
Abstract
This chapter describes the optoelectronic properties of pure and mixed-halide perovskites. It discusses their absorption, carrier recombination, and emission quantum yields under high/low excitation intensities. For absorption, halide composition-dependent band gaps and absorption coefficients are summarized. Differences between various mixed-halide systems are discussed. Photogenerated carrier recombination dynamics are then examined where three principal recombination processes that occur in perovskites are explained and summarized through the development of a kinetic model. An important outcome is a summary of photocarrier behavior under different excitation intensities (e.g. low, medium, and high) with predictions about dominant recombination processes that prevail in each case. Corresponding expressions for emission quantum yields are developed. The chapter ends with a brief description of photoinduced ion migration in perovskites -a phenomenon that adds complexity to the optical response of these materials.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Hybrid perovskite
- Absorption
- Absorption coefficient
- Photoexcitation
- Carrier
- Recombination dynamics
- Carrier lifetimes
- Photoluminescence
- Emission quantum yields
- Recombination
- Generation
- Ion migration
1 Introduction
This chapter focuses on the optoelectronic properties of hybrid and all-inorganic lead halide perovskites. Such materials adopt the chemical stoichiometry ABX3 and are strong contenders for applications in solar photovoltaics. Among leading candidates are systems where the A cation is organic (e.g., methylammonium, CH3NH3+, MA; formamidinium, CH(NH2)2+, FA) or inorganic (e.g., Cs+), the B cation is Pb2+, and the X anion is I−, Br−, or Cl−. Important systems come from the methylammonium lead halide (CH3NH3PbX3, or MAPbX3), formamidinium lead halide [CH(NH2)2PbX3, or FAPbX3], and cesium lead halide (CsPbX3) families. Alloys are also possible and APbX3 materials can be produced as mixed cation, mixed anion, or even mixed cation/mixed anion alloys. Common mixed cation systems include A = FA/Cs or A = FA/MA alloys, e.g., FA1-yCsyPbI3 or FA1-yMAyPbI3. Mixed anion materials are often mixtures of iodine and bromine such as MAPb(I1-xBrx)3 and FAPb(I1-xBrx)3 while mixed cation/anion systems include FAxMAyCs1-x-yPb(I1-zBrz)3 (FAMACs).
Given prior discussion about the unique structural properties of ABX3 materials, we simply recall here that APbX3 perovskites adopt cubic/quasi-cubic structures at room temperature with corner sharing [PbI6]4− octahedra and with A+ cations (MA, FA, Cs) filling octahedral voids. Such structures satisfy the Goldschmidt tolerance factors required of ideal cubic structures (0.9 ≤ t ≤ 1.0) and for structures having tilted octahedra (0.7 < t < 0.9) [1]. The compositional diversity of mixed cation and mixed anion systems is limited by the existence of non-perovskite (δortho- and δhex- phases) phases that appear when A+ ionic radii are insufficient to stabilize interstitial voids in the structure [2, 3]. Although APbX3 perovskites can adopt other (e.g., orthorhombic) crystal structures at different temperatures [4], we focus on the photophysical properties of photovoltaically relevant cubic/pseudo-cubic structures in what follows.
The primary motivation for investigating and ultimately understanding the optical response of APbX3 perovskites stems from their successful implementation in high-efficiency photovoltaics. Today, perovskite solar cell power conversion efficiencies (PCEs) routinely exceed 20%. A maximum PCE of 25.7% has been reported in NREL’s benchmark efficiency chart [5] and will undoubtedly be supplanted shortly. These values collectively represent a remarkable rise of perovskite solar cell efficiencies given their modest starting value of 3.8% in 2009. In short, APbX3 perovskite solar cells are, from a PCE perspective, on par with crystalline silicon.
Responsible for this success are extraordinary and fortuitous perovskite properties. This entails facile solution processability, crystallinity despite low temperature processing, chemical and stoichiometric diversity, and large absorption efficiencies, all simultaneously coupled to low exciton binding energies, large carrier mobilities, and favorable energetics to engender defect tolerance. However, despite extensive research into improving perovskite solar cell PCEs, performance bottlenecks still remain. This prevents them from reaching their full Shockley-Queisser efficiency of ~31% for single-junction devices. A need therefore exists to fully understand the fundamental optical and electrical properties of APbX3 systems to realize their ultimate performance potentials.
2 Absorption
A key feature of lead halide perovskites is their favorable absorption properties. This includes sizable absorption coefficients (α), band edges close to the desired Shockley-Queisser value of 1.55 eV, and tunable absorption edges in mixed halide alloys. Figure 1a highlights these features by showing reported absorption spectra for common APbX3 systems.

In the red, at approximately 1.6 eV lie MAPbI3, FAPbI3, and CsPbI3. Near 2.2 eV are MAPbBr3, FAPbBr3, and CsPbBr3. Further to the blue at ~3.1 eV is MAPbCl3. The figure makes apparent that perovskite band gaps are sensitive to the choice of halide anion, whether I−, Br−, or Cl−. This has previously suggested that the A-site cation plays a lesser role in determining the optical response of these materials. Instead, optical transitions are thought to be primarily established by perovskite’s inorganic [PbI6]4− framework [10,13,14,]. This is supported by electronic structure calculations, which suggest A-site cation-related states being energetically removed from corresponding band edges. Cation-influenced octahedral tilting and lattice contraction [11] do, however, influence band edge energies, as evidenced by measurements on mixed cation perovskites such as MA1-xFAxPbI3 or FA1-xCsxPbI3 where band gaps can be altered using cation stoichiometry [12,13,14,15].
Figure 1b further illustrates the sensitivity of perovskite band gaps to halide stoichiometry by showing how increasing the Br fraction (x) in a MAPb(I1-xBrx)3 alloy causes its Eg to progressively shift towards the MAPbBr3 limit. Analogous trends are observed with MAPb(Cl1-xBrx)3 [16, 17] as well as with FAPb(I1-xBrx)3 [18]. The formation of continuous MAPb(I1-xClx)3 alloys is prevented by large differences in I− and Cl− ionic radii such that little if any Cl incorporation is achieved. Consequently, such systems are denoted MAPbI3(Cl) in what follows [19]. This ability to compositionally tune band gaps makes mixed halide alloys of potential use in tandem (perovskite/silicon) solar cells.
Figure 1 summarizes the absorption coefficients of these materials. Evident are sizable band edge values, which lie between 104 and 105 cm−1. These α-values correspond favorably to those of other photovoltaically relevant semiconductors. To illustrate, GaAs has an absorption coefficient of α ~ 104 cm−1 at its absorption edge. References [6, 20,21,22] highlight this favorable comparison by visually illustrating perovskite α-values relative to those of other semiconductors across a range of energies.
Table 1 summarizes compiled Eg and α-values for the various APbX3 perovskites being discussed. Apart from the general trends noted above, there is a sizable variability in reported values. MAPbI3 band gaps, for instance, range from 1.5 to 1.646 eV. This is also true of MAPbBr3 where Eg-values range from 2.24 to 2.392 eV. In either case, Eg spreads are of order 150 meV.
Associated absorption coefficients are also highly variable, as evident from tabulated α-values compiled at three different energies (2.0 eV, 2.3 eV, and 3.1 eV). In particular, Table 1 shows that MAPbI3 α-values at 2.0 eV range from 0.23 to 1.70 × 105 cm−1. At 2.3 eV, α-values range from 0.47 to 2.88 × 105 cm−1. Analogous variations exist with other perovskites. This variability and lack of accord are summarized visually in References [6, 36, 40, and 58].
Many reasons exist for apparent differences in reported optical parameters. Much has to do with variations in sample quality that stem from the numerous approaches used to prepare perovskite thin films. They include solution deposition (doctor blading, spray coating, slot-die coating, inkjet printing, etc.), solution deposition with solvent recrystallization (two-step spin-coating or antisolvent treatment), hot casting, chemical vapor deposition, and low-pressure vapor-assisted solution processing [73, 74]. Sample quality variability is especially highlighted when thin films are compared to APbX3 single crystals, which possess larger grains, reduced morphological disorder, and correspondingly reduced surface roughness [75].
