
Abstract
Small non-coding RNAs (sRNAs) are key post-transcriptional regulators of gene expression in bacteria, with a diversity of origins, sequences, structures, and modes of action among their members. The variety within these regulatory RNAs makes it difficult to unify this remarkable heterogeneous class of RNA molecules. Structural determinants along with nucleotide sequence play important roles in defining the sRNA ability to interact with their targets. Most sRNAs bind to mRNA targets, either acting as repressors or activators. Nevertheless, the sRNAs themselves are subject to post-transcriptional regulation, either by ribonucleases, RNA chaperones, and other RNA-binding proteins, as well as RNA sponges. In this chapter we summarize the information on structure and function of bacterial sRNAs and provide several examples to better illustrate the broad diversity of this class of regulatory RNAs.
J. P. Sousa and A. F. Q. Silva contributed equally in this chapter.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Bacterial small non-coding RNAs (sRNAs) represent a class of regulatory RNAs, with sRNA-based networks virtually controlling all aspects of cell physiology. sRNAs are powerful regulators of gene expression that may bind to different macromolecules, either DNA, proteins, or other RNA molecules, but mRNAs are by far their most abundant targets (Quendera et al. 2020). Indeed, the vast majority of sRNAs act through an antisense mechanism, binding to their mRNA targets by complementary base pairing, resulting either in inhibition or activation of translation. sRNAs can be divided into two groups according to the location of the sRNA genes in relation to their targets: cis-acting sRNAs are expressed in the same location but in the opposite strand of the target whereas trans-encoded sRNAs are expressed from a different genomic region than their mRNA targets (Fig. 1). As consequence, cis-encoded sRNAs present a perfect base pairing with their mRNA targets, in contrast to the more numerous trans-encoded RNAs that establish short and imperfect antisense base pairing interactions with their targets, resembling the action of eukaryotic microRNAs. The base pairing of individual trans-encoded sRNAs can take place at different sites on the mRNA target, from the 5′ or 3′ untranslated regions (UTRs), as well as in the coding sequence. More often than not, trans-encoded sRNAs seem to be able to bind to more than one target mRNA and may use different regions for interactions with different targets (Andrade et al. 2013). This plasticity of sRNAs contributes to the rapid reprogramming of gene expression and helps to explain the regulatory success of sRNA-based pathways.

Mechanisms of action for cis-acting and trans-acting sRNAs. a A cis-encoded sRNA and its target are located in the same genomic region but on different strands; as result the sRNA-mRNA antisense interaction exhibits a perfect base pairing region. b A trans-encoded sRNA is located in a different genomic region than its target; consequently, the sRNA establishes short and imperfect base pairing interactions with the mRNA target
A plethora of sRNAs act as repressors, acting for example by sequestering the entry of ribosomes to mRNA (e.g., through occlusion of the ribosome binding site (RBS)) or affecting mRNA stability (e.g., by promoting the access to ribonucleases (RNases) that cleave mRNA leading to its inactivation), as it has been described for the sRNA MicA (Udekwu et al. 2005; Viegas et al. 2011). However, some other sRNAs have opposite effects and therefore act as activators of gene expression. For example, the sRNA SraL was shown to upregulate the expression of the transcription termination factor Rho, by interacting with the 5′UTR of rho mRNA and preventing its premature transcription termination (Silva et al. 2019). Broadly, sRNAs can vary in size from 50 to 500 nucleotides (nts), without a common sequence that can be used as signature, making this a very heterogeneous class of RNA molecules; nevertheless, a common feature of these regulatory RNAs is that they are highly structured, with the presence of hairpins and stem-loops. These structures serve as barriers against sRNA degradation by RNases, may act as anchor sites for RNA-binding proteins, and can also affect interaction with mRNA targets. In this chapter we present a comprehensive overview on bacterial sRNAs and how structure is linked to sRNA function, providing several examples to better illustrate the broad diversity of this class of regulatory RNAs.
2 sRNA that Use the 5′ Domain to Interact with mRNA Targets
sRNAs are usually structured molecules, and typically sequences enriched in GC are more structured. The presence of a stem-loop corresponding to the Rho-independent transcriptional terminator followed by a short U-rich sequence seems to be ubiquitous among bacterial small RNAs (Morita et al. 2017), and the presence of additional stems is frequent in many sRNAs (Fig. 2). Nevertheless, even such high structured molecules present linear regions, either located in the body of the sRNA or in the bulge loop position of hairpin stems. These linear domains are highly important since sRNA/mRNA base pairing generally occurs through interactions between these linear regions. Consequently, we can recognize different RNA structural modules within a sRNA (Andrade et al. 2013).

Representative examples of sRNAs structural diversity. a MicA sRNA has a 5′-end linear sequence that is recognized as its main interaction region with mRNA targets. Two stem-loops present in the 3′-end of MicA and the short terminal 3′ poly(U) sequence are important elements for interaction with the RNA chaperone Hfq. b RybB sRNA also shows a 5′-end linear region for interaction with mRNA targets and a structured 3′-end with two stem-loops followed by a terminal poly(U) sequence. c SgrS sRNA is a highly structured sRNA that uses a linear sequence located near the 3′-end for interaction with its mRNA targets. The position of important nucleotides of SgrS for base pairing with ptsG mRNA is indicated with arrows. d OxyS sRNA displays a 5′-end structured region with two stem-loops (SLa and SLb), using a downstream linear region for interaction with its mRNA targets. A third stem-loop (SLc) that corresponds to the Rho-independent transcriptional terminator is located in the 3′-end, followed by the terminal 3′ poly(U) sequence. sRNAs structures were determined using the RNA structure webserver (Bellaousov et al. 2013)
The well-characterized small RNA MicA, firstly identified in Escherichia coli, is commonly used as model of study. MicA is a trans-encoded sRNA that represses several genes, including the expression of major outer membrane proteins, such as OmpA, LamB, Tsx, and EcnB (Gogol et al. 2011). The 5′ linear end of MicA consisting of ~20 nts, located before two strong stem-loops, was identified as the principal target recognition domain (Fig. 2a) (Udekwu et al. 2005; Andrade et al. 2013). Many other sRNAs that preferably use their 5′-end sequence for contact with their targets have also been identified. Interestingly, for a few it was even possible to define a short ‘‘seed’’ sequence responsible for the interaction with multiple targets. This is the case of Salmonella Typhimurium RybB (Fig. 2b) (Papenfort et al. 2010) whose conserved seed domain is also shared with Vibrio cholerae VrrA and MicV sRNAs (Peschek et al. 2019), and E. coli OmrA/B, two seemingly similar sRNAs that use their conserved 5′-end sequence to interact with multiple targets (Guillier and Gottesman 2008). Overall, several studies indicate the importance of the 5′-end domain of many sRNAs as the principal target interaction domain, with these few examples here depicted as illustrative ones. However, research on MicA further identified structural elements present in the 3′-end, namely stem-loops, which could also play a role in target recognition. While one of the stem-loops was more important for the in vivo repression of both ompA and ecnB mRNAs, the other stem-loops were found to be only critical for the regulation of tsx transcript levels (Andrade et al. 2013). This is probably consequence of conformational rearrangements of the sRNA as a result of target interactions with the linear sequence present in the bulge loop of the stems (Fig. 2a). Similar observations have been made in many studies such as the OxyS interaction with the fhlA mRNA (Altuvia et al. 1998).
