
Abstract
Purpose of Review
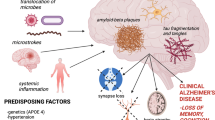
There is an increasing body of evidence from epidemiology and laboratory investigations demonstrating periodontal disease as a risk factor for dementia. In particular, Porphyromonas gingivalis infections in animal models suggest causal associations with Alzheimer’s disease (AD). This review focuses on how P. gingivalis infections promote the incidence of functional loss in AD.
Recent Findings
The risk of the sporadic form of AD doubles when periodontitis persists for ten or more years. AD differs from other forms of dementia in that the clinical signs together with the presence of amyloid-beta (Aβ) plaques and neurofibrillary tangles must be present at autopsy. P. gingivalis oral infections in mice have demonstrated all of the characteristic pathological and clinical features of AD following infection of the brain.
Summary
Multiple factors (inflammation, Aβ oligomers, and bacterial factors) are likely to disrupt neuronal communication channels (synapses) as a plausible explanation for the functional loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Longstanding periodontitis, formerly known as “chronic” periodontitis, has an adverse effect on a number of complex human diseases associated with prolonged inflammation [1,2,3]. Recent research has linked poor oral hygiene to neurological conditions that manifest with dementia. Currently, they include the sporadic form of Alzheimer’s disease (AD), and the Lewy body Parkinson’s disease (dementia) [4••, 5••, 6••]. Amyloid-beta (Aβ) plaques are central to all forms of dementia but are more critical to AD pathology and diagnosis. A significant body of literature considers the Aβ plaques of AD and the α-synuclein of Lewy bodies to be antimicrobial peptides that combat infections of the brain [7••, 8, 9••, 10]. This concept may provide vital clues to the occurrence of these neuropathological lesions and ultimately to its cause.
Porphyromonas gingivalis
Porphyromonas gingivalis is present in the oral cavity of all humans where it may or may not cause pathology. Recent research has implicated P. gingivalis as the keystone pathogen of periodontitis, which is an inflammatory disease constituting a complex dysbiotic microbial community residing below the gum line, within periodontal “pockets.” P. gingivalis appears to translocate from the saliva to the subgingival location using neutrophils as “Trojan horses” in some individuals because clinical observations suggest that not everyone progresses to manifesting periodontal disease.
The mouth harbors a microbiome that in a healthy individual is a diverse collection of commensal bacteria. If the balance shifts towards more pathogenic species, they can cause oral diseases such as periodontitis. P. gingivalis can act as both a commensal and a pathogen, depending on the host’s response and composition of the microbiome.
Outside of the mouth, there is evidence that P. gingivalis can infiltrate the brain. Our group found evidence of P. gingivalis infection in the brains of people with AD [11••]. Further work has reinforced this link and shown evidence that P. gingivalis can replicate the hallmark lesions and functional loss of AD in mice [12••, 13••, 14••, 15••, 16••].
Alzheimer’s Disease
AD is an end-of-life stage and the most common example of dementia. The cardinal clinical signs are cognitive decline with deterioration in memory. The hippocampus is the region of the brain where memory is processed and the functional loss has been associated with the death of neurons in specific regions of the brain related to memory. AD has a long preclinical phase (20 years) with the duration of suffering lasting on average for 8–10 years and longer [17]. At the preclinical stage of the illness, the individual may not seek medical help. Usually, a family member or the caregiver of the person with declining cognition and memory may voice their concern to a health care professional. This may be their general medical practitioner (GP) or a district nurse. The first stage in exploring this health complaint is for the caregiver to take the person (with suspected dementia signs) to his/her GP. The GP will then refer the person on to a memory service to establish a more formal clinical diagnosis and initiate treatment and support. The final diagnosis of AD rests with both the clinical history together with demonstration of the neuropathological occurrence of Aβ plaques and hyperphosphorylated tau protein binding to neurofibrillary tangles (NFTs) in a characteristic pattern in specific regions of the brain. AD neuropathology can co-exist with other neurological and/or vascular pathologies because it is not an isolated disease.
Plausible Cause of Alzheimer’s Disease and Lewy Body Dementia
The cause of the sporadic forms of the neurological diseases under discussion (AD and Parkinson’s disease with Lewy bodies) remains unclear. However, among others, the risk factors include aging and inheritance of the apolipoprotein E gene allele 4 (APOE є4) [18••, 19]. The APOE є4 susceptibility gene links with environmental risk factors that include the host’s dysbiotic oral microbiome [20•]. More recently, scientists found a new function for the APOE є4 in the classical complement cascade as a checkpoint by binding to a key activator, C1q [21••], and this has implications for inappropriate inflammation. P. gingivalis infections of the brain in apolipoprotein E gene knockout mice induced with periodontitis demonstrated excessive oxidative stress and inflammation [13••, 14••, 15••, 22•].
Lewy bodies are intra-neuronal cytoplasmic inclusions composed largely of synuclein lying within the pigmented neurons of the substantia nigra, the limbic system, and the cerebral cortex regions of the brain. The clinical symptoms of Parkinson’s disease in its purest form are tremor, immobility and rigidity of muscles. However, cognitive deficit occurs when Parkinson’s disease co-exists with dementia (Lewy body Parkinsonian dementia); see comment above related to mixed pathologies. Epidemiological investigations [4••, 5••] in a Taiwanese population have linked this to periodontal disease. As mentioned earlier, the Aβ protein of AD plaques and the α-synuclein within Lewy bodies are a form of broad-spectrum antimicrobial peptides, released following infection, including that caused by the periodontal pathogen P. gingivalis [7••, 8, 9••, 10, 14••]. If Aβ and α-synuclein represent the host’s response to a previous infection, it follows that these neurodegenerative diseases have causative associations with microbes during their development. This has given rise to the antimicrobial protection hypothesis [23••] linking infection as a plausible trigger for the sporadic form of AD. If this theory becomes widely accepted, then explaining the existing oxidative stress, the activated complement, the longstanding inflammation, and the defects in the blood-brain barrier (BBB) would be easy in the context of P. gingivalis infection [13••, 22•, 24••]. All of the above-mentioned signaling cascades and others (not included here) would enhance the role of Aβ as an antimicrobial peptide in killing the elusive invader(s) and/or the little understood brain’s own microbiome converting to a pathobiome. In addition, the elderly are unlikely to be immuno-privileged because the BBB defects in the 70+ year’s age group are associated with more rapid cognitive decline [25••] and could have implications for pathogen entry.
