
Abstract
Taste receptors on the tongue communicate information to the brain about the nutrient content or potential toxicity of ingested foods. However, recent research has now shown that taste receptors are also expressed far beyond the tongue, from the airway and gastrointestinal epithelia to the pancreas and brain. The functions of many of these so-called extraoral taste receptors remain unknown, but emerging basic science and clinical evidence suggests that bitter and sweet taste receptors in the airway are important in sensing bacteria and regulating innate immunity. This review focuses on the role of bitter and sweet taste receptors in human airway innate immunity and the potential clinical relevance to airway infections. The T2R38 bitter taste receptor in sinonasal cilia detects bitter bacterial quorum-sensing molecules and activates nitric oxide-dependent innate immune responses. Polymorphisms that underlie T2R38 functionality also appear to be involved in susceptibility to upper respiratory infection and chronic rhinosinusitis (CRS). Bitter and sweet receptors in specialized sinonasal solitary chemosensory cells control antimicrobial peptide secretion, which may have important implications for airway infections in CRS patients as well as patients with diabetes mellitus. Future research on taste receptors in the airway has tremendous potential to identify immune mechanisms involved in host-pathogen interactions and thus reveal novel therapeutic targets.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Understanding the regulation of respiratory innate immunity is critical to elucidating how pathogens evade these mechanisms during infection. The airway is constantly challenged by inhaled microbial pathogens. The “front-line” of respiratory defense is the sinonasal cavity [1, 2]. Failure of sinonasal defenses often results in chronic rhinosinusitis (CRS), a syndrome of complex etiologies involving stasis of sinonasal secretions due to ineffective mucociliary clearance (described below) and subsequent chronic infection and/or persistent inflammation [3]. CRS creates a tremendous economic burden, with direct healthcare costs of ~$6 billion annually in the USA [4–8]. Moreover, patients requiring sinus surgery for CRS report worse quality-of-life scores for physical pain and social functioning than patients suffering from chronic obstructive pulmonary disease, congestive heart failure, or angina [9–11].
Conventional management of CRS often involves antibiotics, with CRS accounting for approximately one in five antibiotic prescriptions in adults [4–7]. Thus, CRS is likely a major contributor to the ongoing crisis of antibiotic resistance [12, 13]. An attractive alternative therapeutic strategy is to eradicate infections by stimulating endogenous host defenses, but this requires a clearer understanding of sinonasal immune mechanisms. Recent studies have linked sinonasal innate immunity with both T2R bitter and T1R sweet taste receptors, identifying them as novel therapeutic targets for treatment of CRS and other respiratory infections.
Extraoral bitter and sweet taste receptors
T2R bitter and T1R sweet taste receptors are G-protein-coupled receptors named because of their roles in type II taste receptor cells on the tongue. T2Rs detect bitter chemicals like toxic plant alkaloids (e.g., strychnine), while T1R sweet receptors detect sugars (e.g., fructose, glucose, sucrose) [14, 15]. However, expression of T1R and T2R receptors extends far beyond the tongue to organs as diverse as the brain, gut, pancreas, bladder, and testes; these have been termed “extraoral” taste receptors [14, 15]. Because many medicinal compounds have a bitter taste [15], extraoral T2Rs may mediate some important off-target drug effects [16], emphasizing the need to understand extraoral taste receptor function, including their physiological roles and ligands. Because many extraoral taste receptors never come into direct contact with ingested food, many known agonists (i.e., bitter products from edible plants) are probably not relevant to their function. It has instead been hypothesized that extraoral T2R receptors detect bitter products from pathogenic bacteria or fungi, which would fit with their widespread distribution throughout the body.
Initial support for this came from studies of solitary chemosensory cells (SCCs) in the mouse nose, which express both T2R bitter and T1R sweet receptors [17–26]. Mouse nasal SCCs respond to acyl-homoserine lactones (AHLs) [21], which are used as quorum-sensing molecules by gram-negative bacteria, including the common airway pathogen Pseudomonas aeruginosa [27, 28]. Many lactone compounds are bitter [29], suggesting that AHLs are relevant ligands for some extraoral T2Rs. The hypothesis that T2Rs play a role in immunity is intriguing due to the large number of naturally occurring T2R polymorphisms [30] that underlie individual taste preferences for bitter foods and beverages, including vegetables [31], coffee [32], scotch [33], and beer [33]. Genetic variation in T1R sweet receptors also plays an important role in sweet taste preference [34–36]. If genetic variation in T2R and/or T1R receptor function also causes variation in how cells from different individuals sense and respond to pathogens, this may partly explain long-standing evidence that there is a genetic basis to respiratory infections [37–39]. Below, we review recent studies that support this hypothesis by demonstrating that (1) at least one human T2R isoform recognizes bacterial products, (2) genetic variation in this T2R correlates with sinonasal infection and CRS, and (3) T1Rs also regulate sinonasal innate immunity.
T2R bitter taste receptors in ciliated cells
Evidence of taste receptor expression in the airway first came in 2009, when researchers demonstrated that human bronchial cells express T2Rs that, when activated with known bitter agonists, stimulate calcium responses to increase ciliary beating [40]. Interestingly, these T2Rs are localized within motile cilia [40], which line much of the airway epithelium and drive mucociliary clearance [3, 41] (Fig. 1). Motile cilia were previously thought to be responsible only for the movement of fluid, as in the airway [42] and during embryonic development [43]. However, this study demonstrated that motile cilia are also “chemosensory organelles”; the authors hypothesized that T2Rs in bronchial cilia protect the airway against inhaled noxious irritants [40].