3 sRNA that Use the 3′ Domain to Interact with mRNA Targets
SgrS is a well-characterized sRNA that is widespread in enteric bacteria (Rice and Vanderpool 2011). In E. coli SgrS is a 227-nt molecule (Fig. 2c) but the size is variable in other species. SgrS levels are upregulated when the balance between sugar uptake and its metabolism is disrupted; the intracellular accumulation of glucose-6-phosphate leads to activation of the transcription factor SgrR, which then induces SgrS expression (from the sgrS gene) to minimize sugar transport (Vanderpool and Gottesman 2007). In the case of glucose excess, SgrS binds reversibly to its main target, the ptsG mRNA encoding the EIICB domain of the glucose-specific phosphotransferase system (PTS), blocking its ribosome binding site and halting the translation of additional glucose importers; the SgrS-mRNA complex is subsequently degraded by RNase E (Rice and Vanderpool 2011). Strikingly, SgrS is also a dual-function molecule since it also encodes SgrT, a small 43-amino acid protein whose regulatory function mechanism acts independently from SgrS-mediated base pairing in response to phosphosugar stress (Wadler and Vanderpool 2007).
In contrast to MicA and other sRNAs, SgrS-mediated target recognition requires a conserved region near its 3′-end rather than a sequence in the 5′-end. SgrS nucleotides 168–187 are complementary to the 5′-UTR of the ptsG mRNA (Kawamoto et al. 2006). Particularly, nucleotides 168–181 have been shown to be enough to downregulate ptsG mRNA levels (Maki et al. 2010). Moreover, SgrS harbors a Hfq-binding motif at the 3′-end, which contains two adjacent stem-loops—the small stem-loop and the U-rich terminator stem-loop—followed by a poly(U) tail (Fig. 2c) (Kawamoto et al. 2006). The length of the poly(U) tail is a key feature of SgrS, not only for the correct biosynthesis of functional sRNAs but also for optimal Hfq binding to the two 3′ stem-loops, and target regulation (Morita et al. 2017).
The sequence and subsequential secondary structure of SgrS have been shown to be highly sensitive to changes, resulting in regulation defects in the cell. It has been shown that point mutations in the region spanning from U175 to G186, encompassed in the base pairing domain of SgrS to ptsG mRNA, led to negative functional consequences in the SgrS-mediated stress response (Poddar et al. 2021). Particularly, mutagenesis of G176 and G178 completely abolishes the ability of SgrS to inhibit the translation of ptsG mRNA (Kawamoto et al. 2006), while point mutations of C174 and G170 only weakly hinders SgrS base pairing (Fig. 2c) (Maki et al. 2008). Likewise, mutations in nucleotides 183–196, 199–219, and 220–227 corresponding to the small hairpin loop, the terminator stem-loop, and the poly(U) tail, respectively, also negatively impact SgrS function in the cell, mainly in the stem region of the terminator stem-loop, specifically in nucleotides C199 to G205, and C213 to G219 (Poddar et al. 2021).
A less specific target of SgrS is the manXYZ mRNA, encoding another PTS sugar transporter—involved mainly in mannose uptake, but also involved in the transport of glucose, glucosamine, and N-acetylglucosamine. SgrS inhibits the expression of this transport system by base pairing with low affinity to two distinct sites of the mRNA: within manX coding sequence and in an UTR between manX and manY (Rice and Vanderpool 2011). As it is the case with ptsG, SgrS annealing halts translation, recruits RNase E causing the degradation of manXYZ mRNA, and therefore minimizes phosphosugar toxicity.
4 sRNA Association with the RNA Chaperone Hfq
Many trans-encoded sRNAs associate with the RNA chaperone Hfq and this can affect their stability and their function (Quendera et al. 2020). Hfq forms an hexamer and it not only protects the sRNAs from degradation, namely from RNase E, RNase III and PNPase cleavage (Udekwu et al. 2005; Andrade and Arraiano 2008; Viegas et al. 2011; Andrade et al. 2012) but also stabilizes the imperfect base pairing between sRNA/mRNA pairs, functioning as an “RNA matchmaker” (Woodson et al. 2018). Indeed, the primary role of Hfq (at least in Gram-negative bacteria) is to promote the annealing of sRNA-mRNA duplexes (dos Santos et al. 2019). However, the role of Hfq in Gram-positive bacteria is more controversial even though extensive Hfq-dependent post-transcriptional regulation was recently identified in the Gram-positive human pathogen Clostridioides difficile (Fuchs et al. 2021). In addition, Hfq may play other functions independently of its association with sRNAs, as was observed with tRNA maturation, ribosome biogenesis and rRNA processing (Lee and Feig 2008; Andrade et al. 2018; dos Santos et al. 2020).
