
Abstract
The template carbonization method was utilized for the production of mesoporous carbon using attapulgite as a template and sucrose as a carbon precursor. Sucrose was polymerized and carbonized in the tubes of natural attapulgite using a sulfuric acid catalyst. The structure of the template and carbon was investigated by powder X-ray diffraction, transmission electron microscopy, and nitrogen adsorption analysis techniques. At the micrometer level, the resultant carbon material templated by natural attapulgite had a similar morphology. Nitrogen adsorption analysis showed that the obtained porous carbon possessed a wide pore-size distribution and a large pore volume, especially in the range of mesopores.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Porous carbon materials are widely used in both traditional areas such as gas separation, water purification, catalyst support, chromatography columns, and storage of natural gas and advanced areas such as electric double-layer capacitors due to the hydrophobic nature of their surfaces, high surface area, large pore volumes, chemical inertness, good mechanical stability, and good thermal stability. In recent years, much attention has been paid to mesopore-rich carbon materials because they can adsorb giant molecules such as dyes, vitamins, polymers, and biomolecules, which cannot be adsorbed by microporous carbons [1–3]. There are mainly three methods to prepare mesoporous carbons: carbonization and activation of carbon precursors, the sol–gel process, and the template method [4]. Compared with other methods, the template approach has been proved to be an efficient way to prepare mesoporous carbons with well-controlled pore sizes and mesopore volume fraction. A lot of studies proved that the highly ordered mesoporous carbon molecular sieves can be synthesized by using ordered mesoporous silica molecular sieves, like MCM and SBA series [5]. However, the high cost of artificial templates leads to the high cost of the resultant mesoporous carbons using this method; thus natural clay minerals are quite good template candidates due to their porous properties and especially low prices. Kyotani et al. [6–8] first synthesized wrinkled, thin carbon films using montmorillonite, taeniolite, and saponite as templates and acrylonitrile, furfuryl alcohol, and vinyl acetate as carbon precursors from the late 1980s. From then on, a lot of minerals have been studied as templates to prepare carbon materials, such as the lithium form of taeniolite [9, 10], Na+-montmorillonite [11], porous clay (montmorillonite) heterostructures containing iron, zinc, or copper [12], and Turkish natural zeolite [13]. However, all of these carbons contain mesopore volume fraction lower than 75 %. Therefore, it is important to prepare mesoporous carbon with a high mesopore volume fraction by choosing an appropriate natural clay template.
Recently, we have synthesized mesopore- and macropore-rich carbons with a mesopore volume fraction of 85 % using halloysite as a template and sucrose/furfuryl alcohol as carbon precursors [4, 14]. Attapulgite, widely distributed in China, is a kind of natural hydrous magnesium–aluminum silicate fibrillar mineral containing ribbons. It has open microchannels of 0.38 × 0.63 nm oriented longitudinally with the fibers, as well as a high proportion of mesopores formed between the nanofibers [15]. Lingmin Shi [16] synthesized mesoporous carbons with a mesopore volume fraction greater than 86 % by using attapulgite as template and furfuryl alcohol as the carbon source through a vapor deposition polymerization (VDP) method. In this chapter, we tried to prepare mesoporous carbon reflecting the morphology and pore structure of the attapulgite template using sucrose as the carbon source by a liquid impregnation method.
2 Materials and Methods
2.1 Materials and Carbon Preparation
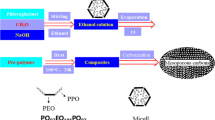
The template used in this study was attapulgite, a natural magnesium–aluminum silicate mineral, with ideal molecular formula of [Si8Mg5O20(OH)2(H2O)4·4H2O], supplied by Jiu Chuan Nami Material Technology Co., Ltd from Xuyi, Jiangsu Province, China, denoted as APT. It was ground and sieved by a 200-mesh sieve and then dried at 110 °C for 24 h before use. Sucrose (AR) and sulfuric acid (98 %) were purchased from Beijing Yili Fine Chemicals Co., Ltd. Sulfuric acid was used as the acid catalyst to polymerize and solidify the sucrose used in the process in order to prepare porous carbons [17].