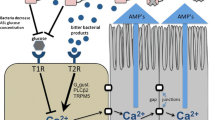
Mucociliary clearance (MCC) and respiratory epithelial innate immunity. Inhaled pathogens, such as viruses, bacteria, and fungal spores, are trapped by mucus created by sticky mucin macromolecules secreted by goblet cells of the respiratory epithelium. These trapped microbes are then removed from the airway by the process of mucociliary transport, the airway’s primary physical defense against inhaled pathogens and irritants. Constant beating of motile cilia drives the pathogen-laden mucus toward the oropharynx, where it is then cleared out of the airway by expectoration or swallowing. Mucociliary clearance is also dependent upon proper regulation of ion and fluid transport by epithelial cells and exocrine glands (not shown), which controls the mucus viscosity. Mucociliary transport is complemented by more direct mechanisms of pathogen inactivation and/or killing, including the generation of reactive oxygen and nitrogen species (ROS and RNS, respectively) and the production of antimicrobial peptides. During more chronic exposure to pathogens, epithelial cells secrete cytokines to activate inflammatory pathways and recruit dedicated immune cells
More recently, it was discovered that sinonasal epithelial cells also express T2Rs. One isoform, T2R38, is likewise localized to motile cilia [44, 45] (Fig. 2). When ciliated epithelial cells were stimulated with known T2R38-specific agonists, such as phenylthiocarbamide (PTC), they exhibited calcium-dependent activation of nitric oxide (NO) synthase (NOS), driving robust NO production [44]. NO production depended on two important components of the canonical taste signal transduction cascade [44], the TRPM5 ion channel [46] and phospholipase C isoform β2 (PLCβ2) [14, 15]. NO production by airway epithelial cells, including ciliated cells, is believed to be important for preventing airway infection [47, 48], because the NO radical and its derivatives can damage bacterial membranes, enzymes, and DNA [49, 50]. The NO produced during T2R38 activation in sinonasal epithelial cells in vitro was found to diffuse into the airway surface liquid (ASL) and be directly bactericidal against P. aeruginosa [44]. Additionally, NO increases ciliary beat frequency by activating guanylyl cyclase and protein kinase G, which can directly phosphorylate ciliary proteins [42].

Confocal immunofluorescence micrograph of T2R38 expression in motile cilia from a fixed human sinonasal tissue explant. T2R38 staining (shown in red) and visualization was carried out as described in [44], with β-tubulin IV (tubIV; shown in green) used as a cilia marker. Yellow “merge” image shows the combination of red and green color channels. Yellow signal indicates overlapping expression of T2R38 with cilia proteins. Image taken by G. Xiong, Children’s Hospital of Philadelphia, Philadelphia, PA USA
This initial T2R38 study also identified two major P. aeruginosa AHLs, N-butyryl-L-homoserine lactone (C4HSL) and N-3-oxo-dodecanoyl-L-homoserine lactone (C12HSL) [28], as T2R38 agonists [44]. Sinonasal epithelial cells stimulated directly with AHLs or with conditioned medium from wild-type P. aeruginosa produced NO in a T2R38-dependent manner. However, conditioned medium from a P. aeruginosa strain that lacks AHLs (ΔlasI, ΔrhlI PAO-JP2; [51]) did not activate NO production. Because many types of gram-negative bacteria secrete AHLs [28], T2R38 is likely part of a general surveillance mechanism to detect invading gram-negative bacteria. A schematic of the T2R38 innate immune pathway is shown in Fig. 3.

The role of the bitter taste receptor T2R38 in human sinonasal epithelial innate immunity. Reading from left to right, gram-negative bacteria secrete acyl-homoserine lactone (AHL) molecules to regulate quorum sensing. AHLs activate T2R38 in sinonasal cilia [44], initiating a calcium (Ca2+) signal that activates nitric oxide synthase (NOS)-dependent nitric oxide (NO) production [44, 45]. NO production has two distinct effects. The first is an increase in mucociliary transport [44, 45] caused by activation of the protein kinase G (PKG) pathway [42] to increase ciliary beating. NO additionally diffuses directly into the airway surface liquid, where it directly permeabilizes and kills bacteria [44]
Human T2R38 functionality is altered by several well-studied polymorphisms in the TAS2R38 gene [34, 52]. Two polymorphisms are common in Caucasian populations. One encodes a functional T2R38 and the other encodes a nonfunctional T2R38. These polymorphisms result in different amino acids at positions 49, 262, and 296. The functional T2R38 contains proline (P), alanine (A), and valine (V) residues and the nonfunctional T2R38 contains alanine (A), valine (V), and isoleucine (I) at these positions, respectively [52]. Loss of the V at the third position of the AVI variant likely prevents receptor activation [34, 52]. Homozygous AVI/AVI individuals (~30 % frequency in Caucasians) are “nontasters” for T2R38-specific agonists such as PTC and 6-propyl-2-thiouracil (PROP) [52]. Homozygous PAV/PAV individuals (~20 % frequency in Caucasians [52]) are “T2R38 supertasters” that perceive PTC and PROP as intensely bitter. Please note that, when used to describe PAV/PAV individuals, the term “supertaster” only refers to T2R38 function, as measured using PTC or PROP. Heterozygote PAV/AVI individuals have varying intermediate taste levels [34, 52]. These natural T2R38 variants were experimentally exploited by growing primary sinonasal cells from genotyped PAV/PAV “supertasters” and AVI/AVI “nontasters.” NO-dependent responses correlated with these polymorphisms. PAV/PAV “supertaster” cells had much higher levels of NO production, increased mucociliary clearance, and more efficient bacterial killing in response to either PTC or AHLs compared with cells from AVI/PAV heterozygote or AVI/AVI “nontaster” individuals [44]. TAS2R38 polymorphisms alter how efficiently sinonasal epithelial cells respond to gram-negative bacteria [44, 45].