In E. coli, interactome studies identified thousands of mRNA-sRNA pairs exhibiting sequence complementarity (Melamed et al. 2016). Hfq promotes sRNA/mRNA interaction as it is able to bind both molecules simultaneously making use of different protein surfaces; the Hfq distal side preferably binds to (ARN)n motifs frequently found on mRNA while the Hfq proximal side preferably binds the short and unstructured poly(U) tails present at the 3′-end of sRNAs (Sauer and Weichenrieder 2011). The basic patched rim surface of Hfq interacts with UA-rich sites present in both RNAs, accelerating the formation of the complex (Panja et al. 2013; Zhang et al. 2013). In addition, the position and structure of Rho-independent terminators located immediately before the 3′ poly(U) stretch of sRNAs are also important for Hfq interaction (Morita et al. 2017). It has been shown that mutations disrupting the terminator stem-loop structure or the poly(U) tail resulted in lower intracellular levels of sRNAs, like MicA and SgrS, due to higher levels of degradation related to defective Hfq interaction (Andrade et al. 2013; Poddar et al. 2021).
The binding and role of Hfq in sRNA function has been elucidated for many sRNAs, as it is the case of the well-studied OxyS, an important factor in the oxidative stress response (Altuvia et al. 1998; Seixas et al. 2022). Depletion of this sRNA results in considerably higher levels of both H2O2 and superoxide in E. coli (González-Flecha and Demple 1999). The overexpression of OxyS negatively affects the expression of the transcription factors RpoS and FlhA by binding to their respective mRNAs, blocking ribosome binding, and then recruiting RNase E for degradation (Fröhlich and Gottesman 2018). The OxyS sRNA is a 118 nt-long molecule, containing two stem-loops near its 5′-end (Fig. 2d): SLa which contains the largest hairpin structure of the molecule and a smaller stem-loop, SLb, immediately downstream. Moreover, this sRNA contains an additional stem-loop, SLc, on its 3′-end, followed by a poly(U) tail (Wang et al. 2015). The binding of Hfq is important for the binding of OxyS to its targets and OxyS contains in its sequence several Hfq-binding motifs: the (AAN)3 motif downstream the second stem-loop (SLb) binds to the distal face of Hfq; the UUUU motif upstream of the third stem-loop (SLc) binds to the lateral surface of Hfq; and the 3′-end poly(U) motif binds to the proximal face of Hfq (Fig. 2d) (Cai et al. 2022). Through the multiple interactions with the RNA-binding surfaces of Hfq, OxyS is able to wrap around the Hfq hexamer, effectively distorting the overall conformation of OxyS from an extended configuration to a more packed and stable “wrapped” structure, which helps to stabilize sRNA regulation (Henderson et al. 2013; Cai et al. 2022). The structural changes of Hfq-bound OxyS better expose its base pairing region to bind to fhlA mRNA resulting in a more stable antisense-target RNA complex which prevents ribosome binding (Altuvia et al. 1998; Hoekzema et al. 2019). Interestingly, depletion of Hfq has not been shown to have a significant effect in OxyS-rpoS mRNA binding, but instead it seems to help to recruit RNase E (Henderson et al. 2013). Hfq binding can be observed in many other sRNAs, for instance, MicA: Hfq was found to bind an AU-rich single stranded region flanked by the two stem-loop structures and the 3′-end poly(U) stretch after the Rho-independent terminator of this sRNA (Andrade et al. 2013). Mutations of these Hfq-binding sequences were also found to disrupt target recognition highlighting the importance of Hfq/sRNA association for MicA function.
5 sRNAs that Bind to Proteins: The Example of CsrB
In addition to mRNA targets, sRNAs can also bind to and regulate the activity of RNA-binding proteins, as can be illustrated by the sRNA CsrB and the protein CsrA, the two main components of the Csr system that acts as the global regulator of carbon storage (Liu et al. 1997). CsrA is a conserved RNA-binding protein that was first discovered in E. coli (Romeo et al. 1993) and participates in the regulation of carbon metabolism, virulence, motility, and biofilm formation (Vakulskas et al. 2015). CsrA can act as a repressor when binding with its mRNA targets occludes the access to the ribosome binding sequence leading thus to translational arrest, as for example was shown to occur with the cstA mRNA (which encodes a transport protein) and glgC mRNA (which encodes glucose-1-phosphate adenylyltransferase) (Dubey et al. 2003). However, CsrA can also act as activator of gene expression, for example, by protecting transcripts from RNase E attack, as it was observed with the flhDC mRNA (which is involved in motility and chemotaxis) (Yakhnin et al. 2013).
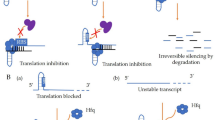
Strikingly, CsrB sRNA inhibits the activity of CsrA by directly binding and sequestering the protein (Vakulskas et al. 2015). CsrB has 18 imperfect repeats of the 5′-CAGGA(U, C, A)G-3′ motif either in the loops or in the linear regions of the secondary structure of CsrB, which are the recognition elements for CsrA (Fig. 3). Thus, CsrA and CsrB assemble in a complex formed by 18 CsrA subunits and a single CsrB sRNA (Liu et al. 1997). As result, CsrA is sequestered and cannot participate in their normal post-transcriptional regulator activity. The sRNAs of the CsrB family participate in the regulation of several metabolic pathways and physiological functions in Gammaproteobacteria (Vakulskas et al. 2015). The expression of CsrB is highly controlled in E. coli by the two-component signal transduction system BarA-UvrY, stringent response factors, as ppGpp and DksA, and two DEAD-box RNA helicases, DeaD (CsdA) and SrmB (Suzuki et al. 2002; Vakulskas et al. 2014). CsrB levels are also controlled by the CsrD protein, which directs this sRNA for degradation by RNase E (Suzuki et al. 2006). In Pseudomonas aeruginosa, there is an homolog to the CsrA-CsrB system, the RsmA-RsmB system, repressor of secondary metabolism, which presents the same type of regulation and interaction (Vakulskas et al. 2015).

