
Abstract
Superior semicircular canal dehiscence (SSCD) syndrome, first described by Minor et al., results from bony dehiscence of the middle fossa overlying the superior semicircular canal, classically leading to symptoms of hearing loss, autophony, and sound-induced vertigo [1]. The mechanism behind this condition involves a pathophysiologic mobile window at the area of dehiscence. The two physiologic mobile windows, the oval and round windows, allow acoustic energy to travel through the cochlear scalae with only limited effect on the vestibular system. The addition of a third mobile window (TMW) provides another route for mechanical energy to traverse the inner ear, thus altering the normal function of both the cochlea and the vestibular organs. In terms of cochlear function, dissipation of acoustic energy through a TMW results in increased air conduction thresholds. Additionally, the impedance differential between the scala tympani and scala vestibuli increases, resulting in decreased bone thresholds. The resulting audiologic effect is a low-to-mid frequency air-bone gap, supranormal bone thresholds, and increased sensitivity to bodily sounds (e.g., autophony, pulsatile tinnitus). In terms of vestibular function, shunting of acoustic energy through the vestibule and superior semicircular canal leads to vestibular symptoms, most classically vertigo with loud sound (Tullio phenomenon). In addition to SSCD, less common areas of dehiscence between the otic capsule and surrounding structures have been reported, including the vestibular aqueduct, internal auditory canal, carotid canal, and facial nerve [2–6]. As with SSCD, these other foci of dehiscence have the potential to create a mobile third window, leading to hearing loss and vestibular dysfunction [2].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Superior canal dehiscence syndrome
- Third mobile window syndrome
- Hearing loss
- Autophony
- Tullio phenomenon
- Vestibular disease
- Vertigo
- Skull base
Background
Superior semicircular canal dehiscence (SSCD) syndrome, first described by Minor et al., results from bony dehiscence of the middle fossa overlying the superior semicircular canal, classically leading to symptoms of hearing loss, autophony, and sound-induced vertigo [1]. The mechanism behind this condition involves a pathophysiologic mobile window at the area of dehiscence. The two physiologic mobile windows, the oval and round windows, allow acoustic energy to travel through the cochlear scalae with only limited effect on the vestibular system. The addition of a third mobile window (TMW) provides another route for mechanical energy to traverse the inner ear, thus altering the normal function of both the cochlea and the vestibular organs. In terms of cochlear function, dissipation of acoustic energy through a TMW results in increased air conduction thresholds. Additionally, the impedance differential between the scala tympani and scala vestibuli increases, resulting in decreased bone thresholds. The resulting audiologic effect is a low-to-mid frequency air-bone gap, supranormal bone thresholds, and increased sensitivity to bodily sounds (e.g., autophony, pulsatile tinnitus). In terms of vestibular function, shunting of acoustic energy through the vestibule and superior semicircular canal leads to vestibular symptoms, most classically vertigo with loud sound (Tullio phenomenon). In addition to SSCD, less common areas of dehiscence between the otic capsule and surrounding structures have been reported, including the vestibular aqueduct, internal auditory canal, carotid canal, and facial nerve [2,3,4,5,6]. As with SSCD, these other foci of dehiscence have the potential to create a mobile third window, leading to hearing loss and vestibular dysfunction [2].
Symptoms of SSCD syndrome are variable and may be nonspecific. A study by Naert et al. aggregating symptoms reported spontaneous dizziness, sound- and pressure-induced vertigo, autophony, and hearing loss as occurring in >35% of affected patients. Other less specific symptoms included aural pressure, pulsatile tinnitus, hyperacusis to bodily or environmental sounds, and spontaneous or pulsatile oscillopsia [7]. Given the broad overlap of SSCD symptomatology with other conditions including Ménière’s disease, vestibular migraine, patulous eustachian tube, conductive hearing loss, idiopathic intracranial hypertension, and other various causes of pulsatile tinnitus, alternative diagnoses should be considered when evaluating a patient with possible SSCD syndrome. Conversely, as growing evidence suggests a complex web of associations among vestibular disorders, including cohort studies showing an association between SSCD and migraine, the possibility of SSCD syndrome coinciding with other vestibular diagnoses should not be ignored [8,9,10].
Physical examination of patients with SSCD may show vertical torsional nystagmus with the fast-phase components directed downward and toward the affected ear with high-intensity acoustic stimuli, positive pressure applied to the tympanic membrane, or Valsalva maneuvers against pinched nostrils. These findings correspond to excitation of the superior canal afferents from ampullofugal deflection of the cupula. Nystagmus with opposite directionality may be seen with negative pressure in the external auditory canal, Valsalva against a closed glottis, and jugular venous compression, all of which cause ampullopetal deflection of the cupola and subsequent inhibition of tonic superior semicircular canal afferent activity. In addition to superior semicircular canal afferent modulation, otolith organ activation and inhibition may also occur, resulting in sound-induced ocular tilt mediated by the utricle and saccule as well as cardiovascular changes mediated by vestibulosympathetic reflexes.
