
Abstract
A biomarker can be defined as a measurable indicator of the presence or severity of a disease state, often present before clinical signs are evident. For the most frequent forms of spinocerebellar ataxia (SCAs), due to expansions of coding CAG repeats SCA1/ATXN1, SCA2/ATXN2, SCA3/ATXN3, SCA6/CACNA1A, SCA7/ATXN7, SCA17/TBP, and DRPLA/ATN1, gene therapies are planned. Reliable biomarkers should indicate the pathological onset or discriminate disease stages that would allow to stratify patients and to monitor drug efficacy. This chapter reviews the available blood and cerebrospinal fluid (CSF) biomarkers. One of the most promising biomarkers is neurofilament light chain (NfL) for which blood and CSF levels accurately correlate. Moreover, NfL concentrations are associated with disease progression, and cerebellum and brainstem atrophy. Specific ataxin bioassays are in development for polyglutamine SCAs, but only ataxin-3 can be measured in blood and CSF. Other biomarkers are related to oxidative stress, inflammation, astrogliosis, and insulin pathway. Others are in development regarding the metabolism of cholesterol, lipids, and amino acids, as well as the micro-RNAs that would be potential biological markers of disease and therapeutic targets.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Autosomal dominant cerebellar ataxias (ADCAs) are a rare cause of cerebellar ataxias. In genetic nomenclature, they referred to spinocerebellar ataxias (SCAs), a group of diseases clinically and genetically heterogeneous (Klockgether et al. 2019). Nowadays, 48 SCAs subtypes have been identified. The most frequent SCAs are due to pathological CAG repeat expansions coding for polyglutamine (polyQ): SCA1/ATXN1, SCA2/ATXN2, SCA3/ATXN3, SCA6/CACNA1A, SCA7/ATXN7, SCA17/TBP and DRPLA/ATN1. Age at onset and disease severity are negatively correlated with the pathological CAG repeat expansion (Durr 2010), and phenotype is clearly associated with CAG repeat size (Stevanin et al. 2000). Pediatric and juvenile forms can also occur, especially for SCA2 and SCA7 (Mao et al. 2002; Bah et al. 2020). PolyQ subtypes clinically share the cerebellar ataxia with gait and balance impairment, limb dysmetria, dysarthria, swallowing difficulties, and oculomotor abnormalities. However, other extra-cerebellar signs are also present: pyramidal syndrome for SCA1, SCA3, SCA7, SCA17, parkinsonism for SCA2, SCA3, SCA7, fasciculations and wasting for SCA2, peripheral neuropathy for SCA2 and SCA3, dystonia for SCA2, SCA3, SCA7, SCA17, choreic movements for SCA17, ophthalmological deficit for SCA7, etc. The Scale for the Assessment and Rating of Ataxia (SARA) (Schmitz-Hübsch et al. 2006), which includes eight items to assess cerebellar syndrome, does not catch these extra-cerebellar signs. This scale is used as the primary outcome in several therapeutic and non-therapeutic trials for SCAs. However, presymptomatic carriers, defined by a SARA score <3 out of 40, can present other non-cerebellar signs and symptoms that are already expression of disease.
Individual variability, even among genetically homogeneous forms due to a same mutation, impedes prediction of progression of the imaging and clinical signs in ataxias. Broadly, a higher number of CAG repeats within the HTT gene predicts earlier onset, but two people with the same repeat length may differ in clinical onset by decades (Lee et al. 2012). This variability has to be tackled using biomarkers that allow to define the state of disease for a single patient and the challenge for the evaluation of potential treatments, particularly in early stages, will rely on longitudinal biomarkers.
Gene therapies have made remarkable progress over the last decade, such as antisense oligonucleotides (ASOs) approach. These are targeted treatment based on the genetic status. The rationale relies on the fact that lowering the burden of mutated protein may improve the disease prognosis. ASOs form a complex with targeted mRNA recruiting an endoribonuclease (Ribonuclease H) that degrades the RNA-DNA hybrid complex (Wild and Tabrizi 2017). Following the impressive results of nusinersen in spinal muscular atrophy (Finkel et al. 2017; Acsadi et al. 2021), major hopes have been put in the development of ASO directed to ATXN1, ATXN2, ATXN3, and ATXN7 mutants even though one recent phase-III clinical trial failed to show that ASOs halted the progression of Huntington disease (HD) (Tabrizi et al. 2019; Kingwell 2021). Promising results have been reported by the ASOs administration in several SCAs mouse models (Friedrich et al. 2018; Scoles et al. 2017; McLoughlin et al. 2018; Niu et al. 2018). Therefore, objective and quantitative biomarkers rather than clinical measures are of critical importance as prognostic or pharmacodynamic markers to monitor drug effects. The aim of this chapter is to review the blood and cerebrospinal fluid (CSF) biomarkers for SCAs (Table 1).
