
Abstract
Adult hippocampal neurogenesis is a remarkable form of brain structural plasticity by which new functional neurons are generated from adult neural stem cells/precursors. Although the precise role of this process remains elusive, adult hippocampal neurogenesis is important for learning and memory and it is affected in disease conditions associated with cognitive impairment, depression, and anxiety. Immature neurons in the adult brain exhibit an enhanced structural and synaptic plasticity during their maturation representing a unique population of neurons to mediate specific hippocampal function. Compelling preclinical evidence suggests that hippocampal neurogenesis is modulated by a broad range of physiological stimuli which are relevant in cognitive and emotional states. Moreover, multiple pharmacological interventions targeting cognition modulate adult hippocampal neurogenesis. In addition, recent genetic approaches have shown that promoting neurogenesis can positively modulate cognition associated with both physiology and disease. Thus the discovery of signaling pathways that enhance adult neurogenesis may lead to therapeutic strategies for improving memory loss due to aging or disease. This chapter endeavors to review the literature in the field, with particular focus on (1) the role of hippocampal neurogenesis in cognition in physiology and disease; (2) extrinsic and intrinsic signals that modulate hippocampal neurogenesis with a focus on pharmacological targets; and (3) efforts toward novel strategies pharmacologically targeting neurogenesis and identification of biomarkers of human neurogenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction: Adult Hippocampal Neurogenesis
Purification of prospective neural progenitor cells, which are characterized by their potential to proliferate and give rise to differentiated neural progeny in vitro, has been successfully achieved from many regions of the adult mammalian central nervous system (CNS). However, despite the widespread distribution of neural precursors throughout the adult brain, adult neurogenesis is maintained in only two discrete regions of the adult mammalian brain: the subventricular zone of the lateral ventricles (Altman 1969; Lois and Alvarez-Buylla 1994; Alvarez-Buylla and Garcia-Verdugo 2002) and the subgranular zone of the dentate gyrus of the hippocampal formation (Kaplan and Hinds 1977; Cameron et al. 1993).
From a neuroscientific perspective, hippocampal adult neurogenesis is of major interest as (1) the generation of new hippocampal neurons contributes to hippocampal function including learning and memory and mood regulation; (2) hippocampal neurogenesis is involved in the pathophysiology of depression, schizophrenia, age-related memory impairment, and multiple neuronal developmental disorders including autism spectrum disorder; and (3) the process of developing newborn neurons in a mainly restrictive environment as the adult brain provides an in vivo model to elucidate the molecular and cellular basis of neural regeneration which ultimately could be harnessed to develop novel therapies for neurodegenerative diseases.
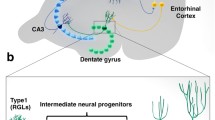
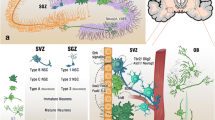
The process of generating new granule neurons from adult neural stem cells is a highly dynamic process and at the same time tightly regulated at multiple developmental stages. Developmental stages include neuronal precursors cell proliferation, differentiation, survival, migration, and integration into preexisting hippocampal networks (see Fig. 1). Hippocampal neural stem cells, commonly referred to as radial Type I cells, have the capacity to self-renew and to differentiate into neurons and astroglia (Bonaguidi et al. 2011; Encinas et al. 2011; Lugert et al. 2010). They are localized in the subgranular zone of the dentate gyrus (DG) with a characteristic radial process spanning through the molecular layer and express the radial-glia marker GFAP (Seri et al. 2001; Kriegstein and Alvarez-Buylla 2009). Active Type I cells give rise to transient amplifying neural precursor cells (NPC), referred to as Type II cells, which then develop into mature granule neurons unless negatively selected by hippocampus resident microglia (Sierra et al. 2010). Morphological maturation of newborn granule neurons includes the development of dendritic trees into the molecular cell layer of the dentate gyrus (DG) and the projection of axons toward CA3 to become functionally integrated (Hastings and Gould 1999; Toni et al. 2007). During development, newly generated cells are characterized by a temporally ordered expression of stage-specific markers and changes in morphological and functional properties. From a morphological standpoint, differentiating progenitors located in the subgranular zone show bipolar horizontal processes; after cell cycle exit they develop the primary apical dendrite toward the molecular cell layer and axons toward the CA3 region and show progressive increase in the complexity of the dendritic tree during maturation. Morphological changes are paralleled by dynamic expression of specific marker proteins. Early in the neurogenic lineage transient amplifying Type II cells show the expression of glial markers including the transcription factor SRY (sex determining region Y)-box2 (Sox2). At later stages differentiating progenitors express neuronal transcription factors like NeuroD (neurogenic differentiation) (Kronenberg et al. 2003; Seki 2002) and immature neurons express the microtubule-associated protein doublecortin (DCX) (Brown et al. 2003b).

Hippocampal neurogenesis in the adult rodent brain follows distinct developmental stages. (Top) Schematic depiction of developmental stages of hippocampal neurogenesis including proliferation, differentiation, maturation, and integration of newborn neurons. (Middle) The morphological development of neurons monitored by GFP expression upon retrovirus-mediated gene transduction in the adult mouse brain. (Proliferation: left panel, 3 days postinjection (dpi); differentiation, middle panel, 7 dpi, differentiation, mature granule neurons, right panel, 28 dpi). Experiments performed by RJ as a fellow in the laboratory of Prof. Dr. Dieter Chichung Lie. (Bottom) Depiction of known and potential CNS receptors/targets modulating the process of adult neurogenesis at the proliferation and differentiation stages in vivo
The trajectory of functional maturation includes formation of synaptic connections and the switch from depolarizing to hyperpolarizing action of the neurotransmitter GABA. In particular, while immature adult-born neurons receive synaptic excitatory GABA inputs (Ge et al. 2006, 2007; Karten et al. 2006), at later stages GABAergic inputs become gradually hyperpolarizing (Ge et al. 2006). Concomitantly, immature neurons form spines and receive glutamatergic excitatory inputs and develop mossy fiber boutons around 4–8 weeks after birth (Faulkner et al. 2008).
Adult hippocampal neurogenesis is not exclusive to rodents, but has repetitively been shown to occur in humans (Knoth et al. 2010; Eriksson et al. 1998). In adult humans around 700 new neurons per day are being integrated per dentate gyrus. This corresponds to an annual turnover rate of 1.75 % of the renewing dentate granule cell population (Spalding et al. 2013). This significant number of new hippocampal granule neurons in humans, together with dynamic regulation under physiological conditions, suggests that adult neurogenesis may be integral to brain functions.
2 Factors Affecting Hippocampal Neurogenesis and Plasticity and Correlation to Cognition
Over the past 15 years, substantial converging evidence has indicated that neurogenesis in the adult hippocampus is functionally relevant to hippocampal-dependent cognition. Many factors that modulate cognition, including pharmacological agents and intrinsic pathways involved in brain plasticity, as well as physiological stimuli such as running, learning, and enriched environment, also modulate hippocampal neurogenesis. Multiple studies impairing proliferation and adult neurogenesis in the dentate gyrus using various approaches such as chemically and irradiation-induced blockade of proliferation, genetic ablations of progenitor cells, and optogenetic physiological silencing have demonstrated that neurogenesis contributes to hippocampus-dependent learning and memory. In addition, specifically increasing the amount of newborn neurons in the dentate gyrus improves cognitive processes associated with the hippocampus. We will highlight the role of adult neurogenesis in cognition and discuss intrinsic and extrinsic factors regulating the process of neurogenesis.
2.1 Cognitive Behaviors Dependent on Hippocampal Function
Early evidence of the role of the hippocampus in memory and learning comes from studies in humans showing that bilateral resection or damage of hippocampus and hippocampal gyrus leads to memory impairment (Scoville and Milner 1957). Detailed analysis in animal models using functional imaging approaches, lesioning of specific areas of the brain, and circuit tracing reveals that the hippocampus is required for several forms of memory including declarative memory (the ability for conscious remembering) (Squire 1992), episodic memory (remembering of autobiographical events) (Wixted et al. 2014), contextual association memory (Rudy and Sutherland 1995; Lee et al. 2014), and spatial navigation. The hippocampus combines information about spatial and non-spatial items coming from inputs from the parahippocampal cortex and medial entorhinal cortex and from the perirhinal and lateral entorhinal areas, respectively. In the hippocampus, the dentate gyrus region receives inputs from the entorhinal cortex and DG granule cells project excitatory mossy fibers to the proximal apical dendrites of pyramidal cells in the CA3 area. The dentate gyrus is characterized by structural and functional heterogeneity along the dorso-ventral axis, with the dorsal region being mainly implicated in the regulation of cognition and memory and the ventral region involved in the modulation of mood and stress (Kheirbek et al. 2013; Tannenholz et al. 2014). One major process supported by the dorsal DG is conjunctive encoding which is the processing of multiple unique sensory spatial and non-spatial inputs from the perirhinal cortex and lateral and medial entorhinal cortex to form metric spatial representations (Kesner 2013). Indeed, lesions in the dorsal DG cause impairments in cue-context associations like the ability to associate environmental cues to specific odors (Morris et al. 2013).
Different circuits and regions in the hippocampus are instrumental in the processes of pattern separation and pattern completion, important mechanisms in declarative memory (Yassa and Stark 2011). Pattern separation is the ability to discriminate between similar overlapping representations by differentially encoding small or weak changes from similar inputs such as between two friend’s faces (Treves et al. 2008). On the other hand, pattern completion allows accurate reconstruction of incomplete representations based on previously stored representations. The dentate gyrus appears to be critical for the process of pattern separation. This process is facilitated by the distributed pattern of firing activity of the DG cells and the sparse mossy fiber connections onto CA3 pyramidal cells, lowering the probability of two CA3 neurons to receive inputs from the same population of DG neurons (Rolls 1996). On the other hand, local axonal inputs of neurons in the CA3 onto dendrites of cells in the same regions (also called recurrent collaterals) appear to mediate the process of pattern completion. The CA3 area of the hippocampus also receives direct inputs from the entorhinal cortex (perforant path) which are critical for memory retrieval, while inactivation of mossy fiber inputs onto CA3 neurons affects encoding and new learning without altering memory recall (Lassalle et al. 2000; Lee and Kesner 2004; Rolls 2007). In humans, fMRI studies performed during incidental encoding tasks show a correlation between level of activity in the CA3/DG and pattern separation tasks, while activity in CA1, the subiculum, the entorhinal, and parahippocampal cortices correlates with pattern completion (Bakker et al. 2008).
2.2 The Role of Hippocampal Neurogenesis in Cognition
How does adult neurogenesis contribute to memory and hippocampal function and why is this so unique to the dentate gyrus? Adult hippocampal neurogenesis has been implicated in several aspects of contextual and spatial memory. For example, in aged outbred rats there is direct correlation between performance in the water maze test, a hippocampus-dependent spatial learning task, and the amount of neurogenesis in the dentate gyrus. Notably, high performers have a significantly higher number of surviving neurons, based on BrdU (bromodeoxyuridine, a synthetic nucleoside analogue of thymidine incorporated in newly synthesized DNA of replicating cells)-positive cell counting, a readout of proliferation, in the dentate gyrus after learning (Drapeau et al. 2003). Spatial learning tasks such as the Morris water maze modulate hippocampal neurogenesis leading to the question of whether these newborn neurons are integrated in the preexisting memory circuit and reactivated during memory recall.
The evidence that newborn neurons are actively integrated in circuits during specific spatial learning tasks comes from studies analyzing the expression of immediate early genes (like c-fos and arc), a molecular correlate of neuronal firing activity, in newborn cells generated before or after exposure to spatial learning and birthdated with specific thymidine analogues. Interestingly, newborn neurons generated during a specific development window before exposure to the learning task are preferentially activated upon reexposure to the same spatial learning paradigm if compared to mature dentate granule neurons. These studies suggest that the newborn neurons are preferentially recruited in the generation of specific memory circuits compared to mature dentate granule neurons (Kee et al. 2007; Ramirez-Amaya et al. 2006; Tashiro et al. 2007). The enhanced plasticity of newborn young granule cells could potentially facilitate the integration into new memory circuits and upon maturation the increase in threshold for induction of synaptic plasticity could render the connectivity more stable. Thus, sustained hippocampal adult neurogenesis and continuous maturation of pools of immature neurons allow the DG network to achieve both stable analysis of “old” features and adaptation to new environments, supporting precise and distinct representations of new memories throughout life.
To address the causal relationship between neurogenesis and cognition, studies have focused on the analysis of the effect of neurogenesis ablation or enhancement on behavioral performances. X-ray irradiation-mediated ablation of neurogenesis, as well as genetic ablation in the GFAP-TK genetic mouse model (in which a modified herpes simplex virus gene encoding thymidine kinase under the control of the GFAP promoter causes dividing cells to die upon administration of the drug ganciclovir), leads to impairment in context fear conditioning tasks (Saxe et al. 2006; Drew et al. 2010). In another mouse model, where neurogenesis is ablated selectively inducing the expression of Bax, a pro-apoptotic protein, in neural precursors, spatial relational memory is strongly impaired (Dupret et al. 2008). In a mouse model that allowed a transient reduction of the number of adult-born DGCs, it has been shown that reduction of immature neurons confers a deficiency in forming robust, long-term spatial memory and leads to impaired performance in extinction tasks. These results further substantiate that the maturing dentate granule neurons are critical in cognition (Deng et al. 2009). These results were largely confirmed in another study where novel object recognition was impaired by the elimination of 4- to 6-week-old immature neurons (Denny et al. 2012). Recent studies looking at the effect of post-training ablation (retrograde effects) of newborn neurons and silencing of adult-generated neurons on hippocampal memory further highlight the importance of this neuronal population in formation of memory. Ablation of newborn neurons using a diphtheria toxin-based strategy after learning leads to degradation of existing contextual fear and water maze memories, even when the ablation is induced 1 month after learning (Arruda-Carvalho et al. 2011). Along the same line, using an optogenetic approach, it has been shown that silencing newborn neurons affects the retrieval of memory after completion of training. Interestingly, silencing specifically 4-weeks-old but not younger or older neurons leads to memory impairment. This strongly suggests a functional role of newly integrated immature neurons in the hippocampal circuit (Denny et al. 2012) and supports the hypothesis that immature adult-born neurons contribute to proper cognitive processing.
As cells in the dentate gyrus possess low firing rates and are only activated in a sparse manner, it has been hypothesized that the dentate gyrus may possess a supportive function in pattern separation. Indeed, the ablation of neurogenesis through X-irradiation or Bax overexpression impairs the ability to discriminate between two contexts with overlapping features (Clelland et al. 2009). Importantly, by specifically enhancing the survival of newborn neurons through the deletion of Bax, it has been shown that increase in adult hippocampal neurogenesis does not affect the ability to distinguish between two different contexts but significantly improves the ability to discriminate between overlapping contextual representations (Sahay et al. 2011).
Newborn neurons integrate in preexisting hippocampal circuitry competing with already established synaptic connections. Thus beyond modulating formation of novel memory, adult hippocampal neurogenesis may affect memories already stored in these circuits. Indeed, in many species including humans, during infancy, when the degree of neurogenesis is highest, the retrieval of hippocampus-dependent memories is impaired at later time points (Rubin 2000). Recently, a link between neurogenesis and the ability to forget previously acquired memories has been provided. In this study, using a combination of genetic, pharmacologic, and behavioral strategies, the authors show that increase in neurogenesis after learning is responsible for forgetting and leads to the hypothesis that reconfiguration of hippocampal circuits by newborn neurons may reduce the ability to retrieve previously acquired patterns of activity (Akers et al. 2014).
In conclusion, although there is a certain variability in the effect of modulation of neurogenesis on specific behavioral tasks, a number of studies have consistently shown the causal relationship between neurogenesis and hippocampal-dependent cognitive processes. In the next section, we will review physiological, pathological, and pharmacological mechanisms which can modulate neurogenesis and behavior.
2.3 Intrinsic Factors Which Regulate Hippocampal Neurogenesis and Implications in Cognition
Neurogenesis is controlled by interaction of neural progenitor cells and newborn neurons with several components of the dentate gyrus microenvironment, including astrocytes, vasculature, mature granule neurons, and GABAergic interneurons (Song et al. 2002; Palmer et al. 2000; Ma et al. 2009; Ge et al. 2006). Moreover, neurogenesis is tightly regulated by several endogenous signaling molecules including hormones and growth factors. In parallel, the activity of the neuronal network and the release of neurotransmitters from afferent projections onto the dentate gyrus can modulate several aspects of neuronal development. The concerted action of these signaling systems ultimately determines the coordinated functional integration of new neurons in preexisting circuitry (Pathania et al. 2010). Below we will highlight key experimental evidence supporting regulatory roles for some of these factors which are relevant in both physiologically and pharmacologically induced neurogenesis.
2.3.1 Neurotransmitters
Neuronal activity strongly modulates various stages of neurogenesis. Lesions of the entorhinal cortex which is one of the major excitatory afferent on granule cells increases DG cells proliferation (Cameron et al. 1995; Nacher et al. 2001). Furthermore, electrical induction of LTP at the perforant path/granule cells synapses promotes proliferation and survival of 1 and 2 weeks old newborn neurons (Bruel-Jungerman et al. 2006; Chun et al. 2006). Glutamatergic neurotransmission and specifically NMDA receptor activity regulates proliferation and correct functional maturation/integration and survival of newborn neurons (Pathania et al. 2010). In tree shrew DG, pharmacological blockade of NMDA receptors leads to increase in the number of BrdU-positive cells (Gould et al. 1997). Along the same line, activation and blockade of NMDA receptors reduce or promote cell proliferation in adult rat DG, respectively. An important question is to what extent this effect is regulated cell-autonomously rather than indirectly via other signals elicited by neuronal activity in the dentate gyrus. Immature neurons have NMDA receptors and express NR1 and NR2B subunits (Nacher and McEwen 2006; Ambrogini et al. 2004). Deletion of the NMDA subunit NR1 in newborn cells reduces the number of properly integrating/surviving newborn neurons. This effect is due to NMDA-dependent regulation of survival during the third week after neuronal birth and it appears to involve a mechanism of competitive survival between the incoming immature neurons and the preexisting neurons. Indeed, global hippocampal reduction in NMDA signaling can rescue the loss of cells. Importantly, maturing neurons (4–6 weeks after neuronal birth) show increased plasticity and reduced threshold for induction of LTP, a process in part mediated by NR2B (Ge et al. 2007). This suggests that glutamatergic signaling plays multiple roles in modulating neurogenesis and controlling the precise integration of newborn neurons into the hippocampal network.
In the SGZ, GABA neurotransmitter, released by specific populations of interneurons, modulates several aspects of neurogenesis, including precursor cells’ proliferation, differentiation, and subsequent neuronal maturation. Like in development, there is a switch between depolarization and hyperpolarization effects of GABA while the newborn cells are maturing and this may alter properties of immature neurons including their synaptic plasticity (Ge et al. 2006). The enhanced synaptic plasticity of immature neurons is likely in part due to a lack of strong GABAergic inhibition (Ge et al. 2008; Markwardt and Overstreet-Wadiche 2008; Pallotto and Deprez 2014). Tonic response of nestin-expressing quiescent radial glia cells to GABA released from parvalbumin interneurons regulates their reactivation and entry into the cell cycle. This is mediated by activation of γ2 containing GABA A receptors since conditional deletion of the subunit induces exit from quiescence and promotes symmetric self-renewal of type I cells (Song et al. 2012). The role of tonic GABA transmission on inhibition of cell proliferation is confirmed in another study upon deletion of the α4 subunit, component of GABA A receptors mediating tonic (extrasynaptic) response (Duveau et al. 2011). Neural progenitors’ proliferation is regulated also by GABA B receptors, metabotropic G-protein-coupled receptors located both on pre- and postsynaptic terminals. Both pharmacological blockage and genetic deletion of the B1 subunit of GABA B receptors promote progenitor cells’ proliferation (Felice et al. 2012; Giachino et al. 2014). GABA-mediated depolarization, due to high concentration of intracellular Cl− in immature neurons, induces neuronal differentiation and NeuroD expression in transient amplifying neuronal progenitor (type 2) cells (Tozuka et al. 2005). Deletion of both the α4 and α2 subunits, a component of GABA A receptors mediating synaptic phasic response, causes reduction of dendritic length and complexity in newborn neurons, which is revealed at different stages of differentiation (Duveau et al. 2011). Altering the GABAergic-dependent depolarization/hyperpolarization switch process by genetically modulating the expression levels of the Cl− importer NKCC1 reveals the key role of this mechanism in the regulation of proper neuronal morphology, differentiation, and synaptic maturation (Jagasia et al. 2009; Ge et al. 2006). The effect of GABA transmission on newborn neurons development is at least in part mediated by activation of downstream signaling events via activity-dependent transcription factors such as CREB (Jagasia et al. 2009).
Loss of cholinergic neurons or blockage of acetylcholine (ACh) receptors in the central nervous system causes learning impairment in experimental and clinical situations in humans (Drachman and Leavitt 1974; Rasmusson and Dudar 1979). Newborn neurons in the dentate gyrus are innervated by forebrain cholinergic fibers (Kaneko et al. 2006) and by septal cholinergic cells as shown using a combination of rabies virus-mediated retrograde tracing and retroviral labeling of new granule cells (Vivar et al. 2012). Neurotoxic and immunotoxic lesion of forebrain cholinergic projections leads to decreased neurogenesis, increased apoptosis and impaired spatial memory (Mohapel et al. 2005; Cooper-Kuhn et al. 2004). Modulation of the cholinergic system using a number of pharmacological approaches further supports the role of the system in regulation of neurogenesis (Veena et al. 2011a). Stimulation of cholinergic receptors with the cholinergic agonist physostigmine and inhibition of acetylcholinesterase using donepezil induces neurogenesis and promotes proliferation and short-term survival (Mohapel et al. 2005; Kaneko et al. 2006; Kotani et al. 2006). On the other hand, scopolamine, a cholinergic muscarinic receptor blocker, decreases the number of BrdU-positive cells in the DG affecting the survival of newborn neurons (Kotani et al. 2006). Early in their development, adult-born neurons express homomeric α7-containing nicotinic acetylcholine receptors and cell autonomous genetic ablation leads to impairment in dendritic maturation and synaptic integration ultimately resulting in reduced survival (Campbell et al. 2010). In global knockout models of the β2 receptor subunit there is significant reduction in cell proliferation culminating in a net reduction in the size of the dentate granule cell layer (Harrist et al. 2004). In vitro, cholinergic stimulation affects proliferation and survival of rat olfactory bulb and cortical neural precursor cells (Coronas et al. 2000; Ma et al. 2000). Acetylcholine neurotransmission appears to be deregulated with age and in Alzheimer’s disease, conditions with reduction in both neurogenesis and cognitive capacity. Notably, pharmacological modulation of the cholinergic activity in aged or stressed animals promotes NSCs’ proliferation and corrects cognitive alterations (Itou et al. 2011; Veena et al. 2011b).