This initial study also found that PAV/PAV “supertasters” had a lower frequency of gram-negative sinonasal infection than PAV/AVI or AVI/AVI patients with lower levels of T2R38 function [44]. Further clinical studies have now validated the relevance of TAS2R38 genotype to sinonasal disease [53–55]. A retrospective study [53] was first carried out in which TAS2R38 genotyping was performed on sinonasal tissue samples from 28 patients who had undergone primary functional endoscopic sinus surgery (FESS) at the Hospital of the University of Pennsylvania and Philadelphia Veterans Affairs Medical Center. Out of these patients, 3.6 % were PAV/PAV, 50 % were PAV/AVI, and 46 % were AVI/AVI, which was significantly different from the approximate expected distributions of 20 % PAV/PAV, 50 % PAV/AVI, and 30 % AVI/AVI (P < 0.043 by χ 2 analysis) [53]. Only one patient out of 28 was a PAV/PAV “supertaster,” suggesting that “supertasters” are less likely to need surgical intervention for CRS. In a subsequent prospective study of TAS2R38 genotype in 70 patients undergoing primary FESS at the same institutions [54], there was a statistically significant difference in the frequency of AVI and PAV alleles compared with the general population. Distributions of AVI/AVI, AVI/PAV, and PAV/PAV alleles were 37, 54, and 8.5 %, respectively, in the CRS patients compared with 29, 51, and 20 %, respectively, in the general Philadelphia population (P < 0.0383 by χ 2 analysis) [54]. No significant differences were found in the allele distribution with respect to allergies, asthma, nasal polyposis, aspirin sensitivity, diabetes, smoking exposure, or other risk factors, suggesting that TAS2R38 is an independent risk factor for CRS requiring FESS.
Another study was carried out with the goal of verifying if taste receptor polymorphisms correlate with CRS. Two separate Canadian CRS populations were compared with one control population [55], using previously collected pooling-based genome-wide association data that was screened for single-nucleotide polymorphisms (SNPs) in taste receptors, including the TAS2R38 I296V (rs10246939) thought to underlie the difference in functionality between PAV and AVI variants. The first population (genetics of chronic rhinosinusitis population 1 [GCRS1]) comprised 206 cases with severe CRS, defined as patients with either a history of more than one FESS for CRS regardless of outcome or persistent signs or symptoms of CRS despite a previous FESS procedure. The GCRS2 population comprised 408 CRS patients with early-onset nasal polyposis but who had had sweat chloride testing to rule out cystic fibrosis. A difference in I296V frequency of ~11 % (GCRS1) and ~15 % (GCRS2) in the two CRS groups compared with control individuals was found, confirming that this SNP is associated with CRS. Three other missense variants in TAS2R genes were associated with CRS (frequency difference ≥10 % between CRS and control), one in TAS2R14 (rs1015443) and two in TAS2R49 (rs12226920 and rs12226919) [55]. Roles for T2R14 or T2R49 in sinonasal immunity require further research.
Continued studies are still ongoing to validate the role of TAS2R38 in CRS, including patient outcomes. However, existing data have already established the T2R38 pathway as a potential therapeutic target to promote immune responses in patients with upper respiratory infections. However, because PAV/AVI and AVI/AVI individuals would be suboptimally responsive to a T2R38 agonist, it is necessary to further define the T2R38-mediated signaling pathway as well as identify other T2Rs that activate similar responses.
T2R bitter and T1R sweet receptors in sinonasal solitary chemosensory cells
Taste receptors are also expressed in specialized solitary chemosensory cells (SCCs), which express both T2Rs and T1Rs and populate the sinonasal cavity at a frequency of approximately 1 in 100 cells [17, 18, 20–26, 56]. The term “solitary chemosensory cell” was first used to describe chemosensory epithelial cells found in fish [57–59], which exhibit an elongated morphology with heavy neuronal innervation. Morphologically similar cells were later discovered in the upper respiratory tracts of the alligators [60], rodents [17, 19–22, 61, 62], and humans [23–25]. These cells are now classified as SCCs based on their morphology as well as expression of chemosensory signal transduction components, including T2R bitter and T1R sweet “taste” receptors, as will be described below.
The responses of mouse nasal SCCs to bitter compounds such as denatonium or bacterial AHLs require similar molecular components important for oral taste receptor signaling, including Gα-gustducin, PLCβ2, and TRPM5 [17, 20–22, 58]. When activated by bitter compounds, mouse nasal SCCs release acetylcholine (ACh) [26] to activate trigeminal nociceptors, stimulating breath-holding [21] and inflammation [26]. Trigeminal nociceptors can also release neuropeptides into the local environment, including vasoactive intestinal peptide and substance P [63, 64]. Thus, mouse nasal SCC activation in vivo may also result in local responses such as enhanced ciliary beating [41] or fluid secretion [65, 66], but this has not yet been experimentally determined.
SCCs have only recently been identified in humans [23–25]. In the human vomeronasal duct, SCC-like cells were observed to express T2R4, T1R1, and T1R2 [23]. More recently, T2R47- and T1R2/3-expressing SCCs were observed in primary cell cultures derived from postsurgical tissues obtained from a variety of sinonasal anatomical regions [25]. A thorough study of sinonasal SCC distribution is yet to be completed, but data so far suggest that SCCs are likely found throughout the human sinonasal cavity.
Human SCC physiology was studied using tissue explants and air-liquid interface (ALI) cell cultures [25], which are a well-validated in vitro model containing ciliated, goblet, basal cells [67–69], and SCCs, as described below. The bitter agonist denatonium, which activates mouse SCCs [17, 20–22], activated an intracellular calcium response in human sinonasal ALIs or inferior turbinate explants that originated from discrete cells, initiating a calcium wave that spread to the surrounding cells through gap junctions [25]. In both mouse and human ALIs, initiation of the calcium signal required known components of taste signaling, including Gα-gustducin, PLCβ2, the inositol 1,4,5-trisphosphate receptor, and TRPM5 [25]. Injection of the human denatonium-responsive cells with a fluorescent tracking dye showed that they have an elongated morphology identical to SCCs [25]. Immunofluorescence revealed that these cells co-express T2R47 (also known as T2R30) and the T1R3 sweet receptor subunit. The pharmacological profile of agonists that activate human SCCs suggests a role for T2R isoforms T2R10, T2R46, and T2R47/T2R30 [25], but not T2R38 as previously shown for mouse SCCs [17, 20–22, 26]. The full range of human SCC T2R isoforms remains to be determined. Human SCCs were not activated by PTC or AHLs, suggesting that human sinonasal T2R38 expression is restricted to cilia, likely reflecting a species-specific difference between human and mouse.