Plausible Cause of Cognitive Deficit
What actually causes the cognitive deficit during dementia onset is unclear, because the individual examples of dementia such as AD are seldom pure. However, the amyloid cascade hypothesis originally focused on Aβ deposits as a possible cause [26]. Subsequent immunological therapy to remove Aβ plaques from the brains of AD patients disproved the notion that insoluble Aβ deposits contribute to cognitive dysfunction [27••]. Prior to the amyloid hypothesis, the synaptic loss hypothesis of Terry et al. [28•] and Masliah et al. [29•] originated from the fact that specific neuronal loss may be due to synaptic loss. The revised version of the amyloid cascade hypothesis has incorporated soluble oligomeric Aβ in the synaptotoxicity and cognitive impairment theory [30••]. It is possible that there is close interplay between the mechanisms underlying these three hypotheses. After all, it is highly plausible that microbial debris, complement activation, inflammatory mediators, oligomeric Aβ, smaller tau peptides released by gingipains, and pathogen-activated inflammasomes [31••] can all act to disrupt synapses and result in cognitive deficit.
Relationship Between Periodontitis and AD
The idea of dementia being a risk factor for periodontitis is undisputable, but then, one would expect all demented individuals to have periodontitis by the time of death. Literature suggests the formerly known “chronic” periodontitis has a clearer relationship with a subgroup of AD cases [32,33,34,35, 36•]. Significant progress will only be made to find the actual direction of this relationship once we better understand the parameters that should be included and/or excluded from the investigation in case control and/or cohort studies. For example, we now understand that periodontitis only becomes a risk factor for AD development some 10 years after it is diagnosed [37••, 38••]. This would imply that studies conducted in less than 10-year cohort analysis would provide inconclusive results [39]. One suggested risk of developing AD is having fewer remaining teeth (loss of up to nine teeth) in early to mid-life due to periodontitis [30••, 40], resulting from longstanding poor oral hygiene. For a more comprehensive discussion on the direction of the relationship between oral health and risk of developing AD, see Daly et al. [41•]. There is agreement that periodontitis doubles the risk for developing late-onset AD with an odds ratio of 2.2 (95% CI 1.1, 4.5) 10 years after its initial diagnosis [37••, 38••]. An interventional study on the periodontal treatment in AD patients [42•] indicated a plausible causal relationship in demented individuals. It is suggested that patients with early-stage dementia (at the time of visiting the memory clinic for initial diagnosis) show worsening oral hygiene [43•], implying that dementia may be the risk factor for periodontal disease in this group of patients. It is also suggested that if dental intervention were provided at the early stage of dementia onset, it would delay the speed of cognitive deterioration. Early intervention is important, and memory clinics should consider taking it on at the time of initial diagnosis [43•]. However, to confirm the direction of the relationship, more studies with larger cohorts are needed in the “at risk” subpopulation of individuals whose periodontitis co-exists with AD cases. In addition, future interventional studies should include participants who suffer from periodontitis approaching the risk age for dementia (pre 65-year age) for maximal impact on delaying the onset of AD.
Relationship of P. gingivalis with AD Development
As mentioned, P. gingivalis is considered a keystone pathogen in periodontitis and it is adept at manipulating the subgingival microbiome and the host’s immune system [44•, 45•, 46,47,48,49]. P. gingivalis is an intracellular pathogen that has been used to develop AD via periodontal infection in mice [13••, 14••]. The infection periodontal model of Ilievski et al. [14••] produced the AD defining hallmark lesions in the mouse brains (Aβ and phosphotau NFTs), a finding reproduced by Dominy et al. [50••]. Since the Ilievski and the Dominy models were of wild-type mice, there is a high probability that Aβ was cleaved from its precursor protein into various oligomer sizes following oxidative stress initiated by P. gingivalis, which in turn activated cathepsin B within the endo/lysosomes [22•, 51••]. This intracellular processing of Aβ agrees with the earlier report of Wu et al. [15••] showing that metabolic processing of the amyloid precursor protein after P. gingivalis lipopolysaccharide (LPS) was administered into cathepsin B sufficient mice. Other studies in which either P. gingivalis or its LPS was introduced supported the development of the AD-like clinical phenotype [15••, 16••, 52••] resulting in impaired spatial learning and memory. All these investigations support a causal relationship of periodontitis with the development of AD.
Mechanisms of Cognitive Deficit by P. gingivalis Infection
Soluble Oligomeric Aβ and BBB defects
In line with Dominy et al. [50••] confirming P. gingivalis genetic footprints (DNA) in the AD brains, in vivo infection models of periodontitis are recapitulating hallmark proteins and the emerging phenotype is supporting cognitive deficit [14••, 15••, 16••, 52••]. P. gingivalis produces two types of cysteine proteases (gingipains). They are the lysine-specific Kgp and the arginine-specific RgpA and RgpB gingipains [53]. A novel finding described by Dominy et al. [50••] is the capacity of these proteases to hydrolyze the biochemical structure of the protein tau. This opens up future avenues for research.
Gingipains activity has the potential to erode endothelial tight junction proteins [24••]. Cognitive deterioration due to BBB defects in the human elderly individuals is also documented [25••], and this may yet be another contributory factor in mice models displaying the AD-like clinical phenotype. In addition, it has been proposed that the soluble form of the olgomeric Aβ can interfere with synapses and contribute to cognitive deficit via complement activation [54••]. If this is the case, P. gingivalis may also contribute to cognitive deficit as it initiates the activation of complement via crosstalk with the Toll-like receptors (TLRs) [13••, 14••, 50••]. Activated complement causes overt synaptic pruning activity in microglia [54••], and this leads to synaptic loss leading to the development of poor memory.