The dopaminergic system has been shown to affect proliferation and differentiation of neural progenitor cells during embryonic development and in both adult neurogenic zones. Lesion and pharmacological studies in the SGZ have yielded discrepancy in results (Veena et al. 2011a). Focusing on the pharmacological approaches, depletion of dopamine in rodents reduces proliferation of SGZ neuronal precursor cells and this is reversed by treatment with a D2-like receptor agonist (Hoglinger et al. 2004). Similarly, activation of D2 receptors using quinpirole promotes NSCs’ proliferation (Yang et al. 2008). However, administration of haloperidol, a D2-like receptor antagonist, has been reported to induce both positive and negative effects on neurogenesis in the SGZ (Wakade et al. 2002; Wang et al. 2004; Keilhoff et al. 2010; Halim et al. 2004). A recent study demonstrates that dopamine increases adult hippocampal NSCs’ proliferation acting on D1-like receptors since the effect is phenocopied by a D1-like receptor agonist but not a D2 agonist (Takamura et al. 2014). On the other hand, stimulating the D3 receptor appears to exert an inhibitory effect on neurogenesis since inhibition of the D3 receptor using the antagonist S33138 increases cell proliferation in the hippocampus and the results are replicated in a D3 KO mouse model (Egeland et al. 2012).
Serotonin and noradrenaline, as well as antidepressant drugs that influence their neurotransmission, play a key role in the regulation of hippocampal neurogenesis and hippocampal-dependent behaviors. This class of molecules will be described more in detail in the section on depression and antidepressant treatments. In Table 1 are listed examples of pharmacological manipulations that have been demonstrated to induce changes in neurogenesis and cognition.
2.3.2 Wnt/Beta-Catenin Pathway
The Wnt signaling pathway is a highly conserved signaling pathway that has been implicated in nervous system development and has multiple functions in the adult brain including a role in hippocampal adult neurogenesis. Disruption of the physiological Wnt signaling pathway has been associated with several CNS pathologies, including schizophrenia, mood disorders, autism, and Alzheimer’s disease. A canonical Wnt ligand inhibits glycogen synthase kinase-3β (GSK-3β), which modulates the degradation of β-catenin. In the presence of extracellular Wnt ligand, and subsequent receptor activation, stabilized β-catenin enters the nucleus and associates with TCF/LEF transcription factors, resulting in transcription of Wnt-target genes (Varela-Nallar and Inestrosa 2013).
Based on in vitro and in vivo results, it has been demonstrated that Wnt/β-catenin signaling regulates adult hippocampal NPC proliferation and differentiation (Lie et al. 2005; Kalani et al. 2008). In the hippocampus, lentivirus-mediated expression of Wnt3 or a dominant-negative form of WNT (dnWNT), respectively, increases and almost abolishes adult neurogenesis. Furthermore, expression of dnWNT impairs both long-term retention of spatial memory in the water maze task and performance in a hippocampus-dependent object recognition task (Jessberger et al. 2009). Importantly, the levels of neurogenesis correlate with the performance on specific memory tasks.
Wnt signaling is modulated in diverse physiological conditions characterized by changes in the rate of hippocampal adult neurogenesis. The relationship between aging, neurogenesis, and cognitive impairment will be described in later sections. For example, Wnt signaling shows a reduction during aging when neurogenesis is decreased and, most importantly, modulation of the pathway can counteract age-related neurogenesis and cognitive declines. Aged astrocytes show a reduced expression of multiple canonical Wnt molecules, which in part results in a reduction of adult hippocampal neurogenesis (Miranda et al. 2012). Moreover, the expression of the Wnt antagonist Dickkopf-1 (Dkk1) increases with age and inducible deletion of Dkk1 enhances neurogenesis. Aged mice with a loss of Dkk1 exhibit enhanced spatial working memory and memory consolidation (Seib et al. 2013). Conversely, activity-dependent induction of neurogenesis using electroconvulsive shock leads to reduction in the expression of secreted frizzled-related protein 3 (sFRP3), a naturally secreted Wnt antagonist, in mature dentate granule neurons (Jang et al. 2013a). Deletion of sFRP3 induces the proliferation of precursor cells and promotes newborn neurons maturation, dendritic growth, and dendritic spine formation in the adult mouse hippocampus.
Modulation of Wnt signaling appears to be of therapeutic relevance also in disease conditions. Indeed, sFRP3 deletion alone is sufficient to induce an antidepressant-like behavioral response on the same magnitude of known antidepressants, whose effect, as we will describe later, is at least in part linked to neurogenesis (Jang et al. 2013b). Moreover, in mouse models of Alzheimer’s disease characterized by impairment in neurogenesis, treatment with lithium, a pharmacological activator of Wnt/β-catenin signaling acting via GSK3-β inhibition, ameliorates memory loss (Toledo and Inestrosa 2010). In a recent study, in vivo administration of both WASP-1, an activator of Wnt/β-catenin signaling, and FOXY-5, an activator of both Wnt/JNK and Wnt/Ca2+ signaling, improves hippocampal-dependent learning and memory processes (Compton et al. 2011). Wnt signaling enhancers would be potentially highly relevant cognitive therapies targeting hippocampal neurogenesis.
2.3.3 Neurotrophic Factors: The Role of BDNF
Neurotrophic factors are extracellular signaling proteins that play critical roles in both the developing nervous system and in adult brain physiology. BDNF and its role in hippocampal neurogenesis have been studied more extensively than any of the other neurotrophins. Chronic infusion of BDNF in the hippocampus of adult rats promotes cell proliferation and neurogenesis (Scharfman et al. 2005). The induction of neurogenesis by BDNF appears to be region specific since it does not affect the process in the SVZ, the other neurogenic niche in the adult rodent brain (Galvao et al. 2008). BDNF affects also later stages of neuronal maturation. Indeed, deletion of TrkB, the receptor of BDNF, in adult neuronal progenitor cells in the hippocampus leads to impairment in dendritic and synaptic growth in newborn neurons and deficits in neurogenesis-dependent LTP (Bergami et al. 2008).
BDNF plays a key role in hippocampus-dependent functions associated with roles in cognition and mood regulation, which will be further discussed in later sections. For example, in pattern separation inhibition of BDNF by infusion of a BDNF-blocking antibody or by antisense oligonucleotide-mediated knockdown impairs the ability to encode and consolidate “pattern separated” memories. On the other hand, acute infusion of recombinant BDNF enhances the separation of representations (Bekinschtein et al. 2013). The effect of BDNF on pattern separation performance is mediated by newborn immature neurons since BDNF infusion has no behavioral effect when neurogenesis is reduced by overexpression of dnWNT (Bekinschtein et al. 2014). As we will describe in more detail later, intact BDNF signaling is critical for learning, exercise, and antidepressants’ treatment-induced increase in neurogenesis and effect on behavior (Rossi et al. 2006; Li et al. 2008). BDNF-based therapies would be highly relevant to increase neurogenesis activity and hippocampal function associated with cognition.
2.4 Physical Exercise and Learning: Effect on Hippocampal Neurogenesis, Synaptic Plasticity, and Cognition
The CNS is known to undergo cellular, molecular, and functional changes in response to external social, cognitive and physical stimuli. Voluntary physical exercise has been shown to have beneficial effects on memory and cognition in physiological and pathological conditions in rodents and in humans (Voss et al. 2013). Interestingly, the cognitive amelioration is paralleled by increase in hippocampal neurogenesis and synaptic plasticity. In this section, we will review the effects of exercise on cognitive performance focusing on hippocampal-dependent behaviors and we will describe the effects on adult neurogenesis and synaptic plasticity.
2.4.1 Exercise and Enriched Environment in Animals: From Cognition to Neurogenesis
In adult rodents, physical exercise and exposure to enriched environment (EE), a complex combination of cognitive, physical and social stimulation, improve cognitive functions. Running and EE ameliorate performance in tasks of contextual fear conditioning, novel object recognition, and passive avoidance learning and in tasks assessing hippocampus-dependent memory like spatial memory in the Morris water maze and pattern separation (Kempermann et al. 1997; Fordyce and Farrar 1991; Falls et al. 2010; O’Callaghan et al. 2007; Creer et al. 2010). Hippocampal neurogenesis is one of the most remarkable changes in cellular and synaptic brain plasticity correlating with cognitive improvements upon exercise and EE. The first study analyzing the correlation between exercise and neurogenesis demonstrated that mice housed in an enriched environment with access to a running wheel exhibited better performance in Morris water maze tasks and have a 15 % increase in granule cell neurons in the dentate gyrus (Kempermann et al. 1997). BrdU birthdating experiments in mice show that cell proliferation peaks after 3 days of running, and the effect is still sustained at 10 days (Kronenberg et al. 2006; van der Borght et al. 2006). Running affects cell cycle kinetics of various subpopulations of newborn neurons. It induces both proliferation and cell cycle exit of DCX-positive type 3 precursors, shortens cell cycle in NeuroD1-positive progenitors, and even activates proliferation of radial type 1 stem cells (Brandt et al. 2010; Farioli-Vecchioli et al. 2014; Lugert et al. 2010). Exercise and EE appear to affect specific stages of the neurogenic process. In a study dissecting the role of learning and exercise on neurogenesis, voluntary exercise increases cell proliferation and integration/survival while exposure to an enriched environment, including access to a running wheel, only affects newborn neurons integration/survival in mice (van Praag et al. 1999b). In another study focusing on the effect of different learning paradigms on neurogenesis, the number of adult-generated neurons doubles in the dentate gyrus of rats trained on hippocampus-dependent associative learning tasks like spatial navigation in a Morris water maze and conditioning of the eye blink response using a trace protocol. Learning tasks that do not require the hippocampus fail in eliciting neurogenesis changes. These results suggest that to affect neurogenesis in the hippocampus, animals need to be trained on learning tasks for which the hippocampus is essential (Gould et al. 1999). The increase in newborn neurons upon learning appears to be due to enhanced survival and or integration rather than proliferation (Gould et al. 1999; Kee et al. 2007). Interestingly, a sequential combination of running and EE in mice leads to a 30 % greater increase in neurons than either stimulus alone. This suggests that coupling a stimulus like running which induces precursor cell proliferation to a survival-promoting stimulus like EE can enhance neurogenic pool and then subsequent integration (Fabel et al. 2009). Moreover, the effect of physical exercise and learning on neurogenesis appears to be region specific since generation of new neurons is not observed in the subventricular zone or in the cortex (Brown et al. 2003a; Gould et al. 1999; Ehninger and Kempermann 2003). Studies aimed at understanding whether neurogenesis is necessary for the beneficial effect of exercise and EE on cognition have yielded contradictory results. Reduction of neurogenesis using the antimitotic agent methylazoxymethanol acetate (MAM) in rats prevents the improvement in long-term recognition memory in a novel object recognition task upon EE (Bruel-Jungerman et al. 2005). In mice, gamma irradiation-mediated reduction in neurogenesis has a behavior-specific effect: while running-induced improvements in motor performance (rotarod) and contextual fear conditioning are not affected, spatial memory amelioration is ablated in the absence of neurogenesis (Clark et al. 2008). Interestingly, in very old mice (22 months old) with physiological reduction of neurogenesis which is no longer induced by running, the improvement in spatial pattern separation by voluntary exercise seen in young mice is lost (Creer et al. 2010). However, another study in mice shows that the improvement in spatial learning and the decrease in anxiety-like behavior upon EE are not affected by irradiation-mediated reduction of neurogenesis (Meshi et al. 2006). While several methodological and species differences might contribute to the discrepancies, this work suggests that cognitive improvement might be mediated also by neurogenesis-independent mechanisms such as increase in neurotrophic factors and induction of neuronal and synaptic plasticity.
Upon exercise several growth factors relevant for neuronal function and plasticity like NGF (Neeper et al. 1996), IGF-1 (Carro et al. 2000; Trejo et al. 2001), FGF2 (Gomez-Pinilla et al. 1997), and BDNF are upregulated. The levels of BDNF are increased upon both short- and long-term exercise paradigms (Molteni et al. 2002; Berchtold et al. 2005; Ding et al. 2011) and the increase is sustained up to 2 weeks after exercise has ended in mice (Berchtold et al. 2005). In rats exposed to voluntary running the expression of BDNF in the hippocampus and neocortex positively correlates with the mean distance run per night (Neeper et al. 1996). BDNF is increased in the dentate gyrus also in response to a forced treadmill-running training and this correlates with improved object recognition learning (O’Callaghan et al. 2007). Interestingly, while the performance in this learning task is improved both upon exercise and EE, only exercise can induce an increase in BDNF expression and cell proliferation (Bechara and Kelly 2013). Importantly, genetic ablation of the BDNF receptor TrkB in hippocampal neural progenitor cells ablates neurogenesis in response to exercise (Li et al. 2008). These results indicate that exercise induces BDNF expression, which results in increased neurogenesis in the DG. Interestingly, peripheral neutralization of VEGF abolishes running-induced neurogenesis potentially affecting angiogenesis, a process required in the modulation of the neurogenic niche to sustain greater cell production (discussed below in more detail) (Fabel et al. 2003).
Exercise-induced increase in BDNF often accompanies changes in synaptic plasticity and expression of genes important for neuronal activity and synaptic function (Tong et al. 2001). Voluntary running in rats induces expression of BDNF, NR2B subunit of NMDA receptor, and glutamate receptor 5 and concomitantly alters the induction threshold for synaptic plasticity leading to enhanced short- and long-term potentiation (LTP) in the dentate gyrus (Farmer et al. 2004). In another study, expression analysis in the whole hippocampus after 3 and 7 days of exercise shows an upregulation of NR2A (Molteni et al. 2002), a subunit shown to be necessary for exercise-induced neurogenesis in a genetic mouse model (Kitamura et al. 2003). Similarly, recordings in hippocampal slices from mice exposed to running show an enhancement of LTP specifically in the dentate gyrus (van Praag et al. 1999a). Synaptic transmission properties in the DG and the CA1 area of the hippocampus are modified also in response to learning and EE. For example, electrophysiological recording in freely moving rats shows increase in fEPSPs and in granule cell excitability in the dentate gyrus upon EE exposure (Irvine et al. 2006).
Exercise and learning also affect the morphological maturation of newborn neurons and the structure of already existing neurons in the hippocampus. In newborn DG neurons, running accelerates the formation of mushroom spines and alters spines motility early during differentiation without affecting the total spine density (Zhao et al. 2006). Spatial learning in the Morris water maze increases dendritic arbor complexity in both immature and mature newborn neurons, between 3 weeks and 4 months after birth (Lemaire et al. 2012). Interestingly, spatial and non-spatial environmental cues affect spine morphogenesis in a layer-specific fashion in the DG. Spatial cues induce mushroom spine formation in the middle molecular layer of newborn neurons that receive inputs from the entorhinal cortex (EC) providing spatial information. Conversely, non-spatial components increase mushroom spine formation in the outer molecular layer receiving inputs from the lateral EC (Zhao et al. 2014). Voluntary exercise affects dendritic complexity and spine density not only in the DG but also in afferent populations like pyramidal neurons in the CA1 and layer III pyramidal neurons of the entorhinal cortex (Redila and Christie 2006; Stranahan et al. 2007).
Another brain structural change that correlates with and indirectly supports the increase in neurogenesis is angiogenesis. The brain vasculature is a key component of the neurogenic niche providing extrinsic signals for progenitor cells that are closely associated with blood vessels. Moreover, angiogenic factors can stimulate neurogenesis. Exercise enhances blood flow and blood vessels growth throughout the brain and in the dentate gyrus (Black et al. 1990; van Praag et al. 2005). The growth is at least in part supported by increased expression of angiogenic factors like IGF-1 and VEGF and correlates with increased neurogenesis (Fabel et al. 2003). Intriguingly, experiments using parabiotic animals have identified blood-derived factors that directly regulate neurogenesis in a positive or negative manner, pointing toward systemic factors influencing neurogenesis in adults (Villeda et al. 2011, 2014; Katsimpardi et al. 2014). Interestingly, MRI studies show that increased cerebral blood volume (CBV) in the dentate gyrus can be used as an in vivo correlate of neurogenesis and it is specifically affected by exercise in mice. These findings are confirmed in human where dentate gyrus CBV correlates with cardiorespiratory fitness and cognitive function (Pereira et al. 2007). These data suggest that CBV measurements could represent a correlative biomarker for neurogenesis in humans.
2.4.2 Human Neurogenesis, Cognition and Exercise
Brain imaging studies support the role of the DG/CA3 subfields of the hippocampus in pattern separation in humans. In a study combining functional MRI and ultrahigh-resolution structural MRI, it has been shown that there is a correspondence between CA3 anatomy and functioning and pattern separation, pattern completion and individual differences in episodic memory recall (Chadwick et al. 2014). In non-demented older adults, changes in the activity measured by fMRI in the CA3/DG region correlate with the performance in pattern separation (Yassa et al. 2011). Moreover, DG/CA3 is also involved in pattern separation of emotional information and in patients affected by depression the severity of depressive symptoms negatively correlates with DG/CA3 activity (Leal et al. 2014). As in animals, hippocampal structural changes appear to correlate with training on tasks dependent on the hippocampus. Indeed, the posterior hippocampus stores spatial representations of the environment and people with high dependence on navigational skills like London taxi drivers show increased posterior hippocampal volume (Maguire et al. 2000). These data support the relevance of hippocampal areas and their dynamic regulation in specific cognitive tasks in humans. Although animal studies demonstrate a key role for neurogenesis in pattern separation, the lack of biomarkers for neurogenesis in humans limits conclusive studies. On the other hand, studies in cancer patients show that systemic treatment with chemotherapy agents often results in cognitive impairment and decline in aspects of memory which require hippocampal function. In animal models it has been shown that neural stem cells proliferation in the DG is reduced by chemotherapy, suggesting this as one of the potential mechanisms underlying some of the cognitive deficits related to the treatment (Wigmore 2013).
Several studies in human suggest that aerobic exercise has a positive effect on cognitive performance in healthy individuals and can counteract cognitive impairment during aging or in pathological conditions (Voss et al. 2013). In the healthy population, aerobic exercise can improve executive functions like task switching, selective attention, working memory updating, and inhibitory control in children and young adults (Guiney and Machado 2013). Cardiovascular fitness positively associates with intelligence assessed using tests for logical, verbal, and technical skills (Aberg et al. 2009; Moore et al. 2014), with increased cognitive flexibility and improved action monitoring process (Themanson et al. 2008; Hillman et al. 2008), and with improvement in academic achievements (Chaddock-Heyman et al. 2013; Chaddock et al. 2012).
Exercise has also been shown to specifically improve performance in cognitive tasks known to critically depend on hippocampal function. Young individuals, exposed to a long-term aerobic exercise regime and experiencing a change in fitness, show better performance in visual pattern separation task, visuospatial memory, and positive affect (Dery et al. 2013; Stroth et al. 2009; Herting and Nagel 2012), relational memory (e.g., children show higher ability to remember pairs of faces and houses studied under relational encoding conditions) (Monti et al. 2012; Chaddock et al. 2010, 2011). Interestingly, magnetic resonance imaging shows that performance in relational and visuospatial memory tasks positively correlates with larger hippocampal volumes, suggesting that structural modifications play a role in improved function (Chaddock et al. 2010; Herting and Nagel 2012).
Physical activity has beneficial effects on cognition also in older adults and in conditions associated with cognitive impairment. Older humans exposed to physical exercise improve executive control processes like planning, scheduling, and working memory (Kramer et al. 1999). Similarly to that observed in younger adults, aerobic fitness correlates with increased hippocampal volume and better spatial memory in older individuals (Erickson et al. 2009). Moreover, a longitudinal study showed that in adults over the age of 65 years physical activity correlates with diminished incidence of Alzheimer’s disease. In conditions of memory problems or cognitive impairment, exercise still improves cognitive function and positive behavior (Heyn et al. 2004).
Overall these studies support a positive effect of exercise on cognition and on hippocampus-dependent processes like spatial and relational memory and pattern separation. Importantly, cognitive improvements correlate with structural changes in the hippocampus both in animals and in humans. However, a direct link to neurogenesis in humans is lacking, as currently there is no possibility to measure neurogenesis in living humans.
2.5 Depression, Stress, and Antidepressants: Effect on Neurogenesis and Behavior
2.5.1 Cognitive Impairment in Depression
Depression is a widespread disorder and it presents with symptoms which include, among others, low mood, feelings of despair, reduced attention capacity, and suicidal ideation. Patients affected by major depressive disorder (MDD) show high prevalence of cognitive dysfunction, including hippocampal-dependent cognitive processes. Among the most common deficits are memory disturbances, difficulty in making decisions, and reduced cognitive flexibility (Fava et al. 2006; Wagner et al. 2012; Jaeger et al. 2006; McCall and Dunn 2003). Morphometric analysis in patients diagnosed with first episode MDD shows structural alterations in hippocampus, amygdala, and corticolimbic regions (Frodl et al. 2002; Zhu et al. 2011). MDD patients show impaired performance in tests assessing hippocampal-dependent declarative memory and functional imaging analysis shows abnormal activation of hippocampus during verbal memory encoding task (Bremner et al. 2004). As we will describe in detail later, alteration of the hypothalamic–pituitary–adrenal (HPA) axis and in glucocorticoids (GCs) levels are hallmarks of depression pathophysiology. Although not confirmed in all studies, cortisol levels are found to correlate with cognitive deficits in MDD patients and acute administration of GCs in healthy individuals can compromise long-term memory retrieval (Schlosser et al. 2010; Wolf et al. 2009).