2 Biological Biomarkers
2.1 Neurofilament Light Chain
Neurofilament light chain (NfL) is a subunit of neuronal cytoskeleton and its level increases in CSF and blood as a result of axonal damage due to different causes (neurodegeneration, infection, traumatic, etc.) (Gaetani et al. 2019). Using the highly sensitive single-molecule array method (Simoa) is possible to measure NfL in blood and CSF more accurately than conventional enzyme-linked immunosorbent assay (ELISA) and electrochemiluminescence-based method (ECL assay) (Kuhle et al. 2016). Close correlation exists between CSF and blood concentrations, making NfL an easily measurable biomarker of neurodegeneration (Gaetani et al. 2019; Khalil 2018). In several neurological disorders, NfL correlates with disease stages, clinical scores, and neuroimaging data. It is the case for amyotrophic lateral sclerosis (ALS) (Lu et al. 2015; Benatar et al. 2018), Alzheimer’s disease (Mattsson et al. 2019; Benedet et al. 2020; Preische et al. 2019), and multiple sclerosis (Bjornevik et al. 2020; Kuhle et al. 2019), HD (Byrne et al. 2017; Johnson et al. 2018; Scahill et al. 2020). This latter disease shares with SCAs the same mutational mechanism, a translated pathological CAG repeat expansion. NfL showed a prognostic value with a significant increase for HD presymptomatic carriers of the pathological expansion close to the age at expected disease onset (Scahill et al. 2020). Moreover, for HD, atrophy of cerebral regions, as the putamen and caudate, is associated with higher NfL concentrations (Scahill et al. 2020).
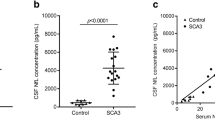
For SCAs, some studies showed the higher NfL levels in carriers than healthy controls. The first pilot study included only 20 SCAs carriers (SCA1, SCA2, SCA3, SCA6) founding elevated Nfl concentrations compared to controls (Wilke et al. 2018). Then, other studies on large cohorts of SCA3 carriers confirmed the correlation between clinical progression and NfL in CSF (Li et al. 2019) and blood (Li et al. 2019; Wilke et al. 2020; Peng et al. 2020). In a longitudinal study with 2-year interval of plasma NfL measurements, NfL confirmed to be a disease biomarker with significant difference between healthy controls (~10 pg/mL) and polyglutamine (polyQ) SCAs carriers [SCA1 (~24 pg/mL), SCA2 (~20 pg/mL), SCA3 (~35 pg/mL), and SCA7 (~26 pg/mL)] (Coarelli et al. 2021). Interestingly, NfL concentrations remained stable at 2-year follow-up despite clinical progression assessed by SARA (Coarelli et al. 2021). Considering all SCAs subtypes, higher plasma NfL levels at baseline predicted a higher SARA score progression as well as a decrease in cerebellar volume at 2-year follow-up (Coarelli et al. 2021). NfL correlated with pons atrophy at baseline and follow-up (Coarelli et al. 2021), confirmed for SCA3 group taken separately. For SCA3, another study also reported significant association between serum NfL and cerebellum and brainstem volumes (Li et al. 2019).
Serum NfL increases already 7.5 years before the expected age at onset for SCA3 carriers (Wilke et al. 2020). NfL levels for SCA3 presymptomatic carriers fall down between controls and symptomatic carriers levels (Li et al. 2019; Wilke et al. 2020; Peng et al. 2020; Coarelli et al. 2021). SCA7 premanifest carriers with non-cerebellar signs at examination present NfL concentration close or above the cut-off level determined to differentiate controls from carriers (Coarelli et al. 2021). Based on presymptomatic carriers’ data (Li et al. 2019; Wilke et al. 2020) and longitudinal data (Coarelli et al. 2021), NfL seems to be a biomarkers that may be used in clinical trials to stratify carriers based on their NfL levels. However, some points remain to be clarified: (i) SCA3 patients present the highest concentration than the other SCAs despite a less severe clinical progression based on SARA score (Coarelli et al. 2021; Jacobi et al. 2015). One possible explanation may be the prominent peripheral nervous system involvement than the other polyQ SCAs; (ii) NfL levels do not change over time in SCAs, similar to ALS, frontotemporal dementia, and atypical parkinsonian syndromes (Gaetani et al. 2019). We may suppose that for these diseases NfL levels reach a plateau that masks the increase due to age. (iii) NfL concentrations for polyQ SCAs fall between the highest levels of ALS or multiple system atrophy and the lowest levels in Friedreich’s ataxia or Parkinson disease (Gaetani et al. 2019; Bridel et al. 2019). It may be due to by either different disease progression rates or different levels of peripheral nervous system dysfunction.
