
Abstract
In eukaryotic organisms, subcellular protein location is critical in defining protein function and understanding sub-functionalization of gene families. Some proteins have defined locations, whereas others have low specificity targeting and complex accumulation patterns. There is no single approach that can be considered entirely adequate for defining the in vivo location of all proteins. By combining evidence from different approaches, the strengths and weaknesses of different technologies can be estimated, and a location consensus can be built. The Subcellular Location of Proteins in Arabidopsis database (http://suba.live/) combines experimental data sets that have been reported in the literature and is analyzing these data to provide useful tools for biologists to interpret their own data. Foremost among these tools is a consensus classifier (SUBAcon) that computes a proposed location for all proteins based on balancing the experimental evidence and predictions. Further tools analyze sets of proteins to define the abundance of cellular structures. Extending these types of resources to plant crop species has been complex due to polyploidy, gene family expansion and contraction, and the movement of pathways and processes within cells across the plant kingdom. The Crop Proteins of Annotated Location database (http://crop-pal.org/) has developed a range of subcellular location resources including a species-specific voting consensus for 12 plant crop species that offers collated evidence and filters for current crop proteomes akin to SUBA. Comprehensive cross-species comparison of these data shows that the sub-cellular proteomes (subcellulomes) depend only to some degree on phylogenetic relationship and are more conserved in major biosynthesis than in metabolic pathways. Together SUBA and cropPAL created reference subcellulomes for plants as well as species-specific subcellulomes for cross-species data mining. These data collections are increasingly used by the research community to provide a subcellular protein location layer, inform models of compartmented cell function and protein–protein interaction network, guide future molecular crop breeding strategies, or simply answer a specific question—where is my protein of interest inside the cell?
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Arabidopsis
- SUBA
- CropPAL
- SUBAcon
- Crops
- Protein localization
- Subcellular location
- System biology
- Protein location
- Subcellular compartment
4.1 Introduction
4.1.1 The Historical Context of Subcellular Location in Proteomics
Subcellular proteomics are an integral part of plant proteomics due to the tight connection between protein location within cells and their function (Weckwerth et al. 2008; Millar et al. 2009; Joshi et al. 2011). The importance of subcellular location of different isozymes of proteins in plants has been long recognized (Gottlieb 1982). However, until recently, most conventional methodologies for determining protein locations in the cell have been labor intensive. Methodologies such as gold particle marking and immunological tagging followed by microscopy were used over decades, but both approaches are very labor and cost intensive. More recently, PCR and cloning techniques, overexpression vectors, fluorescent protein (FP) chimerics and expression in alternative hosts allowed the localization of lowly expressed proteins in difficult to study organisms (Chiu et al. 1996). These studies, although not high throughput, have been a big step forward for rare proteins.
Using mass spectrometry (MS) of tandem mass spectrometry (MS/MS) to build comprehensive subcellular proteome sets came into the picture with improving organellar extractions and MS detection for peptides (Heazlewood et al. 2005). The organelles and compartments that attracted most interest were the major energy organelles such as the mitochondrion (Kruft et al. 2001; Millar et al. 2001) and the chloroplast (Peltier et al. 2001), which had to be physically extracted and purified away from the cytosol, nucleus, and secretory parts of the cells. Following on, research focusing on the secretory system needed to distinguish between endoplasmic reticulum (ER), Golgi apparatus, and tonoplast (vascular membrane) within the cell and the plasma membrane surrounding the cell. Finally, extracellular proteins secreted from cells into the apoplast were studied. With higher resolution of microscopes and better separation techniques, finer points of differentiation began to distinguish suborganellar compartments of these organelles, including protein localizations in the inner and outer membranes, the intracellular space and the matrix of mitochondria (Millar et al. 2001; Werhahn et al. 2001), thylakoids in chloroplasts (Schubert et al. 2002), cytoskeletal structures (Hamada et al. 2013), and specific sections of the secretory system (Drakakaki et al. 2012; Heard et al. 2015). With the improvement of MS/MS sensitivity, over time more proteins could be identified and traced in these purified organellar separations.
The Localization of Organelle Protein by Isotope Tagging (LOPIT) approach offered an alternative for defining the localization of membrane proteins without physical subcellular separation to purity (Dunkley et al. 2004). This method, based on profiling protein abundances in size separated fractions, has been used to map a significant number of proteins from mitochondria, plastids, ER, and secretory compartments (Nikolovski et al. 2012). LOPIT has recently been optimized for subcellular protein mapping and is used in a variety of global subcellular mapping projects spanning plant and disease biology (Mulvey et al. 2017).
When considering all subcellular localization methods as a whole, the majority of data today are derived from proteomic MS methodology due to the high-throughput nature of the approach. A subcellular proteomics mapping study typically produces more than 1000 subcellular localizations (Heard et al. 2015; Nguyen-Kim et al. 2016; Senkler et al. 2017). In contrast, high-throughput fluorescent protein (FP) studies report between 10 and 100 (Cutler et al. 2000; Boruc et al. 2010; Inze et al. 2012) with the largest study to date offering 148 protein localizations (Koroleva et al. 2005). The emerging importance of large data mining requires that subcellular location data are available as a global data set using all available information, making data aggregations for subcellular proteomics increasingly popular resources. A growing number of databases available have generated subcellular proteomics data sets containing over 40,000 experimental localizations spanning different methodologies. These resources often fill gaps in experimental data and compare the advantages as well as disadvantages of each method using computational strategies.
4.1.2 Collation of Arabidopsis Subcellular Data Established Subcellular Proteomics
Subcellular proteomics has been refined and improved through the aggregation of localization data. In plant biology, this first started to take shape for the model plant Arabidopsis. Today, the SUBcellular location database for Arabidopsis proteins (SUBA, http://suba.live) in its fourth generation is a substantial collection of manually curated published data sets of large-scale subcellular proteomics (MS/MS), FP visualization, protein–protein interaction (PPI), and subcellular targeting calls from 22 prediction programs as well as a consensus algorithm (SUBAcon).
The collection started with studies on the mitochondrial proteome more than 10 years ago (Heazlewood et al. 2004) when an MS study of the mitochondrial organelle revealed a large number of low-abundance proteins that had been predicted to localize elsewhere in the cell. This led to the generation of the Arabidopsis Mitochondrial Protein Database (AMPDB) that offered an overview of the detailed MS data sets from 17 published mitochondrial studies as well as predictions from six subcellular location algorithms (Heazlewood and Millar 2005). While similar efforts for the plastid were realized as the Plastid Proteome DataBase (PPDB) at the same time (Friso et al. 2004), it became clear that rapid expansion of data for many organelles required the establishment of a one-stop data collection hub for subcellular compartmentalization data. An initial data collation and categorization showed surprisingly little overlap between data sets from different researchers, and their combination seemed crucial for large-scale data mining (Heazlewood et al. 2005). It was then when the SUBA acronym was introduced and the data aggregation quickly revealed that protein families, subsets, and isoforms with distinct subcellular location patterns existed. The analysis of the data also meta-assessed the reliability of experimental data and indicated that most experimental methods are more variable and error-prone than the wider research community presumed.
Since then, the Arabidopsis subcellular proteome data set and applications associated with the collection have been increasing in terms of gene annotations, experimental, and computational data types (Heazlewood et al. 2007; Tanz et al. 2013a), as well as high-confidence subsets (Arabidopsis Subcellular Reference—ASURE) and location consensus classifications (SUBAcon) (Hooper et al. 2014) to arrive as the current version of SUBA4 that includes a separate section with an interactive analysis toolbox (Hooper et al. 2017a). The core experimental subcellular location data are now more than ten times the volume of the original SUBA1 (Heazlewood et al. 2007). SUBA started out categorizing ten distinct subcellular locations cytosol, cytoskeleton, endoplasmic reticulum (ER), Golgi, mitochondrion, nucleus, plastid, plasma membrane, peroxisome, and vacuole. With improving sensitivity of methods used to pinpoint protein locations, SUBA4 expanded this into sub-organellar compartments including differentiations into distinct membranes and aqueous compartments (Fig. 4.1). Experimental data pinpointing sub-organellar locations are now searchable within SUBA4.

Subcellular and suborganellar structures searchable within SUBA and cropPAL. The subcellular categories previously assessed by SUBA1-3 and cropPAL included 11 major subcellular locations (UPPER CASE). SUBA4 has increased location definition into suborganellar locations (lower case). ER endoplasmic reticulum, SUBA subcellular location database for Arabidopsis proteins (http://suba.live/)
The subcellular proteome data for Arabidopsis has increased from representing information on only 7% of predicted proteins in 2007 to over 32% of predicted proteins in 2017 arriving at the current >40% coverage in 2021 (Fig. 4.2a). Since the foundation of SUBA, Dr JL Heazlewood, Dr SK Tanz, Dr CM Hooper, Dr N Ayamanesh, and Ms Sally Grasso have been the key curators, while Dr J Tonti-Filippini, Dr CM Hooper, and Dr IR Castleden have developed most of the GUI and database services to enable the user experience. A small fraction of manual subcellular curations were independently derived from TAIR, GO, and Swissprot (Lamesch et al. 2012; Croft et al. 2014).

SUBA database expansion and data use in plant biology. (a) Continuous curation and integration of subcellular localization data has increased the number of localizations and the coverage of the Arabidopsis proteome throughout SUBA releases from 2005 to 2017. The size of SUBA4 at the time of writing in 2020 is indicated. (b) The accumulative citation record of published SUBA and SUBA tools indicates increasing importance of subcellular proteomics resources (source: Scopus). (c) The area of research and (d) species studied was determined using keyword-based text mining to show the distribution of fields of research that SUBA was used for in the past decade. A total of 465 studies that cited SUBA were examined. (Figure modified from Hooper et al. 2017a)
4.1.3 The Collation of Plant Subcellular Data Progressed into Crop Plants by Establishing cropPAL
High-throughput genome sequencing technologies, computing, and database management have made the protein sequences available through http://www.gramene.org/ for a range of non-model plant species of economic importance (Gupta et al. 2016; Tello-Ruiz et al. 2018). This has led to the exponential growth in the number of available reference plant genomes in recent years (Monaco et al. 2014). Notably, this includes the improved coverage of the bread wheat genome where researchers tackled a number of significant problems that occur when annotating highly polyploid genome sets (Bolser et al. 2015). Proteins across species share important similarities in their functional motifs, and this has driven linking information on orthologous proteins from model plants to less studied crop plants (Otto et al. 2008). Cross-species comparison highlights the amount of gene and genome duplication and gene loss throughout angiosperm evolution that has led to a huge variation in genome size and proteome composition between even close relatives (Tang et al. 2008). Researching protein specialization and sub-functionalization across and within species provides new insights into why plants differ so extensively in their growth, yield, and response to the environment.
