation with certain tumor genotypes, such as wild-type KIT or PDGFRA, KIT exon 9 mutations and PDGFRA D842V mutation [57–60]. In addition, 50–60 % of the initially responding patients develop disease progression within 2 years, regarded as secondary or acquired resistance [53–56]. The main mechanism of acquired resistance is the acquisition of secondary mutations in the kinase or loop domain of KIT or PDGFRA [61]. Several alternative mechanisms of resistance have been described. Kinase switching is one of them and several kinases have been involved in such mechanism. AXL is an oncogenic tyrosine kinase receptor that regulates the same downstream signaling pathways as KIT. Kinase switching from KIT to AXL was observed in imatinib-resistant GIST cell lines and clinical samples [62]. Besides AXL, a switch from KIT to FAK and FYN activation has also been reported in GIST cells upon acquisition of imatinib resistance, and phosphorylated FAK inhibition can re-sensitize the resistant cells to imatinib-induced cell death [63]. FAK has also been implicated in growth and survival of imatinib-resistant GIST cells [64]. In addition, gene amplification of KIT or PDGFRA was shown as a potential mechanism leading to either primary or secondary resistance [65]. Moreover, microRNAs have also been shown to play a role in imatinib resistance in GIST, as described in the following section.
MicroRNA Deregulation in GIST
MicroRNA signature of GISTs was first described by Subramanian and colleagues in 2008 [66]. The study compared microRNA profiles of 27 sarcomas with different histological types, and demonstrated that GISTs were clearly distinguished from other sarcomas based on their microRNA expressions (Table 4.1). This distinction implicates the role of microRNAs in GIST tumorigenesis and their potential applications as diagnostic markers or therapeutic targets in GIST. Compared to other sarcoma types, miR-221–222 and miR-17–92 clusters were expressed at lower level in GIST [66, 67]. These microRNAs have been shown to target the two key factors KIT and ETV1 in GIST tumorigenesis [67] (Table 4.2), suggesting that lower expression of these microRNAs in GIST could be important for the pathogenesis of this tumor type. The current known microRNAs involved in regulating key genes in GIST development and progression are shown in Fig. 4.2.

MicroRNAs involved in the regulation of GIST development, progression, and imatinib response. In brief, miR-221, miR-222, miR-494, and miR-218 directly target the KIT expression, while miR-17 and miR-20a regulates the survival factor ETV1. miR-133b and miR-137 regulate GIST progression by targeting FSCN1 and TWIST1, respectively. miR-125a-5p regulates imatinib response through the regulation of PTPN18. miR-218 also regulates imatinib response. IM Imatinib, P Phosphate group
MicroRNAs Associated with Clinical and Pathological Features in GIST
Morphology, clinical behavior, and molecular biology of GISTs differ according to their anatomical localization [68]. Likewise, microRNA expression profiles of GISTs located in stomach are distinct from the GISTs found in small intestine [36, 37]. Notably, different sets of microRNAs associated with anatomical location were observed in different studies. For example, Haller et al. showed that gastric GISTs presented higher expressions of miR-504, miR-7-1*, miR-598, and miR-24-1*, while the intestinal GISTs had higher levels of miR-220c, miR-229, miR-370, miR-210, miR-409-3p, miR-376a, and miR-376c [37]. Choi et al. demonstrated higher expressions of miR-383, miR-136, miR-146a, and miR-409a-3p, and lower expressions of miR-124a, miR-199b, miR-451, miR-663, miR-10a, and miR-218 in the intestinal compared to gastric GISTs [36]. The discrepancy is likely due to additional factors (e.g., risk grade and mutation status) that may contribute to differences besides anatomical locations in the tumors analyzed in both studies.
Several microRNA signatures have been described in GIST progression. In terms of tumor-risk group, a number of studies revealed distinct microRNA expression patterns between the high-risk and the low-risk GISTs, and identified a number of tumor-risk associated microRNAs (Table 4.1) [36, 69–71]. In the study of Choi et al., they compared microRNA profiles of 10 high-risk and 4 low-risk GISTs, and identified 28 microRNAs to be expressed at lower level in the high-risk group [36]. Yamamoto et al. reported 24 microRNAs with lower expression in the high-risk GISTs compared to low-to-intermediate risk tumors [70]. Kelly et al. found only miR-150 to be expressed at higher level in the low-risk tumors [71], and Niinuma et al. reported higher miR-196a expression in the high-risk group [69].
Besides tumor risk, several microRNAs are associated with tumor metastasis in GISTs. For example, low expression of miR-150-3p and high expressions of miR-301a-3p and miR-196a are associated with metastasis in GIST [69, 72]. In experimental cell culture systems, two microRNAs have been evaluated for their effect on tumor progression. Overexpression of miR-137 can inhibit cell migration and regulates epithelial-to-mesenchymal transition (EMT) by targeting TWIST1 [73], and inhibition of miR-196a can suppress cell invasion in GIST cells [69].
In terms of survival, low expression of miR-1915 is associated with disease-free and overall survival [72], while higher miR-196a expression is associated with poorer overall survival of GIST patients [69].
