
Abstract
Incontinentia Pigmenti is a complex disease with both systemic and ocular findings. It requires early ophthalmological evaluation given risk of progressive retinal ischemia, neovascularization, and complex tractional and/or rhegmatogenous retinal detachments. Due to its rarity, there are no large-scale clinical trials to establish preferred practice patterns. Nevertheless, management guidelines can be adapted from other proliferative retinopathies that present in infancy. In particular, early identification of retinal ischemia through dilated fundus exams with fluorescein angiography is required to guide treatment course. Laser photocoagulation, cryotherapy, anti-vascular endothelial growth factor medications, scleral buckling, and vitrectomy are all used to curb disease progression. Despite advancements in care, outcomes in Incontinentia Pigmenti can be highly variable.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Bloch-Sulzberger syndrome
- Incontinentia pigmenti
- Neovascularization
- Proliferative retinopathy
- Rhegmatogenous retinal detachment
- Tractional retinal detachment
Introduction
Background
Incontinentia Pigmenti (IP), also known as Bloch-Sulzberger syndrome, is a rare X-linked autosomal dominant disorder that typically manifests at birth or early in childhood. It is a multiorgan syndrome that primarily affects the skin, eyes, central nervous system, and teeth. Initially described in 1906, it was further characterized by Bloch in 1926 and Sulzberger in 1928 [1,2,3].
Epidemiology
The incidence of IP has been reported to be 1 case per 40,000 births, with some estimating an incidence of 0.7 cases per 100,000 births [4, 5]. 90–97% of living patients with IP are female. Because IP is an X-linked dominant disorder, it is typically lethal in hemizygous male embryos. However, a total of at least 120 male patients with IP have been reported in the literature with a few additional case reports published every year since then [6, 7]. Survival in males is mediated through 47, XXY karyotype (Klinfelter syndrome) or somatic mosaicism [8]. Between 65 and 75% of cases are the result of sporadic mutations, while the remainder of the cases are familial [9].
Pathophysiology
In 80% of cases, IP is due to a loss of function, mutation in the IKBKG gene (inhibitor of the nuclear factor KB [NF-kB] kinase subunit gamma), also known as NF-kB Essential Modulator (NEMO), located on the X chromosome at position q28 [10]. NEMO plays a key role in the IK kinase complex; it works to cleave IKB, a protein that inhibits NF-kB.
NF-kB upregulates the immunogenic response and prevents cellular apoptosis. When the IK kinase complex (which includes NEMO) is activated, it cleaves IKB from NF-kB, allowing NF-kB to move to the nucleus to initiate transcription of proteins that prevent cellular apoptosis. Without NF-kB, cells in patients with IP are at an increased risk of apoptosis. These cells may upregulate production of chemotactic factors including eotaxin, a cytokine specific for eosinophils, and likely explains the eosinophilia seen in IP.
Currently, it is postulated that this inflammatory process is the common mechanism in IP. In the retina, the inflammation can result in vaso-occlusion and ischemia. The vaso-occlusion in the retinal arteries leads to areas of under perfused and avascular retina with resulting ischemia. Neovascularization (NV) can then occur as a damaging sequela [11, 12].
Clinical Features
Dermatological findings are typically the first manifestation of IP and are present in almost 100% of patients with IP. The most common dermatologic presentation is macular patches of hyperpigmentation in a swirling or whirling pattern, which are present in 98% of patients and typically fade by early adulthood. A vesicular rash can also be seen in 90% of patients and can be present at birth (Fig. 27.1). This is followed by a verrous rash, which can occur in the first few months of life in approximately 70% of patients (Fig. 27.2). Lastly, about 28% of patients are found to have hypopigmented lesions arranged in streaks or whorls [11].