While duplicated proteins may perform similar functions, small differences in versions that reside in distinct subcellular compartments can allow distinct optima, better suited to individual subcellular compartments. For example, small differences in protein sequence can improve function in differing pH environments (Scheibe et al. 2005). Therefore, the cost of protein or pathway duplication can more than compensate the energy investment in transporting them across membranes (Wu et al. 2006; Cheung et al. 2013). Knowing key turning points between energy budget and protein location during plant evolution has become a crucial consideration for studying plant product yields and determining the energy production of cells. With crop breeding in mind, data from the model plant Arabidopsis has been useful to bridge knowledge gaps for rice and maize through combining information with the independent subcellular proteomics data sets that exist for these species (Natera et al. 2008; Reiland et al. 2011; Majeran et al. 2012; Huang et al. 2013). This led to the generation of two further species-specific subcellular proteomics databases; riceDB for rice (Narsai et al. 2013) and PPDB for maize (Friso et al. 2004; Huang et al. 2013). Nevertheless, many more crops species exists for which Arabidopsis data presented the only resource for subcellular location information (Hooper et al. 2017a).
Subcellular proteomics and other localization data for most crop species exist scattered across published scientific reports and is often linked to obsolete protein accession annotations that are not concurrent with recent genome annotations. In this format a significant body of experimental subcellular proteomics data for barley (Endler et al. 2006; Ploscher et al. 2011) and wheat (Kamal et al. 2012; Suliman et al. 2013) are difficult to access for most researchers. The need for a cross species protein localization database emerged and was formulated in the formation of a new resource, crop Proteins with Annotated Location (cropPAL, https://crop-pal.org/). The cropPAL1 database contained just under 18,000 of the scattered experimental localizations for four mono-cotyledon crops including rice (Oryza sativa), maize (Zea mays), wheat (Triticum aestivum), and barley (Hordeum vulgare) connected to each other and to Arabidopsis (Hooper et al. 2016). CropPAL underwent major upgrades to include the two additional monocotyledon sorghum (Sorghum bicolor) and banana (Musa acuminata) and six additional dicotyledon species granola (Brassica napus), field mustard (Brassica rapa), soybean (Glycine max), tomato (Solanum lycopersicum), potato (Solanum tuberosum), and grape vine (Vitis vinifera). In cropPAL2020, subcellular proteomics data from MS/MS and FP localization data as well as pre-computed subcellular localizations from 11 predictors were collated (Hooper et al. 2020). Aligning to the Ensembl Plants/Gramene identifiers, experimental data in cropPAL was linked to the current genome annotations by a custom semi-automated pipeline. This offers sustainable links of research data that had increasingly obsolete identifiers. Using this system, available experimental data more than tripled to 61,505 localizations and generated large enough data sets for statistical comparisons between mono- and dicotyledon species or cross-species data mining opportunities between legumes and fruiting crops. Altogether, cropPAL2020 collates more than 800 scientific peer-reviewed studies. These data represent the collective work of >700 scientists from 600 organizations in 45 countries showcasing a global effort in elucidating protein subcellular location divergence and conservation across crop species.
The SUBA and cropPAL resources have been used for cultivar discrimination, engineering salt-resistant crops, increasing protein content, as well as improving yield and market value of grains, legumes, palm, mango, and tomato (Bajpai et al. 2018; Lau et al. 2018; Matamoros et al. 2018; Jiang et al. 2019; Schneider et al. 2019; McKenzie et al. 2020). If subcellular protein distributions (subcellulomes) are not cataloged for a species, scientists often fall back on data in Arabidopsis of the nearest species. This raises the questions around the validity of these discoveries if they are based on the assumption that we can borrow cross-species information. On the one hand, homology-linking protein subcellular location data is widely accepted on the basis that the metabolic and biosynthetic pathways in plants are highly conserved. On the other hand, reports exist that highlight the divergence in protein subcellular location between species by mechanisms of dual targeted proteins or protein family expansion (Carrie and Whelan 2013; Xu et al. 2013; One Thousand Plant Transcriptomes 2019). In context of the diversity of species physiology, metabolism capacity, and their ability to adapt to different environments, subcellular location diversification offers a potential starting point for plant performance improvement through biotechnological applications. The combination of data as well as the linking of 12 economically important crop species with Arabidopsis has placed plant subcellular data at the forefront of subcellular proteomics combining the skills of laboratory methodologies, data management and bioinformatics. These comprehensive data resources are now ready to aid current research questions around crop cell compartmentalization and crop biology.
4.2 Research and Technical Approach
4.2.1 Visualization and Separation of Proteins for Subcellular Localization Are Improving
Subcellular compartments and structures in plants were first defined by microscopy; what could be seen inside cells. Their separation and characterization have focused on attempts to recover these observed structures, free of contaminants. Initial separations are often based on the use of empirically derived speeds and times of differential centrifugation to enrich components of specific size ranges. Second, the use of density gradients separates structures based on their isopycnic point (buoyant density) which enables further purification of subcellular structures (for review see Taylor and Millar 2017).
Other physical and chemical properties have been developed as supplemental or even primary methods of isolation of specific structures. Electrical processes to separate organelles started with laminar-flow electrophoresis and lead to development of free-flow electrophoresis to purify subcellular particles like endosomes, lysosomes, peroxisomes, and ER-vesicles based on differences in surface charge. In plants, free-flow electrophoresis has been used to purify Arabidopsis plasma membrane and the tonoplast (Bardy et al. 1998), mitochondria (Eubel et al. 2007), and the Golgi apparatus (Parsons et al. 2012). Solid-phase separations through chromatography can also be used but has been typically limited to smaller sub-cellular structures such as mega Dalton protein complexes, for example, the pyruvate dehydrogenase complex (4–10 MDa), ribosomes (3–4 MDa), and the proteasome (2.5 MDa). Addition of affinity tags to target proteins by chemistry or genetic engineering allows isolation of many structures of interest from cells using the same affinity system. The DYKDDDDK epitope (FLAG) and tandem affinity purification (TAP) tagging are typical approaches performed in plants, for example, for the Arabidopsis proteasome (Book et al. 2010) and cytosolic ribosome (Reynoso et al. 2015). While this is typically expensive compared to other approaches, it can provide access to structures that either cannot be separated or are labile during the sequential physical processes of traditional isolations.
A number of different techniques can then be used for the assessment of organelle and structure contamination including microscopy, the use of marker enzyme activity assays, antibodies raised to marker proteins, selected reaction monitoring (SRM) MS and quantitative MS, or comparisons to literature claims of subcellular protein locations (Taylor et al. 2014; Millar et al. 2009; Taylor and Millar 2017). The use of stable isotope labeling or quantitation tags during MS can help screen out unknown contaminants by ensuring the target proteins are quantitatively enriched during organellar purification or are enriched more than other co-enriched cellular structures (Eubel et al. 2008; Mueller et al. 2014).
MS of compartment-enriched samples remains the most popular large-scale approach for defining subcellular localization of proteins, despite ongoing questions around the purity of these lists (Joshi et al. 2011). The FP-tagging approach is generally more accurate, but labor- and time-intensive, resulting in small study sizes with only a few high-throughput studies (Dunkley et al. 2006; Boruc et al. 2010; Lee et al. 2011). While the low coverage makes FP insufficient as a stand-alone large-scale data set, the collation of such studies over the last 15 years has generated a sizable subcellular proteome data set that remains one of the most widely accepted by biologists. Notably FP studies show both, where targeted proteins are and are not present, providing an internal control to evaluate competing claims of location that is sorely missing from most MS/MS data sets.
4.2.2 Subcellular Proteomics Can Be Supplemented with Homology Gap-Filling and Subcellular Protein Location Predictions
The varying number and type of subcellular proteomic studies performed among each plant species has led to uneven coverage between species and subcellular compartments. Of the 12 crop species, the largest number of subcellular localization experimentation to date has been performed in rice, maize, and soybean with tomato and wheat catching up steadily (Hooper et al. 2020). The proteome coverage of the most comprehensive experimental data sets collated for rice and tomato reached ~18% followed by soybean and maize with 5–10%. High-throughput MS/MS cataloging commonly focused on nucleus, plasma membrane, and extracellular extractions, for assessing proteins induced and secreted during host defense (Shah et al. 2012; Shinano et al. 2013). In contrast, plastidial and mitochondrial purifications for mass spectrometry analyses are often studying biogenesis and metabolic functions (Huang et al. 2009; Barsan et al. 2012; Salvato et al. 2014; Xing et al. 2016). Compartment catalogs existing for Arabidopsis (Reumann et al. 2009; Ito et al. 2011; Parsons et al. 2012; Heard et al. 2015) but not yet in crops include the cytosol, Golgi (Chateigner-Boutin et al. 2015), endoplasmic reticulum (ER) (Komatsu et al. 2017), and peroxisome (Arai et al. 2008). No crop species has been cataloged across all compartments, which means experimental data for any systems biology study is too sparse for downstream applications.
A recent effort pooled all experimental data across 12 crop species and Arabidopsis into biological MapMan categories to reveal that the percentage coverage of biological functions with experimental localizations in crops was similar to that observed in Arabidopsis (Schwacke et al. 2019; Hooper et al. 2020). Thereafter, the integrated data from the 12 crops increases coverage to >70% in most biological categories, showing that this can aid coverage of functional categories for less researched crops. However, the majority of data leading to this coverage derived from the well-researched species rice or maize and thus remains as valid as the two assumptions that (1) experimental error rates are small and that (2) subcellular locations are typically conserved. Since researchers often study proteins from the same compartments and functional categories in crops as well as Arabidopsis, gap-filling across species reaches a limit. To fill gaps beyond experimental data, predictors are necessary to achieve complete subcellulome coverage. A variety of proteome-wide subcellular location predictors have been developed based on sequence properties (Shen et al. 2007; Chou and Shen 2010; Yu et al. 2010). This includes various machine-learning and pattern recognition approaches (Chou and Shen 2007), such as support vector machines (Hua and Sun 2001), k-nearest neighbor (Horton et al. 2007), neural networks (Small et al. 2004), and hidden Markov models (Lin et al. 2011). Similar to different experimental techniques, these individual approaches have their own advantages and shortcomings in terms of the number of required features, the danger of over-fitting, and the ability to handle multiple optima. In order to improve accuracy, single machine-learning approaches have been stacked into multi-layer algorithms (Petsalaki et al. 2006; Pierleoni et al. 2006; Blum et al. 2009). Predictors typically derive their subcellular location calculations using protein sequence features, associated properties and/or gene ontology (GO) (Shen et al. 2007), and curator annotations (Briesemeister et al. 2010). Thereafter, predictors based on protein sequence identify sequence patterns in the primary protein sequence that target to individual organelles (Zybailov et al. 2008; Blum et al. 2009). Using similar or identical inputs, distinct machine-learning algorithms often yield different results that have shown a surprisingly poor overlap (Tanz and Small 2011). This variability is the main reason why experimental data are still seen as the gold standard by most biologists despite the unresolved difficulties associated with the experimental approaches themselves (Millar et al. 2009).
