
Abstract
Cardiac valve calcification (CVC) is characterised by slowly progressive fibro-calcific remodelling of valve leaflets in calcific aortic valve disease or the mitral annulus in mitral annular calcification (MAC). Calcific aortic valve disease is the most prevalent cause of aortic stenosis worldwide and poses a significant disease burden being the third most common cardiovascular problem after coronary artery disease and hypertension. Although MAC has a higher disease prevalence, calcific aortic valve disease is associated with significant morbidity and has important clinical implications. Risk factors of CVC are generally similar to those of atherosclerosis; hypertension, diabetes mellitus and hypercholesterolemia. Historically presumed to be caused by a degenerative process, there is now emerging histopathological and clinical evidence to suggest that the pathophysiology of CVC is an active and multifaceted process that involves chronic inflammation, lipoprotein deposition, extracellular matrix remodelling and osteoblastic transformation of valvular cells. Doppler echocardiography is the principal investigative modality in diagnosing cardiac valve disease, complemented by multi-slice computed tomography (MSCT) and cardiac magnetic resonance imaging (MRI) to quantitate calcification which serves as a surrogate marker of disease severity. To date, there remains no effective pharmacotherapy to delay or halt disease progression and management involves surgical valve replacement or minimally invasive alternatives such as transcatheter aortic/mitral valve insertion in symptomatic patients. The heart valve team plays a crucial role in deciding which patients will benefit most from an intervention by taking into account patient symptoms, cardiac function, coexisting medical conditions and their functional baseline.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Cardiac valve disease
- Cardiac calcification
- Cardiac surgery
- Cardiology
- Surgical valve replacement
- Transcatheter valve implantation
Introduction
In 1663, French physician, Lazare Rivière performed autopsy on a patient with symptoms of progressive shortness of breath, irregular heartbeat and heart palpitations. He identified round caruncle-like masses that obstructed the left ventricular outflow tract (LVOT) associated with an enlarged left ventricle [1]. Physicians in his era also reported similar occurrences and further described an ossifying process of the aortic valve leaflets. These findings were initially presumed to be infective in nature as seen in endocarditis and rheumatic fever [1].
Hasse in 1846, challenged this aetiology and suggested that the calcification process could also be attributed to a degenerative process with ageing [1]. In 1904, Moenckeburg recognised aortic sclerosis as a potential precursor to aortic stenosis and proposed two mechanisms of secondary calcium deposition; ascending and descending. Ascending sclerosis occurs when degeneration within the valve leaflet layers facing the Valsalva sinuses propagates upwards towards the free margin while descending sclerosis occurs with downward sclerotic extension to involve both the cusps and commissures [1].
Cardiac valve calcification (CVC) is characterised by slowly progressive fibro-calcific remodelling of valve leaflets. Rheumatic heart disease is a common cause for CVC in developing countries while in the developed world, the formation of CVC is believed to be a combination of factors including age, gender, genetics, medical comorbidities and cardiovascular risk factors. Compared to mitral annular calcification (MAC), calcific aortic valve disease is associated with significant morbidity and has important clinical implications. Hence, calcific aortic valve disease will be the focus of this chapter with a small subsection to discuss the clinical manifestations and management of mitral annular calcification (MAC).
Epidemiology and Risk Factors of CVC
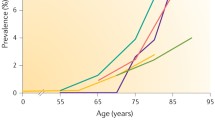
Calcific aortic valve disease is the most prevalent cause of aortic stenosis (AS) worldwide and poses a significant disease burden, with AS being the third most common cardiovascular problem after coronary artery disease and hypertension [2]. Prevalence of aortic sclerosis increases with age and the rate of progression to AS is estimated to be 1.8-1.9% of patients per annum [3]. Calcific AS has an estimated prevalence of 0.4% in the general population and increases to 1.7% in those aged >65 years in developed countries [4].
The calcification process can occur in either a normal trileaflet aortic valve or a congenitally abnormal bicuspid valve. Bicuspid valve is a known risk factor for calcification and accounts for nearly half of all surgically replaced aortic valves [5]. Moreover, these patients tend to develop calcific AS one or two decades earlier compared to those with a tricuspid valve [6]. Other risk factors of calcific aortic valve disease are those of atherosclerosis such as diabetes mellitus, hypertension and hypercholesterolemia [7].
The prevalence of MAC has been reported to be between 8% and 15% and increases with age [8, 9]. Risk factors are generally similar to those in atherosclerosis and calcific aortic valve disease with a few other specific risk factors including female gender, chronic kidney disease and congenital metabolic disorders such as Marfan syndrome and Hurler syndrome [10,11,12].
Anatomy of the Aortic Valve
The aortic valve is an avascular tricuspid structure situated at the LVOT and appended to the aorta by a fibrous annulus. The valve leaflets are named according to their location respective to the coronary arteries; right coronary cusp, left coronary cusp and non-coronary cusp. The leaflets are typically ≤1 mm in thickness and is made up of three layers. The outermost layers, fibrosa and ventricularis, face the aorta and LVOT respectively, with the spongiosa situated between those two layers.
The fibrosa is composed of circumferentially oriented Type 1 and 3 collagen fibres and has a load-bearing function while the ventricularis is made up of elastin-rich fibres in a radial orientation, providing good compliance (ability to expand under pressure) and allowing for the apposition of leaflets during diastole to prevent backflow of blood [13]. The spongiosa layer contains glycosaminoglycans which provides lubrication as the fibrosa and ventricularis layers shear and deform during the cardiac cycle [14, 15].
At a cellular level, these leaflets are defined by three cell types. The vascular endothelial cells (VEC) form the outer layer and is in direct contact with luminal blood flow. These cells regulate valvular homeostasis by controlling permeability, inflammatory cell adhesion and paracrine signalling. Vascular interstitial cells (VIC) are the predominant cell population, interspersed between the fibrosa, spongiosa and ventricularis layers of the valve leaflet. Their function is to secrete extracellular matrix such as elastin, collagen and glycosaminoglycans which provide tensile strength and elastic properties to the valve. Smooth muscle cells (SMC) are the third cell type comprising <5% of the valvular cell population found at the ventricularis [14, 15].
Aetiology and Pathophysiology of CVC
For a long time, CVC was thought to be primarily caused by a degenerative process and passive calcium deposition. There is, however, emerging histopathological and clinical evidence to suggest that the pathophysiology involves an active and multifaceted process that involves chronic inflammation, lipoprotein deposition, extracellular matrix remodelling and osteoblastic transformation of VICs [16].