2.2 Tau
Another biomarker of neuroaxonal damage is Tau protein that promotes microtubule assembly and stability. This protein is an established marker in Creutzfeldt Jakob disease and Alzheimer’s disease (Tumani et al. 2008). In a study including few SCA1, SCA2, and SCA6 patients, Tau levels in CSF were significantly higher in SCA2 carriers than controls (Brouillette et al. 2015). Other proteins were also tested in CSF (α-synuclein, DJ-1, and GFAP) showing a tendency to be higher especially for SCA2 (Brouillette et al. 2015) and indicating the necessity to be reproduced in a larger cohort of patients.
2.3 Astrocytosis and Gliosis
Neuron-specific enolase (NSE) and protein S 100 B (S100B) are markers of neuron damage and gliosis. Serum concentrations of these two proteins are higher in SCA3 patients than controls (Zhou et al. 2011), not tested in other SCA patients. NSE presents a correlations with disease duration and clinical scales (ICARS and SARA), instead of S100B that does not correlate with any clinical parameters (Zhou et al. 2011). In another SCA3 study, only NSE serum level was significantly higher than controls and presented a correlation with depression score (Tort et al. 2005).
2.4 Ataxin-Specific Bioassays
In view of upcoming therapeutic trials that aim to decrease the mutant protein, it seems to be crucial for the development of ataxin-specific assays to monitor the efficacy of these treatments. To date, a time-resolved fluorescence resonance energy transfer (TR-FRET) immunoassay can detect the polyQ-expanded and non-expanded ataxin-3 protein level in blood-derived mononuclear cells from presymptomatic and symptomatic SCA3 carriers (Gonsior et al. 2020). Moreover, polyQ-expanded ataxin-3 protein levels correlated with disease stage and clinical severity assessed by SARA (Gonsior et al. 2020). However, this highly sensitive TR-FRET-based immunoassay cannot measure ataxin-3 level in other fluids such as CSF or plasma and should be validated in other cohorts.
In another study, an electrochemiluminescence immunoassay using the Meso Scale Discovery system detected polyQ-expanded ataxin-3 in CSF and plasma distinguishing controls from SCA3 carriers (Prudencio et al. 2020). In addition, this study showed the strong association between ATXN3 pathological CAG repeat expansion and the rs7158733 SNP located ~132 nucleotides downstream of the CAG repeat (Prudencio et al. 2020) that could facilitate the allele specific ASO treatment.
Another novel single molecule counting (SMC) ataxin-3 immunoassay is able to measure polyQ-expanded ataxin-3 in plasma and CSF (Hübener-Schmid et al. 2021). Clinical correlations (age at onset and SARA score) are reported with plasma polyQ-expanded ataxin-3 levels. Longitudinal data show that plasma levels remain stable over a 1-year period (Hübener-Schmid et al. 2021).
For the other ataxin proteins, specific bioassays are not yet available.
2.5 Oxidative Stress Biomarkers
Oxidative stress has been implicated in several neurodegenerative disorders. Production of abnormally large amounts of reactive oxygen species was reported for SCA3 (Pacheco et al. 2013). This seems to be caused by a dysregulation of major enzymes implicated in antioxidant capacity: superoxide dismutase and glutathione peroxidase (GPx) activities are lower in symptomatic than in pre-symptomatic carriers (de Assis et al. 2017). On the other hand, catalase activity is increased in the serum of SCA3 patients (Pacheco et al. 2013). The correlation of GPx decrease activity with disease severity suggests that GPx may be a reliable biomarker (de Assis et al. 2017).
In SCA2 presymptomatic and symptomatic carriers, glutathione S-transferases (GST) activity is increased by 21.8% and 5.5%, respectively (Almaguer-Gotay et al. 2014). The role of this enzyme is to protect against oxidative stress and prevent apoptosis. GST increase activity supports the role of free radical damage in SCAs physiopathology.
2.6 Inflammation Biomarkers
Inflammatory genes encoding endopeptidase matrix metalloproteinase 2 (MMP-2) and cytokine stromal cell-derived factor 1α (SDF1α) are upregulated in a cell culture model of SCA3 as well as in human SCA3 pons (Evert et al. 2001). Other proteins involved in inflammation process are significantly increased: amyloid β-protein (Aβ), interleukin-1 receptor antagonist (IL-1ra), interleukin-1β (IL-1β), and interleukin-6 (IL-6) (Evert et al. 2001). Activation of microglia and presence of reactive astrocytes are reported in the brains of SCA3 patients (Evert et al. 2006). Based on these data, a large panel of cytokines has been investigated in a large cohort of presymptomatic and symptomatic SCA3 compared to controls (da Silva et al. 2016). No difference in cytokine levels was detected among the groups except for eotaxin. Higher eotaxin concentrations were observed in asymptomatic carriers than in symptomatic carriers (da Silva et al. 2016). In symptomatic carriers, the level dropped after 1 year (da Silva et al. 2016). One possible explanation may be that the levels of eotaxin released by astrocytes are inversely correlated with disease progression (da Silva et al. 2016).