Consequently, what results are thin film/single crystal specimens that possess varying degrees of crystallinity, thicknesses, apparent grain sizes, surface roughness, etc. All lead to measurement variations. Fujiwara [6], for example, attributes reported Eg and α-value variations to surface roughness that introduces significant light scattering to spectroscopic ellipsometry measurements. This degrades subsequent model extraction of perovskite optical constants (i.e., frequency-dependent refractive indices and permittivities) and leads to an underestimation of perovskite band gaps. When such surface roughness variations are explicitly accounted for, a closer agreement between researcher-reported α and Eg values is realized.
Beyond band gaps and absorption coefficients, Fig. 1 reveals other intriguing features of APbX3 materials. For MAPbI3, FAPbI3 and CsPbI3, band edges resemble those of bulk, direct gap semiconductors. No apparent excitonic features are seen. The absence of an excitonic response is corroborated by numerous estimates of their exciton binding energies (Eb), as summarized in Table 1. These estimates arise from magnetoabsorption and temperature-dependent emission and absorption measurements as well as from modeling experimental APbX3 band edge absorption spectra with Elliott’s model [76].
Table 1 shows a spread of reported Eb values. As with Eg and α, large variations can be seen where for MAPbI3 Eb ranges from 1.7 to 55 meV. Despite this, reported binding energies are of order kT and suggest that the optical response of MAPbI3 and FAPbI3 can be described in terms of free carriers. This conclusion is corroborated by time-resolved emission, transient differential absorption, and THz conductivity studies [76]. Note that this is not necessarily true of Br- and Cl-based APbX3 materials such as CsPbBr3 or MAPbCl3 where prominent band edge excitonic features are seen in the linear absorption. The suggestion is supported by their generally larger Eb-values in Table 1.
The specific origin of the optical transitions in lead halide perovskites has been the subject of numerous studies. Most entail density functional theory calculations to varying degrees of approximation [24, 36, 77]. Without delving into specifics, consensus exists that valence to conduction band transitions occurs at the R symmetry point and involves valence bands that originate from the antibonding combination of halide p and Pb(6s) orbitals. Corresponding conduction bands largely arise from Pb(6p) orbitals [77]. These bands are also responsible for above gap transitions and produce apparent structure at higher energies. For example, a feature in the absorption spectrum of MAPbI3 close to 2.5 eV (Fig. 1) is attributed to a valence/conduction band transition at the M symmetry point [24, 76, 77]. The antibonding nature of the APbX3 valence band is supported by apparent increases in perovskite band gap with increasing temperature. This contrasts itself to the response of traditional, tetrahedrally coordinated semiconductors where band gaps decrease (increase) with increasing (decreasing) temperature.
3 Carrier Dynamics
Having briefly summarized the general absorptive properties of APbX3 perovskites, we now provide insight into their carrier recombination processes, following photoexcitation. This is important since the fate of photogenerated carriers is fundamental to device operation and ultimately to their efficiencies. A kinetic model is therefore developed that qualitatively and quantitatively rationalizes the intrinsic photophysics of APbX3 systems [47, 78]. This includes experimental observations of photoluminescence, time-correlated emission decays, and transient differential absorption dynamics. In addition to numerical simulations, analytical approximations are provided to better visualize qualitative trends in both emission intensities and time-correlated decays. Although the model does not explicitly consider device operation, interested readers may refer to Reference [79] for an extension that includes charge extracting interfaces. Such a model has been used to establish the performance bottleneck(s) of a high-efficiency FAMACs solar cell.
In general, the primary recombination processes considered are (a) bimolecular (radiative) electron-hole recombination, (b) carrier trapping, and (c) nonradiative Auger recombination. The latter is nominally only important at high carrier densities, far beyond 1 sun conditions. These processes are summarized in Fig. 2 with the illustration showing photoexcitation creating transient electron and hole populations [n(t) and p(t)] in the perovskite conduction and valence bands. Carriers subsequently recombine via the three processes outlined above. Although the identity of APbX3 trap states remains debated, there appears to be some agreement that such states are electron traps. This is assumed in what follows. Note that other versions of this model exist, which account for exciton dissociation, unintentional doping, and carrier diffusion. The interested reader is therefore referred to References [80], [81], and [49] for details.

Schematic illustration of photophysical processes occurring in lead halide perovskites, following photoexcitation
Kinetic expressions that summarize the model are
where G is the initial charge generation rate (cm−3 s−1), linked to the excitation intensity (Iexc, W cm−2), i.e., \( G=\frac{I_{\textrm{exc}}\alpha }{h\nu} \) with α the absorption coefficient (cm−1) and hν the photon energy (J), n (p) is the electron (hole) concentration (cm−3) in the perovskite conduction (valence) band, kt is an electron trapping rate constant (cm3 s−1), Nt is a corresponding trap density (cm−3), nt is the trap population (cm−3), kb is a bimolecular radiative recombination rate constant (cm3 s−1, referred to as k2 in the literature), and kAuger is the Auger, three-carrier rate constant (cm6 s−1, referred to as k3 in the literature).
Numerous studies now provided estimates for the various rate constants in Eq. (1) and Fig. 2. These literature estimates are summarized in Table 2 across various APbX3 systems. An inspection shows that most work has focused on MAPbI3 and MAPbI3(Cl) thin films with relatively less work carried out on corresponding FA-based materials.
Table 2 also makes apparent that while variations in rate constants exist across systems and even within a given material, there is general consistency in their values. Bimolecular radiative rate constants are of order 10−10 cm3 s−1, while Auger rate constants are of order 10−28 cm6 s−1 [85]. Measured pseudo-first-order rate constants for electron trapping are of order k1 ~ 107 s−1 from where corresponding kt values are of order kt ~ 10−10 cm3 s−1, provided estimated trap densities of order Nt ~ 10−16 cm−3.
The general photogenerated carrier dynamics, predicted by Eq. (1) at different excitation intensities, are now illustrated. Implicit to the discussion is continuous wave (CW) excitation. An identical analysis can be conducted assuming pulsed excitation. However, this is not pursued here since common applications of perovskite materials generally entail CW excitation conditions. Interested readers may refer to References [97] and [98] for details of a pulsed excitation analysis.
4 Low Excitation Intensities
At low excitation intensities, carrier trapping dominates radiative recombination. Low photogenerated carrier densities further mean that nonradiative Auger pathways can be ignored. Consequently, under the assumption that the material is intrinsic, Eq. (1) reduces to
The equations make apparent that at steady state \( n=\frac{G}{k_t{N}_t}\propto G \) and \( p=\sqrt{\frac{G}{k_h}}\propto \sqrt{G} \). Since the emission rate and corresponding intensity, Iem, are proportional to the product of n and p, Iem grows with increasing Iexc (G) in a power law fashion. Its growth exponent is ~3/2, i.e., \( {I}_{\textrm{em}}\propto {G}^{\frac{3}{2}} \).
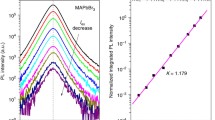
Figure 3a illustrates this for the case where Nt = 1016 cm−3. Employed rate constants for the numerical simulation of Eq. (1) are kb = 10−10 cm3 s−1, kt = 10−9 cm3 s−1, kh = 10−11 cm3 s−1, and kAuger = 10−28 cm6 s−1. The model therefore reveals that Iem grows as G1.6 at low G where recombination is primarily trap-mediated (shaded red region). Figure 3b shows identical behavior for simulations where Nt has been varied between Nt = 1015 and 1018 cm−3. In all cases, fit-extracted growth exponents range from m = 1.5 to 1.6. Of note is the increasing range of G-values where m ~ 1.5. This is rationalized by delayed trap saturation due to larger Nt-values.

(a) Iem versus G for Nt = 1016 cm−3. Dashed lines are linear fits to the data in a given growth regime (shaded regions). Fit-extracted growth exponents shown. G-values are linked to associated Iexc, assuming 405 nm excitation with α = 105 cm−1, for reference purposes. (b) Iem versus G for Nt varying between Nt = 1015–1018 cm−3. Dashed lines are linear fits in the trap-mediated recombination regime. Fit-extracted growth exponents shown. (c) Iem versus G data for a MAPbI3 single crystal and thin film. Data from Reference [78]. Dashed lines are fits to the data at smaller G with fit-extracted growth exponents shown
Figure 3c shows experimental data for a MAPbI3 single crystal and thin film [78] that corroborate these model predictions. Acquired over ~3 orders of magnitude in G, the data reveal m ~ 1.5 power law growth exponents for either material, as established by fits to low G Iem-values (dashed lines with fit-extracted m-values shown). Differences in the range of Iem-values over which m ~ 1.5 qualitatively agree with Fig. 3b and suggest underlying Nt-value variations between MAPbI3 single crystals and thin films. Other data acquired on MAPbI3 and MAPbI3(Cl) over 8 orders of magnitude in G reveal identical m ~ 1.5 power law growth exponents at low G [47]. This further corroborates the analysis and conclusions drawn here. Note that under (low irradiance) pulsed excitation, analogous power law growth of the integrated emission intensity is predicted with an ideal model growth exponent of m = 2.0 [97, 98]. Such quadratic emission growth has previously been observed for MAPbI3 and MAPbI3(Cl) thin films [47].
