
Abstract
Periodontitis and Alzheimer’s disease (AD) exist globally within the adult population. Given that the risk of AD incidence doubles within 10 years from the time of periodontal disease diagnosis, there is a window of opportunity for slowing down or preventing AD by risk-reduction-based intervention. Literature appraisal on the shared risk factors of these diseases suggests a shift to a healthy lifestyle would be beneficial. Generalised (chronic) periodontitis with an established dysbiotic polymicrobial aetiology affects the tooth supporting tissues with eventual tooth loss. The cause of AD remains unknown, however two neurohistopathological lesions – amyloid-beta plaques and neurofibrillary tangles, together with the clinical history, provide AD diagnosis at autopsy. Historically, prominence was given to the two hallmark lesions but now emphasis is placed on cerebral inflammation and what triggers it. Low socioeconomic status promotes poor lifestyles that compromise oral and personal hygiene along with reliance on poor dietary intake. Taken together with advancing age and a declining immune protection, these risk factors may negatively impact on periodontitis and AD. These factors also provide a tangible solution to controlling pathogenic bacteria indigenous to the oral and gastrointestinal tract microbioes in vulnerable subjects. The focus here is on Porphyromonas gingivalis, one of several important bacterial pathogens associated with both periodontitis and AD. Recent research has enabled advances in our knowledge of the armoury of P. gingivalis via reproduction of all clinical and neuropathological hallmark lesions of AD and chronic periodontal disease in vitro and in vivo experimental models, thus paving the way for better future management.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
FormalPara Highlights-
Periodontal disease has been associated with the onset and progression of Alzheimer’s Disease, which causes irreversible cognitive and functional decline
-
This chapter summarises the diverse range of evidence which implicates periodontal disease and its key pathogen, P. gingivalis , as a significant risk factor for AD
-
The potential for prevention is high as the dental team can play an important role in preventing and/or delaying the development of AD by proactive management of periodontitis
-
A drug COR388 targeting the P. gingivalis protease enzyme is a novel treatment modality and may offer promising results, considering the number of failed AD therapeutic clinical trials
-
To deliver personalised preventative advice to patients, not only with regards to oral health, but also to healthy lifestyle choices
-
To maintain and stabilise periodontal disease in a bid to reduce or delay AD progression or development
-
To risk assess each patient and deliver prevention and care accordingly in order to enable ‘successful ageing’ and reduce disease development in their patient base
Dentists understand that maintaining a healthy mouth contributes to maintaining a healthy body. Research suggests that periodontal disease can lead to the development and progression of AD . Patients should be aware of all modifiable risk factors which have been linked to AD , including their oral health and the lifestyle choices they make. The onus comes down to the dental profession to deliver this preventative advice to their patients. It is imperative to maintain gingival health and make healthy lifestyle choices to not only to reduce their risk of AD but also to delay the onset, in susceptible individuals.
1 Introduction
There is a growing body of evidence that supports oral (generalised periodontitis) and gastrointestinal (GI) tract dysbiosis as having a negative impact on mental health. Briefly, the enteric nervous system supplies the brain with “psychobiotics” (serotonin, dopamine and Gamma aminobutyric acid) courtesy of healthy gut bacteria (Strandwitz 2018; Strandwitz et al. 2019). Dysbiosis of the gut bacteria leads to a deficit of these neurotransmitters and causes anxiety and depression (Clapp et al. 2017). Depression, in this context, may either be a risk factor or an early sign of dementia. Alzheimer’s disease (AD) being the most common example of dementia.
Shared features of generalised periodontitis and AD include a progressive, inflammatory disease pathway, and shared disease related risk factors including ageing, infection, immunosuppression, genetic predisposition and socioeconomic factors. This chapter will explain the dental hypothesis surrounding the associations made between periodontal disease and AD . The associations between generalised periodontal disease and AD are evidenced by experimental research data and describe how the hypothesis that “periodontitis is a risk factor for AD ” came about. The evidence to support this hypothesis is substantial, whilst studies supporting a causal relationship are in progress. The aim is to highlight the risk factor involvement of generalised periodontal disease on AD development with a specific focus on Porphyromonas gingivalis , because this microbe is at the heart of current research, which suggests potential for much desired therapy .
2 Periodontal Disease
Periodontitis is an oral disease presenting with a polymicrobial dysbiosis of the subgingival microbiome, which, if left untreated, leads to tooth loss. P. gingivalis is considered a keystone pathogen of periodontal disease alongside its companion bacteria (Treponema denticola, Tannerella forsythia) of the red complex in adult periodontitis (Socransky et al. 1998; Holt and Ebersole 2005; Hajishengallis et al. 2012). Around 50% of all humans in middle age (50 years and over) appear to fall victim to periodontitis (Eke et al. 2015). In the previous classification, the adult form of periodontitis was known as “chronic periodontitis”. However, a new classification (Caton et al. 2018; Dietrich et al. 2019) refers to the formerly known “chronic” periodontitis as generalised periodontitis stages I–IV, grades A–C. This is difficult to integrate into a story that appeals to dental and non-dental professionals so we will refer to “chronic periodontitis” as “generalised periodontitis” to be consistent with the new changes throughout this chapter.
The nature of periodontal disease is episodic with characteristic recurrent periods of active disease progression followed by periods of quiescence in individuals who are unable to prevent commensals (healthy microbiome) converting into pathogens (pathobiome) (Dioguardi et al. 2020). Periodontal disease affects the tooth supporting tissues, and interaction of specific bacteria and consequently the host’s immune responses play a pivotal role (Haffajee et al. 1988). The host’s response to the pathogenic bacteria and their virulence factors such as lipopolysaccharide (LPS), proteases such as gingipains, fimbriae, hemagglutinins, and outer membrane vesicles (OMVs) determine the severity and extent of periodontal disease (Kinane and Marshall 2001; How et al. 2016). Bacterial LPS is located in the outer membrane of Gram-negative bacteria and is a potent stimulator of host innate immune signal transduction pathways (Beutler 2000). The acute bacterial challenge stimulates the junctional pocket epithelium to produce inflammatory mediators to protect against tissue damage via an acute phase receptor-mediated cytokine production and neuropeptide release, resulting in vasodilation of local vessels. Gingival bleeding, swelling and redness together with the presence of neutrophils and macrophages within the inflamed gingival tissues indicate clinical signs of inflammation (Hasturk et al. 2012). In susceptible individuals, the acute phase responses fail to clear infection and chronic inflammatory lesions develop. The subgingival sulcus serves as a niche enabling a cyclic chronic inflammatory process, which facilitates recurrent bacteraemias, enabling micro-organisms and their virulence factors to access the systemic circulation (Forner et al. 2006; Lockhart et al. 2008; Bahrani-Mougeot et al. 2008). Each time we brush or chew on a periodontally-affected tooth, bacteraemia consisting of a spectrum of oral bacteria occurs. In any one day this bacteraemia can last for a total of 3 h (Bahrani-Mougeot et al. 2008; Tomás et al. 2012). In addition, viruses, bacteriophages, and yeasts within the periodontal pocket may follow the bacteria into the blood stream along with inflammatory mediators from the inflamed periodontal tissues (Olsen and Singhrao 2015; Li et al. 2020). From the blood stream these bacteria may be carried to wherever they lodge, potentially providing a nidus for further organ specific inflammatory pathologies. Thus, periodontal disease has been associated with a number of inflammatory pathologies, including cardiovascular disease, diabetes and AD (Makiura et al. 2008; Bale et al. 2017; Demmer et al. 2020; Stein et al. 2007; Poole et al. 2013; Dominy et al. 2019). Indeed, the cause of death in AD cases can result from cerebrovascular diseases such as stroke and pneumonia suggesting a greater risk of dementia for individuals who have suffered multiple co-morbidities in their lives (van Oijen et al. 2007).
