
Abstract
Sphingolipids play key roles in the regulation of several biologic processes that are integral to cancer pathogenesis. Among the sphingolipid metabolites, ceramide and sphingosine-1-phosphate (S1P) have been shown to modulate cancer development and progression. The biologic roles of other metabolites, such as sphingosine, and ceramide 1-phosphate, are also beginning to emerge. In general, ceramide plays a role as a tumor-suppressing lipid-inducing anti-proliferative response such as cell cycle arrest, induction of apoptosis, and senescence whereas S1P plays a role as a tumor-promoting lipid-inducing transformation, cellular proliferation, and inflammation in various cell models. Glycosphingolipids, another emerging class of bioactive sphingolipids, are believed to play anti-apoptotic roles and offer drug resistance to currently used chemotherapeutic drugs. These emerging biologic roles of sphingolipids and its potential usefulness in treating cancer in the form of anticancer therapeutics are discussed in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sphingosine Kinase
- Sphingolipid Metabolism
- Ceramide Level
- A549 Lung Adenocarcinoma Cell
- Ceramide Generation
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
8.1 Sphingolipid Metabolism
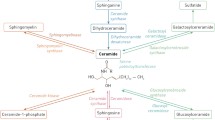
Sphingolipids are a class of lipids with a sphingosine back bone that are formed from non-sphingolipid precursors in the ER and get metabolized further within different sub-cellular compartments thereby giving rise to a plethora of metabolites. Of all these metabolites, ceramide is one of the most widely studied bioactive molecules. It is formed through three distinct pathways (Fig 8.1) (1) de novo synthesis—synthesis from non-sphingolipid precursors; (2) turnover pathways—break down products from complex sphingolipids; and (3) recycling and salvage pathways—The de novo pathway starts with the condensation of serine and palmitoyl-CoA catalyzed by serine palmitoyl transferase (SPT) to generate 3-keto-dihydrosphingosine which is subsequently reduced to form dihydrosphingosine (sphinganine). Ceramide synthases (CerS) then act on dihydrosphingosine (or sphingosine) to form dihydroceramide (or ceramide) [1]. Dihydroceramide is subsequently desaturated by dihydroceramide desaturase (DES) which introduces a 4, 5-trans-double bond, thereby generating ceramide that occurs at the cytosolic face of the endoplasmic reticulum (ER) [2]. Ceramide, thus generated, can be used in biosynthetic reactions for the synthesis of sphingomyelin (SM), glucosylceramide (GluCer), galactosylceramide (GalCer) or ceramide 1-phosphate (C1P) by the attachment of head groups comprised of either phosphocholine, glucose, galactose or phosphate by sphingomyelin synthase(SMS), glucosyl ceramide synthase (GCS), UDP-galactose: ceramide-galactosyltransferase (CGT), or ceramide kinase, respectively (CK) [3, 4, 5]. In the turnover pathways of ceramide generation, sphingomyelinases act by cleaving sphingomyelin as a substrate [6] whereas hydrolases such as β-glucosidases and galactosidases act on glycosphingolipids such as glucosylceramide (GlcCer) and galactosylceramide as substrates to generate ceramide, respectively [7]. In the recycling and salvage pathways, ceramide generated in the lysosomes from the hydrolysis of complex sphingolipids is further broken down to sphingosine by the action of ceramidases [8] which is then re-acylated outside the lysosome by the action of ceramide synthases (CerS) to form ceramide. Since sphingosine derived from ceramide is salvaged to regenerate ceramide, it is referred to as the recycling and salvage pathway. Alternatively, sphingosine can be acted upon by sphingosine kinases (SK1 or SK2) [9] to form sphingosine-1 phosphate (S1P). S1P phosphatases can dephosphorylate S1P to regenerate sphingosine [10]. On the other hand, S1P lyase metabolizes S1P irreversibly to release ethanolamine phosphate and hexadecenal [11].

Ceramide can be formed by de novo pathway from serine and palmitoyl-coA or from hydrolysis of sphingomyelin or cerebrosides (glucosyl or galactosyl ceramide). Ceramide, thus formed, can be phosphorylated by CK to yield ceramide-1-phosphate or serves as a substrate for the synthesis of sphingomyelin or glycosphingolipids. Ceramide can be deacylated by ceramidases to form sphingosine which can be phosphorylated by SKs to generate S1P which can be acted upon by phosphatases to generate sphingosine or by lyase to form ethanolamine-1-phosphate and hexadecanal, an aldehyde. Abbreviations: SPT serine palmitoyl transferase, KDS 3-keto-dihydrosphingosine reductase, DES dihydroceramide desaturase, SPPase Sph-1- phosphate phosphatase, CK cer kinase, C1PP C1P phosphatase, SMS SM synthase, GCS glucosylceramide synthase, GCase glucosyl CDase, CGT UDP-galactose ceramide-galactosyltransferase
The sphingolipid enzymes discussed above are distributed in different intracellular locations. De novo ceramide synthesis takes place on the cytosolic surface of the ER and its associated membranes. Ceramide formed in the ER is transported through ceramide transfer protein (CERT) to the trans-Golgi wherein it serves as a substrate for sphingomyelin synthase for formation of SM [12], or through vesicular transport to the Golgi wherein it serves as substrate for GCS for the formation of glucosylceramide. The transport protein, four-phosphate-adaptor protein 2 (FAPP2) delivers glucosylceramide to appropriate sites in the Golgi for synthesis of more complex glycosphingolipids (GSL) [13]. Subsequently, SM and complex GSLs are transported to the plasma membrane via vesicular trafficking. In the plasma membrane, SM can be hydrolyzed to ceramide and metabolized further by acid sphingomyelinase (aSMase) possibly acting on the outer leaflet, or by neutral sphingomelinase (nSMase) residing on the inner leaflet of the plasma membrane [14, 15].
From the plasma membrane, sphingolipids are recycled through the endosomal pathway. In lysosomes, aSMases and glucosidases metabolize complex sphingolipids (SM and glucosylceramide, respectively) into ceramide which is hydrolyzed by acid ceramidase to form sphingosine. Sphingosine may then traverse the lysosomal membrane to the cytosolic side where it has two cellular fates. Cytosolic sphingosine is either recycled into the sphingolipid pathway in the ER or phosphorylated by SK1 or SK2 [9] (refer to Fig 8.2 [16]).

Ceramide is generated in the ER by de novo pathway. It is transported to the Golgi membranes for SM synthesis in a CERT-dependent way or glucosylceramide (GlcCer) synthesis in FAPP2-dependent way. SM and complex GSLs are transported to the plasma membrane via vesicular trafficking where sphingomyelin gets metabolized by aSMase or neutral SMase to generate ceramide, or they are shuttled to lysosomes where aSMases and glucosidases metabolize SM and glucosylceramide, respectively, into ceramide. It is hydrolyzed by acid ceramidase to form sphingosine. Sphingosine may then traverse the lysosomal membrane to the cytosolic side where it might have two cellular fates. It can either be recycled into sphingolipid pathway in the ER or can be phosphorylated by SK1 or SK2. In mitochondria, ceramide is generated by the activation of n-SMase. Abbreviations: 3KdhSph 3-keto-dihydrosphingosine, dhSph dihydrosphingosine, CERT ceramide transfer protein
8.2 Biological Targets of Ceramide
Ceramide regulates many biologic processes such as cancer cell growth, differentiation, apoptosis, and senescence [17, 18]. Many signals such as cytokines, anticancer drugs, and stress-inducers upregulate ceramide through the de novo or salvage pathways [19, 20]. Ceramide triggers signaling cascades by regulating phosphatases, cathepsin D, or kinase suppressor of RAS (KSR) as described below.
Phosphatases such as protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) are activated by ceramide in vitro. Inhibition of these phosphatases inhibits the ability of ceramide to dephosphorylate (inactivate) several pro-proliferative proteins such as PKCα, Akt/PKB, c-Jun, Bcl-2, Rb, and SR [21, 22].
Many studies have documented the translocation of cathepsin D from lysosomes in response to oxidative stress followed by activation of caspase 3 and cell death [23]. Interestingly, cathepsin D was found to be a ceramide-binding and ceramide-activated protein [24]. Besides, acid sphingomyelinase-derived ceramide has been shown to favor autocatalytic proteolysis of inactive cathepsin D to enzymatically active cathepsin D isoform [25].
Similarly, KSR has been found to be ceramide responsive [26–30]. Mammalian KSR activates Raf, and activation of this pathway results in apoptosis [31, 32]. Also, ceramide has been shown to activate the zeta isoform of protein kinase C (PKCz) by phosphorylation [33]. Ceramide-activation of PKC-zeta has been shown to be necessary for inactivation of Akt-dependent mitogenesis in vascular smooth muscle cells [34] (refer to Fig. 8.3).