2.7 Insulin/Insulin-Like Growth Factor 1 (IGF-1) System
Abnormalities in the signaling pathway of the insulin/insulin-like growth factor 1 (IGF-1) system (IIS), including IGF-1, IGF binding proteins (IGFBPs), and insulin, are thought to play a role in the physiopathological processes of neurodegenerative diseases as Alzheimer’s disease, HD, and polyQ SCAs (Craft and Watson 2004; Cohen and Dillin 2008; Emamian et al. 2003). SCA3 patients show higher serum levels of IGFBP1 and IGF-1/IGFBP-3 ratio than controls (Saute et al. 2011). Inversely, serum levels of IGFBP-3 (that binds more than 80% of peripheral IGF-1 and increases its half-life) and insulin levels are reduced (Saute et al. 2011). β-cell function is preserved in SCA3 patients and the reduction of insulin level is due to an increased peripheral sensitivity to insulin. Higher sensitivity to insulin and lower insulin levels are both related to earlier disease onset (Saute et al. 2011).
IGFBP-1 levels are correlated significantly with CAG repeat expansion (Saute et al. 2011). IGFBP1 may be a biomarker for SCA3 even though its link with expanded ataxin-3 protein remains unclear. One possible explanation could be the endoplasmic reticulum stress induced by mutant ataxin-3 protein that increases IGFBP-1 production in liver (Saute et al. 2011). Even though IGF1 is not significantly higher, it inversely correlates with the volume of medulla oblongata and pons (Saute et al. 2011).
2.8 Co-chaperone Protein
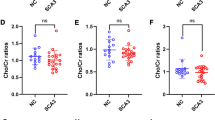
The carboxyl terminus of Hsp-70 interacting protein (CHIP), a co-chaperone protein, is an endogenous binding partner of the mutant ataxin-3. In SCA3 patients, CHIP level is elevated in both serum and CSF, indirectly reflecting mutant ataxin-3 level (Hu et al. 2019). CHIP correlates with disease severity assessed by SARA and ICARS. The main role of CHIP is protein quality control. Ataxin-3 protein directly interacts with CHIP. The affinity between these two proteins increases with CAG expansion causing a cellular homeostasis dysregulation.
3 Biomarkers in Development
3.1 Brain Cholesterol Metabolism
Deregulation of brain cholesterol turnover and metabolism have been associated with several neurodegenerative diseases. 24-hydroxylase (CYP46A1) is the key enzyme of efflux of brain cholesterol, converting the excess cholesterol into 24S-hydroxycholesterol (24OHC) released in systemic circulation (Leoni et al. 2013). Plasma 24OHC is significantly reduced in neurological disorders as Alzheimer’s disease, Parkinson’s disease, Niemann–Pick disease type C, multiple sclerosis, and HD (Papassotiropoulos et al. 2005; Kölsch et al. 2009; Shobab et al. 2005; Solomon et al. 2009; Leoni et al. 2002). For HD, 24OHC levels decrease with disease progression and striatal volume loss (Leoni et al. 2013). In SCA3 cerebellum samples, CYP46A1 is reduced (Nóbrega et al. 2019). The overexpression by an adeno-associated virus (AAV)-mediated expression of CYP46A1 decreases the ATXN-3 aggregates by activation of autophagy and leads to motor improvement in SCA2 mouse model (Nóbrega et al. 2019). Plasma 24OHC may be a potential biomarker for SCAs as reported for HD, therefore further investigations should be carry on.
3.2 Metabolic Profile
The serum metabolomics profile shows a difference between symptomatic SCA3 patients and presymptomatic carriers or controls (Yang et al. 2019). In SCA3 patients, there is a downregulation of branched-chain amino acids including valine and leucine, and aromatic amino acids as tryptophan and tyrosine (Yang et al. 2019). These metabolites are precursors of some neurotransmitters (serotonin, dopamine, GABA) and have a role in energy metabolism. Fatty acid metabolism is also dysregulated in SCA3 patients with decrease of saturated fatty acid and increase of monounsaturated and polyunsaturated fatty acid fatty (Yang et al. 2019).