Next, by assuming above-simplified kinetic expression for n and p, corresponding (normalized) photoluminescence decays take the form
At small G or short times, the numerator in Eq. (2) dominates. Decays are therefore near exponential with an associated pseudo-first-order decay constant of ktNt~107 s−1 (Table 2). Figure 4a illustrates this for the model predicted decay when G = 1015 cm−3. An accompanying (superimposed) dotted line is Eq. (2). This qualitative prediction is supported by numerous studies, which report near exponential decays at low Iexc for various perovskite systems [80, 81, 84, 87, 99].

(a) Predicted PL decays from Eq. (1) for variable G between G = 1015 and 1023 cm−3. Superimposed over the data are analytical predictions from Eqs. (2)–(4) (dotted lines). (b) Predicted decays for G = 1015–7 × 1016 cm−3. (c) MAPbI3 PL decays from Reference [87]. (d) Large G MAPbI3 PL decays from Reference [87] replotted. Dashed lines are linear fits to the data
5 Intermediate Excitation Intensities
As Iexc increases, progressive trap filling (nt → Nt) reduces the electron trapping rate such that n → p. This simplifies Eq. (1) and leads to the following effective rate expressions:
Invoking steady state conditions then means that \( n=\sqrt{\frac{G}{k_b}}\propto \sqrt{G} \) with \( p=\frac{-{k}_t{N}_t+\sqrt{{\left({k}_t{N}_t\right)}^2+4G}}{2}\propto \sqrt{G} \). The emission intensity therefore transitions to power law growth with a growth exponent of m ~ 1. Figure 3a illustrates this transition by showing a fit to the model data, highlighted in the shaded blue region. A fit-extracted power law growth exponent of m = 0.99 is found. Analogous behavior is observed in Fig. 3b when Nt is varied between 1015 and 1018 cm−3. In whole, the model data makes evident that increasing Nt extends the region of trap-mediated recombination (m ~ 1.5) to larger G-values before bimolecular (radiative) recombination (m ~ 1.0) dominates carrier dynamics following photoexcitation.
Using the above simplified rate expressions, associated (normalized) photoluminescence decays adopt the following bimolecular form:
where Fig. 4a shows model-predicted decays for G = 1019 and G = 1022 cm−3 using the same parameters employed earlier at low Iexc. Analytical results from Eq. (3) are superimposed atop the model decays and are in excellent agreement. Model decays also highlight the transition in kinetic response in this Iexc regime. Figure 4b illustrates this for G-values between G = 1016 and 1017 cm−3 where for smaller G-values, near exponential behavior is seen. With increasing G, an apparent near exponential to bimolecular transition occurs. Such PL(t) transitions have previously been reported in the literature [80, 81, 87, 99] and an example from a MAPbI3 thin film is shown in Fig. 4c.
6 High Excitation Intensities
At sufficiently high excitation intensities, trap saturation causes bimolecular (radiative) recombination to dominate. In this limit, Eq. (1) effectively becomes
so that \( n=p=\sqrt{\frac{G}{k_b}}\propto \sqrt{G} \). Iem thus continues to grow in a power law fashion with a growth exponent of m ~ 1. Figure 3 illustrates this for Nt = 1016 cm−3 (Fig. 3a) and across Nt-values between Nt = 1015 and 1018 cm−3 (Fig. 3b). It should be mentioned that a growth exponent of m ~ 1 is common to this intensity regime under pulsed excitation [47, 97, 98].
An associated (normalized) photoluminescence decay takes the bimolecular form
which is near identical to the expression derived earlier for intermediate excitation intensities (Eq. 3). Figure 4a plots a model-predicted decay for G = 1023 cm−3 with Eq. (4) superimposed. Again, there is excellent agreement with the analytical approximation.
Beyond bimolecular fits, predicted bimolecular decays can be confirmed visually by plotting \( \sqrt{\frac{I_{\textrm{em},\max }}{I_{\textrm{em}}(t)}}-1 \) versus time. In this case, linear behavior is expected [100]. Figure 4d illustrates this for the large G experimental data in Fig. 4c. Evident linear behavior is observed, as highlighted by dashed, linear fits.
Above this excitation regime, the onset of Auger-mediated nonradiative recombination causes emission efficiencies to decrease. This stems from competitive, nonradiative recombination of carriers. What results then is sublinear growth of Iem with an associated power law growth exponent m < 1. Figure 3a explicitly illustrates the onset of Auger recombination for Nt = 1016 cm−3 at high G where the simulated data shows m < 1. It can also be shown that in this regime, plotting \( \left[\frac{I_{\textrm{em},\max }}{I_{\textrm{em}}(t)}-1\right] \) versus time yields linear behavior [100].
7 Emission Quantum Yields
Equation (1) simultaneously allows internal emission quantum yields (QYs) to be estimated through the ratio of the bimolecular radiative recombination rate to the initial carrier generation rate, i.e.
The importance of this metric is that high emission efficiencies are requisite for optimizing APbX3 photovoltaic performance. More specifically, it is the associated external quantum efficiency (EQE, EQE = ηeQY where ηe is a photon extraction efficiency) that is fundamentally linked to the maximum open circuit voltage (and PCE) achievable in a solar cell [101]. The seemingly contradictory conclusion that arises then is that a good solar cell must also be a good light emitter [102].
Figure 5 shows model-predicted (internal) QYs plotted as functions of Nt when Nt is varied between 1015 cm−3 and 1018 cm−3. As with Figs. 3 and 4, employed rate constants have been kept constant at their nominal literature values of kb ~ 10−10 cm3 s−1, kt ~ 10−9 cm3 s−1, and kh ~ 10−11 cm3 s−1. Fig. 5 also provides model-predicted EQEs via \( \textrm{EQE}=\frac{\eta_e\left({k}_b np\right)}{G} \).

(a) Predicted (internal) QYs for variable G between G = 1019–1027 cm−3 and Nt between 1015 and 1018 cm−3. For reference purposes, G-values are linked to associated Iexc assuming 405 nm excitation with α = 105 cm−1. (b) Experimental EQE estimates for a MAPbI3 single crystal and thin film. Data from Reference [78]. Fit extracted Nt-values shown
The resulting figure illustrates several things. First, as discussed earlier, at low Iexc, trapping dominates carrier recombination. This leads to low QYs. Attesting to this are experimental perovskite, 1 sun EQEs in Table 3. Values for thin films, (surface) treated thin films, and devices are shown. Inspection of the data reveals that reported 1 sun EQEs are generally on the order of several percent with some notable exceptions. This is consistent with the lower QYs seen at low G in Fig. 5. Of note is that this bulk perovskite data contrasts itself to analogous results, summarized in Reference [115] for perovskite nanocrystals (NCs). In these materials, EQEs regularly approach or attain unity values.
Next, Table 3 shows that treating lead halide perovskite thin films with Lewis bases such as trioctylphosphine oxide (TOPO) or pyridine improves their EQEs. However, they only increase values to numbers of order 10%. This indicates that while defect passivation is possible, significant trap densities still remain. This is again unlike the case of perovskite NCs where effective surface passivation does appear possible and which leads to unity EQEs [116]. Finally, Table 3 shows that device EQEs are all suppressed from corresponding thin film or treated thin film values due to the inevitable competition for carriers by electron and hole extraction interfaces present in working devices.
Figure 5 shows that maximal QYs are achieved upon trap saturation at high excitation intensities. In the case where Nt = 1016 cm−3, near unity internal QYs are realized close to 1 sun conditions. The figure further shows that increasing Nt simply leads to progressively larger Iexc-values required to achieve peak QYs, which themselves become progressively smaller. In all cases, maximum QYs persist until a critical G beyond which the onset of Auger-mediated carrier recombination causes them to fall as discussed earlier.