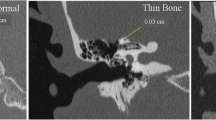
Diagnostic evaluation of SSCD relies on both radiographic and audiologic testing. Assessing the bony integrity of the middle fossa requires high resolution computed tomography (HRCT) with ≤0.5 mm cuts performed perpendicular and parallel to the plane of the superior semicircular canal (Stenver and Poschl views). Audiologic evaluation involves audiogram as well as cervical and/or ocular vestibular evoked myogenic potentials (cVEMP and oVEMP), and electrocochleography (ECOG). Audiogram may show a hearing loss as described above, while cVEMP may show decreased thresholds, and oVEMP may show increased amplitude. ECOG may show an increased SP/AP ratio. Recently, oVEMP has been shown to be the most sensitive and specific test to confirm SSCD syndrome suspected from history, physical exam, audiogram, and HRCT [11,12,13].
Definitive diagnosis of SSCD syndrome can be challenging due to the inherent limitations of current imaging technology and variable patient factors. The incidence of SSCD syndrome in temporal bone histopathologic series is estimated to be between 0.5% and 0.6% [14, 15]. In contrast, radiographic studies suggest a dehiscence rate of 3.9–9% depending on the level of CT resolution and use of Stenver & Poschl views [14]. This discrepancy arises from the inability of available CT technology to reliably detect extremely thin bone (i.e., <0.5 mm), thus raising the risk for false-positive results. Imaging limitations also may lead to false-negative results insofar as it cannot detect areas of increased bony compliance (“near dehiscence”) or pinpoint areas of dehiscence, which have been shown to alter inner ear impedance [16, 17]. Other anatomic and patient factors may also complicate diagnosis. Several series describe patients with clear radiographic and audiovestibular evidence of SSCD who lack clear symptoms of SSCD syndrome [18,19,20]. One hypothesis suggests this is due to a tight dural seal over the area of dehiscence that prevents changes to inner ear impedance [15]. Alternatively, patients may have variable sensitivity to the auditory and vestibular effects of active dehiscence, raising the possibility that large areas of dehiscence may produce no noticeable symptoms for some, while others are exquisitely aware of symptoms produced by radiographically occult lesions. Finally, as noted earlier, SSCD-S may mimic or coincide with other similarly presenting conditions. These points underscore the importance of considering alternative diagnoses when evaluating a patient with possible SSCD-S, obtaining adequate objective testing to support a final diagnosis, and recommending appropriate management options based on the severity of symptoms.
Proposed Etiologies of Superior Canal Dehiscence
The etiology of SSCD syndrome is not fully understood. Early descriptions of the condition included anatomic and histopathologic studies that support an underlying developmental cause. However, other evidence suggests the condition may be acquired later in life due to various factors that cause thinning of the lateral skull base. Some researchers suggest a hybrid, multifactorial etiology for SSCD that incorporates a developmental basis for near dehiscence that later progresses to full-blown SSCD syndrome owing to acquired factors.
Abnormal Development and Congenital Factors
Several findings from histopathologic studies support a developmental etiology for SSCD. First, thinning of the middle fossa tends to be symmetric. Carey et al. showed that extremely thin bone (i.e., ≤0.10 mm) over one superior canal was strongly associated with middle fossa thinning on the contralateral side (0.07 ± 0.05 mm), which was significantly less than the average thickness found in adult controls (0.96 ± 0.61 mm). Similar rates of bilateral skull base attenuation have been described in studies evaluating the association between SSCD and spontaneous tegmen defects [14, 21,22,23]. Second, specimens with canal dehiscence show stable ossification patterns with lamellar bone on the margins of thin or dehiscent areas. Preservation of lamellae deposited during skull base ossification suggests that thinning occurs early in development rather than through a process that erodes through previously deposited bone. Third, samples from pediatric patients show that middle fossa thickness inversely correlates with age. In Anson and Donaldson’s description of otic capsule development from a cartilage precursor, multiple, trilaminar ossification centers grow and fuse between the 15th and 21st weeks of development, eventually encasing the otic capsule in bone. The innermost, endosteal layer shows minimal growth following fusion. The middle layer develops into a dense, petrous layer approximately five months after birth, and the outer layer continues to grow and become pneumatized postnatally [24]. Carey et al. showed that average bone thickness in infants ≤1 year of age was only 0.15 ± 0.15 mm, while in the premature infant, the superior canal is covered only by the thin, inner periosteal layer until as late as ten months postnatally (Fig. 2.1) [15]. In adult specimens with thinning or dehiscence, there is a similar appearance to infant specimens, suggesting a failure in postnatal development (Fig. 2.2) [15, 26].

