
Abstract
Approximately 250,000 people are diagnosed with breast cancer and 40,000 people die from the disease annually in the United States. Significant effort has been put into developing a thorough understanding of the molecular and genetic events that contribute to tumor progression due to mounting evidence that breast cancers are driven by predictable and identifiable genetic mutations, Sphingolipids modulate many of the growth, apoptosis, inflammatory, and angiogenic pathways that breast carcinomas rely on. Recent work in the field of sphingolipidomics has identified many key roles for the sphingolipids in breast tumor progression and identified signaling roles in each of the molecular subtypes of breast cancer. For example, in luminal type tumors, increased expression of ceramide producing enzymes and high levels of complex sphingolipids are associated with poor outcomes and multi-drug resistance. In basal type breast tumors CERT and GCS play a role in resistance to taxanes suggesting that targeting these pathways may be an effective route for the treatment of aggressive triple negative tumors. The role of sphingolipids in Her2-like tumors has not been thoroughly studied. However, CERK has been identified as potential driver of Her2-like tumors. Clearly, sphingolipid signaling in tumor modulation is both complex and vital. By integrating sphingolipid signaling into the molecular characterization of breast tumors, new opportunities for targeted intervention are identified. It is increasingly apparent that modulating sphingolipid levels in tumors will be an effective and powerful method for the treatment of breast cancer.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction to Breast Cancer
Breast cancer results from abnormal growth of the breast tissue. The most common form is ductal carcinoma resulting from hyperplasia and neoplastic change in epithelial cells lining the lactiferous ducts. Breast tumors are a heterogeneous mix of tumor, immune, and mesenchymal cells, and vary widely in their histopathologic make up, aggressiveness, and response to therapy. Annually in the US, roughly 250,000 people are diagnosed with breast cancer and 40,000 die [1].
Breast cancer is one of the earliest malignancies to be identified, and has caused significant morbidity and mortality throughout history. The earliest written accounts of cancer originate in an ancient Egyptian medical treatise known as the Edwin Smith Papyrus. In the text, dated around 2500 BC, the authors describe treatment of 48 medical cases using a scientific rather than mythological approach [2]. Included therein is a description of a palpable breast tumor and the conclusion that it is incurable. Early attempts at treatment were made using a number of remedies such as arsenic paste and cautery [3]. Other civilizations of the same era used various concoctions and salves made from heavy metals or sulfur to treat breast cancer. For the most part, these early treatments were used, with only minimal modification, for several thousand years.
In the eighteenth and nineteenth centuries, the clinicopathalogic signature of breast cancer was becoming clearer. Theodor Schwann (1810–1882) demonstrated that tissues were made up of living cells instead of vacant cell structures as Hook had proposed 175 years earlier. Schwann, together with Johannes Muller (1801–1858), described cancers as new cells aberrantly growing in tissues and causing dysfunction of the organs [4]. Their work was one of the first clinicopathologic correlations of cancer, and subsequent studies by Walter Walshe (1812–1880) suggested that cancer was caused by chronic inflammation, trauma, and smoking [5]. As the study of breast cancer entered the twentieth century, research greatly accelerated as a result of accumulated pathologic knowledge as well as new techniques such as transcutaneous fine needle aspiration [6]. In 1904, Hugo Ribbert (1855–1920) established the theory that breast cancer resulted in the abnormal growth of epithelial cells as a result of chronic inflammation, and scientists began probing the root causes of breast cancer [7].
Despite the growing body of knowledge of the origins and progression of breast cancer, the standard of care had not changed appreciably since the sixteenth century. William Halsted (1852–1922) attempted to standardize mastectomy techniques, and he demonstrated that his technique of deep excision of the breast and pectoral muscle was highly efficacious at preventing local recurrence of breast cancer [7, 8]. In the late nineteenth and early twentieth centuries, aggressive surgical techniques and early diagnosis significantly reduced the number of deaths from breast cancer, and the 3 year survival rate was over 50 % [9].
In the early twentieth century, focus began to shift to finding cures for inoperable breast cancers. In the late 1800s, George Beatson (1848–1933) observed that oophorectomy (removal of the ovaries) reduced the growth and spread of breast tumors in laboratory animals. He attempted oophorectomy combined with treatment with thyroid extracts, and saw reduction in tumor growth in some patients [10]. Oophorectomy came into regular clinical practice, but when Stanley Boyd conducted a national survey of cases of breast cancer with oophorectomy, he found that oophorectomy had limited efficacy in a small number of patients [11]. Nevertheless, this hinted at hormonal regulation of breast tumors. Then, in 1910, one of the most important events in cancer biology occurred. Peyton Rous (1879–1970) published his work demonstrating that sarcomas in hens could be retrovirally induced [12]. His work demonstrated that tumors could be induced by expression of specific genes. Rous’ work further revolutionized cancer research when it was recognized that his virus could immortalize cells and allow them to be grown ex vivo. Tissue culture techniques progressed rapidly, and new models of breast cancer developed rapidly. Explanted tumors were grown in rodents, and the modern era of chemotherapeutic testing began.
These efforts consolidated much of the knowledge of breast cancer, and breast cancer researchers began taking a multidisciplinary approach to understanding the disease. The work of James Ewing (1866–1943) and Albert Broders (1885–1964) produced a histologic grading system for tumors that is still in use today [9]. Their systems were based on cellular atypia, tumor vascularity, and necrosis, as well as size and level of invasion. Based on these parameters, tumor outcome and response to treatment could be reasonably predicted.
Today, treatment of breast cancer is highly complex, multimodal, and interdisciplinary. By utilizing knowledge gained from decades of research, current breast cancer therapies include targeted therapies, radiation treatment, systemic administration of drugs, and surgery to ablate primary early stage tumors. Our current understanding of and approach to treat breast cancer have increased the overall 10 year survival of breast cancer patients to 83 % [1]. However, locally advanced or metastatic tumors have poor outcomes. For example, patients presenting with stage III disease, have a roughly 50 % 5-year survival [1]. Therefore, it is easy to understand the value of early detection, as well as the need to improve therapy for aggressive tumors.
Indeed, breast cancer therapy matured throughout the twentieth century, and several classes of chemotherapeutics were discovered, beginning with the nitrogen mustards and antimetabolites [13]. In the early 1950s Heidelberger and colleagues found that rat hepatomas had increased uptake of uracil. Therefore, they developed drugs targeting uracil metabolism and achieved a durable reduction in tumor size with 5-florouracil. 5-Florouracil was one of the first antimetabolites, followed by its pro-drug capecitabine in the early 2000s [14, 15]. Antimetabolites interfere with metabolic processes, often causing defects in the production of nucleic acids and replication of DNA, leading to replication stress and apoptosis in rapidly dividing or checkpoint deficient cells.
Following the introduction of antimetabolites, were the antitumor antibiotics. This class of chemotherapeutics functions primarily through cytotoxic mechanisms including disruption of DNA structure. The earliest antitumor antibiotic was bleomycin, discovered in the mid 1960s [16]. Shortly after the discovery of bleomycin, adriamycin was introduced [17]. Although the antitumor antibiotics were highly effective, their low therapeutic index limited their use. For example, early Phase I/II trials with Adriamycin highlighted the significant hematologic cytotoxicity and endothelial toxicity of high dose Adriamycin treatment protocols. At doses where tissue and binding factors are saturated (i.e., in patients with high plasma concentrations of Adriamycin), patients experienced catastrophic granulocytopenia, and secondary infections, as well as premature ventricular contractions. Since Adriamycin is rapidly metabolized, achieving therapeutic plasma concentrations requires high dosage schedules that are associated with poor patient outcomes. Despite the fact that significant tumor response was observed in the patients on high dose schedules of Adriamycin, 9 of the 20 patients on high dose schedules died from side effects of the drug [18]. Following the early clinical trials with anti tumor antibiotics, investigators realized the need for targeted therapies and dose limiting combinational therapy to maximize tumor response at lower doses of chemotherapeutic drugs [19].
Drugs targeting estrogen synthesis were developed soon after the discovery of the antimetabolites. Building upon Beatson’s work, estrogen ablative therapy was pursued for many patients throughout the 50s and 60s with varied success. In the early 1970s, Elwood Jensen and colleagues demonstrated that only certain breast cancers expressed estrogen receptors, and that these receptors contributed to tumor growth and predicted response to antiendocrine therapy [20]. Jensen and colleagues went on to develop robust antibodies to the estrogen receptor, and demonstrated a clinically relevant link between ER expression in small tumors and eventual response to antiendocrine therapy [21]. ICI Pharmaceuticals developed Tamoxifen (an antagonist of the estrogen receptor) in the 1960s, but it was not used for breast cancer until the 1970s [22].
In the 1990s and early 2000s, naturally derived taxane based chemotherapeutics were developed. Taxanes hinder cytoskeletal remodeling in dividing cells, and interfere with mitotic spindle formation [23, 24]. The production and distribution of taxanes was a highly politicized and controversial issue in the early 1990s, but the effectiveness of taxanes in the treatment of metastatic disease led to their rapid adoption and widespread use [25, 26]. Therapeutic employment of taxanes (docetaxel or paclitaxel) is currently recommended for treatment of a variety of locally advanced Her2 positive as well as Her2 negative breast tumors.