Taken together, there is increasing evidence supporting a role for hippocampal dysfunction in cognitive deficits in MDD. Recent anatomical and functional evidence indicates a dissociation of the dorsal and ventral regions of the hippocampus with the dorsal region being responsible for memory and cognition and the ventral part playing a key role in regulation of mood. How alterations in cognitive functions contribute to MDD pathogenesis and to the recovery process is still unclear. In this sections, we will review the effects of stress and antidepressant treatment on neurogenesis and will discuss the role of neurogenesis in the onset and recovery of behavioral phenotypes.
2.5.2 Stress and Neurogenesis
Stressors are considered as experiences and events which challenge the ability of the individual to adapt. Reponses which promote adaptation to stressors include the release of hormones and other cellular factors. However, when the response is deregulated it can induce the activation of pathophysiological processes leading to the onset of neuropsychiatric disorders (McEwen 1998).
Stress has been demonstrated to have deleterious effects on multiple stages of hippocampal neurogenesis, newborn neuron maturation, hippocampal neuron plasticity, and dendritic and synaptic density. Exposure of tree shrews to acute conflict to establish a dominant/subordinate relationship leads to reduction of neurogenesis in the dentate gyrus in the subordinate animal (Gould et al. 1997). Negative regulation of neurogenesis is observed also upon chronic stress paradigms in several animal models. For example, cell proliferation in the dentate gyrus shows a significant reduction in mice subjected to repeated intermittent social defeats (Yap et al. 2006) and in young adult marmosets after forced prolonged social isolation (Cinini et al. 2014). Besides affecting cell proliferation, chronic stress exposure impairs newborn neurons’ survival (Tanti et al. 2013; Dagyte et al. 2011) and induces structural abnormalities like dendritic atrophy in CA3 pyramidal neurons (Watanabe et al. 1992; Sousa et al. 2000). At the behavioral level, animal models subjected to chronic stress develop depression-related behaviors. While the effect of stress on neurogenesis has been widely confirmed in several animal models, the role of neurogenesis in the onset of depressive-like behaviors is still a matter of debate. Ablation of neurogenesis in mice via X-ray irradiation of the hippocampus or via pharmacological intervention does not induce behavioral phenotypes relevant to anxiety or depression (Santarelli et al. 2003; David et al. 2009; Bessa et al. 2009). Genetic ablation of neurogenesis via overexpression of the pro-apoptotic protein Bax leads to increase in anxiety-related behaviors while it does not affect depressive behaviors (Revest et al. 2009). Similarly, cyclin D2 knockout mice which lack neurogenesis do not show impaired performance in the forced swimming test (Jedynak et al. 2014). Conversely, in a transgenic model where increase in survival of newborn neurons is achieved by selective deletion of Bax, there are no differences in anxiety and depressive behaviors (Sahay et al. 2011). In contrast to these findings, ablation of radial cell precursors in a GFAP-TK mouse model (expressing under the control of GFAP promoter herpes simplex virus thymidine kinase which renders mitotic cells sensitive to the antiviral drug valganciclovir) leads to depressive behavior in control conditions and increased anxiety in response to stress (Snyder et al. 2011).
2.5.3 Mechanisms Underlying the Effect of Stress on Neurogenesis and Establishment of Depressive Behavior
Several mechanisms have been proposed to mediate the effect of stress on adult neurogenesis and establishment of depressive behavior including alteration in the hypothalamic–pituitary–adrenal (HPA) axis (for a comprehensive review, see Anacker 2014; Bambico and Belzung 2013). Here we will review the evidence supporting a causal link between HPA axis dysfunction and alteration in neurogenesis, synaptic plasticity, and neurotrophic factor signaling in depression.
The hypothalamic–pituitary–adrenal (HPA) axis, part of the neuroendocrine system, plays a crucial role in the response to stress and it is deregulated in depression and chronic stress conditions (Anacker 2014). Upon stress, the hypothalamus releases corticotropin-releasing factor (CRF) which stimulates the pituitary to release adrenocorticotropin (ACTH) which ultimately leads to the synthesis and release of glucocorticoids (GCs) from the adrenal cortex. GCs then bind to their intracellular receptors, namely mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs). Upon chronic stress, the HPA axis is hyperactive and leads to chronically high levels of glucocorticoids (Anacker and Pariante 2012). Recent anatomical studies have shown that projections from the ventral hippocampus mediate an inhibitory effect on the hypothalamus, whereas the dorsal hippocampus projects to cortical areas where cognitive processes are mediated. Studies in preclinical models support a strong causative link between stress, glucocorticoids, and neurogenesis. Exposure of adult rats to predator odor induces rise in adrenal hormones levels and decrease in neurogenesis which is prevented blocking the increase of corticosterone by adrenalectomy (Tanapat et al. 2001). Interestingly, chronic corticosterone treatment induces depressive behavior in rodents (Gourley and Taylor 2009) and affects cell proliferation reducing the number of BrdU-positive cells in the dentate gyrus in adult mice (David et al. 2009). Besides reducing neurogenesis, it has been proposed that stress and corticosterone treatment promote a cell fate switch of nestin-positive neural stem cells in the DG toward increased oligodendrocytes’ differentiation (Chetty et al. 2014). Interestingly, corticosteroid levels and expression of the receptors are also altered during aging, a physiological situation with reduced neurogenesis (Gupta and Morley 2014).
Patients affected by major depressive disorder (MDD) show increased cortisol levels, impaired feedback response to GR activators, and correlation between genetic variation in components of the HPA pathway and clinical manifestations (Young et al. 1991; Belvederi Murri et al. 2014; Schatzberg et al. 2014). Volumetric analysis reveals alterations of several brain regions involved in the control of the HPA axis in MDD patients, like significantly increased volume of adrenal gland and reduced volume of hippocampus (MacMaster et al. 2014; Sheline et al. 1996). At the cellular level, expression of synaptic proteins regulating synapse function and structure as well as glutamate receptor subunits is altered in hippocampi and prefrontal cortex from MDD patients’ brains (Duric et al. 2013; Fatemi et al. 2001; Kang et al. 2012). Interestingly, this correlates with alterations in the expression of neurotrophic factors important for the function and plasticity of synapses. Expression levels of BDNF, NGF, and their relative receptors are decreased in hippocampi and PFC from suicide victims (Banerjee et al. 2013; Pandey et al. 2008) and MDD patients (Dunham et al. 2009). Moreover, serum levels of micro-RNAs associated with the regulation of expression of BDNF are altered in depressed patients (Li et al. 2013). Although the mechanisms which link stress and alterations of neurotrophic factors’ levels are not completely understood, several studies show that antidepressant treatment corrects neurotrophins levels and that normal BDNF signaling is necessary for antidepressant action (Castren et al. 2007; Adachi et al. 2008). This suggests that these factors play an important role in restoring the physiological function of networks involved in mood disorders (Duman and Duman 2015).
2.5.4 Antidepressants, Neurogenesis, and Synaptic Plasticity
For long time, one of the dominant hypothesis to explain the mechanisms underlying MDD has been the monoamine hypothesis, mainly supported by the observation that antidepressants (ADs) exerting a pharmacological action on the central monoamine systems (monoamine oxidase A inhibitors/MAOI, tricyclic compounds/TCA, serotonin and norepinephrine reuptake inhibitors) were effective in relieving depressive symptoms (Delgado 2000). According to this hypothesis, reduced activity of monoamine neurotransmission is at the core of the pathophysiology of depression and correction of this dysfunction alleviates disease symptoms (Asberg 1976). Through different mechanisms, several ADs classes in clinical use enhance serotonergic (5-HT) and noradrenergic neurotransmission.
Many preclinical and some human postmortem studies demonstrate that antidepressant treatment induces an increase in neurogenesis. Malberg and colleagues showed that chronic but not acute fluoxetine treatment increases the number of dividing cells in the dentate gyrus in rats as measured by BrdU incorporation, while the survival of newborn neurons does not appear to be altered by the treatment (Malberg et al. 2000). In humans, analysis of postmortem brains from MDD patients shows that SSRI and TCA chronic treatment increases the number of neural progenitors in the anterior and mid-dentate gyrus (Boldrini et al. 2009, 2012). However, another study could not replicate these findings (Lucassen et al. 2010). The precise role of specific serotonergic receptors in SSRI-induced neurogenesis is still under investigation. Genetic ablation of 5-HT1A receptor in mouse prevents fluoxetine induction of neurogenesis while a partial reduction is observed with 5-HT4 receptor-specific antagonist (Santarelli et al. 2003; Mendez-David et al. 2014). Moreover, pharmacological studies show that 5-HT1A receptors are involved in regulation of proliferation of neuronal precursors, while 5-HT2 receptors influence proliferation and promote neuronal maturation (Klempin et al. 2010). Induction of neurogenesis has been reported also upon treatment with antidepressants which do not act directly through the serotonin system suggesting some potential convergence in mechanisms of action. For example, the endocannabinoid receptor ligand cannabidiol (CBD) exerts anxiolytic and antidepressant effects and induces hippocampal progenitor proliferation and neurogenesis in mice (Campos et al. 2013). Increase in BrdU-positive cells is observed in the dentate gyrus of mice treated with an antidepressant antagonist of group II metabotropic glutamate receptor (Yoshimizu and Chaki 2004). Moreover, non-pharmacological interventions which elicit an antidepressant response like electroconvulsive shock strongly promote hippocampal neurogenesis (Malberg et al. 2000). Altogether these data support a correlation between ADs and neurogenesis, but is this convergent cellular response necessary for these drugs to have an effect on behavioral symptoms? Ablation of neurogenesis through X-irradiation of hippocampus prevented certain behavioral effects of fluoxetine (SSRI) and imipramine (TCA) in mice (Santarelli et al. 2003). The causal role of neurogenesis in certain specific behavioral effects of antidepressants has been confirmed in several studies (Surget et al. 2008, 2011). Fluoxetine treatment corrects the behavioral and neurogenesis defects in mice exposed to chronic corticosterone treatment and ablation of neurogenesis impairs amelioration in novelty suppressed feeding test, while open field and forced swim test performances are neurogenesis independent (David et al. 2009). Recently, the antidepressant action of cannabidiol has been shown to require neurogenesis since its effects are not observed in GFAP-TK mouse models (Campos et al. 2013). On the other hand, the behavioral effects of a vasopressin V1b antagonist and a CRF1 antagonist (corticotropin-releasing factor 1) which reverse stress-induced suppression of neurogenesis in models of depression appear to be neurogenesis independent (Alonso et al. 2004; Surget et al. 2008).
Chronic antidepressant treatment leads to an increase in the expression levels of neurotrophic factors in the hippocampus and cortical regions, including VEGF, FGF2, and BDNF and its receptor trkB which in turn can positively regulate the hippocampal cellular response (Warner-Schmidt and Duman 2007; Mallei et al. 2002). Taking advantage of genetic mouse models designed to have reduced BDNF signaling (heterozygous BDNF knockout or inducible ablation of trkB receptor in NPC), it has been demonstrated that a BDNF response is necessary for modulation of newborn hippocampal neurons upon chronic imipramine treatment (Li et al. 2008; Sairanen et al. 2005). In addition, behavioral response to antidepressant treatment is impaired in mice with altered BDNF signaling and with conditional ablation of BDNF in forebrain regions (Saarelainen et al. 2003; Monteggia et al. 2004). These data suggest that increasing BDNF activity is a key mechanism mediating antidepressant action.
Interestingly, the BDNF signaling pathway is upregulated in the hippocampus and PFC shortly after treatment with fast-acting antidepressants (Zhou et al. 2013b; Yang et al. 2013; Autry et al. 2011) and it is associated with functional synaptic changes (Tizabi et al. 2012). Ketamine, an antagonist of N-methyl-d-aspartate (NMDA) receptors, exerts antidepressant action within few hours after administration with effects lasting up to 2 weeks in depressed patients (Zarate et al. 2006; Price et al. 2009). Administration of NMDA antagonists leads to rapid translation of BDNF in mouse hippocampus and the antidepressant effect is ablated in BDNF conditional knockout mice (Autry et al. 2011). Interestingly, miR-206 regulates the expression of BDNF in response to ketamine (Yang et al. 2014). In light of the role of BDNF in modulation of synaptic plasticity and the increase in BDNF in the hippocampus upon ketamine treatment, it is tempting to speculate that changes in hippocampal synaptic plasticity may contribute, together with other brain regions, to rapid-onset antidepressant effect. However, this hypothesis still needs to be tested.
2.5.5 Antidepressants and Cognitive Impairment
As mentioned above, MDD is associated with alterations in hippocampal-dependent cognitive processes. Since antidepressant treatments have been shown to improve BDNF signaling and neurogenesis, key processes in hippocampus-dependent cognitive functions, antidepressants could potentially ameliorate cognitive symptoms in MDD patients. However, clinical studies have not yielded conclusive results and discrepancies are often attributed to limited study design. Longitudinal studies report persistent cognitive dysfunction in patients after remission from depressive symptomatology (Trivedi and Greer 2014; Hasselbalch et al. 2011; Kuny and Stassen 1995; Weiland-Fiedler et al. 2004; Neu et al. 2005). In one study, cognitive functions have been monitored in patients treated with escitalopram (SSRI) or duloxetine (SNRI); an improvement in working memory, processing speed, and visual episodic memory was best observed with duloxetine (Herrera-Guzman et al. 2009, 2010). To further address the effects of antidepressants on cognitive improvement more studies are needed and understanding this relationship would help design new and more efficacious therapeutic interventions (Goeldner et al. 2013). Along the same line, more research is needed to unravel the role of hippocampal network and neurogenesis in the onset and recovery of cognitive alterations in MDD.
2.6 Cognition and Adult Hippocampal Neurogenic Axis in Physiological and Pathological Aging
Cognitive functions decline with age and are impaired in pathological conditions associated with advanced age like Alzheimer’s disease. Impairment of hippocampus-dependent cognitive processes often correlates with structural and functional changes in this brain region and with alterations in neurogenesis. This sections highlights the relevance of neurogenesis in physiological aging and associated cognitive decline in human and animals. Furthermore, the potential pathophysiological role of neurogenesis in cognitive deficits in Alzheimer’s disease is highlighted.
2.6.1 Aging and Cognitive Decline in Human: Correlation with Hippocampal Changes
In human, aging affects a broad range of cognitive functions like executive functions (task switching, updating, inhibition) associated with the prefrontal cortex, episodic memory (memory for events which include specific temporal and spatial context) associated with prefrontal cortex and medial temporal lobes, information processing speed, specific aspects of language, and visuospatial functions (for a review, see Alexander et al. 2012). Several studies report specific alterations in hippocampus-dependent processes. Older individuals show impairment in spatial navigation tasks designed to specifically assess hippocampus-dependent ability to develop a cognitive map (a mental representation of the landmarks and paths in the environment) and to use it to reach any target location by any route available (Iaria et al. 2009). These cognitive deficits often correlate with functional and structural alterations in the hippocampus. For example, older individuals tested in a virtual spatial navigation task show differential activation of several brain areas, as measured by voxel-based analysis, if compared to younger controls. Among those differences, old participants show a reduced activation of hippocampus and parahippocampal gyrus (Moffat et al. 2006). Similarly, impairment in hippocampus-dependent spatial and non-spatial functions and recognition memory has been shown to correlate with decreased hippocampal volume and neurochemical properties in older individuals (Driscoll et al. 2003). Along the same line, a specific correlation between hippocampal volume and wayfinding skills (generation and use of a cognitive map) has been described in older individuals (Head and Isom 2010). Moreover, older adults show performance decline compared to younger control individuals in pattern separation task and a bias toward pattern completion. High-resolution (1.5 mm isotropic) blood-oxygenation level-dependent fMRI analysis reveals an altered activation in the CA3/dentate gyrus regions during trials (Yassa et al. 2011). However, a meta-analysis of the correlation between hippocampal size and episodic memory in older adults reveals extremely weak correlation (Van Petten 2004). Moreover, while some studies show a progressive change in the volume of several brain regions including shrinkage of the hippocampus (Raz et al. 2010), others show an age-dependent decline in temporal cortex but not in hippocampal volume (Sullivan et al. 2005).
Moving from the neuroanatomical to the cellular level, early analysis with stereological techniques showed no neuronal loss in several areas of the hippocampus during aging (West et al. 1994). On the other hand, neurogenesis appears to decline with age. The expression of neurogenesis markers and the number of DCX-positive cells are decreased in the dentate gyrus in post-mortem human brains from older donors (Knoth et al. 2010). Moreover, birthdating experiments based on measurements of genomic DNA incorporation of 14C, liberated in the atmosphere during atom bomb testing, show that adult hippocampal neurogenesis in the human brain undergoes a modest decline during aging (Spalding et al. 2013). Whether neurogenesis decline contributes to cognitive deficits in elderly humans remains an open question, although studies in rodents suggest that it might have a significant impact.
2.6.2 Age-Related Cognitive and Neurogenesis Decline in Animals
Decline of cognitive functions has been described in aged rodents and non-human primates. Deficits include impaired performance in spatial learning and memory (Barnes 1979; Gage et al. 1984; Rapp et al. 1997; Lazarov and Marr 2013). Structural MRI analysis shows that hippocampal volume does not change with age (Shamy et al. 2006). However, histologic evaluation shows an imbalance in the volume of different areas of the hippocampus in aged rats with a relative reduction of the volume of the middle portion of the molecular layer (Rapp et al. 1999). Interestingly, early studies demonstrated that in rodents neurogenesis in the subgranular zone declines with age (Seki and Arai 1995; Kuhn et al. 1996; Ben Abdallah et al. 2010) with the major decline taking place during adulthood, before aging (Rao et al. 2005; Demars et al. 2013). Whether the decline in neurogenesis correlates with cognitive impairment is a matter of debate. Studies show that aged rats performing better in Morris water maze and hippocampus-dependent tasks have higher number of proliferating cells and newborn neurons (Driscoll et al. 2006; Drapeau et al. 2003). On the other hand, others show no correlation or negative correlation between the number of proliferating cells and performance in Morris Water Maze in aged rats (Merrill et al. 2003; Bizon et al. 2004). Interestingly, in middle-aged (12 months old) rats, despite the massive reduction of progenitor cell proliferation and newborn cells survival, no deficits in trace fear conditioning are observed (Cuppini et al. 2006).
Several hypotheses have been put forward to explain the mechanisms accounting for neurogenesis decrease in aging and they include both cell-autonomous and non-cell-autonomous processes. Aging is accompanied by changes in the hippocampal stem cell niche vasculature (Hattiangady and Shetty 2008) and reduced expression levels of growth factors important for neurogenesis like VEGF, FGF2, BDNF, and WNT signaling activity (Shetty et al. 2005; Hattiangady et al. 2005; Bernal and Peterson 2011). Glucocorticoid signaling is altered in aged animals and decreasing corticosterone levels restores the rate of proliferation of neuronal progenitors (Cameron and McKay 1999; Nichols et al. 2001). A heterochronic parabiosis experiment, in which the circulatory system of two animals is connected, shows that blood-borne factors from old mice can induce impairment in synaptic plasticity and cognitive deficits in young animals. Elevated levels of chemokines, including CCL11, are observed in aged animals and increasing peripheral levels of CCL11 in young mice decreases neurogenesis and induces cognitive decline (Villeda et al. 2011). One of the most robust changes observed during aging is a dramatic reduction in progenitor cells’ proliferation (Olariu et al. 2007; Walter et al. 2011; McDonald and Wojtowicz 2005; Bondolfi et al. 2004), with the most prominent effect in the ventral hippocampus (Jinno 2011). A meta-analysis comparing the reduction in progenitors’ proliferation between different species of rodents, fox, and non-human primates shows that the decline is chronologically equal between species and independent of life span (Amrein et al. 2011). Reduction in neurogenesis in the aged brain has been associated with depletion of the neural stem cells pool as a consequence of their division (Encinas et al. 2011) or to a switch toward a quiescent state (Hattiangady and Shetty 2008; Lugert et al. 2010). Importantly, pro-neurogenic stimuli such as increasing neuronal activity by inducing seizures lead NSCs to reenter the cell cycle and restore proliferation to a level comparable to the one observed in young animals (Lugert et al. 2010). Similarly, exercise and exposure to enriched environment rescue neurogenesis deficits and improve cognitive functions like spatial memory and place recognition memory (van Praag et al. 2005; Kronenberg et al. 2006; Siette et al. 2013). Recently, the chronic administration in aged rats of a blood–brain barrier-permeable peptide derived from the ciliary neurotrophic factor (CNTF), which is known to have neuroprotective properties, restores neurogenesis, synaptic plasticity, and memory (Bolognin et al. 2014). These works suggest that induction of neurogenesis has beneficial effects on cognition during aging. Moreover, targeting the reactivation of neural stem cells in the aged brain could be a promising therapeutic approach to restore cognitive functions. Interestingly, young circulating factors like GDF11, a member of the BMP/TGFβ family, can restore neurogenesis and proper vasculature also in the SVZ, the other neurogenic niche in the adult brain, and improve olfactory discrimination in aged mice (Katsimpardi et al. 2014).
2.6.3 Pathological Aging: Alzheimer’s Disease, Cognitive Impairment, and Neurogenesis
Alzheimer’s disease (AD) is an age-related neurodegenerative disease characterized by progressive memory loss and cognitive decline (Caselli et al. 2006). AD represents the most common cause of dementia in the elderly. Pathological hallmarks of the disease are extracellular deposition of amyloid-beta (Aβ) plaques, excessive phosphorylation of the cytoskeletal protein tau, formation of intraneuronal fibrillary tangles, and neuronal cell loss in the cerebral cortex and hippocampus (Selkoe 2001; Braak and Braak 1991, 1995). The major environmental risk factor for the onset of the disease is aging, while the greatest genetic factor is apolipoprotein E (ApoE) genotype (Ashford 2004). ApoE exists in three isoforms, with isoform E3 being the wt form and the most common in the population, isoform E2 considered to be protective toward AD, and E4 increasing the risk for the disease and anticipating the age of onset. Hereditary autosomal dominant forms of the disease are caused by mutations in genes encoding for amyloid precursor protein (APP) and presenilin-1 and -2, components of the aspartyl protease γ-secretase complex. Cleavage of APP can follow two pathways: in the non-amyloidogenic pathway cleavage by the protease complex α-secretase releases the soluble fragment of APP (sAPPα) and γ-secretase cuts in the intramembrane domain; in the amyloidogenic pathway cleavage by the aspartyl protease β-site APP cleaving enzyme I (BACE1) and by γ-secretase leads to the formation of Aβ (De Strooper and Woodgett 2003; Selkoe and Wolfe 2007).