Plasma lipidomic analysis in a cohort of polyQ SCAs showed that SCA7 patients differentiate from other polyQ SCAs patients for some ceramides and phosphatidylcholines (Garali et al. 2018). These lipids are strongly expressed in retina and their deficit may be linked to the retinal alterations characteristic for SCA7 rather than other polyQ SCAs.
For SCA7 patients, another study has reported the decreased of branched-chain amino acids, leucine and valine, as well as of tyrosine, with a good sensitivity to discriminate from controls (Nambo-Venegas et al. 2020). Moreover, when regarding only SCA7 carriers, methionine level differentiates early onset from late onset patients (Nambo-Venegas et al. 2020).
3.3 Micro-RNAs
Several studies investigated micro-RNAs (miRNAs) levels in SCAs patients reporting different results. Lower levels of miR-25, miR-125b, miR-29a, and miR-34b are found in serum of SCA3 patients compared to controls (Huang et al. 2014; Shi et al. 2014). Reduced concentrations of miR-9 and miR-181a from CSF derived exosomes of SCA3 patients are reported (Hou et al. 2019). Three miRNAs-mir-9, mir-181a, and mir-494 are decreased in SCA3 human neurons (Carmona et al. 2017). These three miRNAs interact with the ATXN3-3′ UTR downregulating its expression (Carmona et al. 2017). In SCA3 mouse model, the overexpression of these miRNAs reduces the mutant ataxin-3 expression by translation inhibition and mRNA degradation (Carmona et al. 2017). For SCA7, the plasma expressions of four miRNAs (hsa-let-7a-5p, hsa-let7e-5p, hsa-miR-18a-5p, and hsa-miR-30b-5p) differentiate carriers from controls and seem to have a prognostic value discriminating between juvenile and adult onset (Borgonio-Cuadra et al. 2019).
These data could suggest miRNAs as potential biological markers of disease and therapeutic targets. However, their use does not seem to be possible in the short term.
3.4 Sirtuin-1
Sirtuin-1 is a NAD+-dependent deacetylase taking part in several cellular functions as chromatin modulation, cell cycle, apoptosis, and autophagy regulation in response to DNA damage. In SCA3 mice and in SCA3 patients’ fibroblasts, sirtuin-1 mRNA levels are lower than controls (Cunha-Santos et al. 2016). In SCA3 mice, the caloric restriction rescues sirtuin-1 with motor improvement (Cunha-Santos et al. 2016). Sirtuin-1 overexpression activates autophagy and increases the mutant protein clearance. This overexpression results in neuropathological changes: activation of autophagy, decrease in neuroinflammation, and reduction in reactive gliosis (Cunha-Santos et al. 2016).
4 Conclusion
This chapter reviews the available biomarkers in blood and CSF for polyQ SCAs. However, the majority of the evidence are reported for SCA3, the most frequent subtype worldwide, and only few longitudinal studies have been conducted with a lower inclusion of presymptomatic carriers. Still many efforts need to obtain an optimal biomarker with diagnostic and prognostic values, reliable to be used in upcoming gene therapy trials. Neurofilaments light chain seems to be currently the best biomarker, already confirmed in several neurological diseases, with a role to monitor drug administration in spinal muscular atrophy (Olsson et al. 2019) and multiple sclerosis (Kuhle et al. 2019). A great interest there is towards the development of ataxin bioassays that are the specific target of ASOs therapy. Other pathways presented in this chapter require validation in larger cohorts.
References
Acsadi G, Crawford TO, Müller-Felber W, Shieh PB, Richardson R, Natarajan N, et al. Safety and efficacy of nusinersen in spinal muscular atrophy: the EMBRACE study. Muscle Nerve. 2021;63(5):668–77.
Almaguer-Gotay D, Almaguer-Mederos LE, Aguilera-Rodríguez R, Estupiñán-Rodríguez A, González-Zaldivar Y, Cuello-Almarales D, et al. Role of glutathione S-transferases in the spinocerebellar ataxia type 2 clinical phenotype. J Neurol Sci. 2014;341(1–2):41–5.
Bah MG, Rodriguez D, Cazeneuve C, Mochel F, Devos D, Suppiej A, et al. Deciphering the natural history of SCA7 in children. Eur J Neurol. 2020;27(11):2267–76.
Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol. 2018;84(1):130–9.
Benedet AL, Leuzy A, Pascoal TA, Ashton NJ, Mathotaarachchi S, Savard M, et al. Stage-specific links between plasma neurofilament light and imaging biomarkers of Alzheimer’s disease. Brain. 2020;143:3793–804.
Bjornevik K, Munger KL, Cortese M, Barro C, Healy BC, Niebuhr DW, et al. Serum neurofilament light chain levels in patients with presymptomatic multiple sclerosis. JAMA Neurol. 2020;77(1):58–64.