For Arabidopsis, SUBA contains the subcellular location outputs of 22 computational predictors including: AdaBoost (Niu et al. 2008), ATP (Mitschke et al. 2009), BaCelLo (Pierleoni et al. 2006), ChloroP 1.1 (Emanuelsson et al. 1999), EpiLoc (Brady and Shatkay 2008), iPSORT (Bannai et al. 2002), MitoPred (Guda et al. 2004), MitoProt (Claros and Vincens 1996), MultiLoc2 (Blum et al. 2009), Nucleo (Hawkins et al. 2007), PCLR 0.9 (Schein et al. 2001), Plant-mPLoc (Chou and Shen 2010), PProwler 1.2 (Hawkins and Boden 2006), Predotar v1.03 (Small et al. 2004), PredSL (Petsalaki et al. 2006), PTS1 (Neuberger et al. 2003), SLPFA (Tamura and Akutsu 2007), SLP-Local (Matsuda et al. 2005), SubLoc (Hua and Sun 2001), TargetP 1.1 (Emanuelsson et al. 2000), WoLF PSORT (Horton et al. 2007), and YLoc (Briesemeister et al. 2010). For Arabidopsis, the targeting predictions were carried out on the full-length protein sequences obtained from TAIR10 (Lamesch et al. 2012) or Ensembl plants (Kersey et al. 2018). The performance of the 22 predictors was tested on a high-confidence subcellular location reference data set (ASURE) that is accessible through SUBA4. The assessment of the predictors indicated that for Arabidopsis some predictors perform better for particular compartments and sometimes even better than experimental data. This is most visual for nuclear proteins where MultiLoc and Yloc outperformed unified contradicting MS and FP data (Fig. 4.3). In the years since their development, some of the above predictors have become unavailable. Hence, for cropPAL, the integration of only 11 out of 22 predictors was suitable or possible (Hooper et al. 2020). In total, predictive data sets in cropPAL span >6 million predictions, but for complete proteomes of all 12 species, only the six predictors MultiLoc2, TargetP, Predotar, YLoc, iPSORT, and WolfPSORT were available. The performance of predictors and experimental data in crop plants remains to be tested when data collections and high-confidence reference proteomes become available. However, a comparative analysis of crop experimental and predictive data to Arabidopsis suggests that the error rates of these methods in crops are similar to those seen in SUBA (Hooper et al. 2020).

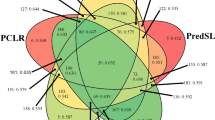
Performance of individual predictors, experimental data, and SUBAcon. The classification performance of six top scoring separate component (lines with symbols) in SUBAcon was compared to the overall SUBAcon consensus classification using all components (gray fill). The comparison of performance indicator MCC indicates superior subcellular location classification of Arabidopsis proteins when all information was used. Individual components generally perform well for some compartments allowing choice of best predictor for target compartments for Arabidopsis proteins. FP fluorescent proteins, MCC Matthews correlation coefficient, MS mass spectrometry, PPI protein-protein interaction. (Figure modified from Hooper et al. 2014)
4.2.3 An Objective Collation and Unification Strategy Can Resolve Varied and Conflicting Subcellular Location Information
Despite significant technological progress, errors in experimental data contribute to overlapping or contradicting data sets (Ito et al. 2011; Elmore et al. 2012; Nikolovski et al. 2012). As experimental data resources grow and the number of predictors increases so do the contradictions, and strategies are needed to integrate this multifaceted information. SUBAcon was developed to integrate the multi- and single-compartment predictor outputs with all available experimental data resources to generate an overall estimation of subcellular location for Arabidopsis proteins (Hooper et al. 2014).
SUBAcon uses FP and MS/MS data as a feature and determines the classification strength on their location calls when trained on a reference data set compared against other available localization features. When searching for other biological data that can be used as features, association data has become a popular choice due to the “guilt by association” principle. Associations like protein–protein interaction (PPI) and transcript co-expression aid the identification of functional clusters within the proteome. Considering that proteins in functional pathways more often co-locate, the location of a PPI partner can be used as indirect experimental evidence for protein location. Co-expression and PPI-associated protein sets are known for containing higher numbers of same-location protein groups than expected by random (Huh et al. 2003; Geisler-Lee et al. 2007). PPI data in particular have been suggested previously to be resources for predicting sub-cellular location of proteins in multiple eukaryotic species (Shin et al. 2009; Jiang and Wu 2012). Less is known about the true value of co-expression data for predicting co-location. These voluminous expression data sets have been used widely for predicting function (Stuart et al. 2003; Heyndrickx and Vandepoele 2012) and are typically the largest data sets available for most species. We showed that co-expression was useful to infer subcellular location for proteins with little experimental evidence, suggesting that such data alone can be highly informative for some compartments, rivaling sequence-based prediction (Hooper et al. 2014).
The lack of a single best method for inferring subcellular location has prompted using all available knowledge about proteins and is an attractive approach for forming a consensus view. Integrating a number of varied data sources has been used in yeast mitochondrial studies for some time, where this approach revealed promising new insights into genes involved in mitochondrial functions (Prokisch et al. 2004). The strategy of SUBAcon was to unify FP, MS, PPI, co-expression, and prediction data objectively to have one output with the highest probability of being correct (Fig. 4.4). This generated a data set where one protein was assigned one location or a set of locations that can be used for downstream omics applications. SUBAcon integrated 22 selected computational predictors into a two-phase naive Bayes classifier, which equaled or surpassed the classification accuracy for most compartments in comparison to single predictors even before integrating subcellular proteomics data (Figs. 4.3 and 4.4). The assessment of single and stepwise integration confirmed that SUBAcon objectively weighs individual predictors and experimental data to assign proteins to a location (or locations) more accurately than any of the input predictors or data did separately (Hooper et al. 2014). The analysis also confirmed a strong influence of experimental data on classification outcome; high proportions of FP protein localizations (~78%) and MS data (~65%) agreed with the ASURE locations. In both methodologies, the proportion of location mismatches was compartment-specific (Fig. 4.3). Consequently, the ongoing collation of experimental localization data will remain a key aspect of up-to-date classification by tools like SUBAcon.

The SUBAcon prediction and unification strategy. The subcellular location information from 22 predictors, fluorescent tagging, mass spectrometry, protein–protein interaction and co-expression (experimental data) and the ASURE standard was used to train a naive Bayes algorithm in two phases. Phase 1 (top) distinguished cytosol, mitochondrion, nucleus, peroxisome, and plastid from secretory proteins. Phase 2 (middle) used six secretory predictors and the experimental data to classify endoplasmic reticulum, extracellular, Golgi, plasma membrane and vacuole proteins. The secretory classifications were combined with the phase 1 locations and present the final SUBAcon output (bottom). ASURE Arabidopsis Subcellular Reference standard. (Figure modified from Hooper et al. 2014)
In order to produce a classifier taking crop species-specific differences as well as error rates of methodologies into account, a cross-species gold standard not biased by experimental and homology inference like ASURE is essential. Such a gold standard does not yet exist for these crops. Therefore, within cropPAL, a winner-takes-all (WTA) uniform location call was derived for each crop protein by vote-counting experimental locations and adding all predictors as one vote with each predictor a fraction of that vote (Hooper et al. 2020). This weights the final location call toward experimental locations when available and allowing predictors to gap-fill when not. When comparing the error rates, the accuracy of both, MS/MS and FP methodologies in the cropPAL collection, was comparable to that previously reported in Arabidopsis (Hooper et al. 2014). As expected, FP localizations were overall more accurate (61–87%), while the accuracy of MS/MS data varied significantly with compartment (22–85%). The overall accuracy of the crop WTA calls when compared to the inferred crop reference was estimated as greater than 67%, supporting the use of the voting system.
Using the WTA, individual species-specific subcellular protein distributions (subcellulomes) were generated that proved to be similar across crops and Arabidopsis. These data resources act as reference distributions of subcellular locations for proteins as well as biological categories in crops (Hooper et al. 2020) marking the beginning of detailed species-specific subcellulome catalogs backed by experimental data. Indeed, the study showed that while close evolutionary relationship between species is evident in the higher agreement of their subcellulomes, there was no obvious difference between monocot and dicot species. The current practice of using Arabidopsis data for dicot research versus rice for monocot research and arguing for species-specific differences is challenging. While most species agree substantially (60–80%) in their protein subcellular locations, the agreement is not equally distributed across biological functions. Underexplored subcellular divergence information was found in particular in metabolic categories. Metabolic diversification has been reported in a number pathways including enzymes of the amino acid metabolism (Schenck and Last 2020) as well as subcellular partitioning of effector or signaling proteins specific to tissues or metabolic changes upon stimuli (Powers et al. 2019). On the proteomics level, the differences between species are often subtle shifts in the distribution of a number of proteins with similar functions between two or more subcellular locations (Hooper et al. 2020). Such shifts are mainly due to alternative splice variants (ASV) or duplications of genes that are more likely to be retained if subcellularly diverse (Avelange-Macherel et al. 2018).
4.2.4 Subcellular Proteomics Data Resources in SUBA Have Contributed to Over 900 Downstream Scientific Reports
SUBA has been cited more than 700 times averaging 30 published studies per year (Fig. 4.2b). The subcellular proteomics data of SUBA has been more commonly used for exploring protein and gene functions and improving the interpretation of transcriptomics, proteomics, genetics, and bioinformatics data (Fig. 4.2c). The use of SUBA has reached beyond Arabidopsis showing application in agricultural hypothesis formation around pressing questions in rice, barley, maize, soybean, and wheat biology (Fig. 4.2d).
In research, SUBA has contributed to the development of widely used organelle marker sets (Nelson et al. 2007), protein family clone collections for functional genomics (Lao et al. 2014), as well as facilitated the functional elucidation of protein families involved in plant growth regulation (Zentella et al. 2007). The latter resources and knowledge were used in over 900 downstream studies. Over the last decade, SUBA has played a pivotal role in estimating plant cell energy budgets (Cheung et al. 2013) and the costs of maintaining the plant proteome in different compartments (Li et al. 2017). In the context of systems biology approaches, knowledge of proteome-wide subcellular locations is an important component for defining functional neighborhoods and deducing metabolic and signaling networks within complex eukaryotic cells (Waese et al. 2017). It has also been used for exploring sugar metabolism networks in barley (Lunn et al. 2014) and demonstrating sub-functionalization of gene family expansions (Tanz et al. 2013b). This shaped our understanding of the subcellular plant metabolism in order to resolve diurnal relationships of plant metabolism (Furtauer et al. 2019) as well as contributed to increasing the resolution and accuracy of mathematical representations of plant cell and tissue metabolism during the last 5 years (Shi and Schwender 2016). The in silico estimation of organellar protein abundance (Hooper et al. 2017b) influenced concepts of subcellular phenotyping that helped achieve cultivar-specific discrimination through rapid estimation of organellar differences (Schneider et al. 2019). The breath of work benefitting from SUBA highlights the importance of ongoing efforts in developing this central subcellular resource.