MicroRNAs Associated with Chromosomal and Genetic Alterations in GIST
As previously described, loss of 14q is common in GIST [31–33]. Downregulation of multiple microRNA clusters located at chromosome 14q (i.e., 14q32.31 and 14q32.33) has been reported in GISTs with 14q loss (Table 4.1) [36, 37]. One of the microRNAs located in this region, i.e., miR-494, was shown to directly target KIT and suppress its expression, and activates downstream signaling components such as AKT and STAT3 [74]. Functionally, inhibition of miR-494 suppresses proliferation and induces apoptosis in GIST cells [74].
Given that KIT and PDGFRA are key factors involved in GIST tumorigenesis, microRNA-mediated regulation of these factors is important for GIST development. As aforementioned, miR-494, miR-221, and miR-222 have been shown to directly regulate KIT expression in GIST cells [67, 74]. Recently, miR-218 was also found directly targeting KIT, and its overexpression suppresses proliferation and invasion, and induces apoptosis in GIST-T1 cells [75]. On the other hand, PDGFRA is known to be regulated by several microRNAs in different cell types, such as miR-126 in osteoblasts [76], miR-34a in gastric cancer [77], lung cancer [78], and glioma [79], and miR-146a/146b-5p in endothelial [80] and hematopoietic cells [81]; however, no microRNA has been experimentally validated to target PDGFRA in GIST.
GISTs show differential microRNA expression patterns according to their mutation status [37]. Several microRNAs are associated with KIT or PDGFRA-mutated GISTs. For example, miR-132, miR-766, miR-652, miR-629, miR-200c, miR-342-3p, miR-185, miR-146b-5p, and miR-150 levels are higher, whereas miR-330-3p is lower in PDGFRA-mutated GISTs as compared to KIT-mutated GISTs [37]. Higher expressions of miR-221 and miR-222 were found in the wild-type tumors compared to the tumors with KIT or PDGFRA mutation [37]. Concordantly, several studies have also revealed distinct mRNA expression profiles between GISTs with KIT and PDGFRA mutations [66, 82]. These findings suggest that, despite the common pathways activated by both mutations (e.g., PI3K/AKT and MAPK) [15], differences exist in the signal transduction networks between GISTs with KIT and PDGFRA mutations. In addition, several microRNAs are differentially expressed between GISTs with a single and double KIT mutations [72], suggesting that these microRNAs may be involved in partly distinct pathways [72].
Besides KIT and PDGFRA, several microRNAs are also associated with SDHB mutation. The SDHB-mutated GISTs show several microRNAs with higher (miR-132, miR-146a, miR-193b, miR-193b*, miR-455-3p, miR-455-5p, miR-484, and miR-886-5p) and lower (miR-125b, miR-450b, miR-488*, miR-542-3p, miR-551b, miR-576-3p and miR-769-5p) expressions compared to non-SDHB-mutated tumors [71].
MicroRNAs in Imatinib Resistance in GIST
MicroRNAs are known to play a role in tyrosine kinase inhibitor resistance [83–88]. The best example is the EGFR-inhibitor resistance in lung cancer. Numerous microRNAs (e.g., miR-205, miR-374a, miR-548b, miR-30b, miR-30c, miR-221, miR-222, and miR-200 family members) have been shown to regulate EGFR-inhibitor response in lung cancer [84, 85, 89, 90]. In chronic myelogenous leukemia (CML), miR-17–19b, miR-30e, miR-203, and miR-138 have been demonstrated to modulate imatinib sensitivity, while miR-30a promotes autophagy that enhances imatinib resistance [91–99].
In GIST, only two microRNAs have been functionally determined to modulate imatinib response [72, 100], despite a number of microRNAs are associated with imatinib resistance [72]. The expression of miR-218 is lower in imatinib-resistant compared to -sensitive GIST cell lines. Overexpression of miR-218 increases imatinib-induced cell death in the imatinib-resistant GIST430 cells. On the other hand, inhibition of miR-218 expression increases cell viability and decreases apoptosis in the imatinib-sensitive GIST882 cells upon imatinib treatment. Although no target gene(s) of miR-218 was identified, the authors propose that the effect might be mediated through PI3K/AKT signaling pathway.
The second microRNA is miR-125a-5p, which was found at higher expression levels in the imatinib-resistant than the -sensitive GISTs [72]. Overexpression of miR-125a-5p increases cell viability in the single KIT-mutated GIST882 cells upon imatinib treatment. However overexpression or suppression of miR-125a-5p in the double KIT-mutated GIST48 cells has no effect on imatinib response, suggesting that microRNA-mediated regulation is an alternative resistance mechanism to secondary KIT mutations in GIST. Ectopic expression of miR-125a-5p suppresses its target gene PTPN18 expression and silencing of PTPN18 increases cell viability in GIST882 cells upon imatinib treatment. The authors also observed an increased expression of miR-125a-5p and a decreased expression of PTPN18 in the imatinib-resistant subclone of GIST882 cells as compared to its sensitive counterpart, providing the functional evidence of miR-125a-5p-mediated regulation in imatinib resistance. PTPN18 is a member of the PEST domain containing protein-tyrosine phosphatase superfamily, which has been shown to dephosphorylate the phosphotyrosine residues of several tyrosine kinases, such as HER2 and SRC [101, 102]. Takahashi et al. recently demonstrated that altered phosphorylation of tyrosine kinases is an alternative mechanism of imatinib resistance in GIST [63]. Further studies have yet to determine whether the tyrosine kinases described by Takahashi et al. could be the substrate(s) of PTPN18.
