
Abstract
The sinonasal mucosa is exposed to large quantities of particulates, antigens, and potential pathogens. To address these possible threats, the human sinonasal epithelium has developed a range of protective functions that effectively clear foreign material with minimal collateral tissue damage. This barrier to the outside world is both physical and immunological in nature. The physical barrier consists of respiratory mucus and mucociliary flow as well as the intact epithelial cell layer. When these structural or clearance mechanisms fail, the epithelium plays an important role in activating a subsequent immune response. The immunologic barrier encompasses the recognition of foreign material via pattern-recognition receptors, which then facilitate the innate and adaptive responses. In chronic rhinosinusitis (CRS), the barrier is breached and a self-perpetuating chronic inflammatory reaction develops. The type of inflammation that results is the basis of the current endotype classification of CRS. Understanding these defense mechanisms provides insight into the pathophysiology of inflammatory disorders such as chronic rhinosinusitis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Sinonasal epithelium
- Mucociliary clearance
- Innate immunity
- Adaptive immunity
- Immune barrier
- Toll-like receptors
- Rhinosinusitis
-
Sinonasal epithelium provides both a physical and immunologic barrier to infection.
-
Intracellular junctions, mucus composition, and mucociliary clearance compose the mechanical barrier to pathogen invasion.
-
Innate and adaptive immune responses form the immunologic barrier.
-
Innate immunity provides the first-line defense to pathogens that circumvent the physical mucosal barrier by recognizing conserved pathogen-associated markers and activating a nonspecific inflammatory response.
-
Adaptive immunity confers memory to particular pathogens, providing a faster response to repeated infections.
-
Sufficient stimulation of the innate immune system results in activation of and directs the type of subsequent adaptive immune response.
-
Fungus and staphylococcal superantigens appear to be disease modifiers in chronic rhinosinusitis rather than the direct cause.
-
Dysregulation in the adaptive immune response is the key factor in the pathogenesis of chronic rhinosinusitis.
-
Understanding of host-specific sinonasal immune defenses will influence future therapies for CRS.
4.1 Overview
The sinonasal epithelium is an important biological point of interface with the external environment, clearing foreign materials without significant collateral tissue inflammation. Multiple components carry out this task, and while they are typically considered separately, they are functionally integrated. The first component is mucus produced by nasal glands and the epithelial goblet cells which trap particulate matter to be swept into the nasopharynx via mucociliary flow. The mucus also contains tonic levels of host defense molecules with antimicrobial properties, limiting microbial survival and proliferation. The next anatomic barrier is the epithelial layer with cells bound together via junctional complexes. Breaching these mechanical barriers brings exogenous agents in contact with receptors that activate the innate response. Secretion of host defense molecules is augmented, and chemokines and cytokines are secreted. The latter initiates inflammation and fosters the accumulation and activation of innate effector cells. If the stimulus is sufficiently strong, dendritic cells are activated to initiate the adaptive response consisting of T and B lymphocytes. The nature of this adaptive response is shaped, in large measure, by input from the epithelial cells. In aggregate, this allows the response to be tailored against the particular pathogen(s) that breach the barrier. Three responses have been broadly defined: Type 1, Type 2, and Type 3 which address viruses, parasites, and bacteria/fungi, respectively. Hence the epithelium plays a pivotal role in maintaining homeostasis as well as initiating and tailoring the immune response across the nasal interface. In CRS, a chronic inflammatory response develops that, in large measure, utilizes these physiological immunodefense mechanisms present in the mucosa. Most commonly, these CRS endotypes are characterized as Type 1, Type 2, and Type 3 immune responses, which trigger the symptoms and remodeling changes associated with these CRS subtypes [1]. The components of this system will be reviewed with a focus on the epithelium, and potential areas of dysfunction will be discussed as these may play a role in the development of CRS.
4.2 Anatomic Barrier
4.2.1 Epithelium Structure
The anterior nasal epithelium consists of stratified squamous epithelium with structural and barrier functions similar to skin. In addition to typical sebaceous and sweat glands, the nasal vestibule contains vibrissa—thick specialized hairs that assist in trapping particles—and many fine hairs for filtering smaller particles [2]. At the internal nasal valves, the sinonasal epithelium transitions to pseudostratified ciliated columnar epithelium; this respiratory-type epithelium is found throughout the remainder of the sinonasal tract [3].
Sinonasal epithelium is respiratory epithelium with the goblet, ciliated, and basal cells bound tightly to each other and the basement membrane.
Sinonasal epithelium consists primarily of ciliated cells, with variable concentrations of mucus-producing goblet cells and glands, and relatively few basal cells. The ciliated cells comprise approximately 70% of the sinonasal epithelium and are responsible for mucous clearance from the sinonasal cavities. Each ciliated cell has hundreds of motile cilia at the apex that beat in unison as well as small, immotile microvilli that increase surface area and prevent drying [4]. Each ciliated cell extends from the basement membrane to the surface of the mucosa with the cilia extending into the overlying mucous layer. Goblet cells comprise approximately 20% of epithelial cells, but their density is irregularly distributed throughout the sinonasal epithelium. Goblet cells have a narrow area of basement membrane attachment and are packed with mucin-containing secretory granules. Their apices extend to the mucosal surface, are also covered with microvilli, and contain a large apical pore for the secretion of mucin. Basal cells comprise less than 5% of the respiratory epithelium. They do not have an epithelial surface component and thus are exposed to fewer inhaled particulates and pathogens. Basal cells contain large numbers of hemidesmosomes that attach them firmly to the basement membrane and function as respiratory cell progenitors [5].
4.2.2 Cilia and Mucus
The sinonasal respiratory epithelium is specialized to effectively trap and remove foreign materials from the nasal cavity before it can cause inflammation or infection. When inspired particulates are removed, a reduction in irritation, allergic response, and pathogen invasion is achieved. The rheologic and immunologic properties of nasal mucous composition combine with ciliary function to produce the phenomenon of mucociliary clearance that maintains a healthy sinus tract.
Respiratory mucus is responsible for trapping and neutralizing inspired environmental particulates. Nasal secretions from the goblet cells and submucous glands are augmented by fluid from the lacrimal glands and transudate from the underlying capillary bed to produce 600–1800 mL of mucous per day. The composition of mucus is designed to trap and bind to particulates and allow for transport by cilia [6]. Nasal mucous exists as a bilayer fluid overlying the epithelium comprised of an outer gel layer and pericellular sol layer. The gel layer is composed primarily of hydrated mucin, a thick mesh of glycoproteins produced by the respiratory goblet cells along with secretions from submucosal mucus glands. As the mucin is secreted, it hydrates and coalesces into mucin rafts overlying the watery pericellular sol layer. The viscous nature of the gel layer allows it to trap inspired pathogens and particulates larger than 0.5 μm [7].
In addition, mucous also contribute to the respiratory host defense system directly. Mucins are known to recognize and bind to microorganism surface adhesins promoting the microorganism–mucin bond and promoting clearance. Mucin is known to bind to Mycoplasma pneumoniae, Streptococcus pneumoniae, Pseudomonas aeruginosa, influenza virus, and Escherichia coli. Mucins may bind and thus concentrate secreted innate immune defense molecules such as lactoferrin and lysozyme, which recognize and kill bacterial pathogens [8].
Mucus contains a variety of innate defense molecules to bind and neutralize pathogens.
Ciliated cells in human respiratory mucosa each contain 50–200 motile cilia extending from the apical surface. In humans, these respiratory cilia are approximately 6 μm in length and are anchored by centriole-like basal bodies [6]. Each cilium consists of an axoneme covered by an extension of the cell membrane. The axoneme is comprised of the “9 + 2” microtubular pattern that is preserved in ciliated cells throughout the body. The nine outer microtubule doublets are each connected radially to the two central microtubules and circumferentially to each other by dynein arms. It is these dynein arms that slide along the microtubules, generating force that is translated into microtubule movement [9].
Ciliary motility generates a forward power stroke with the cilium fully extended, delivering force through the tip into the overlying gel phase of the mucous layer. This is followed by a recovery stroke with the cilium tip bent to sweep through the thin pericellular sol layer as it returns to its starting position [7]. Recovery is followed by a quiescent phase. Coordinated beating of the respiratory cilia creates a metachronous wave that propels the mucin rafts of the gel phase in the direction of the power stroke [10]. The combination of mucus composition and ciliary beating creates the phenomenon of mucociliary transport, which is critical in the maintenance of healthy sinonasal mucosa.
4.2.3 Mucociliary Clearance
The remarkable phenomenon known as mucociliary clearance (MCC) combines the biphasic properties of nasal mucus with coordination of the ciliated cellular beating to effectively clear the mucus blanket with trapped particulates from the nasal cavity. Nasal MCC function responds to environmental factors and is under continual dynamic modulation. Ciliary beat frequency increases as a reaction to stressors including increased breathing, cold air, exercise, or inflammation to accelerate mucus clearance [11]. The mucus blanket is swept out of the sinus cavities in predefined patterns via natural ostia then into the posterior nasopharynx where it is swallowed and neutralized in the stomach.
