
Abstract
We present ContFree-NGS, an open source software that removes reads originating from contaminant organisms in your sequencing dataset. The user has to provide a target taxon, and anything that does not belong to this taxon or its descendants will be labelled as contaminant. In order to achieve this, ContFree-NGS exploits results from a taxonomy assignment engine, like Kraken2 or Kaiju.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Second and third generation DNA sequencing technologies are powerful tools that are revolutionizing biology. However, results from these technologies often present contamination, which could impact their interpretation [1, 2]. A contaminating sequence is one that does not faithfully represent the genetic information from the biological source organism because it contains one or more sequence segments of foreign origin, and they could cause several problems in downstream analyses. The primary consequences of contamination are time and effort wasted on meaningless analyses, erroneous conclusions drawn about the biological significance of the sequence, misassembly of sequence contigs and false sequence clustering, delay in the release of the sequence in public databases and pollution of public databases [3].
Recently, some tools have been made available that aim to remove sequences from contaminating organisms in next generation sequencing (NGS) datasets. DecontaMiner is a tool to unravel the presence of contaminating sequences in the set of reads that do not map to a reference genome [4]. Conterminator removes contaminating sequences from contigs exploiting a taxonomic assignment file [5]. QC-Blind is an automatic tool to do unsupervised assembly and contig binning to identify and remove putative contaminants [6]. These tools have in common that they either require a reference genome of the source organism, or need to perform assembly prior contaminant detection. Our goal was to develop a simpler tool to remove contaminated sequences directly from unassembled reads, without mapping, exploiting fast k-mer analysis implemented in taxonomic assignment engines commonly used in metagenomics. Thus, we developed ContFree-NGS, an open source and very simple filter that removes sequences from contaminating organisms in NGS datasets based on a taxonomic classification file.
2 Implementation
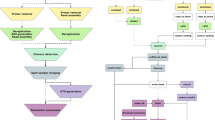
ContFree-NGS was implemented as a single Python v3 (>3.6) script, using the biopython module and the Python Environment for Tree Exploration (ETE). In order to assess contamination, ContFree-NGS exploits a taxonomic assignment file containing the read identifier and a NCBI taxonomic identifier for every sequence in the dataset. This taxonomic classification file can be generated with a taxonomic assignment engine, such as Kraken2 [7] or Kaiju [8]. ContFree-NGS requires that the user provide a target taxon and only reads assigned to this taxon or to its descendants will be regarded as target sequences and further maintained. Sequences not assigned to the target taxon or its descendants will be discarded and sequences that could not be assigned to any taxa will be kept in a separated unclassified file. ContFree-NGS will process the NGS dataset and the taxonomic assignment file in the following way:
-
1.
It creates an indexed database for the sequencing dataset (FastQ format) using the Bio.SeqIO.index_db function with the index stored in a SQLite database;
-
2.
It creates a list with the NCBI identifier for the target taxon and all the identifiers of its descendants according to the NCBI taxonomy database;
-
3.
It iterates over the taxonomy assignment file. If the read was not assigned to any taxa it is saved in a fastq file for unclassified reads. If the read was assigned, it will check if its taxon is found in the list of the target taxon descendants, created in step (ii), if so, will save the read to a fastq file for filtered reads, otherwise the read will be discarded.
As ContFree-NGS exploits the results from a taxonomic assignment engine, users must use the proper switches to achieve an accurate classification, for instance a proper value of the --confidence switch in Kraken2.
3 Evaluation
We evaluated ContFree-NGS on three sugarcane artificially contaminated datasets, A (50.000 paired end reads), B (250.000 paired end reads) and C (1.250.000 paired end reads). In all datasets 80% of the reads came from sugarcane (SRR1774134), 15% came from Acinetobacter baumanii (SRR12763742) and 5% came from Aspergillus fumigatus (DRR289670). We used Kraken2 for taxonomic assignment. To perform that, we built a Kraken2 custom database containing the following reference libraries: archaea, bacteria, viral, human, fungi, plant, protozoa and the NCBI non-redundant nucleotide database. Then, Kraken2 was run with the confidence set to 0.05, resulting in the following number of classified sequences: dataset A: 25.547, dataset B: 128.396, dataset C: 664.270.
At the confidence level of 0.05 set in Kraken2, ContFree-NGS was able to remove over 99% of the known contaminants in the set of classified reads. We run ContFree-NGS on a high performance computing (HPC) cluster and recorded RAM usage and processing wall time for the three datasets (see Fig. 1).

This figure shows the RAM usage and time consuming to remove contaminants of the three sugarcane artificially contaminated datasets. Memory usage is low and independent of the number of classified sequences and wall time scale rapidly with the number of classified sequences. To reduce time consuming, the end user could split the taxonomy assignment file in several files and process them in parallel. Check our GitHub (https://github.com/labbces/ContFree-NGS) page for more details.
4 Conclusion
ContFree-NGS is a very simple filter and useful tool that removes sequences from contaminating organisms in a NGS dataset.
References
Park, S.J., Onizuka, S., Seki, M., et al.: A systematic sequencing-based approach for microbial contaminant detection and functional inference. BMC Biol. 17, 72 (2019). https://doi.org/10.1186/s12915-019-0690-0
Goig, G.A., Blanco, S., Garcia-Basteiro, A.L., et al.: Contaminant DNA in bacterial sequencing experiments is a major source of false genetic variability. BMC Biol. 18, 24 (2020). https://doi.org/10.1186/s12915-020-0748-z
National Center for Biotechnology Information 2016: Contamination in Sequence Databases. https://www.ncbi.nlm.nih.gov/tools/vecscreen/contam/. Accessed 6 Oct 2021
Sangiovanni, M., Granata, I., Thind, A., et al.: From trash to treasure: detecting unexpected contamination in unmapped NGS data. BMC Bioinform. 20, 168 (2019). https://doi.org/10.1186/s12859-019-2684-x
Steinegger, M., Salzberg, S.L.: Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol. 21, 115 (2020). https://doi.org/10.1186/s13059-020-02023-1
Xi, W., Gao, Y., Cheng, Z., et al.: Using QC-blind for quality control and contamination screening of bacteria DNA sequencing data without reference genome. Front. Microbiol. 10, 1560 (2019). https://doi.org/10.3389/fmicb.2019.01560
Wood, D.E., Lu, J., Langmead, B.: Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019). https://doi.org/10.1186/s13059-019-1891-0
Menzel, P., Ng, K., Krogh, A.: Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7, 11257 (2016). https://doi.org/10.1038/ncomms11257
Funding
This work was supported by “Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)” [grant number 2019/24796-5 to F.V.P] and by “Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)” [grant number 310080/2018‐5 to D.M.R-P].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this paper

Cite this paper
Peres, F.V., Riaño-Pachón, D.M. (2021). ContFree-NGS: Removing Reads from Contaminating Organisms in Next Generation Sequencing Data. In: Stadler, P.F., Walter, M.E.M.T., Hernandez-Rosales, M., Brigido, M.M. (eds) Advances in Bioinformatics and Computational Biology. BSB 2021. Lecture Notes in Computer Science(), vol 13063. Springer, Cham. https://doi.org/10.1007/978-3-030-91814-9_6
Download citation
DOI: https://doi.org/10.1007/978-3-030-91814-9_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91813-2
Online ISBN: 978-3-030-91814-9
eBook Packages: Computer ScienceComputer Science (R0)