
Abstract
To detect the expressions of microRNA-218 (miR-218) in an imatinib mesylate-sensitive human gastrointestinal stromal tumor (GIST) cells (GIST882) and an imatinib mesylate-resistant cell line (GIST430) and explore the roles of miR-218 and GIST cells in the sensitivity of gastrointestinal stromal tumor to imatinib mesylate and its potential signaling pathways, with an attempt to provide new insights for the treatment of GIST. The GIST cell lines (GIST882 and GIST430) were cultured in vitro. Quantitative real-time PCR (qRT-PCR) was utilized to determine the expression profiles of miR-218 in both GIST cell lines. Forty-eight hours after the transfection of the miR-218 mimic or miR-218 inhibitor in the GIST cells, the changes in the expression of miR-218 in the GIST cells were detected with qRT-PCR. The effects of the ectopic expression of miR-218 in GIST882 or GIST430 cells on the imatinib mesylate-induced GIST cell viability were determined by MTT. The effects of miR-218 ectopic expression on the apoptosis of imatinib mesylate-induce GIST cells were determined by Annexin V/PI double staining method and flow cytometry. The effects of miR-218 ectopic expression on the AKT and phospho-AKT (p-AKT) expressions of imatinib mesylate-induce GIST cells were determined by Western blot and flow cytometry with the PI3K pathway inhibitor Wortmannin. As shown by qRT-PCR, compared with that in the imatinib mesylate-sensitive GIST882, the expression of miR-218 in imatinib mesylate-resistant GIST430 was significantly decreased (P < 0.01). Compared with the control group, the expression of miR-218 significantly increased in the GIST882 48 h after the transfection of miR-218 mimic (P < 0.01) and significantly declined after the transfection of miR-218 inhibitor (P < 0.01). As shown by MTT and flow cytometry, after the expression of miR-218 was inhibited in GIST882 under the effect of imatinib mesylate, the cell viability significantly increased (P < 0.01) and the number of apoptotic cells significantly decreased (P < 0.05); on the contrary, the over-expression of miR-218 in GIST430 under the effect of imatinib mesylate resulted in the significantly decreased cell viability (P < 0.01) and the significantly increased number of apoptotic cells (P < 0.05). Western blot and flow cytometry showed that, in comparison to the control group, Wortmannin could significantly inhibit the expression of p-AKT in GIST430 cells (P < 0.01) and stimulated apoptosis (P < 0.01). The expression of miR-218 is down-regulated in an imatinib mesylate-resistant GIST cell line (GIST430), whereas miR-218 over-expression can improve the sensitivity of GIST cells to imatinib mesylate, with PI3K/AKT signaling pathway possibly involved in the mechanism.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumor (GIST) originates from the intestinal cell of Cajal (ICC) and is the most common mesenchymal tumors in human gastrointestinal tract [1–3]. It is originally described as leiomyoma or leiomyosarcoma. Recent studies suggest that GIST is an independent clinical solid tumor, which is different from muscle-derived or of neurogenic tumors in the gastrointestinal tract [4, 5]. GIST accounts for about 2.2 % of gastrointestinal malignancies [6]. Notably, 60–70 % of GIST occurs in the stomach, 20–30 % in small intestine, <5 % in the large intestine, and only <5 % in the other parts of the gastrointestinal tract (esophagus, omentum, and mesenterium) [7, 8]. The GIST symptoms are related with the location, size, and growth patterns of the tumors [9, 10].
The pathogenesis of GIST is related with the abnormal activation of type III receptor tyrosine kinase (RTK) KIT/PDGFRA [11, 12]. About 70 % of GIST patients had KIT gene mutations, and over 90 % of GIST patients have KIT gene amplification. The abnormal activation of KIT/PDGFRA can cause the occurrence and development of GIST by activating a series of downstream signaling pathways [13, 14]. The main treatment for GIST remains surgical resection. However, even after complete resection, 70–90 % still may experience recurrence and metastasis [15, 16]. Since GIST is not sensitive to the conventional radiotherapy and chemotherapy; before the availability of molecular targeted drugs, the overall response rate of radiotherapy and chemotherapy will not exceed 5 % [17, 18].