(A) The posterior semicircular canal is not totally covered at gestational age of 24 weeks (arrow). (B) Mastoid development is not complete (arrowhead) in this thin bone from a neonate. (C) The correlation between age and bone thickness overlying the posterior semicircular canal in children (ρ = 0.68, p < 0.01; hematoxylin and eosin staining). D indicates dura, ES endolymphatic sac, P posterior semicircular canal, VA vestibular aqueduct. (a) Shows gestational age of 24 weeks, (b) a neonate, and (c) is a graph illustrating age and bvone thickness in children. (Republished with permission of Wolters Kluwer Health, Inc., from Nomiya S, Cureoglu S, Kariya S, et al. Posterior semicircular canal dehiscence: a histopathologic human temporal bone study. Otol Neurotol. 2010; 31(7):1122-1127. Permission conveyed through Copyright Clearance Center, Inc.) [25]

The dehiscence of posterior semicircular canal in adult (male subject, right ear). The periosteum (arrow) remains between the canal and the dura (hematoxylin and eosin staining). D—indicates dura mater, M—membranous labyrinth, PF—posterior cranial fossa. (Republished with permission of Wolters Kluwer Health, Inc., from Nomiya S, Cureoglu S, Kariya S, et al. Posterior semicircular canal dehiscence: a histopathologic human temporal bone study. Otol Neurotol. 2010; 31(7):1122-1127. Permission conveyed through Copyright Clearance Center, Inc.) [25]
In addition to developmental factors, other congenital comorbidities may play a role in canal dehiscence. Kuhn et al. showed an association between Chiari type I malformation and both posterior and superior canal dehiscence, although rates of posterior dehiscence were higher in these patients [27]. The authors proposed that overcrowding of the posterior fossa and elevated intracranial pressure, both fundamental elements of Chiari malformation, may contribute to bony remodeling or impaired development. Genetic factors have also been implicated, with mutations in the COCH and CDH23 genes being linked to SSCD in some reports [28, 29]. A small number of case reports have also shown a possible familial predisposition to SSCD syndrome [18, 23]. However, a clear inheritable cause has not been identified.
While temporal bone studies support a developmental etiology for SSCD, this theory is not well explained by the sequence of semicircular canal development. As the membranous labyrinth forms from the otocyst, the semicircular canals develop in a predictable sequence, beginning with the superior canal, followed by the posterior and horizontal canals. Ossification then follows this same sequence once the membranous labyrinth approaches adult dimensions [30, 31]. Thus, a purely developmental failure affecting the superior canal would also be expected to affect its posterior and lateral counterparts. However, such associations are rarely seen in patients with SSCD syndrome, with the vast majority showing otherwise normal otic capsule anatomy. One proposed explanation for this discrepancy is that protrusion of the developing superior canal into the cranium exposes this portion of the membranous labyrinth to contact with the dura and/or temporal lobe pulsations, which may lead to adhesion and focally impaired ossification of the superior canal during development [32]. A study by Hadi et al. may provide support for this theory. Authors of this study showed that that 92.3% of surgically confirmed cases of SSCD syndrome showed protrusion of the superior canal into the middle cranial fossa, while only 30% of non-dehiscent cases showed similar protrusion. These authors further reported that 28.6% of non-protruding canals were covered by supralabyrinthine air cells, while the remaining 71.4% were at the level of the tegmen and covered with thick bone [21].
Another major shortcoming of the developmental theory for SSCD syndrome is the tendency for the condition to manifest in mid-to-late adulthood, which suggests an association with age or other longstanding conditions that decrease bony thickness overlying the superior canal [1, 33]. Canal dehiscence has been reported in the pediatric population, although the condition is rare in this age-group [34]. In a series of HRCT scans performed on children with hearing loss, Chen et al. reported a radiographic dehiscence rate of 4% and 11%, respectively [35]. However, these patients lacked other symptoms of SSCD syndrome and the established high rate of false positives using imaging alone makes it difficult to interpret the results of this study. For infants with radiographic dehiscence, the lack of objective findings may be due to limitations associated with newborn hearing screening and audiometric testing coupled with postnatal middle fossa thickening and vestibular maturation that occur before more detailed testing is possible.
Acquired Factors
Age and Gender
The role of age and gender in the development of SSCD syndrome has been evaluated by several studies. Davey et al. evaluated 140 temporal bones from 121 patients ranging from six to 86 years of age. These authors found a statistically significant difference in bone thickness when comparing females 45 years old and younger vs. 45 years old and above. This difference was mostly due to a significant decline in average thickness in females over 70. Similar findings were seen in male patients, although average bone thickness was higher. In addition, a linear regression model using age and gender as independent variables showed a loss of 0.005 mm of bone over the superior canal for every year increase in age [36]. Similarly, in a radiographic study, Nadgir et al. categorized patients in increasing, 20-year age groups. These researchers found a 93% increase in radiographic SSCD with each successively older age category (Fig. 2.3) [37]. Other investigations have shown evidence for progressive thinning with increasing age and even direct observation of radiographic progression [38, 39], although in some studies, significant bone loss was only apparent in female patients [14, 40]. Authors suggested age-related bone demineralization, which is more pronounced in menopausal women, as a possible cause for these findings. The progressive thinning with each successive decade in life is in sharp contrast to the progressive thickening seen during the first four years of life [15]. This suggests that the natural course of bone thickness over the superior semicircular canal is relatively rapid thickening during early childhood with slow progressive thinning over the rest of an individual’s life with perhaps an acceleration of this process in the 7th to 9th decades.