Two-week-old baby girl with incontinentia pigmenti and typical vesicular rash noted on upper and lower extremities

Brownish pigmentation in a reticular pattern A and verrucous rash B noted on a 3-week-old baby with incontinentia pigmenti
While the dermatological findings aid in diagnosis of the disease, dermatologic manifestations are often benign. Patients with IP can also develop dental (54–80%) [11], central nervous system (40%) [13], and ophthalmic manifestations (36.5%) [7], the latter two of which have the most functional impact.
Ocular abnormalities include strabismus, microphthalmia, optic nerve atrophy, congenital cataracts, and retinal anomalies. Retinal anomalies are far more common and often occur in the first year of life [11]. They include incomplete vascularization leading to NV, retinal hemorrhage, and retinal detachment (RD). Foveal hypoplasia and foveal vascularization have also been reported, though there is some disagreement in the literature as to whether true “foveal hypoplasia” occurs [6, 7, 11]. An avascular peripheral retina is considered a classic retinal finding of IP (Fig. 27.3). Macular findings of IP are less obvious on exam but can include a blunted foveal pit and absence of a normal parafoveal vasculature pattern.

Example of stage 1 disease with vessel tortuosity seen on color fundus photo A and incomplete peripheral vascularization without neovascularization on FA B on a 6-month-old female with incontinentia pigmenti
In describing the management of various stages of vitreoretinal disease in IP, we will refer to the staging scheme devised by Montezuma et al. in their systematic review (Table 27.1; Figs. 27.3, 27.4, 27.5 and 27.6) [12].

Example of stage 2 disease with peripheral neovascularization noted on FA on a 2-week-old female with incontinentia pigmenti

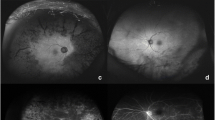
Example of stage 3 disease in a 50-year-old female with late diagnosis of incontinentia pigmenti. Peripheral neovascularization is noted on FA with OCT demonstrating a localized superonasal retinal detachment

Example of stage 4 disease in a 22-year-old female with wide-field photos demonstrating a chronic posterior tractional retinal detachment involving the optic nerve, extensive fibrosis, and abnormalities of the parafoveal vasculature
Medical Management
The main goal of treatment for IP is to prevent the development of retinal NV and subsequent RD. The medical treatment modalities are similar to that of retinopathy of prematurity (ROP), given that both retinopathies are a result of incomplete peripheral vascularization [14]. The two most studied non-surgical treatments for IP are laser photocoagulation therapy and cryotherapy. Recently, intravitreal anti-vascular endothelial growth factor (anti-VEGF) therapy has been employed, however few cases have been described. The timing of when to initiate treatment is debated [15], however many authors agree that early detection and treatment of IP can help prevent complications of retinal ischemia and progression to retinal detachment [12, 13, 16,17,18]. Non-surgical management is typically employed for stage 1 and stage 2 IP, while stage 3 IP and above would necessitate surgical intervention [12].
Laser Photocoagulation Therapy
Laser photocoagulation in treatment of IP has been described as early as the 1970s [19]. Most authors have suggested to start treatment at the first sign of incomplete peripheral vascularization (stage 1) [17, 20], while few have suggested to reserve treatment until fibrovascular proliferation develops (stage 2) [15]. Infants who are treated with laser photocoagulation should be re-evaluated in approximately 6–8 weeks at the office. If response is non-satisfactory, repeated laser photocoagulation should be performed [12].
In general, retinal screening exams should be performed as early as possible with FA (either in the OR using RETeval or in-office examination with non-contact ultra-wide-field Optos imaging), to evaluate peripheral vascular changes and to guide surgeons on where to perform the retina photocoagulation treatment or cryotherapy. During the first year of life the patients should be followed approximately every 3 months at the office with repeated FA at least once a year, or earlier if concerns for persistent NV. Once stable, the recommendation is semiannually until 5 years of age and then annually for the rest of their life [12].