The hippocampus and the afferent entorhinal cortex show early neuronal loss that correlates with memory decline (Van Hoesen et al. 1991; Gomez-Isla et al. 1996; West et al. 1994, 2004). Patients affected by amnesic mild cognitive impairment (aMCI) and mild AD show reduced performance in hippocampus-dependent tasks like pattern separation (Ally et al. 2013). Importantly, impairment in performance correlates with CSF levels of Aβ42 and patients carrying the ApoE4 genotype perform worse than others in difficult pattern separation tasks (Wesnes et al. 2014). As discussed previously, performance in pattern separation appears to require hippocampal neurogenesis in the dentate gyrus and growing evidence supports alterations in the neurogenic process in AD. Several proteins associated with AD and mutated in familial forms of the disease are directly involved in the regulation of neural stem cell proliferation and differentiation. Notch1, a master regulator of neural stem cell physiology, is a substrate of PS1/γ-secretase and it is cleaved in response to ligand binding (Alexson et al. 2006; LaVoie and Selkoe 2003; De Strooper et al. 1999). Deletion of PSEN1, the gene encoding for PS1 in mice, leads to aberrant neurogenesis during development (Shen et al. 1997). In adult animals, lentiviral-mediated knockdown of PS1 in the dentate gyrus leads to enhanced differentiation of neural stem cells (Gadadhar et al. 2011). Moreover, Notch1 and EGF receptor ligands are substrates of ADAM10, a component of the α-secretase complex (Hartmann et al. 2002; LaVoie and Selkoe 2003). Neural stem cells’ proliferation is regulated also by cleavage products of APP, and the intraventricular injection of sAPPα rescues age-dependent decline in neural progenitors’ proliferation (Caille et al. 2004; Demars et al. 2013; Lazarov and Demars 2012).
Studies in several mouse models of AD suggest a strong correlation between alterations in neurogenesis, including neural stem cells’ proliferation, newborn neurons’ survival and maturation, and disease progression. For example, in the transgenic mouse model Tg2576 which overexpresses the human APP isoform with the Swedish double mutation and develops cognitive defects and amyloidosis, aberrant maturation of newborn neurons is observed already in young age (3 months) and this might lead to altered functional integration of new neurons in the hippocampal circuits (Krezymon et al. 2013). Recently, spatial learning and memory impairments have been described in a knock-in mouse model bearing the human pathological domain of ApoE4 gene in young age. Neurogenesis appears to be increased in young animals and decreased with age, while apoptotic markers indicate enhanced cell death. Interestingly, these early cognitive and cellular changes take place in the absence of classical AD pathological hallmarks (Adeosun et al. 2014). In another study, deletion of ApoE in mice results in reduced neurogenesis and knock-in of the human ApoE4 leads to impaired maturation of newborn neurons through a mechanism dependent on GABAergic signaling (Li et al. 2009). Comprehensive reviews of neurogenesis phenotypes in AD models can be found elsewhere (Lazarov and Marr 2010; Verret et al. 2007; Mu and Gage 2011).
Analysis of postmortem brain samples from senile AD patients reveals increased expression of newborn neurons markers including DCX in the subgranular zone, granular layer, and CA1 area. These findings suggest that activation of neurogenesis might represent a mechanism of compensation and replacement of degenerated neurons (Jin et al. 2004). In another study, AD brain samples show altered expression of early progenitor markers in the hippocampus, suggestive of enhanced proliferation, while the number of newborn neurons does not appear to be affected (Perry et al. 2012). Plasma and CSF concentrations of stem cell factor, a growth factor exerting neuroprotective effects and supporting neurogenesis, are decreased in AD patients and the level inversely correlates with the degree of dementia (Laske et al. 2008). On the other hand, in pre-senile AD patients no evidence of altered neurogenesis has been detected, while a proliferative change has been observed in glia and vasculature cells (Boekhoorn et al. 2006).
As described above, the hippocampus and the neurogenic process are strongly altered in AD and this might account for some aspects of cognitive decline in patients. However, growing evidence suggests that molecular and structural prerequisites for activity-dependent hippocampal plasticity are preserved in models of AD. Exposure to enriched environment leads to amyloid plaque load reduction and increased expression of genes involved in protective processes and neurogenesis in a transgenic AD mouse model (Lazarov et al. 2005). Similarly, exposure to EE of mice bearing the Swedish mutation in APP reverses spatial learning and memory deficits and this correlates with restoration of some aspects of the neurogenic process. As opposed to the previous study, amyloid load is not affected by EE in this model (Valero et al. 2011). A study dissecting the role of physical activity and cognitive stimuli on neurogenesis in AD shows that while exposure to EE improves neurogenesis and water maze performance in the APP23 mouse model, exercise alone does not show beneficial effect (Wolf et al. 2006). Besides memory and learning impairment, AD is associated with behavioral symptoms that include anxiety and depression. As described previously, the ventral hippocampus is involved in regulation of the response to stress and depressive behavior and neurogenesis might play a role in this process. In a 3xTgAD mouse model bearing mutations in APP, PS1, and Tau genes, restoration of neurogenesis through chronic overexpression of Wnt3a in the ventral region of the dentate gyrus leads to correction of impairments in danger assessment. The effect is neurogenesis dependent since ablation of neurogenesis via X-irradiation prevents the recovery (Shruster and Offen 2014).
Altogether these works suggest that neurogenesis is pathologically impaired in AD and restoration of the neurogenic process might be a promising therapeutic approach to reverse behavioral and cognitive symptoms in AD patients. Below is highlighted the potential of targeting neurogenesis in disease and drug-discovery approaches for the identification of neurogenic compounds are discussed.
3 Novel Concepts for Targeting Neurogenesis in Cognition and Disease
As described above, compelling preclinical evidence suggests that hippocampal neurogenesis is modulated by a broad range of physiological and pharmacological stimuli, and most excitingly, de novo neurogenesis positively correlates with improved cognitive and emotional states. Three lines of research are emerging that suggest that targeting hippocampal neurogenesis will have translational value in human cognition and warrant future work: (1) recent rodent models demonstrate a cell-autonomous role of neurogenesis in cognition in both physiology and disease; (2) drug screening efforts have led to the identification of novel CNS active compounds with both pro-neurogenic and pro-cognitive effects; and (3) the study of human neurogenesis will potentially lead to the identification of biomarkers to monitor the process in mental illnesses for both diagnostic and therapeutic interventions. Below we highlight discoveries, opportunities, and challenges in these research domains. Research on the role of hippocampal neurogenesis in human physiology and disease will ultimately increase the translational value of this remarkable process of plasticity in the adult human brain.
3.1 Neurogenesis as a Target to Improve Cognition in Physiology and Disease
Recent studies strongly suggest that the discovery of cell-autonomous signaling pathways that enhance adult neurogenesis may lead to therapeutic strategies for improving memory loss due to aging or injury. Here we highlight some of these pathways relevant for the regulation of neurogenesis and implicated in mental disability and we describe the effect of their modulation on cognitive functions. As previously mentioned, the hippocampus plays a key role in declarative memory functioning as a pattern separator. Genetic modulation of the apoptotic pathway in neural stem cells in the DG through ablation of the pro-apoptotic protein Bax leads to increased neurogenesis and improved performance in pattern separation tasks (Sahay et al. 2011). Future studies using this model in disease-relevant backgrounds (e.g., AD models) could potentially elucidate whether the enhancement of neurogenesis changes significantly disease states.
The ERK pathway is important for the modulation of neurogenesis and cognition. ERK5 is a member of the MAP kinase family and it plays a role in prenatal neuronal development and cell fate specification (Cavanaugh et al. 2001; Finegan et al. 2009; Cundiff et al. 2009). ERK5 is expressed in the adult brain in neurogenic regions and inducible conditional ablation of ERK5 in neural progenitors in the adult brain leads to attenuation of hippocampal neurogenesis (Pan et al. 2012a, b). This correlates with alterations in hippocampus-dependent memory tasks like contextual fear conditioning, with defects in learning flexibility and deficits in pattern separation. Conversely, in a genetic mouse model the activation of ERK5 in neuronal progenitors in the DG induces neurogenesis promoting cell survival, neuronal differentiation, and enhanced dendritic structural complexity. The morphological changes are paralleled by improved spatial learning and memory and hippocampus-dependent long-term memory persistence (Wang et al. 2014). The discovery that memory can be prolonged by stimulating adult neurogenesis has important implications for the development of therapeutic strategies to treat memory disorders.
Recent studies show that restoring the function of adult neurogenesis in disease models with clear cognitive dysfunction can reverse the learning deficits. Consequently, treatments directed at this cell population may have a significant impact on disease-relevant cognitive and learning phenotypes. Fragile X syndrome (FXS) is the most common cause of inherited intellectual disability and the leading monogenic cause of autism spectrum disorders. FXS is a trinucleotide repeat disorder caused by a CGG repeat expansion in FMR1, which leads to the loss of its protein product FMRP. Deletion of FMRP in neural stem cells in adult mice affects several aspects of adult hippocampal neurogenesis leading to deficits in proliferation and differentiation of newborn neurons. This is accompanied by reduced performance in hippocampus-dependent learning tasks. Importantly, in FMRP-deficient mice, restoration of FMRP expression specifically in adult neurogenic compartments can rescue these learning deficits (Guo et al. 2011). Although more studies are needed to understand whether postnatal and adult neurogenesis plays a role in the pathogenesis of intellectual disability, these data suggest that FMRP-dependent aberrant neurogenesis could potentially contribute in part to the cognitive deficits observed in Fragile X patients.
Altered neurogenesis appears to contribute to phenotypes observed in DISC1 loss-of-function models. Disrupted-in-schizophrenia 1 (DISC1) is a multifunctional scaffold protein that has been implicated as a susceptibility gene for major mental illness including schizophrenia, bipolar disorders, and autism (Chubb et al. 2008). DISC1 plays an important role in multiple stages of adult neurogenesis in the DG including proliferation, maturation, migration, and synaptic plasticity of newborn neurons. Importantly, the levels of neurogenesis correlate with the performance on specific memory tasks. The cellular phenotypes appear to be in part regulated by disruption in critical signaling pathways modulating adult neurogenesis including the AKT-mTOR, GABA, and GSK3β signaling pathways (Kim et al. 2009, 2012). Indeed, DISC1 has been shown to modulate the proliferation of adult hippocampal precursor cells by inhibiting GSK3β activity (Mao et al. 2009). Pharmacologically targeting these pathways leads to restoration of both neurogenesis and behavioral phenotypes. Notably, in a DISC1 loss-of-function mouse model, treatment with a GSK3β inhibitor, SB-216763, normalizes both the neurogenesis deficits and the schizophrenia- and depression-like behaviors (Mao et al. 2009). In another study, the downregulation of Disc1 specifically during the development of newborn neurons results in defective neuronal maturation, neuronal hyperexcitability, and alteration of neuronal structure. On a molecular level, knockdown of Disc1 results in increased mTOR signaling. Moreover, Disc1 knockdown induces cognitive and affective deficits and behavioral abnormalities are reversed by pharmacological inhibition of the mTOR pathway (Zhou et al. 2013a).
The described roles of adult hippocampal neurogenesis in neurodevelopmental disorders suggest that treatments targeting the adult neural stem cell population may have a significant impact on pathophysiology and endophenotypes and can represent a novel therapeutic approach.
3.2 Potential of Screening for Neurogenic Compounds
Neuro-regenerative approaches aimed at treating pathological cognitive decline could include both the stimulation of endogenous adult neural stem cell populations and the transplantation of exogenous stem cells in the brain. Seminal experiments suggest that both approaches have potential for treating neurological diseases. For example, in a mouse model of hippocampal neurodegeneration that results in significant memory impairment, transplanting neural stem cells improves cognition (Yamasaki et al. 2007). Similarly, transplantation of hippocampal neural stem cells ameliorates complex cognitive deficits in a model of Alzheimer’s disease characterized by abundant amyloid/TAU pathology. Interestingly, the behavioral rescue is achieved despite persistent pathological hallmarks and it is mediated by BDNF-dependent increase in hippocampal synaptic density (Blurton-Jones et al. 2009). On the other hand, the ability of neural stem cells to be reactivated in aging and in disease conditions supports the idea that identification of novel pharmacological targets/molecules promoting adult hippocampal neurogenesis represents a promising therapeutic approach.
A search of Thomas Reuter Integrity suggests that there are well over 50 relevant patents claiming neurogenic molecules. A large number of diverse chemical classes are neurogenic and have been patented for diverse CNS therapeutic indications (Rishton 2008). Reported patents claim that besides CNS acting molecules such as anticonvulsants, cognition modifiers, anxiolytics, and mood-stabilizing agents, also antidiabetic, blood pressure-lowering, and cholesterol-lowering agents have surprisingly shown neurogenic properties (Rishton 2008). For example, metformin, which is widely used to treat type II diabetes and other metabolic syndromes, has been shown to have neurogenic properties (Wang et al. 2012). In vitro work on both mouse and human cell cultures shows that metformin promotes neurogenesis and this translates in vivo in increased neurogenesis and enhanced spatial reversal learning in a water maze task.
A promising route for the identification of novel neurogenesis-increasing compounds is the high-throughput screening of chemical libraries using neural stem cell-based assays. Screens have already demonstrated the potential to elucidate known and novel mechanisms regulating adult hippocampal neurogenesis. In an early study, 1,200 compounds with known pharmacological activity were screened on rat neurospheres for proliferative and differentiation capacity leading to the identification of many CNS-relevant targets. Active compounds included modulators of dopamine, serotonin, opioid, glutamate, and vanilloid signaling (Diamandis et al. 2007). Besides targeting known receptors relevant for neurogenesis, molecule screening can help identify novel relevant signal transduction pathways associated with hippocampal plasticity and cognition. In another screen the small molecule isoxazole 9 [Isx-9; N-cyclopropyl-5-(thiophen-2-yl) isoxazole-3-carboxamide] was identified. Isx-9 robustly induces neuronal differentiation in an in vitro model of adult neural stem cells. In addition, Isx-9’s effects appear to involve myocyte-enhancer factor 2 (Mef2), a family of transcription factors that had never been linked before to adult neurogenesis in vivo (Schneider et al. 2008). In a follow-up study, it has been shown that Isx-9 promotes multiple stages of neurogenesis in vivo and this correlates with enhanced memory in Morris water maze tasks (Petrik et al. 2012). The mechanism of action of Isx-9 on hippocampal neurogenesis and cognition is at least in part cell autonomous since inducible deletion of Mef2 isoforms from neural stem cells and their progeny thwarts the cognitive enhancing effect. The identification of ISX-9 highlights the potential of small molecule screening campaigns to identify novel mechanisms of action relevant to hippocampal neurogenesis and cognition.
A novel in vivo screening campaign has been undertaken identifying a compound called P7C3 with neurogenic properties (Pieper et al. 2010). Further characterization demonstrates that P7C3 can enhance neurogenesis in the dentate gyrus, can modulate mitochondrial physiology and neuronal survival, can increase synaptic activity, and can preserve cognitive capacity in aged rats. The target(s) and pathway(s) of P7C3 remain unknown, which provides both a challenge as well as an interesting opportunity to identify new pathways linked to neurogenesis and pathological cognitive decline. Clearly, the mechanisms mediating the effect of P7C3 on cognition might involve biological processes beyond hippocampal neurogenesis that could regulate broader aspects of brain plasticity. In follow-up studies, modified and more potent analogues of P7C3 demonstrated that the compound class exhibits neuroprotective effect in models of Parkinson’s disease, amyotrophic lateral sclerosis, traumatic brain injury, and age-related cognitive decline (MacMillan et al. 2011; Blaya et al. 2014; Walker et al. 2014). Derivatives appended with immobilizing moieties may reveal the protein targets of the P7C3 class of neuroprotective compounds.
Few classes of neurologically active molecules have been discovered in the last 50 years, at least in part because neuroscience drug discovery efforts have been dominated by target-based assays. Unlike target-based approaches, phenotype-based screens can identify compounds hitting on pathways that capture the complexity of the neurobiological tissue. One impressive example of a large-scale HTS screening campaign on primary cells has been recently reported, where one million small-molecule compounds have been screened on primary rat neurospheres, assessing proliferation and differentiation capacity based on the measurement of ATP levels (Liu et al. 2009). Over 5,000 primary hits have been identified to induce proliferation and differentiation. Further characterization of these compounds and a strategy for target identification will potentially provide novel insight into the mechanisms involved in the regulation of neural stem cells and adult neurogenesis.
As highlighted above, several neurodevelopmental disorders show adult neurogenesis alterations that appear to be partly responsible for cognitive deficits. Recent advances in human-induced pluripotent stem cell (hiPSCs) technologies allow the use of human cells with patient-specific genetic alterations for in vitro neuronal disease modeling. In vitro human neuronal differentiation recapitulates several aspects and stages of in vivo neurogenesis and this system can be used to characterize specific disease-related phenotypes observed in vivo. Moreover, hiPSCs represent an unprecedented tool for development of platforms for phenotypic screening using human tissue- and disease-relevant models. Despite the challenges associated with the novelty of the technology and the limited understanding of human neuronal differentiation in vitro, hiPSC disease models hold great promise as tools for innovative drug discovery (Chailangkarn et al. 2012; Bellin et al. 2012).
3.3 Translational Biomarkers of Human Neurogenesis
In CNS translational research, good translational biomarkers are crucial to identify pre-symptomatic early disease states, to improve dose finding, and to allow patient stratification and responder/nonresponders analysis, significantly affecting the success in drug development. As described above, increasing preclinical evidence in animal models of neurodevelopmental disorders, schizophrenia, Alzheimer’s disease, stress, and depression demonstrates that alterations in neurogenesis correlate with behavioral and cognitive symptoms. Moreover, multiple pharmacological interventions targeting cognition modulate adult hippocampal neurogenesis. Therefore, the identification of a noninvasive biomarker for human neurogenesis is an area of growing interest and intensive scientific research. The lack of biomarker currently represents a key limitation in the ability to ultimately demonstrate the relevance of the neurogenic process in human health and disease and its role in mediating beneficial effects of therapeutic interventions.
Identification of hippocampal neurogenesis-specific biomarkers in blood or CSF could serve as a proxy for the rates of neurogenesis in the brain. Immature neuronal precursors in the adult brain are characterized by specific transcriptomes, cellular and membrane proteomes, secretomes, metabolomes, and lipidomes (Ramm et al. 2009; Knolhoff et al. 2013). Unbiased approaches to identify neurogenesis-specific transcriptomic profiles have led to the identification of several transcripts expressed during specific stages of adult neurogenesis (Miller et al. 2013; Couillard-Despres et al. 2006; Lim et al. 2006). Moreover, expression levels of miRNA involved in the regulation of neurogenesis appear to be altered in brain samples from AD patients. Importantly, miRNA can be released and detected in the CSF, representing a potential biomarker (Cogswell et al. 2008). Similarly, some neurogenesis-specific proteins are secreted into fluids including the CSF. For example, doublecortin (DCX), which encodes for a microtubule-associated protein specifically expressed in newborn neurons, is detected in CSF of both rodents and humans (Kremer et al. 2013).
Another potential strategy to monitor neurogenesis is brain imaging. A possible approach would be to identify a PET ligand binding molecular markers of adult hippocampal neurogenesis. A PET imaging strategy could be feasible based on anatomical considerations. PET resolution is in the order of 1.5–3 mm, the hippocampus volume in human is ~3 cm3, and the dentate gyrus is 90 mm3. It has been reported that around 0.033 % of granular cells are newborn neurons in old primates and 0.07 % in old mice (Aizawa et al. 2009). Considering the sensitivity of PET imaging, shown to reach 30 pM of receptor density for the D2 receptor, the detection of the newborn cells’ fraction would be feasible (Kessler et al. 1993; Farde 1996). The challenge is to find a hippocampal neurogenesis-selective protein with a binding pocket that could serve as a PET tracer. 18F-3′-deoxy-3′-fluoro-l-thymidine (FLT) has been described to be a potential PET tracer to monitor neural stem cell division in the rat hippocampus (Rueger et al. 2010). FLT is a thymidine analogue that accumulates in dividing cells and it is a common marker to monitor cell proliferation in tumors. For measurements in the brain, strategies to improve the signal-to-noise ratio and to reduce radioactive doses would significantly implement the approach (Rueger et al. 2010). Monitoring neurogenesis targeting immature neurons could represent another approach considering that in aged macaque monkeys and mice, immature neurons are 4- to 4.5-fold more abundant than dividing NPCs (Aizawa et al. 2009).
MRI/MRS technologies could be a potential way to identify biomarkers of neurogenesis or at least epiphenomena of the process. A conceptually very appealing study, using MRI-based technologies, suggested relative cerebral blood volume (rCBV) increases specific to the dentate gyrus as a putative biomarker for neurogenesis (Pereira et al. 2007). As described previously, neurogenesis and angiogenesis in the DG are tightly linked processes (Palmer et al. 2000; Louissaint et al. 2002). However, the specificity of this method is challenged by the fact that hippocampus hemodynamics observed by fMRI is sensitive to changes due to disease states and drug treatments (Choi et al. 2006; Littlewood et al. 2006; Gozzi et al. 2008).
Using proton MRS, a very exciting study identified a particular resonance at 1.28 ppm with enhanced prevalence in NPCs in vitro (Maletic-Savatic et al. 2008; Manganas et al. 2007). This marker was demonstrated to be a translational biomarker in both rats and human subjects, and the putative NPC-specific peak strongly decreases during brain development from childhood to adulthood (Manganas et al. 2007). If confirmed, this study provides an unparalleled glimpse into the human brain with the promise to provide a truly noninvasive biomarker of adult neurogenesis. Not unexpectedly, this report has aroused a certain amount of controversy on technical and study design aspects (Friedman 2008; Hoch et al. 2008; Jansen et al. 2008; Dong et al. 2009; Ramm et al. 2009). More studies are needed to demonstrate feasibility and practicality of this approach as a tool to investigate the role of neurogenesis in a wide variety of human brain disorders.