In the first case, 25.0 g of sucrose was dissolved in 50.0 g of deionized water to prepare the sucrose solution. The sucrose solution was introduced into 5.0 g of attapulgite matrix by liquid impregnation at room temperature, under vacuum. Afterward, 2.8 g of sulfuric acid was added to the mixture of attapulgite and sucrose solution. This mixture was stirred for 24 h at room temperature. The mixture was then polymerized at 100 °C for 6 h and subsequently heated at 160 °C for another 6 h. For the carbonization, the polymerized composite was heat treated under a nitrogen atmosphere from room temperature to 700 °C at a heating rate of 5 °C/min and then kept for 3 h in order to complete the carbonization.
After carbonization, the attapulgite–carbon composite (denoted as APT-C) was leached in HF (40 wt%) solution at room temperature and stirred for 3 h, then kept overnight to dissolve the mineral framework, followed by washing with deionized water, and treating with 36 wt% HCl solution under stirring overnight. This demineralization procedure was repeated twice to completely remove the inorganic matrix. The resultant mesoporous carbon was obtained by filtering, washing with deionized water, and drying in an oven at 110 °C overnight. The carbon was denoted as C-700.
2.2 Samples Characterization
2.2.1 X-ray Powder Diffraction
Powder X-ray diffraction pattern of the sample was recorded using X-ray diffractometer (Rigaku, D/Max with CuKa radiation, k = 1.5406 Å) operated at 40 kV and 100 mA. The step width was 0.02° in the range of 5°–70° 2θ. The analyzed samples were spread as thin layers on a glass slide. Low-angle X-ray diffraction pattern of the resultant sample was also recorded with a D/Max diffractometer with a step width of 0.02° in the range of 0.06°–3.0° 2θ.
2.2.2 Scanning Electron Microscopy
The scanning electron microscopy (SEM) images were taken with a Cambridge S-360 microscope equipped with a cold field-emission gun operating at 20 kV and all SEM images were captured by CCD (charge-coupled device) cameras. The specimen was mounted on SEM mount with a carbon tape.
2.2.3 Transmission Electron Microscopy
Transmission electron microscopy (TEM) images of samples were obtained on a Hitachi H9000NAR microscope equipped with a cold field-emission gun. The acceleration voltage was 100 kV, and all TEM images were captured by CCD (charge-coupled device) cameras. The specimens were prepared by the following procedure: the sample was ultrasonically dispersed in ethanol for 15 min, and then a drop of the suspension was placed onto a carbon-coated copper grid, which was left to stand for 10 min before transferred to the microscope.
2.2.4 Surface Area Measurements
N2 adsorption isotherms (at 77 K) were obtained using an Accelerated Surface Area Porosimeter (ASAP 2020) from Micromeritics. The samples were outgassed for 2 h before measurements at 200 °C in a vacuum at about 10−3 Torr. All adsorbed amounts are expressed per gram of outgassed sample. The specific surface area of the samples was calculated by the Brunauer–Emmett–Teller (BET) method using the adsorption data in the relative pressure range of 0.05–0.20 as all materials contained micropores, which prevented a proper determination of their specific surface areas by the BET method. The total pore volume was estimated to be the liquid volume at a relative pressure, P/P 0, of 0.99(V 0.99). The micropore volume (V mic) was calculated using the t-plot method. The mesopore volume (V meso) was determined by subtracting the micropore volume from the total volume. The pore-size distribution (PSD) curves were obtained using the Brunauer–Joyner–Halenda (BJH) method from the desorption branch. Mesopore volume fraction was calculated by V meso/V total.
3 Results
3.1 X-ray Powder Diffraction
The X-ray powder diffraction (XRD) patterns of the template and resultant carbon are shown in Fig. 1. The template used consists mostly of attapulgite, with the typical diffraction peak observed at 2θ = 8.6°, which corresponds to a basal spacing of 10.4 Å, attributed to the primary diffraction of the (001) plane [18]. Other peaks observed at 2θ = 13.6, 16.1, and 19.7° were attributed to the (020), (310), (400) planes, respectively [19]. Quartz and illite impurities were also detected as shown by their most intense peaks situated at 2θ = 26.5° (d = 0.334 nm) and 2θ = 34.9° (d = 0.257 nm), respectively. The XRD pattern of the resultant carbon shows two broad diffraction peaks observed at 2θ = 22.3° and 44.0°, which are characteristic of a disordered carbonaceous structure; the broad peak at 2θ = 22.3° represents the (002) reflection of carbon due to the stacking structure of aromatic layers. The XRD result indicates the resultant carbon has an amorphous structure.