3 Alzheimer’s Disease (AD)
There are two main forms of AD; familial or early-onset form, which involves mutated genes such as amyloid precursor protein (APP) and presenilin 1 and presenilin 2. In the sporadic or late-onset form of AD, Genome-Wide-Association Studies (GWAS ) have identified a number of susceptibility genes expressed by the brain cells. Of these, the apolipoprotein E, allele 4 (APOE є4) is firmly established as the second strongest risk factor after advancing age (Corder et al. 1993; Saunders et al. 1993). AD is a leading neurodegenerative disease with clinical signs of deteriorating memory, which together with its two neuropathological hallmark lesions, amyloid-beta (Aβ) and neurofibrillary tangles (NFTs), complete its post-mortem diagnosis (Hyman et al. 2012). Although not pathognomonic of AD , other lesions which present in AD pathophysiology include neuronal and synaptic loss and gliosis (intracerebral inflammation) (Dugger and Dickson 2017). The origins and the roles of Aβ plaques and NFTs are quite distinct but they both lead to neurodegeneration within the associated regions of the cerebral cortex and medial temporal lobes. The hippocampus, by contrast, typically contains abundant intra-neuronal NFTs composed of abnormally phosphorylated tau protein representing destabilized microtubules that are non–membrane-bound masses of abnormal straight and/or paired helical filaments (PHF) (Grundke-Iqbal et al. 1986; Goedert et al. 2006). The NFTs were thought to first appear in the entorhinal cortex leading to the hippocampus, but this view has changed identifying early involvement in the subcortical nuclei such as the locus coeruleus in the pons (Braak and Braak 1991; Braak et al. 2011).
The AD plaque is composed of an insoluble form of Aβ and continues to form the basis of the influential “Amyloid Cascade Hypothesis” (Hardy and Selkoe 2002). Molecular cloning methodologies identified Aβ as a cleavage fragment of a single membrane-spanning receptor-like glycoprotein known as amyloid precursor protein (APP) that is inserted, in part, in the plasma membrane (Kang et al. 1987). APP also occurs in internal vesicular membranes, including the Golgi apparatus and endosomes (Choi et al. 2012) as explained by Singhrao and Olsen (2019) in intracellular bacterial infections such as P. gingivalis . APP proteolytic cleavage involves three proteases, namely α-, β- and γ-secretases, the β- secretase acts with γ-secretases to release Aβ which becomes insoluble and fibrillar in appearance (Hook et al. 2008) giving rise to the neuritic/senile or Aβ plaques of AD . Activated microglia and astrocytes (gliosis) accompany these plaques and contribute to intracerebral inflammation (Perry et al. 2010; Boche et al. 2013; Heneka et al. 2015).
4 The Emerging Association Between Periodontal Disease and Alzheimer’s Disease Provides a Rationale for Therapy
Researchers have shown that the genetic variant of the APOE є4 increases a person’s risk of developing AD (Genin et al. 2011). An indirect effect of inheriting the APOE є4 gene variant with respect to patients with periodontal disease may be the increased susceptibility of the host to microbial infections (de Bont et al. 1999; Moretti et al. 2005; Watts et al. 2008). This genotype has also been linked to increased inflammatory burdens in terms of cytokines (systemic circulation and brain) in response to LPS (Tsoi et al. 2007; Watts et al. 2008; Hubacek et al. 2010).
Previous research has indicated that those individuals from low socioeconomic status and low levels of education show poorer cognitive functioning in later life, and are at greater risk of developing AD (Brayne and Calloway 1990; Chen and Miller 2013; Russ et al. 2013; Marden et al. 2017). Low socioeconomic status groups have also shown an increased likelihood of engaging in habits (poor dietary choices) and lifestyle choices that are detrimental to their oral and general health. These behavioural traits (for example, smoking and drinking excessive amounts of alcohol) increase the risk for an individual to develop both periodontitis, and AD (Singh et al. 2013). A poor diet contributes to making the immune system less robust in its ability to fight infection. Smoking tobacco has a detrimental impact on general health, increasing the likelihood of developing cardiovascular and cerebrovascular disease, but more importantly increases the severity to which periodontal disease progresses due to decreased oxygen and an increased inflammatory burden (Kamma et al. 1999; Grossi et al. 1995).
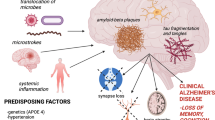
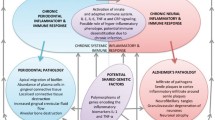
It is important to point out that not everyone who develops clinical AD is comorbid with periodontal disease (Stein et al. 2007; Farhad et al. 2014). The reason for this observation remains unknown. However, a plausible explanation could be that the more virulent strains of P. gingivalis may develop in poorly managed oral health and in cases of periodontitis that eventually lead to sporadic AD . Figure 13.1 illustrates the bidirectional relationship between periodontitis and AD .

The bidirectional relationship between periodontitis and AD
5 The Experimental Journey Towards Risk Factor Identification Between Periodontitis and Alzheimer’s Disease
The journey began in 1994 with the Japanese elderly population based epidemiological study which introduced the concept of fewer teeth in later life as a risk factor for dementia (Kondo et al. 1994). For a significant length of time this concept was left unexplored. Later Chalmers et al. (2002) showed that cognitively impaired institutionalised patients exhibited more retained roots, carious teeth and missing teeth due to poor oral health compared to community-dwelling older adults. A systematic review by Cerutti-Kopplin et al. (2016) concluded that there was evidence to support that tooth loss increased the risk of cognitive impairment and dementia.
It was Stein et al. (2007) who investigated a potential association between the history of oral disease and dementia development in female subjects of a religious order. The study included analysis of the previously collected data from 10 annual cognitive assessments of 144 Milwaukee participants in the “Nun Study”. Participants with fewer teeth directly correlated with an increased risk of prevalence and incidence of dementia (Stein et al. 2007). It also concluded that missing up to nine teeth carried the highest risk for developing late-onset AD with an odds ratio of 2.2 (95% CI 1.1, 4.5), proposing tooth loss due to periodontitis was doubling the risk factor for AD development. Together with other studies, this helped to consolidate the idea that potential neglect of oral health could have a detrimental effect on brain health (Stein et al. 2007).
6 Does Peripheral inflammation Induce Cerebral Inflammation?
In establishing the events leading from poor oral health to AD manifestation, Noble et al. (2009) used a cross-sectional methodology to investigate an association between cognitive impairment and P. gingivalis via serum markers of its infection. It was found that those subjects who were cognitively impaired had higher mean P. gingivalis IgG levels compared to those who were cognitively healthy. Differences in individuals’ humoral responses supported the association of P. gingivalis with cognitive impairment. Also, Holmes et al. (2009) had proposed that peripheral inflammation may be a key determinant of the cognitive decline associated with AD progression. This prompted Kamer et al. (2009) to establish if inflammatory markers such as TNF-α cytokine and antibodies to periodontal bacteria could discriminate between AD and non-AD subjects using case-control methodology. Sparks Stein et al. (2012) also investigated whether or not serum antibodies to periodontal pathogens were a risk factor for AD . They used a case-control methodology with information gathered from a larger cohort study. They found that antibodies to certain periodontal pathogens over 10 years were significantly higher at baseline in those who went on to develop AD (Sparks Stein et al. 2012). A large scale Taiwanese insurance based epidemiological survey also reported that individuals with chronic periodontitis for at least 10 years had a 70% higher risk of developing AD compared to individuals without periodontal disease (Chen et al. 2017). This reinforced the postulated 10-year lag phase from the time of periodontal disease diagnosis to developing AD and lends credence to the bidirectional relationship between the periodontitis-AD brain risk-axis.
The report by Poole et al. (2013) analysed human AD and non-AD post-mortem brains using immunolabelling and immunoblotting and demonstrated the LPS of P. gingivalis exclusively in the AD brains and not the controls. This finding was consolidated by Dominy et al. (2019) who used more advanced molecular techniques identifying P. gingivalis deoxyribonucleic acid (DNA), LPS and gingipain antigens within the brain tissue of AD subjects. These two studies (Poole et al. 2013; Dominy et al. 2019) provided the rationale and the impetus for developing periodontal disease models for AD to better understand the potential of a bacterial aetiology for this neurodegenerative disease occurring via the peripheral (systemic) system.