Ceramide regulates many protein signaling molecules such as cathepsin D and ceramide-activated protein phosphatases (CAPPs), KSR, RAF, MEKK. Proteins that are modulated by these pathways include RB,SR, AKT, PKC-α, c-JUN, Bcl-2,telomerase, c-MYC, caspases, and cyclin-dependent kinases (CDKs) which in turn brings about cellular responses such as growth arrest, apoptosis, and/or senescence. Ceramide that gets metabolised to S1P by ceramidase and SK, regulates proteins such as ERK, NF-κB, Cox-2 which in turn brings about cellular responses such as inhibition of apoptosis, malignant transformation, angiogenesis, and inflammation
In contrast to ceramide, S1P has emerged as a potential regulator of biologic processes such as proliferation, inflammation, vasculogenesis, and tumor promotion [35]. S1P is a soluble molecule, secreted outside the cell which acts in autocrine and paracrine manner to bring about receptor-mediated cellular functions like cell motility and proliferation [35]. S1P acts directly on members of the S1P1-5 receptor family, which are G protein-coupled receptors. Non-receptor-mediated functions of S1P such as activation of TNF receptor–associated factor 2 (TRAF2) in the TNF pathway leading to NF-kB activation have been reported [36]. It has also been shown that interaction of TRAF2 with SK and activation of SK is critical for prevention of TNFα-mediated apoptosis [36] (refer to Fig. 8.3). Thus, these findings underscore the significance of uncovering bioactive sphingolipid-mediated cellular pathways that would help to conceive novel therapeutic strategies for many pathological conditions, importantly cancer.
8.3 Ceramide as a Tumor Suppressor
The involvement of sphingolipids in cancer pathogenesis is brought to light from the observation that the levels of sphingolipid metabolizing enzymes are altered in many tumors. For example, in a study, SK expression has been found to be upregulated in many human tumors originating from tissues such as breast, colon, lung, stomach, uterus, etc., [37]. In another study done by Riboni et al., an inverse correlation between the levels of ceramide and tumor malignancy in glial tumors (astrocytomas) was observed [38]. In general, exogenous ceramide-induced anti-proliferative responses like apoptosis, differentiation and growth inhibition, senescence, and autophagy. Therefore, it is considered as a tumor suppressor lipid. These anti-proliferative roles of ceramide are discussed in detail in the following section.
8.3.1 Ceramide and Apoptosis
Ceramide has been implicated in many cell death paradigms. Birbes et al. [39] showed that selective targeting of bacterial sphingomyelinase (bSMase) to mitochondria and not to any other compartments such as plasma membrane, ER, or Golgi resulted in apoptosis that was associated with generation of ceramide and release of cytochrome c in MCF-7 cells. Overexpression of Bcl-2 prevented the mitochondria-targeted bSMase effects on apoptosis. Dai et al. [40] showed that UV-induced apoptosis was marked by increase in SM in all sub-cellular locations, particularly mitochondria, in HeLa cells. Ceramide levels were found to be elevated in mitochondria at 2–6 h, consistent with the cell death time course. D609, an inhibitor of sphingomyelin synthase to a marked extent and fumonisin B1 (FB1), a ceramide synthase inhibitor to a lesser extent, rescued the cells from increases in SM and ceramide, and consequently cell death. On the other hand, the SPT inhibitor myriocin did not rescue the UV effects on cell death, suggesting the involvement of the turnover pathway-generated ceramide in bringing about UV-triggered cell death.
In another study in Caenorhabditis elegans, upon inactivation of ceramide synthase, somatic apoptosis was unaffected, but ionizing radiation-induced apoptosis of germ cells was obliterated, and this phenotype was reversed by microinjection of long-chain natural ceramide. Radiation-induced ceramide accumulation in mitochondria consequently activated CED-3 caspase and apoptosis [41].
In Ramos B cells, surface B-cell receptor (BcR)-triggered cell death was marked by an early increase in C16 ceramide. Pulse labeling with sphinoglipid precursor, palmitate, in the presence of ceramide synthase inhibitor, FB1, demonstrated that the de novo ceramide-generating pathway was activated following BcR activation. The apoptotic cell death induced by cross-linking of BcR was mediated through mitochondrial cell death pathways followed by caspase activation [42]. In LNCaP prostate cancer cells, androgen ablation, which is considered as one of the therapeutic modalities, was found to increase C16 ceramide level followed by G0/G1 cell cycle arrest and apoptosis. 5alpha-dihydrotestosterone (DHT) or fumonisin B1 treatment rescued LNCaP cells from apoptosis [43].
In another study, ceramide acting via PP1, dephosphorylated SR proteins that regulated the alternate splicing of Bcl-x(L) and caspase 9. In A549 lung adenocarcinoma cell lines, cell-permeable D-e-C(6) ceramide downregulated the mRNA levels of anti-apoptotic Bcl-x(L) and caspase 9b with concomitant increase in the mRNA of pro-apoptotic Bcl-x(s) and caspase 9. The chemotherapeutic agent, gemcitabine, induced de novo generation of ceramide and brought about aforementioned alternate splicing of Bcl-x(L) and caspase 9b and consequent loss of cell viability as measured by MTT assay [44].
In several studies, aSMase was shown to be necessary for radiation-induced apoptosis in endothelial cells and mice lacking aSMase were protected from gastrointestinal and CNS apoptosis [45–47]. In another study, the endolysosomal aspartate protease cathepsin D (CTSD) was identified as a target of ceramide generated by acid sphingomyelinase in response to TNFα. CTSD cleaved pro-apoptotic Bid and activated it in vitro. The lack of Bid activation in cathepsin-deficient fibroblasts suggested Bid is downstream of cathepsin D in bringing about apoptosis as a result of TNFα treatment [48].
In addition to aSMase, neutral sphingomyelinase has been implicated in stress response pathways initiated by TNFα in MCF-7 cells [49]; amyloid-β peptide in neuronal cells [50]; ethanol in HepG2 hepatoma cells [51]; and staurosporine in several neuronal cell lines [52].
In addition to the sphingomyelinases, ceramidases (CDases) were also found to regulate apoptosis. In one study, nitric oxide induced the degradation of nCDase, thereby, enabling ceramide accumulation and cell death [53]. In another study, the degeneration of photoreceptor cells was marked by an increase in ceramide which was rescued by overexpression of CDase that cleared the ceramide and prevented its apoptotic effect [54]. These studies clearly implicate ceramide in apoptosis and in mediating the cellular response to various stress causing stimuli.
8.3.2 Ceramide in Senescence
Ceramide has been implicated in cellular senescence. Their relationship stems from the observation that in WI-38 human diploid fibroblasts (HDF), there was increased neutral sphingomyelinase activity with generation of ceramide when cells entered senescence. These changes were not seen when cells entered quiescence achieved with serum withdrawal or contact inhibition [55]. Exogenous administration of ceramide (15uM) onto young WI-38 cells induced retinoblastoma protein dephosphorylation and inhibited serum-induced AP-1 activation, DNA synthesis, and mitosis, thereby inducing a senescence phenotype [55]. Involvement of ceramide in replicative senescence has been shown in human umbilical vein endothelial cells (HUVEC) as well [56]. In another study, gemcitabine induced senescence in pancreatic cancer cells, and sphingomyelin treatment enhanced chemosensitivity to the drug by reducing the induction of senescence and redirected the cells to enter apoptosis. The authors concluded that ceramide inhibited cell cycle progression at low levels, induced senescence at moderate levels, and apoptosis at high levels [57]. Besides these studies, the yeast aging genes lac l and lag1 were subsequently identified as ceramide synthases, thereby providing a genetic link between ceramide to senescence and aging [58].
Ceramide also regulates senescence by inhibiting telomerase which is the enzyme that prevents the shortening of the telomeres, the long tandem repeats of G-rich sequences (5′-TTAGGG-3′) found at the ends of chromosomes. Telomerase is found to be frequently activated in many immortal cells in culture representing different tissues and malignant tumors, suggesting its role in cellular immortalization and tumorigenesis [59, 60]. In the A549 lung carcinoma cell line, daunorubicin treatment or sphingomyelinase overexpression increased ceramide generation followed by inhibition of telomerase activity. Clearance of ceramide by overexpression of GCS prevented the telomerase inhibition [61]. These studies suggest ceramide is an upstream regulator of senescence and aging.
8.3.3 Ceramide in Cell Differentiation and Growth Inhibition
Historically, the role of ceramide in cellular differentiation was discovered with the observation that vitamin D3-induced monocytic, but not neutrophilic-type cell differentiation in HL-60, and U037 leukemia cells was accompanied by increase in nSMase activity and a concomitant spike in ceramide levels [62]. In turn, exogenous ceramide was found to induce monocytic differentiation of these cells. In neuronal cell lines, ceramide-induced differentiation in T9 glioma cells, purkinje and hippocampal neurons [63].