The laser treatment is similar to other lasers used in pediatrics with the Argon green laser as the most commonly used. The goal is to destroy the tissue that is producing VEGF and to treat the peripheral areas of avascularization and NV. We recommend FA guided laser photocoagulation as peripheral areas of retina avascularity and NV can often be difficult to identify on exam and FA can highlight areas that require treatment (Figs. 27.7 and 27.8). Similar to the laser recommendations for ROP [21], gray-white spots of near-confluent laser treatment are recommended. The laser parameters of laser power and duration should be adjusted for each patient based on the area being treated and the child’s pigmentation.

A FA of right eye of 2-week-old female with peripheral neovascularization prior to photocoagulation treatment. B Fundus photo of right eye post-photocoagulation treatment

A Fundus photo and B FA of a 6-month-old female with incontinentia pigmenti, demonstrating peripheral capillary dropout and panretinal photocoagulation scars in the far periphery prior to additional photocoagulation treatment. C Fundus photo and D FA post-additional photocoagulation treatment
Cryotherapy
Cryotherapy was a more common treatment option in the cases published prior to the late 1990s [22,23,24]. With increasing use of laser photocoagulation in the late 1990s, several studies were conducted comparing efficacy and visual outcomes in patients with threshold ROP. Cryotherapy was found to be associated with worse structural and functional outcomes [25], and leads to more inflammation compared to laser photocoagulation. Based on these studies, cryotherapy is now commonly regarded as the second line treatment for stage 2 IP. Moreover, cicatricial changes including epiretinal membranes or proliferative vitreoretinopathy can result from aggressive cryotherapy (and photocoagulation) [15], and have been associated with retinal tears at the edge of previously treated retina [26]. If cryotherapy is required, a similar follow up period to that of laser photocoagulation should be followed to assess treatment response.
Intravitreal Anti-vascular Endothelial Growth Factor
With the success of intravitreal anti-VEGF injections for retinopathy of prematurity, anti-VEGF therapy has recently been proposed as an additional treatment option of IP. It has been suggested that it could be used as sole first line therapy or as adjunct therapy after failed laser photocoagulation. However, the use of anti-VEGF injection has only been described in several case reports [27,28,29] with limited evidence on treatment outcomes. As such, it is the clinician’s discretion to decide whether to use it based on the clinical context. It is important to note that patients with IP may develop cerebrovascular involvement, including occlusive arteriopathies and infarcts, which are theoretical contraindications for anti-VEGF injections [17, 30].
Surgical Management
When less invasive methods have failed to control retinal ischemia in infants and young children, retinal detachment (RD) may result (stages 3 or 4 IP) [12, 15, 31]. These primarily tractional detachments are complicated because retinal ischemia in IP is not limited to developmental patterns of anterior avascularity and posterior vascularity. Due to the rarity of IP, no controlled trials exist evaluating treatments for retinal ischemia, and practice patterns vary based on physician preference [12]. Nevertheless, knowledge gained from late-stage ROP surgical management can guide surgical decision making in IP.
RD occurs in approximately 31% of patients with IP in a recent systematic review [12]. The RD pathophysiology appears to follow a bimodal age distribution, with tractional detachments occurring in children (median age 1.5 years) and rhegmatogenous detachments occurring in adults (age 31.5 years) [15]. This is not surprising as children are prone to retinal ischemia resulting in fibrovascular proliferations, whereas this ischemic process is more quiescent in adults. Neonates or young children with concurrent ocular pathology on initial examination, particularly ischemic optic neuropathy and/or retinal NV, must be identified because of greater odds of developing RDs [15]. These children should be followed closely due to the progressive nature of IP. The decision to pursue RD repair in IP is multifactorial. Care must be taken to weigh the visual potential of the patient. Coexisting eye disease is often present, including persistent fetal vasculature or macular ischemia, which may limit any visual potential and make repair futile [15, 32,33,34].
It is important to note that retinal detachment can occur late in life and early detection is critical because in some cases prophylactic laser demarcation can be performed in certain cases. This reinforces the concept that long life-term follow up is needed in patients diagnosed with IP (Fig. 27.9).