This QY behavior has previously been modeled by reducing the kinetics in Eq. (1) to [85].
In Eq. (6), N is an effective electron-hole density, A is a generic first-order rate constant that describes nonradiative, trap-mediated recombination of photogenerated carriers, B is a second-order (radiative) rate constant, and C is a third-order rate constant that accounts for nonradiative Auger recombination. The rate constants A, B, and C are effectively k1, kb, and kAuger in Table 2. A corresponding internal QY is then
Equation (7) can be recast in terms of EQE via \( \textrm{EQE}=\frac{\eta_e{\textrm{BN}}^2}{\textrm{AN}+{\eta}_e{\textrm{BN}}^2+{\textrm{CN}}^3} \). In either case, a peak QY can be found by taking the derivative of QY (EQE) with respect to N to find a critical point (i.e., \( \frac{d\textrm{QY}}{dN}=\frac{d\textrm{EQE}}{dN}=0 \)) [85]. A resulting optimal carrier density (Nopt) is then
and is linked to a corresponding maximum internal QY of
with a corresponding maximum EQE of \( {\textrm{EQE}}_{\textrm{max}}=\frac{1}{1+\frac{2\sqrt{AC}}{\eta_eB}}\sim \frac{1}{1+\frac{2\sqrt{k_1{k}_{\textrm{Aug}}}}{\eta_e{k}_2}} \).
Equations (6) and (7) can be used to model experimental EQEs to extract relevant rate constants. They are, however, not predictive in that tabulated rate constants from Table 2 cannot be used to estimate QYs and EQE a priori. This is because Figs. 3–5 show that trap saturation occurs due to nt → Nt. Consequently, the pseudo-first-order trapping rate constant (here A) is Iexc-dependent. Immediate application of Eqs. (8) and (9) therefore leads to predicted Nopt and QYmax smaller than those revealed by full numerical simulations of Eq. (1) (Fig. 5).
Finally, beyond trap-mediated recombination, an important reason for small overall EQE values and for why EQEs are smaller than internal QYs is photon trapping within the perovskite. This originates from refractive index differences with the surrounding medium and is most prominent in APbX3 films and crystals where physical sizes approach the wavelength of light. This leads to estimated photon extraction efficiencies of ηe ~ 10% [104, 105]. Note that such trapping is not significant for NCs as they effectively behave as dipole emitters. This rationalizes why unity/near unity EQEs are readily seen with these materials [115].
8 Ion Migration
Finally, despite all of the notable properties of APbX3 materials, preventing their widespread commercialization is their well-known environmental sensitivities. Addressing this have been a number of studies [117, 118]. Less recognized but equally important are intrinsic instabilities linked to ion migration. Namely, both cations and anions in APbX3 materials are mobile with ion mobilities stemming from vacancy-mediated ion migration under bias or under illumination.
For cations, evidence for bias-induced A+ migration comes from observed device hysteresis thought to stem from cation accumulation at electrodes [119,120,121]. The phenomenon is better illustrated using more direct measurements such as time-of-flight secondary ion mass spectrometry and super-resolution infrared absorption measurements [122, 123], which explicitly reveal cation migration and accumulation near electrodes.
For anions, a well-known phenomenon is light-induced photosegregation whereby shining light on mixed I−/Br− systems [e.g., MAPb(I1-xBrx)3] induces halide segregation. This results in the formation of I-rich inclusions within parent, mixed halide perovskites. Such photoinduced halide segregation is experimentally observed as progressive photoluminescence redshifts due to photogenerated carriers being funneled to I-rich domains. Such domains act as emissive recombination centers because of favorable (valence) band offsets relative to those of parent mixed halide materials. Observed redshifts/photosegregation are reversible when specimens are no longer illuminated with remixing being entropically driven. References [4, 124, and 125] provide more details on the phenomenon.
At this point, an inevitable question that arises is the connection between the earlier photocarrier dynamics and the bias−/light-induced ion migration described here. Since ion migration, whether cation or anion, is thought to be vacancy-mediated and since point defects are likely responsible for carrier trapping, a connection between the two is suggested. Evidence for this can already be found in the literature where References [126, 127, and 128] already suggest that decreasing defect densities mitigates ion migration. Studies linking the two topics, however, are limited. Consequently, more work is required to establish a comprehensive picture of ion-inclusive carrier dynamics that follow photoexcitation of APbX3 systems. As such, linking the two sets the direction for future investigations of these unique materials.
References
Goldschmidt, V. (1926). Die Gesetze der Krystallochemie. Die Naturwissenschaften, 14, 477–485.
Manser, J., Christians, J., & Kamat, P. (2016). Intriguing optoelectronic properties of metal halide perovskites. Chemical Reviews, 116, 12956–13008.
Stoumpos, C., & Kanatzidis, M. (2015). The renaissance of halide perovskites and their evolution as emerging semiconductors. Accounts of Chemical Research, 48, 2791–2802.
Brennan, M., Ruth, A., Kamat, P., & Kuno, M. (2020). Photoinduced anion segregation in mixed halide perovskites. Trends in Chemistry, 2, 282–301.
Best research-cell efficiency chart [Internet]. Nrel.gov. 2022 [cited 26 January 2022]. Available from https://www.nrel.gov/pv/cell-efficiency.html.
Fujiwara, H., Kato, M., Tamakoshi, M., Miyadera, T., & Chikamatsu, M. (2018). Optical characteristics and operational principles of hybrid perovskite solar cells. Physica Status Solidi (a), 215, 1700730.
Fujiwara, H., & Collins, R. W. (2018). Organic-inorganic hybrid perovskites. Spectroscopic Ellipsometry for Photovoltaics, 2, 471–493.
Yan, W., Guo, Y., Beri, D., Dottermusch, S., Chen, H., & Richards, B. S. (2020). Experimental determination of complex optical constants of air-stable inorganic CsPbI3 perovskite thin films. Physica Status Solidi (RRL)–Rapid Research Letters, 14, 2000070.
Yan, W., Mao, L., Zhao, P., Mertens, A., Dottermusch, S., Hu, H., Jin, Z., & Richards, B. S. (2020). Determination of complex optical constants and photovoltaic device design of all-inorganic CsPbBr3 perovskite thin films. Optics Express, 28, 15706–15717.
Chen, H., Lee, M., & Chen, C. (2016). Wavelength-dependent optical transition mechanisms for light-harvesting of perovskite MAPbI3 solar cells using first-principles calculations. Journal of Materials Chemistry C, 4, 5248–5254.
Prasanna, R., Gold-Parker, A., Leijtens, T., Conings, B., Babayigit, A., Boyen, H. G., Toney, M. F., & McGehee, M. D. (2017). Band gap tuning via lattice contraction and octahedral tilting in perovskite materials for photovoltaics. Journal of the American Chemical Society, 139, 11117–11124.
Pellet, N., Gao, P., Gregori, G., Yang, T., Nazeeruddin, M., Maier, J., & Grätzel, M. (2014). Mixed-organic-cation perovskite photovoltaics for enhanced solar-light harvesting. Angewandte Chemie International Edition, 53, 3151–3157.
Ono, L., Juarez-Perez, E., & Qi, Y. (2017). Progress on perovskite materials and solar cells with mixed cations and halide anions. ACS Applied Materials & Interfaces, 9, 30197–30246.
Li, Z., Yang, M., Park, J., Wei, S., Berry, J., & Zhu, K. (2015). Stabilizing perovskite structures by tuning tolerance factor: Formation of formamidinium and cesium lead iodide solid-state alloys. Chemistry of Materials, 28, 284–292.
Weber, O., Charles, B., & Weller, M. T. (2016). Phase behaviour and composition in the formamidinium–methylammonium hybrid lead iodide perovskite solid solution. Journal of Materials Chemistry A, 4, 15375–15382.
Sadhanala, A., Ahmad, S., Zhao, B., Giesbrecht, N., Pearce, P., Deschler, F., Hoye, R. L. Z., Gödel, K. C., Bein, T., Docampo, P., Dutton, S. E., De Volder, M. F. L., & Friend, R. H. (2015). Blue-green color tunable solution processable organolead chloride–bromide mixed halide perovskites for optoelectronic applications. Nano Letters, 15, 6095–6101.