Cellular and Molecular Mechanisms
Valvular homeostasis is regulated by an intricate process involving the interaction between valvular cells and their environment. Under normal circumstances, an insult to the valvular surface activates a passive calcium-phosphate complex deposition process to initiate valve repair. In this process, the VICs transition to osteoblast-like bone-forming cells and the VECs undergo endothelial-to-mesenchymal transformation to form matrix vesicles and microcalcific nodules [17,18,19,20]. This procalcific process is counter-balanced simultaneously by circulating calcification inhibitors including matrix Gla protein (MGP), γ-carboxyglutamic acid-rich protein and Vitamin K-dependent protein, all of which inhibit bone morphogenetic protein (BMP) signalling [21]. Another potent circulating calcification inhibitor is Fetuin-A which binds to calcium and phosphate ions, stabilizing them and preventing cell uptake of the ions [22]. Dysregulation of this mechanism would lead to pathological cardiovascular calcification.
In patients with calcific aortic valve disease, MGP levels have been shown to be significantly depressed compared to patients with normal valves [23]. MGP activity depends on its carboxylation status and vitamin K availability. The use of warfarin, a vitamin K epoxide reductase and γ-carboxylase inhibitor, downregulates MGP activity and has been demonstrated to be a contributing factor to CVC [24]. Furthermore, deficiency in Fetuin-A has also been found to be implicated in aortic valve calcification [17, 22].
The role of BMPs is to stimulate osteoblasts and initiate calcium deposition and bone formation by activating Smad and Wnt/β-catenin signalling and upregulate the expression of Msx2, an osteochondrogenic transcription factor. These signalling pathways ultimately lead to the expression of master osteoblast transcription factor Runx2 [25]. Cells committed to an osteoblastic lineage, as in VICs, will secrete calcification-related protein in response to Runx2, causing valvular calcification [26]. The endothelial-to-mesenchymal transition of VECs can also lead to differentiation to osteoblast-like cells, resulting in a similar response to that of VICs which further contributes to the calcification process. Additionally, the presence of transforming growth factor-β, β-catenin signalling and transcription factor Msx2 are able to stimulate VECs to migrate into surrounding tissues and contribute further to calcification [20, 25].
Progenitor cells have been found to populate normal aortic valves and may also partake in the CVC process [27]. In porcine aortic valves, mesenchymal progenitor cells were found to possess the ability to differentiate into osteoblast-like cells [28]. An environment that favours calcification may be a further driving factor for osteogenic differentiation of these cells, contributing to CVC [29]. Endothelial progenitor cells, on the other hand, plays a role in repairing damaged endothelium by secreting proliferating factors and promoting the migration of resident endothelial cells [30]. Abnormal function of these cells would yield the repair process ineffective and cause abnormal calcification.
Aberrant Immune Response and Inflammation
The pathophysiology of CVC may involve an aberrant immunomodulatory response supported by the observation of leucocyte and macrophage infiltration in explanted calcified human aortic valve compared to the trace amount of macrophages found in normal aortic valves [17]. Inflammatory cell infiltration was observed more frequently at sites where VECs were activated, increasing the concentration of adhesion molecules and facilitating monocyte and macrophage recruitment to the valve [31,32,33]. Enhanced recruitment of inflammatory cells leads to the secretion of pro-inflammatory cytokines and the release of matrix metalloproteinases and cysteine endoproteases. These enzymes break down collagen and elastin causing disruption to the normal valvular architecture [18, 34].
There is also evidence to suggest that lipoprotein recruitment during endothelial injury and the retention of lipids encourage a chronic low-grade inflammatory process and may precede the pathologic mineralisation [35]. Oxidative stress and oxidisation of low-density lipoproteins have been found to be related to the degree of inflammation and fibrocalcific remodelling of the valves by stimulating fibroblasts to release matrix vesicles [36,37,38]. The production of reactive oxygen species in the vicinity of calcified areas also promotes the osteogenic potential of VICs and has the potential to activate the innate immune response [39, 40]. The adaptive immune response may also be activated concurrently during the calcification process evidenced by the presence of activated CD8+ T cells [41]. Hence, it is very likely that both the innate and adaptive immune responses are actively involved in the calcification process.
Matrix Remodelling and Neovascularisation
In patients with CVC, there is evidence to suggest that abnormal matrix deposition and valvular fibrosis contribute to valve calcification. Activated VICs secrete extracellular matrix to maintain valve function and elasticity but the deposition of matrix substances is often haphazard which leads to altered biomechanical properties of the valve [3]. The resultant changes to valve stiffness may further augment phenotypic transition of VICs to osteoblast-like cells [42,43,44]. In addition, experimental models of aortic valve calcification have demonstrated raised pro-fibrotic signalling molecules such as transforming growth factor-β and thrombospondin-2, contributing to fibrocalcific remodelling of the valve leaflets [45, 46].
In contrast to a healthy avascular human aortic valve, calcified valves possess their own tiny vasculature [47]. Histological studies have identified a subgroup of cells that express pro-angiogenic factors Tie-2 and vascular endothelial growth factor (VEGF) receptor 2; these cells may represent activated VECs or VECs that have undergone phenotypic transitions [48]. The downregulation of angiogenic inhibitors also have an equally important role in neovascularisation of these calcified valves.
The presence of mast cells has been identified in calcified valves and plays a pivotal role in the release of VEGF (pro-angiogenic) while also releasing tryptase which degrades endostatin (angiogenesis inhibitor) [47]. Reduced expression of chondromodulin-I, an angiogenic inhibitor, has also been observed and is associated with increased VEGF and periostin. Periostin can stimulate the formation of capillary tube-like structures and have previously been implicated in calcified aortic valves [49]. Once neovascularisation is achieved, the vasculature network expedites the transfer of inflammatory cells and pro-calcifying molecules, further contributing to calcification.
Clinical Characteristics and Diagnosis of AS
Clinical Features
Patient evaluation should always include a thorough patient history and clinical examination, particularly auscultation of the heart sounds and looking for signs of heart failure. Patients with aortic sclerosis or mild to moderate AS are usually asymptomatic and the clinical suspicion for aortic valve disease is usually raised when a systolic murmur is heard on clinical examination.
A classical harsh crescendo-decrescendo systolic murmur is audible on auscultation, loudest at the aortic area (right sternal edge, second intercostal space) with the presence of a single second heart sound. The absence of radiation to the carotid arteries and a wide pulse pressure would suggest AS rather than aortic sclerosis. Symptoms occur particularly when patients have other comorbidities or in cases where there is severe AS leading to left ventricular dysfunction. The described symptoms are usually dyspnoea, syncope or angina.