Borgonio-Cuadra VM, Valdez-Vargas C, Romero-Córdoba S, Hidalgo-Miranda A, Tapia-Guerrero Y, Cerecedo-Zapata CM, et al. Wide profiling of circulating MicroRNAs in spinocerebellar ataxia type 7. Mol Neurobiol. 2019;56(9):6106–20.
Bridel C, van Wieringen WN, Zetterberg H, Tijms BM, Teunissen CE, and the NFL Group, et al. Diagnostic value of cerebrospinal fluid neurofilament light protein in neurology: a systematic review and meta-analysis. JAMA Neurol. 2019;76:1035–48.
Brouillette AM, Öz G, Gomez CM. Cerebrospinal fluid biomarkers in spinocerebellar ataxia: a pilot study. Dis Markers. 2015;2015:413098.
Byrne LM, Rodrigues FB, Blennow K, Durr A, Leavitt BR, Roos RAC, et al. Neurofilament light protein in blood as a potential biomarker of neurodegeneration in Huntington’s disease: a retrospective cohort analysis. Lancet Neurol. 2017;16(8):601–9.
Carmona V, Cunha-Santos J, Onofre I, Simões AT, Vijayakumar U, Davidson BL, et al. Unravelling endogenous MicroRNA system dysfunction as a new pathophysiological mechanism in Machado-Joseph disease. Mol Ther. 2017;25(4):1038–55.
Coarelli G, Darios F, Petit E, Dorgham K, Adanyeguh I, Petit E, et al. Plasma neurofilament light chain predicts cerebellar atrophy and clinical progression in spinocerebellar ataxia. Neurobiol Dis. 2021;153:105311.
Cohen E, Dillin A. The insulin paradox: aging, proteotoxicity and neurodegeneration. Nat Rev Neurosci. 2008;9(10):759–67.
Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169–78.
Cunha-Santos J, Duarte-Neves J, Carmona V, Guarente L, Pereira de Almeida L, Cavadas C. Caloric restriction blocks neuropathology and motor deficits in Machado-Joseph disease mouse models through SIRT1 pathway. Nat Commun. 2016;7:11445.
da Silva Carvalho G, Saute JAM, Haas CB, Torrez VR, Brochier AW, Souza GN, et al. Cytokines in Machado Joseph disease/spinocerebellar ataxia 3. Cerebellum. 2016;15(4):518–25.
de Assis AM, Saute JAM, Longoni A, Haas CB, Torrez VR, Brochier AW, et al. Peripheral oxidative stress biomarkers in spinocerebellar ataxia type 3/Machado-Joseph disease. Front Neurol. 2017;8:485.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885–94.
Emamian ES, Kaytor MD, Duvick LA, Zu T, Tousey SK, Zoghbi HY, et al. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38(3):375–87.
Evert BO, Vogt IR, Kindermann C, Ozimek L, de Vos RA, Brunt ER, et al. Inflammatory genes are upregulated in expanded ataxin-3-expressing cell lines and spinocerebellar ataxia type 3 brains. J Neurosci. 2001;21(15):5389–96.
Evert BO, Schelhaas J, Fleischer H, de Vos RAI, Brunt ER, Stenzel W, et al. Neuronal intranuclear inclusions, dysregulation of cytokine expression and cell death in spinocerebellar ataxia type 3. Clin Neuropathol. 2006;25(6):272–81.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723–32.
Friedrich J, Kordasiewicz HB, O’Callaghan B, Handler HP, Wagener C, Duvick L, et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight. 2018;3(21).
Gaetani L, Blennow K, Calabresi P, Di Filippo M, Parnetti L, Zetterberg H. Neurofilament light chain as a biomarker in neurological disorders. J Neurol Neurosurg Psychiatry. 2019;90(8):870–81.
Garali I, Adanyeguh IM, Ichou F, Perlbarg V, Seyer A, Colsch B, et al. A strategy for multimodal data integration: application to biomarkers identification in spinocerebellar ataxia. Brief Bioinform. 2018;19(6):1356–69.
Gonsior K, Kaucher GA, Pelz P, Schumann D, Gansel M, Kuhs S, et al. PolyQ-expanded ataxin-3 protein levels in peripheral blood mononuclear cells correlate with clinical parameters in SCA3: a pilot study. J Neurol. 2020;268:1304–15.
Hou X, Gong X, Zhang L, Li T, Yuan H, Xie Y, et al. Identification of a potential exosomal biomarker in spinocerebellar ataxia Type 3/Machado-Joseph disease. Epigenomics. 2019;11(9):1037–56.