The smaller fraction of crop research using SUBA highlights the importance of improving the linkage of SUBA across species-specific borders as well as the need to improve linkage of comprehensive subcellular data collections for more crop species. The compendium of cropPAL begins to address this challenge across 12 crop species, and it has provided protein localization data since 2015. Right from the start, cropPAL the subcellular proteomics data contributed to the characterization of protein families across species (Chen et al. 2016a) as well as to a high confidence training set used for a novel plant and effector protein localization prediction algorithm (Sperschneider et al. 2017). Increasing awareness put cropPAL forward as a valuable resource for developing accurate proteomics pathways and network maps in economically important crops (Larrainzar and Wienkoop 2017).
SUBA and cropPAL also hold considerable contributions to molecular breeding concepts for increased crop quality and global food security. Highlights include the recent report listing the use of SUBA4 for unraveling crucial adaptation mechanisms for salinity tolerance in plants that provide promising genetic targets for engineering salt-resistant crops (Jiang et al. 2019) as well as the reviewed importance of SUBA resources for molecular biomarker identification for addressing a variety of diseases, yield quality and sustainability challenges within the palm oil industry (Lau et al. 2018). It is particularly exciting to see the rising influence of cropPAL on agricultural breeding strategies including the identification of genetic breeding targets for improved mango peel features increasing the mango market value (Bajpai et al. 2018) and the identification of molecular targets that expand flowering duration for increasing pollination opportunities and yield in rice (Chen et al. 2016b).
Tackling a serious global concern about nitrogen integration and protein increase in crops, SUBA4 was used in guiding the discovery of proteins regulating the nitrogen metabolism in root nodules (Matamoros et al. 2018) while cropPAL was named one of the “key aspects that need to be strengthened in the future” considering the large number of proteins involved in nitrogen-fixation efficiency (Larrainzar and Wienkoop 2017). These resources will be crucial in the near future for unraveling the complexity of nitrate metabolism in plants with the aim to guide molecular breeding strategies toward securing nutrition of global food crops under changing environments.
4.2.5 The Collation and Integration of Arabidopsis Subcellular Proteomics Data Presents Opportunities for New Approaches for In Silico Analysis
Both SUBA and cropPAL subcellular data collections are data warehouses publicly available through http://suba.live and http://crop-pal.org, respectively, that provide easy GUI-based data search and filtering functions. The web portals enable biologically meaningful subcellular location annotations and integrations by APIs or focused list creation through the web query builder. Users do not need computational expertise to mine the data sets for subcellular locations, methodology, protein properties, gene associations, authorship, or country of data origin. In addition, the interface offers a BLAST function for scientists researching alternative crops that enables to link their sequence of interest to the closest match in the SUBA or cropPAL data set. Within SUBA4, a separate toolbox exists that contains tools that allow immediate access and analysis of the core Arabidopsis subcellular location data in linkage to external user data sources. The toolbox currently offers the Multiple Marker Abundance Profiling tool (MMAP), the Co-expression Adjacency Tool (CAT), and the PPI Adjacency Tool (PAT). Each tool provides a unique link to the subcellular location consensus (SUBAcon).
For Arabidopsis, the co-expression data and PPI data sets were linked to the unified SUBAcon calls. The SUBAcon calls of each protein partner were joined and categorized into proximity relationships according to their biological interpretation, such as location within the same organelle, neighboring organelle, or distant organelles. The subcellular locations, proximity relationships, mutual rank and average correlation coefficient data allow user-lead prioritizing of strong associations. With the CAT tool, the user can assess a list of proteins for their subcellular location in context with their gene expression association, allowing to discover potential relationships between proteins based on the vast amount of expression data available.
The PAT tool uses experimental evidence from 26,327 unique PPI and assigns subcellular location derived from SUBAcon to each protein. This allows the interpretation of protein associations in context of proteins proximity in the cell. Thereafter, proteins located within the same subcellular compartment can interact and proteins on outer membranes or interfaces of one organelle can also interact with proteins from neighboring organelles. The PAT tool allows filtering for location pairings to target specific organellar or inter-organellar interactions for hypotheses formation around PPI networks that influence biological processes.
The newest showcase using the full range of proteomics integrated into subcellular proteomics is the MMAP tool (Hooper et al. 2017b). This tool can estimate the proportion of different subcellular protein structures in a user-provided list of Arabidopsis Gene Identifiers (AGIs). It is based on combining localization information from SUBA and quantitative MS observations of proteins collated in the MASCP gator database (Joshi et al. 2011; Mann et al. 2013). While relative protein quantitation is possible using quantitative MS such approaches are expensive or moderately accurate (Thompson et al. 2003; Cox and Mann 2008; Arike and Peil 2014; Christoforou et al. 2016). Using available quantitative tissue proteome data indicates that such data can be standardized to achieve a more true representation of an Arabidopsis protein observation (Wang et al. 2012, 2015). In order to achieve a subcellular proteome quantitation including low-abundance proteins in specific organelles, data from over 100 publications describing enriched subcellular proteomes, organelles and protein complexes derived from public databases were added and normalized to an in silico protein abundance score (Sun et al. 2009; Ferro et al. 2010; Hooper et al. 2017a). The novel way of using normalized protein abundance scores (NPAS) for 23,191 proteins contained 2602 proteins that had not previously been scored, and it covers a total of 85% of the predicted Arabidopsis proteome (Wang et al. 2012).
The user can submit custom AGI lists to the MMAP tool and receive the number of distinct proteins per each organelle as well as an estimate of relative protein abundance composition compared to expected subcellular abundances. The tool was developed because conventional methods for determining organellar abundance rely on a few marker proteins, which can be hit and miss considering the variety of biological conditions the data are derived from. A high-confidence subcellular marker lists for Arabidopsis was generated using SUBAcon, which can gather a high probability list of proteins for each organelle. Extensive manual curation and cross-examination against experimental data verified these data for use in the in silico tool enabling ad hoc estimates of relative organelle abundance. Thereafter, the tool allows assessment of experimental data before committing to further experimentation. While the latter was the original aim of the developers, the tool offers additional opportunities for subcellular proteomics to drive big data questions in the near future.
While the MMAP tool is relatively new and not yet widely applied, its function has been demonstrated on data derived from Golgi (Parsons et al. 2012), chloroplast (Zybailov et al. 2008), and plasma membrane proteomes (de Michele et al. 2016; Elmore et al. 2012), where the tool output directly corresponds to the changes in organellar protein abundance measured by spectral counting (Hooper et al. 2017b). The MMAP tool was able to retrospectively demonstrate progressive Golgi enrichment in silico equivalent to immunoblotting but was also able to show how other compartments were enriched or depleted at the same time without any further experimentation (Cox and Mann 2008). In better-known organelles, such as chloroplasts (Zybailov et al. 2008), the MMAP tool revealed that current plastidial isolation procedures deplete all other organelle fractions and only plastidial proteins enrich (Uberegui et al. 2015; Yin et al. 2015). Using the MMAP tool to compare two plasma membrane isolations demonstrated that the free-flow electrophoresis was better able to decrease the contamination of plastid, Golgi, and mitochondria (Elmore et al. 2012; de Michele et al. 2016).
Using this tool, a cross analysis of all proteomes in SUBA can be done very rapidly, showing that mitochondrial, plastidial, and peroxisomal protein isolations are among the purest, whereas current plasma membrane, cytosol, and vacuole separation techniques cannot achieve the same purities (Fig. 4.5). The data also highlight the problem that attempts to detect low abundance proteins within organellar extractions lead to a near exponential increase in captured impurities.

Subcellular proteomics purity for Arabidopsis preparations. Published organellar separations during the last 15 years were retrieved from SUBA4 (http://suba.live). The lists of organellar proteins were loaded into the MMAP tool (http://suba.live/toolbox-app.html) and the obtained enrichment data was extrapolated to cover the whole protein list. Obtained fractions were graphed for each organelle as median bar showing purity of individual studies in dots. MMAP multiple marker abundance profiling
One of the unexplored functions of subcellular proteomics and quantitative proteomics is its use in analyzing tissue proteomes and potentially proteomes from different biological conditions in terms of subcellular proteome shifts or relative subcellular structure abundances. Using the MMAP tool, the plasmodesmata proteome (Fernandez-Calvino et al. 2011) showed predominance in the fraction of Golgi, vacuole, peroxisome, mitochondria, cytosolic, and plasma membrane compared to standard values allowing a superior analytical interpretation to the methods used by the authors in the study.
The MMAP tool allows the rapid generation of a holistic overview of relative organelle abundance for different tissues (Fig. 4.6), the same tissue following a treatment or environmental stimuli, or in mutant proteome phenotyping. Such analyses give an insight into how organelle proportions relate to tissue function. Analysis of MS/MS data from different tissues confirmed observed and biological relevant differences in organelles in cotyledons, leaf, root, and pollen tissues that would have taken considerable experimental efforts to otherwise confirm (Dunand et al. 2007; Grobei et al. 2009; Piques et al. 2009; Baerenfaller et al. 2011). The MMAP tool is open access and only requires a list of protein identifiers, thus a broad range of conditions can be queried beyond the ones listed above. It is yet to be seen how this tool can help interpret a variety of biological data including available proteomics, gene expression (Birnbaum et al. 2003) as well as protein turnover rate data (Li et al. 2012, 2017).