Imatinib is a small molecule tyrosine kinase inhibitor (TKI). It is effective in the prevention and treatment of the postoperative recurrence and metastasis of GIST, and currently the drug of choice for GIST patients after their surgical treatment, with a total clinical benefit rate of approximately 84 % [19, 20]. However, some GIST patients who are initially sensitive to imatinib may suffer from recurrence and disease progression during the extended treatment, which is known as drug resistance. It is believed that secondary resistance occurs about 2 years after imatinib treatment [21, 22]. Clinical observations have shown that about 40–50 % of GIST patients experienced the secondary resistance after 2 years of imatinib treatment, which severely affected the therapeutic effectiveness for GIST [23]. Thus, how to deal with the secondary resistance of imatinib has become a hot topic in GIST treatment.
The microRNAs (miRNAs) are a class of endogenous small non-coding single-stranded molecule RNA, with a length of about 22 nucleotides (nt). By complementary pairing with the 3′ untranslated region (3′ UTR) of the target gene mRNA, it can regulate mRNA expression at the post-transcriptional level, and thus further be involved in the physiological processes including the cell proliferation, apoptosis, differentiation, metabolism, and growth and pathological processes including cardiovascular diseases, neurological diseases, and tumors [24–26]. It has been predicted that more than one-third of the human genes are conserved miRNA targets. The roles of miRNA in human disease, especially tumors, have been attracting increasing attention [27, 28].
By using miRNA microarray analysis, Shi et al. screened the differentially expressed miRNA in borderline tumors and malignant tumors in GIST samples. The most significantly expressed miRNAs were then chosen for further validation by real-time PCR, among which miRNA-218 was identified [29]. In their study on glioma, Tu et al. [30] found that miR-218 could inhibit the proliferation and migration of glioma cells by regulating the target gene Bmi1. In a study on osteosarcoma, miR-218 was found to be able to inhibit the invasion and migration of osteosarcoma cells by regulating the target genes TIAM1, MMP2, and MMP9 [31]. In our previous studies, down-regulation of miR-218 expression was observed in human gastric cancer cell lines; miR-218 can negatively regulate survivin protein expression and inhibit GIST cell proliferation and invasion. A recent study has shown that miR-218 could inhibit the growth of cervical cancer and promote its sensitivity to cisplatin-based chemotherapy [32]. However, the relationship between miR-218 and the sensitivity of GIST to chemotherapies has not been established. In order to provide new methods and strategies for the treatment of GIST, we investigated the relationship between miR-218 and the sensitivity of GIST cells to imatinib-based therapy as well as the signaling pathways.
Materials and methods
Major reagents
Fetal bovine serum, DMEM medium, l-glutamine, HEPES, and Lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA, USA). GIST882, as previously described, was established from an untreated human GIST with a homozygous missense mutation in KIT exon 13, encoding a K642E mutant KIT oncoprotein [33]. GIST430 were established from GISTs that had progressed, after initial clinical response, during imatinib therapy.
TaqMan miRNA isolation Kit, TaqMan microRNA assay kit, and TaqMan microRNA Assay and TaqMan Universal PCR Master Mix were purchased from Applied Biosystems (CA, USA). The miR-218 mimic, inhibitor, and non-specific control (NC) were synthesized by Genepharma (Shanghai, China).
The KIT inhibitor (imatinib mesylate) was purchased from Novartis Pharma (Basel, Switzerland). Cell culture plates and dishes were purchased from Corning, USA. MTT (3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-di-phenytetrazoliumromide), trypsin, and phosphate buffer solution (PBS) were purchased from Sigma-Aldrich, USA. PI3K pathway inhibitors Wortmannin was purchased from Axxora (San Diego, CA, USA). Annexin V and propidium iodide (PI) were purchased from Roche. Mouse anti-human β-actin monoclonal antibody was purchased from Abcam (Cambridge, UK). AKT and phospho-AKT (p-AKT, Ser473) primary antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). The secondary antibodies including IRDye 800 conjugated affinity purified goat anti-mouse IgG and IRDye 800 conjugated affinity purified goat anti-rabbit IgG were purchased from Odyssey. Protein extraction, and quantification kits were purchased from Bio-Rad (Hercules, CA, USA).