Tang, S., Xiao, X., Hu, J., Gao, B., Chen, H., Peng, Z., Wen, J., Era, M., & Zou, D. (2020). Solvent-free mechanochemical synthesis of a systematic series of pure-phase mixed-halide perovskites MAPb(IxBr1−x)3 and MAPb(BrxCl1-x)3 for continuous composition and band-gap tuning. ChemPlusChem, 85, 240–246.
Rehman, W., Milot, R. L., Eperon, G. E., Wehrenfennig, C., Boland, J. L., Snaith, H. J., Johnston, M. B., & Herz, L. M. (2015). Charge-carrier dynamics and mobilities in formamidinium lead mixed-halide perovskites. Advanced Materials, 27, 7938–7944.
Colella, S., Mosconi, E., Fedeli, P., Listorti, A., Gazza, F., Orlandi, F., Ferro, P., Besagni, T., Rizzo, A., Calestani, G., Gigli, G., De Angelis, F., Mosca, R., & Gigli, G. (2013). MAPbI3-xClx mixed halide perovskite for hybrid solar cells: The role of chloride as dopant on the transport and structural properties. Chemistry of Materials, 25, 4613–4618.
De Wolf, S., Holovsky, J., Moon, S. J., Löper, P., Niesen, B., Ledinsky, M., Haug, F. J., Yum, J. H., & Ballif, C. (2014). Organometallic halide perovskites: Sharp optical absorption edge and its relation to photovoltaic performance. Journal of Physical Chemistry Letters, 5, 1035–1039.
Ziang, X., Shifeng, L., Laixiang, Q., Shuping, P., Wei, W., Yu, Y., Li, Y., Zhijian, C., Shufeng, W., Honglin, D., Minghui, Y., & Qin, G. G. (2015). Refractive index and extinction coefficient of CH3NH3PbI3 studied by spectroscopic ellipsometry. Optical Materials Express, 5, 29–43.
Xie, Z., Sun, S., Yan, Y., Zhang, L., Hou, R., Tian, F., & Qin, G. G. (2017). Refractive index and extinction coefficient of NH2CH=NH2PbI3 perovskite photovoltaic material. Journal of Physics: Condensed Matter, 29, 245702.
Ghimire, K., Cimaroli, A., Hong, F., Shi, T., Podraza, N., & Yan, Y.. (2015, June 14). Spectroscopic ellipsometry studies of CH3NH3PbX3 thin films and their growth evolution. In 2015 IEEE 42nd Photovoltaic Specialist Conference (PVSC) (pp. 1–5). IEEE.
Leguy, A. M., Azarhoosh, P., Alonso, M. I., Campoy-Quiles, M., Weber, O. J., Yao, J., Bryant, D., Weller, M. T., Nelson, J., Walsh, A., Van Schilfgaarde, M., & Barnes, P. R. F. (2016). Experimental and theoretical optical properties of methylammonium lead halide perovskites. Nanoscale, 8, 6317–6327.
Phillips, L. J., Rashed, A. M., Treharne, R. E., Kay, J., Yates, P., Mitrovic, I. Z., Weerakkody, A., Hall, S., & Durose, K. (2016). Maximizing the optical performance of planar CH3NH3PbI3 hybrid perovskite heterojunction stacks. Solar Energy Materials and Solar Cells, 147, 327–233.
Loper, P., Stuckelberger, M., Niesen, B., Werner, J., Filipic, M., Moon, S. J., Yum, J. H., Topic, M., De Wolf, S., & Ballif, C. (2015). Complex refractive index spectra of CH3NH3PbI3 perovskite thin films determined by spectroscopic ellipsometry and spectrophotometry. Journal Physical Chemistry Letters, 6, 66–71.
Manzoorm, S., Häusele, J., Bush, K. A., Palmstrom, A. F., Carpenter, J., Zhengshan, J. Y., Bent, S. F., Mcgehee, M. D., & Holman, Z. C. (2018). Optical modeling of wide-bandgap perovskite and perovskite/silicon tandem solar cells using complex refractive indices for arbitrary-bandgap perovskite absorbers. Optics Express, 26, 27441–27460.
Ball, J. M., Stranks, S. D., Hörantner, M. T., Hüttner, S., Zhang, W., Crossland, E. J., Ramirez, I., Riede, M., Johnston, M. B., Friend, R. H., & Snaith, H. J. (2015). Optical properties and limiting photocurrent of thin-film perovskite solar cells. Energy & Environmental Science, 8, 602–609.
Marronnier, A., Lee, H., Lee, H., Kim, M., Eypert, C., Gaston, J. P., Roma, G., Tondelier, D., Geffroy, B., & Bonnassieux, Y. (2018). Electrical and optical degradation study of methylammonium-based perovskite materials under ambient conditions. Solar Energy Materials and Solar Cells, 178, 179–185.
Bailey, C. G., Piana, G. M., & Lagoudakis, P. G. (2019). High-energy optical transitions and optical constants of CH3NH3PbI3 measured by spectroscopic ellipsometry and spectrophotometry. Journal of Physical Chemistry C, 123, 28795–28801.
Lin, Q., Armin, A., Nagiri, R. C., Burn, P. L., & Meredith, P. (2015). Electro-optics of perovskite solar cells. Nature Photonics, 9, 106–112.
Leguy, A. M., Hu, Y., Campoy-Quiles, M., Alonso, M. I., Weber, O. J., Azarhoosh, P., Van Schilfgaarde, M., Weller, M. T., Bein, T., Nelson, J., Docampo, P., & Barnes, P. R. F. (2015). Reversible hydration of CH3NH3PbI3 in films, single crystals, and solar cells. Chemistry of Materials, 27, 3397–3407.
Wang, Z., Yuan, S., Li, D., Jin, F., Zhang, R., Zhan, Y., Lu, M., Wang, S., Zheng, Y., Guo, J., Fan, Z., & Chen, L. (2016). Influence of hydration water on CH3NH3PbI3 perovskite films prepared through one-step procedure. Optics Express, 24, A1431–A1443.
Wang, X., Gong, J., Shan, X., Zhang, M., Xu, Z., Dai, R., Wang, Z., Wang, S., Fang, X., & Zhang, Z. (2018). In situ monitoring of thermal degradation of CH3NH3PbI3 films by spectroscopic ellipsometry. Journal of Physical Chemistry C, 123, 1362–1369.
Demchenko, D. O., Izyumskaya, N., Feneberg, M., Avrutin, V., Özgür, Ü., Goldhahn, R., & Morkoç, H. (2016). Optical properties of the organic-inorganic hybrid perovskite CH3NH3PbI3: Theory and experiment. Physical Review B, 94, 075206.
Shirayama, M., Kadowaki, H., Miyadera, T., Sugita, T., Tamakoshi, M., Kato, M., Fujiseki, T., Murata, D., Hara, S., Murakami, T. N., Fujimoto, S., Chikamatsu, M., & Fujiwara, H. (2016). Optical transitions in hybrid perovskite solar cells: Ellipsometry, density functional theory, and quantum efficiency analyses for CH3NH3PbI3. Physical Review Applied, 5, 014012.
Jiang, Y., Soufiani, A. M., Gentle, A., Huang, F., Ho-Baillie, A., & Green, M. A. (2016). Temperature dependent optical properties of CH3NH3PbI3 perovskite by spectroscopic ellipsometry. Applied Physics Letters, 108, 061905.
Guerra, J. A., Tejada, A., Korte, L., Kegelmann, L., Töfflinger, J. A., Albrecht, S., Rech, B., & Weingärtner, R. (2017). Determination of the complex refractive index and optical bandgap of CH3NH3PbI3 thin films. Journal of Applied Physics, 121, 173104.
Ghimire, K., Zhao, D., Cimaroli, A., Ke, W., Yan, Y., & Podraza, N. J. (2016). Optical monitoring of CH3NH3PbI3 thin films upon atmospheric exposure. Journal of Physics D: Applied Physics, 49, 405102.
Green, M. A., Jiang, Y., Soufiani, A. M., & Ho-Baillie, A. (2015). Optical properties of photovoltaic organic–inorganic lead halide perovskites. Journal Physical Chemistry Letters, 6, 4774–4785.
Yamada, Y., Nakamura, T., Endo, M., Wakamiya, A., & Kanemitsu, Y. (2015). Photoelectronic responses in solution-processed perovskite CH3NH3PbI3 solar cells studied by photoluminescence and photoabsorption spectroscopy. IEEE Journal of Photovoltaics, 5, 2156–3381.