Clinical Implications of microRNAs in GIST
MicroRNA expression profiles can distinguish GISTs from other sarcomas, and distinct microRNA expression signatures are associated with clinical, molecular, and histopathological features of GIST. These findings suggest a promising role for microRNAs as diagnostic and prognostic indicators in GIST.
Given their relatively higher stability in clinical samples and robust expression patterns, microRNAs have been suggested to have a greater utility as biomarkers in comparison to mRNAs [103]. Importantly, microRNAs can be released into the body fluids through microvesicles, which gives them a potential value as noninvasive biomarkers [104, 105]. Future studies evaluating the potential of circulating microRNAs as response markers for treatment or as reflective markers of GIST biological outcome would have a clinical benefit. However, there are some obstacles for circulating microRNAs, e.g., identification of an appropriate endogenous control and fluctuations in microRNA expression caused by diet, infection, treatment, trauma, or other factors [106].
Inhibition of KIT and PDGFRA by imatinib is the key therapeutic approach for advanced GISTs beside surgery. However, imatinib resistance is one of the biggest challenges in current GIST clinical practice. Post-transcriptional inhibition of oncogenes by microRNA mimics and activation of tumor suppressor genes by microRNA inhibitors are currently under investigation for their potential as therapeutic agents in cancer. KIT-targeting microRNA mimics (e.g., miR-221, miR-222, miR-494) [67, 74] may be used directly to target GIST cells to enhance the effect of imatinib for the purpose of overcoming resistance. Likewise, microRNA mimics/inhibitors for microRNAs specific to imatinib resistance, metastasis, risk grade or survival may be used for therapeutic purposes. Off-target effects and delivery of these molecules to specific GIST tissues/cells remain as the biggest challenges. Several strategies have been developed for delivery of microRNA-based therapeutics, including the use of nanoparticles, liposomes, antibodies and nucleic acid structure modifications [107].
Conclusion
In the last 20 years, growing knowledge of GIST molecular biology has revolutionized the clinical management of this disease, from a treatment-resistant uncontrolled disease to the development of targeted therapies. Despite tyrosine kinase inhibitors improve the outcome of the majority of patients, they fail to provide a permanent cure and resistant clones are observed in most of the initially responding tumors.
Development of alternative treatment strategies is needed in order to overcome resistance to ATP-competitive kinase inhibitors. Complete understanding of molecular biology in GIST development, progression, and treatment response is necessary to establish a ground for developing effective combinational therapies with a goal of not only to temporarily control the disease, but also to permanently eradicate all tumor cells.
MicroRNAs have been shown to play a role not only in the GIST tumorigenesis, but also in the stratification of patients at risk of developing the disease or therapy response. Although this research area is still relatively understudied, the work reported in the last 3 years is indicative of the excitement in this area. Ongoing and future studies will illuminate effectiveness and safeness of microRNAs as novel agents for GIST treatment and their predictive value as novel biomarkers. This will hopefully turn GIST from a model of targeted therapies that control the disease progression to a model of complete cancer cure.
References
Ducimetiere F, Lurkin A, Ranchere-Vince D, Decouvelaere AV, Peoc’h M, Istier L, Chalabreysse P, Muller C, Alberti L, Bringuier PP, Scoazec JY, Schott AM, Bergeron C, Cellier D, Blay JY, Ray-Coquard I. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One. 2011;6, e20294.
Chan KH, Chan CW, Chow WH, Kwan WK, Kong CK, Mak KF, Leung MY, Lau LK. Gastrointestinal stromal tumors in a cohort of Chinese patients in Hong Kong. World J Gastroenterol. 2006;12:2223–8.
Goettsch WG, Bos SD, Breekveldt-Postma N, Casparie M, Herings RM, Hogendoorn PC. Incidence of gastrointestinal stromal tumours is underestimated: results of a nation-wide study. Eur J Cancer. 2005;41:2868–72.
Nilsson B, Bumming P, Meis-Kindblom JM, Oden A, Dortok A, Gustavsson B, Sablinska K, Kindblom LG. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era—a population-based study in western Sweden. Cancer. 2005;103:821–9.
Tryggvason G, Gislason HG, Magnusson MK, Jonasson JG. Gastrointestinal stromal tumors in Iceland, 1990–2003: the icelandic GIST study, a population-based incidence and pathologic risk stratification study. Int J Cancer. 2005;117:289–93.
Dematteo RP, Heinrich MC, El-Rifai WM, Demetri G. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2002;33:466–77.