Prevalence of patients with SSCD and age (Republished with permission of American Society of Neuroradiology, from Nadgir RN, Ozonoff A, Devaiah AK, Halderman AA, Sakai O. Superior Semicircular Canal Dehiscence: Congenital or Acquired Condition? Am J Neuroradiol. 2011; 32(5):947-949. Permission conveyed through Copyright Clearance Center, Inc) [37]
Chronic Conditions
Several chronic conditions have also been proposed to lead to acquired dehiscence, including idiopathic intracranial hypertension (IIH), and chronic otitis media. These factors have all been implicated more broadly with middle fossa erosion leading to cerebrospinal fluid leak (CSF) and encephalocele formation [41,42,43]. By extension, it is thought they may also contribute to thinning of the otic capsule over the superior canal. This is borne out by the literature, which does seem to support an association between tegmen defects and SSCD syndrome, with rates of coinciding defects ranging from 14 to 76% [14, 21, 38, 44, 45]. A retrospective study by Oh et al. found that patients with lateral skull base encephalocele and CSF leak had a 5.7 times greater likelihood of having SSCD syndrome compared to controls [22].
IIH has been linked to skull base attenuation, encephalocele formation, and CSF leak in multiple studies [14, 15, 46]. Mechanistically, this is thought to be due to increased force of dural pulsations resulting in bony erosion of the middle fossa. However, the association between IIH and SSCD proper is less clear. In their large temporal bone series, Carey et al. found no association between SSCD and a clinical history of elevated intracranial pressure [15]. More recent studies investigating the role of intracranial pressure have been equivocal. Several series have found an association with tegmen thinning and/or dehiscence and a history of IIH or high opening pressure on lumbar puncture [47, 48]. However, while some studies showed associated thinning over the superior canal, others found otic capsule bone to be unaffected [49,50,51]. Obesity, considered a risk factor for IIH and lateral skull base defects, has also been evaluated by several studies, but its association with SSCD specifically is unclear [22, 50, 52, 53]. Obstructive sleep apnea (OSA), which is closely linked to obesity, has been linked to radiographic SSCD [54]. One proposed theory for this association involves dramatic increases in intracranial pressure, with CSF pressures transiently rising between 50 and 750 mm H2O during apneic events [55]. However, as with other studies reporting only rates of radiographic dehiscence, it is difficult to make firm conclusions about an association with SSCD syndrome proper.
The role of chronic inflammation in SCCD has also been evaluated. In a large retrospective study, Cho et al. compared rates of radiologic SSCD in ears with a history of unilateral chronic otitis media (COM), using the healthy ears as controls. The authors found that ears affected by COM had significantly higher rates of both definite and suspicious SSCD compared to ears without COM (3.4% vs. 0.3% and 3.2% vs. 0.9%, respectively). Furthermore, authors found reduced mastoid volumes with intact tympanic membranes in patients with SSCD, suggesting that a past history of otitis media without active inflammation may have a role in the development of dehiscence [56]. Other studies have also shown smaller temporal volumes, as well as reduced pneumatization and density, in patients with SSCD compared to controls [14, 57, 58].
Other Causes for Acquired Dehiscence Associations
Other less common causes of acquired dehiscence have been reported. Temporal bone fracture has been implicated in several reports of SSCD syndrome, as have acute infection, fibrous dysplasia, neoplasm, vascular anomalies, and erosion from the superior petrosal sinus [39, 59,60,61,62,63,64]. Other acquired foci of labyrinthine dehiscence have also been described. Lateral canal dehiscence is a well-known entity, with the most common causes being cholesteatoma, infection, and iatrogenic injury. Additionally, posterior semicircular canal dehiscence has been described in several reports, with symptoms mimicking SSCD syndrome but vestibular findings consistent with a posterior canal lesion [65, 66].