Inflammation and Inflammatory Mediators in General
Numerous studies have shown that LPS from Gram-negative bacteria administered directly into either the peritoneum or the brain induce neuroinflammation in the form of glial cell activation [55] and when measured, the inflammatory response is accompanied by learning and memory impairment [56, 57]. This is a result of IL-1β secretion following peripheral challenge with LPS [58•]. It is in agreement with the Wu et al. [15••] hypothesis that systemic administration of P. gingivalis LPS leads to cognitive deficit following Aβ liberation in an IL-1β receptor-dependent pathway on neurons (also see [20•]). IL-1β cytokine is implicated in synaptic loss [59•, 60] with reduced long-term potentiation. This is a unit of memory [59•], supporting the role of this cytokine in deteriorating cognition.
P. gingivalis, Complement, and Immune Dysbiosis
Gingipains are virulence factors of great importance to the immune subversion activity of P. gingivalis [53]. In the context of the complement cascade, these proteases play a major role. P. gingivalis oral infection of apolipoprotein E−/− mice demonstrated complement activation in their brains [13••]. Activation of complement does take place in AD brains, where Aβ plaques are the suggested trigger [61]. If, according to the novel hypothesis of Allen [62] that Aβ senile plaques are miniature foci of bacterial biofilms, and that the antimicrobial protection theory of Moir et al. [23••] supporting the Aβ antimicrobial peptide idea, then the downstream immune activity triggering complement activation in AD brains does fit. Inappropriately activated complement compromises the function of healthy neurons, because of their inadequate shielding from protective proteins that rescue them from the non-specific mode of activity of this powerful innate immune signaling cascade [63]. During complement activation, release of several small proteins (opsonins) takes place, which then opsonize to neurons [13••]. Depending on the site of opsonin binding to the neuron, e.g., at the synaptic cleft), there remains a potential to disrupt the path of neuronal communication and give way to cognitive dysfunction. In addition, the continuation of this cyclic cascade will generate more cytokines and contribute to cognitive deficit (see above).
P. gingivalis infection continues to cleave complement components (C1-C5) through its gingipains activity and prevents both deposition of C3b on the bacterial surface and capture of the C4b binding protein [64,65,66,67,68]. By hijacking the complement regulator C4bp on the bacterial surface, P. gingivalis prevents assembly of the membrane attack complex and acquires the ability to regulate C3 convertase [66]. Accordingly, the gingipains do not only destroy complement through proteolytic degradation, but they also inhibit activation of complement by binding to the complement inhibitor C4bp [66]. This inhibits complement action and results in a local accumulation of the anaphylatoxin C5a [69•]. P. gingivalis also exerts C5 convertase-like enzymatic activity and exploits complement-TLR crosstalk to subvert host defenses and thus escape elimination from the host [45•]. Zhang et al. [52••] recently demonstrated that the mechanism by which P. gingivalis impairs spatial learning and memory is via TLR crosstalk because inhibiting this pathway rescued memory in their infection mouse model.
As an analogy to TLR signaling, our in-house data clearly showed that CD14, an LPS-binding receptor expressed on healthy IMR32 neurons (also participating in TLR signaling) was completely or partially removed following exposure to endo/exotoxins from P. gingivalis ATCC 33277T and W50, respectively (see Fig. 1). Such mechanisms lead to defective immune surveillance because of their influence in remodeling the periodontal microbiota into a dysbiotic state. P. gingivalis can also reduce the antibacterial and proinflammatory activity of C5a by deiminating its C-terminal arginine residues [70•]. Degradation of complement proteins probably allows colonization and proliferation of bacteria possessing higher sensitivity towards complement killing than found in P. gingivalis itself [47]. Thus, P. gingivalis may support survival of the entire biofilm community by helping bystander bacteria evade complement-mediated killing [46], while neurons survive with compromised function. These activities have consequences for the developing neuropathology. Thus, the neuropathology and the clinical functional loss together constitute the AD diagnosis. P. gingivalis infection under laboratory conditions are supporting both of these characteristics [13••, 14••, 15••, 16••, 22•, 24••, 52••].

Western blot showing CD14 protein on the human neuroblastoma cell line IMR32. a An immunoblot of cell lysate prepared from IMR32 neurons following their standard growth culture medium and incubation conditions, (lane 1). IMR32 neurons cultured in their growth medium to which P. gingivalis sterile growth medium diluted 1:4 from stock was added (lane 2). IMR32 neurons in their growth medium plus P. gingivalis ATCC 33277T conditioned medium diluted 1:4 from stock (lane 3) and strain W50 conditioned medium (diluted 1:4 from stock) (lane 4) for 24 h. The proteins were separated by SDS-PAGE electrophoresis and electro transferred onto the PVDF (polyvinylidene difluoride) membrane. Following incubation of the membrane overnight with mouse anti-CD14 antibody, clear bands around the 55 kDa molecular weight were seen (in the control lanes 1 and 2, long arrow) which indicates that the CD14 receptor protein was expressed by these cells. Upon challenge with P. gingivalis 33277T, the band completely diminished (lane 3, CD14 cleaved from cell membrane). Treatment of the same cells with the W50 strain surprisingly only partially cleaved CD14 (lane 4) as compared with the control lanes 1 and 2. b IMR32 cells grown on coverslips were also incubated with the same anti-CD14 antibody. The green color shows CD14 labeling on the surface membrane of cells confirming that the receptor is intact. The red color indicates the nucleus due to propidium iodide uptake from the mounting medium. c Following exposure to P. gingivalis 33277T cells for 24 h (as for the blot), the green labeling was missing and correlated with the blot data. d Exposure to P. gingivalis W50 demonstrated green labeling on the membranes again correlating with the blot data
Bacterial Factors Disrupting Synapses
Our in-house in vitro studies in which IMR32 (neuroblastoma-derived) neurons challenged with P. gingivalis virulence factors (LPS and gingipains) indicated considerable alterations in their actin cytoskeletal filaments following their detection with fluorescein-phalloidin dye. The LPS binding to cell surface membranes caused blebbing [11••], while the protease caused the cells to withdraw their processes and round up (see Fig. 2). In summary, the structural alteration of the IMR32 neurons, in vitro, could provide the basis for the failure of communication between neighboring cells. In vivo infection of microglia with P. gingivalis has promoted cell migration and an inflammatory response through gingipain-mediated activation of protease-activated receptor-2 [71•]. We need to clarify if and how infectious episodes impair memory at the synaptosomal level, rather than at the synaptic cleft level. Such information may refine our understanding at an earlier stage of deteriorating cognition albeit at the neurotransmitter release and its uptake levels.