Messerklinger classically described the defined mucus clearance patterns of the paranasal sinuses in 1966 [12]. In the maxillary sinuses, mucus is swept upwards against gravity along the walls and roof towards the superomedially based maxillary ostium, into the ethmoid infundibulum, and then into the middle meatus. The anterior ethmoid cells each drain through inferiorly based individual ostia into the middle meatus. The posterior ethmoids drain into the superior meatus and then sphenoethmoid recess. The sphenoid sinus sweeps mucus anteriorly toward its natural ostium and also drains into the sphenoethmoid recess. The frontal sinus has the most complex clearance pattern. Mucus is cleared from the medial portion of the sinus in a retrograde fashion superiorly along the posterior wall away from the natural ostium, laterally along the roof to the anterior wall, and then carried inferiorly and medially toward the ostium [13].
Ciliated cells beat in a coordinated metachronous wave that rapidly clears the overlying mucus blanket.
The basis of modern endoscopic sinus surgery is to maintain functional MCC patterns. Surgical goals include relief of obstruction while utilizing mucosal-sparing techniques and preserving natural drainage pathways. Failure to incorporate natural ostia when opening a sinus can lead to recirculation particularly in the maxillary sinuses [14]. Preservation of normal mucosa promotes restoration of natural mucociliary flow patterns. This is particularly true at the narrow nasofrontal duct where disruption of normal mucosa can lead to circumferential scarring with narrowing of the frontal duct, obstruction, and mucostasis.
4.2.3.1 Diseases Affecting Mucociliary Clearance
The sinonasal epithelium utilizes a mechanical barrier, effective mucociliary clearance, and optimal healing to maintain mucosal health. However, disease processes affecting ciliary function, rheologic properties of mucus, or obstruction of natural ostia results in altered MCC effectiveness and resultant infection [15]. These include genetic conditions, allergy, asthma, and acute and chronic rhinosinusitis.
Mucociliary clearance efficiency declines with changes in mucus rheology, ciliary impairment, or narrowed sinus ostia.
Primary ciliary dyskinesia (PCD) is a disorder of the ciliary dynein arms resulting in systemically immotile cilia. These patients may have infertility and are unable to effectively transport mucus out of the sinorespiratory tract. The resultant mucus stasis results in chronic rhinosinusitis, bronchiectasis, and recurrent respiratory infections [16]. Cystic fibrosis (CF) is an autosomal-recessive disorder of the CFTR gene resulting in abnormal electrolyte transport. Clinically, there is an abnormal function of the goblet cells and highly viscous mucus. CF patients have defective MCC clearance due to the inability of the cilia to adequately transport the thick mucus rafts and are subject to bronchiectasis and severe sinopulmonary infections [17]. Nasal polyps are found in approximately 40% of CF patients and show a neutrophilic rather than eosinophilic pattern [18]. However, these patients still display a wide spectrum in the severity of their sinus disease despite having identical CFTR gene mutations [19].
Nasal irritants, allergy, and acute infection result in inflammation and mucosal edema that negatively affect mucociliary clearance. Acute upper respiratory infection has been shown to alter mucus composition, reduce ciliary motility, and create mucosal edema [20]. When MCC function fails to clear sinuses in key areas such as the osteomeatal complex or eustachian tube orifice, associated sinusitis results.
Allergic challenge has been shown to increase transudate levels in nasal mucus. As a result, the depth of the periciliary layer increases, and the cilia tips cannot reach the overlying gel phase to effectively propel the mucin rafts [21]. Swelling of the nasal mucosa in allergic rhinitis is also thought to obstruct sinus ostia leading to poor ventilation and mucostasis [22]. While the role of allergy in the causal development of chronic rhinosinusitis is unclear, failure to address the allergic component clearly lowers the success of surgical intervention in CRS [23].
Failure to address nasal allergies or smoke exposure leads to worse outcomes in the treatment of CRS.
Exposure to cigarette smoke has been shown to significantly alter ciliary beat frequency and secretions resulting in reduced mucociliary clearance [24]. Long-term exposure has been proposed to cause epithelial and submucosal gland hyperplasia, squamous metaplasia, and increase epithelial permeability [25, 26] and to foster the formation of bacterial biofilms [27]. In addition, cigarette smoking and second-hand smoke have been shown to be independently associated with CRS [28, 29]. As such, exposure to cigarette smoke should obviously be avoided in patients with recurrent or chronic sinus disease.
In addition to relatively rare genetic disorders such as PCD and CF, multiple investigations have also demonstrated significant impairment of sinonasal mucociliary clearance in idiopathic CRS. Inflammatory changes seen in chronic rhinosinusitis with and without nasal polyposis can cause secondary ciliary dyskinesia. Possible pathophysiologic explanations include reduced ciliary beating, changing depth or viscosity of respiratory mucus, or alterations in epithelial integrity. CRS patients exhibit blunted ciliary beat frequency responses to adrenergic and cholinergic stimulation in explanted nasal epithelial cultures [30]. Additional studies showed that when removed from the chronic inflammatory environment, cilia resume normal basal and stimulated beat frequency though recovery takes time. This suggests that a chronic inflammatory nasal environment reversibly suppresses normal ciliary function [31, 32].
4.2.4 Epithelial Integrity
Sinonasal cells contain several types of intercellular junctions that serve both communication and attachment functions. Maintenance of epithelial integrity is important for cellular communication and prevention of pathogen invasion or foreign protein exposure to the underlying tissues. Sloughing of epithelial cells leaves a denuded basement membrane and evokes an inflammatory response.
Gap junctions primarily allow cell–cell transfer of ions and signaling molecules; they do not contribute significantly to adhesion [33]. Respiratory cells are held together on their lateral surfaces with adherens junctions and desmosomes, both of which contain cadherin proteins. Hemidesmosomes, which contain integrin proteins, attach cells tightly to the basement membrane and prevent cellular sloughing. The most important junctions, tight junctions, are critical in epithelial barrier integrity against invasion. Tight junctions attach adjacent epithelial cells in a narrow band just beneath their apical surface [34]. The resultant epithelial surface is a watertight barrier to chemicals and particulates including proteins and pathogens at the surface.
Below the basement membrane lies the lamina propria, a network of blood vessels and loose fibrous stroma that contains immune cells responsible for pathogen detection and initial inflammatory response. Here reside the lymphocytes, plasma cells, macrophages, and dendritic cells which monitor the health of the epithelium. Pathogens that overcome the protective barriers of mucus and epithelium activate the innate and adaptive immune systems. Breakdown of the intracellular connections of the epithelium plays an important role in permitting stimulation of an immune response.
4.2.4.1 Diseases Affecting Epithelial Integrity
Invasion of pathogens into the underlying stroma requires disruption of the epithelium and penetration through the basement membrane. Viral pathogens, the initial cause of many upper respiratory tract infections, have developed mechanisms for epithelial disruption that target tight and adherens junctions, allowing for subsequent invasion into the underlying tissues [35, 36]. Respiratory viruses disrupt the upper airway epithelial barrier, resulting in inflammation and predisposing for additional exposure to foreign material and potential tissue invasion [37]. Nasal epithelial explant studies show that viruses loosen epithelial tight junction bonds and penetrate the basement membrane within 24 h [38]. Rhinoviruses, a major cause of the common cold, bind to intercellular adhesion molecule-1 (ICAM-1) on nasal epithelial cells, allowing RNA entry into the cell [39].
Fungi, bacteria, and allergens all carry significant intrinsic protease and virulence activity with the capacity of weakening tight junctions permitting access into the underlying tissues [40, 41]. Intrinsic alterations in tight junction barrier function may be responsible for the increase in epithelial permeability seen in allergic responses [42]. Penetration of antigenic proteins through the epithelium has been proposed as a major factor in asthma and atopy [43].
Respiratory viruses disrupt epithelial tight junctions and increase susceptibility to infection and inflammation.
Chronic inflammatory disorders may weaken the epithelial barrier function and predispose to infection. CRSwNP with a Type 2 cytokine environment exhibits disruption of normal intercellular junctions including decreased levels of the desmosome cadherin proteins DSG2 and DSG3 [44] as well as tight junction proteins claudin and occludin [45]. Disruption of intercellular tight junction connections is seen in nasal polyposis [46]. Decreased production of protease inhibitors such as LEKT1, encoded by the gene SPINK5, has been found in CRSwNP [41]. This molecule acts as a proteinase inhibitor and regulates the processing of epithelial tight junctions critical to maintaining a barrier to infection. In addition, altered intercellular ion transport is seen in CRS and may interfere with the coordination of ciliated cells leading to ineffective mucociliary clearance [47]. Interestingly, it has been recently suggested that epithelial turnover may be altered by a Type 2 cytokine milieu that created a leaky, immature barrier. Furthermore, this leaky barrier may be responsible for the high rate of recurrence seen with T2 CRS. (EPOS 2020) Substantiating this concept are recent single-cell transcriptomic studies, which have suggested that the epithelial changes are epigenetically programmed and will therefore be durable [48]. Periostin is associated with this process and high levels of this protein in the serum may be a sign of the T2 endotype [49].