Multifactorial Etiology
Competing evidence for a developmental and an acquired etiology for SSCD may be reconciled by a multifactorial model that incorporates both sets of factors. In this model, developmentally thin middle fossa bone is subjected to long-term, progressive thinning that ultimately results in development of SSCD syndrome. This hypothesis could account for the observation that radiographic thinning and dehiscence appears to be present in a subset of patients who lack symptoms or other acquired risk factors for SSCD syndrome. Exposure to such risk factors need only cause sub-millimeter reductions in bone thickness to cause frank dehiscence and development of SSCD symptoms. One line of evidence that may support this theory is the relatively high rate of patients with SSCD syndrome who report a specific, often innocuous, precipitating event prior to developing symptoms. In their original article, which included both surgical and non-surgical patients, Minor et al. reported that 23% experienced a precipitating event leading to onset of symptoms, including minor head trauma, falls without head trauma, lifting, and straining [1]. In a later meta-analysis of surgically managed patients, Watters et al. observed a second event, including acute pressure changes, in 48% of patients [67]. That such common, often low-intensity events could lead to dehiscence suggests that these patients were already predisposed to developing dehiscence, due to developmental thinning, acquired attenuation, or a combination of both. This phenomenon also underscores research showing that even pinpoint areas of dehiscence can alter inner ear impedance, leading to development of a third mobile window [16, 68].
Conclusion
SSCD is a TMW phenomenon with unclear etiology, although there is evidence supporting both developmental and acquired causes. Evidence for a developmental etiology largely comes from histopathologic studies showing a high rate of symmetric middle fossa attenuation, stable bone deposition, and progressive middle fossa thickening in infants. Evidence of SSCD as an acquired phenomenon stems from its manifestation in later life, as well as studies showing associations between middle fossa thinning with factors including advancing age, female gender, and IIH. These competing theories may be reconciled by a multifactorial model wherein developmentally thin bone over the superior canal is subjected to further thinning from acquired causes, ultimately leading to frank dehiscence and development of SSCD syndrome. Regardless of its cause, clear diagnosis may be challenging due to nonspecific symptomatology, limitations of current radiologic technology, and variable patient factors. Thorough patient evaluation and appropriate testing are required to both establish a diagnosis of SSCD and to properly assess symptom burden before recommending treatment.
References
Minor LB. Superior canal dehiscence syndrome. Am J Otol. 2000;21(1):9–19.
Merchant SN, Nakajima HH, Halpin C, et al. Clinical investigation and mechanism of air-bone gaps in large vestibular aqueduct syndrome. Ann Otol Rhinol Laryngol. 2007;116(7):532–41. https://doi.org/10.1177/000348940711600709.
Blake DM, Tomovic S, Vazquez A, Lee HJ, Jyung RW. Cochlear-facial dehiscence–a newly described entity. Laryngoscope. 2014;124(1):283–9. https://doi.org/10.1002/lary.24223.
Karlberg M, Annertz M, Magnusson M. Mondini-like malformation mimicking otosclerosis and superior semicircular canal dehiscence. J Laryngol Otol. 2006;120(5):419–22. https://doi.org/10.1017/S0022215106000934.
Kim HHS, Wilson DF. A third mobile window at the cochlear apex. Otolaryngol Head Neck Surg Off J Am Acad Otolaryngol-Head Neck Surg. 2006;135(6):965–6. https://doi.org/10.1016/j.otohns.2005.04.006.
Lund AD, Palacios SD. Carotid artery-cochlear dehiscence: a review. Laryngoscope. 2011;121(12):2658–60. https://doi.org/10.1002/lary.22391.
Naert L, Berg R, Heyning P, et al. Aggregating the symptoms of superior semicircular canal dehiscence syndrome. Laryngoscope. 2018;128(8):1932–8. https://doi.org/10.1002/lary.27062.
Zhu RT, Van Rompaey V, Ward BK, Van de Berg R, Van de Heyning P, Sharon JD. The interrelations between different causes of dizziness: a conceptual framework for understanding vestibular disorders. Ann Otol Rhinol Laryngol. 2019;128(9):869–78. https://doi.org/10.1177/0003489419845014.
Chung LK, Ung N, Spasic M, et al. Clinical outcomes of middle fossa craniotomy for superior semicircular canal dehiscence repair. J Neurosurg. 2016;125(5):1187–93. https://doi.org/10.3171/2015.8.JNS15391.
Jung DH, Lookabaugh SA, Owoc MS, McKenna MJ, Lee DJ. Dizziness is more prevalent than autophony among patients who have undergone repair of superior canal dehiscence. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2015;36(1):126–32. https://doi.org/10.1097/MAO.0000000000000531.
Zhang L, Creighton FX, Carey JP. A cohort study comparing importance of clinical factors in determining diagnosis and treatment for superior semicircular canal dehiscence syndrome. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2021;42(9):1429–33. https://doi.org/10.1097/MAO.0000000000003274.
Zuniga MG, Janky KL, Nguyen KD, Welgampola MS, Carey JP. Ocular versus cervical VEMPs in the diagnosis of superior semicircular canal dehiscence syndrome. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2013;34(1):121–6. https://doi.org/10.1097/MAO.0b013e31827136b0.
Janky KL, Nguyen KD, Welgampola M, Zuniga MG, Carey JP. Air-conducted oVEMPs provide the best separation between intact and superior canal dehiscent labyrinths. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2013;34(1):127–34. https://doi.org/10.1097/MAO.0b013e318271c32a.