The discovery that tumors are driven by specific and predictable growth and development pathways led to the advancement of targeted therapies. One of the first rationally developed and targeted therapies to come to market, imatinib mesylate (Gleevec), was developed by Ciba-Geigy (Novartis) using in silico computational methods to design inhibitors of bcr-abl, the aberrant tyrosine kinase in chronic myelogenous leukemia [27]. The success of imatinib validated the use of pathway analysis and bioinformatics as a guide for research and development of novel therapeutics, and heralded in a new era in chemotherapy. In a subset of breast tumors, the membrane bound receptor tyrosine kinase Her2 (ERBB2) is overexpressed, and efforts to develop therapies targeting the Her2 receptor were also met with rapid success [28, 29]. One of the best characterized targeted therapies for breast cancer is trastuzumab, a monoclonal antibody that results in blockage of Her2 signaling as well as opsonization of the tumor cells [30]. Opsonization of the tumor cells results in immune mediated clearance of these cells, and future therapeutics may rely more heavily on targeted manipulation of adaptive immunity [31].
2 Molecular Genetic Portraits of Breast Cancer
Traditionally, the choice of treatment protocol has been based on anatomic and histopathologic characteristics of the tumor. The American Joint Committee on Cancer has set forth staging criteria for anatomical classification of tumors based on tumor size (T), node status (N), and presence or absence of distant metastasis (M) [32]. T refers to the size of the primary tumor and local invasion, N denotes the presence or absence of tumor cells in regional lymph nodes, and M indicates distant metastasis of the tumor. These TNM criteria are used to establish the stage of the tumor, and by extension, the likely outcomes of the disease. Generally speaking, early stage tumors include tumors up to T2N1M0. Locally advanced tumors include tumors up to T4N3M0, and metastatic tumors are any T or N status with identifiable metastasis to other organs.
However, it is difficult to predict the biomolecular make up of a tumor vis-à-vis receptor status from the clinical designation (e.g., TNM or grade). Thus, histopathologic characterization of tumors is also used to establish receptor status for the purposes of choosing targeted therapies and predicting the response to endocrine therapy. Tumors generally fall into one of three therapeutically relevant categories: ER+, Her2+, or triple negative. Based on the stage and receptor status, surgical recommendations are made, and chemotherapeutic regimens are chosen. Thus, the presence or absence of progesterone receptors (PR), estrogen receptors (ER), and the human epidermal growth factor receptor (HER2) determines the type and duration of targeted treatment [33].
The last decade has witnessed increasing intensity in molecular profiling and sub-typing of breast cancer based on gene expression profiles and presence of specific mutations in the hope of developing more accurate diagnosis of breast cancer sub-types. Perou et al. performed hierarchical clustering of gene expression data from a large microarray database and established several gene expression patterns [34]. These expression patterns are known as Luminal type/ER+, Basal type (triple negative), and Her2 like. Thus, these and other results have validated the use of receptor status to stratify tumors into molecular subtypes [35]. Subsequent analysis has demonstrated two distinct subsets of Luminal-like tumors, referred to as Luminal A and Luminal B, both ER positive but relying on different growth pathways for survival and proliferation [36]. Outcome analysis further established functional significance of these molecular subtypes with respect to response to treatment and overall survival of patients [37, 38].
In 2012, the Cancer Genome Atlas (TCGA) published a thorough and multiplatform proteomic and genetic characterization of breast carcinomas and described the main signaling pathways that are up- and down-regulated in each tumor subtype. Luminal type tumors were found to have frequent mutations in GATA3, PIK3CA and MAP3K1 genes, and Luminal B tumors in particular have increased incidence of mutations in DNA damage pathway genes, as opposed to Luminal A tumors. Basal type tumors were found to have high expression of proliferation associated genes as well as HIF1-α/ARNT. Her2 like tumors had high expression of Her2 amplicon genes, as well as active PIK3CA signaling and over expression of Cyclin D1 [39]. These signaling pathways, as they relate to sphingolipids, will be discussed thoroughly in the following sections. The general characteristics of each breast cancer subtype, most common histopathologic findings, and outcomes are summarized in Table 1.
3 Sphingolipids in Breast Cancer
It is now recognized that many convergent and divergent pathways drive breast tumor growth and spread. Modulation of these pathways by intracellular signaling is an extremely complex topic. In the 1980s it was discovered that lipids behave as potent signaling molecules in addition to being structural components of the cell. Bioactive lipids include the phosphoinositides, diacylglycerols, eicosanoids, fatty acids, and sphingolipids. Lipids represent a unique class of signaling molecules because they mostly belong in biological membranes and, as a class of biologic molecules, lipids have an extremely wide range of physicochemical properties. The structural and physiochemical diversity of lipids requires a wide range of metabolic enzymes and binding molecules. The resulting multiplicity leads to an extremely diverse set of possible combinations of signaling partners. We are just beginning to understand the diverse and interconnected nature of lipid second messengers, and the sphingolipids represent the forefront of this research.
Sphingolipids are a diverse set of signaling and homeostatic molecules. Head group composition, acyl chain length, and saturation all have significant effects on their properties. Additionally, spatiotemporal distribution of sphingolipid synthesis is highly regulated, and subcellular compartmentalization has drastic effects on the function of individual sphingolipids (thoroughly reviewed in [40] and discussed in the overview on sphingolipid metabolism and signaling of this book). Sphingolipids were shown to be powerful signaling molecules capable of modulating many of the mitogenic pathways that tumors rely on for growth. Glycosphingolipid studies in the 1970s and earlier focused heavily on the gangliosides and their antigenic function in different cancers [41–43]. Work by Hakomori and others demonstrated that the gangliosides were important membrane components that affected cell growth, differentiation, and transformation and also served as tumor antigens [44–46]. Work in the mid 1980s demonstrated a powerful role for sphingolipids in subcellular signaling cascades. The finding that sphingosine is a competitive inhibitor of PKC,[47] a known regulator of several oncogenes [48, 49], sparked the interest in the role of sphingolipids in tumors. Subsequent work in the 1990s identified the tumor suppressors Protein Phosphatases 1 and 2A as direct targets of ceramide [50]. Ever since, the metabolism and signaling functions of sphingolipids, and of ceramide in particular have been the subject of significant research.
The main sources of ceramide are de novo synthesis or catabolism of sphingomyelin and complex sphingolipids (for details, please refer to the overview on sphingolipid metabolism at the beginning of this book). The de novo biosynthetic pathway resides in the ER/Golgi and requires the Serine Palmitoyltransferase complex (SPTLC1-3) and the Ceramide Synthases (CerS1-6). Ceramide derived from the de novo pathway is then shuttled to the Golgi either by vesicular transport or by non-vesicular transport through the action of Ceramide Transfer Protein (CERT) [51]. Ceramide delivered to the Golgi by CERT appears to be more specifically targeted for the synthesis of sphingomyelin through the action of Sphingomyelin Synthases (SGMS1 and 2) [52, 53]. Once ceramide has been delivered to the Golgi, it can also be utilized by several other enzymes, including CERK (ceramide kinase), and Ceramide Glucosyltransferase (UCGC). Thus, ceramide-1-phosphate, sphingomyelin, and glycosphingolipids are the major lipid species exported from the Golgi.
In contrast to the de novo pathway, the hydrolytic (catabolic) pathways occur at the plasma membrane, in the lysosome/endolysosomal compartments, in mitochondria, and in the Golgi, resulting in the formation of ceramide. These pathways involve one or more of the Sphingomyelinases (SMPD1-5), the Ceramidases (ASAH1-2, and ACER1-3), the Glucocerebrosidases (GBA1-3), Sphingosine and Ceramide kinase (SPHK1-2 and CERK) (reviewed in [54]). Catabolism of sphingosine-1-phosphate and ceramide 1-phosphate is carried out by a S1P lyase and the lipid phosphatases. The salvage or recycling pathway involves hydrolysis of complex sphingolipids in the endolysosomal system, resulting in the formation of sphingosine that can then be salvaged for the re-synthesis of ceramide and other sphingolipids.
It is now increasingly appreciated that any perturbations in the strictly regulated sphingolipid metabolic pathway have significant effects on signaling and growth of breast tumors. The following sections will address the changes seen in sphingolipid metabolism, and the functional consequences thereof, in Luminal, Basal, and Her2-like breast cancer subtypes. A number of cell lines are available that recapitulate gene expression patterns and phenotypic characteristics of the molecular subtypes of breast cancers [55, 56]. A few example cell lines are described in Table 2.