Investigations
Electrocardiography (ECG) may be useful in demonstrating the impact of AS on the left ventricle. Although the findings are non-specific, there may be ECG evidence of left ventricular hypertrophy with a strain pattern (increased R wave amplitude in left-sided leads and increased depth of S wave in right-sided leads) and left atrial enlargement. Chest radiograph usually reveals a normal cardiac shadow since the left ventricular hypertrophy in AS is concentric but will manifest as cardiomegaly when systolic failure occurs.
A Doppler echocardiography is a useful modality in assessing the haemodynamic severity of AS by analysing the peak aortic jet velocity, aortic valve area (AVA) and the mean transvalvular pressure gradient (mean gradient). AS may be visualised as thickened valve leaflets with a restrictive opening causing increased peak aortic jet velocity and mean gradient. The resultant impact of AS on cardiac geometry and function, particularly the left ventricle can also be assessed simultaneously and may provide important prognostic information. Where a transthoracic echocardiography (TTE) is suboptimal, a transoesophageal echocardiography (TOE) should be considered. Particularly when performing the valvular procedure, TOE can be used to monitor the function and results of the valve post-implantation or repair [50].
Exercise testing may also be used in patients with non-specific symptoms or those who claim to be asymptomatic. It can also provide useful information for patients regarding appropriate levels of physical activity and participation in sports. In patients with AS and mitral regurgitation, exercise echocardiography may be used to evaluate prognostic impact of the disease [51]. An alternative for stress testing is by using low-dose dobutamine stress echocardiography which can assess coronary flow reserve (ratio of maximum increase in blood flow through the coronary arteries to normal resting flow) and severity of AS, particularly in low-flow low-gradient AS [52, 53].
Imaging modalities with multi-slice computed tomography (MSCT) and cardiac magnetic resonance (CMR) may also be utilised to evaluate severity of valve disease in patients with inadequate echocardiographic quality. The high resolution of MSCT allows calcium load to be quantified and scored using the Agatston modified method, which may be useful in predicting haemodynamic severity and clinical outcomes [54, 55]. CMR is equally useful in predicting severity of disease by evaluating myocardial fibrosis and ventricular volumes and systolic function [50].
Invasive modalities include coronary angiography and cardiac catheterisation. Coronary angiography is indicated in suspected coronary artery disease, left ventricular systolic dysfunction or patients with one or more cardiovascular risk factors within the context of severe valvular disease to determine if concomitant coronary revascularisation is needed [50]. Cardiac catheterisation used to be the modality of choice before the advent of echocardiography. This modality allows the measurement of cardiac pressures and cardiac output to assess ventricular performance and severity of valve disease. It should, however, only be considered in patients where echocardiography is inconclusive or discordant with the clinical findings and where reclassification of the valve disease would change therapeutic management. This is due to its association with serious complications such as bleeding and cerebral embolism [56].
Spectrum of Severity in AS
Aortic sclerosis is the preclinical phase of calcific aortic valve disease. It is defined by echocardiographic evidence of focal areas of leaflet calcification causing thickening, without compromising valve function or cardiac blood flow [57]. Patients with aortic sclerosis are clinically asymptomatic but there is an independent association with increased risk of coronary events and cardiovascular death [58].
Mild to moderate AS is diagnosed on the basis of reduced AVA and increased peak aortic jet velocity and mean gradient across the valve. In severe AS, specific haemodynamic parameters on echocardiography would include a peak aortic jet velocity of ≥4 ms, a transvalvular mean pressure gradient of ≥40 mmHg and a calculated aortic valve area ≤ 1.0 cm2[50]. Patients may be asymptomatic even in severe AS and should undergo stress testing to delineate the disease severity.
While the majority of severe AS would manifest with the haemodynamic parameters previously described, a subgroup of patients may have low peak aortic jet velocity and mean gradient despite a small AVA. The most common cause is a low-flow state (low-flow low-gradient AS), where there is a reduction in stroke volume (≤35 mL/m2) related to left ventricular systolic dysfunction. Two subtypes exist depending on the left ventricular ejection fraction; low-flow, low-gradient AS with reduced ejection fraction (<50%) and low-flow, low-gradient AS with preserved ejection fraction (≥50%). The diagnosis in patients with the latter disease where ejection fraction is paradoxically preserved is challenging and will require MSCT to evaluate the degree of valve calcification which corroborates stenosis severity [54, 55, 59]. Where ejection fraction is reduced, low-dose dobutamine stress echocardiography is recommended to distinguish true severe aortic stenosis from pseudosevere aortic stenosis (defined by increased AVA to >1.0cm2 with flow normalisation) [50].
Finally, another group of patients will have echocardiographic evidence of small AVA (≤1.0cm2) but with normal flow (normal flow, low-gradient AS). These patients generally have only moderate aortic stenosis with better outcomes compared to those with high gradient AS or low-flow, low-gradient AS [54, 60,61,62]. Again, MSCT can be considered to quantify calcium burden to confirm severity of stenosis.
Management of AS
At present, aortic valve replacement (AVR) is the only available treatment for patients with symptomatic severe AS. This procedure may be performed surgically or percutaneously via a catheter, a procedure known as transcatheter aortic valve implantation (TAVI). While some studies and trials have suggested statins and angiotensin converting enzyme inhibitors (ACE-i) to be potential pharmacotherapeutic agents in preventing or slowing the calcification process, the evidence behind medical management remains inconclusive.
The decision for the need of an intervention is dependent on severity of disease and patient symptoms. Patients with symptomatic severe AS with evidence of left ventricular function compromise should be considered for intervention unless it has been deemed that the risks of the intervention outweigh any benefit, and especially so if it is unlikely to be of any benefit. Risk stratification tools such as the European System for Cardiac Operative Risk Evaluation (EuroSCORE) and Society of Thoracic Surgery (STS) risk calculator may be used by the Heart Team in deciding between surgical AVR and TAVI in patients at high surgical risk [50].
Surgery
Since the first successful surgical AVR in 1960, the operative techniques and valve technology have advanced tremendously over the years [63]. Patient outcome and long-term survival have improved significantly despite increasing age and comorbidities of surgically managed patients [64, 65]. The type of valves used include bioprosthetic valves (made from porcine aortic valve or bovine pericardium) and mechanical valves.
Mechanical valves have better durability compared to their bioprosthetic counterpart but with the disadvantage of requiring lifelong anticoagulation due to its propensity for thrombosis. With advancing valve technology, however, durability of bioprosthetic valves has improved remarkably and is nearly comparable to that of mechanical valves. Bioprosthetic valves used to be advocated for patients in older age groups but are now increasingly used in younger patients to avoid anticoagulation. Bioprosthetic valves may be stented or stentless depending on whether the leaflets are mounted to a metal or polymeric ring. Although stentless valves provide better haemodynamics, implantation is more complex and will require longer operative time and duration on cardiopulmonary bypass. Sutureless bioprosthetic valves are also becoming popular as it allows easier and quicker implantation.