IMR32 neurons in culture: fluorescein-phalloidin (5 units/ml final, for 30 min) labeling for actin cytoskeletal protein (green) (nuclei = red due to propidium iodide uptake). a IMR32 monolayer in growth medium shows long processes of the cells extending outwards. b Exposure to P. gingivalis ATCC 33277T, spent medium (diluted 1:4) for 6 h demonstrated the processes thickened while the cell soma enlarged. c As for b, but after 24 h exposure, the cells rounded up and detached. Images taken after examining the cells under the 510 series Zeiss confocal microscope (Carl Zeiss Ltd). Micron bar = 10
Dysbiosis of Immune Defense by Alternative Means
MiRNA has a role in the virulence of P. gingivalis, contributing to modulation of host-cell immune responses in a manner that promotes bacterial survival, and progressively reduces the host’s protective function [49]. Some miRNAs are even associated with P. gingivalis itself [72], while others (miRNA-128, miRNA-146, miRNA-203, and miRNA-584) are host derived for inflammation. Bacterium-associated miRNAs are likely to influence the innate immune response against P. gingivalis, whereas LPS from this bacterium may affect the level of the host’s miRNA–mRNA interactions, thus subverting innate and adaptive immune responses [54••, 69•].
P. gingivalis and Tau Protein Phosphorylation
As mentioned earlier, Ilievski et al. [14••] demonstrated that P. gingivalis infection can lead to tau phosphorylation and NFT formation in mice. The neurons that develop these hallmark lesions in the human AD brain are cells with compromised function and structural change in the nerve cell soma and axons, the latter disrupting their connectivity. The effect of gingipains on the integrity of actin filaments seen with IMR32 neurons (Fig. 2) may be analogous to the NFT-bearing neurons in AD. This structural change is likely to be detrimental to their communications with other brain cells resulting in deteriorated cognition.
Previously, we have suggested that outer membrane vesicles (microbullets) from P. gingivalis [73•] play a role in AD development. P. gingivalis cultures produce them in vast numbers, indicating that they constitute the main superhighway of communication with other bacteria in the biofilm [74]. Since they carry additional arsenals of weapons to access disparate organs, disrupt actin structures, erode epithelial junctional proteins, hijack phagocytosis, destroy tissues, and affect complement-related genes, they may also be responsible for transducing proinflammatory signaling cascades that ultimately lead to lesion development and cognitive decline, typical of AD.
Ilievski et al. [14••] demonstrated a chronic infection with live P. gingivalis strain W83 for 22 weeks with both the hallmark lesions (Aβ and NFTs) that characterize AD with tau protein phosphorylation at the serine396 (ser396) residue. This generated a new concept that an oral infective focus in neurological diseases may result in dementia. Up until now, abnormally phosphorylated tau protein has not featured negatively in the pathophysiology of periodontal disease per se. However, Adamowicz et al. [75••] implicated the role of glycogen synthase kinase 3 (GSK-3) in bacterial-induced periodontitis because its inhibition rescued bone loss. Thus, GSK-3 may be influencing phosphorylation of brain tau via immune responses mediated by P. gingivalis in the Ilievski et al. [14••] study. GSK-3β appears to mediate proinflammatory cytokine production during bacterial infections because inhibition of GSK-3β leads to an innate hypo-reactivity to oral pathogens [76]. Macrophages treated with LPS in vitro suggest that GSK-3β stimulates interferon-β (IFN-β) production via the c-Jun pathway, thus activating a transcription factor (ATF)-2-dependent mechanism [76]. GSK-3β also negatively regulates production of the endogenous IL-1β antagonist, IL-1R, via its ability to regulate the MAPK and ERK 1/2 in LPS-stimulated innate immune cells. There is no doubt that further research will widen investigation of these pathways for more direct causal links with oral disease and dementing diseases with cognitive deterioration.
The Dominy et al. [50••] publication has provided a stronger argument for the role of pathogenic tau in AD development. In their in vitro neuronal culture system, Dominy et al. [50••] demonstrated that tau is a substrate for gingipains and show low-molecular-weight bands corresponding to novel tau peptides. Further research will establish if they are neurotoxic or benefit the bacterium.
P. gingivalis and Lymphocytes
It is possible that T cell entry into the AD brain is restricted, and this somehow influences ineffective clearance of the Aβ by macrophages and the resident microglia. Baek et al. [77] found that Treg cells (subpopulation of T cells) had an effect on cognitive function by decreasing Aβ deposition and inflammatory cytokine secretion in a 3xTg-AD mice model. In contrast, depletion of Tregs increased the onset of cognitive deficit, accelerated the amount of the Aβ burden, enhanced microglia/macrophage responses, and decreased glucose metabolism in 3xTg-AD mice. In patients with atherosclerosis, the Treg population was reduced if they harbored type II fimA of P. gingivalis compared to those with other types of fimbriae [78]. Therefore, P. gingivalis type II fimA could be associated with dysregulation of Tregs in extraoral lesions. Severe immunosuppression seems to favor not only colonization with varying serotypes of periodontopathogenic bacteria, but also with species not commonly found in the subgingival microbiota [79]. In the brain, this may contribute to the establishment of a multispecies microbiota, previously reported in AD patients [80]. In addition, accumulation of insoluble and toxic Aβ42 has detrimental effect on the neighboring neurons and their connections, which may have further implications for neurodegeneration and related cognitive loss.