4 Sphingolipids in Luminal Type Breast Cancers
Luminal A Type breast tumors have favorable outcomes and are the most heterogeneous group of breast tumors in terms of signaling characteristics [37]. Luminal Type A tumors represent about 50 % of breast tumors, and are responsive to endocrine therapy [57] as they have high expression of hormone receptors, and downstream estrogen receptor (ESR1) signaling partners such as FOXA1 and RUNX1 [58]. One of the defining characteristics of luminal type A tumors, that separates them from the other molecular subtypes, is intact p53 and RB1 signaling [39]. Additionally, luminal A type tumors frequently have phosphatidylinositol-3-kinase (PI3K) mutations, especially in the PIK3 catalytic subunit (PIK3CA), even though these mutations are not associated with enhanced activation of the PI3K pathway as evidenced by phospho-AKT status, a key downstream target of this pathway [59]. This may be due to high expression of S6K and resultant feedback inhibition. Luminal type tumors also have high frequencies of inactivating mutations of mitogen activated protein kinases (MAPK). For example, loss-of-function mutations in MAP3K1 and MAP2K4 were found with high frequency in these tumors, suggesting loss of function in the p38-JNK1 signaling cascade [58, 60].
Compared to luminal A breast cancer, Luminal B breast cancer has significantly higher risk of rapid relapse following endocrine therapy [61]. Luminal B tumors are classified as ER+, but have lower expression of ER related genes than luminal A tumors and higher expression of proliferative genes [36, 62]. Luminal B tumors often have increased HER2 expression, but since HER2 status is used to distinguish a very specific clinical subset of tumors, HER2 status is not relevant to classification of luminal type tumors. Nevertheless, luminal B tumors have increased growth factor signaling as compared to luminal A tumors [63]. In contrast to luminal A tumors, the proliferative gene signatures of luminal B tumors seem to stem from increased genomic signaling events secondary to genetic duplication of proliferative genes [39, 64]. This finding is in accordance with a high frequency of p53 inactivating mutations, loss of ATM, and deregulation of MDM2 in luminal B tumors [39, 65]. Therefore, the defining characteristics of luminal B tumors are loss of function of the TP53 signaling pathway and amplification of proliferative genes, such as cyclin D1. For the most part, luminal type breast cancers can be considered ER+, HER2−, and the distinction between luminal A and luminal B is mainly a molecular genetic definition that is not regularly made in a clinical setting [66]. Luminal tumors rely heavily on cyclin D1 and on growth factor signaling for growth. Therefore, the following section will focus on the interplay between sphingolipids and growth factor receptors, and sphingolipids and cell cycle progression.
A number of interesting correlations between estrogen receptor signaling and sphingolipid metabolic enzymes have been published recently. Many of the studies on sphingolipids in breast cancer have been conducted using breast cancer cell lines, which limits their impact, nevertheless several studies with patient samples have also been carried out. Among the studies focusing on ER+ cell lines and tumors, UGCG, CerS, sphingomyelinases, ASAH1, and Sphingosine Kinase 1 and 2 (SK1 and SK2) play prominent roles [67–72].
4.1 Glucosylceramide Transferase
Glucosylceramide transferase (GCS) produces glucocerebrosides from ceramide and glucose. Subsequent action by other glycosyltransferases leads to a wide variety of glycolipids. In a study of 200 human breast cancer samples, the authors found high GCS (UGCG, gene name) expression in luminal A type tumors, and high GCS expression was also associated with poor outcomes in ER+ tumors [67]. Since ceramide is known to induce apoptosis and to act as a stress signal, conversion to other lipids, such as the glycosphingolipids, can reduce cell stress. Indeed, Liu et al. have reported that breast tissue generally has low expression of GCS, as compared to other organs, and that high GCS expression is associated with multi-drug resistance and metastatic disease in ER+, HER2+ tumors [73]. In agreement with this study, several groups have reported on the ability of GCS inhibitors to reverse drug resistance in breast cancer cell lines with high GCS expression [74, 75]. A recent report has even suggested that GCS activity is directly inhibited by tamoxifen, and that high GCS activity can decrease sensitivity to the drug [76]. Besides a potential role in receptor function and endocytosis, several groups have also implicated GCS in resistance to chemotherapy [77, 78]. In fact, GCS may limit the efficacy of several chemotherapeutic drugs by titrating away apoptotic ceramide that accumulates following treatment with drugs such as taxol and Adriamycin, among others [79, 80].
4.2 The Ceramide Synthases
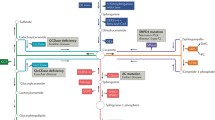
The Ceramide Synthase (CerS) enzymes produce dihydroceramide from dihydrosphingosine, and the rapid desaturation of dihydroceramide produces ceramide. In a large-scale lipidomics study of breast cancers, Schiffmann et al. demonstrated a statistically significant increase in C18:0 and C20:0 ceramide in ER+ tumor samples [81]. The authors also demonstrated increased expression of CerS2, CerS4, and CerS6 in malignant tumor samples. Erez-Roman et al. have also reported elevations in CerS2 and CerS6 in malignant breast tissue samples as compared to normal solid tissue [82]. However, neither group could conclude whether the elevation in ceramide content or CerS expression was a consequence of or a driver of malignancy in breast carcinomas. Continuation of the CerS studies demonstrated opposite roles for long chain and very long chain ceramides. The authors found that CerS2, and by extension the very long chain C24 and C26 ceramides, increased colony formation in MCF7 breast cancer cells whereas CerS4 and CerS6, which produce long chain ceramides (mostly C14 and C16 ceramides) decreased colony formation [83]. These studies concluded that the long chain ceramides induce mitochondrial damage and inhibit cell proliferation whereas the very long chain ceramides decrease contact inhibition in MCF7 cells. Data mining has confirmed robust up-regulation of CerS2, CerS4, and CerS6 in Luminal type tumors (Fig. 1).

Sphingolipid gene expression in molecular subtypes of breast cancer. Publicly available TCGA expression data was used to assess the expression levels of sphingolipid genes in molecular subtypes of breast cancer. The expression levels of the indicated genes were compared to expression in adjacent normal solid tissue, and the fold change was calculated. Hierarchical clustering was carried out as shown below
In line with the above observations, different studies have proposed that long chain ceramides and the enzymes responsible for their production CerS6 and CerS1 have tumor suppressive properties [84, 85]. For instance, CerS6 has been implicated in the response to folate stress, suggesting CerS6 may also play a role in response to antimetabolites [86]. Antimetabolites are employed in the treatment of some early stage breast cancers, and cell culture studies suggest that combined therapy with the antimetabolite pemetrexed and the kinase inhibitor sorafenib can induce increases in the dihydroceramides. The authors showed that the production of dihydroceramide was dependent on the activity of CerS6 and led to apoptosis of luminal type breast cancer cell lines [87]. Other researchers have reported repression of hTERT activity by CerS1 in various cancer cell lines [88, 89], suggesting an inhibitory role for the long chain ceramides in tumor growth.
4.3 The Sphingomyelinases
The Sphingomyelinases hydrolyze sphingomyelin to ceramide and phosphocholine. There are several isoforms of sphingomyelinase (SMPD1-5), and both SMPD1 (Acid Sphingomyelinase, ASMase) and SMPD3 (Neutral Sphingomyelinase 2, NSMase 2) have been implicated in breast cancer progression. As seen in Fig. 1, SMPD3 is down regulated in all 4 subtypes of breast carcinomas. Loss of heterozygosity and epigenetic regulation of this region has been described, and may account for the down regulation of SMPD3. A comparative epigenetic study reported that there is aberrant CpG methylation around the SMPD3 locus in human breast cancer cell lines as well as several primary tumor samples [90]. Additionally, another group reported that there is frequent loss of heterozygosity events at 16q22.1, a chromosomal area that includes the SMPD3 locus [91]. These results suggest NSmase 2 (SMPD3) may play an anti-tumor role, however this hypothesis has never been directly tested in breast cancer.
The role of NSMase 2 in drug response has been characterized in breast cancer. Adriamycin resistant MCF7 (MCF7-ADR) cells were found to have higher SM and lower NSMase levels as compared to MCF7 cells. Furthermore, expression and activity of NSMase was significantly up regulated in MCF7-ADR cells following treatment with a demethylating agent that decreases methylation-dependent silencing of genes. This response was not seen in MCF7 cells, suggesting MCF7-ADR cells acquire epigenetic alterations that contribute to the ADR phenotype [92]. In the MCF7-ADR cells, demethylation of DNA was correlated with the subsequent decrease in plasma membrane sphingomyelin, and increased membrane fluidity, attributable to increased activity of NSMase 2. Up-regulated NSMase 2 activity resulted in increased doxorubicin uptake and apoptosis in MCF7-ADR cells, suggesting that silencing of the SMPD3 locus may be one mechanism by which MCF7 cells acquire resistance to Adriamycin [92]. In line with the work by Vijayaraghavalu et al. is the finding by Ito and colleagues that apoptosis of Adriamycin sensitive MCF7 cells is mediated by NSMase 2 following daunorubicin treatment [93]. Taken together, these observations suggest that re-activation of SMPD3 expression may be a powerful tool for restoration of sensitivity to anthracyclines in resistant tumors.