Interestingly, denatonium-induced SCC responses were observed to be dose-dependently blocked by apical sugars such as glucose and sucrose [25] as well as sucralose, a non-metabolizable sweet agonist [14, 15, 70]. Inhibition was dependent on the T1R sweet receptor, as it was blocked by the antagonists lactisole [71] and amiloride [25, 72]. These anatomical and physiological data show that human nasal SCCs co-express both bitter and sweet taste receptors, functioning in distinct, antagonistic physiological roles, the significance of which will be further discussed below.
T2R receptors in SCCs regulate antimicrobial peptide secretion and local innate immunity
Unlike T2R38 stimulation, SCC stimulation had no effect on NO production [25]. Instead, SCC T2Rs activated robust calcium-dependent secretion of antimicrobial peptides (AMPs), including β-defensins 1 and 2, from the surrounding epithelial cells [25]. This AMP secretion required gap-junction propagation of the calcium wave from the SCC to the surrounding ciliated and nonciliated epithelial cells [25]. These AMPs were active against both gram-positive (Staphylococcus epidermis and methicillin-resistant Staphylococcus aureus) and gram-negative (P. aeruginosa and Klebsiella pneumoniae) bacteria [25]. The majority of antimicrobial peptide secretion during SCC T2R stimulation occurred quickly (~5 min), which contrasts with the enhanced AMP production seen over hours in response to Toll-like receptor (TLR) stimulation [25]. TLRs in sinonasal epithelial cells upregulate messenger RNA for AMPs [73, 74], while T2Rs appear to regulate more rapid release of AMP stores [25].
Comparison of ALI AMP secretion responses with human sinonasal explants suggests that human sinonasal ALIs accurately reflect in vivo physiology. However, SCC-mediated responses are unique to the upper airway, as ALIs derived from bronchial tissue did not exhibit localized SCC-mediated calcium signals or AMP secretion [25]. As previously reported [40], bronchial responses to bitter agonists were more global, reflecting the activation of ciliated cells rather than SCCs [25]. Additionally, while many components of SCC signaling appeared to be similar in human and mouse ALIs, stimulation of mouse nasal SCCs did not result in release of AMPs. As described above, mouse SCC stimulation in vivo was linked to activation of trigeminal neurons mediating breath holding [17, 20–22, 58] and inflammation [26]. So far, human sinonasal SCCs have been linked to more local innate immunity [25], but human SCCs might also activate trigeminal nociceptors, perhaps also involving neurotransmitter-mediated triggering of gland secretion [65], ciliary beating [41], and/or inflammation [26]. ALIs lack neuronal innervation and cannot be used to study SCC interactions with neurons. Mice must be utilized with caution, as the data demonstrate that the localized SCC AMP responses are not present in the mouse, a major species-specific difference. As experiments in living human patients are difficult, clinically relevant in vivo studies of SCCs may require the use of a model that better recapitulates human sinonasal physiology, potentially the rabbit [75], sheep [76], or pig [77].
Potential clinical relevance of T1R receptors in SCCs
The inhibition of T2R responses by T1R sweet receptors in response to glucose [25], as described above is intriguing. Interestingly, normal ASL glucose is ~0.5 mM [25], which is sufficient to activate human airway T1Rs and inhibit SCC-mediated AMP secretion by approximately half [25]. While this concentration is 10–100× lower than concentrations required to activate heterologously expressed T1R receptors in vitro [70] or sweet taste in vivo [78], increased sensitivity of T1Rs has also been observed in pancreatic β-cells [79] and gut endocrine cells [80]. We hypothesize that oral T1R sweet receptor sensitivity is tuned to the appropriate higher sugar concentrations found in foods, while extraoral T1R sweet receptor sensitivity is tuned toward lower sugar concentrations more physiologically relevant to the tissues in which they are found. The mechanism(s) underlying this is unknown, but emerging evidence from β-cells points toward changes in subunit stoichiometry [81].
T1R sweet receptor attenuation of T2R-mediated AMP secretion may have important clinical relevance. We hypothesize that airway T1R sweet receptors function as a “rheostat” to control the magnitude of SCC/T2R-mediated AMP secretion depending on the glucose concentration in the ASL, which may be an indicator of the onset of infection. In vivo, sinonasal T1R2/3 sweet receptors would be activated by the normal ASL glucose (0.5 mM) of healthy individuals [25]. The T1R sweet receptors in SCCs may function to desensitize SCC T2Rs to bitter compounds secreted by some bacteria during low-level colonization in healthy individuals. However, during infection, bacterial consumption of ASL glucose may reduce ASL glucose concentration and allow T2Rs to mediate an appropriate defense response. This model is presented graphically in Fig. 4.

Nasal solitary chemosensory cells (SCCs) use taste receptors to regulate airway innate immunity. From left to right, bitter chemicals secreted by microbes during infection activate T2R receptors in SCCs, activating a calcium (Ca 2+) response that propagates to surrounding cells through gap junctions [25]. In human, but not mouse, sinonasal epithelial cells, this calcium signal causes the surrounding cells to secrete antimicrobial peptides (AMPs), which directly kill bacteria. Airway surface liquid (ASL) glucose (~0.5 mM in healthy individuals [25]) normally attenuates T2R-mediated signaling through activation of T1R sweet receptors. However, during times of infection, when bacteria likely decrease ASL glucose concentration by consuming it, T1R2/3-mediated inhibition of T2R signaling and AMP secretion is relieved [25]. In mice, SCC activation by bitter compounds results in acetylcholine (ACh) release and activation of trigeminal nociceptors [26]. Because AHLs have been shown to activate mouse nasal SCCs [21, 26] but do not appear to activate human SCCs in vitro [25], the bitter molecules that human SCCs respond to in vivo remain to be determined
This hypothesis remains to be fully validated but may have clinical importance in CRS and diabetes. Glucose is present in ASL because it tonically leaks through the epithelium, but the concentration is kept ~10-fold below serum levels by constant uptake via apical glucose transporters on the epithelial cells themselves [82–84]. Steady-state ASL glucose results from the balance of leakage and uptake across the epithelium [82–84]; upsetting this balance can alter ASL glucose concentration. In diabetic patients, hyperglycemia results in increased glucose leak and elevated ASL glucose [84, 85]. CRS patients also have a glucose concentration in nasal secretions that is three to fourfold higher than healthy individuals [25], but this is not correlated with blood glucose [25]. The higher ASL glucose in CRS likely results from increased leak due to breakdown of the epithelial barrier secondary to chronic infection and inflammation [82]. Pro-inflammatory mediators are known to increase paracellular glucose flux in human bronchial cells and disrupt tight junctions in human sinonasal cells in vitro [86, 87].