A Wide-field fundus photo and B FA after FA-guided laser photocoagulation on the 50-year-old female from Fig. 27.5
Tractional RD Repair (see the video, by courtesy of Şengül Özdek)
If repair of tractional RD is feasible, the surgical management in IP is similar to that used for late-stage ROP. The first decision is whether to pursue vitrectomy or scleral buckling (SB) or vitrectomy with SB. There is no consensus on the correct approach in IP. Due to similarities in pathophysiology to late-stage ROP, primary vitrectomy may lead to better outcomes in terms of anatomical and functional success rates, particularly in lens-sparing vitrectomy [35]. Lens-sparing pars plicata vitrectomy is preferred if possible due to the risk of aphakic amblyopia. In this approach, a 3-port vitrectomy is performed through pars plicata rather than pars plana. Pars plicata sclerotomy can be approximated by 1 mm posterior to the limbus and adding 1 mm to the distance from the limbus with each year of age after the first year of life up to 4 years of age [36]. Instruments introduced through pars plana may penetrate the subretinal space or cause iatrogenic retinal tears [37]. Moreover, the trajectory of sclerotomy should be perpendicular rather than oblique to avoid lenticular injury and sutureless sclerotomies are not advised because pediatric sclera is thinner than adult which precludes a watertight seal [36]. Vitrectomy gauges typically used include 23G, 25G, and 27G [35]. Vitrectomy and membranectomy may then be performed taking care to avoid iatrogenic retinal breaks. For patients with retrolental adhesions and limited retrolental space, limbal vitrectomy with lensectomy can be performed [36].
For advanced disease, open-sky vitrectomy has also been described with limited success [31, 38]. Although less commonly performed these days, open-sky vitrectomy allows direct visualization of posterior structures after removal of the cornea and lens. This has the advantage of easier access for surgical instruments to the area of pathology, and anterior structures are more readily accessible and more visible; however, this technique risks intraoperative hypotony, choroidal hemorrhage, and globe instability [39].
Lifting the posterior hyaloid membrane is often difficult and complete PVD might not be achieved, and some recommend limiting attempts at PVD induction to avoid iatrogenic breaks [35]. Triamcinolone acetonide can be used intraoperatively to help visualize residual vitreous and assist in removal of persistent tractional membranes. Enzyme-assistant vitrectomy with tissue plasminogen activator [40], autologous plasmin [41], or Ocriplasmin [42] are still being investigated for pediatric vitrectomy but their utility remains to be elucidated. If full vitrectomy or membranectomy is unable to be achieved, silicone oil may be used for endotamponade with fair results [43], however there is the risk of subretinal migration of silicone oil if retinal breaks are present [36].
SB still is advantageous as a primary treatment or adjunct. Several case reports have described the use of SB in IP to minimize tractional forces anterior to the equator [15, 24, 31]. In stage 4A ROP, SB has been shown to prevent further progression of RD and may limit ischemia in the detached retina [44]. It is unclear whether this would apply to IP but extraordinary efforts are often required for disease control. Unlike in adults, the induced myopic shift from SB in pediatrics is large (−11 diopters) and risks refractive amblyopia [45]. Moreover, buckle division or removal is required typically within 6 months for children below the age of 2 years [36]. The role of SB in IP remains uncertain but may be helpful in select cases.
Anatomic success in repair of tractional RDs in IP is uncommon, estimated between 28 and 50% [12, 31], and multiple surgeries are often required. Moreover, many patients are poor surgical candidates at initial presentation due to advanced disease and poor visual potential, highlighting the need for early diagnosis and frequent monitoring.
Rhegmatogenous RD Repair
Rhegmatogenous RDs generally occur in the older subset (31.5 years of age) [15] and may be amenable to standard repairs. Rhegmatogenous RDs have been described in adult ROP and often require multiple surgeries. Patients with IP have been found to have retinal tears at the border of the vascular and avascular retina and therefore require lifelong follow up [15, 26, 46]. Some cases may even spontaneously resolve on their own without intervention [47], although this is an exception to the rule. As with routine rhegmatogenous RD repair, SBs can be used as well [48].