In addition to NSMase 2, ASMase plays an important role in tumor biology. ASMase has been found to have significant impact on lysosomal stability and in tumor motility. Traditionally, ASMase is associated with Niemann–Pick Disease, and loss of ASMase enzymatic activity results in lysosomal dysfunction. It should come as no surprise that inhibition of ASMase causes significant lysosomal dysfunction in tumor cells as well. In MCF7-Bcl-2 cells, inhibition of ASMase with the receptor tyrosine kinase inhibitor Sunitinib, leads to lysosomal instability and apoptosis [94]. In contrast to lysosomal stability, governance of cell motility seems to be a tumor specific function of ASMase. ASMase was shown to mediate some of the cytotoxic effects of cisplatin on cytoskeletal remodeling in MCF7 cells [95]. The researchers showed that induction of ASMase and the subsequent generation of ceramide reduces ERM phosphorylation and retards cell motility [95, 96]. Besides its role in cell motility and lysosomal stability, ASMase is known to modulate inflammatory signaling in breast carcinoma cells. Studies on ASMase in MCF7 cells showed ASMase was necessary for p38-dependent expression of IL-6 following PKC activation by the cell permeable phorbol ester phorbol 12-myristate 13-acetate (PMA) [97]. However, the role of ASMase-induced IL-6 in these cells remains unclear.
4.4 The Ceramidases
Catabolism of ceramide to other sphingolipids is another potential route by which luminal type breast cancers are able to escape the pro-apoptotic effects of ceramide. The ceramidases rapidly convert ceramide to sphingosine, and then sphingosine is subsequently converted into S1P. Down stream products of the ceramidases, especially sphingosine and S1P, have been shown to be potent signaling molecules. Therefore, conversion of a small amount of ceramide is likely to have profound effects on the cell. Several studies have linked acid ceramidase with growth of ER+ cell lines. Indeed, inhibition of acid ceramidase causes lysosomal disruption that results in rapid cell death in ER+ cell lines [98, 99]. However, the cytotoxic effect of acid ceramidase inhibition is not only linked to its effects on lysosomal permeability. Inhibition of acid ceramidase with non-lysomatrophic compounds still results in cell death, suggesting that the functional consequence of inhibition extends beyond lysosomal homeostasis [70]. The downstream signaling function of ASAH1/Sphingosine was recently delineated by Lucki et al. The authors found that induction of ASAH1 by phytoestrogens in MCF7 cells resulted in cyclin B2 activation and increased cell proliferation [69], and they further showed that induction of ASAH1 relied on c-SRC and ERK activation and subsequent ERα recruitment to the ASAH1 promoter. In accordance with these findings, tumor samples with increased estrogen receptor signaling, i.e., luminal tumors, have high levels of ASAH1 mRNA (Fig. 1).
Paradoxically, high ASAH1 expression has also been correlated with better outcomes following treatment of ER+ breast tumors [71]. A potential reason cell culture studies may not align with in vivo studies is because acid ceramidase has been shown to modulate the tumor microenvironment through regulation of inflammatory cytokines and prostanoid signaling [97, 100]. These inflammatory pathways may play a role in baseline tumor growth, but may make the tumor more prone to apoptosis or immune surveillance following chemotherapy. Therefore, cell culture studies may underestimate the anti-tumor effects of ASAH1 that are mediated through the immune response to tumors following chemotherapy.
Several groups have investigated the interplay between immune modulators and sphingolipid signaling. Signaling events intrinsic to ER+ tumor cells seem to mediate both autocrine and paracrine signaling within the tumor microenvironment. Stress induced cytokines, such as Tumor Necrosis Factor (TNF-α) and Interleukin-1 (IL-1β), activate sphingolipid catabolic pathways [101–103], which in turn have been shown to regulate production of inflammatory mediators in an ASAH1 dependent manner. In breast cancer cell lines, several investigators have demonstrated a requirement for ASAH1 in the production of prostaglandin E2 (PGE2), Chemokine Ligand 5 (CCL5), and Interleukin-6 (IL-6) [97, 100, 104]. Each of these inflammatory mediators can induce tumor progression by creating a permissive tumor microenvironment. Tumor derived PGE2 induces p38/JNK activation in adjacent fibroblasts. The activated p38/JNK signaling cascade resulted in transcription of aromatase and consequent production of estrogen by fibroblasts that promotes tumor growth in estrogen responsive tumors [105]. CCL5 promotes growth and metastasis of ER+ tumors by recruiting macrophages, inducing angiogenesis, and stimulating proliferation of CD44+/CD24− cancer stem like cells [106–108]. The role of IL-6 in breast cancer is multifaceted. In clinical studies on the relationship between IL-6 and breast cancer, high IL-6 is associated with early locally advanced tumors, and good prognosis [109]. Conversely, in late stage tumors, IL-6 was associated with worse prognosis [110]. Cell line studies have demonstrated that ER+ cell lines become growth suppressed by IL-6 and undergo EMT when exposed to IL-6 for extended periods of time. Conversely, ER− cell lines are not responsive to exogenous IL-6 (reviewed in [111]). A possible reason for ER− tumors being unresponsive to IL-6 is the finding that ER− tumor cell lines have a very high level of endogenous IL-6 production, as compared to ER+ cell lines, and therefore ER− tumors already have activated IL-6 signaling pathways and are not responsive to additional IL-6 [112].
4.5 The Sphingosine Kinases
Sphingosine kinase phosphorylates sphingosine to produce sphingosine-1-phosphate (S1P). S1P acts as an intracellular signaling molecule as well as an extracellular signaling molecule that interacts with S1P receptors on the cell surface. In a thorough study of SK1 in human breast cancer samples, Watson et al. found that SK1/S1P participate in non-genomic signaling by ER and cross talk between ER and ERK-1/2. Patients with ER+ tumors displaying high ERK-1/2 and SK1 relapsed nearly 10 years earlier than patients with low SK1 and ERK-1/2. Additionally, the patients with high SK1 and ERK-1/2 were found to be tamoxifen resistant [113]. In accordance with the work by Watson et al., Zhang et al. carried out a large scale, pan-cancer meta-study demonstrating a significant correlation between SK1 expression and worse outcomes in a number of solid tumors. The authors reviewed immunohistochemical staining for SK1 in a range of breast tumor samples and calculated a hazard ratio of 1.86 for tumors with high SK1 expression [114]. Mining of microarray data has revealed luminal type tumors have high SPHK1 and SPHK2 expression (Fig. 1).
SK1 expression in ER+ tumors is modulated by ER signaling. In a study examining expression of micro RNA miR-515-5p, it was noted that in MCF7 cells this miRNA is correlated with SK1 expression. The authors reported that when the ERα receptor is activated by estradiol (E2), the ERα/E2 complex directly interacts with the promoter sequence that regulates miR-515-5p. This interaction leads to a decrease in miR-515-5p expression and de-repression of SK1 expression [115]. The authors went on to report that in a small cohort of 34 tumor samples, ER+ tumors had lower miR-515-5p expression than ER− tumors. However, they do not correlate this finding with SK1 expression in their cohort [115]. In support of the findings by Pinho et al., Takabe and colleagues found that E2 treatment resulted in increased S1P efflux from cultured MCF7 cells, and the observed increase in S1P export was dependent on the ERα receptor suggesting that active ER signaling is necessary for SK1, but not SK2, activation [116]. Taken together, active ERα signaling promotes S1P production in MCF7 cells.
In addition to Sphingosine Kinase activity, S1P receptors (S1PR1-5) seem to be modulated by ER signaling. Indeed, multiple groups have observed ERK activation by S1P/S1P receptor signaling, and correlations between S1PR expression and ERK1/2 expression have been made [117, 118]. In a study of 304 ER+ breast tumor samples, Watson et al. correlated S1P receptor 1 and 3 expression with ERK1/2 expression. The authors found that patients with S1PR1 highly localized in the membrane had shorter time to recurrence of disease, and high levels of cytoplasmic S1PR1 and 3 were associated with worse survival [113]. The authors also noted that TRANSFAC analysis (Biobase) revealed binding sites for ERα, c-Jun, and Sp1 in the S1P3 promoter region [113]. This suggests that tumors with active ER signaling (i.e., tumors with high PR and ER expression [119]) also have high S1P mediated ERK-1/2 activation.
The finding that the SK1/S1P/S1PR axis is up-regulated in ER+ tumors would suggest that targeting this axis would be beneficial in the treatment of breast carcinomas. However, targeting of the SK1/S1P/S1P receptor axis in breast cancer has been met with mixed results. A group from the Department of Chemistry at Amgen utilized the crystal structure of SK1 to generate specific inhibitors of the sphingosine kinases. The inhibitors, termed ‘compound A’ and ‘compound B,’ were potent and reproduced the phenotypes of SK1−/− mice [120]. The Amgen Oncology Research group then carried out a cell line study with compound A and compound B, as well as the inhibitor SKII, and found that their compounds reduced S1P levels, but did not impact viability of tumor cell lines at concentrations that reduced S1P levels in these cells [121]. Additionally, the authors reported that ablation of SK1 or SK2 did not reduce tumor cell viability. The conclusion was that SK1 and SK2 are not high yield targets for cancer chemotherapeutics. On the other hand, as it will be discussed later on, targeting the S1P receptors is a very attractive strategy in basal type tumors.