In another study, incubation of exponentially growing Saccharomyces cerevisiae with short-chain ceramide inhibited cell growth with the involvement of an okadaic acid-sensitive protein phosphatase [64]. Another study uncovered the mechanism of ceramide-induced growth suppression in that serum withdrawal in MOLT-4 cells resulted in significant dephosphorylation of Rb, correlating with the induction of G0/G1 cell cycle arrest [65]. Taken together, these studies implicate a role for ceramide in cell differentiation and cell cycle progression.
8.3.4 Ceramide and Autophagy
In mammalian cells, ceramide and/or dihydroceramide have been shown to induce autophagy. For example, in glioma cells, ceramide has been shown to activate the transcription of death-inducing mitochondrial protein, BNIP3, and subsequent autophagy [66]. In the human colon cancer HT-29 cells, C2 ceramide inhibited activation of protein kinase B, which is a negative regulator of interleukin 13-dependent macroautophic inhibition. In MCF-7 breast cancer cells, ceramide stimulated the expression of Beclin-1 which is an autophagy gene product. This study also showed that tamoxifen-induced autophagy was blocked using the SPT inhibitor myriocin (ISP1) [67]. In another study, ceramide was shown to induce autophagy by regulating calpain in MEFs [68]. In DU145 cells, fenretinide (4HPR) treatment favored autophagic induction possibly due to the increase in endogenous dihydroceramide [69].
In a study by Signorelli et al. [70], resveratrol induced autophagy in HGC-27 cells with an increase in dihydroceramides possibly by inhibition of dihydroceramide desaturase. Inhibitors of dihydroceramide desaturase mimicked the autophagic induction induced by resveratrol.
Mechanistically, Beclin-1 has been shown to be physiologically associated with the mammalian class III phosphatidylinositol 3-kinase (PI 3-kinase) Vps34, and the knockdown of Beclin-1 blunted the autophagic response of the cells to nutrient deprivation or C2-ceramide treatment [71]. Class I PI3K and AKT pathway are known to suppress autophagy and ceramide has been shown to inhibit AKT by activation of PP2A, thereby establishing a possible mechanistic link between ceramide and autophagy induction [67, 72, 73].
Based on the above studies showing ceramide effects on mammalian autophagic regulation, combined with the observation that yeast subjected to heat stress exhibits growth suppression accompanied by upregulation of ceramide synthesis and downregulation of nutrient transporters on their cell surface [74, 75], Edinger and colleagues hypothesized that ceramide-induced mammalian autophagy might be mediated through a yeast-like response to heat stress by downregulation of nutrient transporters. They showed that C2 ceramide produced a profound downregulation of nutrient transporter proteins in mammalian cells. Inhibition of autophagy or acute limitation of extracellular nutrients increased the sensitivity of cells to ceramide. Supplementation of cells with the cell-permeable nutrient methyl pyruvate protected the cells from ceramide-induced cell death and delayed autophagic induction. So the authors concluded that ceramide killed cells (apoptosis) by provoking nutrient limitation via downregulation of nutrient transporters and subsequent autophagy [76]. Taken together, these studies implicate ceramide in the regulation of autophagic response.
8.4 S1P/S1PR as Tumor Promoters
S1P is considered a pro-survival lipid. For example, S1P stimulated the invasiveness of glioblastoma tumor cells [77], promoted estrogen-dependent tumorigenesis of breast cancer cells [78] and conferred resistance to the cytotoxic actions of TNF-α and daunorubicin [10]. A number of studies documented the role of S1P/S1PR in proliferation, inhibition of apoptosis, vasculogenesis/angiogenesis, and inflammation. These topics will be discussed in the following sections.
8.4.1 S1P in Proliferation and Inhibition of Apoptosis
Sphingosine kinase (SK) phosphorylates sphingosine to form S1P. Overexpression of the SK1 isoform induced oncogeneic transformation in NOD/SCID mice. Using inhibitors of SK, investigators implicated SK in the involvement of oncogenic H-Ras-mediated transformation [79]. In another study, addition of exogenous S1P reversed the cell death induced by ceramide [80]. Mechanistically, S1P counteracted ceramide-induced activation of stress-activated protein kinase (SAPK/JNK) and activated the extracellular signal-regulated kinase (ERK) pathway in governing the fate of the cell [80].
Neutralizing antibody to S1P substantially reduced tumor progression in murine xenograft and allograft models. The antibody arrested tumor-associated angiogenesis, neutralized S1P-induced proliferation, attenuated release of pro-angiogenic cytokines, and blocked the ability of S1P to protect tumor cells from apoptosis [81].
In yet another study, S1P-mediated inhibition of apoptosis in C3H10T 1/2 fibroblasts depended on ERK activation and MKP-1, which downregulated SAPK/JNK to bring about inhibition of apoptosis [82]. In male germ cells, S1P inhibited stress-induced cell death, possibly by inhibiting nuclear factor kappa B (NF-kappa B) and AKT phosphorylation [83].
8.4.2 S1P and Vasculogenesis/Angiogenesis
S1P promotes vasculogenesis and angiogenesis. S1P, the natural ligand for S1P3 receptor or KRX-725, a synthetic peptide that mimics S1P action on this receptor, favored angiogenesis, as demonstrated by assessment of vascular sprouting using aortic rings as an ex vivo model of angiogenesis. When S1P or KRX-725 were combined with other growth factors such as basic fibroblast growth factor (b-FGF), stem cell factor, or vascular endothelial growth factor (VEGF), the investigators observed synergistic induction of angiogenesis [84]. In a cultured mouse allantois explant model of blood vessel formation, Argraves et al. [85] showed that S1P, synthesized via the action of SK2, promoted vasculogenesis by promoting migratory activities of angioblasts and early endothelial cells to expand the vascular network.
VEGF has been shown to stimulate SK1 activity with an increase in the production of S1P and activation of H and N Ras oncogenes in T24 bladder tumor cell lines [86]. Endothelial cells undergo morphogenesis into capillary networks in response to S1P involving G protein receptors [87]. S1P has been shown to induce endothelial cell invasion and morphogenesis in physiologically relevant collagen and fibrin matrices [88]. Based on studies employing inhibitors and functional antagonists of S1P receptors, it has been hypothesized that the angiogenic function of S1P is mediated by S1P1 and S1P3 signaling [87, 89, 90]. Knockout of the S1P1 receptor resulted in vascular deficiencies in mice [91]. In a recent study, SphK1–SphK2 double-knockout mice manifested defective neural and vascular systems and exhibited embryonic lethality. The authors inferred that S1P was required for “functionally intertwined pathways of angiogenesis and neurogenesis” [92].
8.4.3 S1P in Inflammation
The SK1/S1P pathway has been implicated in inflammation. For instance, TNF-alpha resulted in activation of SK1/S1P pathway specifically leading to extracellular signal-regulated kinases and NF-kappa B activation [36] and consequently expression of vascular cell adhesion molecule (VCAM) and intercellular adhesion molecule (ICAM) [93]. S1P also induced cyclooxygenase 2 (COX2) and prostaglandin E2 (PGE2) production in L929 fibrosarcoma and A549 lung adenocarcinoma cells and genetic knockdown using siRNA blocked their production. Additionally, preventing S1P clearance using siRNAs against S1P lyase/phosphatase resulted in increased production of COX2 and PGE2, implicating a key role of S1P in this pathway [94]. Microglial activation has been implicated in neuroinflammation. LPS treatment increased SK1 mRNA and protein levels and consequently upregulated expression of proinflammatory cytokines such as TNF-alpha, IL-1beta, and iNOS in microglia. Chronic production of inflammatory cytokines by microglia has been implicated in neuroinflammation [95, 96]. Further, the SK/S1P pathway has been implicated in many other inflammatory disease paradigms such as asthma, rheumatoid arthritis, and inflammatory bowel diseases [97].
In summary, these data collectively demonstrate that S1P regulates cancer cell viability, angiogenesis, and inflammation which favor cancer pathogenesis.
8.5 Role of Other Metabolites of Sphingolipid Metabolism in Tumorigenesis
In addition to ceramide and S1P, ceramide -1 phosphate has been implicated in cell survival pathways. Chemotherapeutic agents changed the alternative splicing of caspase 9 and Bcl-x pre-mRNA into pro-apoptotic forms which was mediated by ceramide-dependent activation of PP1 [98]. C1P has been found to inhibit PP1, and therefore, it might antagonize ceramide-mediated apoptosis, functioning as a pro-survival lipid. In another study, C1P blocked apoptosis in part by activating PI3-K/PKB/NF-κB pathways and production of anti-apoptotic Bcl-XL [99]. CERK might play an important role in regulating the balance between ceramide and C1P and therefore cell death and cell survival similar to S1P.