4.3 Immunologic Barrier Function of the Sinonasal Epithelium
In addition to forming a physical barrier and providing clearance of pathogens from the nasal cavity, sinonasal epithelial cells (SNECs) contribute to the innate and adaptive immune responses. When the various mucosal barrier functions are circumvented by irritant or pathogenic particulates, the mucosal immune system must distinguish between normal commensal organisms and invading pathogens. In addition, the immune system must determine how to mount an appropriate level or intensity of response. The first of these responses, the innate immune system, consists of receptors, defense molecules, and cells that respond to microbial organisms in a relatively nonspecific manner. This innate response is an inborn, germ line-coded defense to generalized pathogen invasion and does not typically exhibit memory. Should the innate immune system be sufficiently challenged, it activates the adaptive arm of the immune system, with a delayed but highly specific T and B cell response capable of memory against specific pathogens. Emerging evidence also indicates that a diminished innate response coupled with a compensatory overactivation of adaptive immunity may play a role in the pathogenesis of the chronic mucosal inflammation seen in CRS.
Pathogen recognition by the innate immune system results in a cascade of cytokines and chemokines that determine the type and strength of the inflammatory response.
4.3.1 Receptor Molecules
Innate immune responses in the sinonasal epithelium are initiated by membrane-bound and cytoplasmic pattern-recognition receptors (PRRs) that recognize highly conserved pathogen-associated molecular patterns (PAMPs) found in various bacteria, mycobacteria, viruses, and parasites as well as necrotic debris from cellular damage [50]. PRRs are also expressed on various types of antigen-presenting cells including dendritic cells, macrophages, and B cells. In addition, SNECs express genes associated with antigen-presenting function [51]. Once the innate immune response is activated, SNECs may also serve as antigen-presenting cells, increasing local tissue inflammatory response. Overall, stimulation of PRRs facilitates both the innate and the adaptive response.
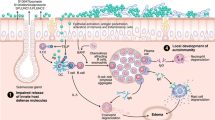
Recognition of PAMPs by PRRs results in the secretion of the endogenous antimicrobial factors that directly aid in pathogen clearance. PRR activation also stimulates SNECs and antigen-presenting cells (APCs) to release inflammatory cytokines and chemokines that attract other innate cellular defenses such as phagocytes. Other receptors detect cellular injury through damage-associated molecular patterns DAMPs [52, 53]. If the combination of cellular damage and PRR activation is sufficiently strong, the resultant innate immune response triggers cytokine patterns that initiate, and determine the nature of, the subsequent adaptive immune response [54] (see Fig. 4.1).

Innate immunity at the epithelial surface involves PAMP activation of epithelial PRRs leading to release of host defense molecules. Exogenous proteases may activate PAR receptors or degrade tight junctional proteins. Secreted IgA, mucins, antiproteases, and host defense molecules are released into the nasal mucus to form a second line of innate defense. Sufficient PRR stimulation activates dendritic cells causing migration to local lymph nodes to present antigen fragments to naïve Th0 lymphocytes. The type and strength of antigen stimulation drive the adaptive immunity through polarized Th1, Th2, Th17, or Treg responses. B cells are stimulated to undergo proliferation, class switch recombination, and differentiation leading to production of IgE, IgA, and other immunoglobulins as well as stimulation of the cells of the adaptive immune system. See text for details
PRRs can be separated into three main classes: endocytic, secreted, and signaling. Endocytic PRRs are found on the surface of phagocytes, which recognize pathogenic PAMPs, engulf the pathogens, and present the antigens to lymphocytes of the acquired immune system. One example is the mannose receptor found on macrophages [55].
Secreted PRRs are used as opsonins that trigger the complement cascade or signal phagocytosis. The most abundant of the secreted antimicrobial peptides in the nasal mucosa include lactoferrin, mannose-binding lectin, secretory leukocyte proteinase inhibitor, and lysozyme [56]. These are released into the mucus by nasal epithelial cells in response to activation of PRRs and confer protection by inhibiting epithelial invasion or directly lysing the microorganism [57].
Signaling PRRs trigger the production of antimicrobial peptides and cytokines by epithelial cells in response to PAMPs [58]. Signaling PRRs found in nasal epithelium include the toll-like receptor (TLR) family, the nucleotide binding and oligomerization domain (NOD)-like receptor family (NLRs), and the retinoic acid-inducible-like receptors (RLRs).
Important pathogen recognition receptors (PRRs) include the TLR, NOD, Bitter Taste receptors, and PAR families.
TLRs are transmembrane receptors expressed on various cell types including SNECs that recognize extracellular or intracellular PAMPs such as bacterial lipopolysaccharide (LPS) [59]. TRL2, TRL3, TRL4, TRL9, and possibly others are expressed in sinonasal epithelium and contribute to the host defense. TLR2 responds to gram-positive bacterial as well as fungal PAMPs, TLR 3 recognizes viral replication products, TLR4 recognizes bacterial endotoxin, and unmethylated CpG areas on pathogenic DNA activate TLR9 [33]. TLR activation triggers intercellular signaling through proteins MyD88 and TRIF, which affects gene expression through transcription factors NF-κβ, AP-1, and IRF3 [60].
The NOD-like receptor family includes NOD-1 and -2, which have been shown to recognize bacterial cell walls including staphylococci [61]. NOD levels were increased in CRSwNP and levels decreased after nasal steroid use [62]. RLRs are intracellular receptors important for recognizing RNA derived from RNA and DNA viruses, though their role in sinonasal epithelial response is still being investigated [63].
Bitter taste receptors (T2Rs) are recently identified group of pathogen recognition molecules also present on epithelial cells, function as non-classical PRRs. They are G protein-coupled receptors (GPCRs), which are widely expressed in host airway epithelial membranes. Airway T2R-mediated immune responses are activated by bacterial quinolones as well as acyl-homoserine lactones [64] secreted by gram-negative bacteria, including Pseudomonas aeruginosa [65]. Linkage studies have demonstrated associations between taste receptor genetics with CRS [66].
The protease-activated receptors (PARs) are a distinct set of receptors found on sinonasal epithelial cells that are activated by endogenous and exogenous proteases including those from bacteria, fungi, and allergens. Once activated, PARs evoke the NF-κβ signaling pathway, the results in chemokine and cytokine production, and phagocytic recruitment and potentially influence the subsequent acquired immune response based on the cytokine milieu [67]. PAR-2 activation by Staphylococcus aureus proteases results in increased levels of cytokine IL-8 [68]. Fungal proteases may drive the eosinophilic as well as neutrophilic response via PARs [69]. SNECs secrete antiproteases such as LEKT1 coded by the Spink 5 gene, which likely acts to protect PARs from both exogenous and endogenous protease stimulation. Reduced levels of LEKT1 have been associated with CRSwNP, suggesting that excessive PAR activation may play a role in polyp pathogenesis [70].
4.3.2 Host Defense Molecules
Sinonasal epithelial cells secrete a multitude of antimicrobial molecules into the surrounding mucus. Enzymes break down pathogen cell walls and include lysozyme, chitinases, and peroxidases. Foreign material is marked for phagocytosis by opsonins such as complement and pentraxin-3. Permeabilizing proteins include defensins and cathelicidins. Defensins are inducible and provide broad antimicrobial activity and inhibit invasion of bacteria and viruses [71]. Cathelicidins are a family of secreted peptides that are active after extracellular cleavage. In humans, cathelicidin LL-37 directly disrupts bacterial membranes [72]. They are chemotactic for effector cells of both the innate and adaptive immune system including neutrophils, monocytes, mast cells, and T cells and may modulate their activity [73]. Collectins include surfactant proteins (SP-A, SP-D) and mannose-binding lectin; these proteins, long studied in lower respiratory mucosa, are also found in sinonasal secretions. They are important in reducing nasal bacterial colonization, inflammation, and infection [74]. Decreased surfactant levels are found in patients with cystic fibrosis, poorly controlled COPD, allergic fungal sinusitis, and CRS [75,76,77]. Binding proteins include mucin, discussed previously, and lactoferrin, which attaches to foreign material and facilitates its removal by MCC. PLUNC, another secreted antimicrobial, has important anti-biofilm properties. Diminished secretion of many but not all of these host defense molecules has been proposed as a common mechanism broadly underlying the etiology and pathogenesis of CRS [41, 78] (see Fig. 4.2). The cause for this selective reduction is unclear but may be [1] a primary defect in the innate response or [2] a downstream secondary effect of the Type 2 cytokine milieu associated with some forms of CRS [79]. Mechanistic studies to evaluate this have not been completed, but IL-22 and the STAT 3 pathway appear to broadly govern innate nasal mucosal host defense and mechanical barrier integrity [80,81,82]. Diminished STAT 3 activity in the sinonasal epithelium has been identified in CRS supporting a primary innate defect [83].