Culture and treatment of human GAST cell lines
The human GIST cell line GIST882 and imatinib mesylate-resistant cell line GIST430 were cultured at 37 °C in DMEM (GIBCO, California, USA) supplemented with 10 % fetal bovine serum (FBS) and 2 % penicillin/streptomycin. Cell growth was observed under an inverted microscope. When cell growth reached 70–80 % confluence, the cells were digested with 0.25 % trypsin and passaged. The culture medium was changed every other day, and the cells were passaged every 2–3 days. Cells in the logarithmic growth phase were collected for experiments.
GIST882 or GIST430 cells that were cultured under normal conditions were inoculated uniformly into 6- or 96-well culture plates at a concentration of 3 × 105 cells/ml. After adherent cell culture, transfections were conducted for miR-218 mimic, non-specific control (NC), and miR-218 inhibitor according to the Lipofectamine 2000 transfection manual. The normal control group was also established. The miR-218 mimic, inhibitor, and non-specific control (NC) were diluted by MEM medium free of serum components, and the liposomes Lipofectamine 2000 was added to the MEM medium. After being mixed mildly and incubation under ambient temperature for 5 min, the diluted Lipofectamine 2000 was mixed with diluted miR-218 mimic, inhibitor, and non-specific control (NC), respectively. The mixture was added to the culture plate with GIST882 or GIST430 cells. The obtained mixture was incubated in 5 % CO2 atmosphere at 37 °C. The culture medium was exchanged to DMEM medium containing 10 % fetal bovine serum after 5 h, and the mixture was incubated for another 48 h.
Detection of expression of miR-218 in GIST882 and GIST430 cell lines
The expressions of miR-218 in GIST cell lines GIST882 and GIST430 were detected using quantitative real-time PCR (qRT-PCR). The two GIST cell lines were cultured in vitro and then collected. The total RNA was extracted using the TaqMan miRNA isolation kit. The expression of mature miR-218 was detected using the TaqMan microRNA assay and the TaqMan Universal PCR Master Mix with U6 as an internal control gene. All reactions were performed in triplicate wells. The cycle threshold (CT) value of the samples in each reaction well was recorded. The experimental results were analyzed using the relative quantification method of qRT-PCR.
Detection of the effects of miR-218 ectopic expression on the viability of imatinib mesylate-induced GIST cells with MTT
GIST882 cells cultured under normal conditions were inoculated uniformly into 6-well culture plates at a concentration of 3 × 105 cells/ml. After adherent cell culture, transfections were conducted for miR-218 mimic, non-specific control (NC), or miR-218 inhibitor according to the Lipofectamine 2000 transfection manual. The normal control group was also established. The RNA of cells in each group was extracted using the TaqMan miRNA Isolation kit, and qRT-PCR was used to determine the changes in expression in GIST882 cells.
GIST882 or GIST430 cells cultured under normal conditions were inoculated uniformly into 96-well culture plates at a concentration of 3 × 105 cells/ml. The transfection procedure was described above. The miR-218 inhibitor and non-specific control (NC) were transfected into GIST882 cells, while miR-228 mimic and non-specific control (NC) were transfected to GIST430 cells. After the transfection, the GIST882 or GIST430 cells were added with imatinib mesylate, yielding a final concentration of 1.0 μM. Forty-eight hours after transfection, 100 μl of MTT (0.5 mg/ml) solution was added to each well, and the plates were placed in a 37 °C/5 % CO2 incubator for 4 h. A total of 100 μl of 20 % sodium dodecyl sulfate (SDS) (cosolvent 50 % dimethyl formamide) was added to each well, and the plates were incubated at 37 °C for 24 h. A microplate reader (Bio-Tek, USA) was used to measure the OD values at 570 nm. Each experimental group contained ten replicate wells, and the experiment was repeated three times.