The first mouse model used genetically modified apolipoprotein E knock-out (ApoE−/−) mice, which were orally infected with P. gingivalis to initiate experimental chronic periodontitis (Velsko et al. 2014). Poole et al. (2015) showed that within 3 months of P. gingivalis oral infection, bacterial DNA and LPS had spread to the ApoE−/− mice brains. In addition, there was evidence of intracerebral inflammatory pathology and complement activation once the bacterium had entered the hippocampus, compromising the health of neurons (Poole et al. 2015). Due to the genetic weaknesses of the ApoE−/− P. gingivalis-mono-infection model, demonstrating the Aβ plaque hallmark of AD (intracerebral pathology) was not possible.
The next stage saw researchers introducing genetic mutations to their periodontitis mouse model. They induced P. gingivalis mono-infection into the APP-transgenic (APP-Tg) model carrying the Swedish and Indiana mutations (Ishida et al. 2017). The aim was to assess the role of chronic periodontitis in the development of AD hallmark pathology. However, Ishida et al. (2017) concluded that their results reflected an overall susceptibility to AD rather than having contributed to the overall Aβ hallmark pathology. In retrospect this interpretation appears to be plausible, as APP in the familial form of AD , harbours a missense mutation, (where a single nucleotide is changed with the substitution of a different amino acid) (Rossor et al. 1993), suggesting that the bacterial and/or host’s β-secretase equivalent digestive enzymes (cathepsin B and gingipains) (Dominy et al. 2019; Hook et al. 2008) may be unable to cleave APP in significant amounts to notice Aβ contribution in this model.
Moving away from genetically modified mice, Ilieviski et al. (2018) chose a wild-type mouse as their model of periodontitis for reproducing AD pathophysiology. They confirmed the spread of P. gingivalis from the oral niche to the brain in their wild-type mice and observed glial cell activation, the same as reported by Poole et al. (2015), however in addition, the infection reproduced the cardinal neuropathological hallmark lesions (Aβ plaques and phosphorylated tau protein at serine396 position) of AD for the first time. This is corroborated by the fact that P. gingivalis has its own β-secretase (in the form of gingipains) activity to cleave APP (Dominy et al. 2019). Both bacterial and host β-secretase (in the form of neuronal cathepsin B) enhances the overall APP cleavage (Hook et al. 2008).
P. gingivalis gingipains and its LPS are also known to activate kinases such as glycogen synthase kinase-3 (GSK-3) which subsequently phosphorylates neuronal tau at two sites (serine 396 and Threonine231), involved with the NFT AD lesion (Haditsch et al. 2020; Ilieviski et al. 2018; Bahar and Singhrao 2021).
Further studies have examined the mechanistic links for cognition with P. gingivalis . The first of these functional testing studies was reported by Ishida et al. (2017) in their APP-Tg mice orally infected with P. gingivalis . They demonstrated that cognitive function was significantly impaired in periodontitis-induced APP-Tg mice compared to the sham-infected group when tested using a water maze (Morris 1984). An explanation for the greater cognitive deficit in the infected APP-Tg group was an increased inflammatory mediator (cytokine ) burden following induction of experimental periodontitis.
Subsequent investigations performed similar behavioural tests on orally mono-infected P. gingivalis mice and supported the mechanism of cognitive deficit due to inflammation resulting from infection in which ageing was also a factor (Ding et al. 2018). These mouse model based studies are a proof of concept that periodontitis detrimentally impacts cognition via release of inflammatory mediators into the blood stream.
6.1 Systemic LPS and Its Effect on Cognition
Wu et al. (2017) were the first to report that chronic exposure to LPS from P gingivalis elicited AD-like phenotypes in middle-aged (12 months old) mice. The phenotypes included learning and memory deficits, intracellular Aβ in neurons, and microglia-induced neuroinflammation in the hippocampus. The suggested mechanism is that Aβ is cleaved indirectly by the action of cathepsin B on the parent APP. APP is initially stimulated by the interaction of the cytokine interleukin (IL)-1β with its cognate receptor (IL-1R) on neurons. Exposure to LPS from P. gingivalis led to a significant increase in microglia implying their activation and subsequent secretion of cytokines and neurons. This indicates that the memory deficit seen following systemic exposure to LPS of P. gingivalis in middle-aged mice is depended on cathepsin B and gingipains.
Following on from this, Zhang et al. (2018) conducted functional testing, but specifically analysing the effect of P. gingivalis LPS on cognitive function. Behavioural changes were assessed with the open field test, Morris water maze, and passive avoidance test. Using immunohistochemistry, they assessed for activation of astrocytes and microglia within the cerebral cortex and hippocampus; and assessed for pro-inflammatory cytokine expression of Interleukins (IL) IL-1β, IL-6, IL-8, Tumour necrosis factor-α (TNFα), toll-like receptors (TLRs) and CD-14 using reverse transcriptase-polymerase chain reaction (RT-PCR), enzyme-linked immunoassay (ELISA) and western blotting. The mice infected with P. gingivalis-LPS showed impairment in spatial learning and memory, along with activation of microglia and astrocytes within both the cerebral cortex and hippocampus. In addition, there was up-regulation of pro-inflammatory cytokines and activation of the TLR4/nuclear factor kappaB (NF-ϰB) signaling pathway. These findings suggested that P. gingivalis-LPS can lead to impaired memory and learning, which would appear to be mediated by the TLR4 signalling pathway. This demonstrates the potential for periodontal pathogen endotoxin as a risk factor for cognitive disorders.
6.2 Systemic Aβ; Does It Contribute to the Intracerebral Burden of This Hallmark in AD?
It has recently been suggested that periodontitis increases the levels of peripheral Aβ from gingival tissue and animal models and inflammatory cell sources (Leira et al. 2019; Nie et al. 2019). This has led to the proposal that in the human scenario, there is a potential for this peripheral source of Aβ to gain access into the brain thereby adding to the Aβ pool in the AD brain. Whether this would be a plausible mechanism in the human AD brain is not known. However, Zeng et al. (2020), in their P. gingivalis infection mouse model, identified advanced glycation end products (AGE) as a plausible receptor for Aβ in cerebral endothelial cells in mice. This implies that AGE products-receptor are a plausible mediator of cerebrovascular-related Aβ accumulation in the brain; supporting the hypothesis that patients harboring the generalized form of periodontal disease may be at risk of developing AD via multiple pathways involving periodontitis.
Limitations to the study included the intraperitoneal administration of P. gingival-LPS (rather than oral infection of whole P. gingivalis as with other research); the route of administration may have had some bearing on the passage into the cerebral tissues. Also, all mice were of the same age, and therefore the effect of ageing was not assessed by this study. However, it provided useful information with regard to the potential effect of periodontal pathogen endotoxin on cognitive function, and that inflammation, which plays an important role in cognitive impairment, was mediated by the TLR4 signaling pathway, giving an indication of possible underlying mechanisms.
These studies have demonstrated major advances that have been made experimentally to investigate the relationship between periodontal pathogens and their associated products, cognitive impairment, and AD pathophysiology. This research has been fundamental in the positive progress of the development of potential therapeutic agents against AD .