Diabetics may be more prone to airway infections than nondiabetics, potentially partly due to higher ASL glucose [82, 88]. Recently, a retrospective study of CRS patients correlated diabetics with a higher frequency of intraoperative microbiology cultures including gram-negative bacteria [89]. Previously, it was assumed that low ASL glucose promotes airway sterility by limiting nutrients available for bacterial growth [82–84]. However, higher ASL glucose in CRS or diabetic patients may also repress T2R-mediated SCC responses to bitter bacterial molecules through over-activation of T1R sweet receptors. If true, topical application of sweet receptor antagonists (e.g., lactisole) may restore normal sinonasal responses in some patients.
Another area requiring further study is whether polymorphisms in TAS1R genes, which can alter T1R responses to sugars [34–36], may play a role in susceptibility to respiratory infection. Increased sugar sensitivity in the airway might lead to increased repression of SCC-mediated AMP secretion. Investigation of this is strongly warranted by a recent study of Canadian CRS patients and healthy individuals showing allele frequency differences of >10 % for 16 different SNPs in TAS1R genes [55].
Taste receptors in other airway cell types
Airway taste receptor expression is not limited to ciliated cells and SCCs. Table 1 contains a list of known taste receptors in the airway. Tracheal chemosensory cells in mice, termed “brush cells” due to their apical microvilli, also express T2Rs and respond to AHLs to mediate breath-holding responses controlled by ACh release and trigeminal neurons [90–93]. A role for tracheal brush cells in humans has not yet been reported. Another major site of expression of T2Rs in the airway is bronchial smooth muscle cells, where they mediate bronchodilation [94–97]. It remains unknown whether bronchial smooth muscle T2Rs respond to pathogens, yet-unidentified endogenous autocrine or paracrine signaling molecules, or both.
Conclusions
It is becoming clear that T2R bitter and T1R sweet taste receptors are part of a novel pathogen detection network in the airway, where they are expressed in a variety of cell types and regulate multiple innate immune responses in both mice [17, 20–22, 26, 45] and humans [25, 44]. Previously, it was proposed that the immune system is the human “sixth sense” [98–100], because it detects invading pathogens similarly to the way the other senses (sight, smell, hearing, touch, and taste) perceive the outside world. From this perspective, it makes intuitive sense that our immune and taste systems utilize some of the same chemosensory receptors. The emerging and exciting field of extraoral taste receptors has tremendous potential to reveal novel insights into host-pathogen interactions and human disease in a variety of organ systems, including the airway.
References
Parker D, Prince A (2011) Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol 45:189–201
Waterer GW (2012) Airway defense mechanisms. Clin Chest Med 33:199–209
Antunes MB, Gudis DA, Cohen NA (2009) Epithelium, cilia, and mucus: their importance in chronic rhinosinusitis. Immunol Allergy Clin N Am 29:631–643
Blackwell DL, Collins JG, Coles R (2002) Summary health statistics for U.S. adults: National Health Interview Survey, 1997. Vital Health Stat 10:1–109
Ly N, McCaig LF (2002) National Hospital Ambulatory Medical Care Survey: 2000 outpatient department summary. Adv Data: 1–27
Ray NF, Baraniuk JN, Thamer M, Rinehart CS, Gergen PJ, Kaliner M et al (1999) Healthcare expenditures for sinusitis in 1996: contributions of asthma, rhinitis, and other airway disorders. J Allergy Clin Immunol 103:408–414
Cherry DK, Woodwell DA (2002) National Ambulatory Medical Care Survey: 2000 summary. Adv Data: 1–32
Fokkens WJ, Lund VJ, Mullol J, Bachert C, Alobid I, Baroody F, Cohen N, Cervin A, Douglas R, Gevaert P, Georgalas C, Goossens H, Harvey R, Hellings P, Hopkins C, Jones N, Joos G, Kalogjera L, Kern B, Kowalski M, Price D, Riechelmann H, Schlosser R, Senior B, Thomas M, Toskala E, Voegels R, Wang de Y, Wormald PJ (2012) European Position Paper on Rhinosinusitis and Nasal Polyps 2012. Rhinology Supplement: 3 p preceding table of contents, 1–298
Gliklich RE, Metson R (1995) The health impact of chronic sinusitis in patients seeking otolaryngologic care. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery 113: 104–109
Khalid AN, Quraishi SA, Kennedy DW (2004) Long-term quality of life measures after functional endoscopic sinus surgery. Am J Rhinol 18:131–136
Toledano A, Rodriguez G, Martin AM, Onrubia T, Galindo N (2011) Quality of life in patients with smell loss due to upper respiratory tract infections. Am J Otolaryngol 32:504–510
Bhattacharyya N, Kepnes LJ (2008) Assessment of trends in antimicrobial resistance in chronic rhinosinusitis. Ann Otol Rhinol Laryngol 117:448–452
Genoway KA, Philpott CM, Javer AR (2011) Pathogen yield and antimicrobial resistance patterns of chronic rhinosinusitis patients presenting to a tertiary rhinology centre. J Otolaryngol Head Neck Surg 40:232–237
Yamamoto K, Ishimaru Y (2013) Oral and extra-oral taste perception. Semin Cell Dev Biol 24:240–246