Ziffer, M., Mohammed, J., & Ginger, D. S. (2016). Electroabsorption spectroscopy measurements of the exciton binding energy, electron–hole reduced effective mass, and band gap in the perovskite CH3NH3PbI3. ACS Photonics, 3, 1060–1068.
Yang, Y., Ostrowski, D., France, R., Zhu, K., van de Lagemaat, J., Luther, J., & Beard, M. C. (2015). Observation of a hot-phonon bottleneck in lead-iodide perovskites. Nature Photonics, 10, 53–59.
Yang, Y., Yang, M., Li, Z., Crisp, R., Zhu, K., & Beard, M. C. (2015). Comparison of recombination dynamics in CH3NH3PbBr3 and CH3NH3PbI3 perovskite films: influence of exciton binding energy. Journal of Physical Chemistry Letters, 6, 4688–4692.
Miyata, A., Mitioglu, A., Plochocka, P., Portugall, O., Wang, J., Stranks, S., Snaith, H. J., & Nicholas, R. J. (2015). Direct measurement of the exciton binding energy and effective masses for charge carriers in organic–inorganic tri-halide perovskites. Nature Physics, 11, 582–587.
Sun, S., Salim, T., Mathews, N., Duchamp, M., Boothroyd, C., Xing, G., Sum, T. S., & Lam, Y. M. (2014). The origin of high efficiency in low-temperature solution-processable bilayer organometal halide hybrid solar cells. Energy & Environmental Science, 7, 399–407.
Saba, M., Cadelano, M., Marongiu, D., Chen, F., Sarritzu, V., Sestu, N., Figus, C., Aresti, M., Piras, R., Lehmann, A. G., Cannas, C., Musinu, A., Quochi, F., Mura, A., & Bongiovanni, G. (2014). Correlated electron–hole plasma in organometal perovskites. Nature Communications, 5, 5049.
Sestu, N., Cadelano, M., Sarritzu, V., Chen, F., Marongiu, D., Piras, R., Mainas, M., Quochi, F., Saba, M., Mura, A., & Bongiovanni, G. (2015). Absorption f-sum rule for the exciton binding energy in methylammonium lead halide perovskites. Journal of Physical Chemistry Letters, 6, 4566–4572.
Savenije, T. J., Ponseca, C. S., Jr., Kunneman, L., Abdellah, M., Zheng, K., Tian, Y., Zhu, Q., Canton, S. E., Scheblykin, I. G., Pullerits, T., Yartsev, A., & Sundström, V. (2014). Thermally activated exciton dissociation and recombination control the carrier dynamics in organometal halide perovskite. Journal of Physical Chemistry Letters, 5, 2189–2194.
Hirasawa, M., Ishihara, T., & Goto, T. (1994). Exciton features in 0-, 2-, and 3-dimensional networks of [PbI6]4−octahedra. Journal of the Physical Society of Japan, 63, 3870–3879.
Zhu, X., Su, H., Marcus, R., & Michel-Beyerle, M. E. (2014). Computed and experimental absorption spectra of the perovskite CH3NH3PbI3. Journal of Physical Chemistry Letters, 5, 3061–3065.
Ishihara, T. (1994). Optical properties of PbI-based perovskite structures. Journal of Luminescence, 60-61, 269–274.
Tanaka, K., Takahashi, T., Ban, T., Kondo, T., Uchida, K., & Miura, N. (2003). Comparative study on the excitons in lead-halide-based perovskite-type crystals CH3NH3PbBr3 CH3NH3PbI3. Solid State Communications, 127(9–10), 619–623.
D’Innocenzo, V., Grancini, G., Alcocer, M., Kandada, A., Stranks, S. D., Lee, M. M., Lanzani, G., Snaith, H. J., & Petrozza, A. (2014). Excitons versus free charges in organo-lead tri-halide perovskites. Nature Communications, 5, 3586.
Kato, M., Fujiseki, T., Miyadera, T., Sugita, T., Fujimoto, S., Tamakoshi, M., Chikamatsu, M., & Fujiwara, H. (2017). Universal rules for visible-light absorption in hybrid perovskite materials. Journal of Applied Physics, 121, 115501.
Park, J. S., Choi, S., Yan, Y., Yang, Y., Luther, J. M., Wei, S. H., Parilla, P., & Zhu, K. (2015). Electronic structure and optical properties of α-CH3NH3PbBr3 perovskite single crystal. Journal of Physical Chemistry Letters, 6, 4304–4308.
Xing, G., Mathews, N., Sun, S., Lim, S., Lam, Y., Grätzel, M., Mhaisalkar, S., & Sum, T. C. (2013). Long-range balanced electron- and hole-transport lengths in organic-inorganic CH3NH3PbI3. Science, 342, 344–347.
Jiang, Y., Green, M. A., Sheng, R., & Ho-Baillie, A. (2015). Room temperature optical properties of organic–inorganic lead halide perovskites. Solar Energy Materials and Solar Cells, 137, 253–257.
Xing, G., Mathews, N., Lim, S. S., Yantara, N., Liu, X., Sabba, D., Grätzel, M., Mhaisalkar, S., & Sum, T. C. (2014). Low-temperature solution-processed wavelength-tunable perovskites for lasing. Nature Materials, 13, 476–480.
Galkowski, K., Mitioglu, A., Miyata, A., Plochocka, P., Portugall, O., Eperon, G., Wang, J., Stergiopoulos, J., Stranks, S., Snaith, H. J., & Nicholas, R. J. (2016). Determination of the exciton binding energy and effective masses for methylammonium and formamidinium lead tri-halide perovskite semiconductors. Energy & Environmental Science, 9, 962–970.
Liu, Y., Wang, J., Zhu, N., Liu, W., Wu, C., Liu, C., Xiao, L., Chen, Z., & Wang, S. (2019). Investigation on binding energy and reduced effective mass of exciton in organic–inorganic hybrid lead perovskite films by a pure optical method. Optics Letters, 44, 3474–3477.
Protesescu, L., Yakunin, S., Bodnarchuk, M., Krieg, F., Caputo, R., Hendon, C., Yang, R. X., Walsh, A., & Kovalenko, M. V. (2015). Nanocrystals of cesium lead halide perovskites (CsPbX3, X = Cl, Br, and I): Novel optoelectronic materials showing bright emission with wide color gamut. Nano Letters, 15, 3692–3696.
Mannino, G., Deretzis, I., Smecca, E., La Magna, A., Alberti, A., Ceratti, D., & Cahen, D. (2020). Temperature-dependent optical band gap in CsPbBr3, MAPbBr3, and FAPbBr3 single crystals. Journal of Physical Chemistry Letters, 11, 2490–2496.
Alias, M. S., Dursun, I., Saidaminov, M. I., Diallo, E. M., Mishra, P., Ng, T. K., Bakr, O. M., & Ooi, B. S. (2016). Optical constants of CH3NH3PbBr3 perovskite thin films measured by spectroscopic ellipsometry. Optics Express, 24, 16586–16594.
Awasthi, K., Du, K., Wang, C., Tsai, C., Hamada, M., Narra, S., Diau, E., & Ohta, N. (2018). Electroabsorption studies of multicolored lead halide perovskite nanocrystalline solid films. ACS Photonics, 5, 2408–2417.
Ruf, F., Aygüler, M., Giesbrecht, N., Rendenbach, B., Magin, A., Docampo, P., Kalt, H., & Hetterich, M. (2019). Temperature-dependent studies of exciton binding energy and phase-transition suppression in (Cs,FA,MA)Pb(I,Br)3 perovskites. APL Materials, 7, 031113.
Koutselas, I., Ducasse, L., & Papavassiliou, G. C. (1996). Electronic properties of three- and low-dimensional semiconducting materials with Pb halide and Sn halide units. Journal of Physics: Condensed Matter, 8, 1217–1227.
Chen, C., Wu, D., Yuan, M., Yu, C., Zhang, J., Li, C., & Duan, Y. (2021). Spectroscopic ellipsometry study of CsPbBr3 perovskite thin films prepared by vacuum evaporation. Journal of Physics D: Applied Physics, 54, 224002.
Zhao, M., Shi, Y., Dai, J., & Lian, J. (2018). Ellipsometric study of the complex optical constants of a CsPbBr3 perovskite thin film. Journal of Materials Chemistry C, 6, 10450–10455.
Chen, X., Wang, Y., Song, J., Li, X., Xu, J., Zeng, H., & Sun, H. (2019). Temperature dependent reflectance and ellipsometry studies on a CsPbBr3 single crystal. Journal of Physical Chemistry C, 123, 10564–10570.