Detection of the effects of miR-218 ectopic expression on the apoptosis of imatinib mesylate-induced GIST cells using flow cytometry
GIST882 or GIST430 cells cultured under normal conditions were inoculated uniformly into 6-well culture plates at a concentration of 3 × 105 cells/ml. The miR-218 inhibitor and non-specific control (NC) were transfected into GIST882 cells, while miR-228 mimic and non-specific control (NC) were transfected to GIST430 cells. After the transfection, the GIST882 or GIST430 cells were added with imatinib mesylate, yielding a final concentration of 1.0 μM. After the transfection for 48 h, the cells were washed with PBS for 1–2 times. After having been added with Annexin V-FITC and PI dye, the solution was labeled at room temperature in the dark for 1 min. After being filtrated with screen cloth, the cells were analyzed using flow cytometry (BD, USA). FCM CellQuest software was used to count the cells, and Macquit software was used to analyze the data.
Effects of miR-218 ectopic expression on the signaling pathways involved in the apoptosis of imatinib mesylate-induced GIST cells
GIST430 cells cultured under normal conditions were inoculated uniformly into 6-well culture plates at a concentration of 3 × 105 cells/ml. The GIST430 cells were transfected with miR-218 mimic or non-specific control. After the transfection, the GIST430 cells were added with imatinib mesylate, yielding a final concentration of 1.0 μM. Finally the transfected GIST 430 cells were added with the PI3K pathway inhibitor Wortmannin, yielding a final concentration of 5 μM. The normal control group was also established. Forty-eight hours after the transfection, the cells were washed with PBS for 1–2 times. Then, 1 ml RIPA lysis buffer was added. The supernatants were collected, and the protein concentration was determined using the bicinchoninic acid method. The proteins were separated by electrophoresis and wet transferred to a polyvinylidene difluoride membrane (Bio-Rad, USA). After blocking in TBST solution containing 5 % nonfat dry milk at room temperature for 1 h, AKT, phospho-AKT (p-AKT, Ser473) primary antibody (1:500 dilution), and mouse anti-human β-actin monoclonal antibody (1:1,000 dilution) and incubated overnight at 4 °C. After washing with TBST, the Odyssey infrared imaging system was used to scan the membranes. The relative content of survivin was represented as the gray scale ratio of survivin/β-actin, and the grayscale was analyzed using the QuantityOne software.
The effect of PI3K pathway inhibitor Wortmannin on the apoptosis of cisplatin-induce gastric cancer cells with miR-imatinib mesylate ectopic expression was determined by flow cytometry.
Statistical Analysis
Data were analyzed using SPSS 17.0 software (SPSS, Inc., Chicago, IL, USA). Comparisons between two groups are performed using the t test, whereas comparisons among three or more groups using analysis of variance. P < 0.05 was considered statistically significant.
Results
Expression of miR-218 in GIST882 and GIST430 cell lines
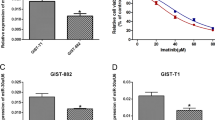
Using qRT-PCR, we determined that the miR-218 expression level in the imatinib-resistant GIST cell line (GIST430) was significantly lower than that in GIST882 (P < 0.01) (Fig. 1).

The expressions of miR-218 in GIST cell lines GIST882 and GIST430 (by quantitative real-time PCR). **P < 0.01
Effects of miR-218 ectopic expression on the viability of imatinib mesylate-induced GIST cell lines
The miR-218 was over-expressed in GIST882 cells by the transfection of miR-218 mimic and its expression was inhibited by the transfection of miR-218 inhibitor. As shown by qRT-PCR, the expression level of miR-218 was significantly higher in the miR-218 mimic transfected group than in the normal control group (P < 0.01) and negative control (non-specific control) group (P < 0.01). In the miR-218 inhibitor transfected group, miR-218 expression was significantly lower than those of normal control group (P < 0.01) and negative control group (NC) (P < 0.01) (Fig. 2).