Conclusions
Dominy et al. [50••] have recently provided robust data linking the main pathogen (P. gingivalis) of periodontitis with the cause of AD. This bacterium appears to migrate from the mouth to the brain of some individuals as they age and a significant proportion of subjects who go onto developing AD. This further highlights the possibility that AD has a microbial infection origin. Ilievski et al. [14••] provide evidence for P. gingivalis infection having causal associations by reproducing the hallmark lesions. Four independent studies carried out in mice infected with P. gingivalis provide causal links through impaired learning and memory. The suggested mechanism is related to the complement-TLR crosstalk, and this may have relevance to the inflammasome formation with the resulting cytokines (mature IL-1β) being linked to memory disturbances.
These studies reinforce the advice that oral hygiene is important in keeping pathogens low and encouraging greater diversity of commensals (health promoting bacteria). This provides a healthy microbiome and better general health. Health authorities need to heed this warning and take research-based evidence seriously. For those who fear the prospects of AD in their later life, the UK NHS England provides a recommendable oral health toolkit for the elderly to maintain better oral hygiene with the aim of delaying/preventing AD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Olsen I. From the Acta Prize Lecture 2014: the periodontal-systemic connection seen from a microbiological standpoint. Acta Odontol Scand. 2015;73(8):563–8. https://doi.org/10.3109/00016357.2015.1007480.
Olsen I, Singhrao SK. Can oral infection be a risk factor for Alzheimer’s disease? J Oral Microbiol. 2015;7:29143. https://doi.org/10.3402/jom.v7.29143.
Olsen I, Singhrao SK, Potempa J. Citrullination as a plausible link to periodontitis, rheumatoid arthritis, atherosclerosis and Alzheimer’s disease. J Oral Microbiol. 2018;10(1):1487742. https://doi.org/10.1080/20002297.2018.1487742.
•• Chen CK, Wu YT, Chang YC. Periodontal inflammatory disease is associated with the risk of Parkinson’s disease: a population-based retrospective matched-cohort study. PeerJ. 2017;5:e3647. https://doi.org/10.7717/peerj.3647. Reducing periodontitis may modify the risk of developing Parkinson’s disease.
•• Chen CK, Huang JY, Wu YT, Chang YC. Dental scaling decreases the risk of Parkinson’s disease: a nationwide population-based nested case-control study. Int J Environ Res Public Health. 2018;15(8). https://doi.org/10.3390/ijerph15081587. First study to show that patients without periodontitis who received dental scaling over 5 consecutive years had a significantly lower risk of developing Parkinson’s disease.
•• Pang S, Li J, Zhang Y, Chen J. Meta-analysis of the relationship between the APOE gene and the onset of Parkinson's disease dementia. Parkinsons Dis. 2018;2018:9497147. https://doi.org/10.1155/2018/9497147. APOE genotypes, ɛ3/4 and ɛ4/4 may be risk factors for Parkinson’s disease dementia.
•• Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. https://doi.org/10.1371/journal.pone.0009505. Abeta is a hitherto unrecognized antimicrobial peptide that may normally act in the innate immune system.
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72. https://doi.org/10.1126/scitranslmed.aaf1059.
•• Park SC, Moon JC, Shin SY, Son H, Jung YJ, Kim NH, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. 2016;478(2):924–8. https://doi.org/10.1016/j.bbrc.2016.08.052. Alpha-synuclein appears to be a natural antimicrobial peptide in addition to having a role in neurotransmitter release.
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s disease-associated β-amyloid is rapidly seeded by Herpesviridae to protect against brain infection. Neuron. 2018;99(1):56–63.e3. https://doi.org/10.1016/j.neuron.2018.06.030.
•• Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis. 2013;36(4):665–77. https://doi.org/10.3233/JAD-121918. The original study confirmed that P. gingivalis lipopolysaccharide reaches the Alzheimer disease brain.
•• Poole S, Singhrao SK, Chukkapalli S, Rivera M, Velsko I, Kesavalu L, et al. Active invasion of Porphyromonas gingivalis and infection-induced complement activation in ApoE−/− mice brains. J Alzheimers Dis. 2015;43(1):67–80. https://doi.org/10.3233/JAD-140315 P. gingivalis. was able to access the ApoE −/− mice brain contributing to complement activation by bystander neuronal injury.
•• Ilievski V, Zuchowska PK, Green SJ, Toth PT, Ragozzino ME, Le K, et al. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and production amyloid beta in wild type mice. PLoS One. 2018;13(10):e0204941. https://doi.org/10.1371/journal.pone.0204941. First study to show neurodegeneration and formation of extracellular Aß42 and phosphotau bound to neurofibrillary tangles in young adult wild type mice after repeated application of P. gingivalis.
•• Wu Z, Ni J, Liu Y, Teeling JL, Takayama F, Collcutt A, et al. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav Immun. 2017;65:350–61. https://doi.org/10.1016/j.bbi.2017.06.002. Chronic systemic exposure to PgLPS induced AD-like phenotypes, including microglia-mediated neuroinflammation, intracellular Aβ accumulation in neurons and impairment of the learning and memory functions in the middle-aged mice in a CatB-dependent manner. Linked IL-1β involvement in memory deterioration.
•• Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. NPJ Aging Mech Dis. 2017;3:15. https://doi.org/10.1038/s41514-017-0015-x. Periodontitis caused by P. gingivalis may exacerbate brain Aβ deposition, leading to increased cognitive impairments, by a mechanism that involves triggering brain inflammation.
•• Ding Y, Ren J, Yu H, Yu W, Zhou Y. Porphyromonas gingivalis, a periodontitis causing bacterium, induces memory impairment and age-dependent neuroinflammation in mice. Immun Ageing. 2018;15:6. https://doi.org/10.1186/s12979-017-0110-7. The learning and memory abilities of the middle-aged mice infected with P. gingivalis were impaired.
Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nat Rev Dis Primers. 2015;1:15056. https://doi.org/10.1038/nrdp.2015.56.