Mennella JA, Spector AC, Reed DR, Coldwell SE (2013) The bad taste of medicines: overview of basic research on bitter taste. Clin Ther 35:1225–1246
Clark AA, Liggett SB, Munger SD (2012) Extraoral bitter taste receptors as mediators of off-target drug effects. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 26:4827–4831
Finger TE, Bottger B, Hansen A, Anderson KT, Alimohammadi H, Silver WL (2003) Solitary chemoreceptor cells in the nasal cavity serve as sentinels of respiration. Proc Natl Acad Sci U S A 100:8981–8986
Sbarbati A, Osculati F (2003) Solitary chemosensory cells in mammals? Cells Tissues Organs 175:51–55
Tizzano M, Merigo F, Sbarbati A (2006) Evidence of solitary chemosensory cells in a large mammal: the diffuse chemosensory system in Bos taurus airways. J Anat 209:333–337
Gulbransen BD, Clapp TR, Finger TE, Kinnamon SC (2008) Nasal solitary chemoreceptor cell responses to bitter and trigeminal stimulants in vitro. J Neurophysiol 99:2929–2937
Tizzano M, Gulbransen BD, Vandenbeuch A, Clapp TR, Herman JP, Sibhatu HM et al (2010) Nasal chemosensory cells use bitter taste signaling to detect irritants and bacterial signals. Proc Natl Acad Sci U S A 107:3210–3215
Tizzano M, Cristofoletti M, Sbarbati A, Finger TE (2011) Expression of taste receptors in solitary chemosensory cells of rodent airways. BMC Pulm Med 11:3
Braun T, Mack B, Kramer MF (2011) Solitary chemosensory cells in the respiratory and vomeronasal epithelium of the human nose: a pilot study. Rhinology 49:507–512
Barham HP, Cooper SE, Anderson CB, Tizzano M, Kingdom TT, Finger TE et al (2013) Solitary chemosensory cells and bitter taste receptor signaling in human sinonasal mucosa. International forum of allergy & rhinology 3:450–457
Lee RJ, Kofonow JM, Rosen PL, Siebert AP, Chen B, Doghramji L et al (2014) Bitter and sweet taste receptors regulate human upper respiratory innate immunity. J Clin Invest 124:1393–1405
Saunders CJ, Christensen M, Finger TE, Tizzano M (2014) Cholinergic neurotransmission links solitary chemosensory cells to nasal inflammation. Proc Natl Acad Sci U S A 111:6075–6080
Pearson JP, Passador L, Iglewski BH, Greenberg EP (1995) A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 92:1490–1494
Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ (2012) The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev 76:46–65
Chadwick M, Trewin H, Gawthrop F, Wagstaff C (2013) Sesquiterpenoids lactones: benefits to plants and people. Int J Mol Sci 14:12780–12805
Kim U, Wooding S, Ricci D, Jorde LB, Drayna D (2005) Worldwide haplotype diversity and coding sequence variation at human bitter taste receptor loci. Hum Mutat 26:199–204
Li D, Zhang J (2014) Diet shapes the evolution of the vertebrate bitter taste receptor gene repertoire. Mol Biol Evol 31:303–309
Hayes JE, Wallace MR, Knopik VS, Herbstman DM, Bartoshuk LM, Duffy VB (2011) Allelic variation in TAS2R bitter receptor genes associates with variation in sensations from and ingestive behaviors toward common bitter beverages in adults. Chem Senses 36:311–319
Lanier SA, Hayes JE, Duffy VB (2005) Sweet and bitter tastes of alcoholic beverages mediate alcohol intake in of-age undergraduates. Physiol Behav 83:821–831
Bachmanov AA, Bosak NP, Lin C, Matsumoto I, Ohmoto M, Reed DR et al (2014) Genetics of taste receptors. Curr Pharm Des 20:2669–2683
Mennella JA, Pepino MY, Reed DR (2005) Genetic and environmental determinants of bitter perception and sweet preferences. Pediatrics 115:e216–e222
Fushan AA, Simons CT, Slack JP, Manichaikul A, Drayna D (2009) Allelic polymorphism within the TAS1R3 promoter is associated with human taste sensitivity to sucrose. Curr Biol 19:1288–1293
Greisner WA 3rd, Settipane GA (1996) Hereditary factor for nasal polyps. Allergy and asthma proceedings : the official journal of regional and state allergy societies 17:283–286
Cohen NA, Widelitz JS, Chiu AG, Palmer JN, Kennedy DW (2006) Familial aggregation of sinonasal polyps correlates with severity of disease. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery 134: 601–604. DOI 10.1016/j.otohns.2005.11.042
Hamilos DL (2007) Chronic rhinosinusitis patterns of illness. Clin Rev Allergy Immunol 20:1–13
Shah AS, Ben-Shahar Y, Moninger TO, Kline JN, Welsh MJ (2009) Motile cilia of human airway epithelia are chemosensory. Science 325:1131–1134
Lee RJ, Chen B, Doghramji L, Adappa ND, Palmer JN, Kennedy DW et al (2013) Vasoactive intestinal peptide regulates sinonasal mucociliary clearance and synergizes with histamine in stimulating sinonasal fluid secretion. FASEB journal :official publication of the Federation of American Societies for Experimental Biology 27:5094–5103
Salathe M (2007) Regulation of mammalian ciliary beating. Annu Rev Physiol 69:401–422
Babu D, Roy S (2013) Left-right asymmetry: cilia stir up new surprises in the node. Open biology 3:130052
Lee RJ, Xiong G, Kofonow JM, Chen B, Lysenko A, Jiang P et al (2012) T2R38 taste receptor polymorphisms underlie susceptibility to upper respiratory infection. J Clin Invest 122:4145–4159