Sphingosine which is the breakdown product of ceramide is also implicated in apoptotic responses. In one study, gamma irradiation along with TNF-α induced sphingosine and S1P levels. The elevation of sphingosine by exogenous administration of sphingosine or by treatment with SK inhibitor induced apoptosis in LNCaP prostate cancer cell lines [100]. In another study, phorbol myristate acetate (PMA) and tumor necrosis factor (TNF) separately induced apoptosis marked by elevation of sphingosine in HL-60 cells and neutrophils, respectively. Exogenous administration of sphingosine or its methylated derivative N, N,-dimethylsphingosine (DMS) also induced apoptosis in cells of both hematopoietic and carcinoma origin [101]. In an attempt to find the mechanism of sphingosine-induced apoptosis, Domae and colleagues found that sphingosine induced c-Jun expression and apoptosis in HL-60 cells and inhibition of protein kinase A (PKA) potentiated this effect [102]. In another study by Houghton and co-workers, rhabdomyosarcoma cell lines were found to be more sensitive to the induction of apoptosis with an increase in the cellular levels of sphingosine. Mechanistically, sphingosine-mediated cell death involved mitochondrial events such as Bax activation and translocation to the mitochondria, release of cytochrome c and Smac/Diablo, but not apoptosis-inducing factor (AIF), endonuclease G, and HtrA2/Omi, from mitochondria and finally activation of caspase-3 and caspase-9 [103].
Gangliosphingolipids have been implicated in the epithelial-to-mesenchymal cell transition (EMT) which is believed to play a role in cancer progression. Pharmacological inhibition of GlcCer synthase has been shown to result in downregulation of E-cadherin, a major epithelial marker, and upregulation of vimentin and N-cadherin, major mesenchymal cell markers, with marked changes in gangliosphingolipids (Gg4 or GM2) and increased motility, implying these specific glycosphingoplipids (Gg4 or GM2) might play a role in inhibition of EMT [104]. Some glycosphingolipids have been recognized as tumor antigens and thus could participate in tumor cell regulation and as immune targets. In many studies, it was shown that GM3 modulated receptor tyrosine kinase activity in cells [105–107]. In a recent study, ganglioside GM3 inhibited autophosphorylation of the EGFR kinase domain, thereby inactivating it in response to ligand binding, and removal of neuraminic acid of the GM3 headgroup or expression of the K642G mutant released this inhibitory effect [108].
8.6 Sphingolipids in Cancer Therapy
Among the bioactive sphingolipids, ceramide and S1P act as pro-apoptotic and anti-apoptotic lipids, respectively, and therefore, modulation of these lipids may be effective as a treatment strategy for cancer (refer to Fig. 8.4 [109]). Such strategies to increase the accumulation of ceramide and attenuation of S1P are discussed in detail in the following section.

Chemotherapeutic agents increases ceramide levels and induces apoptosis through the de novo pathway or through the neutral sphingomyelinase (N-SMase) pathway. Induction of SK1 in colon cancer leads to accumulation of S1P, possibly leading to tumor proliferation, angiogenesis, and inflammation. Clearance of ceramide to Glucosylceramide by GCS in breast cancer cells leads to the development of drug resistance. P-glycoprotein (P-gp) expression might potentiate the chemo-resistant phenotype. Marked in red arrows are modulators of SPL metabolism. B13 is an inhibitor of acid CDase, OGT2378 is GCS inhibitor, D609 is an inhibitor of SMS, FTY720 is a sphingosine analogue, and ceramide analogues mimic endogenous ceramides, and Pyridinium ceramide targets mitochondria and promotes mitochondria mediated apoptosis. Abbreviations: CDase ceramidase, ER endoplasmic reticulum, PP1 protein phosphatase 1, PP2A protein phosphatase 2A, GSL glycosphingolipid, GCS glucosylceramide synthase
8.6.1 Targeting Ceramide Generation
Cytotoxic chemotherapeutic agents such as daunorubicin, etoposide, camptothecin, fludarabine, and gemcitabine were shown to induce de novo ceramide generation, and inhibition of this pathway reduced the cytotoxic responses to these drugs meaning that these drugs manifest their cytotoxic effects partly through ceramide production [110–112]. Besides the de novo pathway, certain drugs, including daunorubicin, also induce ceramide generation through the activation of nSMase which hydrolyzes sphingomyelin to generate ceramide. For instance, in leukemia cells, cytosine arabinoside (Ara-C) induced activation of nSMase [113]. Mechanistically, this induction of nSMase was brought about by generation of reactive oxygen species followed by Jun N-terminal kinase phosphorylation and apoptosis [114]. In another study, actinomycin D and etoposide induced nSMase activity in a p53- and ROS-dependent manner [115].
Alternatively, studies on reagents that inhibit the enzymes that favor the ceramide clearance pathway, leading to accumulation of ceramide and, thereby, potentiating the cytotoxic effects were tested. For instance, compounds such as B13 that inhibited acid CDase or tricyclodecan-9-yl-xanthogenate (D609) that inhibited SMS, induced apoptosis in colon cancer and in U937 human monocytic leukemia cells, respectively [116, 117] (refer to Fig. 8.4 and Table 8.1).
Interestingly, SM was found to potentiate the chemotherapeutic response of gemicitabine in prostate cancer cell lines [118]. In a study involving combination of SM with chemotherapeutic agents, doxorubicin, epirubicin, or topotecan, it was found that the combination therapy increased the cytotoxic effect of the drugs by increasing their bioavailability, possibly by modulation of plasma membrane lipophilicity, facilitating entry of these agents into the various cancer cell lines studied [119].
8.6.2 Mimicking Ceramide Action (Analogues of Ceramide)
Ceramide analogues (such as the soluble short-chain C2- and C6-ceramides) have been shown to bring about cell death in many types of cancer cell lines tested [120]. C16-serinol, 4, 6-diene-ceramide, 5R-OH-3E-C8-ceramide, adamantyl-ceramide, and benzene-C4-ceramide (Table 8.1) are some of the ceramide analogues that induced cell death in cell lines such as neuroblastoma and breast cancer [121–124]. A novel, cationic, water soluble, pyridinium ceramide (Table 8.1) accumulated predominantly in cellular compartments that are negatively charged such as mitochondria and the nucleus and caused changes in mitochondrial structure and function and inhibited growth in various human head and neck cancer cell lines [125], while inducing apoptosis in squamous cell carcinoma (HNSCC) cell lines [126–128].
Experiments to uncover the most efficient means of delivery of these ceramide analogues have been tried extensively. Pegylated liposomes were very effective in bringing about ceramide-mediated cell death in breast cancer cell lines (Table 8.1). Liposomal delivery of ceramide decreased phosphorylated AKT and activation of caspase-3/7 more effectively than non-liposomal ceramide [129]. Vincristine incorporated in SM-liposomes called sphingosomes (Table 8.1) was found to be effective in animal models for treatment of acute lymphocytic leukemia (ALL) to the extent that it is currently in Phase II clinical trials [130]. These studies clearly demonstrate that targeting ceramide generation might be an effective method to bring about cancer cell death.
8.6.3 Attenuation of the S1P Pathway
Since S1P is found to be involved in angiogenesis and proliferation, it is intuitive to think that modulation of this pathway offers hope for the treatment of cancer. In fact, inhibition of SK1 resulted in increased cell death in many forms of cancer [131, 132] and increased the sensitivity to cell death stimuli such as TNFα and FAS ligand [133]. Dihydroxyaurone, an SK1 inhibitor, exhibits anti-tumor activity in mammary tumors [37]. Interestingly, in normal tissues of ovary and testis, exogenous S1P treatment seems to protect cells from chemotherapy-induced apoptosis [134, 135].
FTY720 (Table 8.1), an analogue of sphingosine, has been found to be phosphorylated in vivo, and the resulting FTY720 phosphate functioned as a ligand for sphingosine-1-phosphate receptors. This signaling mechanism enabled sequestration of lymphocytes in lymphoid tissues thereby causing immunosuppression [136–138]. This compound also induced apoptosis in various cancer cell lines such as lymphocyte and bladder cancer (T24, UMUC3 and HT1197), glioma (T98G), and prostate (DU145) cancer [37, 139–142]. Therefore, it is tempting to hypothesize that S1PR1 and S1PR3 antagonists might have similar anticancer effects. In another study, anti-S1P mAb greatly reduced tumor progression in murine xenograft and allograft models by inhibiting capillary formation and angiogenesis [81]. These data strongly support the candidacy of S1P as a potential therapeutic target for the treatment of cancer.