Hu Z-W, Yang Z-H, Zhang S, Liu Y-T, Yang J, Wang Y-L, et al. Carboxyl terminus of Hsp70-interacting protein is increased in serum and cerebrospinal fluid of patients with spinocerebellar ataxia type 3. Front Neurol. 2019;10:1094.
Huang F, Zhang L, Long Z, Chen Z, Hou X, Wang C, et al. miR-25 alleviates polyQ-mediated cytotoxicity by silencing ATXN3. FEBS Lett. 2014;588(24):4791–8.
Hübener-Schmid J, Kuhlbrodt K, Peladan J, Faber J, Santana MM, Hengel H, et al. Polyglutamine-expanded ataxin-3: a target engagement marker for spinocerebellar ataxia type 3 in peripheral blood. Mov Disord. 2021;36:2675–81.
Jacobi H, du Montcel ST, Bauer P, Giunti P, Cook A, Labrum R, et al. Long-term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol. 2015;14(11):1101–8.
Johnson EB, Byrne LM, Gregory S, Rodrigues FB, Blennow K, Durr A, et al. Neurofilament light protein in blood predicts regional atrophy in Huntington disease. Neurology. 2018;90(8):e717–23.
Khalil M. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol [Internet]. 2018;14:577. Available from: https://doi.org/10.1038/s41582-018-0058-z.
Kingwell K. Double setback for ASO trials in Huntington disease. Nat Rev Drug Discov. 2021;20(6):412–3.
Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5(1):24.
Kölsch H, Lütjohann D, Jessen F, Popp J, Hentschel F, Kelemen P, et al. CYP46A1 variants influence Alzheimer’s disease risk and brain cholesterol metabolism. Eur Psychiatry. 2009;24(3):183–90.
Kuhle J, Barro C, Andreasson U, Derfuss T, Lindberg R, Sandelius Å, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med. 2016;54(10):1655–61.
Kuhle J, Kropshofer H, Haering DA, Kundu U, Meinert R, Barro C, et al. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology. 2019;92(10):e1007–15.
Lee J-M, Ramos EM, Lee J-H, Gillis T, Mysore JS, Hayden MR, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78(10):690–5.
Leoni V, Masterman T, Diczfalusy U, De Luca G, Hillert J, Björkhem I. Changes in human plasma levels of the brain specific oxysterol 24S-hydroxycholesterol during progression of multiple sclerosis. Neurosci Lett. 2002;331(3):163–6.
Leoni V, Long JD, Mills JA, Di Donato S, Paulsen JS. Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression. Neurobiol Dis. 2013;55:37–43.
Li Q-F, Dong Y, Yang L, Xie J-J, Ma Y, Du Y-C, et al. Neurofilament light chain is a promising serum biomarker in spinocerebellar ataxia type 3. Mol Neurodegener. 2019;14(1):39.
Lu C-H, Macdonald-Wallis C, Gray E, Pearce N, Petzold A, Norgren N, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology. 2015;84(22):2247–57.
Mao R, Aylsworth AS, Potter N, Wilson WG, Breningstall G, Wick MJ, et al. Childhood-onset ataxia: testing for large CAG-repeats in SCA2 and SCA7. Am J Med Genet. 2002;110(4):338–45.
Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76(7):791–9.
McLoughlin HS, Moore LR, Chopra R, Komlo R, McKenzie M, Blumenstein KG, et al. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann Neurol. 2018;84(1):64–77.
Nambo-Venegas R, Valdez-Vargas C, Cisneros B, Palacios-González B, Vela-Amieva M, Ibarra-González I, et al. Altered plasma acylcarnitines and amino acids profile in spinocerebellar ataxia type 7. Biomol Ther. 2020;10(3):E390.
Niu C, Prakash TP, Kim A, Quach JL, Huryn LA, Yang Y, et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci Transl Med. 2018;10(465):eaap8677.
Nóbrega C, Mendonça L, Marcelo A, Lamazière A, Tomé S, Despres G, et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol. 2019;138(5):837–58.
Olsson B, Alberg L, Cullen NC, Michael E, Wahlgren L, Kroksmark A-K, et al. NFL is a marker of treatment response in children with SMA treated with nusinersen. J Neurol. 2019;266(9):2129–36.
Pacheco LS, da Silveira AF, Trott A, Houenou LJ, Algarve TD, Belló C, et al. Association between Machado-Joseph disease and oxidative stress biomarkers. Mutat Res Genet Toxicol Environ Mutagen. 2013;757(2):99–103.
Papassotiropoulos A, Wollmer MA, Tsolaki M, Brunner F, Molyva D, Lütjohann D, et al. A cluster of cholesterol-related genes confers susceptibility for Alzheimer’s disease. J Clin Psychiatry. 2005;66(7):940–7.