sRNAs can bind to proteins and be sequestered by sponge RNAs. a The sRNA CsrB forms a complex with the protein CsrA. CsrB contains 18 repeated sequences that can bind to and sequester CsrA. b The regulatory RNA SroC derives from the processing of the intergenic region within the small transcript of the gltIJKL operon and acts as a sponge RNA that is able to bind to the sRNA GcvB
6 Sponge RNAs that Regulate sRNA Levels
sRNAs themselves can be post-transcriptionally regulated by the so-called sponge RNAs. These regulatory RNAs modulate sRNA levels through sequestration of the sRNA and/or promotion of degradation of the sRNA or the sponge RNA-sRNA complex (Denham 2020). The term sponge RNA was first used by Ebert and colleagues, when they engineered an RNA molecule capable of controlling microRNA activity by competing with its targets (Ebert et al. 2007). In bacteria, sponge RNAs can be divided into two groups, those that result from the processing of an mRNA and those that are independently transcribed. Most of the sponge RNAs already described were found in Enterobacteriaceae, in part due to their association with Hfq (Denham 2020). The best characterized sponge RNAs, which will be described in more detail, are the intergenic region between chbB and chbC, SroC, and the external transcribed spacer of the tRNALeu (3′ETSLeuZ).
6.1 Intergenic Region of the chbBC Transcript
The first sponge RNA described in bacteria was the intergenic region between chbB and chbC, in Salmonella enterica (Figueroa-Bossi et al. 2009). Immediately after the discovery of this mechanism in Salmonella, it was reported its existence in E. coli (Rasmussen et al. 2009). The translocation of chitin products to the cytosol (chitobiose and chitotriose that can be used as carbon and nitrogen sources) is carried out by the membrane protein porin ChiP which is repressed by the sRNA ChiX (Plumbridge et al. 2014). This sRNA ChiX also inhibits the expression of the chitin PTS encoded by the chbBCARFG operon. However, the chbBCARFG operon gives rise to a polycistronic mRNA that upon binding of ChiX is rapidly cleaved by RNase E, releasing an intercistronic spacer (around 400 nucleotides) between the chbB and chbC genes. This intergenic sequence will then act as sponge RNA capable of sequestering ChiX and directing it to degradation by RNase E (Overgaard et al. 2009; Figueroa-Bossi et al. 2009). This tight regulation allows the controlled expression of the chitin utilization operon: when chitosugars are present, the chb operon is transcriptionally upregulated and the repression of ChiP expression is relieved by the binding of the sRNA ChiX to the intercistronic spacer sequence between chbB and chbC (Denham 2020).
6.2 3′ETSLeuZ
Bacterial tRNAs are derived from a polycistronic transcript, which contains external transcribed spacers (ETS), on the 5′ and 3′-ends, and sometimes internal transcribed spacers (ITS). For the maturation of tRNAs to occur, is necessary the action of endo and exoribonucleases separating the tRNAs and releasing the ITS and ETS, which were initially thought to be immediately degraded (Grüll and Massé 2019). Processing of the polycistronic tRNA transcript glyW-cysT-leuZ is done by RNase E, giving rise to three tRNA precursors and releasing a small fragment corresponding to the 3′ETSLeuZ, a sponge RNA with ~50 nts (Lalaouna et al. 2015). The 3′ETSLeuZ is involved in the regulation of two sRNAs, RyhB and RybB, which are respectively related to the regulation of iron homeostasis and the integrity of the outer membrane (Massé et al. 2005; Salvail et al. 2010). In the absence of stress, 3′ETSLeuZ sequesters RyhB and RybB, with no change in the expression of the targets of these sRNAs. However, in the presence of iron or envelope stress for RyhB or RybB, respectively, sRNA expression increases significantly. In this manner, the excess of sRNAs produced in comparison to the sponge RNA enables them to act on their target mRNAs, triggering the stress response. The regulation of RyhB and RybB by 3′ETSLeuZ establishes a relationship between two essential metabolisms in the bacterial cell, iron homeostasis and the envelope stress response (Lalaouna et al. 2015).
For the interaction between the 3′ETSLeuZ and the target sRNAs to occur, it is necessary the intervention of the RNA chaperone Hfq. 3′ETSLeuZ binds to the distal and proximal faces of Hfq, although it is not yet known whether these interactions occur simultaneously. Since this sponge RNA has a shorter single stranded 3′ poly(U) tail, the interaction with the proximal face of the Hfq is not very strong, serving only to assist in binding to other regions of the protein. Thus, the proximal face of the Hfq seems to have more importance in the stabilization of ternary complexes formed between the sponge RNA and the target, while the distal face binds to the sponge RNA to allow greater exposure of the sequence to base pair with the target. On the other hand, the interaction of RyhB and RybB with Hfq takes place in the proximal face and the rim of Hfq (Małecka et al. 2021). In silico predictions revealed that 3′ETSLeuZ pairs with the central loop of RyhB and with the 5′-end of RybB, which are the regions of interaction between these sRNAs and the target mRNAs. After the discovery of this sponge RNA, it was already suggested that some 3′ETS of certain tRNAs may also perform sponge RNA functions, given their sequence conservation and size to pair with other RNAs (Lalaouna et al. 2015). However, up to now, this mechanism has not yet been found with other tRNA intergenic spacers nor in other species.
6.3 SroC
Two different transcripts are synthesized from the gltIJKL operon in Salmonella: (i) a larger transcript that results from the transcription of the entire operon; (ii) and a smaller one that is formed due to the presence of a Rho-dependent terminator between gltI and gltJ. The full-length transcript encodes for the glutamate/aspartate ATP-binding cassette transporter (Denham 2020). The 3′-end of the smaller transcript is cleaved by RNase E, originating SroC, a regulatory RNA fragment with 150 nts that binds to Hfq (Sittka et al. 2008; Chao et al. 2012). SroC is a sponge RNA which base pairs with the sRNA GcvB, leading to its inhibition and degradation by RNase E (Fig. 3). GcvB is master regulator, controlling the expression of 1% of Salmonella genes, especially in amino acid transport and biosynthesis genes (Sharma et al. 2011). GcvB is highly expressed during faster bacterial growth in a rich medium, suggesting that the main function of this sRNA is the optimization of energy consumption for the synthesis and transport of amino acids (Sharma et al. 2007). The SroC sponge RNA pairs with two distinct regions of GcvB, which are relatively distant from each other (Lalaouna et al. 2018). While the binding sites are only 14 nts apart in SroC, the distance between the binding sequences in GcvB is 137 nts (Sittka et al. 2008).