6.3 Evidence Supporting the Inflammatory Burden of Periodontitis Affecting Memory
An observational cohort study by de Rolim et al. (2014) involved 29 participants with clinically mild AD. Intervention involved a complete evaluation performed by a dental surgeon, and included a clinical questionnaire; research diagnostic criteria for temporomandibular disorders; the McGill pain questionnaire; oral health impact profile; decayed, missing and filled teeth index; and complete periodontal examination before and after the trial, which involved any dental treatment deemed necessary based on the findings of the initial evaluation. The dental treatment most frequently performed included periodontal treatment (scaling), extractions, and application of topical Nystatin (anti-fungal). The study found an increase in orofacial pain, followed by improvement in the mandibular function and periodontal indices in patients with mild AD after treatment. These improvements were maintained until the last evaluation after 6 months, and this was followed by a reduction in the functional cognitive impairment. The limitation of this study is that it lacked a bigger cohort and appropriate (non-AD) controls. The second observational cohort study was performed by Ide et al. (2016) who set out to test the hypothesis that circulating inflammatory cytokines due to periodontal disease bacteria were linked to greater rates of cognitive decline in clinical AD cases. The study recruited 59 participants with mild to moderate AD in which cognition and circulating inflammatory markers were tested. Fifty-two of the participants were followed-up at 6 months when they all underwent repeat assessment of their initial biomarkers for any changes. The findings of the study concluded that the presence of periodontitis at baseline was associated with a six-fold increase in the rate of cognitive decline in participants over the 6-month follow-up period. Periodontitis at baseline was also associated with a relative increase in the pro-inflammatory state over the 6-month follow-up. The authors concluded that periodontitis is associated with an increase in cognitive decline in AD . This study upheld the view linking cognitive decline with the body’s inflammatory responses. The major weakness of the study was the absence of participants with intact cognition as controls. A more recent report by Li et al. (2020) also supports a potential link with inflammatory cell counts as part of systemic inflammation due to periodontal disease and the risk of cognitive decline in the US elderly population. Table 13.1 summarizes the key evidence which supports the association between periodontitis and AD .
7 AD Brain Amyloid-β: A Potentially Shared Pool from Peripheral and Intracerebral Sources Towards a Blood Biomarker for Clinical AD
It has recently been suggested that periodontitis increases the levels of peripheral Aβ within gingival tissues and human serum (Kamer et al. 2015; Gil-Montoya et al. 2017; Nie et al. 2019; Leira et al. 2020). Blood-based biomarkers are very appealing as obtaining cerebrospinal fluid from patients is not ideal. Ashton et al. (2021) recently suggested that blood pTau231 has the potential to indicate early amyloid formation. This residue (Threonine231) phosphorylation in tau protein is also a site that P. gingivalis infection contributes to (Haditsch et al. 2020), and further supports a causative role for periodontitis in AD development.
7.1 The Inflammatory Hypothesis – A Potentially Shared Pathway
Inflammation is the inevitable consequence of an infectious episode in the body. This is better appreciated in the case of generalised periodontitis (due to polymicrobial aetiology). The existence of an intact blood-brain barrier avoids any impact of peripheral inflammation on the brain. Details of the blood-brain barrier will not be described here, as the reader is encouraged to read Singhrao and Olsen (2018) for related information. However, reports (Marques et al. 2013; Montagne et al. 2015, 2016; Halliday et al. 2016) have suggested that during ageing and manifestation of AD , the blood-brain barrier becomes defective. This may be the result of a compromised immune protection coupled with poor oral hygiene and increased circulating inflammatory mediators. These mediators are implicated with age-related defects (cardiovascular health) and with possible microbial components in vulnerable subjects. It is therefore plausible to suggest that pathogenic bacteria from chronic infections and their endotoxins do spread to other anatomical sites (Makiura et al. 2008; Bale et al. 2017; Poole et al. 2013; Rokad et al. 2017; Dominy et al. 2019; Demmer et al. 2020). This invariably impacts on glial cell activation. Subsequent exposure of oral microbial debris or their secondary products to the brain causes the already reactive microglial cells to ferociously up-regulate cytokine secretion. Based on the reviews by Olsen et al. (2017); Olsen and Singhrao (2019, 2020), there is an increasing body of evidence to support complement as a potential shared pathway for both periodontitis and AD . P. gingivalis has developed impressively successful strategies for immune evasion in the periodontal pocket (Hajishengallis 2011). This is because P. gingivalis has to evade immune-mediated killing to survive and yet requires inflammation in order to obtain nutrients to flourish. This may be the reason for how this bacterium has resolved this predicament whilst also benefiting its companion species within the microbial community as a whole, and particularly under inflammatory environments to compete for dominance (Hajishengallis 2011; Singhrao et al. 2015). An imbalance in complement activity may influence dysbiosis of the host’s microbiome. We will not describe the complement system in detail as it is a subject of its own and the reader is directed to excellent reviews (Morgan and Harris 2015; Olsen et al. 2017).
Complement evasion in periodontitis nearly always involves gingipains, the digestive enzymes specific to P. gingivalis . These enzymes break down the host’s protective barriers enabling the pathogen to penetrate periodontal tissues. In doing so, they switch on the host’s immune responses. Gingipains have dual functionality and can exert dose-dependent biphasic effects on complement activation. At low concentrations they can activate complement, which have the advantage of eliminating complement sensitive commensals which may compete with P. gingivalis for niche space and nutrients. In addition, P. gingivalis is relatively resistant to complement-mediated opsonisation and killing, and is intrinsically resistant to the lytic action of complement. Conversely at high concentration, P. gingivalis is likely to be established, and so gingipains can inhibit bactericidal activity of complement thus preventing opsonisation of complement-sensitive bacteria promoting a mixed species biofilm . Sustained complement activation is a potent driver of inflammation in the body, including the brain. Inappropriate complement activity also plays a part in AD pathogenesis.
GWAS has identified four possible genes linking complement to AD . These include Complement and sub-complement 1 s (C1s), Complement receptor 1 (CR1), Clusterin and Complement component 9 (C9). The concern is that the brain does not have a traditional lymphatic drainage system, meaning an efficient complement system (which also acts as a pathway for clearing debris) is essential for the clearance of damaged cerebral tissue. Defective complement genes may disable phagocytic activity of local microglia resulting in ineffective clearance of proteins like Aβ and NFTs. Two clearance systems, unique to the brain, have been described. Firstly, the glymphatic system, a form of brain cleansing system that works better during night sleep (Iliff et al. 2013; Nedergaard 2013). Hence adequate sleep is implicated for better brain function. The second system is the intramural periarterial drainage pathway. This clears solutes from the brain to the periphery along arterial smooth muscle cell basement membranes. This works more efficiently during the daytime. For more information on these clearance pathways in relation to bacterial products see Singhrao and Olsen (2018).
It is becoming clear that the susceptibility gene APOE є4 also exerts its own risk for developing AD via inflammation mediated by the classical complement pathway linked to deregulating C1q to keep the classical complement pathway activated (Yin et al. 2019). This has an impact on further inflammatory responses via cytokine liberation by activated monocytes/macrophages/microglia (Ihara and Yamamoto 2016). Interestingly, a combined action of inappropriate complement activity can instruct microglia to over-prune synapses and this has implications in loss of synapses and memory (Vasek et al. 2016).
It has been demonstrated that susceptibility genes, such as APOE, can exert adverse effects on the host in combination with environmental factors, which are often controllable or modifiable. Hence controlling the environmental factors may exert a protective effect on cognitive functioning. This appears to be the case in the human AD interventional study carried out by Ide et al. (2016). The inflammatory burden is often described as the mechanism of risk; and certainly the impact of any lifestyle modification which has an anti-inflammatory effect will be significant for reducing both AD and periodontitis.
8 Therapeutic Advances – Are We Nearly There Yet?
8.1 Periodontal Therapy
Current therapies for periodontal treatment include non-surgical instrumentation to disrupt the biofilm and remove calculus, root surface instrumentatio and, removal of plaque retention factors (such as overhangs on restorations). Non-surgical therapy must be combined with changing the patient’s behaviour and improving oral hygiene measures for it to succeed. More complex and advanced periodontal therapy include full mouth disinfection, local antimicrobial application, systemic antibiotic therapy , host modulation therapy , and advanced surgical techniques such as flap surgery, soft tissue grafts, bone grafting, guided tissue regeneration and tissue stimulating proteins. By managing periodontal disease, we can keep the numbers of pathogenic bacteria in the periodontal pocket low. This should prevent their spread.