Inferior turbinate and uncinate process tissue from healthy patients were compared for levels of the mucosal innate defense molecules PLUNC, lactoferrin, pentraxin, S100A7, and hBD2. While levels of pentraxin remained the same between the two sites, polarization was noted with the remaining molecules. In nasal uncinate tissue, low levels of PLUNC and lactoferrin but high levels of S100A7 and hBD2 were found. In inferior turbinate tissue, the converse was true, suggesting that host defense molecules show regional specialization in human nasal airways
A decrease in innate host defense molecules contributes to the pathogenesis of CRS
4.3.3 Epithelial Chemokines and Cytokines
In response to PRR and PAR stimulation, sinonasal epithelial cells produce a variety of cytokines, which are small proteins that regulate inflammation and mediate associated pain, swelling, and vascular dilatation [84]. A partial list include IL-1, TNF, IFN, GM-CSF, eotaxins, RANTES, IP-10, IL-6, IL-8, MDC, SCF, TARC, MCP-4, BAFF, osteopontin, IL-25, IL-33, and TSLP [85,86,87]. These cytokines have a diverse array of functions including triggering inflammatory responses, activating and recruiting innate effector cells, and facilitating and shaping the adaptive response. To the last point, IL-1, IL-6, and IL-8 have all been identified as particularly important in modulating the transition between the two arms of the immune response [86]. Newly identified epithelial-derived cytokines, including TSLP, IL-25, IL-33, and BAFF, help shape the local adaptive responses more directly. They foster B cell proliferation with immunoglobulin production and shaping the T helper profile via dendritic cell polarization [88, 89]. Defective regulation of these processes has been proposed as playing a role in the pathogenesis of CRSwNP. It has been noted that corticosteroids, a mainstay for treating inflammation and CRS, act in part via the downregulation of epithelial cytokine secretion but spare or even augment the epithelial secretion of host defense molecules [90, 91]. In this fashion, corticosteroids effectively upregulate the innate immune response and downregulate the adaptive response.
Epithelial-derived molecules directly contribute to the innate immune defense and shape-associated B cell and T cell responses.
4.3.4 Epithelial Co-stimulatory Molecules and Inflammatory Enzymes
SNECs express co-stimulatory molecules, particularly homologs of the B7 family of cell-surface ligands [92]. Expression results in downregulation of T cell-acquired response and is induced by TNF-α and IFN-γ [93]. Increased B7 family expression is induced by viral infection and CRS [94]. Reactive oxygen species (ROS) are important in many of the processes discussed: mucin production, epithelial healing, response to toxins, and innate immunity [95]. They can be beneficial, generating hypothiocyanite and peroxide which aid in the microbial killing. In addition, they may be induced by toxins, requiring neutralization by antioxidants in airway epithelial cells. ROS can also interact with reactive nitrogen species (RNS) to create tissue damage in disease [37]. Of particular interest is nitric oxide (NO), an intracellular messenger that mediates inflammation and antimicrobial effects and regulates apoptosis. In particular, NO is produced in high concentrations in the paranasal sinuses and may limit bacterial colonization [96].
4.3.5 Cells of the Innate Immune System
Important cells of the innate immune response that respond to cytokines secreted by the sinonasal epithelium include innate lymphocytes (ILC), dendritic cells, macrophages, neutrophils, eosinophils, natural killer (NK) cells, basophils, and mast cells. The ILCs are divided into three subsets (ILC 1, 2, and 3) and they help tailor the subsequent innate cellular and adaptive response to the address the particular inciting pathogen [1]. Macrophages and neutrophils are primary phagocytes which recognize and bind to pathogens that have been opsonized, or marked as foreign, by antibodies, complement, or collectins [57]. They then engulf the opsonized pathogens and neutralize them by a variety of methods including nitric oxide and radical oxygen species. In addition, macrophages assist in tissue homeostasis, removal of particulates, and tissue repair and influence the adaptive immune response. Two pathways of macrophage differentiation and activation exist: the M1, or classical, pathway and the M2, or alternative, pathway. The M1 pathway is driven by Type 1 cytokines and fosters macrophages directed against intracellular pathogens. The M2 pathway is driven by Type 2 cytokines and fosters macrophages specialized against helminthes, with additional roles in tissue repair and antibody formation [97]. High levels of M1 macrophages are seen in sinus mucosa and polyps of CF patients, while high levels of M2 macrophages are seen in CRSwNP tissues [98, 99]. Neutrophils are among the first cells to respond to infection and are important in early phagocytosis of extracellular microbes. Mucosal recruitment is triggered by PRR stimulation creating chemokines, particularly IL-8, which is also secreted in response to PAR-2 stimulation [100]. Neutrophils kill using free radicals and many of the same antimicrobial peptides that are secreted in the nasal mucus. Their overactivation may contribute to the pathogenesis of CRS, inflammation related to tobacco smoking, and CF-associated polyposis [27, 101].
High numbers of eosinophils are seen in mucosal surfaces in allergy and asthma. Eosinophils are granulocytes that respond to mucosal inflammation by degranulating and releasing stored cytokines. They are prominent in fungal and parasitic responses and release major basic protein, eosinophil-derived neurotoxin, and neutrophil elastase [102]. Eosinophilia is also an important component of CRS where it contributes to mucosal damage and chronic inflammation [103]. Multiple studies have shown that tissue eosinophilia is correlated with severity of CRS and comorbid asthma [104, 105]. The highest levels of tissue eosinophils are seen in CRSwNP, particularly in Western populations [106]. Eosinophil recruitment and activation is in large measure via epithelial cytokines and chemokines, including eotaxins, RANTES, and MCP [36, 107,108,109]. The regulation of this secretion is in part not only through PRR stimulation but also via Type 2 cytokines IL-4, IL-5, and IL-13 [110, 111]. The primary cellular sources of these Type 2 cytokines include Th 2 and ILC-2 cells. (EPOS 2020) Other factors that may foster eosinophil activation and recruitment include staphylococcal superantigens, IL-25, IL-33, TSLP, SCF, and eicosanoids [112,113,114,115].
Upregulation of eosinophils is seen in allergy, asthma, and CRSwNP.
Mast cells are resident cells in the sinonasal mucosa that not only function in innate immunity and tissue repair but also play key roles in the pathogenesis of allergic rhinitis and possibly nasal polyposis. Stem Cell Factor (SCF), secreted by SNECs, is likely important in mast cell recruitment [85]. Mast cell activation results in the secretion of preformed granules including histamine, prostaglandins, serotonin, and serine proteases. In addition, de novo synthesis and secretion of various cytokines, chemokines, and eicosanoids also takes place. They can be induced to phagocytose pathogens as well, though this is not their primary function. Physiological activation occurs typically through stimulation of PRRs. In nasal disease states such as allergic rhinitis, mast cells are activated strongly via surface IgE bound to antigen [116]. In CRSwNP, both IgE-dependent and IgE-independent pathways for mast cell activation likely contribute to pathogenesis [117, 118]. Specifically, mast cells may be able to induce and maintain eosinophilic inflammation leading to polyposis as well as influence the subsequent adaptive immune response.
Dendritic cells (DCs) are key cells in both the innate and adaptive responses via antigen capture, antigen presentation to immature T cells, and the secretion of soluble mediators. DCs phagocytose and thereby sample commensal organisms and pathogens at the sinonasal epithelial surface. Cytokine cross talk between SNECs and DCs helps determine DC polarization [88]. Polarized DCs then migrate to local lymph nodes and present a fragment of the engulfed pathogen, via major histocompatibility complex (MHC) type II on the cell surface, to immature T cells. DC cytokines strongly influence the subsequent T cell profile, secondarily polarizing the helper response [119]. Overall, DCs act as a bridge from the innate to the adaptive immune response. SNECs acting through cytokines that influence DCs to play a significant upstream role in shaping the subsequent adaptive response, which will be discussed below. Defects in this cross talk pathway may foster the development of CRSwNP.
4.3.6 Adaptive Immunity
The adaptive immune response is mediated by T and B cells and includes immunoglobulin and T effector processes that augment the faster, but less specific innate response. Sinonasal epithelial responses connect with ILCs and together play a key upstream role in the adaptive response including the recruitment of cells of the T and B lineage. In addition, as mentioned above, the type, duration, and intensity of PRR activation by PAMP stimulus in SNECs, ILCs, DCs, and other cell types are believed to shape the resultant T lymphocyte profile and to ensue antibody and cell-mediated response at the mucosal surface [120].