4.6 Ceramide Kinase
In contrast to S1P, comparatively little work has been done on delineating the role of ceramide-1-phosphate (C1P). C1P is produced by CERK mediated phosphorylation of ceramide. In low grade breast cancer, it has been noted that high CERK expression correlates with poor histologic grading of tumors and ER negativity [122]. Additionally, CERK inhibition results in M-phase arrest of MCF7 cells, and sensitized cells to protein kinase inhibitors [123]. Message level of CERK is not significantly different between tumor subtypes (Fig. 1) suggesting that post-translational regulation and subcellular localization of CERK may play a significant role in its function within the tumor cell.
5 Sphingolipids in Basal Type Breast Cancer
The basal-like genetic signature was one of the first genetic portraits of disease to be described. This signature includes expression of mitogenic pathways, as well as high expression of keratins 5, 6, and 17 [34]. Basal type breast carcinomas are mostly triple negative tumors that do not express ER, PR, or HER2 receptors, but roughly 25 % of basal like tumors are not triple negative. Greater than 80 % of basal like tumors have mutant TP53 and loss of p53 signaling, as well as loss of RB1 and BRCA1 [39]. Myc activation, cyclin E1 amplification, and activation of the HIF1-α/ARNT pathways are prominent features of basal like tumors [39, 124]. Another interesting observation from TCGA is that basal-like breast carcinomas share several common features with serous ovarian carcinoma, including Myc activation, cyclin E1 amplification, and inactivation of TP53 and RB1, as well as loss of BRCA1 [39]. This finding suggests that some of the processes driving serous ovarian tumors may be active in basal type breast cancers as well. The lack of hormone receptors and the aggressive nature of basal-like tumors have created a significant need for identification of new therapies.
The approach to surgical treatment of triple negative breast cancers is similar to other breast carcinomas. The choice between adjuvant (treatment given after surgical excision of a primary tumor) and neoadjuvant (treatment given before surgical excision) therapy is made based on tumor size and local spread. Triple negative tumors often respond well to chemotherapy; however, there is a high recurrence rate of tumors [125]. Therefore, there is a significant need for more effective chemotherapeutics. In this regard, ceramide has been used to induce apoptosis of hormone insensitive breast cancer cell lines [126, 127], and some success has been achieved in murine models of breast cancer [128]. Ceramide causes clustering and activation of death receptors, and the apoptotic threshold of cells is modulated by the presence of ceramide at the plasma membrane [129]. Ceramide nanoliposomes can increase the concentration of ceramide at the plasma membrane of target cells and promote pro-apoptotic signaling events in breast cancer cell lines [130].
Beside using ceramide nanoliposomes to indiscriminately increase ceramide levels in cell membranes, ceramide nanoliposomes have also been used to improve the bioavailability and efficacy of breast cancer chemotherapeutics. Nanoliposomal preparations containing ceramide and the tyrosine kinase inhibitor sorafenib were shown to decrease AKT phosphorylation and Cyclin D1 expression, and the presence of ceramide sensitized MDA-MB-231 cells to sorafenib-induced apoptosis [131]. Sorafenib is traditionally used to treat renal carcinoma, and has powerful anti-angiogenic and anti-mitogenic properties, making it an interesting therapeutic for the treatment of basal type breast cancers [132]. Indeed recent clinical trials have demonstrated a potential role for sorafenib in Her2 negative tumors as well as in tamoxifen resistant tumors [133, 134]. Ceramide nanoliposomes have also been used to improve the cellular uptake of doxorubicin in MCF7 and SKBR3 breast cancer cell lines [135]. Ceramide nanoliposomes have been used in vivo and may hold promise in adjuvant or neoadjuvant treatment of breast cancers by increasing the bioavailability, stability, and efficacy of current chemotherapeutics [136].
The success of neoadjuvant treatment of triple negative breast tumors is assessed by the pathologic response of the tumor to chemotherapeutic agents. Achieving a preoperative pathologic complete response (pCR) is the goal of neoadjuvant therapy, but it is difficult to predict what patients will achieve pCR with a given chemotherapy regiment. Roughly 55 % of patients with triple negative tumors do not respond well to multi-agent neoadjuvant therapy [137]. A series of studies has recently uncovered potential roles for GCS, CERT, and the ceramide producing β-Glucosidase enzymes (GBA1 and GBA3) in resistance to taxanes in hormone insensitive breast tumors. The first study demonstrated that down regulation of CERT, or up-regulation of GBA1 or 3, sensitized tumor cells to taxanes [138]. As a follow up to their in vitro functional genomics study, the research group carried out a retrospective meta-study and found that up-regulation of GCS and CERT, and concomitant down-regulation of GBA1 and GBA3, was highly associated with failure of 5-fluorouracil, doxorubicin, cyclophosphamide, paclitaxel (TFAC) treatment of triple negative tumors possibly because of poor accumulation of ceramide [139]. It is important to note though that the majority of basal type tumors have low GCS and CERT expression (Fig. 1), therefore, the association between high expression of these genes and poor response to treatment may make them valuable biomarkers for a subset of triple negative patients. Altogether these results highlight the importance of regulation of ceramide levels (and of ceramide metabolizing enzymes) in triple negative breast carcinoma cells in order to sensitize tumors to adjuvant chemotherapy.
Based on the work by the TCGA and others, triple negative tumors have high expression of the hypoxia inducible genes. This suggests these tumors may be susceptible to anti-vasculogenic therapies. One sphingolipid analog with potent anti-vasculogenic properties is fingolimod (Gilenya). Fingolimod (also known as FTY720) is a sphingosine analog approved for treatment of multiple sclerosis that serves as a precursor for an agonist of the S1P receptors that often results in internalization of the receptor followed by its shut down [140, 141]. In studies with cell lines, fingolimod has been shown to disrupt ERK1/2 and HER2 cross talk in Her2-like breast cancer cells and to decrease the proliferation of luminal type breast cancer cells [142, 143]. In studies using syngeneic mouse models of breast cancer, research has shown fingolimod decreases vascular smooth muscle motility in response to tumor-derived cytokines [144]. Based on its anti-vasculogenic properties, fingolimod may also serve as a potent inhibitor of basal type breast tumor growth in vivo.
6 Sphingolipids in Her2-Like Breast Cancer
Genomic duplication of the Her2 gene results in overexpression of HER2 and HER2-associated genes. In these tumors, HER2 itself as well as FGFR4, EGFR, and several other receptor tyrosine kinases are up regulated. Her2-like tumors have high levels of phospho-SRC and phospho-S6 [39]. Her2-like tumors are largely treated with adjuvant chemotherapy. Effective treatment relies primarily on targeted therapies such as trastuzumab (targeting HER2) in combination with a taxane or anthracycline [145, 146]. Despite the finding that EGFR signaling is up regulated in Her2-like tumors, lapatinib, an inhibitor of the Her2 and EGFR pathways, has not been clinically successful in the adjuvant treatment of Her2-like tumors [147]. On the other hand, adjuvant treatment with trastuzumab has a fairly high disease free survival rate of 76 % [148]. Although there is a small subset of Her2-like tumors that are ER positive, the clinical relevance of combining endocrine therapy with targeted HER2 therapies has not been rigorously evaluated. Also, general clinical practice focuses on targeting the HER2 receptor, and current investigations are underway to evaluate the efficacy of targeting tyrosine kinase activity in Her2 like tumors [149].
Several groups have studied the role of sphingolipids in Her2-like tumors with a focus on CERT, CERK, and sphingomyelin homeostasis. CERT seems to play a prominent role in Her2-like tumors, and has been shown to mediate paclitaxel sensitivity in these tumors. Extending their work with functional metagenomics, the Swanton group found that CERT protein was overexpressed in HER2+ tumors, and they demonstrated that silencing of CERT in HER2+ tumor cell lines sensitized the cells to chemotherapeutics [139, 150]. Inhibition of CERT in these cell lines induced autophagic flux and LAMP2 expression, and increased sensitivity to paclitaxel, doxorubicin, cisplatin and trastuzumab. Therefore, CERT may be of both therapeutic and prognostic value in HER2+ breast tumors that express high levels of CERT. However, mining of TCGA data has revealed that CERT mRNA is not highly expressed in Her2-Like tumors as compared to adjacent normal tissue. The reason for this discrepancy may be due to discordance between CERT protein and mRNA levels, or possibly due to the choice of controls used for comparison.