Yamada, T., Aharen, T., & Kanemitsu, Y. (2018). Near-band-edge optical responses of CH3NH3PbCl3 single crystals: Photon recycling of excitonic luminescence. Physical Review Letters, 120, 057404.
Comin, R., Walters, G., Thibau, E., Voznyy, O., Lu, Z., & Sargent, E. H. (2015). Structural, optical, and electronic studies of wide-bandgap lead halide perovskites. Journal of Materials Chemistry C, 3, 8839–8843.
Abbas, M., Zeng, L., Guo, F., Rauf, M., Yuan, X., & Cai, B. (2020). A critical review on crystal growth techniques for scalable deposition of photovoltaic perovskite thin films. Materials, 13, 4851.
Saki, Z., Byranvand, M., Taghavinia, N., Kedia, M., & Saliba, M. (2021). Solution-processed perovskite thin-films: The journey from lab- to large-scale solar cells. Energy & Environmental Science, 14, 5690–5722.
Trivedi, S., Prochowicz, D., Parikh, N., Mahapatra, A., Pandey, M., Kalam, A., Tavakoli, M. M., & Yadav, P. (2021). Recent progress in growth of single-crystal perovskites for photovoltaic applications. ACS Omega, 6, 1030–1042.
Herz, L. M. (2016). Charge-carrier dynamics in organic-inorganic metal halide perovskites. Annual Review of Physical Chemistry, 67, 65–89.
Even, J., Pedesseau, L., Katan, C., Kepenekian, M., Lauret, J. S., Sapori, D., & Deleporte, E. (2015). Solid-state physics perspective on hybrid perovskite semiconductors. Journal of Physical Chemistry C, 119, 10161–10177.
Draguta, S., Thakur, S., Morozov, Y. V., Wang, Y., Manser, J. S., Kamat, P. V., & Kuno, M. (2016). Spatially non-uniform trap state densities in solution-processed hybrid perovskite thin films. Journal of Physical Chemistry Letters, 7, 715–721.
Draguta, S., Christians, J. A., Morozov, Y. V., Mucunzi, A., Manser, J. S., Kamat, P. V., Luther, J. M., & Kuno, M. (2018). A quantitative and spatially resolved analysis of the performance-bottleneck in high efficiency, planar hybrid perovskite solar cells. Energy & Environmental Science, 11, 960–969.
Hutter, E. M., Eperon, G. E., Stranks, S. D., & Savenije, T. J. (2015). Charge carriers in planar and meso-structured organic–inorganic perovskites: Mobilities, lifetimes, and concentrations of trap states. Journal of Physical Chemistry Letters, 6, 3082–3090.
Stranks, S. D., Burlakov, V. M., Leijtens, T., Ball, J. M., Goriely, A., & Snaith, H. J. (2014). Recombination kinetics in organic-inorganic perovskites: Excitons, free charge, and subgap states. Physical Review Applied, 2, 034007.
DeQuilettes, D. W., Koch, S., Burke, S., Paranji, R. K., Shropshire, A. J., Ziffer, M. E., & Ginger, D. S. (2016). Photoluminescence lifetimes exceeding 8 μs and quantum yields exceeding 30% in hybrid perovskite thin films by ligand passivation. ACS Energy Letters, 1, 438–444.
D’Innocenzo, V., Srimath Kandada, A. R., De Bastiani, M., Gandini, M., & Petrozza, A. (2014). Tuning the light emission properties by band gap engineering in hybrid lead halide perovskite. Journal of the American Chemical Society, 136, 17730–17733.
Wehrenfennig, C., Eperon, G. E., Johnston, M. B., Snaith, H. J., & Herz, L. M. (2014). High charge carrier mobilities and lifetimes in organolead trihalide perovskites. Advanced Materials, 26, 1584–1589.
Johnston, M. B., & Herz, L. M. (2016). Hybrid perovskites for photovoltaics: Charge-carrier recombination, diffusion, and radiative efficiencies. Accounts of Chemical Research, 49, 146–154.
Milot, R. L., Eperon, G. E., Snaith, H. J., Johnston, M. B., & Herz, L. M. (2015). Temperature-dependent charge-carrier dynamics in CH3NH3PbI3 perovskite thin films. Advanced Functional Materials, 25, 6218–6227.
Yamada, Y., Nakamura, T., Endo, M., Wakamiya, A., & Kanemitsu, Y. (2014). Photocarrier recombination dynamics in perovskite CH3NH3PbI3 for solar cell applications. Journal of the American Chemical Society, 136, 11610–11613.
Wehrenfennig, C., Liu, M., Snaith, H. J., Johnston, M. B., & Herz, L. M. (2014). Charge-carrier dynamics in vapour-deposited films of the organolead halide perovskite CH3NH3PbI3−xClx. Energy & Environmental Science, 7, 2269–2275.
Wolff, C. M., Bourelle, S. A., Phuong, L. Q., Kurpiers, J., Feldmann, S., Caprioglio, P., Marquez, J. A., Wolansky, J., Unold, T., Stolterfoht, M., Shoaee, S., Deschler, F., & Neher, D. (2021). Orders of recombination in complete perovskite solar cells-linking time-resolved and steady-state measurements. Advanced Energy Materials, 45, 2101823.
Dobrovolsky, A., Merdasa, A., Li, J., Hirselandt, K., Unger, E. L., & Scheblykin, I. G. (2020). Relating defect luminescence and nonradiative charge recombination in MAPbI3 perovskite films. Journal of Physical Chemistry Letters, 11, 1714–1720.
de Quilettes, D. W., Vorpahl, S. M., Stranks, S. D., Nagaoka, H., Eperon, G. E., Ziffer, M. E., Snaith, H. J., & Ginger, D. S. (2015). Impact of microstructure on local carrier lifetime in perovskite solar cells. Science, 348, 683–686.
Samiee, M., Konduri, S., Ganapathy, B., Kottokkaran, R., Abbas, H. A., Kitahara, A., Joshi, P., Zhang, L., Noack, M., & Dalal, V. (2014). Defect density and dielectric constant in perovskite solar cells. Applied Physics Letters, 105, 153502.
Chen, X., Lu, H., Yang, Y., & Beard, M. C. (2018). Excitonic effects in methylammonium lead halide perovskites. Journal of Physical Chemistry Letters, 9, 2595–2603.
Guo, Z., Manser, J. S., Wan, Y., Kamat, P. V., & Huang, L. (2015). Spatial and temporal imaging of long-range charge transport in perovskite thin films by ultrafast microscopy. Nature Communications, 6, 7471.
Manser, J. S., & Kamat, P. V. (2014). Band filling with free charge carriers in organometal halide perovskites. Nature Photonics, 8, 737–743.
Trinh, M. T., Wu, X., Niesner, D., & Zhu, X. Y. (2015). Many-body interactions in photo-excited lead iodide perovskite. Journal of Materials Chemistry A, 3, 9285–9290.
Vietmeyer, F., Frantsuzov, P. A., Janko, B., & Kuno, M. (2011). Carrier recombination dynamics in individual CdSe nanowires. Physical Review B, 83, 115319.
Morozov, Y. V., Draguta, S., Zhang, S., Cadranel, A., Wang, Y., Janko, B., & Kuno, M. (2017). Defect-mediated CdS nanobelt photoluminescence up-conversion. Journal of Physical Chemistry C, 121, 16607–16616.
Stranks, S. D. (2017). Nonradiative losses in metal halide perovskites. ACS Energy Letters, 2, 1515–1525.
Puthussery, J., Lan, A., Kosel, T. H., & Kuno, M. (2008). Band-filling of solution-synthesized CdS nanowires. ACS Nano, 2, 357–367.
Ross, R. T. (1967). Some thermodynamics of photochemical systems. The Journal of Chemical Physics, 46, 4590–4593.
Miller, O. D., Yablonovitch, E., & Kurtz, S. R. (2012). Strong internal and external luminescence as solar cells approach the Shockley–Queisser limit. IEEE Journal of Photovoltaics, 2, 303–311.
Braly, I. L., de Quilettes, D. W., Pazos-Outón, L. M., Burke, S., Ziffer, M. E., Ginger, D. S., & Hillhouse, H. W. (2018). Hybrid perovskite films approaching the radiative limit with over 90% photoluminescence quantum efficiency. Nature Photonics, 12, 355–361.