B lymphocytes secrete immunoglobulins, or antibodies, to specific antigens, playing an important role in the memory of the adaptive immune response. Immunoglobulins help bind and trap commensal organisms and pathogens, aiding mechanical clearance and facilitating active killing via multiple mechanisms. In the nasal mucosa, B cells respond to antigen presentation by proliferating and undergoing differentiation into mature plasma cells that produce immunoglobulin. In normal mucosal defense, the primary antibody class is secreted IgA from extrafollicular B cells. This response works to limit bacterial colonization with a minimum of inflammation, is T cell independent, and helps maintain mucosal homeostasis. SNECs and other cell types secrete cytokines and chemokines that foster this baseline B cell activity with the capacity for upregulation in response to an immune challenge.
Staphylococcal superantigens and fungal elements act as disease modifiers in CRS.
During frank mucosal infection, secretory IgA is joined by IgG, resulting in the development of a robust inflammatory response. This response exhibits high affinity for the invading pathogens, is T cell dependent, and utilizes immunoglobulins generated by both tissue plasma cells and follicular B cells. Other immunoglobulins play a role in mucosal inflammation including IgM, IgE, and IgD [121]. IgM is an early-response antibody that precedes the development of long-term IgG. IgE is important in allergic response, mast cell activation and survival, and homeostasis as well as defense against pathogens, especially parasitic infections. IgD, though little understood, may influence antigen binding and basophil activation against respiratory bacteria [122].
In chronic inflammatory conditions such as CRS, immunoglobulin profiles are skewed from the normal, apparently in response to bacterial and fungal antigens. CRSwNP appears to show a particularly dysregulated B cell response. Higher levels of IgA, IgE, and IgG are seen in nasal polyp tissues compared to controls and to CRSsNP, and this may have pathophysiological significance. IgE facilitates mast cell degranulation and IgA is a potent activator for eosinophil degranulation [123]. The combined presence of these antibodies with mast cells and eosinophils within nasal polyps may facilitate degranulation and tissue damage. It should be noted that these immunoglobulin levels do not reflect the systemic profile, indicating a localized mucosal response [124]. Not surprisingly, higher levels of immunoglobulin-producing B cells and plasma cells are also found in nasal polyps, and the process of polyp growth may be orchestrated by abnormal local B cell proliferation and recruitment [125]. Evidence suggests that this process may be driven by the epithelial cytokine BAFF, a TNF family member that influences B cell proliferation and class switching [89]. BAFF is found at higher levels in nasal polyps and correlates with the number of B cells within the tissue. In mouse models, excessive BAFF has been associated with the development of autoimmunity. This process has also been documented in recalcitrant CRSwNP with the presence of high levels of local autoantibodies in the polyp tissue [126].
Abnormal B cell proliferation creates inflammation and tissue damage that may lead to polyposis.
Staphylococcal superantigenic toxins (SAGs) have been proposed as disease modifiers of nasal polyposis through the generation of a polyclonal IgE response including IgE directed against the SAGs themselves. The presence of IgE to these toxins within polyp tissue has been correlated with overall increases in polyclonal IgE, eosinophils, asthma, and severity of CRSwNP [126, 127]. It is unclear whether this superantigen-driven process works through BAFF or another, superimposed, pathway.
4.3.7 T Cells and Cytokine Response
Homeostasis across the nasal mucosa is typically maintained via the mechanical barrier, innate immune responses, and tonic IgA secretion. When the mucosal barrier is breached, a protective response is initiated with SNECs, DCs, and other innate immune cells helping to guide the adaptive response and match it to the inciting stimulus. Minor damage is likely handled by activation augmentation of innate responses from SNECs and migrating innate effector cells. A more substantial breach will activate the adaptive response; IL-6 has been proposed as a key cytokine mediating the transition, suppressing innate responses and triggering production of chemokines that promote the adaptive response [89]. At homeostasis, DCs still regularly phagocytize foreign material, but when activated and exposed to sufficient PAMP activation, such as would occur in a mucosal breach, they cease phagocytosis and acquire additional chemokine receptors. Chemokines, stimulated into production during the innate immune response, cause the DCs to migrate to nearby lymph nodes and to secrete the cytokine IL-1 [128]. Antigen from the phagocytosed pathogens is presented to naïve CD4+ T helper (Th) cells in the lymph tissue. These lymphocytes will differentiate into a specific T cell lineage, determining the type of adaptive immune response. This process is further activated by IL-1. The types, duration, and intensity of the PRR activation by PAMP stimulus are believed to influence the resultant cytokine production and shape the resultant T lymphocyte profile [129]. As mentioned above, cytokines from SNECs and other innate cell types play a critical upstream role in this process matching the response to the pathogen.
Signaling cross talk between innate immune cells drives T cell differentiation.
Mature T cells migrate back to the sinonasal mucosa to mediate the adaptive response upon subsequent antigen challenge. T helper lymphocyte responses are divided based on cytokine profiles generated in response to the presented antigen stimulus. Classically, Th1 or Th2 responses were thought to be the primary adaptive sinonasal inflammatory pathways. The Th1 pathway shows high levels of IL-12 and IFN-γ and has a macrophage-rich cellular infiltrate. Th1 responses facilitate defense against intracellular pathogens, particularly viruses and intracellular bacteria including mycobacteria. They appear to be blunted in chronic obstructive pulmonary disease (COPD), psoriasis, Crohn’s disease, and CRSsNP [130]. The Th2 pathway results in high levels of cytokines IL-4, IL-5, and IL-13 and has a more eosinophilic cellular response. Th2 responses are important in parasitic infections and are also seen in frequently allergic and asthmatic responses [79]. They are reduced in asthma, atopic dermatitis, ulcerative colitis, and CRSwNP [131]. More recent data indicate that additional Th profiles are important in mucosal immunity. Th17 responses aid in defense against extracellular bacteria and fungi, particularly Staphylococcus aureus [132]. This response is fostered primarily by IL-17A as well as cytokines IL-6, TGF-β1, and IL-23 and has a neutrophilic cellular response [133]. Tregs are regulatory lymphocytes that foster immune tolerance with the goal of limiting excessive responses from other Th lineages; Treg differentiation is facilitated by TGF-β [131].
4.3.8 T Cell Response Modulation
Differentiation of CD4+ T cells into a specific lineage is determined in part by innate immune response, co-stimulatory signals, and the cytokine profile [134]. Signaling cross talk between local DCs, SNECs, and resident innate immune cells, including eosinophils, mast cells, NK cells, and macrophages, generates the cytokines that drive the T cell differentiation [135, 136]. In addition, circulating innate lymphoid cells (ILCs) migrate to the local site of immune stimulus and also play a role. These cells are presumably responding to chemokine homing signals emanating from resident cells and are termed “innate” because they recognize foreign substances via PRRs rather than immunoglobulin or T cell receptors. Capable of responding rapidly, ILCs bridge innate and adaptive immunity and may play the pivotal role in orchestration of the adaptive response as Th1, Th2, and Th17 ILC subsets have been described [137]. In terms of pathology, exceptionally high levels of ILC2s have been observed in polyp homogenates from Western CRSwNP patients [138, 139] (see Fig. 4.3).

Environmental agents stimulate the immune system inciting an innate response. If strong enough, an adaptive response is recruited as well. Typical protective responses include activation of the T1 and T3 pathways which includes the Th1 and Th17 subsets. If a T2 response (with Th2 or Treg activation) is generated, the innate response may be suppressed
Innate lymphoid cells play a key role in orchestrating Th1, Th2, and Th17 responses.
4.3.9 NKT Cells, NK Cells, Cytotoxic T Cells, and Memory T Cells
In addition to the Th subsets discussed above, other T cell subsets play a role in mucosal immunity. Naïve CD8+ T cells differentiate and proliferate following exposure to antigen presented by DCs. Cytotoxic T cells are generated whose primary function is to eliminate intracellular microbes mainly by killing infected cells. These infected cells display microbial antigens on their surface, which the T cells recognize via their T cell receptors (TCR). Although not technically T cells, NK cells have a function similar to cytotoxic T cells but lack TCRs, recognizing foreign proteins by PRRs on their surface. NKT cells have characteristics of both T cells and NK cells with TCRs but with limited variability. Memory T cells are generated along with the effector T subsets and are numerically the predominant subset in nasal polyps [140]. These cells are present in the mucosa and respond to subsequent antigen challenge.
4.4 Conclusion
The sinonasal mucosal defenses are a highly sophisticated interplay involving the local structural cells, resident innate response cells, and circulating innate and adaptive immune cells. In approximately 10% of the Western population, this system fails in that foreign agents, while still cleared, trigger collateral inflammation of the mucosa of varying types and intensities. The associated clinical syndrome is broadly termed “CRS.” Recent research in the field of CRS has been geared toward a better understanding of the specific pathway defects in the host. These specific genetic and epigenetic defects in the local immunologic pathways should eventually be associated with the various CRS phenotypes. Ultimately, greater understanding of sinonasal immune defenses will lead to more effective therapies for CRS in the future.