Kindblom LG, Remotti HE, Aldenborg F, Meis-Kindblom JM. Gastrointestinal pacemaker cell tumor (GIPACT): gastrointestinal stromal tumors show phenotypic characteristics of the interstitial cells of Cajal. Am J Pathol. 1998;152:1259–69.
Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279:577–80.
Joensuu H, Vehtari A, Riihimaki J, Nishida T, Steigen SE, Brabec P, Plank L, Nilsson B, Cirilli C, Braconi C, Bordoni A, Magnusson MK, Linke Z, Sufliarsky J, Federico M, Jonasson JG, Dei Tos AP, Rutkowski P. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13:265–74.
Corless CL, McGreevey L, Haley A, Town A, Heinrich MC. KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am J Pathol. 2002;160:1567–72.
Heldin CH. Dimerization of cell surface receptors in signal transduction. Cell. 1995;80:213–23.
Hubbard SR, Mohammadi M, Schlessinger J. Autoregulatory mechanisms in protein-tyrosine kinases. J Biol Chem. 1998;273:11987–90.
Roskoski Jr R. Signaling by Kit protein-tyrosine kinase—the stem cell factor receptor. Biochem Biophys Res Commun. 2005;337:1–13.
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78.
Heinrich MC, Corless CL, Duensing A, McGreevey L, Chen CJ, Joseph N, Singer S, Griffith DJ, Haley A, Town A, Demetri GD, Fletcher CD, Fletcher JA. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10.
Hirota S, Ohashi A, Nishida T, Isozaki K, Kinoshita K, Shinomura Y, Kitamura Y. Gain-of-function mutations of platelet-derived growth factor receptor alpha gene in gastrointestinal stromal tumors. Gastroenterology. 2003;125:660–7.
Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, English JM, Greig MJ, He YA, Jacques SL, Lunney EA, McTigue M, Molina D, Quenzer T, Wells PA, Yu X, Zhang Y, Zou A, Emmett MR, Marshall AG, Zhang HM, Demetri GD. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106:1542–7.
Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–25.
Bauer S, Duensing A, Demetri GD, Fletcher JA. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene. 2007;26:7560–8.
Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, Fletcher CD, Demetri GD, Fletcher JA. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23:3999–4006.
Rossi F, Ehlers I, Agosti V, Socci ND, Viale A, Sommer G, Yozgat Y, Manova K, Antonescu CR, Besmer P. Oncogenic Kit signaling and therapeutic intervention in a mouse model of gastrointestinal stromal tumor. Proc Natl Acad Sci U S A. 2006;103:12843–8.
Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, Dahia PL, Liegl B, Ball ER, Raygada M, Lai AH, Kelly L, Hornick JL, O’Sullivan M, de Krijger RR, Dinjens WN, Demetri GD, Antonescu CR, Fletcher JA, Helman L, Stratakis CA. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U S A. 2011;108:314–8.
Pantaleo MA, Astolfi A, Indio V, Moore R, Thiessen N, Heinrich MC, Gnocchi C, Santini D, Catena F, Formica S, Martelli PL, Casadio R, Pession A, Biasco G. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst. 2011;103:983–7.
Hostein I, Faur N, Primois C, Boury F, Denard J, Emile JF, Bringuier PP, Scoazec JY, Coindre JM. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol. 2010;133:141–8.
Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, Shinomura Y, Matsuzawa Y. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol. 2004;202:80–5.
Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom LG. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol. 2005;29:1170–6.
Miranda C, Nucifora M, Molinari F, Conca E, Anania MC, Bordoni A, Saletti P, Mazzucchelli L, Pilotti S, Pierotti MA, Tamborini E, Greco A, Frattini M. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18:1769–76.
Janeway KA, Liegl B, Harlow A, Le C, Perez-Atayde A, Kozakewich H, Corless CL, Heinrich MC, Fletcher JA. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007;67:9084–8.
Agaram NP, Laquaglia MP, Ustun B, Guo T, Wong GC, Socci ND, Maki RG, DeMatteo RP, Besmer P, Antonescu CR. Molecular characterization of pediatric gastrointestinal stromal tumors. Clin Cancer Res. 2008;14:3204–15.
Prakash S, Sarran L, Socci N, DeMatteo RP, Eisenstat J, Greco AM, Maki RG, Wexler LH, LaQuaglia MP, Besmer P, Antonescu CR. Gastrointestinal stromal tumors in children and young adults: a clinicopathologic, molecular, and genomic study of 15 cases and review of the literature. J Pediatr Hematol Oncol. 2005;27:179–87.
Bergmann F, Gunawan B, Hermanns B, Hoer J, Schumpelick V, Fuzesi L. Cytogenetic and morphologic characteristics of gastrointestinal stromal tumors. Recurrent rearrangement of chromosome 1 and losses of chromosomes 14 and 22 as common anomalies. Verh Dtsch Ges Pathol. 1998;82:275–8.