In silico estimation of subcellular proteome distribution in tissues. Arabidopsis protein identifiers were obtained from mass spectrometry experimentation by Baerenfaller and colleagues (Baerenfaller et al. 2011) and submitted to the MMAP tool in the SUBA4 toolbox. The output was extrapolated across the unknown protein fraction and estimated subcellular abundance were graphed as percentage of total protein abundance. MMAP multiple marker abundance profiling, SUBA subcellular localization database for Arabidopsis proteins
4.3 Future Directions in the Field
Despite the reasonable coverage of subcellular proteomics in Arabidopsis, recent tools have pinpointed that there are large gaps in the analysis of subcellular proteomes that require specialized attention. This includes the nucleus where only 22% of the proteome have experimental data attached to it, whereas more than 50% of the plastid and peroxisome proteins have been experimentally observed. Similarly, when using the MMAP tool, the unknown proteins are generally low-expressed proteins that are not easy to measure by MS/MS or FP. The organelle-specific coverage was reflected in the struggle to find enough markers for the MMAP lists (Hooper et al. 2017b). Only two data sets describing ER (Dunkley et al. 2006; Nikolovski et al. 2012) and five data sets describing Golgi or Trans-Golgi network enrichments exist (Dunkley et al. 2006; Drakakaki et al. 2012; Parsons et al. 2012; Nikolovski et al. 2014; Heard et al. 2015), compared to over 30 plastidial and mitochondrial separations that are available in SUBA. Using the MMAP tool to assess the subproteomes has shown that we need to pay particular attention to the nuclear and ER proteomes, as they have the poorest coverage and a poor purity (Figs. 4.5 and 4.6). Targeted subcellular proteome mapping may be a much-needed focus for generating more complete subcellular proteomes for these less covered organelles or low abundance protein families.
While Arabidopsis subcellular proteomics has developed a solid omics presence, for crops this field is only now emerging. This is reflected in the much lower experimental coverage of crop proteomes for subcellular location. Considering that subcellular location has been regarded as crucial for determining protein function and belonging to biological processes and pathways (Cook and Cristea 2019), this is surprising and unsatisfying. The recent development of SUBAcon for Arabidopsis and WTA for crops enabled to fill these gaps and generated large enough data sets to assess the conservation of subcellular locations across 12 crop species and Arabidopsis. Subcellular location divergence of proteins is species-specific and harbors unexplored potential for data-driven agricultural breeding strategies. An increased understanding of how the subcellular location differences influence plant metabolism would be beneficial for designing breeding strategies toward more sustainable varieties. Protein subcellular location shifts have shown to increase plant growth, biosynthesis of secondary metabolites relevant to industrial production and therapeutic application (Shen et al. 2019) and comparable strategies in protein biosynthesis has the potential to achieve crop varieties with higher protein content in crops. Computational modeling approaches are emerging as a promising way to test current hypotheses around crop metabolic traits based on protein subcellular location shifts (Terasawa et al. 2016; Tabbita et al. 2017). However, such metabolic models currently rely on available subcellular data (Vinga et al. 2010) mainly derived from Arabidopsis through SUBA (Mintz-Oron et al. 2012). Other species data has been too sparse causing errors and redundancies in crop metabolic models resulting in the removal of potentially species-specific reactions (Seaver et al. 2012). The growing subcellular location resources SUBA and cropPAL will be an exciting contribution to achieving a better species-specific representation of such models in the near future.
Abbreviations
- AMPDB:
-
Arabidopsis Mitochondrial Protein Database
- ASV:
-
Alternative splice variant
- CAT:
-
Co-expression Adjacency Tool
- cropPAL:
-
Database for crop proteins with annotated locations
- ER:
-
Endoplasmic reticulum
- FLAG:
-
Epitope polypeptide DYKDDDDK
- FP:
-
Fluorescent protein
- GO:
-
Gene Ontology http://www.geneontology.org
- GUI:
-
Graphical user interface
- LOPIT:
-
Localization of Organelle Protein by Isotope Tagging
- MMAP:
-
Multiple Marker Abundance Profiling
- MS:
-
Mass spectrometry
- MS/MS:
-
Tandem mass spectrometry
- NPAS:
-
Normalized protein abundance scores
- PAT:
-
PPI adjacency tool
- PPI:
-
Protein–protein interaction
- SRM:
-
Selected reaction monitoring
- SUBA :
-
Subcellular localization database for Arabidopsis proteins
- TAP:
-
Tandem affinity purification
References
Arai Y, Hayashi M, Nishimura M (2008) Proteomic identification and characterization of a novel peroxisomal adenine nucleotide transporter supplying ATP for fatty acid beta-oxidation in soybean and Arabidopsis. Plant Cell 20:3227–3240
Arike L, Peil L (2014) Spectral counting label-free proteomics. Methods Mol Biol 1156:213–222
Avelange-Macherel MH, Candat A, Neveu M, Tolleter D, Macherel D (2018) Decoding the divergent subcellular location of two highly similar paralogous LEA proteins. Int J Mol Sci 19:1620
Baerenfaller K, Hirsch-Hoffmann M, Svozil J, Hull R, Russenberger D, Bischof S, Lu Q, Gruissem W, Baginsky S (2011) pep2pro: a new tool for comprehensive proteome data analysis to reveal information about organ-specific proteomes in Arabidopsis thaliana. Integr Biol 3:225–237
Bajpai A, Khan K, Muthukumar M, Rajan S, Singh NK (2018) Molecular analysis of anthocyanin biosynthesis pathway genes and their differential expression in mango peel. Genome 61:157–166
Bannai H, Tamada Y, Maruyama O, Nakai K, Miyano S (2002) Extensive feature detection of N-terminal protein sorting signals. Bioinformatics 18:298–305
Bardy N, Carrasco A, Galaud JP, Pont-Lezica R, Canut H (1998) Free-flow electrophoresis for fractionation of Arabidopsis thaliana membranes. Electrophoresis 19:1145–1153
Barsan C, Zouine M, Maza E, Bian W, Egea I, Rossignol M, Bouyssie D, Pichereaux C, Purgatto E, Bouzayen M, Latche A, Pech JC (2012) Proteomic analysis of chloroplast-to-chromoplast transition in tomato reveals metabolic shifts coupled with disrupted thylakoid biogenesis machinery and elevated energy-production components. Plant Physiol 160:708–725
Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN (2003) A gene expression map of the Arabidopsis root. Science 302:1956–1960
Blum T, Briesemeister S, Kohlbacher O (2009) MultiLoc2: integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinformatics 10:274
Bolser DM, Kerhornou A, Walts B, Kersey P (2015) Triticeae resources in ensembl plants. Plant Cell Physiol 56:e3
Book AJ, Gladman NP, Lee SS, Scalf M, Smith LM, Vierstra RD (2010) Affinity purification of the Arabidopsis 26 S proteasome reveals a diverse array of plant proteolytic complexes. J Biol Chem 285:25554–25569
Boruc J, Mylle E, Duda M, De Clercq R, Rombauts S, Geelen D, Hilson P, Inze D, Van Damme D, Russinova E (2010) Systematic localization of the Arabidopsis core cell cycle proteins reveals novel cell division complexes. Plant Physiol 152:553–565
Brady S, Shatkay H (2008) EpiLoc: a (working) text-based system for predicting protein subcellular location. Pac Symp Biocomput 13:604–615
Briesemeister S, Rahnenfuhrer J, Kohlbacher O (2010) YLoc--an interpretable web server for predicting subcellular localization. Nucleic Acids Res 38:W497–W502
Carrie C, Whelan J (2013) Widespread dual targeting of proteins in land plants: when, where, how and why. Plant Signal Behav 8:e25034
Chateigner-Boutin AL, Suliman M, Bouchet B, Alvarado C, Lollier V, Rogniaux H, Guillon F, Larre C (2015) Endomembrane proteomics reveals putative enzymes involved in cell wall metabolism in wheat grain outer layers. J Exp Biol 66:2649–2658
Chen BX, Li WY, Gao YT, Chen ZJ, Zhang WN, Liu QJ, Chen Z, Liu J (2016a) Involvement of polyamine oxidase-produced hydrogen peroxide during coleorhiza-limited germination of rice seeds. Front Plant Sci 7:1219
Chen Y, Ma J, Miller AJ, Luo B, Wang M, Zhu Z, Ouwerkerk PB (2016b) OsCHX14 is involved in the K+ homeostasis in rice (Oryza sativa) flowers. Plant Cell Physiol 57:1530–1543
Cheung CY, Williams TC, Poolman MG, Fell DA, Ratcliffe RG, Sweetlove LJ (2013) A method for accounting for maintenance costs in flux balance analysis improves the prediction of plant cell metabolic phenotypes under stress conditions. Plant J 75:1050–1061
Chiu W, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in plants. Curr Biol 6:325–330
Chou KC, Shen HB (2007) Recent progress in protein subcellular location prediction. Anal Biochem 370:1–16
Chou KC, Shen HB (2010) Plant-mPLoc: a top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS One 5:e11335
Christoforou A, Mulvey CM, Breckels LM, Geladaki A, Hurrell T, Hayward PC, Naake T, Gatto L, Viner R, Martinez Arias A, Lilley KS (2016) A draft map of the mouse pluripotent stem cell spatial proteome. Nat Commun 7:8992
Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241:779–786
Cook KC, Cristea IM (2019) Location is everything: protein translocations as a viral infection strategy. Curr Opin Chem Biol 48:34–43
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372
Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, Jassal B, Jupe S, Matthews L, May B, Palatnik S, Rothfels K, Shamovsky V, Song H, Williams M, Birney E, Hermjakob H, Stein L, D’Eustachio P (2014) The Reactome pathway knowledgebase. Nucleic Acids Res 42:D472–D477