Effect of the transfected miR-218 mimic or miR-218 inhibitor on miR-218 expression in GIST882 cells. **P < 0.01 versus normal group or NC group
Effects of miR-218 ectopic expression on the viability of imatinib mesylate-induced GIST cell lines
As shown by MTT assays, the viability of GIST882 cells transfected with miR-218 inhibitor and incubated with 1.0 μg/ml imatinib mesylate (IM) for 48 h was significantly higher than that of negative control cells (P < 0.01). The viability of GIST430 cells transfected with miR-218 mimic and incubated with 1.0 μM IM for 48 h was significantly lower than that of negative control (NC) cells (P < 0.01) (Fig. 3). These results showed miR-218 over-expression could increase the sensitivity of GIST430 cells to IM.

Effects of miR-218 ectopic expression on the viability of imatinib mesylate-induced GIST cell lines. **P < 0.01 versus non-specific control group
Effects of miR-218 ectopic expression on the apoptosis of imatinib mesylate-induced GIST cells
As shown by flow cytometry, the number of apoptotic GIST882 cells was significantly smaller in the GIST882 cells transfected with miR-218 inhibitor and incubated with 1.0 μg/ml IM for 48 h than that of negative control cells (P < 0.05). The number of apoptotic GIST430 cells transfected with miR-218 mimic and incubated with 1.0 μM IM for 48 h was significantly higher than that of negative control (NC) cells (P < 0.05) (Fig. 4). These results showed miR-218 over-expression could increase the sensitivity of GIST430 cells to IM.

Effects of miR-218 ectopic expression on the apoptosis of imatinib mesylate-induced GIST cells (by flow cytometry). *P < 0.05 versus non-specific control group
Effects of miR-218 ectopic expression on the signaling pathways involved in the apoptosis of imatinib mesylate-induced GIST cells
As shown by MTT and flow cytometry, the inhibition of miR-218 expression contributed to the decreased sensitivity of GIST882 cells to IM, while the over-expression of miR-218 resulted in increased sensitivity of GIST430 cells to IM. Moreover, the pathway of miR-218-mediated mechanism was investigated by using the PI3K pathway inhibitor Wortmannin.
As shown by Western blot, the p-AKT/AKT ratio of GIST430 cells transfected with miR-218 mimic and incubated with 1.0 μM IM for 48 h was significantly lower than that of normal control group (P < 0.01) but was not significantly different from that of Wortmannin treatment group (Fig. 5).

Expression profiles of p-AKT, AKT, and β-actin in GIST430 cells of each group by Western blot analysis. *P < 0.05 and **P < 0.01 versus normal control group
As shown by flow cytometry, the GIST430 cells were transfected with miR-218 mimic and then inoculated at 1.0 μM IM and 5 μM Wortmannin; then, the number of the apoptotic GIST430 cells was significantly higher than that in the normal control group (P < 0.01) (Fig. 6). Thus, miR-218 over-expression might improve the sensitivity of GIST cells to imatinib mesylate, with PI3K/AKT signaling pathway possibly involved in the mechanism.

Apoptosis of GIST cells per group by flow cytometry. **P < 0.01 versus normal group
Discussion
GIST originates from the intestinal cell of Cajal (ICC) and is the most common mesenchymal tumors in human gastrointestinal tract, particularly in stomach and small intestine [1–3]. GIST is mainly caused by the continuous non-ligand-dependent receptor activation and continuous activation of downstream signaling pathways due to the KIT/PDGFRA mutations. IM can be used to treat advanced unresectable GIST, with a clinical response rate of about 75–90 %. However, even in patients who respond well to IM, drug resistance will be inevitably during the treatment [22, 34]. Thus, how to deal with the secondary resistance of imatinib has became a hot topic in GIST treatment.