XRD patterns of the template APT and the resultant carbon C-700
3.2 TEM and SEM Observation
Figures 2, 3, and 4 showed TEM and SEM images of the original attapulgite APT and the resultant carbon C-700. It is shown that the attapulgite exhibited a one-dimensional structure with diameters at the manometer scale under TEM. Most of the nanorods were separated after being treated by ultrasonic wave during TEM sample preparation. The attapulgite nanorods showed the typical dimensions of 100–1,000 nm in length and 20–40 nm in diameter (Fig. 2). Therefore, if the surface of attapulgite was coated with carbon, we can expect the formation of the porous carbon reflecting the morphology when the carbon is extracted from the attapulgite framework.

TEM image of the attapulgite APT

SEM image of the resultant carbon C-700

TEM image of the resultant carbon C-700
Just as expected, the resultant carbon C-700 shows a tubular-like morphology (Fig. 3), with ca. 20–40 nm in diameter tubes as shown in Fig. 4, which is similar to the attapulgite, and demonstrates that sucrose was mainly deposited on the external surface of the template, and the carbon preserved after carbonization and demineralization, thus, the attapulgite shows the templating effect.
3.3 Surface Area Measurements
The nitrogen adsorption–desorption isotherm and pore size distribution (PSD) of attapulgite are shown in Fig. 5. From Fig. 5, we can see that the isotherm shows a type IV isotherm and type H3 hysteresis loops according to IUPAC classification [19], with a high adsorption in the high-pressure section and a pronounced capillary condensation step at relative pressures of 0.7. This indicates that the attapulgite possesses a high proportion of slit-shape mesopores and almost no micropores, and the pores were formed by the packing of flake particles, which is very common in the layered clay porous minerals. The result is in accordance with previous work, which pointed out that the nitrogen adsorption of attapulgite belongs to outer surface adsorption, rather than its channel surface [20]. There are also some macropores as suggested from the final raising tail at relative pressure near 1.0 [11]. The BET-specific surface area value of attapulgite is 126.34 m2 g−1 (see Table 1). The original attapulgite has a large mesoporosity of 99.5 % and a large specific surface area and pore volume. From the BJH pore-size distributions obtained, shown in Fig. 5 (inset), it can be seen that the attapulgite exhibits a relatively broad PSD between 3 and 40 nm with maxima at 3.7 and 31.6 nm.

N2 adsorption isotherms and pore-size distribution (inset) of the attapulgite
The nitrogen adsorption–desorption isotherm and pore-size distribution (PSD) of the resultant carbon C-700 are shown in Fig. 6. The isotherm of the resultant carbon also shows a type IV isotherm and type H3 hysteresis loops. This suggests the existence of a high proportion of mesopores with pores being slit shaped. In addition, the appearance of hysteresis loops at a relative pressure higher than P/ P 0 = 0.44 indicates that the resultant carbon has a broad PSD [21]. In Fig. 6 (inset), it can be seen that the carbon material exhibits a pore structure with maxima at 3.85 and 9.36 nm and a relatively broad PSD between 30 and 70 nm, which is also in accordance with TEM results, indicating that the resultant carbon partially inherits or replicates the morphology of the template. The increase of the adsorbed volume at low relative pressure reveals the presence of a small amount of micropores in the carbon [11]. The BET-specific surface area value of the resultant carbon is 368.80 m2 g−1 (Table 1), with a large mesoporosity of 87.3 %, indicating that it is a mesopore-rich carbon material.

N2 adsorption isotherms and pore-size distribution (inset) of the resultant carbon C-700
4 Discussion
The resultant carbon prepared by using attapulgite as a template and sucrose as a carbon source through liquid impregnation without a high-temperature graphitization can be identified as amorphous carbon from the XRD patterns. The small-angle X-ray diffraction result also shows that carbon does not possess a regular structure, in accordance with other studies [4, 5, 17].
During carbon preparation, the sucrose mainly deposited on the external surface of the template, and the resultant carbon partially inherits or replicates the morphology of the template and exhibits a tubular morphology, as shown in the TEM images, which demonstrates a “template effect.” The same result can be acquired from the N2 adsorption results, which show that the resultant carbon exhibits a pore structure with a relatively broad PSD between 30 and 70 nm and reflects the morphology of the attapulgite.