Cutler SR, Ehrhardt DW, Griffitts JS, Somerville CR (2000) Random GFP::cDNA fusions enable visualization of subcellular structures in cells of Arabidopsis at a high frequency. Proc Natl Acad Sci U S A 97:3718–3723
Drakakaki G, van de Ven W, Pan S, Miao Y, Wang J, Keinath NF, Weatherly B, Jiang L, Schumacher K, Hicks G, Raikhel N (2012) Isolation and proteomic analysis of the SYP61 compartment reveal its role in exocytic trafficking in Arabidopsis. Cell Res 22:413–424
Dunand C, Crevecoeur M, Penel C (2007) Distribution of superoxide and hydrogen peroxide in Arabidopsis root and their influence on root development: possible interaction with peroxidases. New Phytol 174:332–341
Dunkley TP, Watson R, Griffin JL, Dupree P, Lilley KS (2004) Localization of organelle proteins by isotope tagging (LOPIT). Mol Cell Proteomics 3:1128–1134
Dunkley TP, Hester S, Shadforth IP, Runions J, Weimar T, Hanton SL, Griffin JL, Bessant C, Brandizzi F, Hawes C, Watson RB, Dupree P, Lilley KS (2006) Mapping the Arabidopsis organelle proteome. Proc Natl Acad Sci U S A 103:6518–6523
Elmore JM, Liu J, Smith B, Phinney B, Coaker G (2012) Quantitative proteomics reveals dynamic changes in the plasma membrane during Arabidopsis immune signaling. Mol Cell Proteomics 11(M111):014555
Emanuelsson O, Nielsen H, von Heijne G (1999) ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci 8:978–984
Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300:1005–1016
Endler A, Meyer S, Schelbert S, Schneider T, Weschke W, Peters SW, Keller F, Baginsky S, Martinoia E, Schmidt UG (2006) Identification of a vacuolar sucrose transporter in barley and Arabidopsis mesophyll cells by a tonoplast proteomic approach. Plant Physiol 141:196–207
Eubel H, Lee CP, Kuo J, Meyer EH, Taylor NL, Millar AH (2007) Free-flow electrophoresis for purification of plant mitochondria by surface charge. Plant J 52:583–594
Eubel H, Meyer EH, Taylor NL, Bussell JD, O’Toole N, Heazlewood JL, Castleden I, Small ID, Smith SM, Millar AH (2008) Novel proteins, putative membrane transporters, and an integrated metabolic network are revealed by quantitative proteomic analysis of Arabidopsis cell culture peroxisomes. Plant Physiol 148:1809–1829
Fernandez-Calvino L, Faulkner C, Walshaw J, Saalbach G, Bayer E, Benitez-Alfonso Y, Maule A (2011) Arabidopsis plasmodesmal proteome. PLoS One 6:e18880
Ferro M, Brugiere S, Salvi D, Seigneurin-Berny D, Court M, Moyet L, Ramus C, Miras S, Mellal M, Le Gall S, Kieffer-Jaquinod S, Bruley C, Garin J, Joyard J, Masselon C, Rolland N (2010) AT_CHLORO, a comprehensive chloroplast proteome database with subplastidial localization and curated information on envelope proteins. Mol Cell Proteomics 9:1063–1084
Friso G, Giacomelli L, Ytterberg AJ, Peltier JB, Rudella A, Sun Q, Wijk KJ (2004) In-depth analysis of the thylakoid membrane proteome of Arabidopsis thaliana chloroplasts: new proteins, new functions, and a plastid proteome database. Plant Cell 16:478–499
Furtauer L, Kustner L, Weckwerth W, Heyer AG, Nagele T (2019) Resolving subcellular plant metabolism. Plant J 100:438–455
Geisler-Lee J, O’Toole N, Ammar R, Provart NJ, Millar AH, Geisler M (2007) A predicted interactome for Arabidopsis. Plant Physiol 145:317–329
Gottlieb LD (1982) Conservation and duplication of isozymes in plants. Science 216:373–380
Grobei MA, Qeli E, Brunner E, Rehrauer H, Zhang R, Roschitzki B, Basler K, Ahrens CH, Grossniklaus U (2009) Deterministic protein inference for shotgun proteomics data provides new insights into Arabidopsis pollen development and function. Genome Res 19:1786–1800
Guda C, Guda P, Fahy E, Subramaniam S (2004) MITOPRED: a web server for the prediction of mitochondrial proteins. Nucleic Acids Res 32:W372–W374
Gupta P, Naithani S, Tello-Ruiz MK, Chougule K, D’Eustachio P, Fabregat A, Jiao Y, Keays M, Lee YK, Kumari S, Mulvaney J, Olson A, Preece J, Stein J, Wei S, Weiser J, Huerta L, Petryszak R, Kersey P, Stein LD, Ware D, Jaiswal P (2016) Gramene database: navigating plant comparative genomics resources. Curr Plant Biol 7-8:10–15
Hamada T, Nagasaki-Takeuchi N, Kato T, Fujiwara M, Sonobe S, Fukao Y, Hashimoto T (2013) Purification and characterization of novel microtubule-associated proteins from Arabidopsis cell suspension cultures. Plant Physiol 163:1804–1816
Hawkins J, Boden M (2006) Detecting and sorting targeting peptides with neural networks and support vector machines. J Bioinforma Comput Biol 4:1–18
Hawkins J, Davis L, Boden M (2007) Predicting nuclear localization. J Proteome Res 6:1402–1409
Heard W, Sklenar J, Tome DF, Robatzek S, Jones AM (2015) Identification of regulatory and cargo proteins of endosomal and secretory pathways in Arabidopsis thaliana by proteomic dissection. Mol Cell Proteomics 14:1796–1813
Heazlewood JL, Millar AH (2005) AMPDB: the Arabidopsis mitochondrial protein database. Nucleic Acids Res 33:D605–D610
Heazlewood JL, Tonti-Filippini JS, Gout AM, Day DA, Whelan J, Millar AH (2004) Experimental analysis of the Arabidopsis mitochondrial proteome highlights signaling and regulatory components, provides assessment of targeting prediction programs, and indicates plant-specific mitochondrial proteins. Plant Cell 16:241–256
Heazlewood JL, Tonti-Filippini J, Verboom RE, Millar AH (2005) Combining experimental and predicted datasets for determination of the subcellular location of proteins in Arabidopsis. Plant Physiol 139:598–609
Heazlewood JL, Verboom RE, Tonti-Filippini J, Small I, Millar AH (2007) SUBA: the Arabidopsis subcellular database. Nucleic Acids Res 35:D213–D218
Heyndrickx KS, Vandepoele K (2012) Systematic identification of functional plant modules through the integration of complementary data sources. Plant Physiol 159:884–901
Hooper CM, Tanz SK, Castleden IR, Vacher MA, Small ID, Millar AH (2014) SUBAcon: a consensus algorithm for unifying the subcellular localization data of the Arabidopsis proteome. Bioinformatics 30:3356–3364
Hooper CM, Castleden IR, Aryamanesh N, Jacoby RP, Millar AH (2016) Finding the subcellular location of barley, wheat, rice and maize proteins: the compendium of crop proteins with annotated locations (cropPAL). Plant Cell Physiol 57:e9
Hooper CM, Castleden IR, Tanz SK, Aryamanesh N, Millar AH (2017a) SUBA4: the interactive data analysis centre for Arabidopsis subcellular protein locations. Nucleic Acids Res 45:D1064–D1074
Hooper CM, Stevens TJ, Saukkonen A, Castleden IR, Singh P, Mann GW, Fabre B, Ito J, Deery MJ, Lilley KS, Petzold CJ, Millar AH, Heazlewood JL, Parsons HT (2017b) Multiple marker abundance profiling: combining selected reaction monitoring and data-dependent acquisition for rapid estimation of organelle abundance in subcellular samples. Plant J 92:1202–1217
Hooper CM, Castleden IR, Aryamanesh N, Black K, Grasso SV, Millar AH (2020) CropPAL for discovering divergence in protein subcellular location in crops to support strategies for molecular crop breeding. Plant J 104:812
Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K (2007) WoLF PSORT: protein localization predictor. Nucleic Acids Res 35:W585–W587
Hua S, Sun Z (2001) Support vector machine approach for protein subcellular localization prediction. Bioinformatics 17:721–728
Huang S, Taylor NL, Narsai R, Eubel H, Whelan J, Millar AH (2009) Experimental analysis of the rice mitochondrial proteome, its biogenesis, and heterogeneity. Plant Physiol 149:719–734
Huang M, Friso G, Nishimura K, Qu X, Olinares PD, Majeran W, Sun Q, van Wijk KJ (2013) Construction of plastid reference proteomes for maize and Arabidopsis and evaluation of their orthologous relationships; the concept of orthoproteomics. J Proteome Res 12:491–504
Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK (2003) Global analysis of protein localization in budding yeast. Nature 425:686–691
Inze A, Vanderauwera S, Hoeberichts FA, Vandorpe M, Van Gaever T, Van Breusegem F (2012) A subcellular localization compendium of hydrogen peroxide-induced proteins. Plant Cell Environ 35:308–320
Ito J, Batth TS, Petzold CJ, Redding-Johanson AM, Mukhopadhyay A, Verboom R, Meyer EH, Millar AH, Heazlewood JL (2011) Analysis of the Arabidopsis cytosolic proteome highlights subcellular partitioning of central plant metabolism. J Proteome Res 10:1571–1582
Jiang JQ, Wu M (2012) Predicting multiplex subcellular localization of proteins using protein-protein interaction network: a comparative study. BMC Bioinformatics 13(Suppl 10):S20
Jiang ZH, Zhou XP, Tao M, Yuan F, Liu LL, Wu FH, Wu XM, Xiang Y, Niu Y, Liu F, Li CJ, Ye R, Byeon B, Xue Y, Zhao HY, Wang HN, Crawford BM, Johnson DM, Hu CX, Pei C, Zhou W, Swift GB, Zhang H, Vo-Dinh T, Hu ZL, Siedow JN, Pei ZM (2019) Plant cell-surface GIPC sphingolipids sense salt to trigger Ca2+ influx. Nature 572:341
Joshi HJ, Hirsch-Hoffmann M, Baerenfaller K, Gruissem W, Baginsky S, Schmidt R, Schulze WX, Sun Q, van Wijk KJ, Egelhofer V, Wienkoop S, Weckwerth W, Bruley C, Rolland N, Toyoda T, Nakagami H, Jones AM, Briggs SP, Castleden I, Tanz SK, Millar AH, Heazlewood JL (2011) MASCP Gator: an aggregation portal for the visualization of Arabidopsis proteomics data. Plant Physiol 155:259–270
Kamal AH, Cho K, Komatsu S, Uozumi N, Choi JS, Woo SH (2012) Towards an understanding of wheat chloroplasts: a methodical investigation of thylakoid proteome. Mol Biol Rep 39:5069–5083
Kersey PJ, Allen JE, Allot A, Barba M, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Grabmueller C, Kumar N, Liu Z, Maurel T, Moore B, McDowall MD, Maheswari U, Naamati G, Newman V, Ong CK, Paulini M, Pedro H, Perry E, Russell M, Sparrow H, Tapanari E, Taylor K, Vullo A, Williams G, Zadissia A, Olson A, Stein J, Wei S, Tello-Ruiz M, Ware D, Luciani A, Potter S, Finn RD, Urban M, Hammond-Kosack KE, Bolser DM, De Silva N, Howe KL, Langridge N, Maslen G, Staines DM, Yates A (2018) Ensembl Genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res 46:D802–D808