Crovetto M, Whyte J, Rodriguez OM, Lecumberri I, Martinez C, Eléxpuru J. Anatomo-radiological study of the superior semicircular canal dehiscence. Eur J Radiol. 2010;76(2):167–72. https://doi.org/10.1016/j.ejrad.2009.05.038.
Carey JP, Minor LB, Nager GT. Dehiscence or thinning of bone overlying the superior semicircular canal in a temporal bone survey. Arch Otolaryngol Neck Surg. 2000;126(2):137. https://doi.org/10.1001/archotol.126.2.137.
Pisano DV, Niesten MEF, Merchant SN, Nakajima HH. The effect of superior semicircular canal dehiscence on intracochlear sound pressures. Audiol Neurotol. 2012;17(5):338–48. https://doi.org/10.1159/000339653.
Ward BK, Wenzel A, Ritzl EK, et al. Near-dehiscence: clinical findings in patients with thin bone over the superior semicircular canal. Otol Neurotol. 2013;34(8):1421–8. https://doi.org/10.1097/MAO.0b013e318287efe6.
Mikulec AA, McKenna MJ, Ramsey MJ, et al. Superior semicircular canal dehiscence presenting as conductive hearing loss without vertigo. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2004;25(2):121–9. https://doi.org/10.1097/00129492-200403000-00007.
Erdogan N, Songu M, Akay E, et al. Posterior semicircular canal dehiscence in asymptomatic ears. Acta Otolaryngol. 2011;131(1):4–8. https://doi.org/10.3109/00016489.2010.502184.
Berning AW, Arani K, Branstetter BF. Prevalence of superior semicircular canal dehiscence on high-resolution CT imaging in patients without vestibular or auditory abnormalities. AJNR Am J Neuroradiol. 2019;40(4):709–12. https://doi.org/10.3174/ajnr.A5999.
El Hadi T, Sorrentino T, Calmels MN, Fraysse B, Deguine O, Marx M. Spontaneous tegmen defect and semicircular canal dehiscence: same etiopathogenic entity? Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2012;33(4):591–5. https://doi.org/10.1097/MAO.0b013e31824bae10.
Oh MS, Vivas EX, Hudgins PA, Mattox DE. The prevalence of superior semicircular canal dehiscence in patients with mastoid encephalocele or cerebrospinal fluid otorrhea. Otol Neurotol. 2019;40(4):485–90. https://doi.org/10.1097/MAO.0000000000002155.
Brantberg K, Bergenius J, Mendel L, Witt H, Tribukait A, Ygge J. Symptoms, findings and treatment in patients with dehiscence of the superior semicircular canal. Acta Otolaryngol. 2001;121(1):68–75. https://doi.org/10.1080/000164801300006308.
Anson BJ, Donaldson JA. Surgical anatomy of the temporal bone. 3rd ed. Philadelphia: Saunders; 1981.
Nomiya S, Cureoglu S, Kariya S, et al. Posterior semicircular canal dehiscence: a histopathologic human temporal bone study. Otol Neurotol. 2010;31(7):1122–7. https://doi.org/10.1097/MAO.0b013e3181eb3309.
Whyte Orozco J, Martínez C, Cisneros A, Obón J, Gracia-Tello B, Angel CM. Defect of the bony roof in the superior semicircular canal and its clinical implications. Acta Otorrinolaringol Esp. 2011;62(3):199–204. https://doi.org/10.1016/j.otorri.2010.11.009.
Kuhn JJ, Clenney T. The association between semicircular canal dehiscence and Chiari type I malformation. Arch Otolaryngol Neck Surg. 2010;136(10):1009. https://doi.org/10.1001/archoto.2010.169.
Hildebrand MS, Tack D, Deluca A, et al. Mutation in the COCH gene is associated with superior semicircular canal dehiscence. Am J Med Genet A. 2009;149(2):280–5. https://doi.org/10.1002/ajmg.a.32618.
Niesten MEF, Lookabaugh S, Curtin H, et al. Familial superior canal dehiscence syndrome. JAMA Otolaryngol Neck Surg. 2014;140(4):363. https://doi.org/10.1001/jamaoto.2013.6718.
Nemzek WR, Brodie HA, Chong BW, et al. Imaging findings of the developing temporal bone in fetal specimens. AJNR Am J Neuroradiol. 1996;17(8):1467–77.
Satar B, Mukherji SK, Telian SA. Congenital aplasia of the semicircular canals. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2003;24(3):437–46. https://doi.org/10.1097/00129492-200305000-00014.
Takahashi N, Tsunoda A, Shirakura S, Kitamura K. Anatomical feature of the middle cranial fossa in fetal periods: possible etiology of superior canal dehiscence syndrome. Acta Otolaryngol. 2012;132(4):385–90. https://doi.org/10.3109/00016489.2011.637234.