Richter, J. M., Abdi-Jalebi, M., Sadhanala, A., Tabachnyk, M., Rivett, J. P., Pazos-Outón, L. M., Gödel, K. C., Price, M., Deschler, F., & Friend, R. H. (2016). Enhancing photoluminescence yields in lead halide perovskites by photon recycling and light out-coupling. Nature Communications, 7, 13941.
Pazos-Outón, L. M., Szumilo, M., Lamboll, R., Richter, J. M., Crespo-Quesada, M., Abdi-Jalebi, M., Beeson, H. J., Vrućinić, M., Alsari, M., Snaith, H. J., Ehrler, B., Friend, R. H., & Deschler, F. (2016). Photon recycling in lead iodide perovskite solar cells. Science, 351, 1430–1433.
Liu, Z., Krückemeier, L., Krogmeier, B., Klingebiel, B., Márquez, J. A., Levcenko, S., Öz, S., Mathur, S., Rau, U., Unold, T., & Kirchartz, T. (2018). Open-circuit voltages exceeding 1.26 V in planar methylammonium lead iodide perovskite solar cells. ACS Energy Letters, 4, 110–117.
Deschler, F., Price, M., Pathak, S., Klintberg, L. E., Jarausch, D. D., Higler, R., Hüttner, S., Leijtens, T., Stranks, S. D., Snaith, H. J., Atatüre, M., Phillips, R. T., & Friend, R. H. (2014). High photoluminescence efficiency and optically pumped lasing in solution-processed mixed halide perovskite semiconductors. Journal of Physical Chemistry Letters, 5, 1421–1426.
Noel, N. K., Abate, A., Stranks, S. D., Parrott, E. S., Burlakov, V. M., Goriely, A., & Snaith, H. J. (2014). Enhanced photoluminescence and solar cell performance via Lewis base passivation of organic–inorganic lead halide perovskites. ACS Nano, 8, 9815–9821.
Kojima, K., Ikemura, K., Matsumori, K., Yamada, Y., Kanemitsu, Y., & Chichibu, S. F. (2019). Internal quantum efficiency of radiation in a bulk CH3NH3PbBr3 perovskite crystal quantified by using the omnidirectional photoluminescence spectroscopy. APL Materials, 7, 071116.
Droseros, N., Longo, G., Brauer, J., Sessolo, M., Bolink, H., & Banerji, N. (2018). Origin of the enhanced photoluminescence quantum yield in MAPbBr3 perovskite with reduced crystal size. ACS Energy Letters, 3, 1458–1466.
Longo, G., La-Placa, M., Sessolo, M., & Bolink, H. J. (2017). High photoluminescence quantum yields in organic semiconductor-perovskite composite thin films. ChemSusChem, 10, 3788–3793.
Saliba, M., Matsui, T., Domanski, K., Seo, J. Y., Ummadisingu, A., Zakeeruddin, S. M., Correa-Baena, J. P., Tress, W. R., Abate, A., Hagfeldt, A., & Grätzel, M. (2016). Incorporation of rubidium cations into perovskite solar cells improves photovoltaic performance. Science, 354, 206–209.
Abdi-Jalebi, M., Andaji-Garmaroudi, Z., Cacovich, S., Stavrakas, C., Philippe, B., Richter, J. M., Alsari, M., Booker, E. P., Hutter, E. M., Pearson, A. J., Lilliu, S., Savenije, T. J., Rensmo, H., Divitini, G., Ducati, C., Friend, R. H., & Stranks, S. D. (2018). Maximizing and stabilizing luminescence from halide perovskites with potassium passivation. Nature, 555, 497–501.
Kramarenko, M., Ferreira, C. G., Martínez-Denegri, G., Sansierra, C., Toudert, J., & Martorell, J. (2020). Relation between fluorescence quantum yield and open-circuit voltage in complete perovskite solar cells. Solar RRL, 4, 1900554.
Zhang, S., Zhukovskyi, M., Jankó, B., & Kuno, M. (2019). Progress in laser cooling semiconductor nanocrystals and nanostructures. NPG Asia Materials, 11, 1–9.
Koscher, B. A., Swabeck, J. K., Bronstein, N. D., & Alivisatos, A. P. (2017). Essentially trap-free CsPbBr3 colloidal nanocrystals by postsynthetic thiocyanate surface treatment. Journal of the American Chemical Society, 139, 6566–6569.
Mazumdar, S., Zhao, Y., & Zhang, X. (2021). Stability of perovskite solar cells: Degradation mechanisms and remedies. Frontiers in Electronics, 2, 712785.
Wang, R., Mujahid, M., Duan, Y., Wang, Z., Xue, J., & Yang, Y. (2019). A review of perovskites solar cell stability. Advanced Functional Materials, 29, 1808843.
Futscher, M., Lee, J., McGovern, L., Muscarella, L., Wang, T., Haider, M. C., Fakharuddin, A., Schmidt-Mende, L., & Ehrler, B. (2019). Quantification of ion migration in CH3NH3PbI3 perovskite solar cells by transient capacitance measurements. Materials Horizons, 6, 1497–1503.
Calado, P., Telford, A., Bryant, D., Li, X., Nelson, J., O’Regan, B. C., & Barnes, P. R. (2016). Evidence for ion migration in hybrid perovskite solar cells with minimal hysteresis. Nature Communications, 7, 13831.
Singh, R., & Parashar, M. (2020). Origin of hysteresis in perovskite solar cells. In J. Ren & Z. Kan (Eds.), Soft-Matter Thin Film Solar Cells (pp. 1–42). AIP Publishing Books.
Pavlovetc, I. M., Brennan, M. C., Draguta, S., Ruth, A., Moot, T., Christians, J. A., Aleshire, K., Harvey, S. P., Toso, S., Nanayakkara, S. U., Messinger, J., Luther, J. M., & Kuno, M. (2020). Suppressing cation migration in triple-cation lead halide perovskites. ACS Energy Letters, 5, 2802–2810.
Lin, Y., Chen, B., Fang, Y., Zhao, J., Bao, C., Yu, Z., Deng, Y., Rudd, P. N., Yan, Y., Yuan, Y., & Huang, J. (2018). Excess charge-carrier induced instability of hybrid perovskites. Nature Communications, 9, 4981.
Brennan, M. C., Draguta, S., Kamat, P. V., & Kuno, M. (2017). Light-induced anion phase segregation in mixed halide perovskites. ACS Energy Letters, 3, 204–213.
Wang, Y., Quintana, X., Kim, J., Guan, X., Hu, L., Lin, C. H., Jones, B. T., Chen, W., Wen, X., Gao, H., & Wu, T. (2020). Phase segregation in inorganic mixed-halide perovskites: From phenomena to mechanisms. Photonics Research, 8, A56–A71.
Zhang, G., Long, R., Lu, Y., Zhang, S., Zhao, W., Yu, Y., Mi, Q., Qin, P., & Huang, F. (2021). Calcium-assisted in situ formation of perovskite nanocrystals for luminescent green and blue emitters. ACS Applied Nano Materials, 4, 14303–14311.
Zheng, X., Yuan, S., Liu, J., Yin, J., Yuan, F., Shen, W., Yao, K., Wei, M., Zhou, C., Song, K., Zhang, B., Lin, Y., Hedhili, M. N., Wehbe, N., Han, Y., Sun, H., Lu, Z., Anthopoulos, T. D., Mohammed, O. F., Sargent, E. H., Liao, L., & Bakr, O. M. (2020). Chlorine vacancy passivation in mixed halide perovskite quantum dots by organic pseudohalides enables efficient rec. 2020 blue light-emitting diodes. ACS Energy Letters, 5, 793–798.
Wang, F., Bai, S., Tress, W., Hagfeldt, A., & Gao, F. (2018). Defects engineering for high-performance perovskite solar cells. Npj Flexible Electronics, 2, 22.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kuno, M., Gushchina, I. (2023). Photophysics of Hybrid and Inorganic Lead Halide Perovskites. In: Nie, W., Iniewski, K.(. (eds) Metal-Halide Perovskite Semiconductors. Springer, Cham. https://doi.org/10.1007/978-3-031-26892-2_3
Download citation
DOI: https://doi.org/10.1007/978-3-031-26892-2_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-26891-5
Online ISBN: 978-3-031-26892-2
eBook Packages: EnergyEnergy (R0)