4 Conclusions
Adult hippocampal neurogenesis is a complex process that provides an excellent example of the surprising plasticity of the mature brain. Research within the last few years has identified molecular mechanisms that regulate individual steps in neurogenesis and has highlighted its function in various neurological disorders. However, the process of translating basic knowledge into pharmacological interventions is only at the beginning. Key aspects need to be further clarified to support targeting of adult neurogenesis for the treatment of neurological and cognitive disorders in humans: (1) Can results obtained from animal models of neurological disorders be translated to humans?, (2) Can changes in neurogenesis be assessed in humans (i.e., is there a biomarker)?, (3) Is it possible to identify chemical entities or biomolecules to pharmacologically modulate neurogenesis in humans? In this chapter, we have highlighted key studies investigating the role of neurogenesis in cognitive impairment associated with physiological and disease conditions. Moreover, we have described endogenous and pharmacological factors that modulate neurogenesis and have an effect on cognitive functions. Deep understanding of several of these aspects would enable the development of drugs that target and modulate adult neurogenesis in humans to treat patients with neurological disorders.
References
Aberg MA, Pedersen NL, Toren K, Svartengren M, Backstrand B, Johnsson T, Cooper-Kuhn CM, Aberg ND, Nilsson M, Kuhn HG (2009) Cardiovascular fitness is associated with cognition in young adulthood. Proc Natl Acad Sci U S A 106(49):20906–20911. doi:10.1073/pnas.0905307106
Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM (2008) Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry 63(7):642–649. doi:10.1016/j.biopsych.2007.09.019
Adeosun SO, Hou X, Zheng B, Stockmeier C, Ou X, Paul I, Mosley T, Weisgraber K, Wang JM (2014) Cognitive deficits and disruption of neurogenesis in a mouse model of apolipoprotein E4 domain interaction. J Biol Chem 289(5):2946–2959. doi:10.1074/jbc.M113.497909
Aizawa K, Ageyama N, Yokoyama C, Hisatsune T (2009) Age-dependent alteration in hippocampal neurogenesis correlates with learning performance of macaque monkeys. Exp Anim 58(4):403–407
Akers KG, Martinez-Canabal A, Restivo L, Yiu AP, De Cristofaro A, Hsiang HL, Wheeler AL, Guskjolen A, Niibori Y, Shoji H, Ohira K, Richards BA, Miyakawa T, Josselyn SA, Frankland PW (2014) Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 344(6184):598–602. doi:10.1126/science.1248903
Alexander GE, Ryan L, Bowers D, Foster TC, Bizon JL, Geldmacher DS, Glisky EL (2012) Characterizing cognitive aging in humans with links to animal models. Front Aging Neurosci 4:21. doi:10.3389/fnagi.2012.00021
Alexson TO, Hitoshi S, Coles BL, Bernstein A, van der Kooy D (2006) Notch signaling is required to maintain all neural stem cell populations—irrespective of spatial or temporal niche. Dev Neurosci 28(1–2):34–48. doi:10.1159/000090751
Ally BA, Hussey EP, Ko PC, Molitor RJ (2013) Pattern separation and pattern completion in Alzheimer’s disease: evidence of rapid forgetting in amnestic mild cognitive impairment. Hippocampus 23(12):1246–1258. doi:10.1002/hipo.22162
Alonso R, Griebel G, Pavone G, Stemmelin J, Le Fur G, Soubrie P (2004) Blockade of CRF(1) or V(1b) receptors reverses stress-induced suppression of neurogenesis in a mouse model of depression. Mol Psychiatry 9(3):278–286, 224. doi:10.1038/sj.mp.4001464
Altman J (1969) Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J Comp Neurol 137(4):433–457. doi:10.1002/cne.901370404
Alvarez-Buylla A, Garcia-Verdugo JM (2002) Neurogenesis in adult subventricular zone. J Neurosci 22(3):629–634
Ambrogini P, Lattanzi D, Ciuffoli S, Agostini D, Bertini L, Stocchi V, Santi S, Cuppini R (2004) Morpho-functional characterization of neuronal cells at different stages of maturation in granule cell layer of adult rat dentate gyrus. Brain Res 1017(1–2):21–31. doi:10.1016/j.brainres.2004.05.039
Amrein I, Isler K, Lipp HP (2011) Comparing adult hippocampal neurogenesis in mammalian species and orders: influence of chronological age and life history stage. Eur J Neurosci 34(6):978–987. doi:10.1111/j.1460-9568.2011.07804.x
Anacker C (2014) Adult hippocampal neurogenesis in depression: behavioral implications and regulation by the stress system. Curr Top Behav Neurosci. doi:10.1007/7854_2014_275
Anacker C, Pariante CM (2012) New models to investigate complex glucocorticoid receptor functions. Front Behav Neurosci 6:90. doi:10.3389/fnbeh.2012.00090
Arruda-Carvalho M, Sakaguchi M, Akers KG, Josselyn SA, Frankland PW (2011) Posttraining ablation of adult-generated neurons degrades previously acquired memories. J Neurosci 31(42):15113–15127. doi:10.1523/JNEUROSCI.3432-11.2011
Asberg M (1976) Treatment of depression with tricyclic drugs—pharmacokinetic and pharmacodynamic aspects. Pharmakopsychiatr Neuropsychopharmakol 9(1):18–26
Ashford JW (2004) APOE genotype effects on Alzheimer’s disease onset and epidemiology. J Mol Neurosci 23(3):157–165. doi:10.1385/JMN:23:3:157
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, Kavalali ET, Monteggia LM (2011) NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 475(7354):91–95. doi:10.1038/nature10130
Bakker A, Kirwan CB, Miller M, Stark CE (2008) Pattern separation in the human hippocampal CA3 and dentate gyrus. Science 319(5870):1640–1642. doi:10.1126/science.1152882
Bambico FR, Belzung C (2013) Novel insights into depression and antidepressants: a synergy between synaptogenesis and neurogenesis? Curr Top Behav Neurosci 15:243–91. doi:10.1007/7854_2012_234
Banerjee R, Ghosh AK, Ghosh B, Bhattacharyya S, Mondal AC (2013) Decreased mRNA and protein expression of BDNF, NGF, and their receptors in the hippocampus from suicide: an analysis in human postmortem brain. Clin Med Insights Pathol 6:1–11. doi:10.4137/CMPath.S12530
Barnes CA (1979) Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol 93(1):74–104
Bechara RG, Kelly AM (2013) Exercise improves object recognition memory and induces BDNF expression and cell proliferation in cognitively enriched rats. Behav Brain Res 245:96–100. doi:10.1016/j.bbr.2013.02.018
Bekinschtein P, Kent BA, Oomen CA, Clemenson GD, Gage FH, Saksida LM, Bussey TJ (2013) BDNF in the dentate gyrus is required for consolidation of “pattern-separated” memories. Cell Rep 5(3):759–768. doi:10.1016/j.celrep.2013.09.027
Bekinschtein P, Kent BA, Oomen CA, Clemenson GD, Gage FH, Saksida LM, Bussey TJ (2014) Brain-derived neurotrophic factor interacts with adult-born immature cells in the dentate gyrus during consolidation of overlapping memories. Hippocampus. doi:10.1002/hipo.22304
Bellin M, Marchetto MC, Gage FH, Mummery CL (2012) Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol 13(11):713–726. doi:10.1038/nrm3448
Belvederi Murri M, Pariante C, Mondelli V, Masotti M, Atti AR, Mellacqua Z, Antonioli M, Ghio L, Menchetti M, Zanetidou S, Innamorati M, Amore M (2014) HPA axis and aging in depression: systematic review and meta-analysis. Psychoneuroendocrinology 41:46–62. doi:10.1016/j.psyneuen.2013.12.004
Ben Abdallah NM, Slomianka L, Vyssotski AL, Lipp HP (2010) Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiol Aging 31(1):151–161. doi:10.1016/j.neurobiolaging.2008.03.002
Berchtold NC, Chinn G, Chou M, Kesslak JP, Cotman CW (2005) Exercise primes a molecular memory for brain-derived neurotrophic factor protein induction in the rat hippocampus. Neuroscience 133(3):853–861. doi:10.1016/j.neuroscience.2005.03.026
Bergami M, Rimondini R, Santi S, Blum R, Gotz M, Canossa M (2008) Deletion of TrkB in adult progenitors alters newborn neuron integration into hippocampal circuits and increases anxiety-like behavior. Proc Natl Acad Sci U S A 105(40):15570–15575. doi:10.1073/pnas.0803702105
Bernal GM, Peterson DA (2011) Phenotypic and gene expression modification with normal brain aging in GFAP-positive astrocytes and neural stem cells. Aging Cell 10(3):466–482. doi:10.1111/j.1474-9726.2011.00694.x
Bessa JM, Ferreira D, Melo I, Marques F, Cerqueira JJ, Palha JA, Almeida OF, Sousa N (2009) The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry 14(8):764–773, 739. doi:10.1038/mp.2008.119
Bianchi P, Ciani E, Guidi S, Trazzi S, Felice D, Grossi G, Fernandez M, Giuliani A, Calzà L, Bartesaghi R (2010) Early pharmacotherapy restores neurogenesis and cognitive performance in the Ts65Dn mouse model for Down syndrome. J Neurosci 30(26):8769–8779. doi:10.1523/JNEUROSCI. 0534-10.2010
Bizon JL, Lee HJ, Gallagher M (2004) Neurogenesis in a rat model of age-related cognitive decline. Aging Cell 3(4):227–234. doi:10.1111/j.1474-9728.2004.00099.x
Black JE, Isaacs KR, Anderson BJ, Alcantara AA, Greenough WT (1990) Learning causes synaptogenesis, whereas motor activity causes angiogenesis, in cerebellar cortex of adult rats. Proc Natl Acad Sci U S A 87(14):5568–5572
Blaya MO, Bramlett HM, Naidoo J, Pieper AA, Dietrich WD (2014) Neuroprotective efficacy of a proneurogenic compound after traumatic brain injury. J Neurotrauma 31(5):476–486. doi:10.1089/neu.2013.3135
Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Muller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM (2009) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A 106(32):13594–13599. doi:10.1073/pnas.0901402106
Boekhoorn K, Joels M, Lucassen PJ (2006) Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis 24(1):1–14. doi:10.1016/j.nbd.2006.04.017
Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, Arango V (2009) Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 34(11):2376–2389. doi:10.1038/npp.2009.75
Boldrini M, Hen R, Underwood MD, Rosoklija GB, Dwork AJ, Mann JJ, Arango V (2012) Hippocampal angiogenesis and progenitor cell proliferation are increased with antidepressant use in major depression. Biol Psychiatry 72(7):562–571. doi:10.1016/j.biopsych.2012.04.024
Bolognin S, Buffelli M, Puolivali J, Iqbal K (2014) Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiol Aging 35(9):2134–2146. doi:10.1016/j.neurobiolaging.2014.02.017
Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming GL, Song H (2011) In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145(7):1142–1155. doi:10.1016/j.cell.2011.05.024
Bondolfi L, Ermini F, Long JM, Ingram DK, Jucker M (2004) Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiol Aging 25(3):333–340. doi:10.1016/S0197-4580(03)00083-6
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259
Braak H, Braak E (1995) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16(3):271–278, discussion 278–284
Brandt MD, Maass A, Kempermann G, Storch A (2010) Physical exercise increases Notch activity, proliferation and cell cycle exit of type-3 progenitor cells in adult hippocampal neurogenesis. Eur J Neurosci 32(8):1256–1264. doi:10.1111/j.1460-9568.2010.07410.x
Bremner JD, Vythilingam M, Vermetten E, Vaccarino V, Charney DS (2004) Deficits in hippocampal and anterior cingulate functioning during verbal declarative memory encoding in midlife major depression. Am J Psychiatry 161(4):637–645
Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG (2003a) Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci 17(10):2042–2046
Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG (2003b) Transient expression of doublecortin during adult neurogenesis. J Comp Neurol 467(1):1–10. doi:10.1002/cne.10874
Bruel-Jungerman E, Laroche S, Rampon C (2005) New neurons in the dentate gyrus are involved in the expression of enhanced long-term memory following environmental enrichment. Eur J Neurosci 21(2):513–521. doi:10.1111/j.1460-9568.2005.03875.x
Bruel-Jungerman E, Davis S, Rampon C, Laroche S (2006) Long-term potentiation enhances neurogenesis in the adult dentate gyrus. J Neurosci 26(22):5888–5893. doi:10.1523/JNEUROSCI.0782-06.2006
Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, Prochiantz A (2004) Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131(9):2173–2181. doi:10.1242/dev.01103
Cameron HA, McKay RD (1999) Restoring production of hippocampal neurons in old age. Nat Neurosci 2(10):894–897. doi:10.1038/13197
Cameron HA, Woolley CS, McEwen BS, Gould E (1993) Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience 56(2):337–344
Cameron HA, McEwen BS, Gould E (1995) Regulation of adult neurogenesis by excitatory input and NMDA receptor activation in the dentate gyrus. J Neurosci 15(6):4687–4692
Campbell NR, Fernandes CC, Halff AW, Berg DK (2010) Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci 30(26):8734–8744. doi:10.1523/JNEUROSCI.0931-10.2010
Campos AC, Ortega Z, Palazuelos J, Fogaca MV, Aguiar DC, Diaz-Alonso J, Ortega-Gutierrez S, Vazquez-Villa H, Moreira FA, Guzman M, Galve-Roperh I, Guimaraes FS (2013) The anxiolytic effect of cannabidiol on chronically stressed mice depends on hippocampal neurogenesis: involvement of the endocannabinoid system. Int J Neuropsychopharmacol 16(6):1407–1419. doi:10.1017/S1461145712001502
Carro E, Nunez A, Busiguina S, Torres-Aleman I (2000) Circulating insulin-like growth factor I mediates effects of exercise on the brain. J Neurosci 20(8):2926–2933
Caselli RJ, Beach TG, Yaari R, Reiman EM (2006) Alzheimer’s disease a century later. J Clin Psychiatry 67(11):1784–1800
Castren E, Voikar V, Rantamaki T (2007) Role of neurotrophic factors in depression. Curr Opin Pharmacol 7(1):18–21. doi:10.1016/j.coph.2006.08.009
Cavanaugh JE, Ham J, Hetman M, Poser S, Yan C, Xia Z (2001) Differential regulation of mitogen-activated protein kinases ERK1/2 and ERK5 by neurotrophins, neuronal activity, and cAMP in neurons. J Neurosci 21(2):434–443
Chaddock L, Erickson KI, Prakash RS, Kim JS, Voss MW, Vanpatter M, Pontifex MB, Raine LB, Konkel A, Hillman CH, Cohen NJ, Kramer AF (2010) A neuroimaging investigation of the association between aerobic fitness, hippocampal volume, and memory performance in preadolescent children. Brain Res 1358:172–183. doi:10.1016/j.brainres.2010.08.049
Chaddock L, Hillman CH, Buck SM, Cohen NJ (2011) Aerobic fitness and executive control of relational memory in preadolescent children. Med Sci Sports Exerc 43(2):344–349. doi:10.1249/MSS.0b013e3181e9af48
Chaddock L, Hillman CH, Pontifex MB, Johnson CR, Raine LB, Kramer AF (2012) Childhood aerobic fitness predicts cognitive performance one year later. J Sports Sci 30(5):421–430. doi:10.1080/02640414.2011.647706
Chaddock-Heyman L, Erickson KI, Voss MW, Knecht AM, Pontifex MB, Castelli DM, Hillman CH, Kramer AF (2013) The effects of physical activity on functional MRI activation associated with cognitive control in children: a randomized controlled intervention. Front Hum Neurosci 7:72. doi:10.3389/fnhum.2013.00072
Chadwick W, Mitchell N, Caroll J, Zhou Y, Park S-S et al (2011) Amitriptyline-mediated cognitive enhancement in aged 3×Tg Alzheimer’s disease mice is associated with neurogenesis and neurotrophic activity. PLoS One 6(6):e21660. doi:10.1371/journal.pone.0021660
Chadwick MJ, Bonnici HM, Maguire EA (2014) CA3 size predicts the precision of memory recall. Proc Natl Acad Sci U S A. doi:10.1073/pnas.1319641111
Chailangkarn T, Acab A, Muotri AR (2012) Modeling neurodevelopmental disorders using human neurons. Curr Opin Neurobiol 22(5):785–790. doi:10.1016/j.conb.2012.04.004
Chetty S, Friedman AR, Taravosh-Lahn K, Kirby ED, Mirescu C, Guo F, Krupik D, Nicholas A, Geraghty AC, Krishnamurthy A, Tsai MK, Covarrubias D, Wong AT, Francis DD, Sapolsky RM, Palmer TD, Pleasure D, Kaufer D (2014) Stress and glucocorticoids promote oligodendrogenesis in the adult hippocampus. Mol Psychiatry 19(12):1275–1283. doi:10.1038/mp.2013.190
Choi JK, Mandeville JB, Chen YI, Kim YR, Jenkins BG (2006) High resolution spatial mapping of nicotine action using pharmacologic magnetic resonance imaging. Synapse 60(2):152–157. doi:10.1002/syn.20284
Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK (2008) The DISC locus in psychiatric illness. Mol Psychiatry 13(1):36–64. doi:10.1038/sj.mp.4002106
Chun SK, Sun W, Park JJ, Jung MW (2006) Enhanced proliferation of progenitor cells following long-term potentiation induction in the rat dentate gyrus. Neurobiol Learn Mem 86(3):322–329. doi:10.1016/j.nlm.2006.05.005
Cinini SM, Barnabe GF, Galvao-Coelho N, de Medeiros MA, Perez-Mendes P, Sousa MB, Covolan L, Mello LE (2014) Social isolation disrupts hippocampal neurogenesis in young non-human primates. Front Neurosci 8:45. doi:10.3389/fnins.2014.00045
Clark PJ, Brzezinska WJ, Thomas MW, Ryzhenko NA, Toshkov SA, Rhodes JS (2008) Intact neurogenesis is required for benefits of exercise on spatial memory but not motor performance or contextual fear conditioning in C57BL/6 J mice. Neuroscience 155(4):1048–1058. doi:10.1016/j.neuroscience.2008.06.051
Clelland CD, Choi M, Romberg C, Clemenson GD Jr, Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, Bussey TJ (2009) A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325(5937):210–213. doi:10.1126/science.1173215
Cogswell JP, Ward J, Taylor IA, Waters M, Shi Y, Cannon B, Kelnar K, Kemppainen J, Brown D, Chen C, Prinjha RK, Richardson JC, Saunders AM, Roses AD, Richards CA (2008) Identification of miRNA changes in Alzheimer’s disease brain and CSF yields putative biomarkers and insights into disease pathways. J Alzheimers Dis 14(1):27–41
Compton DM, Dietrich KL, Selinger MC, Testa EK (2011) 5-methoxy-N, N-di(iso)propyltryptamine hydrochloride (Foxy)-induced cognitive deficits in rat after exposure in adolescence. Physiol Behav 103(2):203–209. doi:10.1016/j.physbeh.2011.01.021
Contestabile A, Greco B, Ghezzi D, Tucci V, Benfenati F, Gasparini L (2013) Lithium rescues synaptic plasticity and memory in Down syndrome mice. J Clin Invest 123(1):348–361
Cooper-Kuhn CM, Winkler J, Kuhn HG (2004) Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. J Neurosci Res 77(2):155–165. doi:10.1002/jnr.20116
Coronas V, Durand M, Chabot JG, Jourdan F, Quirion R (2000) Acetylcholine induces neuritic outgrowth in rat primary olfactory bulb cultures. Neuroscience 98(2):213–219
Couillard-Despres S, Winner B, Karl C, Lindemann G, Schmid P, Aigner R, Laemke J, Bogdahn U, Winkler J, Bischofberger J, Aigner L (2006) Targeted transgene expression in neuronal precursors: watching young neurons in the old brain. Eur J Neurosci 24(6):1535–1545. doi:10.1111/j.1460-9568.2006.05039.x
Creer DJ, Romberg C, Saksida LM, van Praag H, Bussey TJ (2010) Running enhances spatial pattern separation in mice. Proc Natl Acad Sci U S A 107(5):2367–2372. doi:10.1073/pnas.0911725107
Cundiff P, Liu L, Wang Y, Zou J, Pan YW, Abel G, Duan X, Ming GL, Englund C, Hevner R, Xia Z (2009) ERK5 MAP kinase regulates neurogenin1 during cortical neurogenesis. PLoS One 4(4):e5204. doi:10.1371/journal.pone.0005204
Cuppini R, Bucherelli C, Ambrogini P, Ciuffoli S, Orsini L, Ferri P, Baldi E (2006) Age-related naturally occurring depression of hippocampal neurogenesis does not affect trace fear conditioning. Hippocampus 16(2):141–148. doi:10.1002/hipo.20140
Dagyte G, Crescente I, Postema F, Seguin L, Gabriel C, Mocaer E, Boer JA, Koolhaas JM (2011) Agomelatine reverses the decrease in hippocampal cell survival induced by chronic mild stress. Behav Brain Res 218(1):121–128. doi:10.1016/j.bbr.2010.11.045
David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, Drew M, Craig DA, Guiard BP, Guilloux JP, Artymyshyn RP, Gardier AM, Gerald C, Antonijevic IA, Leonardo ED, Hen R (2009) Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 62(4):479–493. doi:10.1016/j.neuron.2009.04.017
De Strooper B, Woodgett J (2003) Alzheimer’s disease: mental plaque removal. Nature 423(6938):392–393. doi:10.1038/423392a
De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R (1999) A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398(6727):518–522. doi:10.1038/19083
Delgado PL (2000) Depression: the case for a monoamine deficiency. J Clin Psychiatry 61(Suppl 6):7–11
Demars MP, Hollands C, Zhao Kda T, Lazarov O (2013) Soluble amyloid precursor protein-alpha rescues age-linked decline in neural progenitor cell proliferation. Neurobiol Aging 34(10):2431–2440. doi:10.1016/j.neurobiolaging.2013.04.016