Other operative strategies particularly for younger patients include the Ross procedure (also known as the Ross-Yacoub procedure). The procedure involves utilising the patient’s own pulmonary valve to replace the diseased aortic valve followed by replacement of the pulmonary valve with a pulmonary homograft [66,67,68]. Alternatively, an aortic homograft may be implanted to replace the diseased aortic valve. These procedures are performed comparatively less than the traditional AVR with congenital aortic stenosis being the most common indication for the Ross procedure.
A minimally invasive operative strategy has also been developed by using a mini-sternotomy incision to gain access to the aortic valve and is a feasible option in patients who undergo an isolated AVR procedure. Although it is associated with a similar mortality, there is reduction in resource utilisation and post-procedural morbidity [65].
Transcatheter Aortic Valve Implantation (TAVI)
TAVI is a minimally invasive procedure involving the insertion of a bioprosthetic aortic valve by using a catheter. The catheter insertion may be transfemoral, transapical or transaortic to gain access to the native stenosed aortic valve. Most TAVI procedures are performed using the transfemoral approach as it is associated with lower mortality rates and quicker recovery.
Two types of transcatheter valves have been studied rigorously to date; balloon-expandable (BE) and self-expanding (SE) valves. The CHOICE trial, which compared the two valve types in high-risk patients with severe aortic stenosis, demonstrated that BE valves were more successful with less residual aortic regurgitation and conduction disturbances requiring permanent pacing [69].
TAVI is an AVR option particularly in high-risk patients unsuitable for surgery but is now being extended to patients in the intermediate risk groups. Although TAVI is a relatively safe procedure, some of the complications of TAVI include paravalvular aortic regurgitation, cardiac conduction disturbances and heart block requiring permanent pacemaker implantation, bleeding, acute kidney injury and very rarely, stroke, coronary obstruction and aortic rupture. Paravalvular aortic regurgitations remains the most notable complication due to its link to increased mortality with severity of the leak [70, 71]. Generally, TAVI has been a very successful therapy with outcomes comparable to that of surgical AVR and it is possible that this procedure will eventually be advocated to patients in the low risk categories.
Surgical AVR vs TAVI
The choice between surgical AVR and TAVI is becoming more challenging as TAVI is being extended to low-risk patients, and multiple factors including anatomical considerations and performing concomitant revascularisation or valvular procedures should be taken into consideration. The results from Placement of Aortic Transcatheter Valves (PARTNER) trial has greatly evolved the use of TAVI. Particularly in high-risk patients who would otherwise be unsuitable for surgery and intermediate risk patients, there were no significant differences in short and long-term outcomes between surgical AVR and TAVI. However, surgical AVR had the long-term advantage over TAVI by having fewer rehospitalisations and reinterventions, and particularly over transthoracic TAVI with fewer incidences of death or disabling stroke [70].
The use of TAVI in low-risk patients has recently shown superiority over surgical AVR in mortality outcome, stroke and rehospitalisation at 1 year and it is possible that the use of TAVI will continue to gain favour even in the low-risk cohort. Complication rates in this group remain similar to moderate and high-risk groups and the long-term outcomes remain to be evaluated [72]. It is likely with progressive improvement in valve technology, the complication rates will decrease, and it is possible that surgical AVR will only be reserved for a specific group of patients with complicated anatomy or where other concomitant cardiac procedures are being considered.
There is, however, a subgroup of patients where TAVI may be futile or of limited benefit. This may be the case in frail elderly patients where their quality of life and lifespan are limited by their performance status and coexisting medical comorbidities. In this patient group, the heart valve team may decide that the benefit of TAVI may be limited and a palliative care approach may be appropriate, taking into account the values and wishes of patients and family members when making this decision.
Balloon Aortic Valvuloplasty
Balloon aortic valvuloplasty (BAV) is reserved for haemodynamically unstable patients or patients with symptomatic severe AS who require urgent non-cardiac operation. It may also be used as a bridge to surgical AVR or TAVI or even as a diagnostic mean to decide whether AVR is appropriate in patients with multiple contributing factors to the clinical symptoms. The benefits provided by BAV are short-lived and is therefore, not a definitive therapy for AS [50]. BAV may also be used as a palliative approach as there has been previous evidence to suggest that BAV may provide a short-term benefit to quality of life and functional capacity [73].
Mitral Annular Calcification (MAC)
The term “mitral” was first suggested by Walmsley due to its resemblance to a bishop’s mitre [74]. The mitral valve is seated between the left atrium and the left ventricle, preventing backflow of blood to the left atrium during left ventricular contraction. Its function is served by the orchestration of all its components (valve leaflets, papillary muscles, chordae tendinae and fibrous annulus) with the help of the atrial and ventricular musculature [75].
The mitral annulus marks the hinge line for the valvular leaflets and follows a D-shape, with the straight border of the anterior mitral leaflet forming part of the posterior aortic root. Where the aortic valve communicates with the anterior mitral leaflet via expansions of fibrous tissue forms the right and left trigonal structures. The right trigone is a route of passage for the atrioventricular bundle which explains the association between MAC/mitral valve disease with cardiac conduction disturbances [75].
MAC and its association with complete heart block was first described by Bonninger in 1908 [76]. To shed light on the pathophysiology of MAC, Dewitzky performed a detailed pathologic description of 36 cases and found a close resemblance to aortic valve calcification described by Moenckeberg in 1904 [77]. Moreover, MAC was a common autopsy finding in older people and was then considered to be primarily caused by rheumatic heart disease [78, 79].
Clinical Features
Mitral annular calcification (MAC) involves chronic calcification of the mitral valve fibrous annulus and has a tendency to affect the posterior mitral annulus. The anterior mitral annulus and leaflet are usually spared in MAC, in contrast to rheumatic mitral valve disease where the predominant pathology is that of the anterior leaflet and causes commissural fusion [80]. The pathophysiology observed in MAC draws similarity to those previously discussed in calcific aortic valve disease and shares associated atherosclerotic risk factors. Hence, concomitant calcific aortic valve disease and atherosclerotic cardiovascular disease are not uncommon with MAC. Other associated diseases with MAC include stroke, coronary artery disease, cardiac arrhythmias and endocarditis [81,82,83,84,85,86].
Patients with MAC are generally asymptomatic, and the disease is usually diagnosed incidentally. MAC does not typically contribute to haemodynamic disturbances or affect left ventricular or mitral valve function. However, extensive disease may lead to functional mitral stenosis, mitral regurgitation or a mixed disease process where both pathologies are manifested [87, 88].