8.6.4 Dietary SM as a Cancer Therapeutic
Brasitus and co-workers demonstrated that there was a significant increase in SM levels and activity of SMS in rat colonic mucosa in response to 1, 2-dimethylhydrazine (DMH), a chemical colonic carcinogen [143]. Merrill and co-workers found that milk sphingomyelin dietary supplementation reduced the incidence of DMH-induced premalignant lesions of colon tumors in CF1 mice [144]. In addition, mice fed with SM developed fewer adenocarcinomas. These findings suggest that milk SM might suppress advanced malignant tumors in colon [145]. Administration of synthetic SM and ceramide analogues also suppressed colonic crypt foci formation [146, 147]. In another study, azoxymethane (AOM)/dextran sulfate sodium (DSS)-induced colon carcinogenesis was modulated by dietary SM in the early stages by activation of peroxisome proliferator-activated receptor γ (PPAR-γ), but its anti-carcinogenic effect was independent of PPAR-γ [148]. Therefore, dietary SM might modulate the proteins expressed during early stages of colon carcinogenesis and therefore may be a potential therapeutic candidate in the context of colon carcinogenesis.
8.7 Sphingolipids in Drug Resistance
One of the reasons for the failure of chemotherapy in cancer is the development of tumor cell resistance. Part of the basis for chemoresistance might be attributed to a re-wiring of sphingolipid metabolism. For instance, in many cases of leukemia, breast cancer, and melanoma, chemotherapeutic agents increase the activity of GCS which thereby attenuates ceramide levels, resulting in a drug resistance phenotype [20, 149]. Overexpression of GCS offered increased resistance to doxorubicin whereas siRNA knockdown promoted increased sensitivity to doxorubicin, paclitaxel, and etoposide in breast cancer cells [150–152]. Mechanistically, GCS upregulated P-glycoprotein (P-gp) which is an ABC transporter implicated in drug resistance. Knockdown of GCS inhibited MDR1, a gene that encodes P-gp, reversing drug resistance [153, 154].
Based on the above studies, GCS inhibition has been predicted to improve the effectiveness of chemotherapeutic drugs. Some studies suggest that this hypothesis is in fact true. For instance, OGT2378 (Table 8.1), an inhibitor of GCS, inhibited melanoma growth in a syngeneic orthotopic murine model [155]. In separate studies, combination of fenretinide, a compound that induces accumulation of dihydroceramide [156] through direct inhibition of dihydroceramide desaturase [157], with GCS inhibitors resulted in synergistic suppression of the growth of various tumors. Additionally, fenretinide combined with SK inhibitors such as PPMP or safingol caused growth inhibition [158, 159].
Sphingosine kinase and S1P have also been implicated in drug resistance phenotypes. For instance, it has been brought to light that certain drug-resistant melanoma cell lines such as Mel-2a and M221 are resistant to Fas-induced cell death due to a decrease in ceramide and an increase in S1P compared with Fas-sensitive counterparts such as A-375 and M186. Downregulation of SK1 with siRNA decreased the resistance of Mel-2a cells to apoptosis [160]. Similar inference was made in camptothecin resistant prostate cancer cell lines [161]. In a recent study, SK1 was found to be upregulated in imatinib-resistant chronic myeloid leukemia cell line concomitant with increased BCR-ABL mRNA and protein levels. The PI3K/AKT/mTOR pathway was also found to be upregulated. Knocking down SK1 expression using siRNA reversed the imatinib resistance to apoptosis and returned BCR-ABL to normal levels [162], suggesting a role for SK1 in conferring drug resistance.
8.8 Conclusions
There is compelling evidence to suggest that sphingolipid metabolism plays an integral part of cancer pathogenesis and therapeutic response. Ceramide is a bioactive lipid that activates signaling pathways to induce apoptosis of various cancer cell lines. S1P, on the other hand, is emerging as a pro-proliferative lipid that is frequently upregulated in tumors. Therapeutic regimens targeting the balance of ceramide and S1P may prove useful in the treatment of many carcinomas.
In spite of our understanding of sphingolipid metabolism, and its relevance in cancer models, we still have a long way to go in understanding the intricacies of sphingolipid metabolism, how the metabolism proceeds in different cancer sub-types, how compartmentalization of metabolism may offer unique regulatory roles, and how different species of individual lipid molecules provide unique signaling functions. With the advent of sophisticated lipidomics and bioinformatic approaches, more and more sphingolipid functions/signaling mechanisms are being uncovered, and this area of research holds and will continue to hold promise as a potential avenue of therapy.
References:
Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K (1992) Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J Biol Chem 267(16):11144–11148
Michel C, van Echten-Deckert G (1997) Conversion of dihydroceramide to ceramide occurs at the cytosolic face of the endoplasmic reticulum. FEBS Lett 416(2):153–155
Tafesse FG, Ternes P, Holthuis JC (2006) The multigenic sphingomyelin synthase family. J Biol Chem 281(40):29421–29425
Ichikawa S, Hirabayashi Y (1998) Glucosylceramide synthase and glycosphingolipid synthesis. Trends Cell Biol 8(5):198–202
Shinghal R, Scheller RH, Bajjalieh SM (1993) Ceramide 1-phosphate phosphatase activity in brain. J Neurochem 61(6):2279–2285
Marchesini N, Hannun YA (2004) Acid and neutral sphingomyelinases: roles and mechanisms of regulation. Biochem Cell Biol Biochimie et Biologie Cellulaire 82(1):27–44
Tettamanti G (2004) Ganglioside/glycosphingolipid turnover: new concepts. Glycoconj J 20(5):301–317
Mao C, Obeid LM (2008) Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta 1781(9):424–434
Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S (2006) Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta 1758(12):2016–2026
Johnson KR, Johnson KY, Becker KP, Bielawski J, Mao C, Obeid LM (2003) Role of human sphingosine-1-phosphate phosphatase 1 in the regulation of intra- and extracellular sphingosine-1-phosphate levels and cell viability. J Biol Chem 278(36):34541–34547
Bandhuvula P, Saba JD (2007) Sphingosine-1-phosphate lyase in immunity and cancer: silencing the siren. Trends Mol Med 13(5):210–217
Hanada K, Kumagai K, Tomishige N, Kawano M (2007) CERT and intracellular trafficking of ceramide. Biochim Biophys Acta 1771(6):644–653
Yamaji T, Kumagai K, Tomishige N, Hanada K (2008) Two sphingolipid transfer proteins, CERT and FAPP2: their roles in sphingolipid metabolism. IUBMB Life 60(8):511–518
Tani M, Hannun YA (2007) Analysis of membrane topology of neutral sphingomyelinase 2. FEBS Lett 581(7):1323–1328
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9(2):139–150
Bartke N, Hannun YA (2009) Bioactive sphingolipids: metabolism and function. J Lipid Res 50:S91–S96 Suppl
Hannun YA (1996) Functions of ceramide in coordinating cellular response to stress. Science 274:1855–1859
Hannun YA, Obeid LM (2002) The ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem 277:25847–25850
Andrieu-Abadie N (2001) Ceramide in apoptosis signaling: relationship with oxidative stress. Free Radic Biol Med 31:717–718
Ogretmen B, Hannun YA (2001) Updates on functions of ceramide in chemotherapy-induced cell death and in multidrug resistance. Drug Resist Updat 4:368–377
Hannun YA (1996) Functions of ceramide in coordinating cellular responses to stress. Science 274(5294):1855–1859
Chalfant CE, Ogretmen B, Galadari S, Kroesen BJ, Pettus BJ, Hannun YA (2001) FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem 276(48):44848–44855
Kagedal K, Johansson U, Ollinger K (2001) The lysosomal protease cathepsin D mediates apoptosis induced by oxidative stress. FASEB J: Official Publ Fed Am Soc Exp Biol 15(9):1592–1594
Heinrich M, Wickel M, Winoto-Morbach S, Schneider-Brachert W, Weber T, Brunner J, Saftig P, Peters C, Kronke M, Schutze S (2000) Ceramide as an activator lipid of cathepsin D. Adv Exp Med Biol 477:305–315
Heinrich M, Wickel M, Schneider-Brachert W, Sandberg C, Gahr J, Schwandner R, Weber T, Saftig P, Peters C, Brunner J et al (1999) Cathepsin D targeted by acid sphingomyelinase-derived ceramide. EMBO J 18(19):5252–5263