Lee RJ, Chen B, Redding KM, Margolskee RF, Cohen NA (2014) Mouse nasal epithelial innate immune responses to Pseudomonas aeruginosa quorum-sensing molecules require taste signaling components. Innate immunity 20:606–617
Perez CA, Margolskee RF, Kinnamon SC, Ogura T (2003) Making sense with TRP channels: store-operated calcium entry and the ion channel Trpm5 in taste receptor cells. Cell Calcium 33:541–549
Haight JS, Djupesland PG, Qjan W, Chatkin JM, Furlott H, Irish J et al (1999) Does nasal nitric oxide come from the sinuses? The Journal of otolaryngology 28:197–204
Maniscalco M, Sofia M, Pelaia G (2007) Nitric oxide in upper airways inflammatory diseases. Inflammation research: official journal of the European Histamine Research Society [et al] 56:58–69
Fang FC (1997) Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Investig 99:2818–2825
Marcinkiewicz J (1997) Nitric oxide and antimicrobial activity of reactive oxygen intermediates. Immunopharmacology 37:35–41
Pearson JP, Pesci EC, Iglewski BH (1997) Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J Bacteriol 179:5756–5767
Bufe B, Breslin PA, Kuhn C, Reed DR, Tharp CD, Slack JP et al (2005) The molecular basis of individual differences in phenylthiocarbamide and propylthiouracil bitterness perception. Curr Biol 15:322–327
Adappa ND, Howland TJ, Palmer JN, Kennedy DW, Doghramji L, Lysenko A et al (2013) Genetics of the taste receptor T2R38 correlates with chronic rhinosinusitis necessitating surgical intervention. International forum of allergy & rhinology 3:184–187
Adappa ND, Zhang Z, Palmer JN, Kennedy DW, Doghramji L, Lysenko A et al (2014) The bitter taste receptor T2R38 is an independent risk factor for chronic rhinosinusitis requiring sinus surgery. International forum of allergy & rhinology 4:3–7
Mfuna Endam L, Filali-Mouhim A, Boisvert P, Boulet LP, Bosse Y, Desrosiers M (2014) Genetic variations in taste receptors are associated with chronic rhinosinusitis: a replication study. International forum of allergy & rhinology 4:200–206
Osculati F, Bentivoglio M, Castellucci M, Cinti S, Zancanaro C, Sbarbati A (2007) The solitary chemosensory cells and the diffuse chemosensory system of the airway. Eur J Histochem 51(1):65–72
Kotrschal K (2000) Taste (s) and olfaction (s) in fish: a review of specialized sub-systems and central integration. Pflugers Arch 439:R178–R180
Tizzano M, Finger TE (2013) Chemosensors in the nose: guardians of the airways. Physiology (Bethesda) 28:51–60
Whitear M (1992) Solitary chemoreceptor cells. In: Hara TJ (ed) Chemoreception in fishes Chapman and Hall, London, pp. 103–125
Hansen A (2007) Olfactory and solitary chemosensory cells: two different chemosensory systems in the nasal cavity of the American alligator, Alligator mississippiensis. BMC Neurosci 8:64
Gulbransen B, Silver W, Finger TE (2008) Solitary chemoreceptor cell survival is independent of intact trigeminal innervation. J Comp Neurol 508:62–71
Lin W, Ogura T, Margolskee RF, Finger TE, Restrepo D (2008) TRPM5-expressing solitary chemosensory cells respond to odorous irritants. J Neurophysiol 99:1451–1460
Baraniuk JN, Kaliner MA (1990) Neuropeptides and nasal secretion. J Allergy Clin Immunol 86:620–627
Mosimann BL, White MV, Hohman RJ, Goldrich MS, Kaulbach HC, Kaliner MA (1993) Substance P, calcitonin gene-related peptide, and vasoactive intestinal peptide increase in nasal secretions after allergen challenge in atopic patients. J Allergy Clin Immunol 92:95–104
Lee RJ, Foskett JK (2010) cAMP-activated Ca2+ signaling is required for CFTR-mediated serous cell fluid secretion in porcine and human airways. J Clin Invest 120:3137–3148
Lee RJ, Foskett JK (2012) Why mouse airway submucosal gland serous cells do not secrete fluid in response to cAMP stimulation. J Biol Chem 287:38316–38326
Lai Y, Chen B, Shi J, Palmer JN, Kennedy DW, Cohen NA (2011) Inflammation-mediated upregulation of centrosomal protein 110, a negative modulator of ciliogenesis, in patients with chronic rhinosinusitis. J Allergy Clin Immunol 128(1207–1215):e1201
Ramanathan M Jr, Lane AP (2007) A comparison of experimental methods in molecular chronic rhinosinusitis research. Am J Rhinol 21:373–377
Dimova S, Brewster ME, Noppe M, Jorissen M, Augustijns P (2005) The use of human nasal in vitro cell systems during drug discovery and development. Toxicol in Vitro 19:107–122
Li X, Staszewski L, Xu H, Durick K, Zoller M, Adler E (2002) Human receptors for sweet and umami taste. Proc Natl Acad Sci U S A 99:4692–4696
Jiang P, Cui M, Zhao B, Liu Z, Snyder LA, Benard LM et al (2005) Lactisole interacts with the transmembrane domains of human T1R3 to inhibit sweet taste. J Biol Chem 280:15238–15246
Imada T, Misaka T, Fujiwara S, Okada S, Fukuda Y, Abe K (2010) Amiloride reduces the sweet taste intensity by inhibiting the human sweet taste receptor. Biochem Biophys Res Commun 397:220–225
Ooi EH, Wormald PJ, Tan LW (2008) Innate immunity in the paranasal sinuses: a review of nasal host defenses. Am J Rhinol 22:13–19
Ramanathan M, Jr., Lane AP (2007) Innate immunity of the sinonasal cavity and its role in chronic rhinosinusitis. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery 136: 348–356. DOI 10.1016/j.otohns.2006.11.011