Research concerning the relationship between miRNAs and tumor chemosensitivity has gradually gained attention from researchers in this field, and this research has become an important research topic in recent years. Previous studies have confirmed that miR-218 is a tumor suppressor microRNA. A recent study conducted by Li et al. [32] has shown that miR-218 could inhibit the growth of cervical cancer and promote its sensitivity to cisplatin-based chemotherapy. In our previous studies, down-regulation of miR-218 expression was observed in human GIST cell lines; miR-218 can negatively regulate survivin protein expression and inhibit GIST cell proliferation and invasion. In this study, by using IM-resistant GIST cell line (GIST430) and the normal GIST cell line (GIST882), we explored the relationship between the miR-218 and the GIST cells’ sensitivity to IM. Moreover, the effects of miR-218 ectopic expression on the sensitivity of IM-induced GIST cells were also investigated by transfecting miR-218 inhibitor into GIST882 cells and transfecting miR-218 mimic into GIST430 cells.
As shown by MTT and flow cytometry, the viability of GIST882 cells transfected with miR-218 inhibitor and incubated with 1.0 μM IM for 48 h was significantly higher than that of negative control (NC) cells (P < 0.01), and the number of apoptotic GIST882 cells was significantly lower than that of negative control (NC) group (P < 0.05). In contrast, the viability of GIST430 cells transfected with miR-218 mimic and incubated with 1.0 μM IM for 48 h was significantly lower than that of negative control (NC) cells (P < 0.01), but the percentage of apoptotic GIST430 cells was significantly higher than that of NC group (P < 0.05). As a result, the inhibition of miR-218 expression contributed to the decreased sensitivity of GIST882 cells to IM, and the miR-218 over-expression increased the sensitivity of GIST430 cells to IM.
Phosphatidylinositol-3-kinases (PI3K) is an intracellular phosphatidylinositol kinase, among which the class Ia PI3Ks are heterodimers composed of regulatory subunits (p85α, p55α, p50α, p85β, and p55γ) and catalytic subunits (p110α, p110β, and p110δ) [35]. The class Ia PI3Ks are closely related with the RTK signaling pathway, in which the regulatory subunit p85 plays a key role. The activity of PI3K was enhanced in all the RTK-linked tumors, and such activation is strictly regulated by RTK. An effective RTK inhibitor must be able to exert marked inhibitory effect on the PI3K pathway [36]. In some tumors, multiple RTKs may exist simultaneously, together with the activated PDK pathway. These tumors can eventually become resistant to single-target RTK inhibitors [37].
As the downstream of KIT, the PI3K/AKT pathway also exerts its roles via the regulatory subunit p85. In their in vivo and in vitro experiments, Blume-Jensen et al. [38, 39] confirmed that the phosphorylated KIT (Tyr719) can be bound with the regulatory subunit p85 and then activate the typical PI3K/AKT pathway. In GIST patients who are sensitive to IM, this pathway is remarkably suppressed during the IM treatment [4, 40]. When the GIST becomes resistant to IM, the KIT gene experiences a second mutation, causing the massive KIT amplification; as a result, the PI3K/AKT pathway will be reactivated [41]. In this study, miR-218 over-expression has been demonstrated to improve the sensitivity of GIST430 cells to IM and this effect was considered to be mediated via the PI3K/AKT/survivin signaling pathway.
In summary, the expression of miR-218 is down-regulated in IM-resistant GIST430 cells. Thus, miR-218 over-expression may improve the sensitivity of GIST cells to IM, with PI3K/AKT signaling pathway possibly involved in the mechanism.