Debiec-Rychter M, Lasota J, Sarlomo-Rikala M, Kordek R, Miettinen M. Chromosomal aberrations in malignant gastrointestinal stromal tumors: correlation with c-KIT gene mutation. Cancer Genet Cytogenet. 2001;128:24–30.
Fukasawa T, Chong JM, Sakurai S, Koshiishi N, Ikeno R, Tanaka A, Matsumoto Y, Hayashi Y, Koike M, Fukayama M. Allelic loss of 14q and 22q, NF2 mutation, and genetic instability occur independently of c-kit mutation in gastrointestinal stromal tumor. Jpn J Cancer Res. 2000;91:1241–9.
El-Rifai W, Sarlomo-Rikala M, Andersson LC, Miettinen M, Knuutila S. High-resolution deletion mapping of chromosome 14 in stromal tumors of the gastrointestinal tract suggests two distinct tumor suppressor loci. Genes Chromosomes Cancer. 2000;27:387–91.
Assamaki R, Sarlomo-Rikala M, Lopez-Guerrero JA, Lasota J, Andersson LC, Llombart-Bosch A, Miettinen M, Knuutila S. Array comparative genomic hybridization analysis of chromosomal imbalances and their target genes in gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2007;46:564–76.
Choi HJ, Lee H, Kim H, Kwon JE, Kang HJ, You KT, Rhee H, Noh SH, Paik YK, Hyung WJ. MicroRNA expression profile of gastrointestinal stromal tumors is distinguished by 14q loss and anatomic site. Int J Cancer. 2010;126:1640–50.
Haller F, von Heydebreck A, Zhang JD, Gunawan B, Langer C, Ramadori G, Wiemann S, Sahin O. Localization- and mutation-dependent microRNA (miRNA) expression signatures in gastrointestinal stromal tumours (GISTs), with a cluster of co-expressed miRNAs located at 14q32.31. J Pathol. 2010;220:71–86.
Kim NG, Kim JJ, Ahn JY, Seong CM, Noh SH, Kim CB, Min JS, Kim H. Putative chromosomal deletions on 9P, 9Q and 22Q occur preferentially in malignant gastrointestinal stromal tumors. Int J Cancer. 2000;85:633–8.
Perrone F, Tamborini E, Dagrada GP, Colombo F, Bonadiman L, Albertini V, Lagonigro MS, Gabanti E, Caramuta S, Greco A, Torre GD, Gronchi A, Pierotti MA, Pilotti S. 9p21 locus analysis in high-risk gastrointestinal stromal tumors characterized for c-kit and platelet-derived growth factor receptor alpha gene alterations. Cancer. 2005;104:159–69.
Ricci R, Arena V, Castri F, Martini M, Maggiano N, Murazio M, Pacelli F, Potenza AE, Vecchio FM, Larocca LM. Role of p16/INK4a in gastrointestinal stromal tumor progression. Am J Clin Pathol. 2004;122:35–43.
Sabah M, Cummins R, Leader M, Kay E. Loss of heterozygosity of chromosome 9p and loss of p16INK4A expression are associated with malignant gastrointestinal stromal tumors. Mod Pathol. 2004;17:1364–71.
Schneider-Stock R, Boltze C, Lasota J, Miettinen M, Peters B, Pross M, Roessner A, Gunther T. High prognostic value of p16INK4 alterations in gastrointestinal stromal tumors. J Clin Oncol. 2003;21:1688–97.
El-Rifai W, Sarlomo-Rikala M, Andersson LC, Knuutila S, Miettinen M. DNA sequence copy number changes in gastrointestinal stromal tumors: tumor progression and prognostic significance. Cancer Res. 2000;60:3899–903.
O’Leary T, Ernst S, Przygodzki R, Emory T, Sobin L. Loss of heterozygosity at 1p36 predicts poor prognosis in gastrointestinal stromal/smooth muscle tumors. Lab Invest. 1999;79:1461–7.
Ylipaa A, Hunt KK, Yang J, Lazar AJ, Torres KE, Lev DC, Nykter M, Pollock RE, Trent J, Zhang W. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117:380–9.
Hohenberger P, Ronellenfitsch U, Oladeji O, Pink D, Strobel P, Wardelmann E, Reichardt P. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumour. Br J Surg. 2010;97:1854–9.
Buchdunger E, Cioffi CL, Law N, Stover D, Ohno-Jones S, Druker BJ, Lydon NB. Abl protein-tyrosine kinase inhibitor STI571 inhibits in vitro signal transduction mediated by c-kit and platelet-derived growth factor receptors. J Pharmacol Exp Ther. 2000;295:139–45.
Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA, Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000;96:925–32.
Gordon PM, Fisher DE. Role for the proapoptotic factor BIM in mediating imatinib-induced apoptosis in a c-KIT-dependent gastrointestinal stromal tumor cell line. J Biol Chem. 2010;285:14109–14.
Liu Y, Tseng M, Perdreau SA, Rossi F, Antonescu C, Besmer P, Fletcher JA, Duensing S, Duensing A. Histone H2AX is a mediator of gastrointestinal stromal tumor cell apoptosis following treatment with imatinib mesylate. Cancer Res. 2007;67:2685–92.