8.2
8.3 AD Treatment Based on Cholinesterase Inhibitors
For AD, potential treatments were developed based on the rationale that neurodegeneration led to neurotransmitter deficit, in particular of the cholinergic system. Cholinesterase inhibitors approved for treatment of mild to moderate AD include tacrine (First Horizon Pharmaceuticals), which causes liver toxicity and is therefore rarely prescribed, donepezil (Pfizer) (also approved for severe AD ); rivastigmine (Novartis) and galantamine (Janssen). Certainly, cholinesterase inhibitors are currently still the main drug administered to AD patients. Unfortunately, this treatment is proving to be inadequate as cholinesterase inhibitors do not slow the progression of AD symptoms (Massoud and Gauthier 2010).
8.4 AD Treatment Based on Targeting Aβ
According to the Amyloid Cascade Hypothesis (Hardy and Selkoe 2002) the Aβ plaque lesions of AD were considered to be the cause of AD . Although this has subsequently been shown to be untrue, this hypothesis still remains influential in the field of AD research because of its link with the Aβ diagnostic burden and disease progression. Therapies were based on two main aspects of the amyloid cascade hypothesis. Firstly, the idea of inhibiting α-, β- and γ-secretases was explored (Nunan and Small 2000). This proved not to be successful, but formed the basis for exploration of anti-Aβ immunotherapy to solubilise aggregated Aβ, thus reducing it in the AD brain. Two anti-Aβ drugs, gantenerumab and solanezumab, were designed to help remove excess Aβ in the brain. For a more comprehensive read on the anti-Aβ based therapies the reader should consult (Kozin et al. 2018; van Dyck 2018). What is clear is that altering the course of AD by protease inhibitors of the amyloid cascade and/or mopping up the already aggregated Aβ in the form of plaques has not been successful in humans. This has opened the field of therapy to other related strategies.
8.5 AD Treatment Based on Targeting Tau Protein
NFT formation and its spread in the AD brain correlate with symptom severity and neurodegeneration, thus providing the rationale for developing therapies that target tau in its abnormal state. This formed the basis for developing inhibitors for kinases that phosphorylate tau and/or immunotherapy to control aggregation of abnormally phosphorylated tau, or to stabilise microtubule assembly within neurons. Of these, free cytosolic hyperphosphorylated tau fragments are implicated in the spreading of tau pathology. Researchers have shown that extracts of cytosolic tau from AD brains reproduce tau pathology when injected into mice brains. Therapeutically, these approaches also have their challenges in terms of being toxic and/or a lack of efficacy. Drugs based on tau toxicity are also being developed but this is not our niche. However, the interested reader is directed to some comprehensive references on this subject (Congdon and Sigurdsson 2018; Sayas 2020).
8.6 AD Treatment Based on Targeting Neuroinflammation
Based on the observation of neuroinflammation within the pathogenesis of AD, it became of great interest to assess the impact of Non-Steroidal Inflammatory Drugs (NSAIDs) on the development and progresion of AD . Development of specific NSAIDs along with discovery of NSAID targets in diseased brains led to a multitude of animal and human trials. Many NSAIDs have been found to cross the blood-brain barrier (Parepally et al. 2006) and cyclo-oxygenase (COX)-mediated prostaglandin (PG) synthesis has led to an increase in the understanding of healthy and diseased brain function. However, it has been shown that neuroinflammation and neurodegenerative disease are complex and NSAID efficacy on pathology and behaviour vary according to the specific disease, the region of the brain, and study design (Moore et al. 2010).
Trials assessing the effect of COX-2 inhibitors on the cognitive decline seen in AD, showed initial evidence that indomethacin might have beneficial effects (Rogers et al. 1993). Unfortunately, large scale clinical trials assessing cognitive outcomes following NSAID administration have been disappointing (de Jong et al. 2008; Pasqualetti et al. 2009) and the general consensus was that NSAID treatment becomes ineffective once memory declines and the associated pathologies have already developed (McGeer and McGeer 2007; Townsend and Practico 2005).
8.7 AD Treatment Based on Targeting P. gingivalis
The research by Dominy et al. (2019) highlighted the potential importance of gingipains which have been shown to mediate the toxicity of P. gingivalis in epithelial and endothelial cells (Stathopoulou et al. 2009; Sheets et al. 2005; Kinane et al. 2012). They may also be implicated as narrow spectrum virulence targets (Guo et al. 2010; Travis and Potempa 2000; Clatworthy et al. 2007). Blocking gingipain activity with short peptide analogues reduces the virulence of P. gingivalis (Kadowaki et al. 2004).
Dominy et al. (2019) hypothesised that P. gingivalis acts in AD pathogenesis via gingipain secretion to promote neuronal damage. This was supported with experimental evidence demonstrating that gingipain immunoreactivity in AD human brain was significantly greater than in non-AD brains; and correlated with the presence of gingipains to the increase in tau load and tau NFTs. As a result of this evidence, a potent, selective, brain-penetrating, small-molecule gingipain inhibitor was developed which, in vivo, shows promise with the potential to act as a disease modifier for AD (Ryder 2020).
Based on the ground-breaking research from Poole et al. (2013) and Dominy et al. (2019), the US pharmaceutical company have developed COR388 (Cortexyme), a novel virulence factor inhibitor, that targets gingipains from P. gingivalis . This drug is in phase 2/3 of clinical trials with promising results (Ryder 2020). It is thought to be able to block brain infiltration of P. gingivalis along with the subsequent downstream pathology of AD , including Aβ production, neuroinflammation and neurodegeneration. If effective, it is unclear how this drug will be administered on daily basis. However, speculation suggests it may be an adjunctive management.
8.8 AD Treatment Based on Managing Lifestyles
The Lancet Commission on Dementia Prevention, Intervention and Care have recognised elements of lifestyle such as smoking, obesity, excessive alcohol consumption, physical inactivity, depression and low social contact as potentially modifiable risk factors for dementia (Ngandu et al. 2015). Thus, making better lifestyle choices could prevent or delay dementia. This was supported by findings from the large-scale randomised controlled trial; the Finnish Geriatric Intervention Study to Prevent Cognitive Impairment and Disability (FINGER) which investigated multi-domain intervention in the at-risk segment of the general elderly population. The intervention group, which received risk reducing modifications to lifestyle patterns including diet, exercise, cognitive training and vascular risk monitoring, showed maintenance of, or improved cognitive functioning and a reduction in cognitive decline after the 2-year follow-up. This was the first robust large-scale trial which showed that modifying lifestyle factors could be beneficial to cognition (Ngandu et al. 2015). Although this may not be directly extrapolated to AD patients, the study population, being 60–77 year olds, could be in a pre-dementia state. Therefore, further research involving multi-modal intervention, designed to track participants over longer periods; assessing the level of impact of each factor on disease progression would be useful. Potential therapeutic effects can certainly be observed in pre-symptomatic and pre-dementia stages.
In summary, the importance of lifestyle factors is being increasingly recognized and investigated. Table 13.2 highlights current approaches to treating AD, aimed at maintaining mental function, managing behavioural symptoms, and slowing down the onset of symptoms of the disease.
Finally, poor sleep has been implicated with AD risk (Sprecher et al. 2017). Adequate sleep in advancing age is necessary for retaining memory and for effective functioning of the glymphatic system (Iliff et al. 2013; Nedergaard 2013). In 2019, The World Health Organisation has recognised poor sleep as a risk factor for general health and made recommendations of minimum hours of sleep per night for specific age groups.
The question as to whether we are there yet remains open, despite great efforts being made towards developing fundamental treatments and a push to adopting better lifestyles. In the absence of an adequate treatment for AD, the World Dementia Council (https://worlddementiacouncil.org/), National Institutes of Health (NIH) and Alzheimer’s Research UK (ARUK) (www.alzheimersresearchuk.org/ARUK), lend their support towards prevention by modifiable risk factor identification and implementation during life to stay cognitively healthy for a happier and longer life.