Komatsu S, Wang X, Yin X, Nanjo Y, Ohyanagi H, Sakata K (2017) Integration of gel-based and gel-free proteomic data for functional analysis of proteins through Soybean Proteome Database. J Proteome 163:52–66
Koroleva OA, Tomlinson ML, Leader D, Shaw P, Doonan JH (2005) High-throughput protein localization in Arabidopsis using Agrobacterium-mediated transient expression of GFP-ORF fusions. Plant J 41:162–174
Kruft V, Eubel H, Jansch L, Werhahn W, Braun HP (2001) Proteomic approach to identify novel mitochondrial proteins in Arabidopsis. Plant Physiol 127:1694–1710
Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, Karthikeyan AS, Lee CH, Nelson WD, Ploetz L, Singh S, Wensel A, Huala E (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40:D1202–D1210
Lao J, Oikawa A, Bromley JR, McInerney P, Suttangkakul A, Smith-Moritz AM, Plahar H, Chiu TY, Gonzalez Fernandez-Nino SM, Ebert B, Yang F, Christiansen KM, Hansen SF, Stonebloom S, Adams PD, Ronald PC, Hillson NJ, Hadi MZ, Vega-Sanchez ME, Loque D, Scheller HV, Heazlewood JL (2014) The plant glycosyltransferase clone collection for functional genomics. Plant J 79:517–529
Larrainzar E, Wienkoop S (2017) A proteomic view on the role of legume symbiotic interactions. Front Plant Sci 8:1267
Lau BYC, Othman A, Ramli US (2018) Application of proteomics technologies in oil palm research. Protein J 37:473–499
Lee J, Lee H, Kim J, Lee S, Kim DH, Kim S, Hwang I (2011) Both the hydrophobicity and a positively charged region flanking the C-terminal region of the transmembrane domain of signal-anchored proteins play critical roles in determining their targeting specificity to the endoplasmic reticulum or endosymbiotic organelles in Arabidopsis cells. Plant Cell 23:1588–1607
Li L, Nelson CJ, Solheim C, Whelan J, Millar AH (2012) Determining degradation and synthesis rates of arabidopsis proteins using the kinetics of progressive 15N labeling of two-dimensional gel-separated protein spots. Mol Cell Proteomics 11(M111):010025
Li L, Nelson CJ, Trosch J, Castleden I, Huang S, Millar AH (2017) Protein degradation rate in arabidopsis thaliana leaf growth and development. Plant Cell 29:207–228
Lin TH, Murphy RF, Bar-Joseph Z (2011) Discriminative motif finding for predicting protein subcellular localization. IEEE/ACM Trans Comput Biol Bioinformatics 8:441–451
Lunn JE, Delorge I, Figueroa CM, Van Dijck P, Stitt M (2014) Trehalose metabolism in plants. Plant J 79:544–567
Mann GW, Calley PC, Joshi HJ and Heazlewood JL (2013) MASCP gator: an overview of the Arabidopsis proteomic aggregation portal. Front. Plant Sci. 4:411. https://doi.org/10.3389/fpls.2013.00411
Majeran W, Friso G, Asakura Y, Qu X, Huang M, Ponnala L, Watkins KP, Barkan A, van Wijk KJ (2012) Nucleoid-enriched proteomes in developing plastids and chloroplasts from maize leaves: a new conceptual framework for nucleoid functions. Plant Physiol 158:156–189
Matamoros MA, Kim A, Penuelas M, Ihling C, Griesser E, Hoffmann R, Fedorova M, Frolov A, Becana M (2018) Protein carbonylation and glycation in legume nodules. Plant Physiol 177:1510–1528
Matsuda S, Vert JP, Saigo H, Ueda N, Toh H, Akutsu T (2005) A novel representation of protein sequences for prediction of subcellular location using support vector machines. Protein Sci 14:2804–2813
McKenzie SD, Ibrahim IM, Aryal UK, Puthiyaveetil S (2020) Stoichiometry of protein complexes in plant photosynthetic membranes. BBA-Bioenergetics 1861:148141
de Michele R, McFarlane HE, Parsons HT, Meents MJ, Lao J, Gonzalez Fernandez-Nino SM, Petzold CJ, Frommer WB, Samuels AL, Heazlewood JL (2016) Free-flow electrophoresis of plasma membrane vesicles enriched by two-phase partitioning enhances the quality of the proteome from arabidopsis seedlings. J Proteome Res 15:900–913
Millar AH, Sweetlove LJ, Giege P, Leaver CJ (2001) Analysis of the Arabidopsis mitochondrial proteome. Plant Physiol 127:1711–1727
Millar AH, Carrie C, Pogson B, Whelan J (2009) Exploring the function-location nexus: using multiple lines of evidence in defining the subcellular location of plant proteins. Plant Cell 21:1625–1631
Mintz-Oron S, Meir S, Malitsky S, Ruppin E, Aharoni A, Shlomi T (2012) Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc Natl Acad Sci U S A 109:339–344
Mitschke J, Fuss J, Blum T, Hoglund A, Reski R, Kohlbacher O, Rensing SA (2009) Prediction of dual protein targeting to plant organelles. New Phytol 183:224–235
Monaco MK, Stein J, Naithani S, Wei S, Dharmawardhana P, Kumari S, Amarasinghe V, Youens-Clark K, Thomason J, Preece J, Pasternak S, Olson A, Jiao Y, Lu Z, Bolser D, Kerhornou A, Staines D, Walts B, Wu G, D’Eustachio P, Haw R, Croft D, Kersey PJ, Stein L, Jaiswal P, Ware D (2014) Gramene 2013: comparative plant genomics resources. Nucleic Acids Res 42:D1193–D1199
Mueller SJ, Lang D, Hoernstein SN, Lang EG, Schuessele C, Schmidt A, Fluck M, Leisibach D, Niegl C, Zimmer AD, Schlosser A, Reski R (2014) Quantitative analysis of the mitochondrial and plastid proteomes of the moss Physcomitrella patens reveals protein macrocompartmentation and microcompartmentation. Plant Physiol 164:2081–2095
Mulvey CM, Breckels LM, Geladaki A, Britovsek NK, Nightingale DJH, Christoforou A, Elzek M, Deery MJ, Gatto L, Lilley KS (2017) Using hyperLOPIT to perform high-resolution mapping of the spatial proteome. Nat Protoc 12:1110–1135
Narsai R, Devenish J, Castleden I, Narsai K, Xu L, Shou H, Whelan J (2013) Rice DB: an Oryza Information Portal linking annotation, subcellular location, function, expression, regulation, and evolutionary information for rice and Arabidopsis. Plant J 76:1057–1073
Natera SH, Ford KL, Cassin AM, Patterson JH, Newbigin EJ, Bacic A (2008) Analysis of the Oryza sativa plasma membrane proteome using combined protein and peptide fractionation approaches in conjunction with mass spectrometry. J Proteome Res 7:1159–1187
Nelson BK, Cai X, Nebenfuhr A (2007) A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J 51:1126–1136
Neuberger G, Maurer-Stroh S, Eisenhaber B, Hartig A, Eisenhaber F (2003) Prediction of peroxisomal targeting signal 1 containing proteins from amino acid sequence. J Mol Biol 328:581–592
Nguyen-Kim H, San Clemente H, Balliau T, Zivy M, Dunand C, Albenne C, Jamet E (2016) Arabidopsis thaliana root cell wall proteomics: increasing the proteome coverage using a combinatorial peptide ligand library and description of unexpected Hyp in peroxidase amino acid sequences. Proteomics 16:491–503
Nikolovski N, Rubtsov D, Segura MP, Miles GP, Stevens TJ, Dunkley TP, Munro S, Lilley KS, Dupree P (2012) Putative glycosyltransferases and other plant Golgi apparatus proteins are revealed by LOPIT proteomics. Plant Physiol 160:1037–1051
Nikolovski N, Shliaha PV, Gatto L, Dupree P, Lilley KS (2014) Label-free protein quantification for plant Golgi protein localization and abundance. Plant Physiol 166:1033–1043
Niu B, Jin YH, Feng KY, Lu WC, Cai YD, Li GZ (2008) Using AdaBoost for the prediction of subcellular location of prokaryotic and eukaryotic proteins. Mol Divers 12:41–45
One Thousand Plant Transcriptomes, I (2019) One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574:679–685
Otto TD, Guimaraes AC, Degrave WM, de Miranda AB (2008) AnEnPi: identification and annotation of analogous enzymes. BMC Bioinformatics 9:544
Parsons HT, Christiansen K, Knierim B, Carroll A, Ito J, Batth TS, Smith-Moritz AM, Morrison S, McInerney P, Hadi MZ, Auer M, Mukhopadhyay A, Petzold CJ, Scheller HV, Loque D, Heazlewood JL (2012) Isolation and proteomic characterization of the arabidopsis golgi defines functional and novel components involved in plant cell wall biosynthesis. Plant Physiol 159:12
Peltier JB, Ytterberg J, Liberles DA, Roepstorff P, van Wijk KJ (2001) Identification of a 350-kDa ClpP protease complex with 10 different Clp isoforms in chloroplasts of Arabidopsis thaliana. J Biol Chem 276:16318–16327
Petsalaki EI, Bagos PG, Litou ZI, Hamodrakas SJ (2006) PredSL: a tool for the N-terminal sequence-based prediction of protein subcellular localization. Genom Proteom Bioinformatics 4:48–55
Pierleoni A, Martelli PL, Fariselli P, Casadio R (2006) BaCelLo: a balanced subcellular localization predictor. Bioinformatics 22:e408–e416
Piques M, Schulze WX, Hohne M, Usadel B, Gibon Y, Rohwer J, Stitt M (2009) Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol Syst Biol 5:314
Ploscher M, Reisinger V, Eichacker LA (2011) Proteomic comparison of etioplast and chloroplast protein complexes. J Proteome 74:1256–1265
Powers SK, Holehouse AS, Korasick DA, Schreiber KH, Clark NM, Jing HW, Emenecker R, Han S, Tycksen E, Hwang I, Sozzani R, Jez JM, Pappu RV, Strader LC (2019) Nucleo-cytoplasmic partitioning of ARF proteins controls auxin responses in Arabidopsis thaliana. Mol Cell 76:177
Prokisch H, Scharfe C, Camp DG II, Xiao W, David L, Andreoli C, Monroe ME, Moore RJ, Gritsenko MA, Kozany C, Hixson KK, Mottaz HM, Zischka H, Ueffing M, Herman ZS, Davis RW, Meitinger T, Oefner PJ, Smith RD, Steinmetz LM (2004) Integrative analysis of the mitochondrial proteome in yeast. PLoS Biol 2:e160
Reiland S, Grossmann J, Baerenfaller K, Gehrig P, Nunes-Nesi A, Fernie AR, Gruissem W, Baginsky S (2011) Integrated proteome and metabolite analysis of the de-etiolation process in plastids from rice (Oryza sativa L.). Proteomics 11:1751–1763
Reumann S, Quan S, Aung K, Yang P, Manandhar-Shrestha K, Holbrook D, Linka N, Switzenberg R, Wilkerson CG, Weber AP, Olsen LJ, Hu J (2009) In-depth proteome analysis of Arabidopsis leaf peroxisomes combined with in vivo subcellular targeting verification indicates novel metabolic and regulatory functions of peroxisomes. Plant Physiol 150:125–143