7 3′-UTR-Derived sRNAs
While the firstly identified sRNAs were monocistronic units transcribed from their own promoters, we now know that sRNAs can also derive from mRNA processing. Consequently, sRNAs can also originate from the processing of intergenic regions (Argaman et al. 2001), 5′-UTRs (Vanderpool and Gottesman 2004), 3′-UTRs (Chao et al. 2012), protein coding sequences (Dar and Sorek 2018) and pre-tRNAs (Lalaouna et al. 2015). An emerging class of regulatory RNAs includes those that come from fragments of the 3′-UTRs of bacterial mRNAs, and we will present this in more detail. The 3′-UTR-derived sRNAs are widely distributed in bacteria and control a broad variety of biological processes, they can act either in trans, directed to one or multiple mRNAs, or in cis, regulating the synthesis of the parental mRNA, usually blocking translation (Hoyos et al. 2020; Menendez-Gil and Toledo-Arana 2021).
The 3′-UTR-derived sRNAs represent an example of the differentiation between the regulatory and coding functions through the formation of two different RNA species (Mediati et al. 2021). sRNAs that derive from 3′-UTR regions can be classified into two categories based on their origin. Type I or monocistronic sRNAs are generated due to the presence of an internal promoter positioned in the coding sequence or immediately downstream. On the other hand, type II or polycistronic sRNAs result from the processing of the 3′-UTR region of an mRNA (Miyakoshi et al. 2015; Hoyos et al. 2020). Chemically, it is possible to distinguish the two types due to the presence of a 5′ triphosphate in type I sRNAs and a 5′ monophosphate in type II sRNAs (Miyakoshi et al. 2015). The majority of the 3′-UTR-derived sRNAs described were found in Gram-negative bacteria and belong to type II, reinforcing the important riboregulatory role of ribonucleases, for instance, RNase E in Gram-negative bacteria (Hoyos et al. 2020). However, RNase E is not conserved in all bacteria, being absent in most Gram-positive bacteria and, therefore, biogenesis of 3′-UTR-derived sRNAs may occur through a different mechanism in these bacteria (Ponath et al. 2022). A prominent endoribonuclease in Gram-positive bacteria that can process these sRNAs is RNase III. In fact, the role of RNase III was already described for the synthesis of RsaC in Staphylococcus aureus. This type II sRNA is generated from the mntABC operon mRNA, which encodes the largest manganese importer, hence it is only upregulated under manganese limiting conditions (Lioliou et al. 2012). One possible explanation for the smaller number of 3′-UTR-derived sRNAs in Gram-positive bacteria is that these bacteria have enzymes with 5′-3′ exoribonucleolytic activity, as RNase J1. Thus, these sRNAs are rapidly degraded unless a hairpin form at the 5′-end to stabilize it (Desgranges et al. 2022). In addition, there are also not as many processing sites near the stop codon described in Gram-positive bacteria when compared with Gram-negative bacteria (Mediati et al. 2021).
Despite the variability of 3′-UTR-derived sRNAs, it is possible to predict the formation of a sRNA from a 3′-UTR, due to some common characteristics. One of them is the formation of a stem-loop structure followed by a uridine tail, a preferred binding site of Hfq (Sauer and Weichenrieder 2011). Another is the identification of cleavage sites for RNases and conserved seed sequences that allow to base pair with the target mRNA (Miyakoshi et al. 2015). Besides, a differential expression pattern between the fragments produced from the 3′-UTRs and the corresponding mRNAs, also suggests different functions.
8 CRISPRs
A novel RNA-based system in prokaryotes has been discovered, revolutionizing gene editing systems (Jansen et al. 2002; Brouns et al. 2008). Clustered regularly interspaced short palindromic repeats (CRISPRs) constitute the key components of an adaptive immune system that can be found widespread in many archaea and bacteria (Jinek et al. 2012; Makarova et al. 2013). In association with specific Cas proteins, this system operates in a similar manner to RNA interference modules, using structured small RNAs to recognize and silence target nucleic acids, such as viral genetic material, plasmids and other mobile genetic elements (Barrangou et al. 2007). CRISPR-Cas systems have been divided into three major CRISPR-Cas types (Type I and Type III present in both archaea and bacteria and Type II just in the latter; then divided into additional subtypes) due to their variety in cas gene composition and consequent proteins necessary for the immune response to take effect (Makarova et al. 2011). Standard CRISPR loci are comprised of a CRISPR array, i.e., short direct repeats, interspersed with spacer sequences (invader-derived short variable DNA sequences) and a leader side directly upstream of the first repeat; these loci then tend to be flanked by cas genes (Jansen et al. 2002). All known CRISPR-Cas systems share a modular mechanism for gene silencing: mature CRISPR RNAs (crRNAs) that contain a unique spacer sequence responsible for recognizing complementary invading genetic material and guiding Cas proteins to degrade it (Chylinski et al. 2014; Charpentier et al. 2015).