9 Conclusions
We set out to understand how one common, clinical condition such as generalised periodontitis becomes a potential risk factor for the development of AD and discussed the factors related to oral microbial infection and the behavioural component. The GAIN trials will demonstrate if this is a causative relationship.
The onus is now on both the individual and the dental profession and emphasises the key roles every individual and dentist have in the prevention or delay of AD development. This gives added importance to the need for adequate control and management of periodontal disease, as well as the prevention message that patients receive. Oral health monitoring should complement other lifestyle management commissions such as those conducted in the FINGER trial. Periodontal disease, its pathogens and their virulence factors, have now been linked to many systemic diseases, and our role in maintaining not only the dental health, but now the general health of our patient base has become a key factor in enabling our patients to age ‘successfully’ and enjoy a healthy life.
Abbreviations
- AD:
-
Alzheimer’s disease
- AGE:
-
advanced glycation end products
- APOE є4:
-
apolipoprotein E, allele 4
- ApoE−/−:
-
apolipoprotein knockout
- APP:
-
amyloid precursor protein
- APP-Tg:
-
APP-transgenic
- ARUK:
-
Alzheimer’s Research UK
- Aβ:
-
Amyloid-beta
- C9/C1q and C1s:
-
complement component/sub-complement
- CR1:
-
complement receptor 1
- DNA:
-
deoxyribonucleic acid
- ELISA:
-
enzyme-linked immunoassay
- GI:
-
gastrointestinal tract
- GWAS :
-
Genome-Wide-Association Studies
- IL:
-
interleukins
- IL-1R:
-
interleukin-1 receptor
- LPS:
-
lipopolysaccharide
- NFTs:
-
neurofibrillary tangles
- NF-ϰB:
-
nuclear factor kappaB
- NIH:
-
National Institutes of Health
- NSAIDs:
-
Non-Steroidal Inflammatory Drugs
- OMVs:
-
outer membrane vesicles
- PG:
-
prostaglandin
- PHF:
-
paired helical filaments
- RT-PCR:
-
reverse transcriptase-polymperase chain reaction
- TLRs:
-
toll-like receptors
- TNFα:
-
tumour necrosis factor-α
References
Adaikkan C, Middleton SJ, Marco A et al (2019) Gamma entrainment binds higher-order brain regions and offers neuroprotection. Neuron. https://doi.org/10.1016/j.neuron.2019.04.011
Ashton NJ, Pascoal TA, Karikari TK et al (2021) Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. https://doi.org/10.1007/s00401-021-02275-6
Bahar B, Singhrao SK (2021) An evaluation of the molecular mode of action of trans-resveratrol in the Porphyromonas gingivalis lipopolysaccharide challenged neuronal cell model. Mol Biol Rep 48(1):147–156
Bahrani-Mougeot FK, Paster BJ, Coleman S et al (2008) Diverse and novel oral bacterial species in blood following dental procedures. J Clin Microbiol 46:2129–2132
Bale BF, Doneen AL, Vigerust DJ (2017) High-risk periodontal pathogens contribute to the pathogenesis of atherosclerosis. Postgrad Med J 93:215–220
Beutler B (2000) Endotoxin, toll-like receptor 4, and the afferent limb of innate immunity. Curr Opin Microbiol 3(1):23–28
Beydoun MA, Beydoun HA, Hossain S et al (2020) Clinical and bacterial markers of periodontitis and their association with incident all-cause and Alzheimer’s disease dementia in a large national survey. J Alzheimers Dis 75(1):157–172
Boche D, Perry VH, Nicoll JAR (2013) Activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39:3–18
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Thal DR, Ghebremedhin E et al (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960–969
Brayne C, Calloway P (1990) The association of education and socioeconomic status with the mini mental state examination and the clinical diagnosis of dementia in elderly people. Age Ageing 19:91–96
Caton JG, Armitage G, Berglundh T et al (2018) A new classification scheme for periodontal and peri-implant diseases and conditions – introduction and key changes from the 1999 classification. J Clin Periodontol 45(20):S1–S8. https://doi.org/10.1111/jcpe.12935
Cerutti-Kopplin D, Feine J, Padilha DM et al (2016) Tooth loss increases the risk of diminished cognitive function: a systematic review and meta-analysis. JDR Clin Trans Res 1(1):10–19
Chalmers JM, Hodge C, Fuss JM et al (2002) The prevalence and experience of oral diseases in Adelaide nursing home residents. Aust Dent J 47(2):123–130
Chen E, Miller GE (2013) Socioeconomic status and health: mediating and moderating factors. Annu Rev Clin Psychol 9:723–749
Chen C-K, Wu Y-T, Chang Y-C (2017) Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population based, matched-cohort study. Alzheimers Res Ther 9:56. https://doi.org/10.1186/s13195-017-0282-6
Choi RW, Cheng Z, Schekman R (2012) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci U S A 109(30):E2077–E2082
Clapp M, Aurora N, Herrera L et al (2017) Gut microbiota’s effect on mental health: the gut-brain axis. Clin Pract 7(4):987. https://doi.org/10.4081/cp.2017.987
Clatworthy AE, Pierson E, Hung DT (2007) Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol 3:541–548
Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14(7):399–415
Corder EH, Saunders AM, Strittmatter WJ et al (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123):921–923
Cummings J, Lee G, Ritter A et al (2020) Alzheimer’s disease drug development pipeline. Alzheimers Dement (N Y) 6(1):e12050. https://doi.org/10.1002/trc2.12050
de Bont N, Netea MG, Demacker PN (1999) Apolipoprotein E knock-out mice are highly susceptible to endotoxemia and Klebsiella pneumoniae infection. J Lipid Res 40:680–685
de Jong D, Jansen R, Hoefnagels W et al (2008) No effect of one-year treatment with indomethacin on Alzheimer’s disease progression: a randomized controlled trial. PLoS One 3(1):e1475. https://doi.org/10.1371/journal.pone.0001475
de Rolim TS, Fabri GM, Nitrini R et al (2014) Evaluation of patients with Alzheimer’s disease before and after dental treatment. Arq Neuropsiquiatr 72(12):919–924
Demmer RT, Norby FL, Lakshminarayan K et al (2020) Periodontal disease and incident dementia: The Atherosclerosis Risk in Communities Study (ARIC). Neurology 95(12):e1660–e1671. https://doi.org/10.1212/WNL.0000000000010312
Dietrich T, Ower P, Tank M et al (2019) Periodontal diagnosis in the context of the 2017 classification system of periodontal diseases and conditions - implementation in clinical practice. Br Dent J 226(1):16–22
Ding Y, Ren J, Yu H et al (2018) Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing 215:6. https://doi.org/10.1186/s12979-017-0110-7
Dioguardi M, Crincoli V, Laino L et al (2020) The role of periodontitis and periodontal bacteria in the onset and progression of Alzheimer’s disease: a systematic review. J Clin Med 9(2):495. https://doi.org/10.3390/jcm9020495
Dominy SS, Lynch C, Ermini F et al (2019) Porphorymonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small molecule inhibitors. Sci Adv 5(1). https://doi.org/10.1126/sciadv.aau3333
Dugger BN, Dickson DW (2017) Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol 9(7):a028035. https://doi.org/10.1101/cshperspect.a028035
Eke PI, Dye BA, Wei L et al (2015) Update on prevalence of periodontitis in adults in the United States: NHANES 2009 to 2012. J Periodontol 86(5):611–622. https://doi.org/10.1902/jop.2015.140520
Emery DC, Shoemark DK, Batstone TE et al (2017) 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front Aging Neurosci 9:195. https://doi.org/10.3389/fnagi.2017.00195
Farhad SZ, Amini S, Khalilian A et al (2014) The effect of chronic periodontitis on serum levels of tumor necrosis factor-alpha in Alzheimer disease. Dent Res J (Isfahan) 11(5):549–552
Forner L, Larsen T, Kilian M et al (2006) Incidence of bacteremia after chewing, tooth brushing, and scaling in individuals with periodontal inflammation. J Clin Periodontol 33:401–407