Balachandran VP, Cavnar MJ, Zeng S, Bamboat ZM, Ocuin LM, Obaid H, Sorenson EC, Popow R, Ariyan C, Rossi F, Besmer P, Guo T, Antonescu CR, Taguchi T, Yuan J, Wolchok JD, Allison JP, DeMatteo RP. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094–100.
MetaGIST. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–53.
Blanke CD, Rankin C, Demetri GD, Ryan CW, von Mehren M, Benjamin RS, Raymond AK, Bramwell VH, Baker LH, Maki RG, Tanaka M, Hecht JR, Heinrich MC, Fletcher CD, Crowley JJ, Borden EC. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J Clin Oncol. 2008;26:626–32.
Verweij J, Casali PG, Zalcberg J, LeCesne A, Reichardt P, Blay JY, Issels R, van Oosterom A, Hogendoorn PC, Van Glabbeke M, Bertulli R, Judson I. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364:1127–34.
Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5.
van Oosterom AT, Judson IR, Verweij J, Stroobants S, Dumez H, Donato di Paola E, Sciot R, Van Glabbeke M, Dimitrijevic S, Nielsen OS. Update of phase I study of imatinib (STI571) in advanced soft tissue sarcomas and gastrointestinal stromal tumors: a report of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2002;38 Suppl 5:S83–7.
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, Blay JY, Leyvraz S, Stul M, Casali PG, Zalcberg J, Verweij J, Van Glabbeke M, Hagemeijer A, Judson I. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–103.
Debiec-Rychter M, Wasag B, Stul M, De Wever I, Van Oosterom A, Hagemeijer A, Sciot R. Gastrointestinal stromal tumours (GISTs) negative for KIT (CD117 antigen) immunoreactivity. J Pathol. 2004;202:430–8.
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, McGreevey LS, Chen CJ, Van den Abbeele AD, Druker BJ, Kiese B, Eisenberg B, Roberts PJ, Singer S, Fletcher CD, Silberman S, Dimitrijevic S, Fletcher JA. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, Benjamin RS, Bramwell VH, Demetri GD, Bertagnolli MM, Fletcher JA. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–7.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CD, Sandau K, McDougall K, Ou WB, Chen CJ, Fletcher JA. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74.
Mahadevan D, Cooke L, Riley C, Swart R, Simons B, Della Croce K, Wisner L, Iorio M, Shakalya K, Garewal H, Nagle R, Bearss D. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene. 2007;26:3909–19.
Takahashi T, Serada S, Ako M, Fujimoto M, Miyazaki Y, Nakatsuka R, Ikezoe T, Yokoyama A, Taguchi T, Shimada K, Kurokawa Y, Yamasaki M, Miyata H, Nakajima K, Takiguchi S, Mori M, Doki Y, Naka T, Nishida T. New findings of kinase switching in gastrointestinal stromal tumor under imatinib using phosphoproteomic analysis. Int J Cancer. 2013;133:2737–43.
Sakurama K, Noma K, Takaoka M, Tomono Y, Watanabe N, Hatakeyama S, Ohmori O, Hirota S, Motoki T, Shirakawa Y, Yamatsuji T, Haisa M, Matsuoka J, Tanaka N, Naomoto Y. Inhibition of focal adhesion kinase as a potential therapeutic strategy for imatinib-resistant gastrointestinal stromal tumor. Mol Cancer Ther. 2009;8:127–34.
Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, Vranckx H, Wasag B, Prenen H, Roesel J, Hagemeijer A, Van Oosterom A, Marynen P. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–9.
Subramanian S, Lui WO, Lee CH, Espinosa I, Nielsen TO, Heinrich MC, Corless CL, Fire AZ, van de Rijn M. MicroRNA expression signature of human sarcomas. Oncogene. 2008;27:2015–26.
Gits CM, van Kuijk PF, Jonkers MB, Boersma AW, van Ijcken WF, Wozniak A, Sciot R, Rutkowski P, Schoffski P, Taguchi T, Mathijssen RH, Verweij J, Sleijfer S, Debiec-Rychter M, Wiemer EA. MiR-17-92 and miR-221/222 cluster members target KIT and ETV1 in human gastrointestinal stromal tumours. Br J Cancer. 2013;109:1625–35.
Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23:70–83.
Niinuma T, Suzuki H, Nojima M, Nosho K, Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki Y, Nishida T, Bamba T, Kanda T, Ajioka Y, Taguchi T, Okahara S, Takahashi H, Nishida Y, Hosokawa M, Hasegawa T, Tokino T, Hirata K, Imai K, Toyota M, Shinomura Y. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 2012;72:1126–36.
Yamamoto H, Kohashi K, Fujita A, Oda Y. Fascin-1 overexpression and miR-133b downregulation in the progression of gastrointestinal stromal tumor. Mod Pathol. 2013;26:563–71.