The role of CERK in HER2+ tumors has recently been investigated using a mouse model of breast cancer. Utilizing a doxycycline inducible HER2/neu mouse model of breast cancer, Payne et al. demonstrated that CERK was required for tumor cell survival following loss of Her2 upon doxycycline withdrawal. When expression of Her2 was decreased and cells were treated with shRNA to CERK, tumor cells expressed high levels of pro-apoptotic markers [151]. This result suggests that following deprivation of Her2 signalling, tumors become reliant on CERK for survival. The authors also tested the role of CERK in BT474 (ER+, HER2+, p53 mut) and SKBR3 (ER−, HER2+, p53 mut) cells treated with the tyrosine kinase inhibitor lapatinib. As expected, based on the in vivo studies, Her2 cell lines were dependent on CERK expression for survival following Her2 inhibition by lapatinib. The authors extended their study to analyze microarray data from 2200 patient samples, and found significant correlation between CERK expression and aggressive tumors such as HER2+ tumors [151]. The association between CERK and HER2+ tumor progression makes CERK inhibition an attractive target for combination chemotherapy with Her2 inhibitors.
The role of other sphingolipids in Her2-like tumors has not been rigorously studied. However, a study investigating lipid levels in several patient samples from Her2-like tumors found elevations in the level of sphingomyelin [152]. This finding is interesting and helps validate the work by Lee et al. because overexpression of CERT is known to increase sphingomyelin levels in cells [150, 153]. Our mining of TGCA expression data sets reveals that there are a number of alterations in the expression of several sphingolipid genes that are specific to Her2-Like tumors. Members of the SPT complex, acid sphingomyelinase, DEGS1, and GBA are all up regulated in Her2-Like tumors relative to other tumor types (Fig. 1). But, the functional roles of these enzymes in Her2-like tumors remain to be elucidated.
7 Conclusions
The function of sphingolipids in breast cancer is an area of intense investigation. Sphingolipids modulate many of the growth, apoptosis, inflammatory, and perhaps even angiogenic pathways that breast carcinomas rely on. In luminal type breast cancers, conversion of ceramide to complex sphingolipids is associated with drug resistance and worse patient outcomes. The somewhat paradoxical finding by Schiffmann et al. that luminal type tumors have up-regulated synthesis of several ceramide species highlights the different functional outcomes of the ceramides with different molecular composition. Ceramide is rapidly converted to other lipid species, and several groups have demonstrated the role of sphingosine kinase 1 in tumor growth, while the role of acid ceramidase in luminal type tumors is complex. Acid ceramidase in fact promotes a number of cell-cell signaling pathways, and some of the subtleties of these signaling pathways may be lost in over simplified experimental conditions.
Basal type breast tumors are highly aggressive, and have poor response to treatment. Novel therapeutics based on ceramide, as well as novel drug delivery methods have recently shown promise in the treatment of these tumors. The studies showing potential roles for CERT and GCS suggest that targeting these pathways may be an effective route for treating triple negative tumors. Many current chemotherapeutics, such as doxorubicin and etoposide, cause cell death through generation of ceramide. Therefore, directly modulating ceramide metabolic pathways may increase the effectiveness of current chemotherapeutics and increase their therapeutic index.
The role of sphingolipids in Her2-like tumors has not been thoroughly studied. This is despite the fact that robust murine models of Her2-like tumors are readily available. In one of the only studies on sphingolipids in Her2 driven breast cancers, Payne et al. showed reliance on CERK. In addition to the role of CERK, non-vescicular transport of ceramide seems to be an important process in Her2-like tumors, and inhibition of CERT results in autophagy of HER2+ cell lines.
Sphingolipid research in other systems has demonstrated the power of multimodal bioinformatics studies focused on sphingolipid metabolic pathways [154]. Application of cross platform informatics to breast cancer will reveal new opportunities for targeted therapy focused on sphingolipid metabolic enzymes. It is obvious that more studies are needed into the specific roles of individual enzymes and pathways of sphingolipid metabolism in breast cancer and, as these roles of sphingolipids become clearer, more specific rationales will emerge for targeting sphingolipid metabolism as an attractive therapeutic modality.
References
American Cancer Society (2013) Cancer facts & figures 2013. American Cancer Society, Atlanta
Hajdu SI (2011) A note from history: landmarks in history of cancer, part 1. Cancer 117(5):1097–1102
Vargas A et al (2012) [The Edwin Smith papyrus in the history of medicine]. Rev Med Chil 140(10):1357–1362
Aszmann OC (2000) The life and work of Theodore Schwann. J Reconstr Microsurg 16(4):291–295
Triolo VA (1965) Nineteenth century foundations of cancer research advances in tumor pathology, nomenclature, and theories of oncogenesis. Cancer Res 25:75–106
Hajdu SI, Darvishian F (2013) A note from history: landmarks in history of cancer, part 5. Cancer 119(8):1450–1466
Hajdu SI (2012) A note from history: landmarks in history of cancer, part 4. Cancer 118(20):4914–4928
Sistrunk WE, Maccarty WC (1922) Life expectancy following radical amputation for carcinoma of the breast: a clinical and pathologic study of 218 cases. Ann Surg 75(1):61–69
Hajdu SI, Vadmal M (2013) A note from history: Landmarks in history of cancer, Part 6. Cancer 119(23):4058–4082
Beatson G (1896) On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment with illustrative cases. Lancet 148(3803):162–165
Love RR, Philips J (2002) Oophorectomy for breast cancer: history revisited. J Natl Cancer Inst 94(19):1433–1434
Rous P (1911) A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J Exp Med 13(4):397–411
DeVita VT, Chu E (2008) A history of cancer chemotherapy. Cancer Res 68(21):8643–8653
Umeda M, Heidelberger C (1968) Comparative studies of fluorinated pyrimidines with various cell lines. Cancer Res 28(12):2529–2538
Oshaughnessy JA et al (2001) Randomized, open-label, phase II trial of oral capecitabine (Xeloda) vs. a reference arm of intravenous CMF (cyclophosphamide, methotrexate and 5-fluorouracil) as first-line therapy for advanced/metastatic breast cancer. Ann Oncol 12(9):1247–1254
Kunimoto T, Hori M, Umezawa H (1967) Modes of action of phleomycin, bleomycin and formycin on HeLa S3 cells in synchronized culture. J Antibiot (Tokyo) 20(5):277–281
Di Marco A, Gaetani M, Scarpinato B (1969) Adriamycin (NSC-123,127): a new antibiotic with antitumor activity. Cancer Chemother Rep 53(1):33–37
Creasey WA et al (1976) Clinical effects and pharmacokinetics of different dosage schedules of adriamycin. Cancer Res 36(1):216–221
Moore FD et al (1967) Carcinoma of the breast. A decade of new results with old concepts. N Engl J Med 277(7):343–350
Jensen EV (1975) Estrogen receptors in hormone-dependent breast cancers. Cancer Res 35(11 Pt. 2):3362–3364
Block GE et al (1978) Correlation of estrophilin content of primary mammary cancer to eventual endocrine treatment. Ann Surg 188(3):372–376
Ward HW (1973) Anti-oestrogen therapy for breast cancer: a trial of tamoxifen at two dose levels. Br Med J 1(5844):13–14
Slichenmyer WJ, Von Hoff DD (1991) Taxol: a new and effective anti-cancer drug. Anticancer Drugs 2(6):519–530
Hanauske AR et al (1992) Effects of Taxotere and taxol on in vitro colony formation of freshly explanted human tumor cells. Anticancer Drugs 3(2):121–124
Seidman AD et al (2008) Randomized phase III trial of weekly compared with every-3-weeks paclitaxel for metastatic breast cancer, with trastuzumab for all HER-2 overexpressors and random assignment to trastuzumab or not in HER-2 nonoverexpressors: final results of Cancer and Leukemia Group B protocol 9840. J Clin Oncol 26(10):1642–1649
Sparano JA et al (2008) Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med 358(16):1663–1671
Druker BJ et al (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2(5):561–566
Pegram M et al (1999) Inhibitory effects of combinations of HER-2/neu antibody and chemotherapeutic agents used for treatment of human breast cancers. Oncogene 18(13):2241–2251
Pegram MD, Konecny G, Slamon DJ (2000) The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat Res 103:57–75
Baselga J et al (2001) Mechanism of action of trastuzumab and scientific update. Semin Oncol 28(5 Suppl 16):4–11
Hardy NM, Fowler DH, Bishop MR (2006) Immunotherapy of metastatic breast cancer: phase I trail of reduced-intensity allogeneic hematopoietic stem cell transplantation with Th2/Tc2 T-cell exchange. Clin Breast Cancer 7(1):87–89
Edge S, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A (2010) AJCC cancer staging manual, 7th edn. Springer, New York, p 649