Chiu AG, Antunes MB, Feldman M, Cohen NA (2007) An animal model for the study of topical medications in sinusitis. Am J Rhinol 21:5–9
Ha KR, Psaltis AJ, Tan L, Wormald PJ (2007) A sheep model for the study of biofilms in rhinosinusitis. Am J Rhinol 21:339–345
Wang JC, Hathorn I, Habib AR, Chang E, Javer AR (2013) Evaluation of domestic and Yucatan swine nasal sinus anatomy as models for future sinonasal research of medications delivered by standard instruments used in functional endoscopic sinus surgery. International forum of allergy & rhinology 3:150–156
Schiffman SS, Booth BJ, Sattely-Miller EA, Graham BG, Gibes KM (1999) Selective inhibition of sweetness by the sodium salt of +/−2-(4-methoxyphenoxy) propanoic acid. Chem Senses 24:439–447
Kyriazis GA, Soundarapandian MM, Tyrberg B (2012) Sweet taste receptor signaling in beta cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A 109:E524–E532
Jang HJ, Kokrashvili Z, Theodorakis MJ, Carlson OD, Kim BJ, Zhou J et al (2007) Gut-expressed gustducin and taste receptors regulate secretion of glucagon-like peptide-1. Proc Natl Acad Sci U S A 104:15069–15074
Kojima I, Nakagawa Y, Ohtsu Y, Medina A, Nagasawa M (2014) Sweet taste-sensing receptors expressed in pancreatic beta-cells: sweet molecules act as biased agonists. Endocrinol Metab 29:12–19
Garnett JP, Baker EH, Baines DL (2012) Sweet talk: insights into the nature and importance of glucose transport in lung epithelium. Eur Respir J 40:1269–1276
Kalsi KK, Baker EH, Fraser O, Chung YL, Mace OJ, Tarelli E et al (2009) Glucose homeostasis across human airway epithelial cell monolayers: role of diffusion, transport and metabolism. Pflugers Arch 457:1061–1070
Pezzulo AA, Gutierrez J, Duschner KS, McConnell KS, Taft PJ, Ernst SE et al (2011) Glucose depletion in the airway surface liquid is essential for sterility of the airways. PLoS ONE 6:e16166
Baker EH, Clark N, Brennan AL, Fisher DA, Gyi KM, Hodson ME et al (1985) Hyperglycemia and cystic fibrosis alter respiratory fluid glucose concentrations estimated by breath condensate analysis. J Appl Physiol 102:1969–1975
Rogers GA, Den Beste K, Parkos CA, Nusrat A, Delgaudio JM, Wise SK (2011) Epithelial tight junction alterations in nasal polyposis. International forum of allergy & rhinology 1:50–54
Soyka MB, Wawrzyniak P, Eiwegger T, Holzmann D, Treis A, Wanke K et al (2012) Defective epithelial barrier in chronic rhinosinusitis: the regulation of tight junctions by IFN-gamma and IL-4. J Allergy Clin Immunol 130(1087–1096):e1010
Koziel H, Koziel MJ (1995) Pulmonary complications of diabetes mellitus. Pneumonia. Infect Dis Clin N Am 9:65–96
Zhang Z, Adappa ND, Lautenbach E, Chiu AG, Doghramji L, Howland TJ et al (2014) The effect of diabetes mellitus on chronic rhinosinusitis and sinus surgery outcome. International forum of allergy & rhinology. doi:10.1002/alr.21269
Krasteva G, Canning BJ, Hartmann P, Veres TZ, Papadakis T, Muhlfeld C et al (2011) Cholinergic chemosensory cells in the trachea regulate breathing. Proc Natl Acad Sci U S A 108:9478–9483
Saunders CJ, Reynolds SD, Finger TE (2013) Chemosensory brush cells of the trachea. A stable population in a dynamic epithelium. Am J Respir Cell Mol Biol 49:190–196
Sbarbati A, Osculati F (2005) A new fate for old cells: brush cells and related elements. J Anat 206:349–358
Krasteva G, Canning BJ, Papadakis T, Kummer W (2012) Cholinergic brush cells in the trachea mediate respiratory responses to quorum sensing molecules. Life Sci 91:992–996
Deshpande DA, Wang WC, McIlmoyle EL, Robinett KS, Schillinger RM, An SS et al (2010) Bitter taste receptors on airway smooth muscle bronchodilate by localized calcium signaling and reverse obstruction. Nat Med 16:1299–1304
An SS, Wang WC, Koziol-White CJ, Ahn K, Lee DY, Kurten RC et al (2012) TAS2R activation promotes airway smooth muscle relaxation despite beta (2)-adrenergic receptor tachyphylaxis. Am J Physiol Lung Cell Mol Physiol 303:L304–L311
Grassin-Delyle S, Abrial C, Fayad-Kobeissi S, Brollo M, Faisy C, Alvarez JC et al (2013) The expression and relaxant effect of bitter taste receptors in human bronchi. Respir Res 14:134
Robinett KS, Koziol-White CJ, Akoluk A, An SS, Panettieri RA Jr, Liggett SB (2014) Bitter taste receptor function in asthmatic and nonasthmatic human airway smooth muscle cells. Am J Respir Cell Mol Biol 50:678–683
Blalock JE (2005) The immune system as the sixth sense. J Intern Med 257:126–138
Blalock JE, Smith EM (2007) Conceptual development of the immune system as a sixth sense. Brain Behav Immun 21:23–33
Bedford FL (2011) The missing sense modality: the immune system. Perception 40:1265–1267
Acknowledgments
Some of the research described here was supported by a grant from the Flight Attendants Medical Research Institute (082478), a philanthropic contribution from the RLG Foundation Inc., and USPHS grant R01DC013588 to N.A.C.
Conflict of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lee, R.J., Cohen, N.A. Bitter and sweet taste receptors in the respiratory epithelium in health and disease. J Mol Med 92, 1235–1244 (2014). https://doi.org/10.1007/s00109-014-1222-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-014-1222-6