Deng W, Saxe MD, Gallina IS, Gage FH (2009) Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci 29(43):13532–13542. doi:10.1523/JNEUROSCI.3362-09.2009
Denny CA, Burghardt NS, Schachter DM, Hen R, Drew MR (2012) 4- to 6-week-old adult-born hippocampal neurons influence novelty-evoked exploration and contextual fear conditioning. Hippocampus 22(5):1188–1201. doi:10.1002/hipo.20964
Dery N, Pilgrim M, Gibala M, Gillen J, Wojtowicz JM, Macqueen G, Becker S (2013) Adult hippocampal neurogenesis reduces memory interference in humans: opposing effects of aerobic exercise and depression. Front Neurosci 7:66. doi:10.3389/fnins.2013.00066
Diamandis P, Wildenhain J, Clarke ID, Sacher AG, Graham J, Bellows DS, Ling EK, Ward RJ, Jamieson LG, Tyers M, Dirks PB (2007) Chemical genetics reveals a complex functional ground state of neural stem cells. Nat Chem Biol 3(5):268–273. doi:10.1038/nchembio873
Ding Q, Ying Z, Gomez-Pinilla F (2011) Exercise influences hippocampal plasticity by modulating brain-derived neurotrophic factor processing. Neuroscience 192:773–780. doi:10.1016/j.neuroscience.2011.06.032
Dong Z, Dreher W, Leibfritz D, Peterson BS (2009) Challenges of using MR spectroscopy to detect neural progenitor cells in vivo. AJNR Am J Neuroradiol 30(6):1096–1101. doi:10.3174/ajnr.A1557
Drachman DA, Leavitt J (1974) Human memory and the cholinergic system. A relationship to aging? Arch Neurol 30(2):113–121
Drapeau E, Mayo W, Aurousseau C, Le Moal M, Piazza PV, Abrous DN (2003) Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc Natl Acad Sci U S A 100(24):14385–14390. doi:10.1073/pnas.2334169100
Drew MR, Denny CA, Hen R (2010) Arrest of adult hippocampal neurogenesis in mice impairs single- but not multiple-trial contextual fear conditioning. Behav Neurosci 124(4):446–454. doi:10.1037/a0020081
Driscoll I, Hamilton DA, Petropoulos H, Yeo RA, Brooks WM, Baumgartner RN, Sutherland RJ (2003) The aging hippocampus: cognitive, biochemical and structural findings. Cereb Cortex 13(12):1344–1351
Driscoll I, Howard SR, Stone JC, Monfils MH, Tomanek B, Brooks WM, Sutherland RJ (2006) The aging hippocampus: a multi-level analysis in the rat. Neuroscience 139(4):1173–1185. doi:10.1016/j.neuroscience.2006.01.040
Duman CH, Duman RS (2015) Spine synapse remodeling in the pathophysiology and treatment of depression. Neurosci Lett. doi:10.1016/j.neulet.2015.01.022
Dunham JS, Deakin JF, Miyajima F, Payton A, Toro CT (2009) Expression of hippocampal brain-derived neurotrophic factor and its receptors in Stanley consortium brains. J Psychiatr Res 43(14):1175–1184. doi:10.1016/j.jpsychires.2009.03.008
Dupret D, Revest JM, Koehl M, Ichas F, De Giorgi F, Costet P, Abrous DN, Piazza PV (2008) Spatial relational memory requires hippocampal adult neurogenesis. PLoS One 3(4):e1959. doi:10.1371/journal.pone.0001959
Duric V, Banasr M, Stockmeier CA, Simen AA, Newton SS, Overholser JC, Jurjus GJ, Dieter L, Duman RS (2013) Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int J Neuropsychopharmacol 16(1):69–82. doi:10.1017/S1461145712000016
Duveau V, Laustela S, Barth L, Gianolini F, Vogt KE, Keist R, Chandra D, Homanics GE, Rudolph U, Fritschy JM (2011) Spatiotemporal specificity of GABAA receptor-mediated regulation of adult hippocampal neurogenesis. Eur J Neurosci 34(3):362–373. doi:10.1111/j.1460-9568.2011.07782.x
Egeland M, Zhang X, Millan MJ, Mocaer E, Svenningsson P (2012) Pharmacological or genetic blockade of the dopamine D3 receptor increases cell proliferation in the hippocampus of adult mice. J Neurochem 123(5):811–823. doi:10.1111/jnc.12011
Ehninger D, Kempermann G (2003) Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb Cortex 13(8):845–851
ElBeltagy M et al (2010) Fluoxetine improves the memory deficits caused by the chemotherapy agent 5-fluorouracil. Behav Brain Res 208(1):112–117
Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, Enikolopov G (2011) Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell 8(5):566–579. doi:10.1016/j.stem.2011.03.010
Erickson KI, Prakash RS, Voss MW, Chaddock L, Hu L, Morris KS, White SM, Wojcicki TR, McAuley E, Kramer AF (2009) Aerobic fitness is associated with hippocampal volume in elderly humans. Hippocampus 19(10):1030–1039. doi:10.1002/hipo.20547
Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4(11):1313–1317. doi:10.1038/3305
Fabel K, Fabel K, Tam B, Kaufer D, Baiker A, Simmons N, Kuo CJ, Palmer TD (2003) VEGF is necessary for exercise-induced adult hippocampal neurogenesis. Eur J Neurosci 18(10):2803–2812
Fabel K, Wolf SA, Ehninger D, Babu H, Leal-Galicia P, Kempermann G (2009) Additive effects of physical exercise and environmental enrichment on adult hippocampal neurogenesis in mice. Front Neurosci 3:50. doi:10.3389/neuro.22.002.2009
Falls WA, Fox JH, MacAulay CM (2010) Voluntary exercise improves both learning and consolidation of cued conditioned fear in C57 mice. Behav Brain Res 207(2):321–331. doi:10.1016/j.bbr.2009.10.016
Farde L (1996) The advantage of using positron emission tomography in drug research. Trends Neurosci 19(6):211–214
Farioli-Vecchioli S, Mattera A, Micheli L, Ceccarelli M, Leonardi L, Saraulli D, Costanzi M, Cestari V, Rouault JP, Tirone F (2014) Running rescues defective adult neurogenesis by shortening the length of the cell cycle of neural stem and progenitor cells. Stem Cells 32(7):1968–1982. doi:10.1002/stem.1679
Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR (2004) Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 124(1):71–79. doi:10.1016/j.neuroscience.2003.09.029
Fatemi SH, Earle JA, Stary JM, Lee S, Sedgewick J (2001) Altered levels of the synaptosomal associated protein SNAP-25 in hippocampus of subjects with mood disorders and schizophrenia. Neuroreport 12(15):3257–3262
Faulkner RL, Jang MH, Liu XB, Duan X, Sailor KA, Kim JY, Ge S, Jones EG, Ming GL, Song H, Cheng HJ (2008) Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci U S A 105(37):14157–14162. doi:10.1073/pnas.0806658105
Fava M, Graves LM, Benazzi F, Scalia MJ, Iosifescu DV, Alpert JE, Papakostas GI (2006) A cross-sectional study of the prevalence of cognitive and physical symptoms during long-term antidepressant treatment. J Clin Psychiatry 67(11):1754–1759
Felice D, O’Leary OF, Pizzo RC, Cryan JF (2012) Blockade of the GABA(B) receptor increases neurogenesis in the ventral but not dorsal adult hippocampus: relevance to antidepressant action. Neuropharmacology 63(8):1380–1388. doi:10.1016/j.neuropharm.2012.06.066
Finegan KG, Wang X, Lee EJ, Robinson AC, Tournier C (2009) Regulation of neuronal survival by the extracellular signal-regulated protein kinase 5. Cell Death Differ 16(5):674–683. doi:10.1038/cdd.2008.193
Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F (2010) Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PLoS One 5(12):e14382. doi:10.1371/journal.pone.0014382
Fordyce DE, Farrar RP (1991) Enhancement of spatial learning in F344 rats by physical activity and related learning-associated alterations in hippocampal and cortical cholinergic functioning. Behav Brain Res 46(2):123–133
Friedman SD (2008) Comment on “Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321(5889):640. doi:10.1126/science.1153484, author reply 640
Frodl T, Meisenzahl EM, Zetzsche T, Born C, Groll C, Jager M, Leinsinger G, Bottlender R, Hahn K, Moller HJ (2002) Hippocampal changes in patients with a first episode of major depression. Am J Psychiatry 159(7):1112–1118
Gadadhar A, Marr R, Lazarov O (2011) Presenilin-1 regulates neural progenitor cell differentiation in the adult brain. J Neurosci 31(7):2615–2623. doi:10.1523/JNEUROSCI.4767-10.2011
Gage FH, Dunnett SB, Bjorklund A (1984) Spatial learning and motor deficits in aged rats. Neurobiol Aging 5(1):43–48
Galvao RP, Garcia-Verdugo JM, Alvarez-Buylla A (2008) Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J Neurosci 28(50):13368–13383. doi:10.1523/JNEUROSCI.2918-08.2008
Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, Song H (2006) GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439(7076):589–593. doi:10.1038/nature04404
Ge S, Yang CH, Hsu KS, Ming GL, Song H (2007) A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 54(4):559–566. doi:10.1016/j.neuron.2007.05.002
Ge S, Sailor KA, Ming GL, Song H (2008) Synaptic integration and plasticity of new neurons in the adult hippocampus. J Physiol 586(16):3759–3765. doi:10.1113/jphysiol.2008.155655
Giachino C, Barz M, Tchorz JS, Tome M, Gassmann M, Bischofberger J, Bettler B, Taylor V (2014) GABA suppresses neurogenesis in the adult hippocampus through GABAB receptors. Development 141(1):83–90. doi:10.1242/dev.102608
Goeldner C, Ballard TM, Knoflach F, Wichmann J, Gatti S, Umbricht D (2013) Cognitive impairment in major depression and the mGlu2 receptor as a therapeutic target. Neuropharmacology 64:337–346. doi:10.1016/j.neuropharm.2012.08.001
Gomez-Isla T, Price JL, McKeel DW Jr, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer’s disease. J Neurosci 16(14):4491–4500
Gomez-Pinilla F, Dao L, So V (1997) Physical exercise induces FGF-2 and its mRNA in the hippocampus. Brain Res 764(1–2):1–8
Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E (1997) Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci 17(7):2492–2498
Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ (1999) Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2(3):260–265. doi:10.1038/6365
Gourley SL, Taylor JR (2009) Recapitulation and reversal of a persistent depression-like syndrome in rodents. Curr Protoc Neurosci Chapter 9:Unit 9.32. doi:10.1002/0471142301.ns0932s49. Editorial board, Jacqueline N Crawley (et al.)
Gozzi A, Herdon H, Schwarz A, Bertani S, Crestan V, Turrini G, Bifone A (2008) Pharmacological stimulation of NMDA receptors via co-agonist site suppresses fMRI response to phencyclidine in the rat. Psychopharmacology (Berl) 201(2):273–284. doi:10.1007/s00213-008-1271-z
Guiney H, Machado L (2013) Benefits of regular aerobic exercise for executive functioning in healthy populations. Psychon Bull Rev 20(1):73–86. doi:10.3758/s13423-012-0345-4
Guo W, Allan AM, Zong R, Zhang L, Johnson EB, Schaller EG, Murthy AC, Goggin SL, Eisch AJ, Oostra BA, Nelson DL, Jin P, Zhao X (2011) Ablation of Fmrp in adult neural stem cells disrupts hippocampus-dependent learning. Nat Med 17(5):559–565. doi:10.1038/nm.2336
Guo W, Murthy AC, Zhang L et al (2012) Inhibition of GSK3β improves hippocampus-dependent learning and rescues neurogenesis in a mouse model of fragile X syndrome. Hum Mol Genet 21(3):681–691
Gupta D, Morley JE (2014) Hypothalamic-pituitary-adrenal (HPA) axis and aging. Compr Physiol 4(4):1495–1510. doi:10.1002/cphy.c130049
Halim ND, Weickert CS, McClintock BW, Weinberger DR, Lipska BK (2004) Effects of chronic haloperidol and clozapine treatment on neurogenesis in the adult rat hippocampus. Neuropsychopharmacology 29(6):1063–1069. doi:10.1038/sj.npp.1300422
Han X, Tong J, Zhang J, Farahvar A, Wang E, Yang J, Samadani U, Smith DH, Huang JH (2011) Imipramine treatment improves cognitive outcome associated with enhanced hippocampal neurogenesis after traumatic brain injury in mice. J Neurotrauma 28(6):995–1007. doi:10.1089/neu.2010.1563, PMCID: PMC3113418
Harrist A, Beech RD, King SL, Zanardi A, Cleary MA, Caldarone BJ, Eisch A, Zoli M, Picciotto MR (2004) Alteration of hippocampal cell proliferation in mice lacking the beta 2 subunit of the neuronal nicotinic acetylcholine receptor. Synapse 54(4):200–206. doi:10.1002/syn.20081
Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, Saftig P (2002) The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet 11(21):2615–2624
Hasselbalch BJ, Knorr U, Kessing LV (2011) Cognitive impairment in the remitted state of unipolar depressive disorder: a systematic review. J Affect Disord 134(1–3):20–31. doi:10.1016/j.jad.2010.11.011
Hastings NB, Gould E (1999) Rapid extension of axons into the CA3 region by adult-generated granule cells. J Comp Neurol 413(1):146–154
Hattiangady B, Shetty AK (2008) Aging does not alter the number or phenotype of putative stem/progenitor cells in the neurogenic region of the hippocampus. Neurobiol Aging 29(1):129–147. doi:10.1016/j.neurobiolaging.2006.09.015
Hattiangady B, Rao MS, Shetty GA, Shetty AK (2005) Brain-derived neurotrophic factor, phosphorylated cyclic AMP response element binding protein and neuropeptide Y decline as early as middle age in the dentate gyrus and CA1 and CA3 subfields of the hippocampus. Exp Neurol 195(2):353–371. doi:10.1016/j.expneurol.2005.05.014
Head D, Isom M (2010) Age effects on wayfinding and route learning skills. Behav Brain Res 209(1):49–58. doi:10.1016/j.bbr.2010.01.012
Herrera-Guzman I, Gudayol-Ferre E, Herrera-Guzman D, Guardia-Olmos J, Hinojosa-Calvo E, Herrera-Abarca JE (2009) Effects of selective serotonin reuptake and dual serotonergic-noradrenergic reuptake treatments on memory and mental processing speed in patients with major depressive disorder. J Psychiatr Res 43(9):855–863. doi:10.1016/j.jpsychires.2008.10.015
Herrera-Guzman I, Gudayol-Ferre E, Herrera-Abarca JE, Herrera-Guzman D, Montelongo-Pedraza P, Padros Blazquez F, Pero-Cebollero M, Guardia-Olmos J (2010) Major Depressive Disorder in recovery and neuropsychological functioning: effects of selective serotonin reuptake inhibitor and dual inhibitor depression treatments on residual cognitive deficits in patients with Major Depressive Disorder in recovery. J Affect Disord 123(1–3):341–350. doi:10.1016/j.jad.2009.10.009
Herting MM, Nagel BJ (2012) Aerobic fitness relates to learning on a virtual Morris Water Task and hippocampal volume in adolescents. Behav Brain Res 233(2):517–525. doi:10.1016/j.bbr.2012.05.012
Heyn P, Abreu BC, Ottenbacher KJ (2004) The effects of exercise training on elderly persons with cognitive impairment and dementia: a meta-analysis. Arch Phys Med Rehabil 85(10):1694–1704
Hillman CH, Erickson KI, Kramer AF (2008) Be smart, exercise your heart: exercise effects on brain and cognition. Nat Rev Neurosci 9(1):58–65. doi:10.1038/nrn2298
Hoch JC, Maciejewski MW, Gryk MR (2008) Comment on “Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321(5889):640. doi:10.1126/science.1153058, author reply 640
Hoglinger GU, Rizk P, Muriel MP, Duyckaerts C, Oertel WH, Caille I, Hirsch EC (2004) Dopamine depletion impairs precursor cell proliferation in Parkinson disease. Nat Neurosci 7(7):726–735. doi:10.1038/nn1265
Hou JG, Xue JJ, Lee MR, Sun MQ, Zhao XH, Zheng YN, Sung CK (2013) Compound K is able to ameliorate the impaired cognitive function and hippocampal neurogenesis following chemotherapy treatment. Biochem Biophys Res Commun 436(1):104–109. doi:10.1016/j.bbrc.2013.05.087
Iaria G, Palermo L, Committeri G, Barton JJ (2009) Age differences in the formation and use of cognitive maps. Behav Brain Res 196(2):187–191. doi:10.1016/j.bbr.2008.08.040
Irvine GI, Logan B, Eckert M, Abraham WC (2006) Enriched environment exposure regulates excitability, synaptic transmission, and LTP in the dentate gyrus of freely moving rats. Hippocampus 16(2):149–160. doi:10.1002/hipo.20142
Itou Y, Nochi R, Kuribayashi H, Saito Y, Hisatsune T (2011) Cholinergic activation of hippocampal neural stem cells in aged dentate gyrus. Hippocampus 21(4):446–459. doi:10.1002/hipo.20761
Jaeger J, Berns S, Uzelac S, Davis-Conway S (2006) Neurocognitive deficits and disability in major depressive disorder. Psychiatry Res 145(1):39–48. doi:10.1016/j.psychres.2005.11.011
Jagasia R, Steib K, Englberger E, Herold S, Faus-Kessler T, Saxe M, Gage FH, Song H, Lie DC (2009) GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci 29(25):7966–7977. doi:10.1523/JNEUROSCI.1054-09.2009
Jang MH, Bonaguidi MA, Kitabatake Y, Sun J, Song J, Kang E, Jun H, Zhong C, Su Y, Guo JU, Wang MX, Sailor KA, Kim JY, Gao Y, Christian KM, Ming GL, Song H (2013a) Secreted frizzled-related protein 3 regulates activity-dependent adult hippocampal neurogenesis. Cell Stem Cell 12(2):215–223. doi:10.1016/j.stem.2012.11.021
Jang MH, Kitabatake Y, Kang E, Jun H, Pletnikov MV, Christian KM, Hen R, Lucae S, Binder EB, Song H, Ming GI (2013b) Secreted frizzled-related protein 3 (sFRP3) regulates antidepressant responses in mice and humans. Mol Psychiatry 18(9):957–958. doi:10.1038/mp.2012.158
Jansen JF, Gearhart JD, Bulte JW (2008) Comment on “Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain”. Science 321(5889):640. doi:10.1126/science.1153997, author reply 640
Jedynak P, Kos T, Sandi C, Kaczmarek L, Filipkowski RK (2014) Mice with ablated adult brain neurogenesis are not impaired in antidepressant response to chronic fluoxetine. J Psychiatr Res 56:106–111. doi:10.1016/j.jpsychires.2014.05.009
Jessberger S, Clark RE, Broadbent NJ, Clemenson GD Jr, Consiglio A, Lie DC, Squire LR, Gage FH (2009) Dentate gyrus-specific knockdown of adult neurogenesis impairs spatial and object recognition memory in adult rats. Learn Mem 16(2):147–154. doi:10.1101/lm.1172609
Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA (2004) Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A 101(1):343–347. doi:10.1073/pnas.2634794100
Jinno S (2011) Decline in adult neurogenesis during aging follows a topographic pattern in the mouse hippocampus. J Comp Neurol 519(3):451–466. doi:10.1002/cne.22527
Kalani MY, Cheshier SH, Cord BJ, Bababeygy SR, Vogel H, Weissman IL, Palmer TD, Nusse R (2008) Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A 105(44):16970–16975. doi:10.1073/pnas.0808616105
Kaneko N, Okano H, Sawamoto K (2006) Role of the cholinergic system in regulating survival of newborn neurons in the adult mouse dentate gyrus and olfactory bulb. Genes Cells 11(10):1145–1159. doi:10.1111/j.1365-2443.2006.01010.x
Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, Licznerski P, Lepack A, Majik MS, Jeong LS, Banasr M, Son H, Duman RS (2012) Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med 18(9):1413–1417. doi:10.1038/nm.2886
Kaplan MS, Hinds JW (1977) Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 197(4308):1092–1094
Karten YJ, Jones MA, Jeurling SI, Cameron HA (2006) GABAergic signaling in young granule cells in the adult rat and mouse dentate gyrus. Hippocampus 16(3):312–320. doi:10.1002/hipo.20165
Katsimpardi L, Litterman NK, Schein PA, Miller CM, Loffredo FS, Wojtkiewicz GR, Chen JW, Lee RT, Wagers AJ, Rubin LL (2014) Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344(6184):630–634. doi:10.1126/science.1251141
Kee N, Teixeira CM, Wang AH, Frankland PW (2007) Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat Neurosci 10(3):355–362. doi:10.1038/nn1847
Keilhoff G, Grecksch G, Bernstein HG, Roskoden T, Becker A (2010) Risperidone and haloperidol promote survival of stem cells in the rat hippocampus. Eur Arch Psychiatry Clin Neurosci 260(2):151–162. doi:10.1007/s00406-009-0033-1
Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386(6624):493–495. doi:10.1038/386493a0
Kesner RP (2013) An analysis of the dentate gyrus function. Behav Brain Res 254:1–7. doi:10.1016/j.bbr.2013.01.012
Kessler RM, Votaw JR, de Paulis T, Bingham DR, Ansari MS, Mason NS, Holburn G, Schmidt DE, Votaw DB, Manning RG et al (1993) Evaluation of 5-[18 F]fluoropropylepidepride as a potential PET radioligand for imaging dopamine D2 receptors. Synapse 15(3):169–176. doi:10.1002/syn.890150302
Kheirbek MA, Drew LJ, Burghardt NS, Costantini DO, Tannenholz L, Ahmari SE, Zeng H, Fenton AA, Hen R (2013) Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron 77(5):955–968. doi:10.1016/j.neuron.2012.12.038
Kim JY, Duan X, Liu CY, Jang MH, Guo JU, Pow-anpongkul N, Kang E, Song H, Ming GL (2009) DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron 63(6):761–773. doi:10.1016/j.neuron.2009.08.008
Kim JY, Liu CY, Zhang F, Duan X, Wen Z, Song J, Feighery E, Lu B, Rujescu D, St Clair D, Christian K, Callicott JH, Weinberger DR, Song H, Ming GL (2012) Interplay between DISC1 and GABA signaling regulates neurogenesis in mice and risk for schizophrenia. Cell 148(5):1051–1064. doi:10.1016/j.cell.2011.12.037