References
Fokkens W, Desrosiers M, Harvey R, Hopkins C, Mullol J, Philpott C, et al. EPOS2020: development strategy and goals for the latest European position paper on rhinosinusitis. Rhinology. 2019;57(3):162–8.
Yee KK, Pribitkin EA, Cowart BJ, Vainius AA, Klock CT, Rosen D, et al. Neuropathology of the olfactory mucosa in chronic rhinosinusitis. Am J Rhinol Allergy. 2010;24(2):110–20.
Wagenmann M, Naclerio R. Anatomic and physiologic considerations in sinusitis. J Allergy Clin Immunol. 1992;90(3):419–23.
Busse W, Holgate ST. Asthma and rhinitis. 2nd Ed. Oxford: Blackwell Science Ltd; 2000.
Shami SG, Martinez LA, Evans MJ. The role of migrating inflammatory cells in proliferation of lung interstitium and epithelium. Chest. 1986;89(3):170S–3S.
Sleigh MA, Blake JR, Liron N. The propulsion of mucus by cilia. Am Rev Respir Dis. 1988;137(3):726–41.
Antunes MB, Gudis DA, Cohen NA. Epithelium, cilia, and mucus: their importance in chronic rhinosinusitis. Immunol Allergy Clin N Am. 2009;29(4):631–43.
Houdret N, Ramphal R, Scharfman A, Perini J-M, Filliat M, Lamblin G, et al. Evidence for the in vivo degradation of human respiratory mucins during Pseudomonas aeruginosa infection. Biochim Biophys Acta Gen Subj. 1989;992(1):96–105.
Satir P, Sleigh MA. The physiology of cilia and mucociliary interactions. Annu Rev Physiol. 1990;52(1):137–55.
Gheber L, Priel Z. Synchronization between beating cilia. Biophys J. 1989;55(1):183–91.
Braiman A, Priel Z. Efficient mucociliary transport relies on efficient regulation of ciliary beating. Respir Physiol Neurobiol. 2008;163(1–3):202–7.
Messerklinger W. On the drainage of the human paranasal sinuses under normal and pathological conditions. 1. Monatsschr Ohrenheilkd Laryngorhinol. 1966;100(1–2):56–68.
Stucker FJ. Rhinology and facial plastic surgery. Berlin: Springer; 2009. p. xxix, 946 pp.
Gutman M, Houser S. Iatrogenic maxillary sinus recirculation and beyond. Ear Nose Throat J. 2003;82(1):61–3.
Cohen NA. Sinonasal mucociliary clearance in health and disease. Ann Otol Rhinol Laryngol Suppl. 2006;115(9_suppl):20–6.
Noone PG, Leigh MW, Sannuti A, Minnix SL, Carson JL, Hazucha M, et al. Primary ciliary dyskinesia. Am J Respir Crit Care Med. 2004;169(4):459–67.
Wine JJ. The genesis of cystic fibrosis lung disease. J Clin Investig. 1999;103(3):309–12.
Hadfield PJ, Rowe-Jones JM, Mackay IS. The prevalence of nasal polyps in adults with cystic fibrosis. Clin Otolaryngol. 2000;25(1):19–22.
Cimmino M, Cavaliere M, Nardone M, Plantulli A, Orefice A, Esposito V, et al. Clinical characteristics and genotype analysis of patients with cystic fibrosis and nasal polyposis. Clin Otolaryngol Allied Sci. 2003;28(2):125–32.
Houtmeyers E, Gosselink R, Gayan-Ramirez G, Decramer M. Regulation of mucociliary clearance in health and disease. Eur Respir J. 1999;13(5):1177.
Baumgarten CR, Togias AG, Naclerio RM, Lichtenstein LM, Norman PS, Proud D. Influx of kininogens into nasal secretions after antigen challenge of allergic individuals. J Clin Investig. 1985;76(1):191–7.
Stammberger H, Posawetz W. Functional endoscopic sinus surgery. Eur Arch Otorhinolaryngol. 1990;247(2):63–76.
Lane AP, Pine HS, Pillsbury HC. Allergy testing and immunotherapy in an academic otolaryngology practice: a 20-year review. Otolaryngol Head Neck Surg. 2001;124(1):9–15.
Cohen NA, Zhang S, Sharp DB, Tamashiro E, Chen B, Sorscher EJ, et al. Cigarette smoke condensate inhibits transepithelial chloride transport and ciliary beat frequency. Laryngoscope. 2009;119(11):2269–74.
Dye JA, Adler KB. Effects of cigarette smoke on epithelial cells of the respiratory tract. Thorax. 1994;49(8):825–34.
Karaman M, Tek A. Deleterious effect of smoking and nasal septal deviation on mucociliary clearance and improvement after septoplasty. Am J Rhinol Allergy. 2009;23(1):2–7.
Goldstein-Daruech N, Cope EK, Zhao K-Q, Vukovic K, Kofonow JM, Doghramji L, et al. Tobacco smoke mediated induction of sinonasal microbial biofilms. PLoS One. 2011;6(1):e15700.
García-Rodríguez JF, Corominas M, Fernández-Viladrich P, Monfort JL, Dicenta M. Rhinosinusitis and atopy in patients infected with HIV. Laryngoscope. 1999;109(6):939–44.
Tomassen P, Newson RB, Hoffmans R, Lötvall J, Cardell LO, Gunnbjörnsdóttir M, et al. Reliability of EP3OS symptom criteria and nasal endoscopy in the assessment of chronic rhinosinusitis - a GA2LEN study. Allergy. 2010;66(4):556–61.
Chen B, Shaari J, Claire SE, Palmer JN, Chiu AG, Kennedy DW, et al. Altered sinonasal ciliary dynamics in chronic rhinosinusitis. Am J Rhinol. 2006;20(3):325–9.
Al-Rawi MM, Edelstein DR, Erlandson RA. Changes in nasal epithelium in patients with severe chronic sinusitis: a clinicopathologic and electron microscopic study. Laryngoscope. 1998;108(12):1816–23.
Chen B, Antunes MB, Claire SE, Palmer JN, Chiu AG, Kennedy DW, et al. Reversal of chronic rhinosinusitis-associated sinonasal ciliary dysfunction. Am J Rhinol. 2007;21(3):346–53.
Yeh T-h, Su M-c, Hsu C-j, Chen Y-h, Lee S-y. Epithelial cells of nasal mucosa express functional gap junctions of Connexin 43. Acta Otolaryngol. 2003;123(2):314–20.
Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422(6929):322–6.
Takano K-i, Kojima T, Go M, Murata M, Ichimiya S, Himi T, et al. HLA-DR- and CD11c-positive dendritic cells penetrate beyond well-developed epithelial tight junctions in human nasal mucosa of allergic rhinitis. J Histochem Cytochem. 2005;53(5):611–9.
Yeo N-K, Jang YJ. Rhinovirus infection-induced alteration of tight junction and adherens junction components in human nasal epithelial cells. Laryngoscope. 2009;120(2):346–52.
Pedersen M, Sakakura Y, Winther B, Brofeldt S, Mygind N. Nasal mucociliary transport, number of ciliated cells, and beating pattern in naturally acquired common colds. Eur J Respir Dis Suppl. 1983;128(Pt 1):355–65.
Glorieux S, Bachert C, Favoreel HW, Vandekerckhove AP, Steukers L, Rekecki A, et al. Herpes simplex virus type 1 penetrates the basement membrane in human nasal respiratory mucosa. PLoS One. 2011;6(7):e22160.
Rossmann MG. Viral cell recognition and entry. Protein Sci. 1994;3(10):1712–25.
Zulianello L, Canard C, Köhler T, De C, Lacroix J-S, Meda P. Rhamnolipids are virulence factors that promote early infiltration of primary human airway epithelia by Pseudomonas aeruginosa. Infect Immun. 2006;74(6):3134–47.
Tieu DD, Kern RC, Schleimer RP. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124(1):37–42.
Jeffery PK, Haahtela T. Allergic rhinitis and asthma: inflammation in a one-airway condition. BMC Pulm Med. 2006;6(S1):S5.
Xiao C, Puddicombe SM, Field S, Haywood J, Broughton-Head V, Puxeddu I, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128(3):549–56.e12.
Zuckerman JD, Lee WY, DelGaudio JM, Moore CE, Nava P, Nusrat A, et al. Pathophysiology of nasal polyposis: the role of Desmosomal junctions. Am J Rhinol. 2008;22(6):589–97.
Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. Tight junction proteins Claudin-1 and Occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol. 2008;83(4):2011–4.