Investigation and Diagnosis
Echocardiography is considered to be the principal imaging modality in diagnosing and characterising mitral valve diseases. MAC appears as an echo-dense, irregular, lumpy shelf-like structure affecting the posterior mitral valve annulus with acoustic shadowing on echocardiography. Occasionally, the anterior annulus or interannular fibrosa are also affected [89]. A rare variant of MAC known as caseous calcification is less echo-dense than the typical MAC and appears as a central echolucent area without acoustic shadowing.
Severity is generally divided into mild, moderate and severe depending on the echodensity and the extent of disease to involve the left ventricular inflow tract due to restricted mobility of the affected leaflet [89]. Due to its low specificity in distinguishing calcification from dense collagen, the use of echocardiography should be complemented by MSCT and CMR to quantitate the severity of the calcification [89].
Management of MAC
MAC does not usually require any intervention unless there is evidence of symptomatic concomitant severe mitral stenosis and mitral regurgitation. In fact, surgery should be avoided in patients with severe MAC due to an increased risk of complications such as left ventricular rupture and injury to the circumflex artery [90, 91]. Another indication for valve intervention may include recurrent thromboembolism despite anticoagulation or documented calcific emboli.
In patients with symptomatic severe mitral stenosis or severe mitral regurgitation, mitral valve surgery should be performed. The surgical approach involves decalcification of the mitral annulus followed by reconstruction and if possible, conservation and repair of the mitral valve or otherwise replaced with a prosthetic valve [90, 91]. The benefits of the operation should be carefully weighed against its risks as these patients tend to be older with multiple comorbidities. The use of percutaneous mitral commissurotomy (PMC) is not indicated in MAC since there is no commissural fusion and should be reserved for patients with rheumatic mitral valve disease.
Transcatheter mitral valve insertion (TMVI) may be considered in patients at very high-risk for surgery and deemed unsuitable for surgical intervention. Characteristics of MAC should be taken into consideration when performing this procedure. Circumferential calcification is preferred since it provides good anchorage for the prosthesis. The lack of this can lead to potential displacement of the anterior leaflet into the LVOT, increasing the risk of periprosthetic leak [89]. A heavy calcium burden also increases the risk of annular rupture and calcium embolization and stroke during the procedure [89]. At present, there is limited data to evaluate the outcome and safety of TMVI and more studies are needed to compare its outcomes against surgical mitral valve replacement.
AF is a common complication in mitral valve diseases and MAC and predisposes patients to left atrial thrombosis and potential for embolism. Classically, warfarin is the only medication licensed for use in valvular AF with specific International Normalised Ratio (INR) targets depending on the valvular pathology. The direct oral anticoagulants (DOACs) have been gaining favour in recent years as no INR monitoring is required and there is emerging evidence to suggest these medications are safe to use in valvular heart disease. In fact, there is some evidence to suggest that it may reduce calcium deposition and progression compared to warfarin [92]. However, larger studies will be required to validate this finding.
Future Research
It remains challenging to decide which patients will benefit most from an early therapeutic intervention. The use of blood biomarkers such as B-natriuretic peptide (BNP) has previously been suggested to evaluate left ventricular function or left ventricular strain as an indirect measure of disease severity, particularly in asymptomatic patients [93, 94]. However, the cut-off value to identify patients at high risk of progression of disease is unclear with a previous study suggesting the use of BNP ratio (age and sex-adjusted measured BNP divided by expected value) instead. BNP ratio > 1 may be an independent predictor of mortality in AS, even in asymptomatic patients [94]. The limitations of the use of these blood biomarkers, however, are that they are often non-specific and should be used in conjunction with current investigative modalities. Further research is required to validate the use of these blood biomarkers in clinical practice.
Improved cardiac imaging with magnetic resonance is also promising in risk-stratifying patients. In severe AS, myocardial fibrosis has been documented on CMR and the quantification of myocardial fibrosis may be useful in recommending early therapeutic intervention, particularly in asymptomatic patients [6]. Further studies are needed to standardise CMR findings and their relationship to severity of disease and establishing a threshold at which valve intervention would be most beneficial in preventing further myocardial dysfunction. At present, the use of CMR is also limited by its cost and low availability but this will likely change in the foreseeable future.
While valve replacement is the mechanical solution to a calcified valve, strategies to improve clinical outcomes post-valve replacement are given little attention. Often, there is evidence of left ventricular dysfunction from chronic remodelling in response to valvular disease, and left ventricular function usually improves minimally after valve replacement. Research into adjunctive medical therapies to help improve left ventricular function and reverse the remodelling process could potentially reduce symptom experience and improve quality of life.
As previously discussed, there remains no effective pharmacotherapy to delay or halt the progression of calcification. While medications such as ACE-i and statins have previously been suggested, the evidence is weak and non-conclusive. In addition, the use of statins in randomised-controlled trials has previously shown no benefit [95,96,97]. The disease burden of CVC will continue to increase, and current research should, therefore, focus on effective prophylactic pharmacotherapy.
Conclusion
The disease burden of CVC will continue to increase globally due to better life expectancy and an ageing population. A pharmacotherapy to prevent or slow the progression of calcification has yet to be discovered and valve replacement remains the only effective treatment modality, particularly in calcific AS. Minimally invasive techniques with TAVI are increasingly being utilised and progressively replacing surgical interventions. The role of the heart valve team is crucial in deciding which patients will benefit most from an intervention by taking into account patient symptoms, cardiac function, coexisting medical conditions and their functional baseline. The future of TAVI is promising and by reducing the complications related to the procedure, it will eventually be an option for low-risk patients.
Funding
The authors received no specific funding for this work.
Disclosures
No potential conflict of interest was reported by the authors.
Author Contributions
All authors contributed to the design and implementation of the research and to the writing of the manuscript.
References
Vaslef SN, Roberts WC. Early descriptions of aortic valve stenosis. Am Heart J. 1993 May;125(5 Pt 1):1465–74.
Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Executive summary: heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013 Jan 1;127(1):143–52.
Coffey S, Cox B, Williams MJ. The prevalence, incidence, progression, and risks of aortic valve sclerosis: a systematic review and meta-analysis. J Am Coll Cardiol. 2014 Jul 1;63(25 Pt A):2852–61.
Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006 Sep 16;368(9540):1005–11.
Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005 Feb 22;111(7):920–5.
Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, et al. Calcific aortic stenosis. Nat Rev Dis Primers. 2016 Mar 3;2:16006.