Conclusion
IP is an aggressive disease that leads to progressive retinal ischemia which may be blinding. Management of IP ultimately relies on early screening, frequent monitoring, and low threshold to treat retinal ischemia with laser photocoagulation, anti-VEGF agents or cryotherapy early during the course of the disease. Should these methods fail, and tractional RD develop, surgical management can be undertaken but outcomes are generally poor.
Review Questions
1. A 2-month-old male infant presents with a vesicular rash, suspicious for IP. What is the likely genetic karyotype of this patient?
-
a.
46, XX or somatic mosaicism
-
b.
47, XXY or somatic mosaicism
-
c.
Somatic mosaicism
-
d.
None of the above: the patient does not have IP as all male carriers die in utero
2. An infant is referred for ophthalmic evaluation of incontinentia pigmenti. Fundus exam reveals stage 1 IP. What is the recommended management approach?
-
a.
360° panretinal photocoagulation
-
b.
FA guided laser photocoagulation
-
c.
Cryotherapy
-
d.
Prophylactic scleral buckle
3. What type of retinal detachment is most commonly seen in adult patients with incontinentia pigmenti?
-
a.
Tractional
-
b.
Rhegmatogenous
-
c.
Both
-
d.
Neither are seen
Answers
1. (B) Incontinentia pigmenti is an autosomal dominant X-linked disorder. 90–97% of patients with this disorder are female, however, at least 120 reports of males with incontinentia pigmenti have been reported. Male infants with incontinentia pigmenti typically have Klinefelter’s syndrome (47, XXY) or somatic mosaicism.
2. (B) Stage 1 incontinentia pigmenti, per the grading scheme devised by Montezuma et al. is incomplete peripheral vascularization, abnormal arteriovenous connections, tortuosity and/or engorgement of retinal vessels. These patients benefit from laser photocoagulation. However, FA guided laser therapy is recommended because peripheral areas of retina avascularity and neovascularization can often be difficult to identify.
3. (B) 31% of patients with IP develop retinal detachments. The pathophysiology of the retinal detachment appears to follow a bimodal distribution with tractional detachments occurring more commonly in children (median age 1.5 years) and rhegmatogenous detachments occurring more commonly in adults (median age 31.5).
References
Garrod A. Peculiar pigmentation of the skin of an infant. 1906;39:216.
B B. Eigentumliche bischer nicht beschriebene pigmentaffektion (incontinentia pigmenti). Schweiz Med Wochenschr 1926;7:404–5.
Sulzberger M. Uber eine bischer nicht beschriebene congenitale pigmentanomalie (incontinentia pigmenti). Arch Dermatol Syph (Berl). 1928; v. 154.
Buinauskiene J, Buinauskaite E, Valiukeviciene S. Incontinentia pigmenti (Bloch-Sulzberger syndrome) in neonates. Medicina. 2005;41(6):496–9.
Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
Carney RG. Incontinentia pigmenti. A world statistical analysis. Arch Dermatol. 1976;112(4):535–42.
Minić S, Obradović M, Kovacević I, Trpinac D. Ocular anomalies in incontinentia pigmenti: literature review and meta-analysis. Srpski arhiv za celokupno lekarstvo;138(7–8):408–13.
Adam MP, Ardinger HH, Pagon RA, et al. GeneReviews. 1993.
Cammarata-Scalisi F, Fusco F, Ursini MV. Incontinentia Pigmenti. Actas Dermosifiliogr (Engl Ed). 2019;110(4):273–8.
Fusco F, Pescatore A, Bal E, et al. Alterations of the IKBKG locus and diseases: an update and a report of 13 novel mutations. Hum Mutat. 2008;29(5):595–604.