Karimnejad K, Czerny MS, Lookabaugh S, Lee DJ, Mikulec AA. Gender and laterality in semicircular canal dehiscence syndrome. J Laryngol Otol. 2016;130(8):712–6. https://doi.org/10.1017/S0022215116008185.
Lagman C, Ong V, Chung LK, et al. Pediatric superior semicircular canal dehiscence: illustrative case and systematic review. J Neurosurg Pediatr. 2017;20(2):196–203. https://doi.org/10.3171/2017.3.PEDS1734.
Chen EY, Paladin A, Phillips G, et al. Semicircular canal dehiscence in the pediatric population. Int J Pediatr Otorhinolaryngol. 2009;73(2):321–7. https://doi.org/10.1016/j.ijporl.2008.10.027.
Davey S, Kelly-Morland C, Phillips JS, Nunney I, Pawaroo D. Assessment of superior semicircular canal thickness with advancing age: SSC thickness and age. Laryngoscope. 2015;125(8):1940–5. https://doi.org/10.1002/lary.25243.
Nadgir RN, Ozonoff A, Devaiah AK, Halderman AA, Sakai O. Superior semicircular canal dehiscence: congenital or acquired condition? Am J Neuroradiol. 2011;32(5):947–9. https://doi.org/10.3174/ajnr.A2437.
Lookabaugh S, Niesten MEF, Owoc M, Kozin ED, Grolman W, Lee DJ. Audiologic, cVEMP, and radiologic progression in superior canal dehiscence syndrome. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2016;37(9):1393–8. https://doi.org/10.1097/MAO.0000000000001182.
Bae JS, Lim HW, An YS, Park HJ. Acquired superior semicircular canal dehiscence confirmed by sequential CT scans. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2013;34(6):45–6. https://doi.org/10.1097/MAO.0b013e31828d6753.
Sood D, Rana L, Chauhan R, Shukla R, Nandolia K. Superior semicircular canal dehiscence: a new perspective. Eur J Radiol Open. 2017;4:144–6. https://doi.org/10.1016/j.ejro.2017.10.003.
Kemink JL, Graham MD, Kartush JM. Spontaneous encephalocele of the temporal bone. Arch Otolaryngol Head Neck Surg. 1986;112(5):558–61. https://doi.org/10.1001/archotol.1986.03780050082015.
Stucken EZ, Brown K, Selesnick SH. Clinical and diagnostic evaluation of acoustic neuromas. Otolaryngol Clin N Am. 2012;45(2):269–84, vii. https://doi.org/10.1016/j.otc.2011.12.001.
Sdano MT, Pensak ML. Temporal bone encephaloceles. Curr Opin Otolaryngol Head Neck Surg. 2005;13(5):287–9. https://doi.org/10.1097/01.moo.0000179247.51476.f5.
Nadaraja GS, Gurgel RK, Fischbein NJ, et al. Radiographic evaluation of the tegmen in patients with superior semicircular canal dehiscence. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2012;33(7):1245–50. https://doi.org/10.1097/MAO.0b013e3182634e27.
Teixido M, Kung B, Rosowski JJ, Merchant SN. Histopathology of the temporal bone in a case of superior canal dehiscence syndrome. Ann Otol Rhinol Laryngol. 2012;121(1):7–12. https://doi.org/10.1177/000348941212100102.
Yu A, Teich DL, Moonis G, Wong ET. Superior semicircular canal dehiscence in East Asian women with osteoporosis. BMC Ear Nose Throat Disord. 2012;12:8. https://doi.org/10.1186/1472-6815-12-8.
Kutz JW, Husain IA, Isaacson B, Roland PS. Management of spontaneous cerebrospinal fluid otorrhea. Laryngoscope. 2008;118(12):2195–9. https://doi.org/10.1097/MLG.0b013e318182f833.
Brainard L, Chen DA, Aziz KM, Hillman TA. Association of benign intracranial hypertension and spontaneous encephalocele with cerebrospinal fluid leak. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2012;33(9):1621–4. https://doi.org/10.1097/MAO.0b013e318271c312.
Berkiten G, Gürbüz D, Akan O, et al. Dehiscence or thinning of bone overlying the superior semicircular canal in idiopathic intracranial hypertension. Eur Arch Otorhinolaryngol. 2021;279(6):2899–904. https://doi.org/10.1007/s00405-021-07020-z.
Kuo P, Bagwell KA, Mongelluzzo G, et al. Semicircular canal dehiscence among idiopathic intracranial hypertension patients: SSCD among IIH patients. Laryngoscope. 2018;128(5):1196–9. https://doi.org/10.1002/lary.26795.
Handzel O, Brenner-Ullman A, Niry D, et al. Tegmen attenuation in patients with idiopathic intracranial hypertension is progressive. Laryngoscope. 2020;130(12):28490. https://doi.org/10.1002/lary.28490.