Schott A (2014) Systemic treatment of metastatic breast cancer in women: Chemotherapy
Perou CM et al (2000) Molecular portraits of human breast tumours. Nature 406(6797):747–752
Howlader N et al (2014) US incidence of breast cancer subtypes defined by joint hormone receptor and HER2 status. J Natl Cancer Inst 106(5):pii: dju055
Sørlie T et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci 98(19):10869–10874
Parker JS et al (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167
Voduc KD et al (2010) Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol 28(10):1684–1691
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Hannun YA, Obeid LM (2002) The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem 277(29):25847–25850
Snyder RA, Brady RO, Kornblith PL (1970) Ganglioside patterns of cultured human glioma cells. Neurology 20(4):412
Keenan TW et al (1975) Exogenous glycosphingolipids suppress growth rate of transformed and untransformed 3 T3 mouse cells. Exp Cell Res 92(2):259–270
Manuelidis L, Yu RK, Manuelidis EE (1977) Ganglioside content and pattern in human gliomas in culture. Correlation of morphological changes with altered gangliosides. Acta Neuropathol 38(2):129–135
Bremer EG, Schlessinger J, Hakomori S (1986) Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J Biol Chem 261(5):2434–2440
Okada Y, Matsuura H, Hakomori S (1985) Inhibition of tumor cell growth by aggregation of a tumor-associated glycolipid antigen: a close functional association between gangliotriaosylceramide and transferrin receptor in mouse lymphoma L-5178Y. Cancer Res 45(6):2793–2801
Kim YS et al (1986) Expression of LeY and extended LeY blood group-related antigens in human malignant, premalignant, and nonmalignant colonic tissues. Cancer Res 46(11):5985–5992
Hannun YA, Bell RM (1989) Regulation of protein kinase C by sphingosine and lysosphingolipids. Clin Chim Acta 185(3):333–345
Chen Z et al (2014) Protein kinase C-delta inactivation inhibits the proliferation and survival of cancer stem cells in culture and in vivo. BMC Cancer 14:90
Kim J et al (2011) Sustained inhibition of PKCalpha reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene 30(3):323–333
Chalfant CE et al (1999) Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem 274(29):20313–20317
Hanada K et al (2003) Molecular machinery for non-vesicular trafficking of ceramide. Nature 426(6968):803–809
Charruyer A et al (2008) Decreased ceramide transport protein (CERT) function alters sphingomyelin production following UVB irradiation. J Biol Chem 283(24):16682–16692
Blom T et al (2012) Tracking sphingosine metabolism and transport in sphingolipidoses: NPC1 deficiency as a test case. Traffic 13(9):1234–1243
Fyrst H, Saba JD (2010) An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol 6(7):489–497
Bertucci F et al (2005) Gene expression profiling identifies molecular subtypes of inflammatory breast cancer. Cancer Res 65(6):2170–2178
Prat A et al (2013) Characterization of cell lines derived from breast cancers and normal mammary tissues for the study of the intrinsic molecular subtypes. Breast Cancer Res Treat 142(2):237–255
Schnitt SJ (2010) Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol 23(S2):S60–S64
Bange J, Zwick E, Ullrich A (2001) Molecular targets for breast cancer therapy and prevention. Nat Med 7(5):548–552
Stemke-Hale K et al (2008) An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 68(15):6084–6091
Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9(8):537–549
Tran B, Bedard PL (2011) Luminal-B breast cancer and novel therapeutic targets. Breast Cancer Res 13(6):221
Wirapati P et al (2008) Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res 10(4):R65
Sotiriou C, Pusztai L (2009) Gene-expression signatures in breast cancer. N Engl J Med 360(8):790–800
Troester MA et al (2006) Gene expression patterns associated with p53 status in breast cancer. BMC Cancer 6:276
Jiang Z et al (2010) Rb deletion in mouse mammary progenitors induces luminal-B or basal-like/EMT tumor subtypes depending on p53 status. J Clin Invest 120(9):3296–3309
Neve RM et al (2006) A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10(6):515–527
Ruckhaberle E et al (2009) Prognostic relevance of glucosylceramide synthase (GCS) expression in breast cancer. J Cancer Res Clin Oncol 135(1):81–90
Zhang X et al (2012) Doxorubicin influences the expression of glucosylceramide synthase in invasive ductal breast cancer. PLoS One 7(11), e48492
Lucki NC, Sewer MB (2011) Genistein stimulates MCF-7 breast cancer cell growth by inducing acid ceramidase (ASAH1) gene expression. J Biol Chem 286(22):19399–19409
Bai A et al (2009) Synthesis and bioevaluation of omega-N-amino analogs of B13. Bioorg Med Chem 17(5):1840–1848
Ruckhaberle E et al (2009) Acid ceramidase 1 expression correlates with a better prognosis in ER-positive breast cancer. Climacteric 12(6):502–513
Ruckhaberle E et al (2008) Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat 112(1):41–52
Liu YY et al (2011) Glucosylceramide synthase, a factor in modulating drug resistance, is overexpressed in metastatic breast carcinoma. Int J Oncol 39(2):425–431
Sun YL et al (2005) [Construction of glucosylceramide synthase-specific siRNA expression vector and its efficiency in reversal of drug resistance in breast carcinoma cells]. Zhonghua Yi Xue Za Zhi 85(8):518–521
Gouaze V et al (2005) Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res 65(9):3861–3867
Lavie Y et al (1997) Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J Biol Chem 272(3):1682–1687
Bose R et al (1995) Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 82(3):405–414
Lucci A et al (1999) Multidrug resistance modulators and doxorubicin synergize to elevate ceramide levels and elicit apoptosis in drug-resistant cancer cells. Cancer 86(2):300–311
Charles AG et al (2001) Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother Pharmacol 47(5):444–450
Gewirtz DA (2000) Growth arrest and cell death in the breast tumor cell in response to ionizing radiation and chemotherapeutic agents which induce DNA damage. Breast Cancer Res Treat 62(3):223–235
Schiffmann S et al (2009) Ceramide synthases and ceramide levels are increased in breast cancer tissue. Carcinogenesis 30(5):745–752
Erez-Roman R, Pienik R, Futerman AH (2010) Increased ceramide synthase 2 and 6 mRNA levels in breast cancer tissues and correlation with sphingosine kinase expression. Biochem Biophys Res Commun 391(1):219–223
Hartmann D et al (2012) Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int J Biochem Cell Biol 44(4):620–628
Walker T et al (2009) Sorafenib and vorinostat kill colon cancer cells by CD95-dependent and -independent mechanisms. Mol Pharmacol 76(2):342–355
Min J et al (2007) (Dihydro)ceramide synthase 1 regulated sensitivity to cisplatin is associated with the activation of p38 mitogen-activated protein kinase and is abrogated by sphingosine kinase 1. Mol Cancer Res 5(8):801–812
Hoeferlin LA et al (2013) Folate stress induces apoptosis via p53-dependent de novo ceramide synthesis and up-regulation of ceramide synthase 6. J Biol Chem 288(18):12880–12890
Bareford MD et al (2012) Sorafenib and pemetrexed toxicity in cancer cells is mediated via SRC-ERK signaling. Cancer Biol Ther 13(9):793–803
Wooten-Blanks LG et al (2007) Mechanisms of ceramide-mediated repression of the human telomerase reverse transcriptase promoter via deacetylation of Sp3 by histone deacetylase 1. FASEB J 21(12):3386–3397
Koybasi S et al (2004) Defects in cell growth regulation by C18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J Biol Chem 279(43):44311–44319
Demircan B et al (2009) Comparative epigenomics of human and mouse mammary tumors. Genes Chromosomes Cancer 48(1):83–97
Chalmers IJ et al (2001) Mapping the chromosome 16 cadherin gene cluster to a minimal deleted region in ductal breast cancer. Cancer Genet Cytogenet 126(1):39–44
Vijayaraghavalu S et al (2012) Epigenetic modulation of the biophysical properties of drug-resistant cell lipids to restore drug transport and endocytic functions. Mol Pharm 9(9):2730–2742
Ito H et al (2009) Transcriptional regulation of neutral sphingomyelinase 2 gene expression of a human breast cancer cell line, MCF-7, induced by the anti-cancer drug, daunorubicin. Biochim Biophys Acta 1789(11-12):681–690
Ellegaard AM et al (2013) Sunitinib and SU11652 inhibit acid sphingomyelinase, destabilize lysosomes, and inhibit multidrug resistance. Mol Cancer Ther 12(10):2018–2030
Zeidan YH, Jenkins RW, Hannun YA (2008) Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J Cell Biol 181(2):335–350
Canals D et al (2010) Differential effects of ceramide and sphingosine 1-phosphate on ERM phosphorylation: probing sphingolipid signaling at the outer plasma membrane. J Biol Chem 285(42):32476–32485
Perry DM et al (2014) Defining a role for acid sphingomyelinase in the p38/interleukin-6 pathway. J Biol Chem 10(6):515–527
Ullio C et al (2012) Sphingosine mediates TNFalpha-induced lysosomal membrane permeabilization and ensuing programmed cell death in hepatoma cells. J Lipid Res 53(6):1134–1143