Swinney CC, Han DP, Karth PA. Incontinentia Pigmenti: a comprehensive review and update. Ophthalmic Surg Lasers Imaging Retina. 2015;46(6):650–7.
Huang NT, Summers CG, McCafferty BK, et al. Management of Retinopathy in Incontinentia Pigmenti: a systematic review and update. Los Angeles, CA: Sage; 2018. v. 2.
O’Doherty M, Mc Creery K, Green AJ, et al. Incontinentia pigmenti–ophthalmological observation of a series of cases and review of the literature. Br J Ophthalmol. 2011;95(1):11–6.
Hahn P, Kim JE, Stinnett S, et al. Aflibercept-related sterile inflammation. Ophthalmology. 2013;120(5):1100-1.e1.
Chen CJ, Han IC, Tian J, et al. Extended follow-up of treated and untreated retinopathy in Incontinentia Pigmenti: analysis of peripheral vascular changes and incidence of retinal detachment. JAMA Ophthalmol. 2015;133(5):542–8.
Catalano RA, Lopatynsky M, Tasman WS. Treatment of proliferative retinopathy associated with incontinentia pigmenti. Am J Ophthalmol. 1990;110(6):701–2.
Michel S, Reynaud C, Daruich A, et al. Early management of sight threatening retinopathy in incontinentia pigmenti. Orphanet J Rare Dis. 2020;15(1):223.
Nguyen JK, Brady-Mccreery KM. Laser photocoagulation in preproliferative retinopathy of incontinentia pigmenti. J AAPOS. 2001;5(4):258–9.
Watzke RC, Stevens TS, Carney RG. Retinal vascular changes of incontinentia pigmenti. Arch Ophthalmol. 1976;94(5):743–6.
Jandeck C. Successful treatment of severe retinal vascular abnormalities in incontinentia pigmenti. Retina. 2004;24(4):631.
Jalali S, Azad R, Trehan HS, et al. Technical aspects of laser treatment for acute retinopathy of prematurity under topical anesthesia. Indian journal of ophthalmology;58(6):509–15.
Rahi J, Hungerford J. Early diagnosis of the retinopathy of incontinentia pigmenti: successful treatment by cryotherapy. Br J Ophthalmol. 1990;74(6):377–9.
Yu YS, Cho BJ. Cryotherapy for retinopathy of incontinentia pigmenti. Korean journal of ophthalmology: KJO. 1991;5(1):47–50.
Ann C. Incontinentia pigmenti: a florid case with a fulminant clinical course in a newborn. Retina. 2000;20(5):558.
Ng EYJ, Connolly BP, McNamara JA, et al. A comparison of laser photocoagulation with cryotherapy for threshold retinopathy of prematurity at 10 years: part 1. Visual function and structural outcome. Ophthalmology 2002;109(5):928–34; discussion 35.
Greven CM, Tasman W. Rhegmatogenous retinal detachment following cryotherapy in retinopathy of prematurity. Arch Ophthalmol. 1989;107(7):1017–8.
Ho M, Yip WWK, Chan VCK, Young AL. Successful treatment of refractory proliferative retinopathy of incontinentia pigmenti by intravitreal ranibizumab as adjunct therapy in a 4-year-old child. Retinal cases & brief reports;11(4):352–5.
Shah PK, Bachu S, Narendran V, et al. Intravitreal bevacizumab for incontinentia pigmenti. Journal of pediatric ophthalmology and strabismus 2013;50 Online:e52-e4.
Ni Y, Huang X, Ruan L, et al. Intravitreal injection of ranibizumab in severe retinopathy of incontinentia pigmenti. J AAPOS. 2018;22(4):325-7.e3.
Avery RL. What is the evidence for systemic effects of intravitreal anti-VEGF agents, and should we be concerned? Br J Ophthalmol. 2014;98(Suppl 1):i7-10.