Genin E, Hannequin D, Wallon D et al (2011) APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 16:903–907
Gil-Montoya JA, Barrios R, Santana S et al (2017) Association between periodontitis and amyloid β peptide in elderly people with and without cognitive impairment. J Periodontol 88:1051–1058
Goedert M, Klug A, Crowther RA (2006) Tau protein, the paired helical filament and Alzheimer’s disease. J Alzheimers Dis 9:195–207
Grossi SG, Genco RI, Machtei EE et al (1995) Assessment of risk for periodontal disease II. Risk indicators for alveolar bone loss. J Periodontol 66:23–29
Grundke-Iqbal I, Iqbal K, Quinlan M et al (1986) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261:6084–6089
Guo Y, Nguyen KA, Potempa J (2010) Dichotomy of gingipains action as virulence factors: from cleaving substrates with the precision of a surgeon’s knife to a meat chopper-like brutal degradation of proteins. Periodontol 2000 54:15–44
Haditsch U, Roth T, Rodriguez L et al (2020) Alzheimer’s disease-like neurodegeneration in Porphyromonas gingivalis infected neurons with persistent expression of active gingipains. J Alzheimers Dis 75:1361–1376
Haffajee AD, Socransky SS, Dzink JL et al (1988) Clinical, microbiological and immunological features of subjects with destructive periodontal diseases. J Clin Periodontol 15:240–246
Hajishengallis G (2011) Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosci 53(3):233–240
Hajishengallis G, Darveau RP, Curtis MA (2012) The keystone-pathogen hypothesis. Nat Rev Microbiol 10(10):717–725
Halliday MR, Rege SV, Ma Q et al (2016) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36:216–227
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297(5580):353–356
Hasturk H, Kantarci A, Van Dyke TE (2012) Oral inflammatory diseases and systemic inflammation: role of the macrophage. Front Immunol 3:1–11
Heneka MT, Carson MJ, El Khoury J et al (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14(4):388–405
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH (2009) Systemic inflammation and disease progression in Alzheimer’s disease. Neurology 73:768–774
Holt SC, Ebersole JL (2005) Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia: the “red complex”, a prototype polybacterial pathogenic consortium in periodontitis. Periodontol 2000 38:72–122
Hook V, Schechter I, Demuth HU et al (2008) Alternative pathways for production of beta-amyloid peptides of Alzheimer’s disease. J Biol Chem 389:993–1006
How KY, Song KP, Chan KG (2016) Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol 7:53. https://doi.org/10.3389/fmicb.2016.00053
Hubacek JA, Peasey A, Pikhart H et al (2010) APOE polymorphism and its effect on plasma C-reactive protein levels in a large general population sample. Hum Immunol 71(3):304–308
Hyman BT, Phelps CH, Beach TG et al (2012) National Institute on Aging–Alzheimer’s association guidelines for the Neuropathologic assessment of Alzheimer’s disease. Alzheimer’s & Dement 8(1):1–13
Ide M, Harris M, Stevens A et al (2016) Periodontitis and cognitive decline in Alzheimer’s disease. PLoS One 11(3):e0151081. https://doi.org/10.1371/journal.pone.0151081
Ihara M, Yamamoto Y (2016) Emerging evidence for pathogenesis of sporadic cerebral small vessel disease. Stroke 47:554–560
Ilieviski V, Zuchowska PK, Green SJ et al (2018) Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One. https://doi.org/10.1371/journal.pone.02049418
Iliff JJ, Lee H, Yu M et al (2013) Brain wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123:1299–1309
Ishida N, Ishihara Y, Ishida K et al (2017) Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis 6(3):15. https://doi.org/10.1038/s41514-017-0015-x
Kadowaki T, Baba A, Abe N et al (2004) Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol Pharmacol 66:1599–1606
Kamer AR, Craig RG, Pirraglia E et al (2009) TNFα and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J Immunol 216(1–2):92–97
Kamer AR, Pirraglia E, Tsui W et al (2015) Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging 36(2):627–633
Kamma JJ, Nakou M, Baehni PC (1999) Clinical and microbiological characteristics of smokers with early onset periodontitis. J Periodontal Res 34:25–33
Kang J, Lemaire HG, Unterbeck A et al (1987) The precursor of Alzheimer’s disease amyloid A4protein resembles a cell-surface receptor. Nature 325:733–736
Kinane DF, Marshall GJ (2001) Periodontal manifestations of systemic disease. Aust Dent J 46:2–12
Kinane JA, Benakanakere MR, Zhao J et al (2012) Porphyromonas gingivalis influences actin degradation within epithelial cells during invasion and apoptosis. Cell Microbiol 14:1085–1096
Kondo K, Niino M, Shido K (1994) A case-control study of Alzheimer’s disease in Japan-significance of life-styles. Dementia 5:314–326
Kozin SA, Barykin EP, Mitkevich VA et al (2018) Anti-amyloid therapy of Alzheimer’s disease: current state and prospects. Biochemistry (Mosc) 83(9):1057–1067
Leira Y, Iglesias-Rey R, Gómez-Lado N et al (2019) Porphyromonas gingivalis lipopolysaccharide-induced periodontitis and serum amyloid-beta peptides. Arch Oral Biol 99:120–125
Leira Y, Carballo Á, Orlandi M et al (2020) Periodontitis and systemic markers of neurodegeneration: a case-control study. J Clin Periodontol 47(5):561–571
Li A, Chen Y, van der Sluis LWM et al (2020) Mediation analysis of white blood cell count on the association between periodontal inflammation and digit symbol substitution test scoring cognitive function among older U.S. Adults. J Gerontol A Biol Sci Med Sci 76:glaa223. https://doi.org/10.1093/gerona/glaa223
Lin JW, Chang CH, Caffrey JL (2020) Feature article: examining the association between oral health status and dementia: a nationwide nested case-controlled study. Exp Biol Med (Maywood) 245(3):231–244
Lockhart PB, Brennan MT, Sasser HC et al (2008) Bacteremia associated with tooth brushing and dental extraction. Circulation 117:3118–3125
Makiura N, Ojima M, Kou Y et al (2008) Relationship of Porphyromonas gingivalis with glycemic level in patients with type 2 diabetes following periodontal treatment. Oral Microbiol Immunol 23(4):348–351
Marden JR, Tchetgen EJ, Kawachi I et al (2017) Contribution of socioeconomic status at three life course periods to late life memory function and decline: early and late predictors of dementia risk. Am J Epidemiol 186:805–814
Marques F, Sousa JC, Sousa N et al (2013) Blood-brain-barriers in aging and in Alzheimer’s disease. Mol Neurodegener 8:38. https://doi.org/10.1186/1750-1326-8-38
Massoud F, Gauthier S (2010) Update on the pharmacological treatment of Alzheimer’s disease. Curr Neuropharmacol 8(1):69–80
McGeer PL, McGeer EG (2007) NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiol Aging 28:639–647
Montagne A, Barnes SR, Sweeney MD (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302
Montagne A, Nation DA, Pa J et al (2016) Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol (Berl) 131:687–707
Moore AH, Bigbee MJ, Boynton GE et al (2010) Non-steroidal anti-inflammatory drugs in Alzheimer’s disease and Parkinson’s disease: reconsidering the role of neuroinflammation. Pharmaceuticals (Basel) 3(6):1812–1841
Moretti EW, Morris RW, Podgoreanu M et al (2005) APOE polymorphism is associated with risk of severe sepsis in surgical patients. Perioperative Genetics and Safety Outcomes Study (PEGASUS) Investigative Team. Crit Care Med 33(11):2521–2526
Morgan BP, Harris CL (2015) Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov 14(12):857–877
Morris R (1984) Developments of a water-maze procedure for studying spatial learning in the rat. J Neurosci Methods 11:47–60
Nedergaard M (2013) Neuroscience. Garbage truck of the brain. Science 340(6140):1529–1530