Boon A, Cheriex E, Lodder J, Kessels F. Cardiac valve calcification: characteristics of patients with calcification of the mitral annulus or aortic valve. Heart. 1997 Nov;78(5):472–4.
Allison MA, Cheung P, Criqui MH, Langer RD, Wright CM. Mitral and aortic annular calcification are highly associated with systemic calcified atherosclerosis. Circulation. 2006 Feb 14;113(6):861–6.
Fox CS, Vasan RS, Parise H, Levy D, O’Donnell CJ, D’Agostino RB, et al. Mitral annular calcification predicts cardiovascular morbidity and mortality: the Framingham Heart Study. Circulation. 2003 Mar 25;107(11):1492–6.
Correia J, Rodrigues D, da Silva AM, Sáe Melo A, Providência LA. Massive calcification of the mitral valve annulus in an adolescent with Marfan syndrome. A case report. Rev Port Cardiol. 2006 Oct;25(10):921–6.
Schieken RM, Kerber RE, Ionasescu VV, Zellweger H. Cardiac manifestations of the mucopolysaccharidoses. Circulation. 1975 Oct;52(4):700–5.
Sharma R, Pellerin D, Gaze DC, Mehta RL, Gregson H, Streather CP, et al. Mitral annular calcification predicts mortality and coronary artery disease in end stage renal disease. Atherosclerosis. 2007 Apr;191(2):348–54.
Schoen FJ. Evolving concepts of cardiac valve dynamics: the continuum of development, functional structure, pathobiology, and tissue engineering. Circulation. 2008 Oct 28;118(18):1864–80.
Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011 Jun 10;108(12):1510–24.
Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, et al. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011 Oct 18;124(16):1783–91.
Leopold JA. Cellular mechanisms of aortic valve calcification. Circ Cardiovasc Interv. 2012 Aug 1;5(4):605–14.
Kaden JJ, Dempfle CE, Grobholz R, Fischer CS, Vocke DC, Kiliç R, et al. Inflammatory regulation of extracellular matrix remodeling in calcific aortic valve stenosis. Cardiovasc Pathol. 2005 Mar-Apr;14(2):80–7.
Wylie-Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler Thromb Vasc Biol. 2011 Mar;31(3):598–607.
Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009 Aug 28;105(5):408–21.
Piera-Velazquez S, Li Z, Jimenez SA. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am J Pathol. 2011;179:1074–80.
Yao Y, Bennett BJ, Wang X, Rosenfeld ME, Giachelli C, Lusis AJ, et al. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ Res. 2010 Aug 20;107(4):485–94.
Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res. 2011 Jun 10;108(12):1494–509.
Koos R, Krueger T, Westenfeld R, Kühl HP, Brandenburg V, Mahnken AH, et al. Relation of circulating Matrix Gla-Protein and anticoagulation status in patients with aortic valve calcification. Thromb Haemost. 2009 Apr;101(4):706–13.
Lerner RG, Aronow WS, Sekhri A, Palaniswamy C, Ahn C, Singh T, et al. Warfarin use and the risk of valvular calcification. J Thromb Haemost. 2009 Dec;7(12):2023–7.
Boström KI, Rajamannan NM, Towler DA. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ Res. 2011 Aug 19;109(5):564–77.
Johnson RC, Leopold JA, Loscalzo J. Vascular calcification: pathobiological mechanisms and clinical implications. Circ Res. 2006 Nov 10;99(10):1044–59.
Hajdu Z, Romeo SJ, Fleming PA, Markwald RR, Visconti RP, Drake CJ. Recruitment of bone marrow-derived valve interstitial cells is a normal homeostatic process. J Mol Cell Cardiol. 2011 Dec;51(6):955–65.
Chen JH, Yip CY, Sone ED, Simmons CA. Identification and characterization of aortic valve mesenchymal progenitor cells with robust osteogenic calcification potential. Am J Pathol. 2009 Mar;174(3):1109–19.
Leskelä HV, Satta J, Oiva J, Eriksen H, Juha R, Korkiamäki P, et al. Calcification and cellularity in human aortic heart valve tissue determine the differentiation of bone-marrow-derived cells. J Mol Cell Cardiol. 2006 Oct;41(4):642–9.
Richardson MR, Yoder MC. Endothelial progenitor cells: quo vadis? J Mol Cell Cardiol. 2011 Feb;50(2):266–72.
Davies MJ, Treasure T, Parker DJ. Demographic characteristics of patients undergoing aortic valve replacement for stenosis: relation to valve morphology. Heart. 1996 Feb;75(2):174–8.
Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, et al. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007 Jan 23;115(3):377–86.
Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, et al. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007 Dec 11;116(24):2841–50.
Rabkin E, Aikawa M, Stone JR, Fukumoto Y, Libby P, Schoen FJ. Activated interstitial myofibroblasts express catabolic enzymes and mediate matrix remodeling in myxomatous heart valves. Circulation. 2001 Nov 20;104(21):2525–32.
Abdelbaky A, Corsini E, Figueroa AL, Subramanian S, Fontanez S, Emami H, et al. Early aortic valve inflammation precedes calcification: a longitudinal FDG-PET/CT study. Atherosclerosis. 2015 Feb;238(2):165–72.
Mohty D, Pibarot P, Després JP, Côté C, Arsenault B, Cartier A, et al. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler Thromb Vasc Biol. 2008 Jan;28(1):187–93.
Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol. 1999 May;19(5):1218–22.
Côté C, Pibarot P, Després JP, Mohty D, Cartier A, Arsenault BJ, et al. Association between circulating oxidised low-density lipoprotein and fibrocalcific remodelling of the aortic valve in aortic stenosis. Heart. 2008 Sep;94(9):1175–80.
Miller JD, Chu Y, Brooks RM, Richenbacher WE, Peña-Silva R, Heistad DD. Dysregulation of antioxidant mechanisms contributes to increased oxidative stress in calcific aortic valvular stenosis in humans. J Am Coll Cardiol. 2008 Sep 2;52(10):843–50.
Liberman M, Bassi E, Martinatti MK, Lario FC, Wosniak J Jr, Pomerantzeff PM, et al. Oxidant generation predominates around calcifying foci and enhances progression of aortic valve calcification. Arterioscler Thromb Vasc Biol. 2008 Mar;28(3):463–70.
Arsenault BJ, Boekholdt SM, Dubé MP, Rhéaume E, Wareham NJ, Khaw KT, et al. Lipoprotein(a) levels, genotype, and incident aortic valve stenosis: a prospective Mendelian randomization study and replication in a case-control cohort. Circ Cardiovasc Genet. 2014 Jun;7(3):304–10.