Kelly L, Bryan K, Kim SY, Janeway KA, Killian JK, Schildhaus HU, Miettinen M, Helman L, Meltzer PS, van de Rijn M, Debiec-Rychter M, O’Sullivan M. Post-transcriptional dysregulation by miRNAs is implicated in the pathogenesis of gastrointestinal stromal tumor [GIST]. PLoS One. 2013;8, e64102.
Akcakaya P, Caramuta S, Ahlen J, Ghaderi M, Berglund E, Ostman A, Branstrom R, Larsson C, Lui WO. microRNA expression signatures of gastrointestinal stromal tumours: associations with imatinib resistance and patient outcome. Br J Cancer. 2014;111:2091–102.
Liu S, Cui J, Liao G, Zhang Y, Ye K, Lu T, Qi J, Wan G. miR-137 regulates epithelial-mesenchymal transition in gastrointestinal stromal tumor. Tumour Biol. 2014;35:9131–8.
Kim WK, Park M, Kim YK, Tae YK, Yang HK, Lee JM, Kim H. MicroRNA-494 downregulates KIT and inhibits gastrointestinal stromal tumor cell proliferation. Clin Cancer Res. 2011;17:7584–94.
Fan R, Zhong J, Zheng S, Wang Z, Xu Y, Li S, Zhou J, Yuan F. MicroRNA-218 inhibits gastrointestinal stromal tumor cell and invasion by targeting KIT. Tumour Biol. 2014;35:4209–17.
Schmidt Y, Simunovic F, Strassburg S, Pfeifer D, Stark GB, Finkenzeller G. miR-126 regulates platelet-derived growth factor receptor-alpha expression and migration of primary human osteoblasts. Biol Chem. 2015;396:61–70.
Peng Y, Guo JJ, Liu YM, Wu XL. MicroRNA-34A inhibits the growth, invasion and metastasis of gastric cancer by targeting PDGFR and MET expression. Biosci Rep. 2014;34.
Garofalo M, Jeon YJ, Nuovo GJ, Middleton J, Secchiero P, Joshi P, Alder H, Nazaryan N, Di Leva G, Romano G, Crawford M, Nana-Sinkam P, Croce CM. MiR-34a/c-Dependent PDGFR-alpha/beta Downregulation Inhibits Tumorigenesis and Enhances TRAIL-Induced Apoptosis in Lung Cancer. PLoS One. 2013;8, e67581.
Silber J, Jacobsen A, Ozawa T, Harinath G, Pedraza A, Sander C, Holland EC, Huse JT. miR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis. PLoS One. 2012;7:e33844.
Zhu K, Pan Q, Zhang X, Kong LQ, Fan J, Dai Z, Wang L, Yang XR, Hu J, Wan JL, Zhao YM, Tao ZH, Chai ZT, Zeng HY, Tang ZY, Sun HC, Zhou J. MiR-146a enhances angiogenic activity of endothelial cells in hepatocellular carcinoma by promoting PDGFRA expression. Carcinogenesis. 2013;34:2071–9.
Zhai PF, Wang F, Su R, Lin HS, Jiang CL, Yang GH, Yu J, Zhang JW. The regulatory roles of microRNA-146b-5p and its target platelet-derived growth factor receptor alpha (PDGFRA) in erythropoiesis and megakaryocytopoiesis. J Biol Chem. 2014;289:22600–13.
Kang HJ, Nam SW, Kim H, Rhee H, Kim NG, Hyung WJ, Noh SH, Kim JH, Yun CO, Liu ET. Correlation of KIT and platelet-derived growth factor receptor alpha mutations with gene activation and expression profiles in gastrointestinal stromal tumors. Oncogene. 2005;24:1066–74.
Bryant JL, Britson J, Balko JM, Willian M, Timmons R, Frolov A, Black EP. A microRNA gene expression signature predicts response to erlotinib in epithelial cancer cell lines and targets EMT. Br J Cancer. 2012;106:148–56.
Garofalo M, Romano G, Di Leva G, Nuovo G, Jeon YJ, Ngankeu A, Sun J, Lovat F, Alder H, Condorelli G, Engelman JA, Ono M, Rho JK, Cascione L, Volinia S, Nephew KP, Croce CM. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat Med. 2012;18:74–82.
Wang Y, Xia H, Zhuang Z, Miao L, Chen X, Cai H. Axl-altered microRNAs regulate tumorigenicity and gefitinib resistance in lung cancer. Cell Death Dis. 2014;5:e1227.
Wang YS, Wang YH, Xia HP, Zhou SW, Schmid-Bindert G, Zhou CC. MicroRNA-214 regulates the acquired resistance to gefitinib via the PTEN/AKT pathway in EGFR-mutant cell lines. Asian Pac J Cancer Prev. 2012;13:255–60.
Weiss GJ, Bemis LT, Nakajima E, Sugita M, Birks DK, Robinson WA, Varella-Garcia M, Bunn Jr PA, Haney J, Helfrich BA, Kato H, Hirsch FR, Franklin WA. EGFR regulation by microRNA in lung cancer: correlation with clinical response and survival to gefitinib and EGFR expression in cell lines. Ann Oncol. 2008;19:1053–9.