•• Singhrao SK, Harding A, Chukkapalli S, Olsen I, Kesavalu L, Crean S. Apolipoprotein E related co-morbidities and Alzheimer’s disease. J Alzheimers Dis. 2016;51(4):935–48. https://doi.org/10.3233/JAD150690. The apolipoprotein E gene allele 4 is the plausible commonality for the etiology of co-morbidities that eventually result from periodontal diseases and ultimately progress to mixed pathologies and dementia.
Li J, Luo J, Liu L, Fu H, Tang L. The genetic association between apolipoprotein E gene polymorphism and Parkinson disease: a meta-analysis of 47 studies. Medicine (Baltimore). 2018;97(43):e12884. https://doi.org/10.1097/MD.0000000000012884.
• Singhrao SK, Olsen I. Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J Oral Microbiol. 2019;11:1563405. https://doi.org/10.1080/20002297.2018.1563405. Summation of animal studies supporting the P. gingivalis infection link with AD risk factor development.
•• Yin C, Ackermann S, Ma Z, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med. 2019;25(3):496–506 Publisher correction: Nat Med. 2019; 25(3): 529. Original study suggesting a new function for the APOE gene as a checkpoint of classical complement activation in AD.
• Rokad F, Moseley R, Hardy RS, Chukkapalli S, Crean S, Kesavalu L, et al. Cerebral oxidative stress and microvasculature defects in TNF-α expressing transgenic and Porphyromonas gingivalis-infected ApoE-/- mice. J Alzheimers Dis. 2017;60(2):359–69. https://doi.org/10.3233/JAD-170304. The hippocampal microvascular structure of P. gingivalis -infected ApoE −/− mice possesses elevated oxidative stress levels, resulting in the associated tight junction proteins being susceptible to increased oxidative/proteolytic degradation, leading to hydrolysis of the amyloid precursor protein to generate Aβ in wild type mice infected with P. gingivalis .
•• Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimers Dement. 2018;14(12):1602–14. https://doi.org/10.1016/j.jalz.2018.06.3040. The new Antimicrobial Protection Hypothesis reveals brain microbial burden may directly generate β-amyloid deposits, inflammation, and AD progression. This study underpins the role of microbes in the pathogenesis of AD.
•• Singhrao SK, Chukkapalli S, Poole S, Velsko I, Crean SJ, Kesavalu L. Chronic Porphyromonas gingivalis infection accelerates the occurrence of age-related granules in ApoE−/− mice brains. J Oral Microbiol. 2017;9(1):1270602. https://doi.org/10.1080/20002297.2016.1270602 ApoE −/− mice showing the earliest inflammation-mediated tissue injury, accompanied by cerebral blood-brain barrier breach.
•• Bowman GL, Dayon L, Kirkland R, Wojcik J, Peyratout G, Severin IC, et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimers Dement. 2018;14(12):1640–50. https://doi.org/10.1016/j.jalz.2018.06.2857. Blood-brain-barrier breakdown is associated with cognitive decline and inflammation in non-demented elders.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6 Erratum in: Science 2002; 297(5590): 2209.
•• Boche D, Donald J, Love S, Harris S, Neal JW, Holmes C, et al. Reduction of aggregated Tau in neuronal processes but not in the cell bodies after Abeta42 immunisation in Alzheimer’s disease. Acta Neuropathol. 2010;120(1):13–20. Removal of Abeta from the brain via immunisation does not improve memory.
• Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80. Origins of the synaptic loss theory to explain cognitive deficit.
• Masliah E, Hansen L, Albright T, Mallory M, Terry RD. Immunoelectron microscopic study of synaptic pathology in Alzheimer’s disease. Acta Neuropathol. 1991;81(4):428–33. Provides evidence to support the synaptic loss theory.
•• Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid-β oligomer hypothesis: beginning of the third decade. J Alzheimers Dis. 2018;64(s1):S567–610. https://doi.org/10.3233/JAD-179941. Revised Amyloid-β oligomer theory. Soluble Aβ oligomers accumulate in an AD-dependent manner in human and animal model brain tissue and, experimentally, to impair learning and memory and instigate major facets of AD neuropathology, including tau pathology, synapse deterioration and loss, inflammation, and oxidative damage.
•• Olsen I, Singhrao SK. Inflammasome involvement in Alzheimer’s disease. J Alzheimers Dis. 2016;54(1):45–53. This review supports the mechanism of P. gingivalis infection promoting cognitive deficit (see ref 52).
Gatz M, Mortimer JA, Fratiglioni L, Johansson B, Berg S, Reynolds CA, et al. Potentially modifiable risk factors for dementia in identical twins. Alzheimers Dement. 2006;2:110–7.
Stein PS, Desrosiers M, Donegan SJ, Yepes JF, Kryscio RJ. Tooth loss, dementia and neuropathology in the Nun study. J Am Dent Assoc. 2007;138(10):1314–22, quiz 1381-2.
Stein PS, Kryscio RJ, Desrosiers M, Donegan SJ, Gibbs MB. Tooth loss, apolipoprotein E, and decline in delayed word recall. J Dent Res. 2010;89(5):473–7. https://doi.org/10.1177/0022034509357881.
Kamer AR, Craig RG, Pirraglia E, Dasanayake AP, Norman RG, Boylan RJ, et al. TNF-alpha and antibodies to periodontal bacteria discriminate between Alzheimer’s disease patients and normal subjects. J Neuroimmunol. 2009;216(1–2):92–7. https://doi.org/10.1016/j.jneuroim.2009.08.013.
• Farhad SZ, Amini S, Khalilian A, Barekatain M, Mafi M, Barekatain M, et al. The effect of chronic periodontitis on serum levels of tumor necrosis factor-alpha in Alzheimer disease. Dent Res J (Isfahan). 2014;11(5):549–52. Not all of the AD cases they examined at the clinical level co-existed with periodontitis.