El Husseini D, Boulanger MC, Mahmut A, Bouchareb R, Laflamme MH, Fournier D, et al. P2Y2 receptor represses IL-6 expression by valve interstitial cells through Akt: implication for calcific aortic valve disease. J Mol Cell Cardiol. 2014 Jul;72:146–56.
Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006 Jan;12(1):17–25.
Kaden JJ, Bickelhaupt S, Grobholz R, Haase KK, Sarikoç A, Kiliç R, et al. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulate aortic valve calcification. J Mol Cell Cardiol. 2004 Jan;36(1):57–66.
Nagy E, Andersson DC, Caidahl K, Eriksson MJ, Eriksson P, Franco-Cereceda A, et al. Upregulation of the 5-lipoxygenase pathway in human aortic valves correlates with severity of stenosis and leads to leukotriene-induced effects on valvular myofibroblasts. Circulation. 2011 Mar 29;123(12):1316–25.
Mathieu P, Bouchareb R, Boulanger MC. Innate and adaptive immunity in calcific aortic valve disease. J Immunol Res. 2015;2015:851945.
Capoulade R, Mahmut A, Tastet L, Arsenault M, Bédard É, Dumesnil JG, et al. Impact of plasma Lp-PLA2 activity on the progression of aortic stenosis: the PROGRESSA study. JACC Cardiovasc Imaging. 2015 Jan;8(1):26–33.
Tellis CC, Tselepis AD. The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochim Biophys Acta. 2009 May;1791(5):327–38.
Weiss RM, Lund DD, Chu Y, Brooks RM, Zimmerman KA, El Accaoui R, et al. Osteoprotegerin inhibits aortic valve calcification and preserves valve function in hypercholesterolemic mice. PLoS One. 2013 Jun 6;8(6):e65201.
Baumgartner H, Falk V, Bax JJ, De Bonis M, Hamm C, Holm PJ, et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur Heart J. 2017 Sep 21;38(36):2739–91.
Picano E, Pibarot P, Lancellotti P, Monin JL, Bonow RO. The emerging role of exercise testing and stress echocardiography in valvular heart disease. J Am Coll Cardiol. 2009 Dec 8;54(24):2251–60.
Monin JL, Quéré JP, Monchi M, Petit H, Baleynaud S, Chauvel C, et al. Low-gradient aortic stenosis: operative risk stratification and predictors for long-term outcome: a multicenter study using dobutamine stress hemodynamics. Circulation. 2003 Jul 22;108(3):319–24.
Clavel MA, Magne J, Pibarot P. Low-gradient aortic stenosis. Eur Heart J. 2016 Sep 7;37(34):2645–57.
Clavel MA, Messika-Zeitoun D, Pibarot P, Aggarwal SR, Malouf J, Araoz PA, et al. The complex nature of discordant severe calcified aortic valve disease grading: new insights from combined Doppler echocardiographic and computed tomographic study. J Am Coll Cardiol. 2013 Dec 17;62(24):2329–38.
Clavel MA, Pibarot P, Messika-Zeitoun D, Capoulade R, Malouf J, Aggarval S, et al. Impact of aortic valve calcification, as measured by MDCT, on survival in patients with aortic stenosis: results of an international registry study. J Am Coll Cardiol. 2014 Sep 23;64(12):1202–13.
Omran H, Schmidt H, Hackenbroch M, Illien S, Bernhardt P, von der Recke G, et al. Silent and apparent cerebral embolism after retrograde catheterisation of the aortic valve in valvular stenosis: a prospective, randomised study. Lancet. 2003 Apr 12;361(9365):1241–6.
Nishimura RA, Otto CM, Bonow RO, Carabello BA, Erwin JP 3rd, Guyton RA, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2014 Jun 10;63(22):e57–185.
Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999 Jul 15;341(3):142–7.
Cueff C, Serfaty JM, Cimadevilla C, Laissy JP, Himbert D, Tubach F, et al. Measurement of aortic valve calcification using multislice computed tomography: correlation with haemodynamic severity of aortic stenosis and clinical implication for patients with low ejection fraction. Heart. 2011 May;97(9):721–6.
Mehrotra P, Jansen K, Flynn AW, Tan TC, Elmariah S, Picard MH, et al. Differential left ventricular remodelling and longitudinal function distinguishes low flow from normal-flow preserved ejection fraction low-gradient severe aortic stenosis. Eur Heart J. 2013 Jul;34(25):1906–14.
Jander N, Minners J, Holme I, Gerdts E, Boman K, Brudi P, et al. Outcome of patients with low-gradient “severe” aortic stenosis and preserved ejection fraction. Circulation. 2011 Mar 1;123(8):887–95.
Tribouilloy C, Rusinaru D, Maréchaux S, Castel AL, Debry N, Maizel J, et al. Low-gradient, low-flow severe aortic stenosis with preserved left ventricular ejection fraction: characteristics, outcome, and implications for surgery. J Am Coll Cardiol. 2015 Jan 6;65(1):55–66.
Harken DE, Soroff HS, Taylor WJ, Lefemine AA, Gupta SK, Lunzer S. Partial and complete prostheses in aortic insufficiency. J Thorac Cardiovasc Surg. 1960 Dec;40:744–62.
Lee R, Li S, Rankin JS, O’Brien SM, Gammie JS, Peterson ED, et al. Fifteen-year outcome trends for valve surgery in North America. Ann Thorac Surg. 2011 Mar;91(3):677–84; discussion p 684.
Brown ML, McKellar SH, Sundt TM, Schaff HV. Ministernotomy versus conventional sternotomy for aortic valve replacement: a systematic review and meta-analysis. J Thorac Cardiovasc Surg. 2009 Mar;137(3):670–679.e5.
David TE, Woo A, Armstrong S, Maganti M. When is the Ross operation a good option to treat aortic valve disease? J Thorac Cardiovasc Surg. 2010 Jan;139(1):68–73; discussion 73–5.
Stulak JM, Burkhart HM, Sundt TM 3rd, Connolly HM, Suri RM, Schaff HV, et al. Spectrum and outcome of reoperations after the Ross procedure. Circulation 2010 Sep 21;122(12):1153-1158.
David TE. Reoperations after the Ross procedure. Circulation. 2010 Sep 21;122(12):1139–40.
Abdel-Wahab M, Mehilli J, Frerker C, Neumann FJ, Kurz T, Tölg R, et al. Comparison of balloon-expandable vs self-expandable valves in patients undergoing transcatheter aortic valve replacement: the CHOICE randomized clinical trial. JAMA. 2014 Apr 16;311(15):1503–14.