Basu S, Bayoumy S, Zhang Y, Lozano J, Kolesnick R (1998) BAD enables ceramide to signal apoptosis via Ras and Raf-1. J Biol Chem 273(46):30419–30426
Mathias S, Dressler KA, Kolesnick RN (1991) Characterization of a ceramide-activated protein kinase: stimulation by tumor necrosis factor alpha. Proc Nat Acad Sci USA 88(22):10009–10013
Yao B, Zhang Y, Delikat S, Mathias S, Basu S, Kolesnick R (1995) Phosphorylation of Raf by ceramide-activated protein kinase. Nature 378(6554):307–310
Liu J, Mathias S, Yang Z, Kolesnick RN (1994) Renaturation and tumor necrosis factor-alpha stimulation of a 97-kDa ceramide-activated protein kinase. J Biol Chem 269(4):3047–3052
Joseph CK, Byun HS, Bittman R, Kolesnick RN (1993) Substrate recognition by ceramide-activated protein kinase. Evidence that kinase activity is proline-directed. J Biol Chem 268(27):20002–20006
Xing HR, Kolesnick R (2001) Kinase suppressor of Ras signals through Thr269 of c-Raf-1. J Biol Chem 276(13):9733–9741
Basu S, Kolesnick R (1998) Stress signals for apoptosis: ceramide and c-Jun kinase. Oncogene 17(25):3277–3285
Fox TE, Houck KL, O’Neill SM, Nagarajan M, Stover TC, Pomianowski PT, Unal O, Yun JK, Naides SJ, Kester M (2007) Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem 282(17):12450–12457
Bourbon NA, Sandirasegarane L, Kester M (2002) Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J Biol Chem 277(5):3286–3292
Payne SG, Milstien S, Spiegel S (2002) Sphingosine-1-phosphate: dual messenger functions. FEBS Lett 531:54–57
Xia P, Wang L, Moretti PA, Albanese N, Chai F, Pitson SM, D’Andrea RJ, Gamble JR, Vadas MA (2002) Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem 277(10):7996–8003
French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD (2003) Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res 63(18):5962–5969
Rylova SN, Somova OG, Dyatlovitskaya EV (1998) Comparative investigation of sphingoid bases and fatty acids in ceramides and sphingomyelins from human ovarian malignant tumors and normal ovary. Biochemistry 63:1057–1060
Birbes H, El Bawab S, Hannun YA, Obeid LM (2001) Selective hydrolysis of a mitochondrial pool of sphingomyelin induces apoptosis. FASEB J: Official Publ Fed Am Soc Exp Biol 15(14):2669–2679
Dai Q, Liu J, Chen J, Durrant D, McIntyre TM, Lee RM (2004) Mitochondrial ceramide increases in UV-irradiated HeLa cells and is mainly derived from hydrolysis of sphingomyelin. Oncogene 23(20):3650–3658
Deng X, Yin X, Allan R, Lu DD, Maurer CW, Haimovitz-Friedman A, Fuks Z, Shaham S, Kolesnick R (2008) Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science 322(5898):110–115
Kroesen BJ, Pettus B, Luberto C, Busman M, Sietsma H, de Leij L, Hannun YA (2001) Induction of apoptosis through B-cell receptor cross-linking occurs via de novo generated C16-ceramide and involves mitochondria. J Biol Chem 276(17):13606–13614
Eto M, Bennouna J, Hunter OC, Hershberger PA, Kanto T, Johnson CS, Lotze MT, Amoscato AA (2003) C16 ceramide accumulates following androgen ablation in LNCaP prostate cancer cells. Prostate 57(1):66–79
Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA (2002) De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem 277(15):12587–12595
Santana P, Pena LA, Haimovitz-Friedman A, Martin S, Green D, McLoughlin M, Cordon-Cardo C, Schuchman EH, Fuks Z, Kolesnick R (1996) Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 86(2):189–199
Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, Haimovitz-Friedman A, Cordon-Cardo C, Kolesnick R (2001) Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 293(5528):293–297
Pena LA, Fuks Z, Kolesnick RN (2000) Radiation-induced apoptosis of endothelial cells in the murine central nervous system: protection by fibroblast growth factor and sphingomyelinase deficiency. Cancer Res 60(2):321–327
Heinrich M, Neumeyer J, Jakob M, Hallas C, Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W, Trauzold A, Hethke A et al (2004) Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 11(5):550–563
Luberto C, Hassler DF, Signorelli P, Okamoto Y, Sawai H, Boros E, Hazen-Martin DJ, Obeid LM, Hannun YA, Smith GK (2002) Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J Biol Chem 277(43):41128–41139
Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, Chen S, Hsu CY (2004) Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol 164(1):123–131
Liu JJ, Wang JY, Hertervig E, Cheng Y, Nilsson A, Duan RD (2000) Activation of neutral sphingomyelinase participates in ethanol-induced apoptosis in Hep G2 cells. Alcohol Alcohol 35(6):569–573
Testai FD, Landek MA, Dawson G (2004) Regulation of sphingomyelinases in cells of the oligodendrocyte lineage. J Neurosci Res 75(1):66–74
Franzen R (2002) Nitric oxide induces degradation of the neutral ceramidase in rat renal mesangial cells and is counterregulated by protein kinase C. J Biol Chem 277:46184–46190
Acharya U (2003) Modulating sphingolipid biosynthetic pathway rescues photoreceptor degeneration. Science 299:1740–1743
Venable ME, Lee JY, Smyth MJ, Bielawska A, Obeid LM (1995) Role of ceramide in cellular senescence. J Biol Chem 270(51):30701–30708
Venable ME, Yin X (2009) Ceramide induces endothelial cell senescence. Cell Biochem Funct 27(8):547–551
Modrak DE, Leon E, Goldenberg DM, Gold DV (2009) Ceramide regulates gemcitabine-induced senescence and apoptosis in human pancreatic cancer cell lines. Mol Cancer Res: MCR 7(6):890–896
Guillas I (2001) C26-CoA-dependent ceramide synthesis of Saccharomyces cerevisiae is operated by Lag1p and Lac1p. EMBO J 20:2655–2665
Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266(5193):2011–2015
Shay JW, Bacchetti S (1997) A survey of telomerase activity in human cancer. Eur J Cancer 33(5):787–791
Ogretmen B, Schady D, Usta J, Wood R, Kraveka JM, Luberto C, Birbes H, Hannun YA, Obeid LM (2001) Role of ceramide in mediating the inhibition of telomerase activity in A549 human lung adenocarcinoma cells. J Biol Chem 276(27):24901–24910
Okazaki T, Bell RM, Hannun YA (1989) Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J Biol Chem 264(32):19076–19080
Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA (1994) Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science 265(5178):1596–1599
Fishbein JD, Dobrowsky RT, Bielawska A, Garrett S, Hannun YA (1993) Ceramide-mediated growth inhibition and CAPP are conserved in Saccharomyces cerevisiae. J Biol Chem 268(13):9255–9261
Dbaibo GS (1995) Retinoblastoma gene product as a downstream target for a ceramide-dependent pathway of growth arrest. Proc Natl Acad Sci USA 92:1347–1351
Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S (2004) Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 64(12):4286–4293
Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P (2004) Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem 279(18):18384–18391
Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, Schneider C (2006) Calpain is required for macroautophagy in mammalian cells. J Cell Biol 175(4):595–605
Bedia C, Triola G, Casas J, Llebaria A, Fabrias G (2005) Analogs of the dihydroceramide desaturase inhibitor GT11 modified at the amide function: synthesis and biological activities. Org Biomol Chem 3(20):3707–3712
Signorelli P, Munoz-Olaya JM, Gagliostro V, Casas J, Ghidoni R, Fabrias G (2009) Dihydroceramide intracellular increase in response to resveratrol treatment mediates autophagy in gastric cancer cells. Cancer Lett 282(2):238–243
Zeng X, Overmeyer JH, Maltese WA (2006) Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci 119(Pt 2):259–270
Zhou H, Summers SA, Birnbaum MJ, Pittman RN (1998) Inhibition of Akt kinase by cell-permeable ceramide and its implications for ceramide-induced apoptosis. J Biol Chem 273(26):16568–16575
Schubert KM, Scheid MP, Duronio V (2000) Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J Biol Chem 275(18):13330–13335
Dickson RC (2008) Thematic review series: sphingolipids. New insights into sphingolipid metabolism and function in budding yeast. J Lipid Res 49(5):909–921
Cowart LA, Obeid LM (2007) Yeast sphingolipids: recent developments in understanding biosynthesis, regulation, and function. Biochim Biophys Acta 1771(3):421–431
Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL (2008) Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Nat Acad Sci USA 105(45):17402–17407
Van Brocklyn JR, Young N, Roof R (2003) Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett 199(1):53–60