Zhong M, Ma X, Sun C, Chen L. MicroRNAs reduce tumor growth and contribute to enhance cytotoxicity induced by gefitinib in non-small cell lung cancer. Chem Biol Interact. 2010;184:431–8.
Park KS, Raffeld M, Moon YW, Xi L, Bianco C, Pham T, Lee LC, Mitsudomi T, Yatabe Y, Okamoto I, Subramaniam D, Mok T, Rosell R, Luo J, Salomon DS, Wang Y, Giaccone G. CRIPTO1 expression in EGFR-mutant NSCLC elicits intrinsic EGFR-inhibitor resistance. J Clin Invest. 2014;124:3003–15.
Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL, Hashida S, Maki Y, Ichihara E, Asano H, Tsukuda K, Takigawa N, Kiura K, Gazdar AF, Lam WL, Miyoshi S. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Res. 2013;73:3051–61.
Hershkovitz-Rokah O, Modai S, Pasmanik-Chor M, Toren A, Shomron N, Raanani P, Shpilberg O, Granot G. MiR-30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR-ABL protein. Cancer Lett. 2014;356(2 Pt B):597–605.
Joshi D, Chandrakala S, Korgaonkar S, Ghosh K, Vundinti BR. Down-regulation of miR-199b associated with imatinib drug resistance in 9q34.1 deleted BCR/ABL positive CML patients. Gene. 2014;542:109–12.
Li Y, Yuan Y, Tao K, Wang X, Xiao Q, Huang Z, Zhong L, Cao W, Wen J, Feng W. Inhibition of BCR/ABL protein expression by miR-203 sensitizes for imatinib mesylate. PLoS One. 2013;8, e61858.
Liu L, Wang S, Chen R, Wu Y, Zhang B, Huang S, Zhang J, Xiao F, Wang M, Liang Y. Myc induced miR-144/451 contributes to the acquired imatinib resistance in chronic myelogenous leukemia cell K562. Biochem Biophys Res Commun. 2012;425:368–73.
Lopotova T, Zackova M, Klamova H, Moravcova J. MicroRNA-451 in chronic myeloid leukemia: miR-451-BCR-ABL regulatory loop? Leuk Res. 2011;35:974–7.
Shibuta T, Honda E, Shiotsu H, Tanaka Y, Vellasamy S, Shiratsuchi M, Umemura T. Imatinib induces demethylation of miR-203 gene: an epigenetic mechanism of anti-tumor effect of imatinib. Leuk Res. 2013;37:1278–86.
Venturini L, Battmer K, Castoldi M, Schultheis B, Hochhaus A, Muckenthaler MU, Ganser A, Eder M, Scherr M. Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood. 2007;109:4399–405.
Xu C, Fu H, Gao L, Wang L, Wang W, Li J, Li Y, Dou L, Gao X, Luo X, Jing Y, Chim CS, Zheng X, Yu L. BCR-ABL/GATA1/miR-138 mini circuitry contributes to the leukemogenesis of chronic myeloid leukemia. Oncogene. 2014;33:44–54.
Zimmerman EI, Dollins CM, Crawford M, Grant S, Nana-Sinkam SP, Richards KL, Hammond SM, Graves LM. Lyn kinase-dependent regulation of miR181 and myeloid cell leukemia-1 expression: implications for drug resistance in myelogenous leukemia. Mol Pharmacol. 2010;78:811–7.
Fan R, Zhong J, Zheng S, Wang Z, Xu Y, Li S, Zhou J, Yuan F. microRNA-218 increase the sensitivity of gastrointestinal stromal tumor to imatinib through PI3K/AKT pathway. Clin Exp Med. 2014;15(2):137–44.
Gensler M, Buschbeck M, Ullrich A. Negative regulation of HER2 signaling by the PEST-type protein-tyrosine phosphatase BDP1. J Biol Chem. 2004;279:12110–6.
Rubbi L, Titz B, Brown L, Galvan E, Komisopoulou E, Chen SS, Low T, Tahmasian M, Skaggs B, Muschen M, Pellegrini M, Graeber TG. Global phosphoproteomics reveals crosstalk between Bcr-Abl and negative feedback mechanisms controlling Src signaling. Sci Signal. 2011;4:ra18.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8.
Schwarzenbach H, Nishida N, Calin GA, Pantel K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol. 2014;11:145–56.
Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–8.
Jarry J, Schadendorf D, Greenwood C, Spatz A, van Kempen LC. The validity of circulating microRNAs in oncology: five years of challenges and contradictions. Mol Oncol. 2014;8:819–29.
Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nat Rev Drug Discov. 2014;13:622–38.
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Akçakaya, P., Lui, WO. (2015). MicroRNAs and Gastrointestinal Stromal Tumor. In: Santulli, G. (eds) microRNA: Cancer. Advances in Experimental Medicine and Biology, vol 889. Springer, Cham. https://doi.org/10.1007/978-3-319-23730-5_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-23730-5_4
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23729-9
Online ISBN: 978-3-319-23730-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)