References
Datar M, Khanna R (2012) Inpatient burden of gastrointestinal stromal tumors in the United States. J Gastrointest Oncol 3(4):335–341
Hodges K, Kennedy L, Meng F, Alpini G, Francis H (2012) Mast cells, disease and gastrointestinal cancer: a comprehensive review of recent findings. Transl Gastrointest Cancer 1(2):138–150
Halpern J, Kim YJ, Sultana R, Villani G (2012) Effectiveness of radiation therapy in GIST: a case report. J Gastrointest Oncol 3(2):143–146
McDaniel K, Correa R, Zhou T, Johnson C, Francis H, Glaser S, Venter J, Alpini G, Meng F (2013) Functional role of microvesicles in gastrointestinal malignancies. Ann Transl Med 1(1):4
Kanda T (2013) Criminal or bystander: imatinib and second primary malignancy in GIST patients. Chin J Cancer Res 25(5):490–492
Kinross KM, Sheppard KE, Pearson RB, Phillips WA (2012) Targeting cancer with PI3K pathway inhibitors: who to aim at? Transl Cancer Res 1(2):119–121
Sun X, Wang J, Yang G (2012) Surgical treatment of esophageal leiomyoma larger than 5 cm in diameter: a case report and review of the literature. J Thorac Dis 4(3):323–326
Zhao X, Yue C (2012) Gastrointestinal stromal tumor. J Gastrointest Oncol 3(3):189–208
Eisenberg BL, Pipas JM (2012) Gastrointestinal stromal tumor—background, pathology, treatment. Hematol Oncol Clin N Am 26(6):1239–1259
Kee D, Zalcberg JR (2012) Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther Adv Med Oncol 4(5):255–270
Nannini M, Biasco G, Astolfi A et al (2013) An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J Med Genet 50(10):653–661
Wang C, Jin MS, Zou YB et al (2013) Diagnostic significance of DOG-1 and PKC-theta expression and c-Kit/PDGFRA mutations in gastrointestinal stromal tumours. Scand J Gastroenterol 48(9):1055–1065
O’Brien KM, Orlow I, Antonescu CR et al (2013) Gastrointestinal stromal tumors, somatic mutations and candidate genetic risk variants. PLoS One 8(4):e62119
Tan CJ, Yang JL, Crowe P, Goldstein D (2013) Targeted therapy in soft tissue sarcoma: a novel direction in therapeutics. Chin Clin Oncol 2(3):22
Rutkowski P, Bylina E, Klimczak A et al (2012) The outcome and predictive factors of sunitinib therapy in advanced gastrointestinal stromal tumors (GIST) after imatinib failure-one institution study. BMC Cancer 12:107
Joensuu H (2013) Gastrointestinal stromal tumors: risk assessment and adjuvant therapy. Hematol Oncol Clin N Am 27(5):889–904
Tirumani SH, Jagannathan JP, Krajewski KM et al (2013) Imatinib and beyond in gastrointestinal stromal tumors: a radiologist’s perspective. AJR Am J Roentgenol 201(4):801–810
An HJ, Ryu MH, Ryoo BY et al (2013) The effects of surgical cytoreduction prior to imatinib therapy on the prognosis of patients with advanced GIST. Ann Surg Oncol 20(13):4212–4218
Fujimoto Y, Akiyoshi T, Konishi T et al (2013) Laparoscopic sphincter-preserving surgery (intersphincteric resection) after neoadjuvant imatinib treatment for gastrointestinal stromal tumor (GIST) of the rectum. Int J Colorectal Dis 29(1):111–116
Das D, Ganguly S, Deb AR et al (2013) Neoodjuvant imatinib mesylate for advanced primary and metastactic/recurrent gastro-intestinal stromal tumour (GIST). J Indian Med Assoc 111(1):21–23
Falor A, Arrington AK, Luu C et al (2013) Massive intra-abdominal imatinib-resistant gastrointestinal stromal tumor in a 21-year-old male. Case Rep Med 2013:373981
Linch M, Claus J, Benson C (2013) Update on imatinib for gastrointestinal stromal tumors: duration of treatment. Onco Targets Ther 6:1011–1023