Rogers GA, Beste KD, Parkos CA, Nusrat A, DelGaudio JM, Wise SK. Epithelial tight junction alterations in nasal polyposis. Int Forum Allergy Rhinol. 2011;1(1):50–4.
Dejima K, Randell SH, Stutts MJ, Senior BA, Boucher RC. Potential role of abnormal ion transport in the pathogenesis of chronic sinusitis. Arch Otolaryngol Head Neck Surg. 2006;132(12):1352.
Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, et al. Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature. 2018;560(7720):649–54.
Lehmann AE, Scangas GA, Bergmark RW, El Rassi E, Stankovic KM, Metson R. Periostin and inflammatory disease: implications for chronic rhinosinusitis. Otolaryngol Head Neck Surg. 2019;160(6):965–73.
Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216.
Lane AP, Saatian B, Yu X-Y, Spannhake EW. mRNA for genes associated with antigen presentation are expressed by human middle meatal epithelial cells in culture. Laryngoscope. 2004;114(10):1827–32.
Claeys S, de Belder T, Holtappels G, Gevaert P, Verhasselt B, van Cauwenberge P, et al. Human beta-defensins and toll-like receptors in the upper airway. Allergy. 2003;58(8):748–53.
Bianchi ME, Manfredi AA. IMMUNOLOGY: dangers in and out. Science. 2009;323(5922):1683–4.
Meylan E, Tschopp J, Karin M. Intracellular pattern recognition receptors in the host response. Nature. 2006;442(7098):39–44.
Fraser IP, Koziel H, Ezekowitz RAB. The serum mannose-binding protein and the macrophage mannose receptor are pattern recognition molecules that link innate and adaptive immunity. Semin Immunol. 1998;10(5):363–72.
Cole AM, Liao H-I, Stuchlik O, Tilan J, Pohl J, Ganz T. Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol. 2002;169(12):6985–91.
Ooi EH, Psaltis AJ, Witterick IJ, Wormald P-J. Innate immunity. Otolaryngol Clin N Am. 2010;43(3):473–87.
Medzhitov R, Janeway C. Innate immunity. N Engl J Med. 2000;343(5):338–44.
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95.
Vroling AB, Fokkens WJ, van Drunen CM. How epithelial cells detect danger: aiding the immune response. Allergy. 2008;63(9):1110–23.
Bnd F, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005;18(3):521–40.
Månsson A, Bogefors J, Cervin A, Uddman R, Cardell LO. NOD-like receptors in the human upper airways: a potential role in nasal polyposis. Allergy. 2011;66(5):621–8.
Schröder M, Bowie AG. An arms race: innate antiviral responses and counteracting viral strategies. Biochem Soc Trans. 2007;35(6):1512–4.
Freund JR, Mansfield CJ, Doghramji LJ, Adappa ND, Palmer JN, Kennedy DW, et al. Activation of airway epithelial bitter taste receptors by Pseudomonas aeruginosa quinolones modulates calcium, cyclic-AMP, and nitric oxide signaling. J Biol Chem. 2018;293(25):9824–40.
Lee RJ, Cohen NA. Bitter and sweet taste receptors in the respiratory epithelium in health and disease. J Mol Med (Berl). 2014;92(12):1235–44.
Cohen NA. The genetics of the bitter taste receptor T2R38 in upper airway innate immunity and implications for chronic rhinosinusitis. Laryngoscope. 2017;127(1):44–51.
Hershenson MB. Proteases and protease-activated receptors signalling: at the crossroads of acquired and innate immunity. Clin Exp Allergy. 2007;37(7):963–6.
Rudack C, Steinhoff M, Mooren F, Buddenkotte J, Becker K, von Eiff C, et al. PAR-2 activation regulates IL-8 and GRO-? synthesis by NF-?B, but not RANTES, IL-6, eotaxin or TARC expression in nasal epithelium. Clin Exp Allergy. 2007;37(7):1009–22.
Shin S-H, Lee Y-H, Jeon C-H. Protease-dependent activation of nasal polyp epithelial cells by airborne fungi leads to migration of eosinophils and neutrophils. Acta Otolaryngol. 2006;126(12):1286–94.
Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, et al. Kallikrein 5 induces atopic dermatitis–like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206(5):1135–47.
van Wetering S, Tjabringa GS, Hiemstra PS. Interactions between neutrophil-derived antimicrobial peptides and airway epithelial cells. J Leukoc Biol. 2004;77(4):444–50.
Turner J, Cho Y, Dinh N-N, Waring AJ, Lehrer RI. Activities of LL-37, a Cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother. 1998;42(9):2206–14.
Yang D, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al. Ll-37, the neutrophil granule–and epithelial cell–derived cathelicidin, utilizes formyl peptide receptor–like 1 (Fprl1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192(7):1069–74.
Crouch E, Hartshorn K, Ofek I. Collectins and pulmonary innate immunity. Immunol Rev. 2000;173(1):52–65.
Postle AD, Mander A, Reid KBM, Wang J-Y, Wright SM, Moustaki M, et al. Deficient hydrophilic lung surfactant proteins A and D with normal surfactant phospholipid molecular species in cystic fibrosis. Am J Respir Cell Mol Biol. 1999;20(1):90–8.
Ooi EH, Wormald P-J, Carney AS, James CL, Tan LW. Surfactant protein D expression in chronic rhinosinusitis patients and immune responses in vitro to Aspergillus and Alternaria in a nasal explant model. Laryngoscope. 2007;117(1):51–7.
Sims MW, Tal-Singer RM, Kierstein S, Musani AI, Beers MF, Panettieri RA, et al. Chronic obstructive pulmonary disease and inhaled steroids alter surfactant protein D (SP-D) levels: a cross-sectional study. Respir Res. 2008;9(1):13.
Kern RC, Conley DB, Walsh W, Chandra R, Kato A, Tripathi-Peters A, et al. Perspectives on the etiology of chronic rhinosinusitis: an immune barrier hypothesis. Am J Rhinol. 2008;22(6):549–59.
Ramanathan M, Lee W-K, Spannhake EW, Lane AP. Th2 cytokines associated with chronic rhinosinusitis with polyps Down-regulate the antimicrobial immune function of human sinonasal epithelial cells. Am J Rhinol. 2008;22(2):115–21.
Wolk K, Witte E, Wallace E, Döcke W-D, Kunz S, Asadullah K, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36(5):1309–23.
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nat Med. 2008;14(3):275–81.
Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med. 2009;206(7):1465–72.
Peters AT, Kato A, Zhang N, Conley DB, Suh L, Tancowny B, et al. Evidence for altered activity of the IL-6 pathway in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2010;125(2):397–403.e10.
Bachert C, Wagenmann M, Rudack C, Höpken K, Hiltebrandt M, Wang D, et al. The role of cytokines in infectious sinusitis and nasal polyposis. Allergy. 1998;53(1):2–13.
Kowalski ML, Lewandowska-Polak A, Wozniak J, Ptasinska A, Jankowski A, Wagrowska-Danilewicz M, et al. Association of stem cell factor expression in nasal polyp epithelial cells with aspirin sensitivity and asthma. Allergy. 2005;60(5):631–7.
Lu X, Zhang XH, Wang H, Long XB, You XJ, Gao QX, et al. Expression of osteopontin in chronic rhinosinusitis with and without nasal polyps. Allergy. 2009;64(1):104–11.
Nishi Y. Glucocorticoids suppress NF-kB activation induced by LPS and PGN in paranasal sinus epithelial cells. Rhinology. 2009;47(4):413–8.
Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8(3):193–204.
Kato A, Peters A, Suh L, Carter R, Harris KE, Chandra R, et al. Evidence of a role for B cell–activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2008;121(6):1385–92.e2.
Schleimer RP. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc. 2004;1(3):222–30.
Bobic S, van Drunen CM, Callebaut I, Hox V, Jorissen M, Fokkens WJ, et al. Dexamethasone-induced apoptosis of freshly isolated human nasal epithelial cells concomitant with abrogation of IL-8 production. Rhinology. 2010;48(4):401–7.
Kurosawa S, Myers AC, Chen L, Wang S, Ni J, Plitt JR, et al. Expression of the costimulatory molecule B7-H2 (inducible costimulator ligand) by human airway epithelial cells. Am J Respir Cell Mol Biol. 2003;28(5):563–73.
Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran Q-A, Lane AP, et al. Constitutive and inducible expression of B7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol. 2005;33(3):280–9.
Heinecke L, Proud D, Sanders S, Schleimer RP, Kim J. Induction of B7-H1 and B7-DC expression on airway epithelial cells by the toll-like receptor 3 agonist double-stranded RNA and human rhinovirus infection: in vivo and in vitro studies. J Allergy Clin Immunol. 2008;121(5):1155–60.
Avila PC, Schleimer RP. Airway epithelium. Allergy and allergic diseases. Hoboken: Wiley-Blackwell; 2009. p. 366–97.