Canonical CRISPR-Cas-based immunity consists of three distinctive stages: adaptation, expression, and interference. The adaptation stage is triggered by the presence of exogenous genetic material inside the bacteria. Viral or plasmid DNA is recognized, processed into small fragments known as protospacers and incorporated into the CRISPR array as new DNA spacers (Safari et al. 2019). In most cases, an initial viral attack results in the integration of a single 30 base pairs unique resistance-conferring spacer immediately downstream of the leader side of a CRISPR locus, followed by the duplication of the repeat in order to originate a new spacer–repeat unit (Makarova et al. 2011). The subsequent step is expression, and it consists in the expression and processing of the CRISPR RNA (crRNA or guide RNA), which is essential for the correct function of the CRISPR-Cas system. crRNA maturation can be divided into three distinct events (Safari et al. 2019): (i) an approximately 60–70-nucleotide long initial primary transcript of the CRISPR array is produced, named precursor crRNA (pre-crRNA). This RNA is transcribed from a promoter harbored in leader sequence upstream repeat-spacer array; (ii) pre-crRNA cleavage by nucleases at specific recognition sites in the repeats, resulting in a mature crRNA made up of the complete spacer sequence between two partial repeat sequences; (iii) occasionally, some transcripts undergo additional secondary processing to generate mature crRNA (Carte et al. 2008; Charpentier et al. 2015). While in CRISPR-Cas systems classified as Type I and III, a specific RNase belonging to the Cas6 family cleaves the pre-crRNA, in Type II systems an auxiliary trans-acting small RNA (tracrRNA) independently base pairs with each repeat sequence of the pre-crRNA and associates with the Cas9 protein, resulting in a dual-RNA which is subsequently cleaved by RNase III (Fig. 4) (Deltcheva et al. 2011). The third and final event of CRISPR-based immunity is interference. crRNA, in association with Cas effector endonucleases, recognizes and base pairs to cognate invader DNA sequences that match the prokaryote’s spacers. This process is then followed by recognition of the target DNA by Cas effectors and cleaving of the nucleic acids within the protospacer sequence, leading to double-stranded DNA breaks and subsequent degradation of the invader DNA (Jinek et al. 2012). Different Cas proteins can have a role in one or several steps of CRISPR–Cas gene silencing, acting in most cases in association with other proteins.

CRISPR RNA processing mechanisms in Type I and Type II CRISPR-Cas systems. a In most Type I CRISPR-Cas systems, the short palindromic repeats form hairpin structures can be recognized and cleaved by nucleases Cas6 or Cas5, depending on the system subtype; the cleaved CRISPR RNA (crRNA) continues in association with the nuclease, and additional Cas subunits (forming the Cascade complex) bind to the 5′-end and spacer regions of the crRNA, which can consequently interact with and silence their specific target genetic element. b Type II CRISPR-Cas systems can be distinguished by the formation of duplexes with the pre-crRNA and the trans-acting tracrRNA; the latter helps in the recognition of the Cas9 protein, further stabilizing the RNA duplex which is then processed by RNase III cleavage. An additional processing step by still unknown endo- or exoribonucleases then generates the mature crRNAs (adapted from Charpentier et al. 2015)
CRISPR-based immunity is highly specific, as it has been demonstrated that a single mismatch between the spacer sequence and the target exogenous DNA completely nullified phage resistance in bacteria (Barrangou et al. 2007). Beyond primary nucleic acid sequence the repeat sequences in the flanking region usually adopt secondary structures, more specifically a partial duplex at the 5′-end and a hairpin structure at the 3′-end (Jore et al. 2011; Gu et al. 2019).
9 Conclusions
Small RNAs are an abundant and diverse group of non-coding RNAs present in bacteria. Diversity is the keyword that better describes these regulatory RNA molecules. As we have shown, sRNAs can act as repressors or activators of gene expression; they can be synthesized as independent transcriptional units or derive from mRNA processing; they can make use of different domains to interact with multiple targets; and they can associate with different RNA-binding proteins, among many other features. Importantly, sRNAs rely extensively on their sequence and structure for their function(s), as nicely illustrated in the various examples here provided. Overall, sRNA networks are widely generalized in bacteria. It is anticipated that research in this exciting field will continue to reveal additional players and new RNA features in the years to come.
References
Altuvia S, Zhang A, Argaman L et al (1998) The Escherichia coli OxyS regulatory RNA represses fhlA translation by blocking ribosome binding. EMBO J 17:6069–6075
Andrade JM, Arraiano CM (2008) PNPase is a key player in the regulation of small RNAs that control the expression of outer membrane proteins. RNA 14:543–551
Andrade JM, Pobre V, Arraiano CM (2013) Small RNA modules confer different stabilities and interact differently with multiple targets. PLoS ONE 8:e52866
Andrade JM, Pobre V, Matos AM et al (2012) The crucial role of PNPase in the degradation of small RNAs that are not associated with Hfq. RNA 18:844–855
Andrade JM, Santos RF, Chelysheva I et al (2018) The RNA-binding protein Hfq is important for ribosome biogenesis and affects translation fidelity. EMBO J 37:e97631
Argaman L, Hershberg R, Vogel J et al (2001) Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr Biol 11:941–950
Barrangou R, Fremaux C, Deveau H et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712
Bellaousov S, Reuter JS, Seetin MG et al (2013) RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 41:W471–W474
Brouns SJJ, Jore MM, Lundgren M et al (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964