Reynoso MA, Juntawong P, Lancia M, Blanco FA, Bailey-Serres J, Zanetti ME (2015) Translating Ribosome Affinity Purification (TRAP) followed by RNA sequencing technology (TRAP-SEQ) for quantitative assessment of plant translatomes. Methods Mol Biol 1284:185–207
Salvato F, Havelund JF, Chen M, Rao RS, Rogowska-Wrzesinska A, Jensen ON, Gang DR, Thelen JJ, Moller IM (2014) The potato tuber mitochondrial proteome. Plant Physiol 164:637–653
Scheibe R, Backhausen JE, Emmerlich V, Holtgrefe S (2005) Strategies to maintain redox homeostasis during photosynthesis under changing conditions. J Exp Bot 56:1481–1489
Schein AI, Kissinger JC, Ungar LH (2001) Chloroplast transit peptide prediction: a peek inside the black box. Nucleic Acids Res 29:E82
Schenck CA, Last RL (2020) Location, location! cellular relocalization primes specialized metabolic diversification. FEBS J 287:1359–1368
Schneider S, Harant D, Bachmann G, Nagele T, Lang I, Wienkoop S (2019) Subcellular phenotyping: using proteomics to quantitatively link subcellular leaf protein and organelle distribution analyses of pisum sativum cultivars. Front Plant Sci 10:638
Schubert M, Petersson UA, Haas BJ, Funk C, Schroder WP, Kieselbach T (2002) Proteome map of the chloroplast lumen of Arabidopsis thaliana. J Biol Chem 277:8354–8365
Schwacke R, Ponce-Soto GY, Krause K, Bolger AM, Arsova B, Hallab A, Gruden K, Stitt M, Bolger ME, Usadel B (2019) MapMan4: a refined protein classification and annotation framework applicable to multi-omics data analysis. Mol Plant 12:879–892
Seaver SM, Henry CS, Hanson AD (2012) Frontiers in metabolic reconstruction and modeling of plant genomes. J Exp Biol 63:2247–2258
Senkler J, Senkler M, Eubel H, Hildebrandt T, Lengwenus C, Schertl P, Schwarzlander M, Wagner S, Wittig I, Braun HP (2017) The mitochondrial complexome of Arabidopsis thaliana. Plant J 89:1079–1092
Shah P, Powell AL, Orlando R, Bergmann C, Gutierrez-Sanchez G (2012) Proteomic analysis of ripening tomato fruit infected by Botrytis cinerea. J Proteome Res 11:2178–2192
Shen HB, Yang J, Chou KC (2007) Euk-PLoc: an ensemble classifier for large-scale eukaryotic protein subcellular location prediction. Amino Acids 33:57–67
Shen BR, Wang LM, Lin XL, Yao Z, Xu HW, Zhu CH, Teng HY, Cui LL, Liu EE, Zhang JJ, He ZH, Peng XX (2019) Engineering a new chloroplastic photorespiratory bypass to increase photosynthetic efficiency and productivity in rice. Mol Plant 12:199–214
Shi H, Schwender J (2016) Mathematical models of plant metabolism. Curr Opin Biotechnol 37:143–152
Shin CJ, Wong S, Davis MJ, Ragan MA (2009) Protein-protein interaction as a predictor of subcellular location. BMC Syst Biol 3:28
Shinano T, Yoshimura T, Watanabe T, Unno Y, Osaki M, Nanjo Y, Komatsu S (2013) Effect of phosphorus levels on the protein profiles of secreted protein and root surface protein of rice. J Proteome Res 12:4748–4756
Small I, Peeters N, Legeai F, Lurin C (2004) Predotar: a tool for rapidly screening proteomes for N-terminal targeting sequences. Proteomics 4:1581–1590
Sperschneider J, Catanzariti AM, DeBoer K, Petre B, Gardiner DM, Singh KB, Dodds PN, Taylor JM (2017) LOCALIZER: subcellular localization prediction of both plant and effector proteins in the plant cell. Sci Rep 7:44598
Stuart JM, Segal E, Koller D, Kim SK (2003) A gene-coexpression network for global discovery of conserved genetic modules. Science 302:249–255
Suliman M, Chateigner-Boutin AL, Francin-Allami M, Partier A, Bouchet B, Salse J, Pont C, Marion J, Rogniaux H, Tessier D, Guillon F, Larre C (2013) Identification of glycosyltransferases involved in cell wall synthesis of wheat endosperm. J Proteome 78:508–521
Sun Q, Zybailov B, Majeran W, Friso G, Olinares PD, van Wijk KJ (2009) PPDB, the plant proteomics database at Cornell. Nucleic Acids Res 37:D969–D974
Tabbita F, Pearce S, Barneix AJ (2017) Breeding for increased grain protein and micronutrient content in wheat: ten years of the GPC-B1 gene. J Cereal Sci 73:183–191
Tamura T, Akutsu T (2007) Subcellular location prediction of proteins using support vector machines with alignment of block sequences utilizing amino acid composition. BMC Bioinformatics 8:466
Tang H, Bowers JE, Wang X, Ming R, Alam M, Paterson AH (2008) Synteny and collinearity in plant genomes. Science 320:486–488
Tanz SK, Small I (2011) In silico methods for identifying organellar and suborganellar targeting peptides in Arabidopsis chloroplast proteins and for predicting the topology of membrane proteins. Methods Mol Biol 774:243–280
Tanz SK, Castleden I, Hooper CM, Vacher M, Small I, Millar HA (2013a) SUBA3: a database for integrating experimentation and prediction to define the SUBcellular location of proteins in Arabidopsis. Nucleic Acids Res 41:D1185–D1191
Tanz SK, Castleden I, Small ID, Millar AH (2013b) Fluorescent protein tagging as a tool to define the subcellular distribution of proteins in plants. Front Plant Sci 4:214
Taylor NL, Millar AH (2017) Isolation of plant organelles and structures: methods and protocols. In: Methods in molecular biology: Springer protocols. Springer, New York, NY
Taylor NL, Fenske R, Castleden I, Tomaz T, Nelson CJ, Millar AH (2014) Selected reaction monitoring to determine protein abundance in Arabidopsis using the Arabidopsis proteotypic predictor. Plant Physiol 164:525–536
Tello-Ruiz MK, Naithani S, Stein JC, Gupta P, Campbell M, Olson A, Wei S, Preece J, Geniza MJ, Jiao Y, Lee YK, Wang B, Mulvaney J, Chougule K, Elser J, Al-Bader N, Kumari S, Thomason J, Kumar V, Bolser DM, Naamati G, Tapanari E, Fonseca N, Huerta L, Iqbal H, Keays M, Munoz-Pomer Fuentes A, Tang A, Fabregat A, D’Eustachio P, Weiser J, Stein LD, Petryszak R, Papatheodorou I, Kersey PJ, Lockhart P, Taylor C, Jaiswal P, Ware D (2018) Gramene 2018: unifying comparative genomics and pathway resources for plant research. Nucleic Acids Res 46:D1181–D1189
Terasawa Y, Ito M, Tabiki T, Nagasawa K, Hatta K, Nishio Z (2016) Mapping of a major QTL associated with protein content on chromosome 2B in hard red winter wheat (Triticum aestivum L.). Breed Sci 66:471–480
Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75:1895–1904
Uberegui E, Hall M, Lorenzo O, Schroder WP, Balsera M (2015) An Arabidopsis soluble chloroplast proteomic analysis reveals the participation of the Executer pathway in response to increased light conditions. J Exp Bot 66:2067–2077
Vinga S, Neves AR, Santos H, Brandt BW, Kooijman SA (2010) Subcellular metabolic organization in the context of dynamic energy budget and biochemical systems theories. Phil Trans R Soc Lond B Biol Sci 365:3429–3442
Waese J, Fan J, Pasha A, Yu H, Fucile G, Shi R, Cumming M, Kelley LA, Sternberg MJ, Krishnakumar V, Ferlanti E, Miller J, Town C, Stuerzlinger W, Provart NJ (2017) ePlant: visualizing and exploring multiple levels of data for hypothesis generation in plant biology. Plant Cell 29:1806–1821
Wang M, Weiss M, Simonovic M, Haertinger G, Schrimpf SP, Hengartner MO, von Mering C (2012) PaxDb, a database of protein abundance averages across all three domains of life. Mol Cell Proteomics 11:492–500
Wang M, Herrmann CJ, Simonovic M, Szklarczyk D, von Mering C (2015) Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15:3163–3168
Weckwerth W, Baginsky S, van Wijk K, Heazlewood JL, Millar H (2008) The multinational Arabidopsis steering subcommittee for proteomics assembles the largest proteome database resource for plant systems biology. J Proteome Res 7:4209–4210
Werhahn W, Niemeyer A, Jansch L, Kruft V, Schmitz UK, Braun H (2001) Purification and characterization of the preprotein translocase of the outer mitochondrial membrane from Arabidopsis. Identification of multiple forms of TOM20. Plant Physiol 125:943–954
Wu S, Schalk M, Clark A, Miles RB, Coates R, Chappell J (2006) Redirection of cytosolic or plastidic isoprenoid precursors elevates terpene production in plants. Nat Biotechnol 24:1441–1447
Xing S, Meng X, Zhou L, Mujahid H, Zhao C, Zhang Y, Wang C, Peng Z (2016) Proteome profile of starch granules purified from rice (Oryza sativa) endosperm. PLoS One 11:e0168467
Xu L, Carrie C, Law SR, Murcha MW, Whelan J (2013) Acquisition, conservation, and loss of dual-targeted proteins in land plants. Plant Physiol 161:644–662
Yin L, Vener AV, Spetea C (2015) The membrane proteome of stroma thylakoids from Arabidopsis thaliana studied by successive in-solution and in-gel digestion. Physiol Plant 154:433–446
Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FS (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26:1608–1615
Zentella R, Zhang ZL, Park M, Thomas SG, Endo A, Murase K, Fleet CM, Jikumaru Y, Nambara E, Kamiya Y, Sun TP (2007) Global analysis of della direct targets in early gibberellin signaling in Arabidopsis. Plant Cell 19:3037–3057
Zybailov B, Rutschow H, Friso G, Rudella A, Emanuelsson O, Sun Q, van Wijk KJ (2008) Sorting signals, N-terminal modifications and abundance of the chloroplast proteome. PLoS One 3:e1994
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Hooper, C.M., Castleden, I.R., Tanz, S.K., Grasso, S.V., Millar, A.H. (2021). Subcellular Proteomics as a Unified Approach of Experimental Localizations and Computed Prediction Data for Arabidopsis and Crop Plants. In: Vischi Winck, F. (eds) Advances in Plant Omics and Systems Biology Approaches. Advances in Experimental Medicine and Biology(), vol 1346. Springer, Cham. https://doi.org/10.1007/978-3-030-80352-0_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-80352-0_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-80351-3
Online ISBN: 978-3-030-80352-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)