Rizk HG, Hatch JL, Stevens SM, Lambert PR, Meyer TA. Lateral skull base attenuation in superior semicircular canal dehiscence and spontaneous cerebrospinal fluid otorrhea. Otolaryngol Head Neck Surg Off J Am Acad Otolaryngol. 2016;155(4):641–8. https://doi.org/10.1177/0194599816651261.
Jan TA, Cheng YS, Landegger LD, et al. Relationship between surgically treated superior canal dehiscence syndrome and body mass index. Otolaryngol Head Neck Surg Off J Am Acad Otolaryngol-Head Neck Surg. 2017;156(4):722–7. https://doi.org/10.1177/0194599816686563.
Schutt CA, Neubauer P, Samy RN, et al. The correlation between obesity, obstructive sleep apnea, and superior semicircular canal dehiscence: a new explanation for an increasingly common problem. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2015;36(3):551–4. https://doi.org/10.1097/MAO.0000000000000555.
Sugita Y, Iijima S, Teshima Y, et al. Marked episodic elevation of cerebrospinal fluid pressure during nocturnal sleep in patients with sleep apnea hypersomnia syndrome. Electroencephalogr Clin Neurophysiol. 1985;60(3):214–9. https://doi.org/10.1016/0013-4694(85)90033-1.
Cho YW, Shim BS, Kim JW, et al. Prevalence of radiologic superior canal dehiscence in normal ears and ears with chronic otitis media: radiologic SCD in normal versus COM ears. Laryngoscope. 2014;124(3):746–50. https://doi.org/10.1002/lary.24281.
Shim BS, Kang BC, Kim CH, Kim TS, Park HJ. Superior canal dehiscence patients have smaller mastoid volume than age- and sex-matched otosclerosis and temporal bone fracture patients. Korean J Audiol. 2012;16(3):120. https://doi.org/10.7874/kja.2012.16.3.120.
de Jong MA, Carpenter DJ, Kaylie DM, Piker EG, Frank-Ito DO. Temporal bone anatomy characteristics in superior semicircular canal dehiscence. J Otolaryngol. 2017;12(4):185–91. https://doi.org/10.1016/j.joto.2017.08.003.
Crane BT, Carey JP, McMenomey S, Minor LB. Meningioma causing superior canal dehiscence syndrome. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2010;31(6):1009–10. https://doi.org/10.1097/MAO.0b013e3181a32d85.
Goddard JC, Go JL, Friedman RA. Imaging case of the month: fibrous dysplasia causing superior canal dehiscence. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2013;34(1):1–2. https://doi.org/10.1097/MAO.0b013e3182355642.
Koo JW, Hong SK, Kim DK, Kim JS. Superior semicircular canal dehiscence syndrome by the superior petrosal sinus. J Neurol Neurosurg Psychiatry. 2010;81(4):465–7. https://doi.org/10.1136/jnnp.2008.155564.
Parlea E, Georgescu M, Calarasu R. Superior canal dehiscence syndrome – case report. Romanian J Neurol. 2012;11:142–6.
Peng KA, Ahmed S, Yang I, Gopen Q. Temporal bone fracture causing superior semicircular canal dehiscence. Case Rep Otolaryngol. 2014;2014:1–4. https://doi.org/10.1155/2014/817291.
Aladham Y, Ahmed O, Hassan SAS, Francis-Khoury E. Traumatic superior semicircular canal dehiscence syndrome: case report and literature review. J Surg Case Rep. 2021;2021(1):592. https://doi.org/10.1093/jscr/rjaa592.
Cremer PD, Migliaccio AA, Pohl DV, et al. Posterior semicircular canal nystagmus is conjugate and its axis is parallel to that of the canal. Neurology. 2000;54(10):2016–20. https://doi.org/10.1212/wnl.54.10.2016.
Krombach GA, DiMartino E, Schmitz-Rode T, et al. Posterior semicircular canal dehiscence: a morphologic cause of vertigo similar to superior semicircular canal dehiscence. Eur Radiol. 2003;13(6):1444–50. https://doi.org/10.1007/s00330-003-1828-5.
Watters KF, Rosowski JJ, Sauter T, Lee DJ. Superior semicircular canal dehiscence presenting as postpartum vertigo. Otol Neurotol. 2006;27(6):756–68. https://doi.org/10.1097/01.mao.0000227894.27291.9f.
Merchant SN, Rosowski JJ. Conductive hearing loss caused by third-window lesions of the inner ear. Otol Neurotol Off Publ Am Otol Soc Am Neurotol Soc Eur Acad Otol Neurotol. 2008;29(3):282–9. https://doi.org/10.1097/mao.0b013e318161ab24.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Doerfer, K.W., Hong, R.S. (2022). Etiology. In: Gianoli, G.J., Thomson, P. (eds) Third Mobile Window Syndrome of the Inner Ear. Springer, Cham. https://doi.org/10.1007/978-3-031-16586-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-031-16586-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-16585-6
Online ISBN: 978-3-031-16586-3
eBook Packages: MedicineMedicine (R0)