Cai H, Roca J, Zhao Y-F et al (2022) Dynamic refolding of OxyS sRNA by the Hfq RNA chaperone. J Mol Biol 434:167776
Carte J, Wang R, Li H et al (2008) Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev 22:3489–3496
Chao Y, Papenfort K, Reinhardt R et al (2012) An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31:4005–4019
Charpentier E, Richter H, van der Oost J et al (2015) Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev 39:428–441
Chylinski K, Makarova KS, Charpentier E et al (2014) Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res 42:6091–6105
Dar D, Sorek R (2018) High-resolution RNA 3′-ends mapping of bacterial Rho-dependent transcripts. Nucleic Acids Res 46:6797–6805
Deltcheva E, Chylinski K, Sharma CM et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607
Denham EL (2020) The Sponge RNAs of bacteria – How to find them and their role in regulating the post-transcriptional network. Biochim Biophys Acta Gene Regul Mech 1863:194565
Desgranges E, Barrientos L, Herrgott L et al (2022) The 3′UTR-derived sRNA RsaG coordinates redox homeostasis and metabolism adaptation in response to glucose-6-phosphate uptake in Staphylococcus aureus. Mol Microbiol 117:193–214
dos Santos RF, Andrade JM, Pissarra J et al (2020) Hfq and RNase R mediate rRNA processing and degradation in a novel RNA quality control process. Mbio 11:e02398-e2420
dos Santos RF, Arraiano CM, Andrade JM (2019) New molecular interactions broaden the functions of the RNA chaperone Hfq. Curr Genet 65:1313–1319
Dubey AK, Baker CS, Suzuki K et al (2003) CsrA Regulates translation of the escherichia coli carbon starvation gene, cstA, by blocking ribosome access to the cstA transcript. J Bacteriol 185:4450–4460
Ebert MS, Neilson JR, Sharp PA (2007) MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Meth 4:721–726
Figueroa-Bossi N, Valentini M, Malleret L et al (2009) Caught at its own game: regulatory small RNA inactivated by an inducible transcript mimicking its target. Genes Dev 23:2004–2015
Fröhlich KS, Gottesman S (2018) Small regulatory RNAs in the enterobacterial response to envelope damage and oxidative stress. Microbiol Spectr 6:211–228
Fuchs M, Lamm-Schmidt V, Sulzer J et al (2021) An RNA-centric global view of Clostridioides difficile reveals broad activity of Hfq in a clinically important gram-positive bacterium. Proc Natl Acad Sci USA 118:e2103579118
Gogol EB, Rhodius VA, Papenfort K et al (2011) Small RNAs endow a transcriptional activator with essential repressor functions for single-tier control of a global stress regulon. Proc Natl Acad Sci USA 108:12875–12880
González-Flecha B, Demple B (1999) Role for the oxyS Gene in Regulation of Intracellular Hydrogen Peroxide in Escherichia coli. J Bacteriol 181:3833–3836
Grüll MP, Massé E (2019) Mimicry, deception and competition: the life of competing endogenous RNAs. Wires RNA 10:e1525
Gu D-H, Ha SC, Kim J-S (2019) A CRISPR RNA is closely related with the size of the cascade nucleoprotein complex. Front Microbiol 10:2458
Guillier M, Gottesman S (2008) The 5′ end of two redundant sRNAs is involved in the regulation of multiple targets, including their own regulator. Nucleic Acids Res 36:6781–6794
Henderson CA, Vincent HA, Casamento A et al (2013) Hfq binding changes the structure of Escherichia coli small noncoding RNAs OxyS and RprA, which are involved in the riboregulation of rpoS. RNA 19:1089–1104
Hoekzema M, Romilly C, Holmqvist E et al (2019) Hfq-dependent mRNA unfolding promotes sRNA-based inhibition of translation. EMBO J 38:e101199
Hoyos M, Huber M, Förstner KU et al (2020) Gene autoregulation by 3’ UTR-derived bacterial small RNAs. Elife 9:e58836
Jansen R, van Embden JDA, Gaastra W et al (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science (80-) 337:816–821
Jore MM, Lundgren M, van Duijn E et al (2011) Structural basis for CRISPR RNA-guided DNA recognition by Cascade. Nat Struct Mol Biol 18:529–536
Kawamoto H, Koide Y, Morita T et al (2006) Base-pairing requirement for RNA silencing by a bacterial small RNA and acceleration of duplex formation by Hfq. Mol Microbiol 61:1013–1022
Lalaouna D, Carrier M-C, Semsey S et al (2015) A 3′ external transcribed spacer in a tRNA transcript acts as a sponge for small RNAs to prevent transcriptional noise. Mol Cell 58:393–405
Lalaouna D, Eyraud A, Devinck A et al (2018) GcvB small RNA uses two distinct seed regions to regulate an extensive targetome. Mol Microbiol 111:303–551
Lee T, Feig AL (2008) The RNA binding protein Hfq interacts specifically with tRNAs. RNA 14:514–523
Lioliou E, Sharma CM, Caldelari I et al (2012) Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet 8:e1002782
Liu MY, Gui G, Wei B et al (1997) The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 272:17502–17510
Makarova KS, Haft DH, Barrangou R et al (2011) Evolution and classification of the CRISPR–Cas systems. Nat Rev Microbiol 9:467–477
Makarova KS, Wolf YI, Koonin EV (2013) Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res 41:4360–4377
Maki K, Morita T, Otaka H, Aiba H (2010) A minimal base-pairing region of a bacterial small RNA SgrS required for translational repression of ptsG mRNA. Mol Microbiol 76:782–792
Maki K, Uno K, Morita T et al (2008) RNA, but not protein partners, is directly responsible for translational silencing by a bacterial Hfq-binding small RNA. Proc Natl Acad Sci USA 105:10332–10337
Małecka EM, Sobańska D, Olejniczak M (2021) Bacterial chaperone protein Hfq facilitates the annealing of sponge RNAs to small regulatory RNAs. J Mol Biol 433:167291
Massé E, Vanderpool CK, Gottesman S (2005) Effect of RyhB small RNA on global iron use in Escherichia coli. J Bacteriol 187:6962–6971
Mediati DG, Lalaouna D, Tree JJ (2021) Burning the candle at both ends: have exoribonucleases driven divergence of regulatory RNA mechanisms in bacteria? Mbio 12:e01041-e1121
Melamed S, Peer A, Faigenbaum-Romm R et al (2016) Global mapping of small RNA-target interactions in bacteria. Mol Cell 63:884–897
Menendez-Gil P, Toledo-Arana A (2021) Bacterial 3′UTRs: a useful resource in post-transcriptional regulation. Front Mol Biosci 7:617633