Lundberg JON, Farkas-Szallasi T, Weitzberg E, Rinder J, Lidholm J, Änggåard A, et al. High nitric oxide production in human paranasal sinuses. Nat Med. 1995;1(4):370–3.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27(1):451–83.
Claeys S, Van Hoecke H, Holtappels G, Gevaert P, De Belder T, Verhasselt B, et al. Nasal polyps in patients with and without cystic fibrosis: a differentiation by innate markers and inflammatory mediators. Clin Exp Allergy. 2005;35(4):467–72.
Krysko O, Holtappels G, Zhang N, Kubica M, Deswarte K, Derycke L, et al. Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy. 2010;66(3):396–403.
Marseglia GL, Pagella F, Klersy C, Barberi S, Licari A, Ciprandi G. The 10-day mark is a good way to diagnose not only acute rhinosinusitis but also adenoiditis, as confirmed by endoscopy. Int J Pediatr Otorhinolaryngol. 2007;71(4):581–3.
Polzehl D, Moeller P, Riechelmann H, Perner S. Distinct features of chronic rhinosinusitis with and without nasal polyps. Allergy. 2006;61(11):1275–9.
Lee JJ, Jacobsen EA, McGarry MP, Schleimer RP, Lee NA. Eosinophils in health and disease: the LIAR hypothesis. Clin Exp Allergy. 2010;40(4):563–75.
Harlin S, Ansel D, Lane S, Myers J, Kephart G, Gleich G. A clinical and pathologic study of chronic sinusitis: the role of the eosinophil. J Allergy Clin Immunol. 1988;81(5):867–75.
Bhattacharyya N, Vyas DK, Fechner FP, Gliklich RE, Metson R. Tissue eosinophilia in chronic sinusitis. Arch Otolaryngol Head Neck Surg. 2001;127(9):1102.
Szucs E, Ravandi S, Goossens A, Beel M, Clement PAR. Eosinophilia in the ethmoid mucosa and its relationship to the severity of inflammation in chronic rhinosinusitis. Am J Rhinol. 2002;16(3):131–4.
Jankowski R, Bene MC, Moneret-Vautrin AD, Haas F, Faure G, Simon C, et al. Immunohistological characteristics of nasal polyps. A comparison with healthy mucosa and chronic sinusitis. Rhinol Suppl. 1989;8:51–8.
Jahnsen FL, Haye R, Gran E, Brandtzaeg P, Johansen FE. Glucocorticosteroids inhibit mRNA expression for eotaxin, eotaxin-2, and monocyte-chemotactic protein-4 in human airway inflammation with eosinophilia. J Immunol. 1999;163(3):1545–51.
Meyer JE, Bartels J, Görögh T, Sticherling M, Rudack C, Ross DA, et al. The role of RANTES in nasal polyposis. Am J Rhinol. 2005;19(1):15–20.
Mullol J, Roca-Ferrer J, Alobid I, Pujols L, Valero A, Xaubet A, et al. Effect of desloratadine on epithelial cell granulocyte-macrophage colony-stimulating factor secretion and eosinophil survival. Clin Exp Allergy. 2006;36(1):52–8.
Matsukura S, Stellato C, Plitt JR, Bickel C, Miura K, Georas SN, et al. Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J Immunol. 1999;163(12):6876–83.
Kuperman D, Schleimer R. Interleukin-4, Interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8(5):384–92.
Pérez-Novo CA, Claeys C, Van Zele T, Holtapples G, Van Cauwenberge P, Bachert C. Eicosanoid metabolism and eosinophilic inflammation in nasal polyp patients with immune response to Staphylococcus aureus enterotoxins. Am J Rhinol. 2006;20(4):456–60.
Van Zele T, Coppieters F, Gevaert P, Holtappels G, Van Cauwenberge P, Bachert C. Local complement activation in nasal polyposis. Laryngoscope. 2009;119(9):1753–8.
Buysschaert ID, Grulois V, Eloy P, Jorissen M, Rombaux P, Bertrand B, et al. Genetic evidence for a role of IL33 in nasal polyposis. Allergy. 2010;65(5):616–22.
Foreman A, Holtappels G, Psaltis AJ, Jervis-Bardy J, Field J, Wormald PJ, et al. Adaptive immune responses in Staphylococcus aureus biofilm-associated chronic rhinosinusitis. Allergy. 2011;66(11):1449–56.
Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125(2):S73–80.
Pawankar R, Lee KH, Nonaka M, Takizawa R. Role of mast cells and basophils in chronic rhinosinusitis. Chronic Rhinosinusitis. CRC Press; 2007. p. 109–18.
Balzar S, Strand M, Rhodes D, Wenzel SE. IgE expression pattern in lung: relation to systemic IgE and asthma phenotypes. J Allergy Clin Immunol. 2007;119(4):855–62.
Pérez Novo CA, Jedrzejczak-Czechowicz M, Lewandowska-Polak A, Claeys C, Holtappels G, Van Cauwenberge P, et al. T cell inflammatory response, Foxp3 and TNFRS18-L regulation of peripheral blood mononuclear cells from patients with nasal polyps-asthma after staphylococcal superantigen stimulation. Clin Exp Allergy. 2010;40(9):1323–32.
Hammad H, Lambrecht BN. Dendritic cells and airway epithelial cells at the interface between innate and adaptive immune responses. Allergy. 2011;66(5):579–87.
Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. 2011;29(1):273–93.
Chen K, Cerutti A. New insights into the enigma of immunoglobulin D. Immunol Rev. 2010;237(1):160–79.
Pleass RJ, Lang ML, Kerr MA, Woof JM. IgA is a more potent inducer of NADPH oxidase activation and degranulation in blood eosinophils than IgE. Mol Immunol. 2007;44(6):1401–8.
Pant H, Beroukas D, Kette FE, Smith WB, Wormald PJ, Macardle PJ. Nasal polyp cell populations and fungal-specific peripheral blood lymphocyte proliferation in allergic fungal sinusitis. Am J Rhinol Allergy. 2009;23(5):453–60.
Chen K, Xu W, Wilson M, He B, Miller NW, Bengtén E, et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell–stimulating programs in basophils. Nat Immunol. 2009;10(8):889–98.
Smurthwaite L, Durham SR. Local ige synthesis in allergic rhinitis and asthma. Curr Allergy Asthma Rep. 2002;2(3):231–8.
Gevaert P, Holtappels G, Johansson SGO, Cuvelier C, Cauwenberge P, Bachert C. Organization of secondary lymphoid tissue and local IgE formation to Staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy. 2005;60(1):71–9.
Bachert C, Claeys SEM, Tomassen P, van Zele T, Zhang N. Rhinosinusitis and asthma: a link for asthma severity. Curr Allergy Asthma Rep. 2010;10(3):194–201.
Delves PJ, Roitt IM. The immune system. N Engl J Med. 2000;343(1):37–49.
Jones SA. Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol. 2005;175(6):3463–8.
Schleimer RP, Kato A, Peters A, Conley D, Kim J, Liu MC, et al. Epithelium, inflammation, and immunity in the upper airways of humans: studies in chronic rhinosinusitis. Proc Am Thorac Soc. 2009;6(3):288–94.
Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2011;11(8):505–18.
Tato CM, O'Shea JJ. What does it mean to be just 17? Nature. 2006;441(7090):166–7.
Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology. 2012;50(1):1–12.
Miller SA, Weinmann AS. Common themes emerge in the transcriptional control of T helper and developmental cell fate decisions regulated by the T-box, GATA and ROR families. Immunology. 2009;126(3):306–15.
Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations. Annu Rev Immunol. 2010;28(1):445–89.
Spits H, Di Santo JP. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol. 2010;12(1):21–7.
Allakhverdi Z, Comeau MR, Smith DE, Toy D, Endam LM, Desrosiers M, et al. CD34+ hemopoietic progenitor cells are potent effectors of allergic inflammation. J Allergy Clin Immunol. 2009;123(2):472–8.e1.
Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12(11):1055–62.
Sanchez-Segura A, Brieva JA, Rodriguez C. T lymphocytes that infiltrate nasal polyps have a specialized phenotype and produce a mixed TH1/TH2 pattern of cytokines. J Allergy Clin Immunol. 1998;102(6 Pt 1):953–60.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kern, R.C., Decker, J.R. (2023). Functional Defense Mechanisms of the Nasal Respiratory Epithelium. In: Celebi, Ö.Ö., Önerci, T.M. (eds) Nasal Physiology and Pathophysiology of Nasal Disorders. Springer, Cham. https://doi.org/10.1007/978-3-031-12386-3_4
Download citation
DOI: https://doi.org/10.1007/978-3-031-12386-3_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-12385-6
Online ISBN: 978-3-031-12386-3
eBook Packages: MedicineMedicine (R0)