•• Sparks Stein P, Steffen MJ, Smith C, Jicha G, Ebersole JL, Abner E, et al. Serum antibodies to periodontal pathogens are a risk factor for Alzheimer’s disease. Alzheimers Dement. 2012;8(3):196–203. https://doi.org/10.1016/j.jalz.2011.04.006. A laboratory study that agreed with the epidemiological study of Chen et al. (ref 38) for the 10 year timeline in risk development following periodontitis diagnosis.
•• Chen CK, Wu YT, Chang YC. Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched-cohort study. Alzheimers Res Ther. 2017;9(1):56. https://doi.org/10.1186/s13195-017-0282-6. A 10-year CP exposure was associated with a 1,707-fold increase in the risk of developing AD.
Stewart R, Weyant RJ, Garcia ME, Harris T, Launer LJ, Satterfield S, et al. Adverse oral health and cognitive decline: the health, aging and body composition study. J Am Geriatr Soc. 2013;61(2):177–84. https://doi.org/10.1111/jgs.12094.
Kaye EK, Valencia A, Baba N, Spiro A 3rd, Dietrich T, Garcia RI. Tooth loss and periodontal disease predict poor cognitive function in older men. J Am Geriatr Soc. 2010;58(4):713–8. https://doi.org/10.1111/j.1532-5415.2010.02788.x.
• Daly B, Thompsell A, Sharpling J, Rooney YM, Hillman L, Wanyonyi KL, et al. Evidence summary: the relationship between oral health and dementia. Br Dent J. 2018;223(11):846–53. https://doi.org/10.1038/sj.bdj.2017.992. Poor oral hygiene is associated with dementia, and more so amongst people in advanced stages of the disease.
• de Souza RT, Fabri GM, Nitrini R, Anghinah R, Teixeira MJ, de Siqueira JT, et al. Oral infections and orofacial pain in Alzheimer’s disease: a case-control study. J Alzheimers Dis. 2014;38(4):823–9. https://doi.org/10.3233/JAD-131283. An interventional study on the periodontal treatment in AD patients indicated a plausible causal relationship in demented individuals.
• Emanuel R, Sorensen A. A study of oral health prevention behaviours for patients with early stage dementia. Br Dent J. 2018;224(1):38–42. https://doi.org/10.1038/sj.bdj.2018.5. There was clearly scope for improving oral health education and prevention for dementia patients.
• Hajishengallis G. Immune evasion strategies of Porphyromonas gingivalis. J Oral Biosci. 2011;53(3):233–40. Exploitation of Toll-like receptor-2, complement receptor 3, C5a anaphylatoxin receptor, and CXC-chemokine receptor 4 by P. gingivalis allows the pathogen to escape elimination, obtain nutrients, and collaterally inflict periodontal tissue injury.
• Hajishengallis G, Lambris JD. Complement and dysbiosis in periodontal disease. Immunobiology. 2012;217(11):1111–6. https://doi.org/10.1016/j.imbio.2012.07.007. P. gingivalis expresses C5 convertase-like enzymatic activity and adeptly exploits complement-TLR crosstalk to subvert host defenses and escape elimination.
Hajishengallis G, Abe T, Maekawa T, Hajishengallis E, Lambris JD. Role of complement in host-microbe homeostasis of the periodontium. Semin Immunol. 2013;25(1):65–72. https://doi.org/10.1016/j.smim.2013.04.004.
Hussain M, Stover CM, Dupont A. P. gingivalis in periodontal disease and atherosclerosis - scenes of action for antimicrobial peptides and complement. Front Immunol. 2015;6:45. https://doi.org/10.3389/fimmu.2015.00045.
Olsen I, Hajishengallis G. Major neutrophil functions subverted by Porphyromonas gingivalis. J Oral Microbiol. 2016;8:30936. https://doi.org/10.3402/jom.v8.30936.
Olsen I, Singhrao SK, Osmundsen H. Periodontitis, pathogenesis and progression: miRNA-mediated cellular responses to Porphyromonas gingivalis. J Oral Microbiol. 2017;9(1):1333396. https://doi.org/10.1080/20002297.2017.1333396.
•• Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5:eaau3333. This study has provided the strongest evidence for causal links of P. gingivalis infection with AD. It has raised the vital awareness in periodontitis and AD developmental links. It offers hope for a future treatment via gingipain inhibiting drugs.
•• Llorente P, Kristen H, Sastre I, Toledano-Zaragoza A, Aldudo J, Recuero M, et al. A free radical-generating system regulates amyloid oligomers: involvement of cathepsin B. J Alzheimers Dis. 2018;66:1397–408. https://doi.org/10.3233/JAD-170159. Cathepsin B participates in the changes of amyloid oligomer induced by mild oxidative stress. This supports the oxidative stress related damage following P. gingivalis infection as reported in ref 22.
•• Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, et al. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation. 2018;15(1):37. https://doi.org/10.1186/s12974-017-1052-x. Offer a mechanism of how P. gingivalis -LPS-induced neuroinflammation plays an important role in cognitive impairment. This reference complements the review by Olsen and Singhrao ref 31.
Imamura T. The role of gingipains in the pathogenesis of periodontal disease. J Periodontol. 2003;74(1):111–8.
•• Vasek MJ, Garber C, Dorsey D, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534:538–43. Original study implicating classical complement activation in AD through overt pruning activity in microglia. A novel mechanistic viewpoint of cognitive deficit.
Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–84.
Tanaka S, Ide M, Shibutani T, Ohtaki H, Numazawa S, Shioda S, et al. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res. 2006;83:557–66.
Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW. Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun. 2008;18:223–30.
• Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23(3):309–17. Implicates LPS with release of IL-1β and intracerebral inflammation.
• Bellinger FP, Madamba S, Siggins GR. Interleukin 1 beta inhibits synaptic strength and long-term potentiation in the rat CA1 hippocampus. Brain Res. 1993;628:227–34. Implicate IL-1β in cognitive deficit.
Mishra A, Kim HJ, Shin AH, Thayer SA. Synapse loss induced by interleukin-1beta requires pre- and post-synaptic mechanisms. J NeuroImmune Pharmacol. 2012;7(3):571–8. https://doi.org/10.1007/s11481-012-9342-7.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21(3):383–421.