Miyakoshi M, Chao Y, Vogel J (2015) Regulatory small RNAs from the 3′ regions of bacterial mRNAs. Curr Opin Microbiol 24:132–139
Morita T, Nishino R, Aiba H (2017) Role of the terminator hairpin in the biogenesis of functional Hfq-binding sRNAs. RNA 23:1419–1431
Overgaard M, Johansen J, Møller-Jensen J et al (2009) Switching off small RNA regulation with trap-mRNA. Mol Microbiol 73:790–800
Panja S, Schu DJ, Woodson SA (2013) Conserved arginines on the rim of Hfq catalyze base pair formation and exchange. Nucleic Acids Res 41:7536–7546
Papenfort K, Bouvier M, Mika F et al (2010) Evidence for an autonomous 5′ target recognition domain in an Hfq-associated small RNA. Proc Natl Acad Sci USA 107:20435–20440
Peschek N, Hoyos M, Herzog R et al (2019) A conserved RNA seed-pairing domain directs small RNA-mediated stress resistance in enterobacteria. EMBO J 38:e101650
Plumbridge J, Bossi L, Oberto J et al (2014) Interplay of transcriptional and small RNA-dependent control mechanisms regulates chitosugar uptake in Escherichia coli and Salmonella. Mol Microbiol 92:648–658
Poddar A, Azam MS, Kayikcioglu T et al (2021) Effects of individual base-pairs on in vivo target search and destruction kinetics of bacterial small RNA. Nat Commun 12:874
Ponath F, Hör J, Vogel J (2022) An overview of gene regulation in bacteria by small RNAs derived from mRNA 3′ ends. FEMS Microbiol Rev 46:fuac017
Quendera AP, Seixas AF, dos Santos RF et al (2020) RNA-binding proteins driving the regulatory activity of small non-coding RNAs in bacteria. Front Mol Biosci 7:78
Rasmussen AA, Johansen J, Nielsen JS et al (2009) A conserved small RNA promotes silencing of the outer membrane protein YbfM. Mol Microbiol 72:566–577
Rice JB, Vanderpool CK (2011) The small RNA SgrS controls sugar–phosphate accumulation by regulating multiple PTS genes. Nucleic Acids Res 39:3806–3819
Romeo T, Gong M, Liu MY et al (1993) Identification and molecular characterization of csrA, a pleiotropic gene from Escherichia coli that affects glycogen biosynthesis, gluconeogenesis, cell size, and surface properties. J Bacteriol 175:4744–4755
Safari F, Zare K, Negahdaripour M et al (2019) CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci 9:36
Salvail H, Lanthier-Bourbonnais P, Sobota JM et al (2010) A small RNA promotes siderophore production through transcriptional and metabolic remodeling. Proc Natl Acad Sci USA 107:15223–15228
Sauer E, Weichenrieder O (2011) Structural basis for RNA 3′-end recognition by Hfq. Proc Natl Acad Sci U S A 108:13065–13070
Seixas AF, Quendera AP, Sousa JP et al (2022) Bacterial response to oxidative stress and RNA oxidation. Front Genet 12:821535
Sharma CM, Darfeuille F, Plantinga TH et al (2007) A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev 21:2804–2817
Sharma CM, Papenfort K, Pernitzsch SR et al (2011) Pervasive post-transcriptional control of genes involved in amino acid metabolism by the Hfq-dependent GcvB small RNA. Mol Microbiol 81:1144–1165
Silva IJ, Barahona S, Eyraud A et al (2019) SraL sRNA interaction regulates the terminator by preventing premature transcription termination of rho mRNA. Proc Natl Acad Sci USA 116:3042–3051
Sittka A, Lucchini S, Papenfort K et al (2008) Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. Plos Genet 4:e1000163
Suzuki K, Babitzke P, Kushner SR et al (2006) Identification of a novel regulatory protein (CsrD) that targets the global regulatory RNAs CsrB and CsrC for degradation by RNase E. Genes Dev 20:2605–2617
Suzuki K, Wang X, Weilbacher T et al (2002) Regulatory circuitry of the CsrA/CsrB and BarA/UvrY systems of Escherichia coli. J Bacteriol 184:5130–5140
Udekwu KI, Darfeuille F, Vogel J et al (2005) Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev 19:2355–2366
Vakulskas CA, Pannuri A, Cortés-Selva D et al (2014) Global effects of the DEAD-box RNA helicase DeaD (CsdA) on gene expression over a broad range of temperatures. Mol Microbiol 92:945–958
Vakulskas CA, Potts AH, Babitzke P et al (2015) Regulation of bacterial virulence by Csr (Rsm) systems. Microbiol Mol Biol Rev 79:193–224
Vanderpool CK, Gottesman S (2004) Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol 54:1076–1089
Vanderpool CK, Gottesman S (2007) The novel transcription factor SgrR coordinates the response to glucose-phosphate stress. J Bacteriol 189:2238–2248
Viegas SC, Silva IJ, Saramago M et al (2011) Regulation of the small regulatory RNA MicA by ribonuclease III: a target-dependent pathway. Nucleic Acids Res 39:2918–2930
Wadler CS, Vanderpool CK (2007) A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci U S A 104:20454–20459
Wang L, Wang W, Li F et al (2015) Structural insights into the recognition of the internal A-rich linker from OxyS sRNA by Escherichia coli Hfq. Nucleic Acids Res 43:2400–2411
Woodson SA, Panja S, Santiago-Frangos A (2018) Proteins that chaperone RNA regulation. Microbiol Spectr 6:21
Yakhnin AV, Baker CS, Vakulskas CA et al (2013) CsrA activates flhDC expression by protecting flhDC mRNA from RNase E-mediated cleavage. Mol Microbiol 87:851–866
Zhang A, Schu DJ, Tjaden BC et al (2013) Mutations in interaction surfaces differentially Impact E. coli Hfq association with small RNAs and their mRNA targets. J Mol Biol 425:3678–3697
Acknowledgements
This work was supported by FCT—Fundação para a Ciência e a Tecnologia, I.P., through: MOSTMICRO-ITQB R&D Unit (UIDB/04612/2020, UIDP/04612/2020); LS4FUTURE Associated Laboratory (LA/P/0087/2020); Grant PTDC/BIA-MIC/32525/2017 to JMA; Doctoral Fellowships to AFQS (UI/BD/153390/2022) and JPS (2022.12475.BD).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Sousa, J.P., Silva, A.F.Q., Arraiano, C.M., Andrade, J.M. (2023). Bacterial Small RNAs: Diversity of Structure and Function. In: Barciszewski, J. (eds) RNA Structure and Function. RNA Technologies, vol 14. Springer, Cham. https://doi.org/10.1007/978-3-031-36390-0_12
Download citation
DOI: https://doi.org/10.1007/978-3-031-36390-0_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-36389-4
Online ISBN: 978-3-031-36390-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)