Kitamura T, Mishina M, Sugiyama H (2003) Enhancement of neurogenesis by running wheel exercises is suppressed in mice lacking NMDA receptor epsilon 1 subunit. Neurosci Res 47(1):55–63
Klempin F, Babu H, De Pietri Tonelli D, Alarcon E, Fabel K, Kempermann G (2010) Oppositional effects of serotonin receptors 5-HT1a, 2, and 2c in the regulation of adult hippocampal neurogenesis. Front Mol Neurosci 3:14. doi:10.3389/fnmol.2010.00014
Knolhoff AM, Nautiyal KM, Nemes P, Kalachikov S, Morozova I, Silver R, Sweedler JV (2013) Combining small-volume metabolomic and transcriptomic approaches for assessing brain chemistry. Anal Chem 85(6):3136–3143. doi:10.1021/ac3032959
Knoth R, Singec I, Ditter M, Pantazis G, Capetian P, Meyer RP, Horvat V, Volk B, Kempermann G (2010) Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS One 5(1):e8809. doi:10.1371/journal.pone.0008809
Kotani S, Yamauchi T, Teramoto T, Ogura H (2006) Pharmacological evidence of cholinergic involvement in adult hippocampal neurogenesis in rats. Neuroscience 142(2):505–514. doi:10.1016/j.neuroscience.2006.06.035
Kramer AF, Hahn S, Cohen NJ, Banich MT, McAuley E, Harrison CR, Chason J, Vakil E, Bardell L, Boileau RA, Colcombe A (1999) Ageing, fitness and neurocognitive function. Nature 400(6743):418–419. doi:10.1038/22682
Kremer T, Jagasia R, Herrmann A, Matile H, Borroni E, Francis F, Kuhn HG, Czech C (2013) Analysis of adult neurogenesis: evidence for a prominent “non-neurogenic” DCX-protein pool in rodent brain. PLoS One 8(5):e59269. doi:10.1371/journal.pone.0059269
Krezymon A, Richetin K, Halley H, Roybon L, Lassalle JM, Frances B, Verret L, Rampon C (2013) Modifications of hippocampal circuits and early disruption of adult neurogenesis in the tg2576 mouse model of Alzheimer’s disease. PLoS One 8(9):e76497. doi:10.1371/journal.pone.0076497
Kriegstein A, Alvarez-Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32:149–184. doi:10.1146/annurev.neuro.051508.135600
Kronenberg G, Reuter K, Steiner B, Brandt MD, Jessberger S, Yamaguchi M, Kempermann G (2003) Subpopulations of proliferating cells of the adult hippocampus respond differently to physiologic neurogenic stimuli. J Comp Neurol 467(4):455–463. doi:10.1002/cne.10945
Kronenberg G, Bick-Sander A, Bunk E, Wolf C, Ehninger D, Kempermann G (2006) Physical exercise prevents age-related decline in precursor cell activity in the mouse dentate gyrus. Neurobiol Aging 27(10):1505–1513. doi:10.1016/j.neurobiolaging.2005.09.016
Kuhn HG, Dickinson-Anson H, Gage FH (1996) Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 16(6):2027–2033
Kuny S, Stassen HH (1995) Cognitive performance in patients recovering from depression. Psychopathology 28(4):190–207
Laske C, Stellos K, Stransky E, Seizer P, Akcay O, Eschweiler GW, Leyhe T, Gawaz M (2008) Decreased plasma and cerebrospinal fluid levels of stem cell factor in patients with early Alzheimer’s disease. J Alzheimers Dis 15(3):451–460
Lassalle JM, Bataille T, Halley H (2000) Reversible inactivation of the hippocampal mossy fiber synapses in mice impairs spatial learning, but neither consolidation nor memory retrieval, in the Morris navigation task. Neurobiol Learn Mem 73(3):243–257. doi:10.1006/nlme.1999.3931
LaVoie MJ, Selkoe DJ (2003) The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem 278(36):34427–34437. doi:10.1074/jbc.M302659200
Lazarov O, Demars MP (2012) All in the family: how the APPs regulate neurogenesis. Front Neurosci 6:81. doi:10.3389/fnins.2012.00081
Lazarov O, Marr RA (2010) Neurogenesis and Alzheimer’s disease: at the crossroads. Exp Neurol 223(2):267–281. doi:10.1016/j.expneurol.2009.08.009
Lazarov O, Marr RA (2013) Of mice and men: neurogenesis, cognition and Alzheimer’s disease. Front Aging Neurosci 5:43. doi:10.3389/fnagi.2013.00043
Lazarov O, Robinson J, Tang YP, Hairston IS, Korade-Mirnics Z, Lee VM, Hersh LB, Sapolsky RM, Mirnics K, Sisodia SS (2005) Environmental enrichment reduces Abeta levels and amyloid deposition in transgenic mice. Cell 120(5):701–713. doi:10.1016/j.cell.2005.01.015
Leal SL, Tighe SK, Jones CK, Yassa MA (2014) Pattern separation of emotional information in hippocampal dentate and CA3. Hippocampus. doi:10.1002/hipo.22298
Lee I, Kesner RP (2004) Encoding versus retrieval of spatial memory: double dissociation between the dentate gyrus and the perforant path inputs into CA3 in the dorsal hippocampus. Hippocampus 14(1):66–76. doi:10.1002/hipo.10167
Lee KJ, Park SB, Lee I (2014) Elemental or contextual? It depends: individual difference in the hippocampal dependence of associative learning for a simple sensory stimulus. Front Behav Neurosci 8:217. doi:10.3389/fnbeh.2014.00217
Lemaire V, Tronel S, Montaron MF, Fabre A, Dugast E, Abrous DN (2012) Long-lasting plasticity of hippocampal adult-born neurons. J Neurosci 32(9):3101–3108. doi:10.1523/JNEUROSCI.4731-11.2012
Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, Parada LF (2008) TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59(3):399–412. doi:10.1016/j.neuron.2008.06.023
Li G, Bien-Ly N, Andrews-Zwilling Y, Xu Q, Bernardo A, Ring K, Halabisky B, Deng C, Mahley RW, Huang Y (2009) GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5(6):634–645. doi:10.1016/j.stem.2009.10.015
Li YJ, Xu M, Gao ZH, Wang YQ, Yue Z, Zhang YX, Li XX, Zhang C, Xie SY, Wang PY (2013) Alterations of serum levels of BDNF-related miRNAs in patients with depression. PLoS One 8(5):e63648. doi:10.1371/journal.pone.0063648
Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH (2005) Wnt signalling regulates adult hippocampal neurogenesis. Nature 437(7063):1370–1375. doi:10.1038/nature04108
Lim DA, Suarez-Farinas M, Naef F, Hacker CR, Menn B, Takebayashi H, Magnasco M, Patil N, Alvarez-Buylla A (2006) In vivo transcriptional profile analysis reveals RNA splicing and chromatin remodeling as prominent processes for adult neurogenesis. Mol Cell Neurosci 31(1):131–148. doi:10.1016/j.mcn.2005.10.005
Littlewood CL, Jones N, O’Neill MJ, Mitchell SN, Tricklebank M, Williams SC (2006) Mapping the central effects of ketamine in the rat using pharmacological MRI. Psychopharmacology (Berl) 186(1):64–81. doi:10.1007/s00213-006-0344-0
Liu Y, Lacson R, Cassaday J, Ross DA, Kreamer A, Hudak E, Peltier R, McLaren D, Munoz-Sanjuan I, Santini F, Strulovici B, Ferrer M (2009) Identification of small-molecule modulators of mouse SVZ progenitor cell proliferation and differentiation through high-throughput screening. J Biomol Screen 14(4):319–329. doi:10.1177/1087057109332596
Lois C, Alvarez-Buylla A (1994) Long-distance neuronal migration in the adult mammalian brain. Science 264(5162):1145–1148
Louissaint A Jr, Rao S, Leventhal C, Goldman SA (2002) Coordinated interaction of neurogenesis and angiogenesis in the adult songbird brain. Neuron 34(6):945–960
Lucassen PJ, Stumpel MW, Wang Q, Aronica E (2010) Decreased numbers of progenitor cells but no response to antidepressant drugs in the hippocampus of elderly depressed patients. Neuropharmacology 58(6):940–949. doi:10.1016/j.neuropharm.2010.01.012
Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Gotz M, Haas CA, Kempermann G, Taylor V, Giachino C (2010) Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 6(5):445–456. doi:10.1016/j.stem.2010.03.017
Ma W, Maric D, Li BS, Hu Q, Andreadis JD, Grant GM, Liu QY, Shaffer KM, Chang YH, Zhang L, Pancrazio JJ, Pant HC, Stenger DA, Barker JL (2000) Acetylcholine stimulates cortical precursor cell proliferation in vitro via muscarinic receptor activation and MAP kinase phosphorylation. Eur J Neurosci 12(4):1227–1240
Ma DK, Kim WR, Ming GL, Song H (2009) Activity-dependent extrinsic regulation of adult olfactory bulb and hippocampal neurogenesis. Ann N Y Acad Sci 1170:664–673. doi:10.1111/j.1749-6632.2009.04373.x
MacMaster FP, Carrey N, Langevin LM, Jaworska N, Crawford S (2014) Disorder-specific volumetric brain difference in adolescent major depressive disorder and bipolar depression. Brain Imaging Behav 8(1):119–127. doi:10.1007/s11682-013-9264-x
MacMillan KS, Naidoo J, Liang J, Melito L, Williams NS, Morlock L, Huntington PJ, Estill SJ, Longgood J, Becker GL, McKnight SL, Pieper AA, De Brabander JK, Ready JM (2011) Development of proneurogenic, neuroprotective small molecules. J Am Chem Soc 133(5):1428–1437. doi:10.1021/ja108211m
Maguire EA, Gadian DG, Johnsrude IS, Good CD, Ashburner J, Frackowiak RS, Frith CD (2000) Navigation-related structural change in the hippocampi of taxi drivers. Proc Natl Acad Sci U S A 97(8):4398–4403. doi:10.1073/pnas.070039597
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20(24):9104–9110
Maletic-Savatic M, Vingara LK, Manganas LN, Li Y, Zhang S, Sierra A, Hazel R, Smith D, Wagshul ME, Henn F, Krupp L, Enikolopov G, Benveniste H, Djuric PM, Pelczer I (2008) Metabolomics of neural progenitor cells: a novel approach to biomarker discovery. Cold Spring Harb Symp Quant Biol 73:389–401. doi:10.1101/sqb.2008.73.021
Mallei A, Shi B, Mocchetti I (2002) Antidepressant treatments induce the expression of basic fibroblast growth factor in cortical and hippocampal neurons. Mol Pharmacol 61(5):1017–1024
Manganas LN, Zhang X, Li Y, Hazel RD, Smith SD, Wagshul ME, Henn F, Benveniste H, Djuric PM, Enikolopov G, Maletic-Savatic M (2007) Magnetic resonance spectroscopy identifies neural progenitor cells in the live human brain. Science 318(5852):980–985. doi:10.1126/science.1147851
Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK, Tassa C, Berry EM, Soda T, Singh KK, Biechele T, Petryshen TL, Moon RT, Haggarty SJ, Tsai LH (2009) Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 136(6):1017–1031. doi:10.1016/j.cell.2008.12.044
Markwardt S, Overstreet-Wadiche L (2008) GABAergic signalling to adult-generated neurons. J Physiol 586(16):3745–3749. doi:10.1113/jphysiol.2008.155713
Martínez-Cué C, Martínez P, Rueda N, Vidal R, García S, Vidal V, Corrales A, Montero JA, Pazos Á, Flórez J, Gasser R, Thomas AW, Honer M, Knoflach F, Trejo JL, Wettstein JG, Hernández MC (2013) Reducing GABAA α5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome. J Neurosci 33(9):3953–3966
McCall WV, Dunn AG (2003) Cognitive deficits are associated with functional impairment in severely depressed patients. Psychiatry Res 121(2):179–184
McDonald HY, Wojtowicz JM (2005) Dynamics of neurogenesis in the dentate gyrus of adult rats. Neurosci Lett 385(1):70–75. doi:10.1016/j.neulet.2005.05.022
McEwen BS (1998) Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci 840:33–44
Mendez-David I, David DJ, Darcet F, Wu MV, Kerdine-Romer S, Gardier AM, Hen R (2014) Rapid anxiolytic effects of a 5-HT(4) receptor agonist are mediated by a neurogenesis-independent mechanism. Neuropsychopharmacology 39(6):1366–1378. doi:10.1038/npp.2013.332
Merrill DA, Karim R, Darraq M, Chiba AA, Tuszynski MH (2003) Hippocampal cell genesis does not correlate with spatial learning ability in aged rats. J Comp Neurol 459(2):201–207. doi:10.1002/cne.10616
Meshi D, Drew MR, Saxe M, Ansorge MS, David D, Santarelli L, Malapani C, Moore H, Hen R (2006) Hippocampal neurogenesis is not required for behavioral effects of environmental enrichment. Nat Neurosci 9(6):729–731. doi:10.1038/nn1696
Miller JA, Nathanson J, Franjic D, Shim S, Dalley RA, Shapouri S, Smith KA, Sunkin SM, Bernard A, Bennett JL, Lee CK, Hawrylycz MJ, Jones AR, Amaral DG, Sestan N, Gage FH, Lein ES (2013) Conserved molecular signatures of neurogenesis in the hippocampal subgranular zone of rodents and primates. Development 140(22):4633–4644. doi:10.1242/dev.097212
Miranda CJ, Braun L, Jiang Y, Hester ME, Zhang L, Riolo M, Wang H, Rao M, Altura RA, Kaspar BK (2012) Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell 11(3):542–552. doi:10.1111/j.1474-9726.2012.00816.x
Moffat SD, Elkins W, Resnick SM (2006) Age differences in the neural systems supporting human allocentric spatial navigation. Neurobiol Aging 27(7):965–972. doi:10.1016/j.neurobiolaging.2005.05.011
Mohapel P, Leanza G, Kokaia M, Lindvall O (2005) Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiol Aging 26(6):939–946. doi:10.1016/j.neurobiolaging.2004.07.015
Molteni R, Ying Z, Gomez-Pinilla F (2002) Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur J Neurosci 16(6):1107–1116
Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ (2004) Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A 101(29):10827–10832. doi:10.1073/pnas.0402141101
Monti JM, Hillman CH, Cohen NJ (2012) Aerobic fitness enhances relational memory in preadolescent children: the FITKids randomized control trial. Hippocampus 22(9):1876–1882. doi:10.1002/hipo.22023
Moore RD, Drollette ES, Scudder MR, Bharij A, Hillman CH (2014) The influence of cardiorespiratory fitness on strategic, behavioral, and electrophysiological indices of arithmetic cognition in preadolescent children. Front Hum Neurosci 8:258. doi:10.3389/fnhum.2014.00258
Morris AM, Weeden CS, Churchwell JC, Kesner RP (2013) The role of the dentate gyrus in the formation of contextual representations. Hippocampus 23(2):162–168. doi:10.1002/hipo.22078
Mu Y, Gage FH (2011) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6:85. doi:10.1186/1750-1326-6-85
Nacher J, McEwen BS (2006) The role of N-methyl-D-asparate receptors in neurogenesis. Hippocampus 16(3):267–270. doi:10.1002/hipo.20160
Nacher J, Rosell DR, Alonso-Llosa G, McEwen BS (2001) NMDA receptor antagonist treatment induces a long-lasting increase in the number of proliferating cells, PSA-NCAM-immunoreactive granule neurons and radial glia in the adult rat dentate gyrus. Eur J Neurosci 13(3):512–520
Neeper SA, Gomez-Pinilla F, Choi J, Cotman CW (1996) Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res 726(1–2):49–56
Neu P, Bajbouj M, Schilling A, Godemann F, Berman RM, Schlattmann P (2005) Cognitive function over the treatment course of depression in middle-aged patients: correlation with brain MRI signal hyperintensities. J Psychiatr Res 39(2):129–135. doi:10.1016/j.jpsychires.2004.06.004
Nichols NR, Zieba M, Bye N (2001) Do glucocorticoids contribute to brain aging? Brain Res Brain Res Rev 37(1–3):273–286
O’Callaghan RM, Ohle R, Kelly AM (2007) The effects of forced exercise on hippocampal plasticity in the rat: a comparison of LTP, spatial- and non-spatial learning. Behav Brain Res 176(2):362–366. doi:10.1016/j.bbr.2006.10.018
Olariu A, Cleaver KM, Cameron HA (2007) Decreased neurogenesis in aged rats results from loss of granule cell precursors without lengthening of the cell cycle. J Comp Neurol 501(4):659–667. doi:10.1002/cne.21268
Pallotto M, Deprez F (2014) Regulation of adult neurogenesis by GABAergic transmission: signaling beyond GABAA-receptors. Front Cell Neurosci 8:166. doi:10.3389/fncel.2014.00166
Palmer TD, Willhoite AR, Gage FH (2000) Vascular niche for adult hippocampal neurogenesis. J Comp Neurol 425(4):479–494
Pan YW, Chan GC, Kuo CT, Storm DR, Xia Z (2012a) Inhibition of adult neurogenesis by inducible and targeted deletion of ERK5 mitogen-activated protein kinase specifically in adult neurogenic regions impairs contextual fear extinction and remote fear memory. J Neurosci 32(19):6444–6455. doi:10.1523/JNEUROSCI.6076-11.2012
Pan YW, Zou J, Wang W, Sakagami H, Garelick MG, Abel G, Kuo CT, Storm DR, Xia Z (2012b) Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. J Biol Chem 287(28):23306–23317. doi:10.1074/jbc.M112.344762
Pandey GN, Ren X, Rizavi HS, Conley RR, Roberts RC, Dwivedi Y (2008) Brain-derived neurotrophic factor and tyrosine kinase B receptor signalling in post-mortem brain of teenage suicide victims. Int J Neuropsychopharmacol 11(8):1047–1061. doi:10.1017/S1461145708009000
Pathania M, Yan LD, Bordey A (2010) A symphony of signals conducts early and late stages of adult neurogenesis. Neuropharmacology 58(6):865–876. doi:10.1016/j.neuropharm.2010.01.010
Pereira AC, Huddleston DE, Brickman AM, Sosunov AA, Hen R, McKhann GM, Sloan R, Gage FH, Brown TR, Small SA (2007) An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A 104(13):5638–5643. doi:10.1073/pnas.0611721104
Perry EK, Johnson M, Ekonomou A, Perry RH, Ballard C, Attems J (2012) Neurogenic abnormalities in Alzheimer’s disease differ between stages of neurogenesis and are partly related to cholinergic pathology. Neurobiol Dis 47(2):155–162. doi:10.1016/j.nbd.2012.03.033
Petrik D, Jiang Y, Birnbaum SG, Powell CM, Kim MS, Hsieh J, Eisch AJ (2012) Functional and mechanistic exploration of an adult neurogenesis-promoting small molecule. FASEB J 26(8):3148–3162. doi:10.1096/fj.11-201426
Pieper AA, Xie S, Capota E, Estill SJ, Zhong J, Long JM, Becker GL, Huntington P, Goldman SE, Shen CH, Capota M, Britt JK, Kotti T, Ure K, Brat DJ, Williams NS, MacMillan KS, Naidoo J, Melito L, Hsieh J, De Brabander J, Ready JM, McKnight SL (2010) Discovery of a proneurogenic, neuroprotective chemical. Cell 142(1):39–51. doi:10.1016/j.cell.2010.06.018
Price RB, Nock MK, Charney DS, Mathew SJ (2009) Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry 66(5):522–526. doi:10.1016/j.biopsych.2009.04.029
Ramirez-Amaya V, Marrone DF, Gage FH, Worley PF, Barnes CA (2006) Integration of new neurons into functional neural networks. J Neurosci 26(47):12237–12241. doi:10.1523/JNEUROSCI.2195-06.2006
Ramm P, Couillard-Despres S, Plotz S, Rivera FJ, Krampert M, Lehner B, Kremer W, Bogdahn U, Kalbitzer HR, Aigner L (2009) A nuclear magnetic resonance biomarker for neural progenitor cells: is it all neurogenesis? Stem Cells 27(2):420–423. doi:10.1634/stemcells. 2008-0816
Rao MS, Hattiangady B, Abdel-Rahman A, Stanley DP, Shetty AK (2005) Newly born cells in the ageing dentate gyrus display normal migration, survival and neuronal fate choice but endure retarded early maturation. Eur J Neurosci 21(2):464–476. doi:10.1111/j.1460-9568.2005.03853.x
Rapp PR, Kansky MT, Roberts JA (1997) Impaired spatial information processing in aged monkeys with preserved recognition memory. Neuroreport 8(8):1923–1928
Rapp PR, Stack EC, Gallagher M (1999) Morphometric studies of the aged hippocampus: I. Volumetric analysis in behaviorally characterized rats. J Comp Neurol 403(4):459–470
Rasmusson DD, Dudar JD (1979) Effect of scopolamine on maze learning performance in humans. Experientia 35(8):1069–1070
Raz N, Ghisletta P, Rodrigue KM, Kennedy KM, Lindenberger U (2010) Trajectories of brain aging in middle-aged and older adults: regional and individual differences. Neuroimage 51(2):501–511. doi:10.1016/j.neuroimage.2010.03.020
Redila VA, Christie BR (2006) Exercise-induced changes in dendritic structure and complexity in the adult hippocampal dentate gyrus. Neuroscience 137(4):1299–1307. doi:10.1016/j.neuroscience.2005.10.050
Revest JM, Dupret D, Koehl M, Funk-Reiter C, Grosjean N, Piazza PV, Abrous DN (2009) Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol Psychiatry 14(10):959–967. doi:10.1038/mp.2009.15
Rishton GM (2008) Small molecules that promote neurogenesis in vitro. Recent Pat CNS Drug Discov 3(3):200–208
Rolls ET (1996) A theory of hippocampal function in memory. Hippocampus 6(6):601–620. doi:10.1002/(SICI)1098-1063(1996)6:6<601::AID-HIPO5>3.0.CO;2-J
Rolls ET (2007) An attractor network in the hippocampus: theory and neurophysiology. Learn Mem 14(11):714–731. doi:10.1101/lm.631207
Rossi C, Angelucci A, Costantin L, Braschi C, Mazzantini M, Babbini F, Fabbri ME, Tessarollo L, Maffei L, Berardi N, Caleo M (2006) Brain-derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur J Neurosci 24(7):1850–1856. doi:10.1111/j.1460-9568.2006.05059.x
Rubin DC (2000) The distribution of early childhood memories. Memory 8(4):265–269. doi:10.1080/096582100406810
Rudy JW, Sutherland RJ (1995) Configural association theory and the hippocampal formation: an appraisal and reconfiguration. Hippocampus 5(5):375–389. doi:10.1002/hipo.450050502
Rueger MA, Backes H, Walberer M, Neumaier B, Ullrich R, Simard ML, Emig B, Fink GR, Hoehn M, Graf R, Schroeter M (2010) Noninvasive imaging of endogenous neural stem cell mobilization in vivo using positron emission tomography. J Neurosci 30(18):6454–6460. doi:10.1523/JNEUROSCI.6092-09.2010