Allen HB. Alzheimer’s disease: assessing the role of spirochetes, biofilms, the immune system, and amyloid-β with regard to potential treatment and prevention. J Alzheimers Dis. 2016;53(4):1271–6. https://doi.org/10.3233/JAD-160388.
Singhrao SK, Neal JW, Rushmere NK, Morgan BP, Gasque P. Spontaneous classical pathway activation and deficiency of membrane regulators render human neurons susceptible to complement lysis. Am J Pathol. 2000;157(3):905–18.
Wingrove JA, DiScipio RG, Chen Z, Potempa J, Travis J, Hugli TE. Activation of complement components C3 and C5 by a cysteine proteinase (gingipain-1) from Porphyromonas (Bacteroides) gingivalis. J Biol Chem. 1992;267(26):18902–7.
Popadiak K, Potempa J, Riesbeck K, Blom AM. Biphasic effect of gingipains from Porphyromonas gingivalis on the human complement system. J Immunol. 2007;178(11):7242–50.
Potempa M, Potempa J, Okroj M, Popadiak K, Eick S, Nguyen KA, et al. Binding of complement inhibitor C4b-binding protein contributes to serum resistance of Porphyromonas gingivalis. J Immunol. 2008;181(8):5537–44.
Slaney JM, Curtis MA. Mechanisms of evasion of complement by Porphyromonas gingivalis. Front Biosci. 2008;13:188–96.
Wang M, Krauss JL, Domon H, Hosur KB, Liang S, Magotti P, et al. Microbial hijacking of complement-toll-like receptor crosstalk. Sci Signal. 2010;3(109):ra11. https://doi.org/10.1126/scisignal.2000697.
• Olsen I, Singhrao SK. Genetic defects, complement and Porphyromonas gingivalis immune subversion in Alzheimer’s disease. J Oral Microbiol. 2019, submitted. P. gingivalis is a keystone pathogen for periodontitis and its co-morbidities because of the mastery of this bacterium at subverting selective steps in the immune system for effective dysbiosis, which in turn amplifies altered gene functions in Alzheimer’s disease.
• Bielecka E, Scavenius C, Kantyka T, Jusko M, Mizgalska D, Szmigielski B, et al. Peptidyl arginine deiminase from Porphyromonas gingivalis abolishes anaphylatoxin C5a activity. J Biol Chem. 2014;289(47):32481–7. https://doi.org/10.1074/jbc.C114.617142. In the context of previous studies, which showed crosstalk between C5aR and Toll-like receptors, and enhanced arthritis development in mice infected with PPAD-expressing P. gingivalis , a crucial role of PPAD in the virulence of P. gingivalis was supported.
• Liu Y, Wu Z, Nakanishi Y, Ni J, Hayashi Y, Takayama F, et al. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep. 2017;7(1):11759. https://doi.org/10.1038/s41598-017-12173-1. The authors provide the first evidence that Rgp and Kgp cooperatively contribute to the P. gingivalis -induced cell migration and expression of proinflammatory mediators through the activation of protease-activated receptor 2.
Choi JW, Kim SC, Hong SH, Lee HJ. Secretable small RNAs via outer membrane vesicles in periodontal pathogens. J Dent Res. 2017;96:458–66. https://doi.org/10.1177/0022034516685071.
• Singhrao SK, Olsen I. Are Porphyromonas gingivalis outer membrane vesicles, microbullets for sporadic Alzheimer’s disease manifestation? J Alzheimers Dis Rep. 2018;1:1–10. https://doi.org/10.3233/ADR-180080. Bacterial cultures and established oral biofilms generate vast numbers of microvesicles and P. gingivalis outer membrane vesicles encase key virulence factors (LPS, gingipains, capsule, fimbriae) as though they are complete destructive "microbullets" when shed in the host.
Schertzer JW, Whiteley M. Microbial communication superhighways. Cell. 2011;144(4):469–70. https://doi.org/10.1016/j.cell.2011.02.001.
•• Adamowicz K, Wang H, Jotwani R, Zeller I, Potempa J, Scott DA. Inhibition of GSK3 abolishes bacterial-induced periodontal bone loss in mice. Mol Med. 2012;18:1190–6. https://doi.org/10.2119/molmed.2012.00180. This study confirms the relevance of prior in vitro phenomena and establish GSK3 as a novel, efficacious therapeutic preventing periodontal disease progression in a susceptible host.
Wang H, Brown J, Martin M. Glycogen synthase kinase 3: a point of convergence for the host inflammatory response. Cytokine. 2011;53:130–40.
Baek H, Ye M, Kang GH, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget. 2016;7(43):69347–57. https://doi.org/10.18632/oncotarget.12469.
Yang J, Wu J, Liu Y, Huang J, Lu Z, Xie L, et al. Porphyromonas gingivalis infection reduces regulatory T cells in infected atherosclerosis patients. PLoS One. 2014;9(1):e86599. https://doi.org/10.1371/journal.pone.0086599.
Gonçalves LS, Ferreira SM, Silva A Jr, Villoria GE, Costinha LH, Souto R, et al. Association of T CD4 lymphocyte levels and subgingival microbiota of chronic periodontitis in HIV-infected Brazilians under HAART. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97(2):196–203.
Emery DC, Shoemark DK, Batstone TE, Waterfall CM, Coghill JA, Cerajewska TL, et al. 16S rRNA next generation sequencing analysis shows bacteria in Alzheimer’s post-mortem brain. Front Aging Neurosci. 2017;9:195. https://doi.org/10.3389/fnagi.2017.0019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ingar Olsen and Sim K. Singhrao each declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Microbiology
Rights and permissions
About this article

Cite this article
Olsen, I., Singhrao, S.K. Poor Oral Health and Its Neurological Consequences: Mechanisms of Porphyromonas gingivalis Involvement in Cognitive Dysfunction. Curr Oral Health Rep 6, 120–129 (2019). https://doi.org/10.1007/s40496-019-0212-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40496-019-0212-8