Nava VE, Hobson JP, Murthy S, Milstien S, Spiegel S (2002) Sphingosine kinase type 1 promotes estrogen-dependent tumorigenesis of breast cancer MCF-7 cells. Exp Cell Res 281(1):115–127
Xia P, Gamble JR, Wang L, Pitson SM, Moretti PA, Wattenberg BW, D’Andrea RJ, Vadas MA (2000) An oncogenic role of sphingosine kinase. Curr Biol: CB 10(23):1527–1530
Cuvillier O, Pirianov G, Kleuser B, Vanek PG, Coso OA, Gutkind S, Spiegel S (1996) Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381(6585):800–803
Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS et al (2006) Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 9(3):225–238
Castillo SS, Teegarden D (2003) Sphingosine-1-phosphate inhibition of apoptosis requires mitogen-activated protein kinase phosphatase-1 in mouse fibroblast C3H10T 1/2 cells. J Nutr 133(11):3343–3349
Suomalainen L, Pentikainen V, Dunkel L (2005) Sphingosine-1-phosphate inhibits nuclear factor kappaB activation and germ cell apoptosis in the human testis independently of its receptors. Am J Pathol 166(3):773–781
Licht T, Tsirulnikov L, Reuveni H, Yarnitzky T, Ben-Sasson SA (2003) Induction of pro-angiogenic signaling by a synthetic peptide derived from the second intracellular loop of S1P3 (EDG3). Blood 102(6):2099–2107
Argraves KM, Wilkerson BA, Argraves WS, Fleming PA, Obeid LM, Drake CJ (2004) Sphingosine-1-phosphate signaling promotes critical migratory events in vasculogenesis. J Biol Chem 279(48):50580–50590
Wu W, Shu X, Hovsepyan H, Mosteller RD, Broek D (2003) VEGF receptor expression and signaling in human bladder tumors. Oncogene 22(22):3361–3370
Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha’afi RI, Hla T (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99(3):301–312
Bayless KJ, Davis GE (2003) Sphingosine-1-phosphate markedly induces matrix metalloproteinase and integrin-dependent human endothelial cell invasion and lumen formation in three-dimensional collagen and fibrin matrices. Biochem Biophys Res Commun 312(4):903–913
Yonesu K, Kawase Y, Inoue T, Takagi N, Tsuchida J, Takuwa Y, Kumakura S, Nara F (2009) Involvement of sphingosine-1-phosphate and S1P1 in angiogenesis: analyses using a new S1P1 antagonist of non-sphingosine-1-phosphate analog. Biochem Pharmacol 77(6):1011–1020
LaMontagne K, Littlewood-Evans A, Schnell C, O’Reilly T, Wyder L, Sanchez T, Probst B, Butler J, Wood A, Liau G et al (2006) Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res 66(1):221–231
Liu Y, Wada R, Yamashita T, Mi Y, Deng CX, Hobson JP, Rosenfeldt HM, Nava VE, Chae SS, Lee MJ et al (2000) Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Investig 106(8):951–961
Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL (2005) Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol 25(24):11113–11121
Xia P, Gamble JR, Rye KA, Wang L, Hii CS, Cockerill P, Khew-Goodall Y, Bert AG, Barter PJ, Vadas MA (1998) Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Nat Acad Sci USA 95(24):14196–14201
Pettus BJ, Bielawski J, Porcelli AM, Reames DL, Johnson KR, Morrow J, Chalfant CE, Obeid LM, Hannun YA (2003) The sphingosine kinase 1/sphingosine-1-phosphate pathway mediates COX-2 induction and PGE2 production in response to TNF-alpha. FASEB J: Official Publ Fed Am Soc Exp Biol 17(11):1411–1421
Nayak D, Huo Y, Kwang WX, Pushparaj PN, Kumar SD, Ling EA, Dheen ST (2010) Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 166(1):132–144
Hammad SM, Crellin HG, Wu BX, Melton J, Anelli V, Obeid LM (2008) Dual and distinct roles for sphingosine kinase 1 and sphingosine 1 phosphate in the response to inflammatory stimuli in RAW macrophages. Prostaglandins Other Lipid Mediat 85(3–4):107–114
Snider AJ (2010) Orr Gandy KA, Obeid LM: Sphingosine kinase: Role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 92(6):707–715
Massiello A, Salas A, Pinkerman RL, Roddy P, Roesser JR, Chalfant CE (2004) Identification of two RNA cis-elements that function to regulate the 5′ splice site selection of Bcl-x pre-mRNA in response to ceramide. J Biol Chem 279(16):15799–15804
Gomez-Munoz A, Kong JY, Parhar K, Wang SW, Gangoiti P, Gonzalez M, Eivemark S, Salh B, Duronio V, Steinbrecher UP (2005) Ceramide-1-phosphate promotes cell survival through activation of the phosphatidylinositol 3-kinase/protein kinase B pathway. FEBS Lett 579(17):3744–3750
Nava VE, Cuvillier O, Edsall LC, Kimura K, Milstien S, Gelmann EP, Spiegel S (2000) Sphingosine enhances apoptosis of radiation-resistant prostate cancer cells. Cancer Res 60(16):4468–4474
Sweeney EA, Sakakura C, Shirahama T, Masamune A, Ohta H, Hakomori S, Igarashi Y (1996) Sphingosine and its methylated derivative N, N-dimethylsphingosine (DMS) induce apoptosis in a variety of human cancer cell lines. Int J Cancer J Int du Cancer 66(3):358–366
Sawai H, Okazaki T, Domae N (2002) Sphingosine-induced c-jun expression: differences between sphingosine- and C2-ceramide-mediated signaling pathways. FEBS Lett 524(1–3):103–106
Phillips DC, Martin S, Doyle BT, Houghton JA (2007) Sphingosine-induced apoptosis in rhabdomyosarcoma cell lines is dependent on pre-mitochondrial Bax activation and post-mitochondrial caspases. Cancer Res 67(2):756–764
Guan F, Handa K, Hakomori SI (2009) Specific glycosphingolipids mediate epithelial-to-mesenchymal transition of human and mouse epithelial cell lines. Proc Nat Acad Sci USA 106(18):7461–7466
Bremer EG, Schlessinger J, Hakomori S (1986) Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J Biol Chem 261(5):2434–2440
Zhou Q, Hakomori S, Kitamura K, Igarashi Y (1994) GM3 directly inhibits tyrosine phosphorylation and de-N-acetyl-GM3 directly enhances serine phosphorylation of epidermal growth factor receptor, independently of receptor–receptor interaction. J Biol Chem 269(3):1959–1965
Meuillet EJ, Kroes R, Yamamoto H, Warner TG, Ferrari J, Mania-Farnell B, George D, Rebbaa A, Moskal JR, Bremer EG (1999) Sialidase gene transfection enhances epidermal growth factor receptor activity in an epidermoid carcinoma cell line, A431. Cancer Res 59(1):234–240
Coskun U, Grzybek M, Drechsel D, Simons K (2011) Regulation of human EGF receptor by lipids. Proc Nat Acad Sci USA 108(22):9044–9048
Ogretmen B, Hannun YA (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 4(8):604–616
Bose R (1995) Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell 82:405–414
Perry DK (2000) Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem 275:9078–9084
Chauvier D, Morjani H, Manfait M (2002) Ceramide involvement in homocamptothecin- and camptothecin-induced cytotoxicity and apoptosis in colon HT29 cells. Int J Oncol 20:855–863
Strum JC (1994) 1-[beta]-D-Arabinofuranosylcytosine stimulates ceramide and digylceride formation in HL-60 cells. J Biol Chem 269:15493–15497
Bezombes C (2001) Oxidative stress-induced activation of Lyn recruits sphingomyelinase and is requisite for its stimulation by Ara-C. FASEB J 15:1583–1585
Dbaibo GS (1998) p53-dependent ceramide response to genotoxic stress. J Clin Invest 102:329–339
Selzner M, Bielawska A, Morse MA, Rudiger HA, Sindram D, Hannun YA, Clavien PA (2001) Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res 61(3):1233–1240
Meng A, Luberto C, Meier P, Bai A, Yang X, Hannun YA, Zhou D (2004) Sphingomyelin synthase as a potential target for D609-induced apoptosis in U937 human monocytic leukemia cells. Exp Cell Res 292(2):385–392
Modrak DE, Cardillo TM, Newsome GA, Goldenberg DM, Gold DV (2004) Synergistic interaction between sphingomyelin and gemcitabine potentiates ceramide-mediated apoptosis in pancreatic cancer. Cancer Res 64(22):8405–8410
Veldman RJ, Zerp S, van Blitterswijk WJ, Verheij M (2004) N-hexanoyl-sphingomyelin potentiates in vitro doxorubicin cytotoxicity by enhancing its cellular influx. Br J Cancer 90(4):917–925
Radin NS (2003) Killing tumours by ceramide-induced apoptosis: a critique of available drugs. Biochem J 371(Pt 2):243–256
Bieberich E, Kawaguchi T, Yu RK (2000) N-acylated serinol is a novel ceramide mimic inducing apoptosis in neuroblastoma cells. J Biol Chem 275(1):177–181
Bieberich E (2002) Synthesis and characterization of novel ceramide analogs for induction of apoptosis in human cancer cells. Cancer Lett 181:55–64
Struckhoff AP (2004) Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. J Pharmacol Exp Ther 309:523–532
Crawford KW (2003) Novel ceramide analogs display selective cytotoxicity in drug-resistant breast tumor cell lines compared to normal breast epithelial cells. Cell Mol Biol 49:1017–1023