Bhabak KP et al (2013) Effective inhibition of acid and neutral ceramidases by novel B-13 and LCL-464 analogues. Bioorg Med Chem 21(4):874–882
Jenkins RW et al (2011) Regulation of CC ligand 5/RANTES by acid sphingomyelinase and acid ceramidase. J Biol Chem 286(15):13292–13303
Cain BS et al (1999) Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med 27(7):1309–1318
Mathias S et al (1993) Activation of the sphingomyelin signaling pathway in intact EL4 cells and in a cell-free system by IL-1 beta. Science 259(5094):519–522
Hannun YA (1994) The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem 269(5):3125–3128
Zeidan YH et al (2006) Acid ceramidase but not acid sphingomyelinase is required for tumor necrosis factor-{alpha}-induced PGE2 production. J Biol Chem 281(34):24695–24703
Chen D et al (2007) Prostaglandin E(2) induces breast cancer related aromatase promoters via activation of p38 and c-Jun NH(2)-terminal kinase in adipose fibroblasts. Cancer Res 67(18):8914–8922
Soria G, Ben-Baruch A (2008) The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett 267(2):271–285
Pinilla S et al (2009) Tissue resident stem cells produce CCL5 under the influence of cancer cells and thereby promote breast cancer cell invasion. Cancer Lett 284(1):80–85
Zhang Y et al (2009) Role of CCL5 in invasion, proliferation and proportion of CD44+/CD24- phenotype of MCF-7 cells and correlation of CCL5 and CCR5 expression with breast cancer progression. Oncol Rep 21(4):1113–1121
Karczewska A et al (2000) Expression of interleukin-6, interleukin-6 receptor, and glycoprotein 130 correlates with good prognoses for patients with breast carcinoma. Cancer 88(9):2061–2071
Salgado R et al (2003) Circulating interleukin-6 predicts survival in patients with metastatic breast cancer. Int J Cancer 103(5):642–646
Dethlefsen C, Hojfeldt G, Hojman P (2013) The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res Treat 138(3):657–664
Asgeirsson KS et al (1998) The effects of IL-6 on cell adhesion and e-cadherin expression in breast cancer. Cytokine 10(9):720–728
Watson C et al (2010) High expression of sphingosine 1-phosphate receptors, S1P1 and S1P3, sphingosine kinase 1, and extracellular signal-regulated kinase-1/2 is associated with development of tamoxifen resistance in estrogen receptor-positive breast cancer patients. Am J Pathol 177(5):2205–2215
Zhang Y et al (2014) Sphingosine kinase 1 and cancer: a systematic review and meta-analysis. PLoS One 9(2), e90362
Pinho FG et al (2013) Downregulation of microRNA-515-5p by the estrogen receptor modulates sphingosine kinase 1 and breast cancer cell proliferation. Cancer Res 73(19):5936–5948
Takabe K et al (2010) Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem 285(14):10477–10486
Kim ES et al (2014) Inflammatory lipid sphingosine-1-phosphate upregulates C-reactive protein via C/EBPbeta and potentiates breast cancer progression. Oncogene 33(27):3583–3593
Rutherford C et al (2013) Regulation of cell survival by sphingosine-1-phosphate receptor S1P1 via reciprocal ERK-dependent suppression of Bim and PI-3-kinase/protein kinase C-mediated upregulation of Mcl-1. Cell Death Dis 4, e927
Hu X et al (2009) Genetic alterations and oncogenic pathways associated with breast cancer subtypes. Mol Cancer Res 7(4):511–522
Gustin DJ et al (2013) Structure guided design of a series of sphingosine kinase (SphK) inhibitors. Bioorg Med Chem Lett 23(16):4608–4616
Rex K et al (2013) Sphingosine kinase activity is not required for tumor cell viability. PLoS One 8(7), e68328
Ruckhaberle E et al (2009) Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J Cancer Res Clin Oncol 135(8):1005–1013
Pastukhov O et al (2014) The ceramide kinase inhibitor NVP-231 inhibits breast and lung cancer cell proliferation by inducing M phase arrest and subsequent cell death. Br J Pharmacol 171(24):5829–5844
Chandriani S et al (2009) A core MYC gene expression signature is prominent in basal-like breast cancer but only partially overlaps the core serum response. PLoS One 4(8), e6693
Liedtke C et al (2008) Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol 26(8):1275–1281
Stover T, Kester M (2003) Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther 307(2):468–475
Shabbits JA, Mayer LD (2003) Intracellular delivery of ceramide lipids via liposomes enhances apoptosis in vitro. Biochim Biophys Acta 1612(1):98–106
Stover TC et al (2005) Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res 11(9):3465–3474
Miyaji M et al (2005) Role of membrane sphingomyelin and ceramide in platform formation for Fas-mediated apoptosis. J Exp Med 202(2):249–259
Watters RJ et al (2012) Development and use of ceramide nanoliposomes in cancer. Methods Enzymol 508:89–108
Tran MA et al (2008) Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin Cancer Res 14(11):3571–3581
Wilhelm SM et al (2008) Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther 7(10):3129–3140
Tan QX et al (2014) Sorafenib-based therapy in HER2-negative advanced breast cancer: Results from a retrospective pooled analysis of randomized controlled trials. Exp Ther Med 7(5):1420–1426
Pedersen AM et al (2014) Sorafenib and nilotinib resensitize tamoxifen resistant breast cancer cells to tamoxifen treatment via estrogen receptor alpha. Int J Oncol 45(5):2167–2175
Pedrosa LR et al (2013) Improving intracellular doxorubicin delivery through nanoliposomes equipped with selective tumor cell membrane permeabilizing short-chain sphingolipids. Pharm Res 30(7):1883–1895
Zolnik BS et al (2008) Rapid distribution of liposomal short-chain ceramide in vitro and in vivo. Drug Metab Dispos 36(8):1709–1715
Sirohi B et al (2008) Platinum-based chemotherapy in triple-negative breast cancer. Ann Oncol 19(11):1847–1852
Swanton C et al (2007) Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell 11(6):498–512
Juul N et al (2010) Assessment of an RNA interference screen-derived mitotic and ceramide pathway metagene as a predictor of response to neoadjuvant paclitaxel for primary triple-negative breast cancer: a retrospective analysis of five clinical trials. Lancet Oncol 11(4):358–365
Hla T et al (2000) Sphingosine-1-phosphate signaling via the EDG-1 family of G-protein-coupled receptors. Ann N Y Acad Sci 905:16–24
Mousseau Y et al (2012) Fingolimod inhibits PDGF-B-induced migration of vascular smooth muscle cell by down-regulating the S1PR1/S1PR3 pathway. Biochimie 94(12):2523–2531
Long JS et al (2010) Sphingosine 1-phosphate receptor 4 uses HER2 (ERBB2) to regulate extracellular signal regulated kinase-1/2 in MDA-MB-453 breast cancer cells. J Biol Chem 285(46):35957–35966
Nagaoka Y et al (2008) Effects of phosphorylation of immunomodulatory agent FTY720 (fingolimod) on antiproliferative activity against breast and colon cancer cells. Biol Pharm Bull 31(6):1177–1181
Azuma H et al (2002) Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res 62(5):1410–1419
Slamon D et al (2011) Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 365(14):1273–1283
Joensuu H et al (2006) Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N Engl J Med 354(8):809–820
Goss PE et al (2013) Adjuvant lapatinib for women with early-stage HER2-positive breast cancer: a randomised, controlled, phase 3 trial. Lancet Oncol 14(1):88–96
Goldhirsch A et al (2013) 2 years versus 1 year of adjuvant trastuzumab for HER2-positive breast cancer (HERA): an open-label, randomised controlled trial. Lancet 382(9897):1021–1028
Agrawal A et al (2005) Overview of tyrosine kinase inhibitors in clinical breast cancer. Endocr Relat Cancer 12(Suppl 1):S135–S144
Lee AJ et al (2012) CERT depletion predicts chemotherapy benefit and mediates cytotoxic and polyploid-specific cancer cell death through autophagy induction. J Pathol 226(3):482–494
Payne AW et al (2014) Ceramide kinase promotes tumor cell survival and mammary tumor recurrence. Cancer Res 74(21):6352–6363
Kim IC et al (2013) Lipid profiles for HER2-positive breast cancer. Anticancer Res 33(6):2467–2472
Sano O et al (2007) Sphingomyelin-dependence of cholesterol efflux mediated by ABCG1. J Lipid Res 48(11):2377–2384
Montefusco DJ et al (2013) Distinct signaling roles of ceramide species in yeast revealed through systematic perturbation and systems biology analyses. Sci Signal 6(299):p. rs14
Acknowledgements
The authors would like to acknowledge the SUNY Stony Brook Bioinformatics Facility. The results presented in Fig. 1 are based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. This work is partly supported by NCI grant P01 CA097132. We thank Dr. Christopher J. Clarke for careful review of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Newcomb, B., Hannun, Y.A. (2015). Sphingolipids as Mediators of Breast Cancer Progression, Metastasis, Response and Resistance to Chemotherapy. In: Hannun, Y., Luberto, C., Mao, C., Obeid, L. (eds) Bioactive Sphingolipids in Cancer Biology and Therapy. Springer, Cham. https://doi.org/10.1007/978-3-319-20750-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-20750-6_4
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-20749-0
Online ISBN: 978-3-319-20750-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)