Leon MB, Smith CR, Mack MJ, Makkar RR, Svensson LG, Kodali SK, et al. Transcatheter or surgical aortic-valve replacement in intermediate-risk patients. N Engl J Med. 2016 Apr 28;374(17):1609–20.
Makkar RR, Thourani VH, Mack MJ, Kodali SK, Kapadia S, Webb JG, et al. Five-year outcomes of transcatheter or surgical aortic-valve replacement. N Engl J Med. 2020 Jan 29;382(9):799–809.
Mack MJ, Leon MB, Thourani VH, Makkar R, Kodali SK, Russo M, et al. Transcatheter aortic-valve replacement with a balloon-expandable valve in low-risk patients. N Engl J Med. 2019 May 2;380(18):1695–705.
Kapadia S, Stewart WJ, Anderson WN, Babaliaros V, Feldman T, Cohen DJ, et al. Outcomes of inoperable symptomatic aortic stenosis patients not undergoing aortic valve replacement: insight into the impact of balloon aortic valvuloplasty from the PARTNER trial (Placement of AoRtic TraNscathetER Valve trial). JACC Cardiovasc Interv. 2015 Feb;8(2):324–33.
Walmsley T. The heart. In: Sharpey-Schafer E, Symington J, Bryce TH, eds. Quain’s elements of anatomy, 11th edn, vol 4, pt 3. London: Longmans, Greens & Co, 1929:42.
Ho SY. Anatomy of the mitral valve. Heart. 2002 Nov;88 Suppl 4(Suppl 4):iv5–10.
Bonninger M. Dtsch Med Wochenschr. 1908(34):2292. (a) Bluttransfusion bei pernizioser anamie: (b) Zwei Falle von Herzblock. Dtsch Med Wochenschr. 1908. 34: 2292.
Dewitzky W. Uber den Bau und die Enstehung verschiedener Formen der chronischen Veraderungen in der Herzklappen [German]. Virchow Arch Path Anat Physiol. 1910;199:273.
Korn D, Desanctis RW, Sell S. Massive calcification of the mitral annulus. A clinicopathological study of fourteen cases. N Engl J Med. 1962 Nov 1;267:900–9.
Roberts WC. Morphologic features of the normal and abnormal mitral valve. Am J Cardiol. 1983 Mar 15;51(6):1005–28.
Fulkerson PK, Beaver BM, Auseon JC, Graber HL. Calcification of the mitral annulus: etiology, clinical associations, complications and therapy. Am J Med. 1979 Jun;66(6):967–77.
Kizer JR, Wiebers DO, Whisnant JP, Galloway JM, Welty TK, Lee ET. Mitral annular calcification, aortic valve sclerosis, and incident stroke in adults free of clinical cardiovascular disease: the Strong Heart Study. Stroke. 2005 Dec.;36(12):2533–7.
Acarturk E, Bozkurt A, Cayli M, Demir M. Mitral annular calcification and aortic valve calcification may help in predicting significant coronary artery disease. Angiology. 2003 Sep–Oct;54(5):561–7.
Mellino M, Salcedo EE, Lever HM, Vasudevan G, Kramer JR. Echographic-quantified severity of mitral anulus calcification: prognostic correlation to related hemodynamic, valvular, rhythm, and conduction abnormalities. Am Heart J. 1982 Feb;103(2):222–5.
Nair CK, Sketch MH, Desai R, Mohiuddin SM, Runco V. High prevalence of symptomatic bradyarrhythmias due to atrioventricular node-fascicular and sinus node-atrial disease in patients with mitral anular calcification. Am Heart J. 1982 Feb;103(2):226–9.
Mainigi SK, Chebrolu LH, Romero-Corral A, Mehta V, Machado RR, Konecny T. Prediction of significant conduction disease through noninvasive assessment of cardiac calcification. Echocardiography. 2012 Oct;29(9):1017–21.
Watanakunakorn C. Staphylococcus aureus endocarditis on the calcified mitral annulus fibrosus. Am J Med Sci. 1973 Sep;266(3):219–23.
Ramirez J, Flowers NC. Severe mitral stenosis secondary to massive calcification of the mitral annulus with unusual echocardiographic manifestations. Clin Cardiol. 1980 Aug;3(4):284–7.
Aronow WS, Kronzon I. Correlation of prevalence and severity of mitral regurgitation and mitral stenosis determined by Doppler echocardiography with physical signs of mitral regurgitation and mitral stenosis in 100 patients aged 62 to 100 years with mitral anular calcium. Am J Cardiol. 1987 Nov 15;60(14):1189–90.
Abramowitz Y, Jilaihawi H, Chakravarty T, Mack MJ, Makkar RR. Mitral annulus calcification. J Am Coll Cardiol. 2015 Oct 27;66(17):1934–41.
Okada Y. Surgical management of mitral annular calcification. Gen Thorac Cardiovasc Surg. 2013 Nov;61(11):619–25.
Carpentier AF, Pellerin M, Fuzellier JF, Relland JY. Extensive calcification of the mitral valve anulus: pathology and surgical management. J Thorac Cardiovasc Surg. 1996 Apr;111(4):718–729; discussion 729–30.
Di Lullo L, Tripepi G, Ronco C, D’Arrigo G, Barbera V, Russo D, et al. Cardiac valve calcification and use of anticoagulants: preliminary observation of a potentially modifiable risk factor. Int J Cardiol. 2019 Mar 1;278:243–9.
Bergler-Klein J, Klaar U, Heger M, Rosenhek R, Mundigler G, Gabriel H, et al. Natriuretic peptides predict symptom-free survival and postoperative outcome in severe aortic stenosis. Circulation. 2004 May 18;109(19):2302–8.
Clavel MA, Malouf J, Michelena HI, Suri RM, Jaffe AS, Mahoney DW, et al. B-type natriuretic peptide clinical activation in aortic stenosis: impact on long-term survival. J Am Coll Cardiol. 2014 May 20;63(19):2016–25.
Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, et al. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005 Jun 9;352(23):2389–97.
Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008 Sep 25;359(13):1343–56.
Chan KL, Teo K, Dumesnil JG, Ni A, Tam J, ASTRONOMER Investigators. Effect of Lipid lowering with rosuvastatin on progression of aortic stenosis: results of the aortic stenosis progression observation: measuring effects of rosuvastatin (ASTRONOMER) trial. Circulation. 2010 Jan 19;121(2):306–14.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Kho, J., Petrou, M. (2022). Valve Calcification (Aortic and Mitral). In: Henein, M. (eds) Cardiovascular Calcification. Springer, Cham. https://doi.org/10.1007/978-3-030-81515-8_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-81515-8_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-81514-1
Online ISBN: 978-3-030-81515-8
eBook Packages: MedicineMedicine (R0)