Peng Y, Zhang Y, Chen Z, Peng H, Wan N, Zhang J, et al. Association of serum neurofilament light (sNfL) and disease severity in patients with spinocerebellar ataxia type 3. Neurology. 2020;95:e2977–87.
Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat Med. 2019;25(2):277–83.
Prudencio M, Garcia-Moreno H, Jansen-West KR, Al-Shaikh RH, Gendron TF, Heckman MG, et al. Toward allele-specific targeting therapy and pharmacodynamic marker for spinocerebellar ataxia type 3. Sci Transl Med. 2020;12(566):eabb7086.
Saute JAM, da Silva ACF, Muller AP, Hansel G, de Mello AS, Maeda F, et al. Serum insulin-like system alterations in patients with spinocerebellar ataxia type 3. Mov Disord. 2011;26(4):731–5.
Scahill RI, Zeun P, Osborne-Crowley K, Johnson EB, Gregory S, Parker C, et al. Biological and clinical characteristics of gene carriers far from predicted onset in the Huntington’s disease Young Adult Study (HD-YAS): a cross-sectional analysis. Lancet Neurol. 2020;19(6):502–12.
Schmitz-Hübsch T, du Montcel ST, Baliko L, Berciano J, Boesch S, Depondt C, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717–20.
Scoles DR, Meera P, Schneider MD, Paul S, Dansithong W, Figueroa KP, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. 2017;544(7650):362–6.
Shi Y, Huang F, Tang B, Li J, Wang J, Shen L, et al. MicroRNA profiling in the serums of SCA3/MJD patients. Int J Neurosci. 2014;124(2):97–101.
Shin H-R, Moon J, Lee W-J, Lee HS, Kim EY, Shin S, et al. Serum neurofilament light chain as a severity marker for spinocerebellar ataxia. Sci Rep. 2021;11(1):13517.
Shobab LA, Hsiung G-YR, Feldman HH. Cholesterol in Alzheimer’s disease. Lancet Neurol. 2005;4(12):841–52.
Solomon A, Leoni V, Kivipelto M, Besga A, Oksengård AR, Julin P, et al. Plasma levels of 24S-hydroxycholesterol reflect brain volumes in patients without objective cognitive impairment but not in those with Alzheimer’s disease. Neurosci Lett. 2009;462(1):89–93.
Stevanin G, Dürr A, Brice A. Clinical and molecular advances in autosomal dominant cerebellar ataxias: from genotype to phenotype and physiopathology. Eur J Hum Genet. 2000;8(1):4–18.
Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting huntingtin expression in patients with Huntington’s disease. N Engl J Med. 2019;380(24):2307–16.
Tort ABL, Portela LVC, Rockenbach IC, Monte TL, Pereira ML, Souza DO, et al. S100B and NSE serum concentrations in Machado Joseph disease. Clin Chim Acta. 2005;351(1):143–8.
Tumani H, Teunissen C, Süssmuth S, Otto M, Ludolph AC, Brettschneider J. Cerebrospinal fluid biomarkers of neurodegeneration in chronic neurological diseases. Expert Rev Mol Diagn. 2008;8(4):479–94.
Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017;16(10):837–47.
Wilke C, Bender F, Hayer SN, Brockmann K, Schöls L, Kuhle J, et al. Serum neurofilament light is increased in multiple system atrophy of cerebellar type and in repeat-expansion spinocerebellar ataxias: a pilot study. J Neurol. 2018;265(7):1618–24.
Wilke C, Haas E, Reetz K, Faber J, Garcia-Moreno H, Santana MM, et al. Neurofilaments in spinocerebellar ataxia type 3: blood biomarkers at the preataxic and ataxic stage in humans and mice. EMBO Mol Med. 2020;12(7):e11803.
Yang Z, Shi C, Zhou L, Li Y, Yang J, Liu Y, et al. Metabolic profiling reveals biochemical pathways and potential biomarkers of spinocerebellar ataxia 3. Front Mol Neurosci. 2019;12:159.
Zhou J, Lei L, Shi Y, Wang J, Jiang H, Shen L, et al. Serum concentrations of NSE and S100B in spinocerebellar ataxia type 3/Machado-Joseph disease. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2011;36(6):504–10.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Coarelli, G., Durr, A. (2023). Blood and CSF Biomarkers in Autosomal Dominant Cerebellar Ataxias. In: Soong, Bw., Manto, M., Brice, A., Pulst, S.M. (eds) Trials for Cerebellar Ataxias. Contemporary Clinical Neuroscience. Springer, Cham. https://doi.org/10.1007/978-3-031-24345-5_13
Download citation
DOI: https://doi.org/10.1007/978-3-031-24345-5_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-24344-8
Online ISBN: 978-3-031-24345-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)