Wald KJ, Mehta MC, Katsumi O, et al. Retinal detachments in incontinentia pigmenti. Arch Ophthalmol. 1993;111(5):614–7.
Fard AK, Goldberg MF. Persistence of fetal vasculature in the eyes of patients with incontinentia pigment. Arch Ophthalmol. 1998;116(5):682–4.
Goldberg MF. The blinding mechanisms of incontinentia pigmenti. Ophthalmic Genet. 1994;15(2):69–76.
Goldberg MF. Macular vasculopathy and its evolution in incontinentia pigmenti. Trans Am Ophthalmol Soc 1998;96:55–65; discussion-72.
Roohipoor R, Karkhaneh R, Riazi-Esfahani M, et al. Surgical management in advanced stages of retinopathy of prematurity; our experience. J Ophthalmic Vis Res. 2009;4(3):185–90.
Gan NY, Lam W-C. Special considerations for pediatric vitreoretinal surgery. Taiwan J Ophthalmol;8(4):237–42.
Lightfoot D, Irvine AR. Vitrectomy in infants and children with retinal detachments caused by cicatricial retrolental fibroplasia. Am J Ophthalmol. 1982;94(3):305–12.
Hirose T, Katsumi O, Mehta MC, Schepens CL. Vision in stage 5 retinopathy of prematurity after retinal reattachment by open-sky vitrectomy. Arch Ophthalmol. 1993;111(3):345–9.
Schepens CL. Clinical and research aspects of subtotal open-sky vitrectomy. XXXVII Edward Jackson Memorial Lecture. American journal of ophthalmology 1981;91(2):143–71.
Chuang CC, Chen SN. Induction of posterior vitreous detachment in pediatric vitrectomy by preoperative intravitreal injection of tissue plasminogen activator. J Pediatr Ophthalmol Strabismus. 2016;53(2):113–8.
Wu WC, Drenser KA, Lai M, et al. Plasmin enzyme-assisted vitrectomy for primary and reoperated eyes with stage 5 retinopathy of prematurity. Retina. 2008;28(3 Suppl):S75-80.
Drenser K, Girach A, Capone A. A randomized, placebo-controlled study of intravitreal ocriplasmin in pediatric patients scheduled for vitrectomy. Retina. 2016;36(3):565–75.
Scott IU, Flynn HW, Azen SP, et al. Silicone oil in the repair of pediatric complex retinal detachments: a prospective, observational, multicenter study. Ophthalmology 1999;106(7):1399–407; discussion 407.
Hinz BJ, de Juan E, Repka MX. Scleral buckling surgery for active stage 4A retinopathy of prematurity. Ophthalmology. 1998;105(10):1827–30.
Chow DR, Ferrone PJ, Trese MT. Refractive changes associated with scleral buckling and division in retinopathy of prematurity. Arch Ophthalmol. 1998;116(11):1446–8.
Equi RA, Bains HS, Jampol L, Goldberg MF. Retinal tears occurring at the border of vascular and avascular retina in adult patients with incontinentia pigmenti. Retina. 2003;23(4):574–6.
Fekrat S, Humayun MS, Goldberg MF. Spontaneous retinal reattachment in incontinentia pigmenti. Retina. 1998;18(1):75–7.
Chen CJ, Han IC, Goldberg MF. Variable expression of retinopathy in a pedigree of patients with incontinentia pigmenti. Retina. 2015;35(12):2627–32.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file1 (MOV 139202 kb)
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kohler, J., Mundae, R., Naravane, A., Montezuma, S.R. (2023). Incontinentia Pigmenti. In: Özdek, Ş., Berrocal, A., Spandau, U. (eds) Pediatric Vitreoretinal Surgery. Springer, Cham. https://doi.org/10.1007/978-3-031-14506-3_27
Download citation
DOI: https://doi.org/10.1007/978-3-031-14506-3_27
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-14505-6
Online ISBN: 978-3-031-14506-3
eBook Packages: MedicineMedicine (R0)