5 Conclusions
Mesoporous carbon with high mesoporosity (87 %) and a large total pore volume can be prepared by using two cheap, eco-friendly materials: one-dimensional nanorod-like attapulgite as a template and sucrose as a carbon precursor. The method proved to be an efficient, simple way to prepare carbon with a high mesoporosity and tubular structures, and it may be an appropriate method for the mass production of porous carbons.
References
Sakintuna B, Yurum Y (2005) Templated porous carbons: a review article. Ind Eng Chem Res 44:2893–2902
Xu DP, Yoon SH, Mochida I (2008) Synthesis of mesoporous carbon and its adsorption property to biomolecules. Micropor Mesopor Mater 115:461–468
Lee JW, Yoon SH, Hyeon TH (1999) Synthesis of a new mesoporous carbon and its application to electrochemical double-layer capacitors. Chem Commun 21:2177–2178
Wang AP, Kang FY, Huang ZH (2008) Synthesis of mesoporous carbon nanosheets using tubular halloysite and furfuryl alcohol by a template-like method. Micropor Mesopor Mater 108:318–324
Lu AH, Schüth F (2006) Nanocasting: a versatile strategy for creating nanostructured porous materials. Adv Mater 18:1793–1805
Kyotani T, Sonobe N, Tomita A (1988) Formation of highly oriented graphite from polyacrylonitrile by using a two-dimensional space between montmorllonite lamellae. Nature 28:331–333
Sonobe N, Kyotani T, Tomita A (1990) Carbonization of polyfurfuryl alcohol and polyvinyl acetate between the lamellae of montmorllonite. Carbon 28:483–488
Kyotani T, Yamada H, Sonobe N (1994) Heat-treatment of polyfurfuryl alcohol prepared between taeniolite lamellae. Carbon 32:627
Bandosz TJ, Jagiello J, Putyera K (1995) Sieving properties of carbons obtained by template carbonization of polyfurfuryl alcohol within mineral matrices. Langmuir 11:3964–3969
Bandosz TJ, Jagiello J, Putyera K (1996) Pore structure of carbon-mineral nanocomposites and derived carbons obtained by template carbonization. Chem Mater 8:2023–2029
Bakandritsos A, Steriotis T, Petridis D (2004) High surface area montmorillonite-carbon composites and derived carbons. Chem Mater 16:1551–1559
Nguyen-Thanh D, Bandosz TJ (2006) Metal-loaded carbonaceous adsorbents templated from porous clay heterostructures. Micropor Mesopor Mater 92:47–55
Sakintuna B, Yürüm Y (2006) Preparation and characterization of mesoporous carbons using a Turkish natural zeolitic template/furfuryl alcohol system. Micropor Mesopor Mater 93:304–312
Liu GY, Kang FY, Li BH (2006) Characterization of the porous carbon prepared by using halloysite as template and its application to EDLC. J Phys Chem Solid 67:1186–1189
Bradley WF (1940) The structural scheme of attapulgite. Am Mineral 25:405–410
Shi LM, Yao JF, Jiang JL (2009) Preparation of mesopore-rich carbons using attapulgite as templates and furfuryl alcohol as carbon source through a vapor deposition polymerization method. Micropor Mesopor Mater 122:294–300
Joo SH, Jun S, Ryoo R (2001) Synthesis of ordered mesoporous carbon molecular sieves CMK-1. Micropor Mesopor Mater 44–45:153–158
Smith DK, Mrose ME, Berry LG (1974) Selected powder diffraction data for minerals. Joint Committee on Powder Diffraction Standards
Sing KSW, Everett DH, Haul RAW (1985) Reporting physisorption data for gas solid systems with special reference to the determination of surface-area and porosity (Recommendations 1984). Pure Appl Chem 57:603–619
Galan E (1996) Properties and applications of Palygorskite-sepiolite clays. Clay Miner 31:443–453
Böhme K, Einicke WD, Klepel O (2005) Templated synthesis of mesoporous carbon from sucrose—the way from the silica pore filling to the carbon material. Carbon 43:1918
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this paper
Cite this paper
Cao, X., Chuan, X. (2015). Synthesis and Characterization of Mesoporous Carbon Using Attapulgite Template. In: Dong, F. (eds) Proceedings of the 11th International Congress for Applied Mineralogy (ICAM). Springer Geochemistry/Mineralogy. Springer, Cham. https://doi.org/10.1007/978-3-319-13948-7_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-13948-7_4
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-13947-0
Online ISBN: 978-3-319-13948-7
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)