Dindo D, Dahm F, Szulc Z, Bielawska A, Obeid LM, Hannun YA, Graf R, Clavien PA (2006) Cationic long-chain ceramide LCL-30 induces cell death by mitochondrial targeting in SW403 cells. Mol Cancer Ther 5(6):1520–1529
Senkal CE, Ponnusamy S, Rossi MJ, Sundararaj K, Szulc Z, Bielawski J, Bielawska A, Meyer M, Cobanoglu B, Koybasi S et al (2006) Potent antitumor activity of a novel cationic pyridinium-ceramide alone or in combination with gemcitabine against human head and neck squamous cell carcinomas in vitro and in vivo. J Pharmacol Exp Ther 317(3):1188–1199
Rossi MJ, Sundararaj K, Koybasi S, Phillips MS, Szulc ZM, Bielawska A, Day TA, Obeid LM, Hannun YA, Ogretmen B (2005) Inhibition of growth and telomerase activity by novel cationic ceramide analogs with high solubility in human head and neck squamous cell carcinoma cells. Otolaryngol Head Neck Surg 132(1):55–62
Novgorodov SA, Szulc ZM, Luberto C, Jones JA, Bielawski J, Bielawska A, Hannun YA, Obeid LM (2005) Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J Biol Chem 280(16):16096–16105
Stover T, Kester M (2003) Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J Pharmacol Exp Ther 307:468–475
Thomas DA, Sarris AH, Cortes J, Faderl S, O’Brien S, Giles FJ, Garcia-Manero G, Rodriguez MA, Cabanillas F, Kantarjian H (2006) Phase II study of sphingosomal vincristine in patients with recurrent or refractory adult acute lymphocytic leukemia. Cancer 106(1):120–127
Shirahama T, Sweeney EA, Sakakura C, Singhal AK, Nishiyama K, Akiyama S, Hakomori S, Igarashi Y (1997) In vitro and in vivo induction of apoptosis by sphingosine and N, N-dimethylsphingosine in human epidermoid carcinoma KB-3-1 and its multidrug-resistant cells. Clinical cancer research : an official journal of the American Association for Cancer Research 3(2):257–264
Sweeney EA (1996) Sphingosine and its methylated derivative N, N-dimethylsphingosine (DMS) induce apoptosis in a variety of human cancer cell lines. Int J Cancer 66:358–366
Cuvillier O, Levade T (2001) Sphingosine 1-phosphate antagonizes apoptosis of human leukemia cells by inhibiting release of cytochrome c and Smac/DIABLO from mitochondria. Blood 98(9):2828–2836
Tilly JL, Kolesnick RN (2002) Sphingolipids, apoptosis, cancer treatments and the ovary: investigating a crime against female fertility. Biochim Biophys Acta 1585(2–3):135–138
Suomalainen L, Hakala JK, Pentikainen V, Otala M, Erkkila K, Pentikainen MO, Dunkel L (2003) Sphingosine-1-phosphate in inhibition of male germ cell apoptosis in the human testis. J Clin Endocrinol Metab 88(11):5572–5579
Graler MH, Goetzl EJ (2004) The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G protein-coupled receptors. FASEB J 10
Billich A (2003) Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem 278:47408–47415
Paugh SW (2003) The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2. FEBS Lett 554:189–193
Azuma H (2002) Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res 62:1410–1419
Wang JD (1999) Early induction of apoptosis in androgen-independent prostate cancer cell line by FTY720 requires caspase-3 activation. Prostate 40:50–55
Sonoda Y (2001) FTY720, a novel immunosuppressive agent, induces apoptosis in human glioma cells. Biochem Biophys Res Commun 281:282–288
Azuma H (2003) Induction of apoptosis in human bladder cancer cells in vitro and in vivo caused by FTY720 treatment. J Urol 169:2372–2377
Dudeja PK, Dahiya R, Brasitus TA (1986) The role of sphingomyelin synthetase and sphingomyelinase in 1, 2-dimethylhydrazine-induced lipid alterations of rat colonic plasma membranes. Biochim Biophys Acta 863(2):309–312
Dillehay DL, Webb SK, Schmelz EM, Merrill AH Jr (1994) Dietary sphingomyelin inhibits 1, 2-dimethylhydrazine-induced colon cancer in CF1 mice. J Nutr 124(5):615–620
Schmelz EM, Dillehay DL, Webb SK, Reiter A, Adams J, Merrill AH Jr (1996) Sphingomyelin consumption suppresses aberrant colonic crypt foci and increases the proportion of adenomas versus adenocarcinomas in CF1 mice treated with 1, 2-dimethylhydrazine: implications for dietary sphingolipids and colon carcinogenesis. Cancer Res 56(21):4936–4941
Schmelz EM, Bushnev AS, Dillehay DL, Sullards MC, Liotta DC, Merrill AH Jr (1999) Ceramide-beta-D-glucuronide: synthesis, digestion, and suppression of early markers of colon carcinogenesis. Cancer Res 59(22):5768–5772
Schmelz EM, Bushnev AS, Dillehay DL, Liotta DC, Merrill AH Jr (1997) Suppression of aberrant colonic crypt foci by synthetic sphingomyelins with saturated or unsaturated sphingoid base backbones. Nutr Cancer 28(1):81–85
Mazzei JC, Zhou H, Brayfield BP, Hontecillas R, Bassaganya-Riera J, Schmelz EM (2011) Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: importance of peroxisome proliferator-activated receptor gamma expression. J Nutr Biochem 22(12):1160–1171
Bleicher RJ, Cabot MC (2002) Glucosylceramide synthase and apoptosis. Biochim Biophys Acta 1585:172–178
Liu X, Ryland L, Yang J, Liao A, Aliaga C, Watts R, Tan SF, Kaiser J, Shanmugavelandy SS, Rogers A et al (2010) Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood 116(20):4192–4201
Liu YY (2001) Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J 15:719–730
Liu YY (2004) Oligonucleotides blocking glucosylceramide synthase expression selectively reverse drug resistance in cancer cells. J Lipid Res 45:933–940
Gouaze V, Liu YY, Prickett CS, Yu JY, Giuliano AE, Cabot MC (2005) Glucosylceramide synthase blockade down-regulates P-glycoprotein and resensitizes multidrug-resistant breast cancer cells to anticancer drugs. Cancer Res 65(9):3861–3867
Gouaze-Andersson V, Yu JY, Kreitenberg AJ, Bielawska A, Giuliano AE, Cabot MC (2007) Ceramide and glucosylceramide upregulate expression of the multidrug resistance gene MDR1 in cancer cells. Biochim Biophys Acta 1771(12):1407–1417
Weiss M (2003) Inhibition of melanoma tumor growth by a novel inhibitor of glucosylceramide synthase. Cancer Res 63:3654–3658
Wang H, Maurer BJ, Liu YY, Wang E, Allegood JC, Kelly S, Symolon H, Liu Y, Merrill AH Jr, Gouaze-Andersson V et al (2008) N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Mol Cancer Ther 7(9):2967–2976
Rahmaniyan M, Curley RW Jr, Obeid LM, Hannun YA, Kraveka JM (2011) Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. J Biol Chem 286(28):24754–24764
Maurer BJ (1999) Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)-retinamide in neuroblastoma cell lines. J Natl Cancer Inst 91:1138–1146
Maurer BJ (2000) Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J Natl Cancer Inst 92:1897–1909
Bektas M, Jolly PS, Muller C, Eberle J, Spiegel S, Geilen CC (2005) Sphingosine kinase activity counteracts ceramide-mediated cell death in human melanoma cells: role of Bcl-2 expression. Oncogene 24(1):178–187
Akao Y, Banno Y, Nakagawa Y, Hasegawa N, Kim TJ, Murate T, Igarashi Y, Nozawa Y (2006) High expression of sphingosine kinase 1 and S1P receptors in chemotherapy-resistant prostate cancer PC3 cells and their camptothecin-induced up-regulation. Biochem Biophys Res Commun 342(4):1284–1290
Marfe G, Di Stefano C, Gambacurta A, Ottone T, Martini V, Abruzzese E, Mologni L, Sinibaldi-Salimei P, de Fabritis P, Gambacorti-Passerini C et al (2011) Sphingosine kinase 1 overexpression is regulated by signaling through PI3K, AKT2, and mTOR in imatinib-resistant chronic myeloid leukemia cells. Exp Hematol 39(6):653–665 e656
Acknowledgments
We thank Benjamin Newcomb for his critical review of this chapter. We also thank the members of Yusuf Hannun and Lina Obeid laboratory for their helpful discussion. We apologize to those investigators whose important works were not included in this chapter because of the space limitations. The Yusuf Hannun laboratory is supported by research grants from the National Institutes of Health, USA.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media New York
About this chapter
Cite this chapter
Rajagopalan, V., Hannun, Y.A. (2013). Sphingolipid Metabolism and Signaling as a Target for Cancer Treatment. In: Johnson, D. (eds) Cell Death Signaling in Cancer Biology and Treatment. Cell Death in Biology and Diseases. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4614-5847-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-5847-0_8
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4614-5846-3
Online ISBN: 978-1-4614-5847-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)