Ngandu T, Lehtisalo J, Solomon A et al (2015) A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): a randomised controlled trial. Lancet 385(9984):2255–2263
Nie R, Wu Z, Ni J et al (2019) Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J Alzheimers Dis 72:479–494
Noble JM, Borrell LN, Papapnou PN et al (2009) Periodontitis is associated with cognitive impairment among older adults: analysis of NHSNES-III. J Neurol Neurosurg Psychiatr 80:1206–1211
Nunan J, Small DH (2000) Regulation of APP cleavage by alpha-, beta- and gamma-secretases. FEBS Lett 483(1):6–10
Olsen I, Singhrao SK (2015) Can oral infection be a risk factor for Alzheimer’s disease? J Oral Microbiol 7:29143
Olsen I, Singhrao SK (2019) Is there a link between genetic defects in the complement cascade and Porphyromonas gingivalis in Alzheimer’s disease? J Oral Microbiol 12:167648
Olsen I, Singhrao SK (2020) Interaction between genetic factors, Porphyromonas gingivalis and microglia to promote Alzheimer’s disease. J Oral Microbiol 12(1):1820834
Olsen I, Lambris JD, Hajishengallis G (2017) Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J Oral Microbiol 9(1):1340085
Parepally JMR, Mandula H, Smith QR (2006) Brain uptake of nonsteroidal anti-inflammatory drugs: ibuprofen, flurbiprofen, and indomethacin. Pharm Res 23:873–881
Pasqualetti P, Bonomini C, Dal Forno G et al (2009) A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin Exp Res 21:102–110
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6:193–201
Poole S, Singhrao SK, Kesavalu L et al (2013) Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis 36:665–677
Poole S, Singhrao SK, Chukkapalli S et al (2015) Active infection of Porphorymonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J Alzheimers Dis 43(1):67–80
Riviere GR, Riviere KH, Smith KS (2002) Molecular and immunological evidence of oral Treponema in the human brain and their association with Alzheimer’s disease. Oral Microbiol Immunol 17:113–118
Rogers J, Kirby LC, Hempelman SR et al (1993) Clinical trial of indomethacin in Alzheimer’s disease. Neurology 43:1609–1611
Rokad F, Moseley R, Hardy RS et al (2017) Cerebral oxidative stress and microvasculature defects in TNF-α expressing transgenic and Porphyromonas gingivalis-infected ApoE−/− mice. J Alzheimers Dis 60:359–369
Rossor MN, Newman S, Frackowiak RS et al (1993) Alzheimer’s disease families with amyloid precursor protein mutations. Ann N Y Acad Sci 695:198–202
Russ TC, Stamatakis E, Hamer M et al (2013) Socioeconomic status as a risk factor for dementia death: individual participant meta-analysis of 86,508 men and women from UK. Br J Psychiatry 203:10–17
Ryder MI (2020) Porphyromonas gingivalis and Alzheimer disease: recent findings and potential therapies. J Periodontol 91(Suppl 1):S45–S49. https://doi.org/10.1002/JPER.20-0104
Saunders AM, Strittmatter WJ, Schmechel D et al (1993) Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43(8):1467–1472
Sayas CL (2020) Chapter 10: Tau-based therapies for Alzheimer’s disease: promising novel neuroprotective approaches. In Neuroprotection in Autism, Schizophrenia and Alzheimer’s Disease. Elsevier Inc., pp 245–272. https://doi.org/10.1016/B978-0-12-814037-6.00005-7
Sheets SM, Potempa J, Travis J et al (2005) Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect Immun 73:1543–1552
Singh A, Rouxel P, Watt RG et al (2013) Social inequalities in clustering of oral health related behaviors in a national sample of British adults. Prev Med 57:102–106
Singhrao SK, Olsen I (2018) Are Porphyromonas gingivalis outer membrane vesicles microbullets for sporadic Alzheimer’s disease manifestation? J Alzheimers Dis Rep 2(1):219–228
Singhrao SK, Olsen I (2019) Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J Oral Microbiol 11(1):1563405
Singhrao SK, Harding A, Poole S, et al (2015) Porphyromonas gingivalis periodontal infection and its putative links with Alzheimer’s disease. Mediators Inflamm 2015:137357. https://doi.org/10.1155/2015/137357
Socransky SS, Haffajee AD, Cugini MA et al (1998) Microbial complexes in subgingival plaque. J Clin Periodontol 25(2):134–144
Sparks Stein P, Steffen MJ, Smith C et al (2012) Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimer’s Dementia 8(3):196–203
Sprecher KE, Koscik RL, Carlsson CM et al (2017) Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology 89:445–453
Stathopoulou PG, Galicia JC, Benakanakere MR et al (2009) Porphyromonas gingivalis induce apoptosis in human gingival epithelial cells through a gingipain-dependent mechanism. BMC Microbiol 9:107
Stein PS, Desrosiers M, Donegan SJ et al (2007) Tooth loss, dementia and neuropathy in the Nun study. J Am Dent Assoc 138(10):1314–1322
Strandwitz P (2018) Neurotransmitter modulation by the gut microbiota. Brain Res 1693(Pt B):128–133. https://doi.org/10.1016/j.brainres.2018.03.015
Strandwitz P, Kim KH, Terekhova D et al (2019) GABA-modulating bacteria of the human gut microbiota. Nat Microbiol 4(3):396–403. https://doi.org/10.1038/s41564-018-0307-3
Tomás I, Diz P, Tobias A et al (2012) Periodontal health status and bacteraemia from daily oral activities: systematic review/meta-analysis. J Clin Periodontol 39:213–228
Townsend KP, Practico D (2005) Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J 19(12):1592–1601
Travis J, Potempa J (2000) Bacterial proteinases as targets for the development of second-generation antibiotics. Biochim Biophys Acta 1477:35–50
Tsoi LM, Wong KY, Liu YM et al (2007) Apoprotein E isoform-dependent expression and secretion of pro-inflammatory cytokines TNF-α and IL-6 in macrophages. Arch Biochem Biophys 460:33–40
Tzeng NS, Chung CH, Yeh CB et al (2016) Are chronic periodontitis and gingivitis associated with dementia? A nationwide, retrospective, matched-cohort study in Taiwan. Neuroepidemiology 47:82–93
van Dyck CH (2018) Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: pitfalls and promise. Biol Psychiatry 83(4):311–319
van Oijen M, de Jong FJ, Witteman JC et al (2007) Atherosclerosis and risk for dementia. Ann Neurol 61:403–410
Vasek MJ, Garber C, Dorsey D et al (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534(7608):538–543
Velsko IM, Chukkapalli SS, Rivera MF et al (2014) Active invasion of oral and aortic tissues by Porphyromonas gingivalis in mice causally links periodontitis and atherosclerosis. PLoS One 9:e97811. https://doi.org/10.1371/journal.pone.0097811
Watts A, Crimmins EM, Gatz M (2008) Inflammation as a potential mediator for the association between periodontal disease and Alzheimer’s disease. Neuropsychiatr Dis Treat 4:865–876
Wu Z, Ni J, Liu Y et al (2017) Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun 65:350–361
Yin C, Ackermann S, Ma Z et al (2019) ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med 25(3):496–506
Zeng F, Liu Y, Huang W et al (2020) Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection. J Neurochem. https://doi.org/10.1111/jnc.15096
Zhang J, Yu C, Zhang X et al (2018) Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signalling pathway in C57BL/6 mice. J Neuroinflammation 15(1):37. https://doi.org/10.1186/s12974-017-1052-x
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Harding, A., Kanagasingam, S., Welbury, R., Singhrao, S.K. (2022). Periodontitis as a Risk Factor for Alzheimer’s Disease: The Experimental Journey So Far, with Hope of Therapy. In: Santi-Rocca, J. (eds) Periodontitis. Advances in Experimental Medicine and Biology, vol 1373. Springer, Cham. https://doi.org/10.1007/978-3-030-96881-6_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-96881-6_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-96880-9
Online ISBN: 978-3-030-96881-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)