Blay JY, Rutkowski P (2013) Adherence to imatinib therapy in patients with gastrointestinal stromal tumors. Cancer Treat Rev 40(2):242–247
Wang Q, Wei L, Guan X et al (2013) Briefing in family characteristics of microRNAs and their applications in cancer research. Biochim Biophys Acta 1844(1 Pt B):191–197
Kaplan BB, Kar AN, Gioio AE et al (2013) MicroRNAs in the axon and presynaptic nerve terminal. Front Cell Neurosci 7:126
Li M, Fu W, Wo L et al (2013) miR-128 and its target genes in tumorigenesis and metastasis. Exp Cell Res 319(20):3059–3064
Leite-Moreira AM, Lourenco AP, Falcao-Pires I et al (2013) Pivotal role of microRNAs in cardiac physiology and heart failure. Drug Discov Today 18(23–24):1243–1249
Piva R, Spandidos DA, Gambari R (2013) From microRNA functions to microRNA therapeutics: novel targets and novel drugs in breast cancer research and treatment (Review). Int J Oncol 43(4):985–994
Shi Y, Wang CZ, Hou YY et al (2013) Screening of differentially expressed microRNAs in borderline and malignant gastrointestinal stromal tumors. Zhonghua Bing Li Xue Za Zhi 42(1):20–25
Tu Y, Gao X, Li G et al (2013) MicroRNA-218 inhibits glioma invasion, migration, proliferation and cancer stem-like cell self-renewal by targeting the polycomb group gene Bmi1. Cancer Res 73(19):6046–6055
Jin J, Cai L, Liu ZM et al (2013) miRNA-218 inhibits osteosarcoma cell migration and invasion by down-regulating of TIAM1, MMP2 and MMP9. Asian Pac J Cancer Prev 14(6):3681–3684
Li J, Ping Z, Ning H (2012) MiR-218 impairs tumor growth and increases chemo-sensitivity to cisplatin in cervical cancer. Int J Mol Sci 13(12):16053–16064
Tuveson DA, Willis NA, Jacks T et al (2001) STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene 20(36):5054–5058
Reichardt P, Joensuu H, Blay JY (2013) New fronts in the adjuvant treatment of GIST. Cancer Chemother Pharmacol 72(4):715–723
Kwong LN, Davies MA (2013) Navigating the therapeutic complexity of PI3K pathway inhibition in melanoma. Clin Cancer Res 19(19):5310–5319
Kitagishi Y, Matsuda S (2013) Diets involved in PPAR and PI3K/AKT/PTEN pathway may contribute to neuroprotection in a traumatic brain injury. Alzheimers Res Ther 5(5):42
Siegfried Z, Bonomi S, Ghigna C et al (2013) Regulation of the Ras-MAPK and PI3K-mTOR signalling pathways by alternative splicing in cancer. Int J Cell Biol 2013:568931
Blume-Jensen P, Jiang G, Hyman R et al (2000) Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3′-kinase is essential for male fertility. Nat Genet 24(2):157–162
Gibbons SJ, Rich A, Distad MA et al (2003) Kit/stem cell factor receptor-induced phosphatidylinositol 3′-kinase signalling is not required for normal development and function of interstitial cells of Cajal in mouse gastrointestinal tract. Neurogastroenterol Motil 15(6):643–653
Rajendra R, Pollack SM, Jones RL (2013) Management of gastrointestinal stromal tumors. Future Oncol 9(2):193–206
Floris G, Wozniak A, Sciot R et al (2013) A potent combination of the novel PI3K inhibitor, GDC-0941, with imatinib in gastrointestinal stromal tumor xenografts: long-lasting responses after treatment withdrawal. Clin Cancer Res 19(3):620–630
Acknowledgments
This study was supported by Key Project of Science and Technology Commission of Shanghai Municipality (No. 13411950900).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fan, R., Zhong, J., Zheng, S. et al. microRNA-218 increase the sensitivity of gastrointestinal stromal tumor to imatinib through PI3K/AKT pathway. Clin Exp Med 15, 137–144 (2015). https://doi.org/10.1007/s10238-014-0280-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10238-014-0280-y