Saarelainen T, Hendolin P, Lucas G, Koponen E, Sairanen M, MacDonald E, Agerman K, Haapasalo A, Nawa H, Aloyz R, Ernfors P, Castren E (2003) Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci 23(1):349–357
Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, Hen R (2011) Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472(7344):466–470. doi:10.1038/nature09817
Sairanen M, Lucas G, Ernfors P, Castren M, Castren E (2005) Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J Neurosci 25(5):1089–1094. doi:10.1523/JNEUROSCI.3741-04.2005
Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, Weisstaub N, Lee J, Duman R, Arancio O, Belzung C, Hen R (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301(5634):805–809. doi:10.1126/science.1083328
Saxe MD, Battaglia F, Wang JW, Malleret G, David DJ, Monckton JE, Garcia AD, Sofroniew MV, Kandel ER, Santarelli L, Hen R, Drew MR (2006) Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc Natl Acad Sci U S A 103(46):17501–17506. doi:10.1073/pnas.0607207103
Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S (2005) Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol 192(2):348–356. doi:10.1016/j.expneurol.2004.11.016
Schatzberg AF, Keller J, Tennakoon L, Lembke A, Williams G, Kraemer FB, Sarginson JE, Lazzeroni LC, Murphy GM (2014) HPA axis genetic variation, cortisol and psychosis in major depression. Mol Psychiatry 19(2):220–227. doi:10.1038/mp.2013.129
Schlosser N, Wolf OT, Fernando SC, Riedesel K, Otte C, Muhtz C, Beblo T, Driessen M, Lowe B, Wingenfeld K (2010) Effects of acute cortisol administration on autobiographical memory in patients with major depression and healthy controls. Psychoneuroendocrinology 35(2):316–320. doi:10.1016/j.psyneuen.2009.06.015
Schneider JW, Gao Z, Li S, Farooqi M, Tang TS, Bezprozvanny I, Frantz DE, Hsieh J (2008) Small-molecule activation of neuronal cell fate. Nat Chem Biol 4(7):408–410. doi:10.1038/nchembio.95
Scoville WB, Milner B (1957) Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry 20(1):11–21
Seib DR, Corsini NS, Ellwanger K, Plaas C, Mateos A, Pitzer C, Niehrs C, Celikel T, Martin-Villalba A (2013) Loss of Dickkopf-1 restores neurogenesis in old age and counteracts cognitive decline. Cell Stem Cell 12(2):204–214. doi:10.1016/j.stem.2012.11.010
Seki T (2002) Hippocampal adult neurogenesis occurs in a microenvironment provided by PSA-NCAM-expressing immature neurons. J Neurosci Res 69(6):772–783. doi:10.1002/jnr.10366
Seki T, Arai Y (1995) Age-related production of new granule cells in the adult dentate gyrus. Neuroreport 6(18):2479–2482
Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81(2):741–766
Selkoe DJ, Wolfe MS (2007) Presenilin: running with scissors in the membrane. Cell 131(2):215–221. doi:10.1016/j.cell.2007.10.012
Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A (2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21(18):7153–7160
Shamy JL, Buonocore MH, Makaron LM, Amaral DG, Barnes CA, Rapp PR (2006) Hippocampal volume is preserved and fails to predict recognition memory impairment in aged rhesus monkeys (Macaca mulatta). Neurobiol Aging 27(10):1405–1415. doi:10.1016/j.neurobiolaging.2005.07.019
Sheline YI, Wang PW, Gado MH, Csernansky JG, Vannier MW (1996) Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci U S A 93(9):3908–3913
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S (1997) Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89(4):629–639
Shetty AK, Hattiangady B, Shetty GA (2005) Stem/progenitor cell proliferation factors FGF-2, IGF-1, and VEGF exhibit early decline during the course of aging in the hippocampus: role of astrocytes. Glia 51(3):173–186. doi:10.1002/glia.20187
Shruster A, Offen D (2014) Targeting neurogenesis ameliorates danger assessment in a mouse model of Alzheimer’s disease. Behav Brain Res 261:193–201. doi:10.1016/j.bbr.2013.12.028
Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M (2010) Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7(4):483–495. doi:10.1016/j.stem.2010.08.014
Siette J, Westbrook RF, Cotman C, Sidhu K, Zhu W, Sachdev P, Valenzuela MJ (2013) Age-specific effects of voluntary exercise on memory and the older brain. Biol Psychiatry 73(5):435–442. doi:10.1016/j.biopsych.2012.05.034
Snyder JS, Soumier A, Brewer M, Pickel J, Cameron HA (2011) Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 476(7361):458–461. doi:10.1038/nature10287
Song HJ, Stevens CF, Gage FH (2002) Neural stem cells from adult hippocampus develop essential properties of functional CNS neurons. Nat Neurosci 5(5):438–445. doi:10.1038/nn844
Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, Deisseroth K, Luscher B, Christian KM, Ming GL, Song H (2012) Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489(7414):150–154. doi:10.1038/nature11306
Sousa N, Lukoyanov NV, Madeira MD, Almeida OF, Paula-Barbosa MM (2000) Reorganization of the morphology of hippocampal neurites and synapses after stress-induced damage correlates with behavioral improvement. Neuroscience 97(2):253–266
Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Bostrom E, Westerlund I, Vial C, Buchholz BA, Possnert G, Mash DC, Druid H, Frisen J (2013) Dynamics of hippocampal neurogenesis in adult humans. Cell 153(6):1219–1227. doi:10.1016/j.cell.2013.05.002
Squire LR (1992) Declarative and nondeclarative memory: multiple brain systems supporting learning and memory. J Cogn Neurosci 4(3):232–243. doi:10.1162/jocn.1992.4.3.232
Stranahan AM, Khalil D, Gould E (2007) Running induces widespread structural alterations in the hippocampus and entorhinal cortex. Hippocampus 17(11):1017–1022. doi:10.1002/hipo.20348
Stroth S, Hille K, Spitzer M, Reinhardt R (2009) Aerobic endurance exercise benefits memory and affect in young adults. Neuropsychol Rehabil 19(2):223–243. doi:10.1080/09602010802091183
Sullivan EV, Marsh L, Pfefferbaum A (2005) Preservation of hippocampal volume throughout adulthood in healthy men and women. Neurobiol Aging 26(7):1093–1098. doi:10.1016/j.neurobiolaging.2004.09.015
Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, Hen R, Belzung C (2008) Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry 64(4):293–301. doi:10.1016/j.biopsych.2008.02.022
Surget A, Tanti A, Leonardo ED, Laugeray A, Rainer Q, Touma C, Palme R, Griebel G, Ibarguen-Vargas Y, Hen R, Belzung C (2011) Antidepressants recruit new neurons to improve stress response regulation. Mol Psychiatry 16(12):1177–1188. doi:10.1038/mp.2011.48
Takamura N, Nakagawa S, Masuda T, Boku S, Kato A, Song N, An Y, Kitaichi Y, Inoue T, Koyama T, Kusumi I (2014) The effect of dopamine on adult hippocampal neurogenesis. Prog Neuropsychopharmacol Biol Psychiatry 50:116–124. doi:10.1016/j.pnpbp.2013.12.011
Tanapat P, Hastings NB, Rydel TA, Galea LA, Gould E (2001) Exposure to fox odor inhibits cell proliferation in the hippocampus of adult rats via an adrenal hormone-dependent mechanism. J Comp Neurol 437(4):496–504
Tannenholz L, Jimenez JC, Kheirbek MA (2014) Local and regional heterogeneity underlying hippocampal modulation of cognition and mood. Front Behav Neurosci 8:147. doi:10.3389/fnbeh.2014.00147
Tanti A, Westphal WP, Girault V, Brizard B, Devers S, Leguisquet AM, Surget A, Belzung C (2013) Region-dependent and stage-specific effects of stress, environmental enrichment, and antidepressant treatment on hippocampal neurogenesis. Hippocampus 23(9):797–811. doi:10.1002/hipo.22134
Tashiro A, Makino H, Gage FH (2007) Experience-specific functional modification of the dentate gyrus through adult neurogenesis: a critical period during an immature stage. J Neurosci 27(12):3252–3259. doi:10.1523/JNEUROSCI.4941-06.2007
Themanson JR, Pontifex MB, Hillman CH (2008) Fitness and action monitoring: evidence for improved cognitive flexibility in young adults. Neuroscience 157(2):319–328. doi:10.1016/j.neuroscience.2008.09.014
Tizabi Y, Bhatti BH, Manaye KF, Das JR, Akinfiresoye L (2012) Antidepressant-like effects of low ketamine dose is associated with increased hippocampal AMPA/NMDA receptor density ratio in female Wistar-Kyoto rats. Neuroscience 213:72–80. doi:10.1016/j.neuroscience.2012.03.052
Toledo EM, Inestrosa NC (2010) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer’s disease. Mol Psychiatry 15(3):272–285, 228. doi:10.1038/mp.2009.72
Tong L, Shen H, Perreau VM, Balazs R, Cotman CW (2001) Effects of exercise on gene-expression profile in the rat hippocampus. Neurobiol Dis 8(6):1046–1056. doi:10.1006/nbdi.2001.0427
Toni N, Teng EM, Bushong EA, Aimone JB, Zhao C, Consiglio A, van Praag H, Martone ME, Ellisman MH, Gage FH (2007) Synapse formation on neurons born in the adult hippocampus. Nat Neurosci 10(6):727–734. doi:10.1038/nn1908
Tozuka Y, Fukuda S, Namba T, Seki T, Hisatsune T (2005) GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron 47(6):803–815. doi:10.1016/j.neuron.2005.08.023
Trejo JL, Carro E, Torres-Aleman I (2001) Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci 21(5):1628–1634
Treves A, Tashiro A, Witter MP, Moser EI (2008) What is the mammalian dentate gyrus good for? Neuroscience 154(4):1155–1172. doi:10.1016/j.neuroscience.2008.04.073
Trivedi MH, Greer TL (2014) Cognitive dysfunction in unipolar depression: implications for treatment. J Affect Disord 152–154:19–27. doi:10.1016/j.jad.2013.09.012
Valero J, Espana J, Parra-Damas A, Martin E, Rodriguez-Alvarez J, Saura CA (2011) Short-term environmental enrichment rescues adult neurogenesis and memory deficits in APP(Sw, Ind) transgenic mice. PLoS One 6(2):e16832. doi:10.1371/journal.pone.0016832
van der Borght K, Ferrari F, Klauke K, Roman V, Havekes R, Sgoifo A, van der Zee EA, Meerlo P (2006) Hippocampal cell proliferation across the day: increase by running wheel activity, but no effect of sleep and wakefulness. Behav Brain Res 167(1):36–41. doi:10.1016/j.bbr.2005.08.012
Van Hoesen GW, Hyman BT, Damasio AR (1991) Entorhinal cortex pathology in Alzheimer’s disease. Hippocampus 1(1):1–8. doi:10.1002/hipo.450010102
Van Petten C (2004) Relationship between hippocampal volume and memory ability in healthy individuals across the lifespan: review and meta-analysis. Neuropsychologia 42(10):1394–1413. doi:10.1016/j.neuropsychologia.2004.04.006
van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999a) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A 96(23):13427–13431
van Praag H, Kempermann G, Gage FH (1999b) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat Neurosci 2(3):266–270. doi:10.1038/6368
van Praag H, Shubert T, Zhao C, Gage FH (2005) Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci 25(38):8680–8685. doi:10.1523/JNEUROSCI.1731-05.2005
Varela-Nallar L, Inestrosa NC (2013) Wnt signaling in the regulation of adult hippocampal neurogenesis. Front Cell Neurosci 7:100. doi:10.3389/fncel.2013.00100
Veena J, Rao BS, Srikumar BN (2011a) Regulation of adult neurogenesis in the hippocampus by stress, acetylcholine and dopamine. J Nat Sci Biol Med 2(1):26–37. doi:10.4103/0976-9668.82312
Veena J, Srikumar BN, Mahati K, Raju TR, Shankaranarayana Rao BS (2011b) Oxotremorine treatment restores hippocampal neurogenesis and ameliorates depression-like behaviour in chronically stressed rats. Psychopharmacology (Berl) 217(2):239–253. doi:10.1007/s00213-011-2279-3
Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C (2007) Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J Neurosci 27(25):6771–6780. doi:10.1523/JNEUROSCI.5564-06.2007
Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Despres S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477(7362):90–94. doi:10.1038/nature10357
Villeda SA, Plambeck KE, Middeldorp J, Castellano JM, Mosher KI, Luo J, Smith LK, Bieri G, Lin K, Berdnik D, Wabl R, Udeochu J, Wheatley EG, Zou B, Simmons DA, Xie XS, Longo FM, Wyss-Coray T (2014) Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med 20(6):659–663. doi:10.1038/nm.3569
Vivar C, Potter MC, Choi J, Lee JY, Stringer TP, Callaway EM, Gage FH, Suh H, van Praag H (2012) Monosynaptic inputs to new neurons in the dentate gyrus. Nat Commun 3:1107. doi:10.1038/ncomms2101
Voss MW, Vivar C, Kramer AF, van Praag H (2013) Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn Sci 17(10):525–544. doi:10.1016/j.tics.2013.08.001
Wagner S, Doering B, Helmreich I, Lieb K, Tadic A (2012) A meta-analysis of executive dysfunctions in unipolar major depressive disorder without psychotic symptoms and their changes during antidepressant treatment. Acta Psychiatr Scand 125(4):281–292. doi:10.1111/j.1600-0447.2011.01762.x
Wakade CG, Mahadik SP, Waller JL, Chiu FC (2002) Atypical neuroleptics stimulate neurogenesis in adult rat brain. J Neurosci Res 69(1):72–79. doi:10.1002/jnr.10281
Walker AK, Rivera PD, Wang Q, Chuang JC, Tran S, Osborne-Lawrence S, Estill SJ, Starwalt R, Huntington P, Morlock L, Naidoo J, Williams NS, Ready JM, Eisch AJ, Pieper AA, Zigman JM (2014) The P7C3 class of neuroprotective compounds exerts antidepressant efficacy in mice by increasing hippocampal neurogenesis. Mol Psychiatry. doi:10.1038/mp.2014.34
Walter J, Keiner S, Witte OW, Redecker C (2011) Age-related effects on hippocampal precursor cell subpopulations and neurogenesis. Neurobiol Aging 32(10):1906–1914. doi:10.1016/j.neurobiolaging.2009.11.011
Wang HD, Dunnavant FD, Jarman T, Deutch AY (2004) Effects of antipsychotic drugs on neurogenesis in the forebrain of the adult rat. Neuropsychopharmacology 29(7):1230–1238. doi:10.1038/sj.npp.1300449
Wang J, Gallagher D, DeVito LM, Cancino GI, Tsui D, He L, Keller GM, Frankland PW, Kaplan DR, Miller FD (2012) Metformin activates an atypical PKC-CBP pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 11(1):23–35. doi:10.1016/j.stem.2012.03.016
Wang W, Pan YW, Zou J, Li T, Abel GM, Palmiter RD, Storm DR, Xia Z (2014) Genetic activation of ERK5 MAP kinase enhances adult neurogenesis and extends hippocampus-dependent long-term memory. J Neurosci 34(6):2130–2147. doi:10.1523/JNEUROSCI.3324-13.2014
Warner-Schmidt JL, Duman RS (2007) VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci U S A 104(11):4647–4652. doi:10.1073/pnas.0610282104
Watanabe Y, Gould E, McEwen BS (1992) Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res 588(2):341–345
Weiland-Fiedler P, Erickson K, Waldeck T, Luckenbaugh DA, Pike D, Bonne O, Charney DS, Neumeister A (2004) Evidence for continuing neuropsychological impairments in depression. J Affect Disord 82(2):253–258. doi:10.1016/j.jad.2003.10.009
Wesnes KA, Annas P, Basun H, Edgar C, Blennow K (2014) Performance on a pattern separation task by Alzheimer’s patients shows possible links between disrupted dentate gyrus activity and apolipoprotein [element of]4 status and cerebrospinal fluid amyloid-03B242 levels. Alzheimers Res Ther 6(2):20. doi:10.1186/alzrt250
West MJ, Coleman PD, Flood DG, Troncoso JC (1994) Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer’s disease. Lancet 344(8925):769–772
West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC (2004) Hippocampal neurons in pre-clinical Alzheimer’s disease. Neurobiol Aging 25(9):1205–1212. doi:10.1016/j.neurobiolaging.2003.12.005
Wigmore P (2013) The effect of systemic chemotherapy on neurogenesis, plasticity and memory. Curr Top Behav Neurosci 15:211–240. doi:10.1007/7854_2012_235
Wixted JT, Squire LR, Jang Y, Papesh MH, Goldinger SD, Kuhn JR, Smith KA, Treiman DM, Steinmetz PN (2014) Sparse and distributed coding of episodic memory in neurons of the human hippocampus. Proc Natl Acad Sci U S A 111(26):9621–9626. doi:10.1073/pnas.1408365111
Wolf SA, Kronenberg G, Lehmann K, Blankenship A, Overall R, Staufenbiel M, Kempermann G (2006) Cognitive and physical activity differently modulate disease progression in the amyloid precursor protein (APP)-23 model of Alzheimer’s disease. Biol Psychiatry 60(12):1314–1323. doi:10.1016/j.biopsych.2006.04.004
Wolf OT, Minnebusch D, Daum I (2009) Stress impairs acquisition of delay eyeblink conditioning in men and women. Neurobiol Learn Mem 91(4):431–436. doi:10.1016/j.nlm.2008.11.002
Yamasaki TR, Blurton-Jones M, Morrissette DA, Kitazawa M, Oddo S, LaFerla FM (2007) Neural stem cells improve memory in an inducible mouse model of neuronal loss. J Neurosci 27(44):11925–11933. doi:10.1523/JNEUROSCI.1627-07.2007
Yang P, Arnold SA, Habas A, Hetman M, Hagg T (2008) Ciliary neurotrophic factor mediates dopamine D2 receptor-induced CNS neurogenesis in adult mice. J Neurosci 28(9):2231–2241. doi:10.1523/JNEUROSCI.3574-07.2008
Yang C, Hu YM, Zhou ZQ, Zhang GF, Yang JJ (2013) Acute administration of ketamine in rats increases hippocampal BDNF and mTOR levels during forced swimming test. Ups J Med Sci 118(1):3–8. doi:10.3109/03009734.2012.724118
Yang X, Yang Q, Wang X, Luo C, Wan Y, Li J, Liu K, Zhou M, Zhang C (2014) MicroRNA expression profile and functional analysis reveal that miR-206 is a critical novel gene for the expression of BDNF induced by ketamine. Neuromolecular Med 16(3):594–605. doi:10.1007/s12017-014-8312-z
Yap JJ, Takase LF, Kochman LJ, Fornal CA, Miczek KA, Jacobs BL (2006) Repeated brief social defeat episodes in mice: effects on cell proliferation in the dentate gyrus. Behav Brain Res 172(2):344–350. doi:10.1016/j.bbr.2006.05.027
Yassa MA, Stark CE (2011) Pattern separation in the hippocampus. Trends Neurosci 34(10):515–525. doi:10.1016/j.tins.2011.06.006
Yassa MA, Lacy JW, Stark SM, Albert MS, Gallagher M, Stark CE (2011) Pattern separation deficits associated with increased hippocampal CA3 and dentate gyrus activity in nondemented older adults. Hippocampus 21(9):968–979. doi:10.1002/hipo.20808
Yoshimizu T, Chaki S (2004) Increased cell proliferation in the adult mouse hippocampus following chronic administration of group II metabotropic glutamate receptor antagonist, MGS0039. Biochem Biophys Res Commun 315(2):493–496. doi:10.1016/j.bbrc.2004.01.073
Young EA, Haskett RF, Murphy-Weinberg V, Watson SJ, Akil H (1991) Loss of glucocorticoid fast feedback in depression. Arch Gen Psychiatry 48(8):693–699
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, Manji HK (2006) A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63(8):856–864. doi:10.1001/archpsyc.63.8.856
Zhao C, Teng EM, Summers RG Jr, Ming GL, Gage FH (2006) Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci 26(1):3–11. doi:10.1523/JNEUROSCI.3648-05.2006
Zhao C, Jou J, Wolff LJ, Sun H, Gage FH (2014) Spine morphogenesis in newborn granule cells is differentially regulated in the outer and middle molecular layers. J Comp Neurol 522(12):2756–2766. doi:10.1002/cne.23581
Zhou M, Li W, Huang S, Song J, Kim JY, Tian X, Kang E, Sano Y, Liu C, Balaji J, Wu S, Zhou Y, Zhou Y, Parivash SN, Ehninger D, He L, Song H, Ming GL, Silva AJ (2013a) mTOR Inhibition ameliorates cognitive and affective deficits caused by Disc1 knockdown in adult-born dentate granule neurons. Neuron 77(4):647–654. doi:10.1016/j.neuron.2012.12.033
Zhou W, Wang N, Yang C, Li XM, Zhou ZQ, Yang JJ (2013b) Ketamine-induced antidepressant effects are associated with AMPA receptors-mediated upregulation of mTOR and BDNF in rat hippocampus and prefrontal cortex. Eur Psychiatry. doi:10.1016/j.eurpsy.2013.10.005
Zhu X, Wang X, Xiao J, Zhong M, Liao J, Yao S (2011) Altered white matter integrity in first-episode, treatment-naive young adults with major depressive disorder: a tract-based spatial statistics study. Brain Res 1369:223–229. doi:10.1016/j.brainres.2010.10.104
Acknowledgments
VC and SL are supported by the Roche Postdoctoral Fellowship Program. We thank M. Iglesias for artwork.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Costa, V., Lugert, S., Jagasia, R. (2015). Role of Adult Hippocampal Neurogenesis in Cognition in Physiology and Disease: Pharmacological Targets and Biomarkers. In: Kantak, K., Wettstein, J. (eds) Cognitive Enhancement. Handbook of Experimental Pharmacology, vol 228. Springer, Cham. https://doi.org/10.1007/978-3-319-16522-6_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-16522-6_4
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16521-9
Online ISBN: 978-3-319-16522-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)